

Abstract
For decades, variability in clinical efficacy of the widely used inflammatory bowel disease (IBD) drug, 5-aminosalicylic acid (5-ASA), has been attributed in part to its acetylation and inactivation by gut microbes. Identification of the responsible microbes and enzyme(s), however, has proved elusive. To uncover the source of this metabolism, we developed a multi-omics workflow combining gut microbiome metagenomics, metatranscriptomics, and metabolomics from the longitudinal IBDMDB cohort of 132 controls and patients with IBD. This associated 12 previously uncharacterized microbial acetyltransferases with 5-ASA inactivation, belonging to two protein superfamilies, thiolases and acyl-CoA N-acyltransferases. In vitro characterization of representatives from both families confirmed the ability of these enzymes to acetylate 5-ASA. A cross-sectional analysis within the discovery cohort and subsequent prospective validation within the independent SPARC IBD cohort (n=208) found three of these microbial thiolases and one acyl-CoA N-acyltransferase to be epidemiologically associated with an increased risk of treatment failure among 5-ASA users. Together, these data address a long-standing challenge in IBD management, outline a method for the discovery of previously uncharacterized gut microbial activities, and advance the possibility of microbiome-based personalized medicine.
Inflammatory bowel disease (IBD) is a chronic, debilitating gastrointestinal disorder with high rates of treatment failure.1 At present, there is no systematic means to predict response to IBD therapy. The anti-inflammatory drug mesalamine, also known as 5-aminosalicylic acid (5-ASA), is among the most commonly prescribed therapies available for IBD,2,3 and is usually formulated to be active within the colon; however, over half of IBD patients fail to respond or eventually lose response to 5-ASA over time.4,5 There is thus a need to identify and eliminate the causes of such treatment failure.
Apropos, experimental data suggest that the gut microbiome plays a role in the pharmacokinetics of several medications.6–9 For example, gut microbial enzymes were recently identified and characterized in vitro to metabolize digoxin and L-dopa, medications for heart failure and Parkinson’s disease, respectively.10–12 In humans, association studies suggest a role of the microbiome in modulating drug efficacy of anti-cytokine biologics and cardiometabolic drugs,13,14 although these generally lack mechanistic explanations. As a result, few such examples of gut microbial drug metabolism have been linked both mechanistically and with clinical outcomes in humans, in the case of 5-ASA or other drugs.
Prior anaerobic stool culture experiments suggest that up to a third of 5-ASA can be metabolized by gut bacteria into N-acetyl 5-ASA,15,16 a compound that lacks anti-inflammatory activity in placebo-controlled trials.17,18 This is at least in part due to the metabolite’s diminished (<5%) bioavailability to colonic epithelial cells.19 Experimental work has identified several bacteria capable of 5-ASA metabolism,20,21 but the majority of these are typically absent or of low abundance in patients with IBD. In any case, the specific microbial enzyme(s) responsible for the inactivation of 5-ASA in the gut microbiome have remained elusive for the last forty years. Additionally, prior to this work, the clinical implications of the microbial enzyme(s) which inactivate 5-ASA were unclear.
With this motivation, we identified and biochemically validated gut microbial enzymes capable of generating the clinically ineffective 5-ASA derivative, N-acetyl 5-ASA. In turn, we show that the presence of these microbial acetyltransferase genes in gut metagenomes is associated with an increased risk for 5-ASA treatment failure. These findings were driven and validated by metagenomics (MGX), metatranscriptomics (MTX), and metabolomics (MBX) from >1,000 stool samples collected over a one-year period from patients with IBD and controls,22 as well as subsequent phylogenetics, heterologous expression, and chemical characterization of the target proteins. The process is generalizable to other endpoints, chemicals, and microbiome multi-omics, improving our ability to understand gut microbial metabolism of drugs in patients and, therefore, opening new avenues for targeted or adjuvant therapies.
Results:
Multi-omics from patients with IBD identify 5-ASA use
To study the role of the gut microbiome in metabolizing 5-ASA and modulating its efficacy, we leveraged data from the Integrative Human Microbiome Project (iHMP or HMP2) Multi-omics Database (IBDMDB, http://ibdmdb.org), a multi-center cohort of 132 individuals with and without IBD, who each provided repeated medication, dietary, and symptom assessments along with serial stool and blood samples over one year (Fig. 1 and Methods).22 Stool samples from 79 participants with Crohn’s Disease (CD) or ulcerative colitis (UC) were newly profiled by the bioBakery 3 suite,23 yielding 1,036 metagenomes (MGX), 440 metatranscriptomes (MTX), and 508 untargeted metabolomes (MBX), with 283 MBX-MGX pairs and 213 MGX-MTX pairs.
Figure 1: Identification of microbial 5-ASA inactivating enzymes from IBD microbiome population multi-omics.
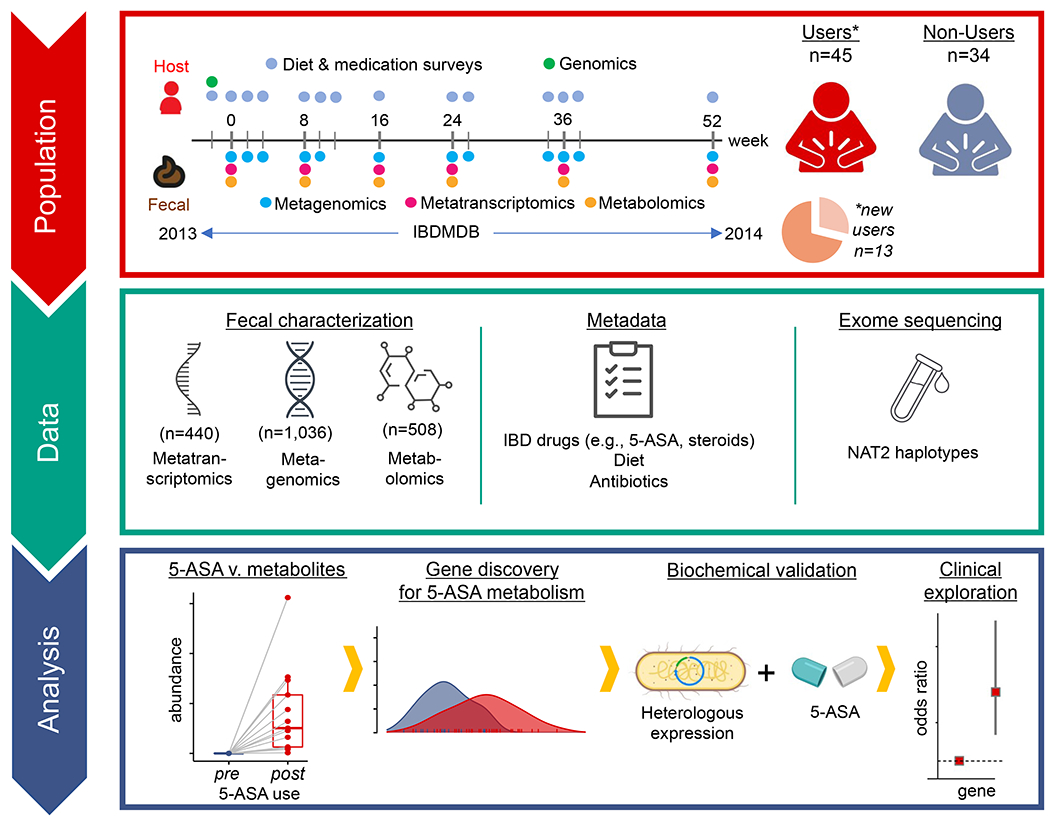
As part of the HMP2 IBDMDB (timeline, upper left),22 132 participants with and without IBD were followed for 1 year, each completing multiple dietary and medication questionnaires, and each providing stool every two weeks and blood samples approximately quarterly. After excluding participants without IBD or without metabolomics data, we identified 45 verified users of 5-ASA in the cohort and 34 non-users. Among 5-ASA users, we found 13 individuals who started or resumed using the drug during the cohort follow-up. Stool from >1,000 samples was then profiled through metagenomics, metatranscriptomics, and/or metabolomics; blood was analyzed by exome sequencing which was ultimately leveraged to determine human NAT2 acetylation phenotypes (“fast” vs. “slow”) for our clinical exploration. In the analysis phase, we first studied the impact of 5-ASA on the fecal metabolome and then identified gut bacterial enzymes involved in inactivating 5-ASA to N-acetyl 5-ASA. Finally, we related these enzymes to risk of disease relapse.
Concordance between self-reported use of 5-ASA (Methods) and detection of fecal 5-ASA was 80.3% (Extended Fig. 1), a rate that is consistent with other prescribed medications in population-based studies.24 To avoid misclassification in self-reported data, we assigned 5-ASA user status according to detection of drug levels in stool drug (Methods). Users had 5-ASA levels which were ~10,000x greater than non-users. Consistent with clinical practice,25 participants on 5-ASA were more likely to have UC than CD, and less than 10% were users of bonded 5-ASA (sulfasalazine, olsalazine, or balsalazide) (Extended table 1). Of note, 13 individuals were found to start or resume 5-ASA therapy during their sampling time courses, termed “new users” – providing an opportunity to examine the direct impact of the drug on the microbiome. Samples pre- and post-5-ASA administration were collected an average of 13.0 (± 8.7) weeks apart.
5-ASA derivatives in the fecal metabolome
Fecal metabolomic profiles segregated significantly according to 5-ASA user status (Fig. 2a, PERMANOVA R2=6.8%, p<0.001). Interestingly, this separation was greater than that due to baseline IBD diagnosis (e.g., CD vs. UC, R2=2.2%, p <0.001). Here, 5-ASA status may serve as a surrogate for metabolic differences driven by disease severity: “ever-users” vs. “never-users” of the drug were less likely to be hospitalized during the study and trended towards lower rates of bowel surgery (Extended table 1).”
Figure 2: 5-ASA directly impacts the fecal metabolome and undergoes biotransformation by the microbiome.
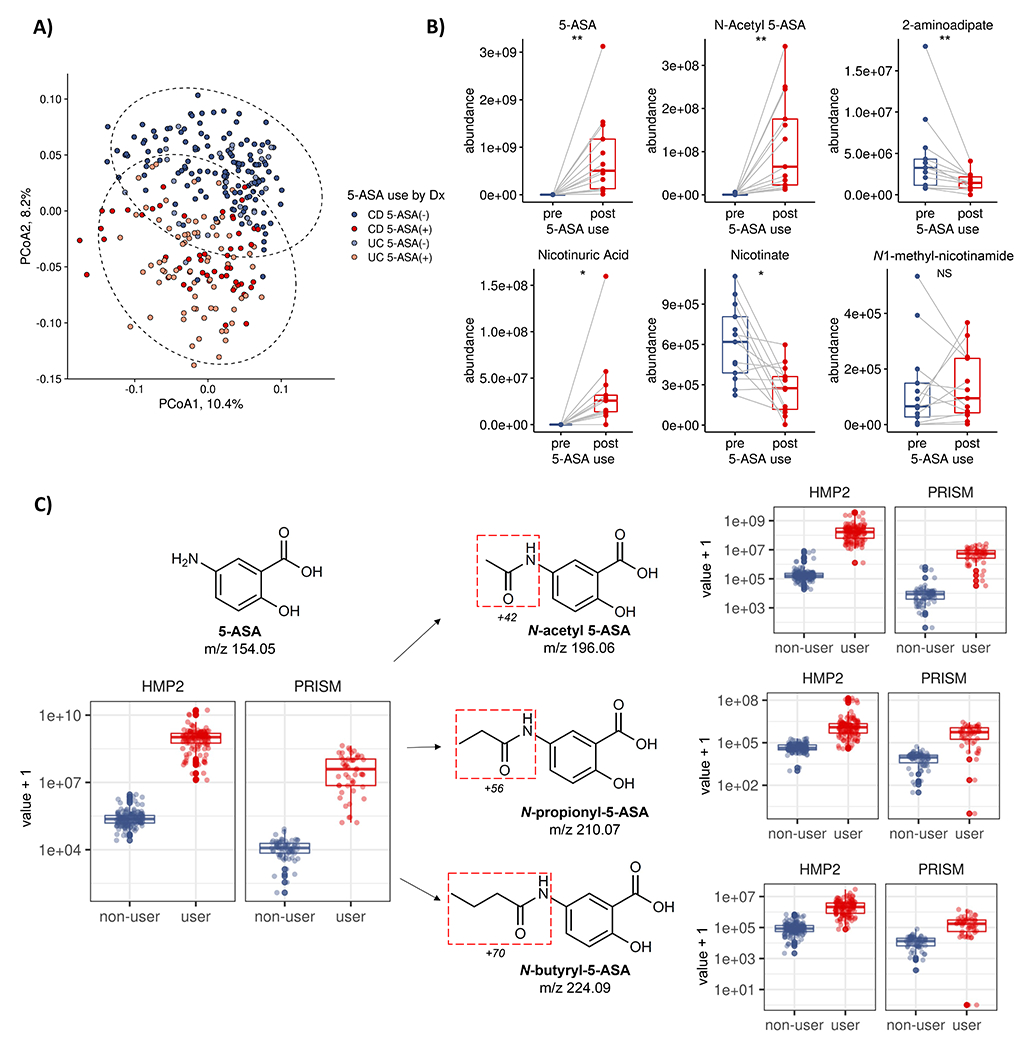
(A) The IBD fecal metabolome segregates by 5-ASA status (PERMANOVA R2=6.8%, p<0.001) more than by UC or CD diagnosis (R2=2.2%), suggesting a substantial role of medication in modulating the fecal biochemical environment of IBD patients (95% bivariate normal confidence ellipses shown, Methods). (B) Initiation of 5-ASA among a subset of participants (n=13) reveals 2,306 (total n=81,868, 2.8%) altered metabolomic features when comparing profiles pre- and post- 5-ASA administration (paired two-sided Wilcoxon, FDR q < 0.25), collected an average of 13.0 (± 8.7) weeks apart. Only 17 were assigned Human Metabolome Database (HMDB) identifiers, including the known 5-ASA metabolite, N-acetyl 5-ASA, as well as potential off-target effects - including shifts in vitamin B3 metabolism and bacterial products implicated in oxidative stress (23, 24). **, q<0.05; *, q<0.25; NS, not significant. Boxplots show median and lower/upper quartiles; whiskers show inner fences. (C). Examining the remaining 2,293 unannotated metabolomic features, we identified two promising candidates in the IBDMDB and independently profiled PRISM datasets using mass differences and retention time matching as likely N-propionyl 5-ASA and N-butyryl 5-ASA that also discriminated 5-ASA users from non-users (c-statistic >0.95) that were not initially annotated by the HMDB (Methods). Boxplots show median and lower/upper quartiles; whiskers show inner fences.
To identify specific fecal metabolites directly modulated by 5-ASA use, we focused our attention on the natural experiment of 13 new users of 5-ASA, defined as those patients for whom there were pre- and post-treatment stool samples. 2,306 metabolomic features (2.8%, n=81,868) were significantly differentially abundant when comparing stool pre- and post-5-ASA (paired Wilcoxon, FDR q < 0.25) (Extended table 2, Supplementary Fig. 1), of which only 17 were confidently assigned to Human Metabolome Database (HMDB) identifiers. As expected, these included 5-ASA and N-acetyl 5-ASA, but also potential microbial mediators of the anti-inflammatory effects of the drug. For example, the abundance of 2-aminoadipate, a bacterial metabolite in the lysine synthesis pathway26 linked to greater oxidative stress,27 was reduced following 5-ASA treatment, which may represent another way 5-ASA exerts its anti-inflammatory effects (Fig. 2b). In addition, we found that 5-ASA induced large shifts in niacin metabolism, known to occur in anaerobic gut bacteria,28 and previously seen to modulate inflammation in ulcerative colitis29 (Fig. 2b; Extended Fig. 2). These effects were highly specific to 5-ASA use and could not be attributed simply to suppression of inflammation: in a subset of 9 participants in the IBDMDB who started biologic drugs, there was no change in nicotinic acid levels, and nicotinuric acid levels were undetectable in non-users of 5-ASA (Extended Fig. 2).
We next sought to estimate the relative contributions of microbial, host, and other factors to these differentially abundant metabolites, given that many of these factors will co-vary during 5-ASA use (Extended Table 3; Methods). In models containing fecal 5-ASA levels; microbiome taxonomic data; host variable data, including diet, disease type, age, other medications, and sex; and an other/unexplained term, we quantified variance explained (EV) in each 5-ASA-shifted metabolite. As expected, by definition, 5-ASA drug levels had the largest predictive power (mean ~29% EV) in determining 5-ASA-modulated metabolite levels, followed by microbiome features (median ~19% EV) and other host factors (Extended Fig. 3, Methods). As an example, N-acetyl 5-ASA, moderately correlated with 5-ASA levels in stool (ρ 0.50, p=4e-9), with 35% of EV by fecal 5-ASA levels, 15% by the microbiome, and 7% by other host features.
We then wanted to determine if any of the remaining 2,293 unannotated metabolic features represented unrecognized biotransformations of 5-ASA. Specifically, we calculated differences between the masses of significantly altered metabolites and 5-ASA, and then mapped these against mass shifts of known microbial biotransformations.8 As a positive control, we started with N-acetyl 5-ASA. As expected, the mass shift corresponded to acetylation (+42), and this peak distinguished 5-ASA users from non-users in the IBDMDB essentially as well as the parent compound (c-statistic 0.99) (Fig. 2c). This finding was further replicated in another independent cohort of IBD patients (PRISM)30 (Supplementary Fig. 2), as was the identification of two previously unannotated likely 5-ASA derivatives, N-propionyl 5-ASA and N-butyryl 5-ASA (Fig. 2c, Extended table 4). Thus, in combination with large-scale impacts of 5-ASA on the gut metabolome, we consistently identified several 5-ASA acyl-derivatives in stool metabolomes and found evidence that their variance can be explained, at least in part, by the gut microbiome.
Identification of 5-ASA-metabolizing gut microbial enzymes
Next, we sought to identify which gut microbial enzymes were involved in generating the clinically ineffective metabolite N-acetyl 5-ASA, as well as the other putative biotransformations of 5-ASA observed above. Notably, typical homology-based approaches6,9,11 were not able to provide any candidates, even using very generous thresholds over more than a thousand metagenomes. Specifically, using two arylamine N-acetyltransferase (NAT) sequences from Salmonella enterica serovar typhimurium LT2 (nhoA) and Pseudomonas aeruginosa, previously shown to metabolize 5-ASA,20,21 as well as sequences from 105 microbial NAT or N-hydroxyarylamine O-acetyltransferase enzymes predicted to metabolize 5-ASA (Methods, Extended Table 5), we queried the complete set of gut microbial proteins in the IBDMDB HUMAnN 3-formatted UniRef90 database at a minimum of 25% full-length homology. This matched 5,685 unique gene clusters. Only 16 of these gene families were also detected in the IBDMDB gut metagenomic profiles, 14 of which mapped to E. coli and 2 of which were NATs. Notably, none were present in our metatranscriptomic data. As expected, there was no detectable Salmonella or Pseudomonas aeruginosa found in the gut of IBD patients. Furthermore, bacterial species previously found to acetylate 5-ASA were absent or of low abundance and/or prevalence in the gut microbiome (Extended Fig 4) and did not express NAT genes.
This prompted the development of our novel multi-omic criteria, combining 1) metatranscriptomics- and 2) metabolomics-based strategies. To first derive candidates from MTX profiles, we performed multivariate testing for differentially expressed microbial transcript families between 5-ASA users and non-users, accounting for DNA copy number31 (Methods). We identified two significantly overexpressed UniRef90 gene clusters with putative acetyltransferase function: 1) a GNAT family N-acetyltransferase (UniRef90 ID: C7H1G6) and 2) an acetyl-CoA acetyltransferase (UniRef90 ID: R6TIX3) (linear mixed effects models, β 0.0003, FDR q 0.24 and β 0.0003, FDR q 0.14, respectively) (Fig. 3a). We then re-extended our search using sequence similarity to these two novel hits, identifying proteins sharing >80% amino acid identity to retain conserved function.32 We identified four additional candidates with putative acetyltransferase domains for the acetyl-CoA acetyltransferase hit (UniRef90 IDs: R6CZ24, R5CY66, T5S060, A0A1C6JPG6) that were nominally enriched at a metatranscriptomic level between 5-ASA users vs. non-users (p=0.05, p=0.02, p=0.04, p=0.006 respectively), but not significant in our first search. There were no expanded candidates for the GNAT family N-acetyltransferase.
Figure 3: Microbial genes metatranscriptomically implicated in generation of N-acetyl 5-ASA cluster into thiolase and acyl CoA N-acyltransferase superfamilies.
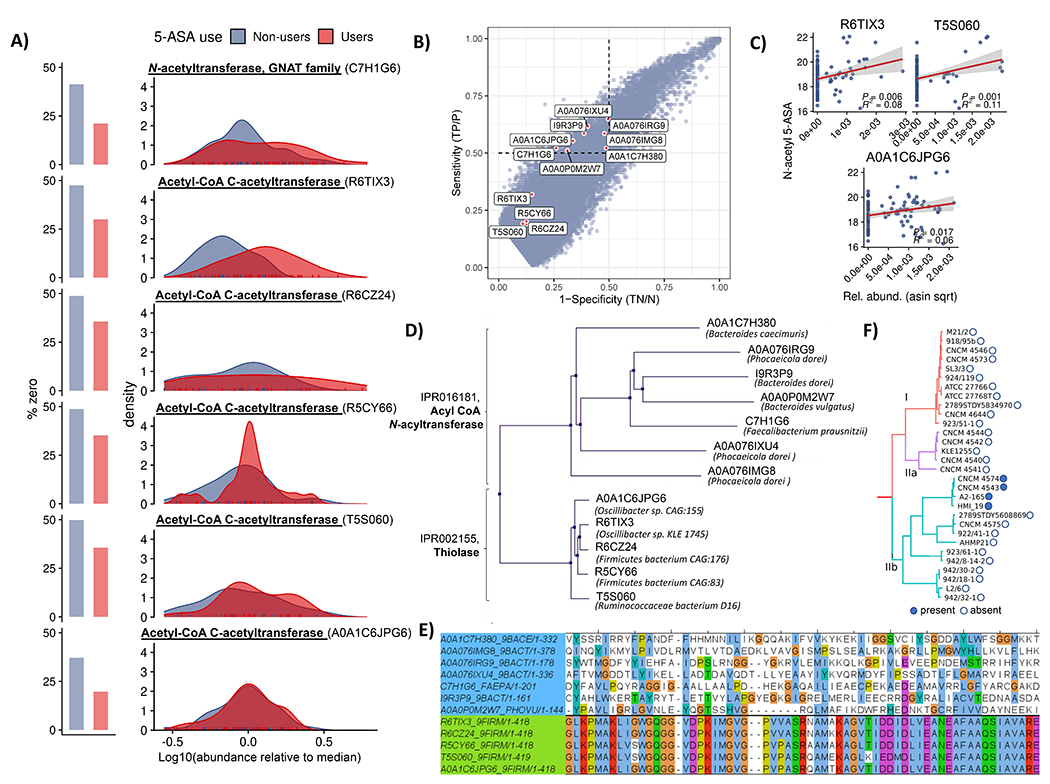
(A) MTX abundance distributions for six putative 5-ASA-acetylating gene families in 5-ASA users vs non-users. Multivariate linear mixed effects models adjusted for DNA copy number (27) identified two significantly overexpressed gene clusters with putative acetyltransferase function in 5-ASA users vs. non-users: 1) a GNAT family N-acetyltransferase (UniRef90 ID: C7H1G6) and 2) an acetyl-CoA acetyltransferase (UniRef90 ID: R6TIX3) (FDR q 0.24 and 0.14, respectively). Searching for any additional sequences with at least 80% full-length sequence similarity yielded three additional hits, all from the first acetyl-CoA acetyltransferase, each nominally enriched in 5-ASA users compared to non-users. (B) Specificity and sensitivity for microbial transcripts with respect to presence/absence of fecal N-acetyl 5-ASA across samples from 5-ASA users identifies seven additional putative 5-ASA acetylating gene families. In our second criteria, we estimated sensitivity and specificity for how each metatranscriptomic gene cluster (presence/absence) detected dichotomized N-acetyl 5-ASA (high/low). Using a 50% cutoff (hashed line) for each test characteristic revealed an additional 7 putative acetyltransferase gene clusters. Shown outside of these bounds are results from the first criteria, highlighting high degrees of specificity, but lower sensitivity. (C) MTX relative abundances for three of these MBX-based candidates identified in the second criteria also correlated with fecal N-acetyl 5-ASA levels (R2 and p values inset). Error bands represent 95% confidence intervals. (D) Pooled metatranscriptomic families from parts A and B proved to cluster into two protein superfamilies – thiolase and acyl-CoA N-acyltransferases – which are carried primarily by Bacteroides and Firmicutes phylum members, respectively. (E). Multiple sequence alignment of these twelve candidate enzymes shows highly conserved sequences among the thiolase enzymes (green) but greater diversity among acyltransferases (light blue). (F) In genomes from isolate strains, the only acyl-CoA N-acyltransferase gene carried by a Firmicutes member was in a subset of F. prausnitzii strains. When hierarchically clustered according to a prior schema33 (repurposed with permission), these appeared to originate from only one of the clade’s major phylogroups, suggestive of acquisition via a horizontal transfer event.
In our second, MBX-based strategy, we correlated the presence and/or absence of microbial transcripts with fecal N-acetyl 5-ASA levels across samples from 5-ASA users. Specifically, first we represented each metatranscriptomic gene family as present or absent, based on detection in the IBDMDB (relative abundance > 0). Next, we classified stool samples as N-acetyl 5-ASA high (≥ median) or N-acetyl 5-ASA low/negative (< median, including undetectable levels). Then we calculated the sensitivity and specificity with which each metatranscriptomic gene cluster associated with the dichotomized N-acetyl 5-ASA. At a 50% cutoff for sensitivity and specificity (as used previously, Methods), we uncovered an additional 7 putative acetyltransferase gene clusters (Fig. 3b). One of these clusters that met our criteria overlapped with the GNAT N-acetyltransferase (C7H1G6) detected in our first step. Although they did not meet the sensitivity and specificity cutoffs used here, three of the acetyltransferase transcripts from the first criteria positively correlated with N-acetyl 5-ASA levels in users when considered as continuous values (p < 0.05, Fig. 3c).
We finally pooled all candidate gene families that met either of these two MTX or MBX criteria (summarized in Extended Fig. 5), yielding 12 candidate acetyltransferase gene clusters, which proved to bin (ClustalW, Methods) into two groups according to sequence similarity and membership in two large protein families: thiolases and acyl-CoA N-acyltransferases (Fig. 3d). The acyl-CoA N-acyltransferase family showed greater sequence heterogeneity (less strict conservation) than the thiolase family (mean alignment score 0.09 ± 0.04 vs. 0.84 ± 0.03) (Fig. 3e). The taxonomic origins of these protein families were also consistent. The acyl-CoA N-acyltransferase enzymes were almost all derived from the Bacteroidetes phylum, whereas the thiolase enzymes originated from the Firmicutes phylum, although many of these were from unclassified bacteria. One notable exception to this pattern was F. prausnitzii, which is a major member of the Firmicutes phylum. Exploring strain-level genomes, we found that only a subset of F. prausnitzii strains encoded the acetyltransferase of interest, and when hierarchically clustered according to a prior schema,33 these appeared to originate from only one of the clade’s major phylogroups, which could suggest possible horizontal gene transfer from a Bacteroidetes sp. to a common F. prausnitzii ancestor (Fig. 3f).”
Biochemical characterization of putative 5-ASA-inactivating enzymes
Having identified thiolase and acyl-CoA N-acetyltransferase superfamilies with potential 5-ASA-inactivating capabilities, we sought to biochemically confirm these predicted activities in vitro. Since examining the genomic context of each candidate using assembled complete genomes revealed no obviously related regulatory genes (Extended table 6), we synthesized codon-optimized DNA sequences for each candidate gene according to their UniRef90 amino acid sequence and heterologously expressed them in E. coli (Methods). We expressed the known NAT from Salmonella enterica, the candidate thiolase from Firmicutes CAG:176 (UniRef90 ID R6CZ24), and the acyl-CoA N-acetyltransferase from F. prausnitzii (UniRef90 ID C7H1G6) for further biochemical characterization (Extended Fig. 6a). An in vitro mass spectrometry assay for 5-ASA acetylation confirmed the ability of the Firmicutes CAG:176 thiolase and the F. prausnitzii acyl-CoA N-acetyltransferase to acetylate 5-ASA using acetyl-CoA, generating product at a level consistent with >25% conversion in a physiologic time frame (Fig. 4a). The known S. enterica enzyme served as a positive control (Extended Fig. 6b).
Figure 4: Heterologous expression and purification of a gut microbial acetyltransferase confirms 5-ASA acetylation activity in vitro.
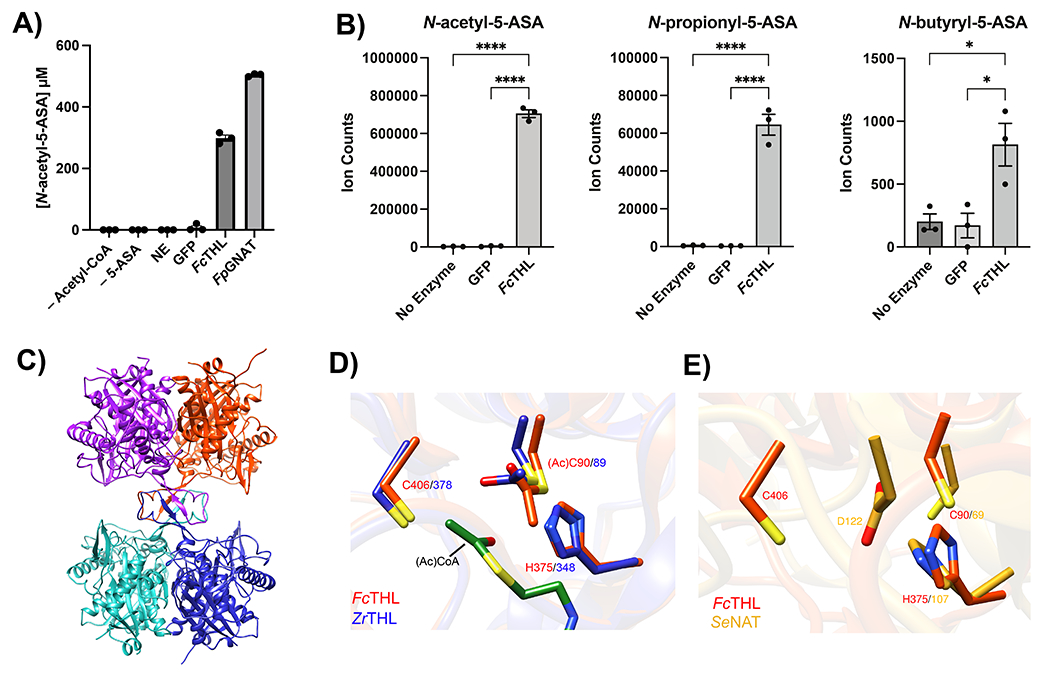
(A) A predicted thiolase (R6CZ24) from an uncultured Firmicutes (FcTHL) and a predicted acyl CoA N-acyltransferase (C7H1G6) from F. prausnitzii (FpGNAT) convert 5-ASA to N-acetyl-5-ASA in the presence of acetyl-CoA. Reactions were carried out for 6 hr at 37°C with 1 mM of each substrate and 50 μM enzyme. N=3 biologically independent samples per enzyme/condition. (B) An in vitro pooled acyl-CoA competition assay with 1 mM of each acyl-CoA species, confirms varying length acyl groups can be donated to 5-ASA, as detected in our analysis in patients with IBD (Fig 2c). N=3 biologically independent samples per enzyme/condition. (****=p<0.0001, *=p= 0.02, one-way ANOVA followed by Tukey’s multiple comparisons test.) (C) Tetrameric crystal structure of FcTHL, shown as a ribbon diagram with one dimer in purple and orange and the other dimer in green and blue. Two tightly interacted dimers form a tetramer through an L-domain. D). Alignment of an acetylated monomer from the FcTHL with an acetylated biosynthetic thiolase monomer (1DM3) in complex with acetyl CoA from Zoogloea ramigera (ZrTHL) highlights good agreement and reveals an overlapping Cys-His-Cys catalytic triad poised to acetylate a substrate. (E) Using a similar method of protein structure superimposition, the crystallized Salmonella enterica typhimurium NAT (PDB ID: 1E2T, SeNAT, known to acetylate 5-ASA) and the Firmicutes thiolase have very different overall structures, yet both enzymes’ active sites contain cysteine and histidine residues in similar positions and perform similar reactions (Extended Fig11a-b). Where applicable, data are presented as mean values +/− SEM.
We focused our next efforts on the thiolase superfamily due to its combination of phylogenetic consistency and substantially greater sequence conservation. The thiolase enzyme also accepted longer chain acyl-CoA donors in a pooled assay (Fig. 4b), which supports our predicted assignment of the secondary biotransformation products of 5-ASA observed in patient samples (Fig. 2). We then tested the activity of the thiolase for acetylation of other xenobiotics containing amine groups, including the 5-ASA isomer, 4-ASA, and other common clinical compounds, such as isoniazid, procainamide, and hydralazine. In contrast to 5-ASA, we did not observe any acetylation of these compounds, which suggests some level of substrate selectivity (Extended Fig. 7a). Kinetics analysis revealed a Km of 1.88 mM +/− 0.46 mM, well below the observed intraluminal stool concentrations of 5-ASA (median of 30 mM)34 and a kcat of 0.0647 min-1 +/− 0.0199, similar to that reported for the S. enterica NAT20 (Extended Fig 7b). Finally, we noted that a live culture of Oscillibacter sp., strain KLE 1745 encoding a second predicted thiolase gene (R6TIX3) was also capable of acetylation of 5-ASA to N-acetyl 5-ASA (Extended Fig 7c).
To gain insight into how the Firmicutes CAG:176 thiolase (FcTHL) acetylates 5-ASA, we generated an acetylated unliganded crystal structure of FcTHL refined at 1.9 Å resolution (Methods, Extended Table 7). As seen in other biosynthetic thiolases,35–37 protein subunits join to form dimers, which then link via four interacting loops (one from each subunit) to form a tetramer (Figure 4c). The four predicted active sites reside near these loops, projecting from the dimers, in the center of the tetramer.35 To better understand the canonical thiolase mechanism, we then superimposed FcTHL on an acetylated thiolase crystal structure in complex with acetyl CoA from the well-characterized gram-negative Zoogloea ramigera (ZrTHL, PDB ID: 1DM3). ZrTHL is known to condense short-chain acyl-CoA molecules via a two-step “ping pong” mechanism (Extended Fig 8a) to generate acetoacetyl-CoA, used to synthesize polyhydroxybutyrate, a major bacterial energy storage molecule. Alignment of the two structures yielded a root mean square deviation (RMSD) of 1.652 Å (Supplementary Fig. 3). The Cys-His-Cys triad in the known ZrTHL active site was conserved in FcTHL (Fig 4d). In both enzymes, the first catalytic cysteine (ZrTHL:Cys89/ FcTHL:90) is acetylated, forming a covalent acyl-enzyme intermediate, which is then poised to acetylate its substrate. In ZrTHL, when in complex with acetyl-CoA, this Cys89 rotates slightly to be parallel to the acetyl group to complete the condensation reaction. We next asked if this putative active site of the Firmicutes CAG:176 thiolase was similar to that of the S. enterica NAT, which is known to acetylate arylamines via a similar two-step “ping pong” mechanism (Extended Fig. 8b). To do so, we aligned FcTHL with the crystal structure of the NAT (PDB ID: 2PFR). Intriguingly, the comparison also revealed a cysteine and histidine dyad in the active site region (Fig. 4e).
Metagenomic carriage of 5-ASA-metabolizing enzymes predicts treatment failure
Having identified human gut microbial acetyltransferases capable of converting 5-ASA to the clinically ineffective N-acetyl 5-ASA, we next examined whether the presence of these twelve enzymes was associated with 5-ASA treatment failure. Consistent with the primary outcome in prior clinical trials of 5-ASA, we defined this as initiation of corticosteroid treatments within our HMP2 patient subpopulation38 (Methods). While the subcohort to which this was applicable was small, 39 individuals in the HMP2 who were treated with 5-ASA at any point and who provided longitudinal information on steroid use (prednisone, budesonide, methylprednisolone) contributed 609 stool samples across the entire year-long cohort.22 Using multivariate logistic regression models, we adjusted for age, sex, smoking status, and IBD subtype, each of which are thought to be linked to risk of disease flare.39 We also were potentially able to account for host genetics, through inclusion of each participant’s NAT2 phenotype (e.g. “fast” vs “slow” acetylator, Methods), as conflicting data suggest that NAT2 may or may not be implicated in 5-ASA metabolism.40–42
We found that the presence of any of four acetyltransferase genes in stool samples was significantly associated with an increased risk of steroid initiation (Fig. 5a) – three from the thiolase superfamily (R5CY66 OR 2.88, 95% CI, [1.66-5.00] ; and T5S060 OR 3.24, 95% CI [1.63-6.42]), including the Firmicutes CAG:176 enzyme characterized above (R6CZ24 OR 2.58, 95% CI [1.40-4.77]) and one from the acyl-CoA superfamily (C7H1G6, odds ratio [OR] 2.81, 95% confidence interval [CI] [1.68–4.68]). As expected, earlier age of onset and CD vs UC status were similarly associated with greater risk of steroid initiation25,43 (Extended table 8). Importantly, we found no association between risk for steroid use and metagenomic presence of the E. coli NAT, identified from our initial homology search using the Salmonella NAT as a template sequence.
Figure 5. Gut microbial 5-ASA-inactivating acetyltransferases are associated with greater risk of treatment failure in 5-ASA users.
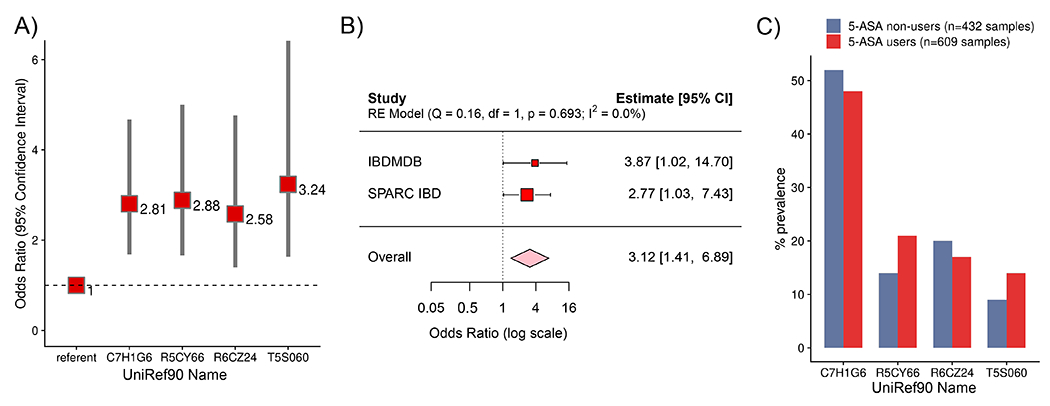
(A) Bacterial acetyltransferase genes are associated with an increased risk of steroid use. Red squares with labels represent odds ratios, with 95% confidence intervals represented by gray bars. N=609 independent stool samples collected from 39 5-ASA users over the year-long HMP2 IBDMDB. (B) Metagenomic carriage of 3-4 acetyltransferases (identified in panel A) compared to those with 0-2 have an increased risk of steroid use in the IBDMDB. In an independent, prospective validation cohort, SPARC IBD (n=250 independent stool samples collected from 5-ASA users), baseline carriage of the same 3-4 acetyltransferases confers a similarly increased risk of future steroid use. Red squares with labels represent odds ratios. Pooled analysis (random effects meta-analysis) demonstrates consistent effect size across the two cohorts (pink diamond represents odds ratio) Studies are inversely weighted according to their variance. (C) Gene prevalence varies across participants with IBD but does not vary by 5-ASA status (GLMM, p≥0.07), arguing for effect modification of the effects of 5-ASA on prevention of disease relapse.
In sensitivity analyses, when considering each of these genes as part of a risk score, an increasing number of microbial acetyltransferase genes was significantly associated with a greater risk of steroid initiation (ptrend <0.0001), and models mutually adjusted for the other genes were essentially unchanged. Importantly, those with scores of ≥3 genes were similar in distribution by CD and age to those with 2 or fewer genes. When performing the same analyses among IBD participants who were never-users of 5-ASA (who instead were treated with other drugs e.g., on 6-MP, infliximab, or methotrexate), we found no positive association of these gene families with use of steroids. Further, we considered that participants’ samples were not independent and thus we used mixed effects models adjusting for participant and found that presence of 3 or more acetyltransferase genes (compared to 2 or fewer) was also associated with an increased risk of steroid use, albeit with wider confidence intervals given the small sample size (OR 3.87, 95% CI 1.02 - 14.70) (Fig. 5b).
To test the reproducibility of our findings and to provide clarity about the temporal sequence between carriage of these microbial acetyltransferases and the transition from 5-ASA use to steroids, we validated our results in the independent Study of a Prospective Adult Research Cohort with IBD (SPARC IBD) (Methods). We note that we were blinded to these validation data, which were not available to us during the initial identification process for genes linked with steroid use in the IBDMDB. Among 208 users of 5-ASA who were steroid-free at study entry, we identified 60 new cases of corticosteroid use (Extended Fig 9, Extended Table 9). Consistent with our analysis in the IBDMDB, we found that metagenomic carriage of 3 or more microbial acetyltransferase genes in SPARC IBD (compared to 2 or fewer) was associated with treatment failure (OR 2.77, 95% CI 1.03–7.43). In a pooled analysis of the IBDMDB and SPARC IBD results, the overall effect size suggested more than a three-fold increased risk of drug failure (OR 3.12, 95% CI 1.41–6.89) (Fig. 5b).
Finally, we observed that these acetyltransferase genes were variably prevalent across individuals, but also that their presence did not differ between 5-ASA users and non-users (Fig. 5c), supporting the notion that these genes are not merely a biomarker of medication status. Indeed, in further support of a microbiome-environment interaction – i.e. when microbial genes present at baseline become exposed to 5-ASA, they are overexpressed, which in turn leads to medication inactivation – we found overall similar prevalence rates of these enzymes among participants without IBD (Extended table 10).
Discussion
Nearly forty years ago, researchers observed the conversion of 5-ASA to its inactive form (N-acetyl 5-ASA) in human stool cultures, leading to the hypothesis that gut bacteria may be responsible for this transformation.16 Fifteen years later, several bacteria, including S. typhimurium that contains a known homologue to the human NAT, were shown to inactivate 5-ASA.20 However, the identity of the specific enzymes responsible for this conversion in healthy adults or in those with IBD remained unknown. Here, we offer evidence that the culprits are “moonlighting” gut commensal acetyltransferases44 – which canonically condense short chain acyl-CoAs as part of an intracellular energy storage strategy45 – and that they may have a direct, deleterious impact on host health.
Intriguingly, these findings highlight both the strengths and weaknesses of enzyme identification by sequence homology alone, as compared with the multi-omic methods we employed. While no transcribed gut microbiome sequences shared even remote homology with the known Salmonella NAT sequence, the predicted structural similarity between its active site residues and those of the Firmicutes thiolase could suggest an analogous and near-convergent enzymatic mechanism. Both enzymes form acyl-enzyme intermediates via cysteine residues that are poised to donate an acetyl group to a nucleophile, and both rely upon stabilizing histidine residues, providing a possible explanation (Extended Fig 8c) for the presumed off-target metabolism of 5-ASA by thiolases. Alternatively, the lack of concordance at the likely third active site residue could suggest a distinct mechanism either involving this second cysteine or facilitated by additional residues outside of the hypothesized active site. Further biochemical characterization, including crystallography with 5-ASA bound, will be necessary to unequivocally assess how these thiolases inactivate 5-ASA, and why this subset of four enzymes were found to be clinically relevant. It also remains to be seen if thiolase-mediated inactivation of 5-ASA confers any fitness advantage to microbial communities, or whether this is a truly off-target effect of the enzyme’s natural activity.
More broadly, while some medications are known to influence the microbiome,46 the converse effect of gut bacteria on drug safety and efficacy has even greater translational potential. One of the major challenges to discovering microbe-drug interactions is that their specificity can vary widely. In our study and recent case-examples,7,9–11 drug-metabolizing enzymes are restricted to very few microbial strains and sequences. Others, such as the microbial beta glucuronidases affecting cancer therapies,47 can be taxonomically and phylogenetically widespread. In either case, such microbial enzymes are likely present in the host gut prior to any encounters with these non-antibiotic medications. Thus, the biomarker of interest for precision medicine is the baseline presence of relevant genes, which can often be identified by transcriptional responses to a compound, not the differential abundance of their microbial carriers. This necessitates novel and agnostic discovery methods such as those developed here – beginning with population-scale multi-omics (metatranscriptomics, metabolomics, and metagenomics) in the complex anaerobic nutritional and chemical environment of a natural human host – and ending with biochemical validation for the hundreds of health-relevant drug-microbe interactions that remain to be discovered.
Finally, our findings provide the first direct link between specific gut metabolic enzymes and 5-ASA treatment failure in IBD, thus yielding immediate clinical potential. 5-ASA is the most commonly prescribed medication for UC. To remain effective, it must remain available in unmodified form in the lumen of the colon, and thus, unlike other drugs, it is designed to be poorly absorbed in the small bowel.48 Individuals in which 5-ASA therapy fails are typically progressed to riskier immunosuppressive treatments, and the ability to do so only when necessary (i.e. when 5-ASA-modifying thiolase sequences are present in the gut microbiome) would provide a valuable biomarker for precision medicine. Furthermore, the elucidation of the enzymatic mechanisms by which microbes inactivate 5-ASA may lead to microbiome-specific inhibitors of enzymes to enhance 5-ASA efficacy in the future. Here, our data are observational; additional interventional studies are needed to strengthen our findings, whether through clinical trials in patients with IBD or through monocolonized mice. For now, these findings open a conceptual window into personalized microbiome-based medicine for IBD.
Methods
Study population and biospecimen sample collection
As previously published,22 132 IBDMDB participants were recruited between 2013-2014 from five major medical centers: Cincinnati Children’s Hospital, Emory University Hospital, Massachusetts General Hospital, Massachusetts General Hospital for Children, and Cedars-Sinai Medical Center, and had an initial colonoscopy upon enrollment to determine study phenotype (UC, CD, or non-IBD). Based on a combination of endoscopic and histopathologic findings as well as gastrointestinal (GI) symptoms or positive imaging (for example, colonic wall thickening or ileal inflammation), 104 participants were classified as having UC or CD. Of these, 79 participants provided 1,036 stool samples every two weeks (either in person or by mail, deposited in 5 ml of molecular biology grade 100% ethanol22) which were characterized by MGX, MBX, and/or MTX (see below). Due to restrictions such as available sample mass and missing samples, there were minor differences in the sampling pattern and in ability to perform MGX, MBX, and MTX on all samples. Blood draws (whole blood) occurred at approximately quarterly follow-up visits at the clinic.22 The study was reviewed by the Institutional Review Boards at each sampling site.22 All participants provided informed consent.
Medication assessment and covariate measurement
Throughout the study, participants completed serial questionnaires with information collected on diet, symptoms, and medication use.22 At baseline and with each collection of stool samples, participants completed a food frequency questionnaire, the former long-format and the latter short. Collection questionnaires also reported use of antibiotics, chemotherapy, or immunosuppressants such as steroids. More detailed medication information was collected three times at blood draws, at approximately the start, in the middle, and at the end of the study. Using this detailed data, we generated the following medication classes for mesalamine/5-ASA (oral: Asacol, Pentasa, Lialda, and Apriso; per-rectum: Rowasa enemas, Canasa suppositories; and bonded: Dipentum, Colazol, and Azulfidine). Steroids were grouped as use of Entecort, Medrol, or prednisone for the purposes of progression from 5-ASA in our case-cohort study (see ‘Case-cohort study’ below). Biologics were grouped as use of infliximab, adalimumab, certilizumab, and natalizumab. In the case of missing data, last observation carried forward (LOCF) was employed. Lifestyle and demographic data including age at consent, age at diagnosis, smoking status, and BMI were assessed at the start of the study. All questionnaires, as well as detailed protocols (including product numbers), can be found on the IBDMDB data portal at http://ibdmdb.org/protocols. Responses and metadata are available at http://ibdmdb.org/results. Dysbiosis is a term previously defined22 as microbial excursions from the non-IBD microbiome that is considered to be both a surrogate of disease severity as well as activity.
As above, to maximize accuracy of 5-ASA user classification, use was determined according to detection of drug levels in stool (see section on ‘Metabolomics measurements and identification’). Based on two distinct groupings of 5-ASA levels in stool (Extended Fig. 1), use of 5-ASA was defined biochemically, as containing levels of >107 in the IBDMDB MBX. Concordance was measured by statistical accuracy, according to the formula with TP as true positives and TN as true negatives:
In PRISM cohort metabolomics (Supplementary Fig. 2), which was used as a validation cohort for discovery of unannotated compounds, given lower metabolomic abundance values overall due to normalization, use of 5-ASA was defined as levels >105.
Sequencing assays
DNA and RNA isolation for metagenomics and metatranscriptomics
Total nucleic acid was extracted from an aliquot of each stool sample Chemagic DNA Blood Kit-96 from Perkin Elmer. 22 This combines chemical and mechanical lysis with magnetic bead-based purification, with full details available from the IBDMDB. DNA samples were quantified using a fluorescence-based PicoGreen assay; RNA samples were quantified using a fluorescence-based RiboGreen assay (see below). RNA quality was assessed via smear analysis on the Caliper LabChip GX.
Metagenome sequencing
In brief, metagenomic DNA was quantified using Quant-iT PicoGreen dsDNA Assay (Life Technologies) and normalized to a concentration of 50 pg/ul. Illumina sequencing libraries were prepared from 100–250 pg DNA using the Nextera XT DNA Library Preparation kit (Illumina). Prior to shotgun sequencing, libraries were pooled by collecting equal volumes of each library from batches of 96 samples. Insert sizes and concentrations for each pooled library were determined using an Agilent Bioanalyzer DNA 1000 kit (Agilent Technologies). Libraries were sequenced on HiSeq2000 or 2500 2x101 to yield ~10 million paired end reads. Post-sequencing de-multiplexing and generation of BAM and FASTQ files were generated using the Picard suite (https://broadinstitute.github.io/picard).
Metatranscriptome sequencing
Illumina cDNA libraries were generated using a modified version of the RNAtag-seq protocol.49 In brief, 500 ng–1 μg of total RNA was fragmented, depleted of genomic DNA, dephosphorylated, and ligated to DNA adapters carrying 5′-AN8-3′ barcodes of known sequence with a 5′ phosphate and a 3′ blocking group. Barcoded RNAs were pooled and depleted of rRNA using the RiboZero rRNA depletion kit (Epicentre). Pools of barcoded RNAs were converted to Illumina cDNA libraries and then sequenced as above.
Exome sequencing
Although only host NAT2 genotypes were used in this analysis (see below), whole-exome libraries were originally constructed and sequenced on an Illumina HiSeq 4000 sequencer with 151-bp paired end reads.22 Output from Illumina software was processed by the Picard pipeline to yield BAM files containing calibrated, aligned reads. Host genetic exome sequence data were processed using the Broad Institute sequencing pipeline by the Data Sciences Platform (Broad Institute).
Metabolomics
Stool processing
A portion of each selected stool sample (40–100 mg) and the entire volume of originating ethanol preservative were stored in 15-ml centrifuge tubes at −80 °C until all samples were collected. Samples were then thawed on ice and centrifuged (4 °C, 5,000g) for 5 min. Ethanol was evaporated using a nitrogen evaporator (TurboVap LV; Biotage) and stored at −80 °C until all samples in the study had been dried. After a homogenization and vortexing process, the mixture was aliquoted and stored at −80 C until LC–MS analyses.
Untargeted assays
A combination of four untargeted LC–MS methods were used to profile metabolites in the fecal homogenates;22 1) HILIC-pos (positive ion mode MS analyses of polar metabolites), 2) HILIC-neg (negative ion mode MS analysis of polar metabolites), 3) C18-neg (negative ion mode analysis of metabolites of intermediate polarity), and 4) C8-pos. Lipids (polar and nonpolar). Additionally, pairs of pooled reference samples were inserted into the queue at intervals of approximately 20 samples for quality control and data standardization. Samples were prepared for each method using extraction procedures that are matched for use with the chromatography conditions. Data were acquired using LC–MS systems composed of Nexera X2 U-HPLC systems (Shimadzu Scientific Instruments) coupled to Q Exactive/Exactive Plus orbitrap mass spectrometers (Thermo Fisher Scientific).
Taxonomic and functional profiling of metagenomes and metatranscriptomes
Taxonomic and functional profiles from the HMP2 were newly generated with the updated bioBakery3 meta’omics workflow using default parameters (http://huttenhower.sph.harvard.edu/biobakery_workflows) (20) (provenance log: https://ibdmdb.org/tunnel/cb/document/Public/HMP2/Metadata/anadama_run.log.gz). In brief, reads mapping to the human genome were first filtered out using KneadData v0.7.0. Taxonomic profiles of shotgun metagenomes were generated using MetaPhlAn 3.0, (http://huttenhower.sph.harvard.edu/metaphlan3),. Functional profiling was performed by HUMAnN v3.0.0.alpha.1 (http://huttenhower.sph.harvard.edu/humann3). Relative abundance data was filtered to remove features with no variance or >90% zeroes and then arc-sine square-root transformed to reduce effects from zero-inflation.
Metabolomic measurements and identification
Peaks of unknown ID were tracked by method, m/z and retention time. Identification of nontargeted metabolite LC–MS peaks was conducted by: i) matching measured retention times and masses to mixtures of reference metabolites analyzed in each batch; and ii) matching an internal database of >600 compounds that have been characterized using the Broad Institute methods. Temporal drift was monitored and normalized with the intensities of features measured in the pooled reference samples.
For the identification of 5-ASA, two separate standards (SIGMA catalog PHR1060 and 18858) confirmed the identity of the 5-ASA peak in the IBDMDB (m/z: 154.0502, RT: 3.83 min) through retention time and spectral matching (Extended Fig. 10).
Statistical analysis
Impact of 5-ASA of the fecal metabolome and microbial contributions to 5-ASA metabolism
To assess association of overall metabolome structure with 5-ASA use, omnibus testing was performed on Bray-Curtis dissimilarity matrices from MBX measurements. Profiles were first log-transformed and filtered to exclude features with more than 90% missing values before calculation of dissimilarities. Quantification of variance explained for the metabolomics data was calculated using PERMANOVA with the adonis function in the R v4.0.1 package “vegan” 2.5–6.51 The total variance explained by each variable was calculated independently of other variables to avoid issues related to ordering, as done previously.22 PCoA ordination in Fig. 2a was generated with the cmdscale function on Bray-Curtis distances, with unsupervised bivariate ellipses drawn at the 95% confidence level using the stat_ellipse function in the R package “ggplot2” v3.3.5.52
To identify individual significantly differentially abundant metabolomic features before and after 5-ASA administration in Fig 2b, we used paired two-sided Wilcoxon tests on data collected among 13 participants starting or resuming 5-ASA (“new users”), with an FDR threshold of 0.25. Box-plots elements shown here and throughout include medians as the center line, upper and lower quartiles as box limits; 1.5x interquartile range as whiskers; and outliers as points.
To identify possible biotransformations of 5-ASA, and specifically gain insights into the chemistry of microbial 5-ASA metabolism, we performed a four-step search, replicated in an independent cohort. First, we calculated mass differences (Δm/z) between the mass of each unannotated metabolite significantly altered by 5-ASA use (identified above, among the subpopulation of “new users”) and the expected parent compound (i.e. 5-ASA, a mass of 154.05). Second, we mapped these against known mass shifts from microbial biotransformations (6). Third, we calculated a c-statistic for each candidate metabolite, to determine how well it discriminated 5-ASA users from non-users in the entire study population, and subset these features to 95% predictive value (c-statistic >0.95). Finally, we further filtered this subset of annotated metabolomic features to those enriched among 5-ASA users compared to non-users (as opposed to enriched in non-users), given that drug derivatives should be more common among users. We then replicated this same four-step process in an independent cohort of 220 participants with and without IBD, the Prospective Registry in IBD Study at MGH (PRISM), which had fecal metabolomics generated using the same platform (26). The features from HMP2 and PRISM then were overlaid, yielding the proposed 5-ASA derivatives in Fig 2c.
As further confirmation of our proposed derivatives, we pursued an independent metabolomics identification approach, where the known MS2 spectra of N-acetyl 5-ASA (MassBank of North America) were passed to the GNPS workflow MASST,53 then input to the METABOLOMICS SNETS-V2 workflow, and finally passed to a second GNPS workflow, Network Annotation Propagation (NAP_CCMS v1.2.5), all with default parameters.54 The results were exported for visualization using the GNPS workflow MolNetEnhancer (v15). Nodes that were adjacent to (and thus co-occurring with) N-acetyl 5-ASA were inspected, and their m/z ratios were compared to the proposed candidates.
Identification of contribution of the microbiome to 5-ASA linked metabolites
In order to identify relative contributions of microbial, host, and other factors to the subset of differentially abundant metabolomic features altered by 5-ASA in Fig S5a, we built linear regression models to predict log-transformed levels of each metabolite with four terms and an intercept. In an analysis restricted to 5-ASA users alone, these four terms comprised: 1) The “host data” feature group, comprising age, sex, disease type (CD, UC), use of antibiotics, and a Western dietary pattern (built from the per-sample food frequency questionnaires described above, where responses about dietary intake were converted to servings per day and separated into two well-established dietary patterns, a “Western” and “prudent”, using factor analysis with a varimax rotation as previously;55 2) The “drug data” group, represented by 5-ASA levels in stool; 3) The “microbiome data” group, including the first 10 principal components computed over the arc-sin square-root transformed relative abundance of taxonomic data as previously described;56 and 4) An unknown/unexplained term. We then extracted the total variance explained by each model, and partitioned the relative contribution of each feature group to the overall R2, using the Lindemann, Merenda and Gold method in the R package “relaimpo” v2.2–3,57 which considers the order of the sequence of predictors appearing in each model.
Differentially abundant microbial species between 5-ASA users and non-users were identified using arcsine square-root transformed taxonomic data with an FDR q < 0.25. Abundances were fit with the following per-feature linear mixed-effects model:
Using the significant results of this differential abundance testing among species and the significantly altered metabolites identified among new users, we then conducted, hierarchical all-against-all association testing using Spearman associations using HAllA 0.8.20 for Fig S5b-d, with an FDR q threshold < 0.05, (http://huttenhower.sph.harvard.edu/halla).
Identification of candidate acetyltransferases
We first performed an amino acid homology search (blastp function, Diamond v0.9.24.125) of the Human Microbiome Project (HMP) reference isolate gut microbial genomes (UniRef90 version 2019_01) using an e-value of 10, >25% identity, “sensitive” mode, and 107 input microbial protein sequences (Extended Table 5). Two came from arylamine N-acetyltransferases (NAT) from Salmonella enterica serovar typhimurium LT2 (nhoA) (UniProt accession, Q00267) and Pseudomonas aeruginosa (UniProt accession, Q9HUY3), previously shown to metabolize 5-ASA.20,21 105 additional sequences were also included (that majority of which were NATs or N-hydroxyarylamine O-acetyltransferases), after having been generated by “Similarity to Identify MicrobioMe Enzymatic Reactions” (SIMMER),58 a computational tool designed to make informed hypotheses about microbial enzymes predicted to metabolize xenobiotics, in part informed by prior experimental knowledge. For this tool, we provided the SMILES substrate input as 5-ASA and the product as N-acetyl 5-ASA.
We requested to report all alignments that were found and then overlapped the resultant list of 5,206 unique UniRef90 IDs with the entire catalog of UniRef90 IDs provided by the HMP2 MGX and MTX datasets.
In our MTX-based strategy, we used differential abundance testing to identify significantly overexpressed acetyltransferases based on paired MTX and MGX profiles. After an arcsine square-root transformation was applied to both MTX and MGX datasets, abundances were fit with the following per-feature linear mixed-effects model, which adjusted for DNA copy number, which allows for biological and technical zero values while also controlling for underlying DNA levels (27):
as prior studies have shown that RNA levels can be a function of mere abundance of DNA.59 Fitting was performed with the lme function from the R package “nlme” package v3.1–149,60 where significance of the association was assessed using Wald’s test. Nominal P values were adjusted for multiple hypothesis testing with a target FDR q of 0.25. In order to reduce the effect of zero inflation in microbiome data, features with a relative abundance of less than 1e-8 in at least 10% of samples were excluded. Among the significant hits, we then sought to further characterize those with putative acyl transfer function, as defined by containing an acetyltransferase domain as annotated on UniProt.
In our MBX-based strategy, we dichotomized each metatranscriptomic gene family as present (relative abundance > 0) or absent. Next, we classified stool samples as N-acetyl 5-ASA high (≥ median) or N-acetyl 5-ASA low/negative (< median, including undetectable levels). Then we calculated the sensitivity and specificity with which each metatranscriptomic gene cluster associated with the dichotomized N-acetyl 5-ASA. Filtering gene clusters with greater than 50% sensitivity and specificity (as used previously),61 we then sought to further characterize those with putative acyl transfer function, as defined by those containing an acetyltransferase domain as annotated on UniProt.62 For continuous correlations, abundance of the seven additional acetyltransferase gene clusters were then arc-sine square-root transformed and associated with log-transformed N-acetyl 5-ASA with linear regression, with an FDR q < 0.25 threshold applied (95% CI shown in Fig 3c).
The pooled twelve amino acid sequences were aligned with clustalW v2 (default parameters).63 Candidate enzymes were then grouped in an average distance tree using neighbor-joining with Blosum62 (Jalview v2.11.1.3),64 which were then mapped to the InterPro database 85.065 to identify protein superfamilies. Amino acid residues in the multiple sequence alignment were colored by clustalX v2.63 Taxonomic lineages of each candidate enzyme was then determined using the UniProt database.62
For gene context analysis, we downloaded the assembled reference genomes of the strains of interest from the NCBI assembly database66 in July 2019. We then queried the proteins of interest (our candidate UniRef90s) against the proteins encoded by the reference genomes using DIAMOND v0.9.24,67 and identified homologous loci by requiring identity ≥90% and coverage ≥80%. Next, we examined up to 10 genes that were upstream and downstream from these acetyltransferase gene loci to explore genomic neighborhoods. These neighborhood genes’ UniRef accessions (from NCBI) were then mapped to the UniProt62 to assess their approximate functions, and in particular, were screened for any glosses indicating transcriptional regulators and/or promoters.
Finally, using previously defined phylogroups generated for F. prausnitzii,33 based on a comparative analysis of genomes, in which a phylogenetic tree of the family Ruminococcaceae was constructed based on concatenated alignments of 245 highly conserved proteins. Within this hierarchy, F. prausnitzii was represented as a monophyletic group of strains, and within this, there were three clear and statistically significant splits into species/subspecies level groups (Fig 3f). We explored all available F. prausnitzii genomes and annotated all strains containing and not containing the UniRef90 of interest.
Case-cohort study
We examined the association of candidate acetyltranferases with risk of disease of relapse among a convenience sample of 39 “ever users” of 5-ASA -- defined as any use of 5-ASA in the cohort. Participants with missing corticosteroid data were excluded. First, we discretized each of the 12 candidate protein families as present/absent based on MGX abundance per sample >0. We estimated multivariate odds ratios (ORs) for relapse with 95% confidence intervals (CIs) using logistic regression, adjusting for potential confounders, including smoking status (never vs ever), age at consent (continuous), disease type (CD vs UC), and host acetylation phenotype (defined below). We also tested for trend by considering each protein as an independent risk factor, and creating an additive score. In a sensitivity analyses, we tested the association between presence of the E. coli NAT identified in our homology search with steroid use, as well as the association between these acetyltransferases and risk of steroid use among 5-ASA non-users. Finally, as shown above in the results, our results were similar when we used fixed effects models and mixed effects models (including a random effects term for individual effects). In light of this similarity, and the fact that steroid use can truly occur multiple times throughout a year, we focused on the results of fixed effects models.
Validation of treatment failure based on microbial acetyltransferase genes in SPARC IBD
Data Source
We validated the association between metagenomic carriage of gut microbial acetyltransferases with risk of 5-ASA treatment failure in an ongoing independent cohort, Study of a Prospective Adult Research Cohort with IBD (SPARC IBD), from the IBD Plexus platform of the Crohn’s & Colitis Foundation. As previously published,68,69 3,029+ well-phenotyped adult patients with UC, CD, or unclassified IBD have been recruited since 2015 from 17 major academic medical centers with no overlap with those in the IBDMDB.. The study protocol was approved by the University of Pennsylvania’s institutional review board. All participants provided informed consent.
Study population
The study population consisted of 240 patients enrolled in SPARC IBD between 2016 and 2020 who 1) provided a stool sample at time of consent and 2) were on 5-ASA at cohort entry (Extended Fig. 9). To ensure a prospective design, participants who were on steroids at baseline were excluded from this analysis (n=26). Additionally, we excluded participants who withdrew consent from the study after enrollment (n=6). Thus, the remaining 208 individuals formed the analytic sample. Follow up was through October 19, 2021.
Stool sampling
At the time of consent, samples were collected by the patient at home and shipped directly to the biobank through a courier service as previously described.69 A minority of individuals (n=33) provided more than one sample during the cohort. Although the SPARC IBD protocol does not include a prespecified schedule for repeated sampling after enrollment, participants are able to provide additional biosamples at time of usual care sigmoidoscopy or colonoscopy. Additional stool samples may also be obtained approximately 3 months after a change in therapy if the patient has a follow-up office visit during that time.
Medication Data
At enrollment current medications were captured through the SPARC electronic data capture tool. Subsequent medication data in SPARC IBD were collected from an IBD Smartform and more broadly from the Epic electronic health record system. In addition, electronic surveys were delivered to patients every 3 months to capture IBD-related symptoms and current IBD therapies to track patient-reported disease activity between office visits and to further confirm medications. Use of 5-ASA was defined identically as in the IBDMDB by use any of the following, which includes bonded formulations: oral mesalamine, mesalamine suppository, mesalamine enema, olsalazine, balsalazide, and sulfasalazine. Use of corticosteroids was defined identically as in the IBDMDB by use any of the following: budesonide, prednisone, or methylprednisolone.
DNA extraction
Genomic DNA extraction was performed via the Mag Attract Power Soil kit (Qiagen, Cat# 27000-4-EP) following the manufacturer’s instructions. Total nucleic acids were extracted using the Qiagen MagAttract PowerMicrobiome kit (Qiagen, Catalog No. 27500-4-EP).
Metagenomic sequencing
Metagenomic sequencing was performed by Diversigen. Genomic DNA was prepared into libraries for sequencing by the Nextera DNA Flex library preparation kit (Illumina, Catalog No. 20018705) with Nextera Index Kit (Illumina, Catalog No. 20027213). Library size estimation and quantification were determined with the fragment analyzer (Advanced Analytical Technologies, Inc.) electrophoresis system. The prepared libraries were sequenced via the NovaSeq 6000, 2x150 bp sequencing platform (Illumina).
Statistical Analysis
Data from the validation cohorts were not available to us while developing the original prediction models in the HMP2. Sequencing data from SPARC IBD went through the exact same analysis pipeline (biobakery3) as in the IBDMDB to extract the metagenomic data that our prediction models were based on. Only microbial acetyltransferases that were significantly predicted to be linked with steroid use in our discovery cohort (R6CZ24, T5S060, R5CY66, C7H1G6) were considered for further analysis. As in the IBDMDB, we discretized each of the four candidate protein families as present/absent based on abundance per sample >0. To mitigate sampling bias, samples were not included if they were provided fewer than 30 days after the previous. Similarly, to avoid sampling bias, participants were censored when they reported using steroids.
The primary exposure was defined as metagenomic carriage of 3-4 acetyltransferases compared to 0-2 acetyltransferases, just as performed in the IBDMDB. We were not powered to examine the association between each acetyltransferase with risk of steroids. Given the possibility of correlation within samples, we calculated the odds ratios (ORs) and 95% confidence intervals (CIs) of use of steroids using multivariable generalized estimating equations in the in the R v4.0.1 package “gee” v. 4.1370 with adjustment for age and sex. In a sensitivity analysis, we limited our analysis to the single baseline sample, and related metagenomic carriage of these genes with future steroid risk using multivariable logistic regression with adjustment for age, sex, and IBD disease type (Extended Fig. 9). Random effects meta-analysis was performed using the R v4.0.1 package “metafor” 1.4-0.71
Assignment of host acetylation phenotype
Human NAT2 acetylator genotype status has previously been linked with increased risk of bladder cancer due to decreased ability to detoxify carcinogens,72 as well in predicting drug-induced liver injury from isoniazid.73 Although studies attempting to link NAT2 acetylation phenotypes with 5-ASA response have not been fruitful,40 many continue to speculate if NAT2 could play a role in inactivating 5-ASA.41 Therefore, using a simple panel of two SNPs identified in participants’ exome sequencing data (rs1041983, rs1801280) previously shown to accurately predict acetylation phenotypes as “fast” vs. “slow”,74 we first calculated the sum of variant alleles in each patient. As previously, any participant who had two or more variant alleles could then be categorized as a slow acetylators, inferring that they had either the NAT2*5 or NAT2*6 haplotype.
Expression, purification and in vitro assessment of candidate acetyltransferase enzymes
Candidate 5-ASA acetyltransferase genes (as well as control GFP and known Salmonella typhymirium gene) were synthesized according to the E. coli codon-optimized amino acid sequence on UniProt (GenScript) and then cloned into pET28b inducible expression vectors using Gibson assembly (including an in-frame either N- or C-terminal polyhistidine sequence). Identities of the constructs were confirmed with DNA sequencing and then were transformed into E. coli BL21 strains for expression (according to the standardized protocols from GenScript, ordered sequences are listed in Extended Table 11). All E. coli expression constructs were grown anaerobically in sealed Hungate tubes in Terrific Broth (VWR) supplemented with 5 mM MgSO4 and kanamycin (50 μg/mL) and were incubated at 37 °C overnight before dilution at 1:100 into fresh media the following morning. These diluted cultures were grown anaerobically in sealed flasks at 37 °C to an OD600 ~0.6 at which point protein expression was induced by the addition of 100 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG, TEKNOVA) followed by aerobic culture overnight at 16 °C. The following morning, E. coli were pelleted by centrifugation and then lysed in 20 mM HEPES pH 8.0 buffer containing 30 mM imidazole and 300 mM NaCl and supplemented with 0.5% Octyl-B-D-thrioglucopyranoside (Chem-Impex), 0.5 mg/mL lysozyme (Sigma), and SIGFAST protease inhibitor cocktail (Sigma). After lysis and clarification by centrifugation, lysates were incubated for 1 hour at 4 °C with His-Pure Cobalt Purification beads (Thermo). After incubation, beads were washed with 6 column volumes of 20 mM HEPES pH 8.0 buffer containing 30 mM imidazole and 300 mM NaCl before elution with 1 column volume of 20 mM HEPES pH 8.0 containing 300 mM imidazole and 300 mM NaCl. Eluted protein was then buffer exchanged into 20 mM HEPES pH 8.0 with 300 mM NaCl and 10% glycerol for storage at −80 °C prior to further experiments.
Enzyme concentrations were calculated according to Beer’s Law with extinction coefficients calculated by Benchling software based on the amino acid sequences of the putative enzymes. Assays mixtures contained 20 mM HEPES pH 7.5 with 50 mM NaCl with 50 μM enzyme for the end-point assay and 5 μM for promiscuity assays; 1 mM of the specified substrates (5-ASA, 4-ASA, dapsone, isoniazid, procainamide, hydralazine), and 1 mM of the indicated acyl-CoAs (acetyl-CoA, propionyl-CoA, butyryl-CoA, CoALA Biosciences) supplemented with 1 mM MgCl2 and 1 mM Tris-(2-carboxyethyl)phosphine (TCEP, Sigma). Reactions were conducted at 37 °C for 6 hours for the initial endpoint assay and room temperature for 1 hour for the promiscuity assay. Quenching/extraction for the promiscuity assay was with equal volumes of acetonitrile and methanol. Quenching/extraction for the end-point was one part sample and nine-parts extraction mix (75% acetonitrile : 25% methanol v/v with 10 μM 1,2-13C2-taurine from Cambridge Isotopes as an internal standard for quantification). Samples were vortexed and cooled to −20 °C and then centrifuged prior to LC-MS analysis.
For the kinetic assay, assay conditions were the same as above (20 mM HEPES pH 7.5 with 50 mM NaCl, 1 mM MgCl2, and 1 mM TCEP, and 5 μM enzyme). Concentration of 5-ASA substrate was varied as indicated with 1 mM acetyl-CoA held constant. Reactions were carried out at 37 ºC for 0, 10, 20, 30, 45, 60, 90, and 120 min in triplicate and quenched/extracted with one part sample and nine-parts extraction mix (75% acetonitrile : 25% methanol v/v with 10 μM 1,2-13C2-taurine from Cambridge Isotopes as an internal standard for quantification). Samples were vortexed and cooled to −20 °C and then centrifuged prior to LC-MS analysis.
For the promiscuity assays, mass spectrometry analyses were conducted using an LC–MS system composed of an Agilent 1260 Infinity HPLC (Agilent Technologies) coupled to a 6530 Accurate-Mass QTOF LC-MS (Agilent Technologies). The samples were injected into a 150x3 mm Hypersil GOLD aQ column (Thermo Scientific) at 30 °C. The column was eluted isocratically at a flow rate of 500 μL/min with 99% mobile phase A (0.1% formic acid in water) for two minutes followed by a linear gradient for 16 minutes to 99% mobile phase B (acetonitrile with 0.1% formic acid). This ratio was continued for 5 minutes before returning to 99% mobile phase A over 1 minute and continuing at that ratio for additional 6 minutes. MS analyses were performed in negative ion mode (N-acetyl-5-ASA, N-propionyl-5-ASA, N-butyryl-5-ASA, and N-acetyl-4-ASA) and positive ion mode (N-acetyldapsone, N-acetylprocainamide, N-acetylhydralazine, and N’-acetylisoniazid) using electrospray ionization, full scan MS acquisition over 100 to 3000 m/z, a resolution setting of 10,000, and centroided masses. Other MS settings were as follows: heater temperature 300 °C, ESI nebulizer 35 psi, spray voltage 3.5kV, 2 spectra/s. The identities of N-acetyl-5-ASA (Cayman Chemical, Ann Arbor, MI) and N-acetyl-4-ASA (Santa Cruz Biotechnology, Dallas, TX), were confirmed using authentic mass standards. Remaining identities were inferred using calculated masses with a mass detection tolerance of 10 ppm. Raw data from the LC-MS were analyzed using Agilent MassHunter Qualitative Analysis 10.0 software.
For end-point and kinetic assays, mass spectrometry analyses were conducted using an LC–MS system composed of an Agilent 1290 Infinity II UHPLC (capable of column switching) coupled to a Agilent 6470A Triple Quadrupole LC/MS. The samples were injected into an Infinity Lab Poroshell120 Hilic column (2.1 x 100 mm 2.7 μm) at 25°C. The column was eluted isocratically at a flow rate of 600 μL/min with 5% mobile phase A (10 mM ammonium formate with 0.1% formic acid in water) for 18 seconds followed by a linear gradient for 132 seconds to 60% mobile phase B (acetonitrile with 0.1% formic acid). This was followed by a 3 second gradient to 40% mobile phase B. This was then followed by a 27 second gradient returning to 5% mobile phase A at a flow rate of 1200 μL/min. This flow rate and ratio was held for an additional 48 seconds. Additional column equilibration was carried out on a secondary pump for 117 seconds at 5% mobile phase A and a flow rate of 1000 μL/min. MS was conducted in negative ion mode using electrospray ionization. Data were collected via MRM (N-acetyl 5-ASA MRM 194 -> 150, 1,2–13C2-taurine MRM 125.9 -> 79.9). Other MS settings were as follows: heater temp 300°C, ESI nebulizer 45 psi, spray voltage 3.5 kV, acquisition time 100 ms/spectrum. Raw data from the LC–MS were analyzed using Agilent MassHunter Quantitative Analysis Version 10.1 Software. For absolute quantification, all samples were normalized to the taurine internal standard and concentrations calculated against a standard curve.
Oscillibacter culture
Oscillibacter sp., strain KLE 1745 was obtained through BEI resources, the NIH Institute of Allergy and Infectious Diseases (NIAID) as part of the Human Microbiome Project. This strain was cultured anaerobically directly from freezer stocks for three days in reinforced Clostridial media (BD Difco) at 37 °C before the addition of 5-ASA to a final concentration of 1 mM (stock concentration 100 mM in DMSO, sterile filtered with 0.22 μm filter) for an additional 24 hours prior to harvesting. Spent media was centrifuged at 10,000xg for 3 minutes to pellet bacteria and cell debris. 10 μL of media was then extracted with 90 μL of acetonitrile:methanol (75%:25% v/v) and measured via LC-QTOF as described elsewhere in the Methods section.
Crystal Structure Analysis
Protein expression and purification
The C-terminal non-cleavable His tag construct of the thiolase from the Firmicutes CAG:176 thiolase (FcTHL) was overexpressed in E. coli BL21 (DE3) and purified using affinity chromatography and size-exclusion chromatography. Briefly, cells were grown at 37°C in TB medium in the presence of 50 μg/ml of kanamycin to an OD of 0.8, cooled to 17°C, induced with 500 μM isopropyl-1-thio-D-galactopyranoside (IPTG), incubated overnight at 17°C, collected by centrifugation, and stored at −80°C. Cell pellets were lysed in buffer A (25 mM HEPES, pH 7.5, 500 mM NaCl, 0.5 mM TCEP, and 20 mM Imidazole) using Microfluidizer (Microfluidics), and the resulting lysate was centrifuged at 30,000g for 40 min. Ni-NTA beads (Qiagen) were mixed with cleared lysate for 30 min and washed with buffer A. Beads were transferred to an FPLC-compatible column, and the bound protein was washed further with buffer A for 10 column volumes and eluted with buffer B (25 mM HEPES, pH 7.5, 500 mM NaCl, 0.5 mM TCEP, and 400 mM Imidazole). The eluted sample was concentrated and purified further using a Superdex 200 16/600 column (Cytiva) in buffer C containing 20 mM HEPES, pH 7.5, 200 mM NaCl, and 10 μM TCEP. FcTHL containing fractions were concentrated to ~50mg/mL and stored in −80°C.
Crystallization
FcTHL at 800 μM was crystallized in 100 mM sodium acetate, pH 4.9, 55% MPD, and 20 mM CaCl2 by sitting-drop vapor diffusion at 20°C. Crystals were transferred briefly into crystallization buffer containing 25% glycerol prior to flash-freezing in liquid nitrogen.
Determination
Diffraction data were collected at beamline NE-CAT-24ID-E at the Advanced Photon Source (Argonne National Laboratory). Data sets were integrated and scaled using XDS.75 Structures were solved by molecular replacement using the program Phaser and the search model PDB entry 4XL2. Iterative manual model building and refinement using Phenix76 and Coot77 led to a model with excellent statistics.
Visualization
Protein structures were visualized in ChimeraX v1.15.78 The predicted structure was then overlaid using the MatchMaker tool (default settings) on monomers from the PDB entries 1DM3 and 1E2T. To compare active sites, residues were pre-specified, and then overlaid using the match tool (default settings) which performs least-squares fitting of active site atoms, first moving the one set of atoms onto the second, followed by the remaining model containing the atoms.
Extended Data
Extended Figure 1. Fecal 5-ASA levels according to self-reported 5-ASA use in the IBDMDB.
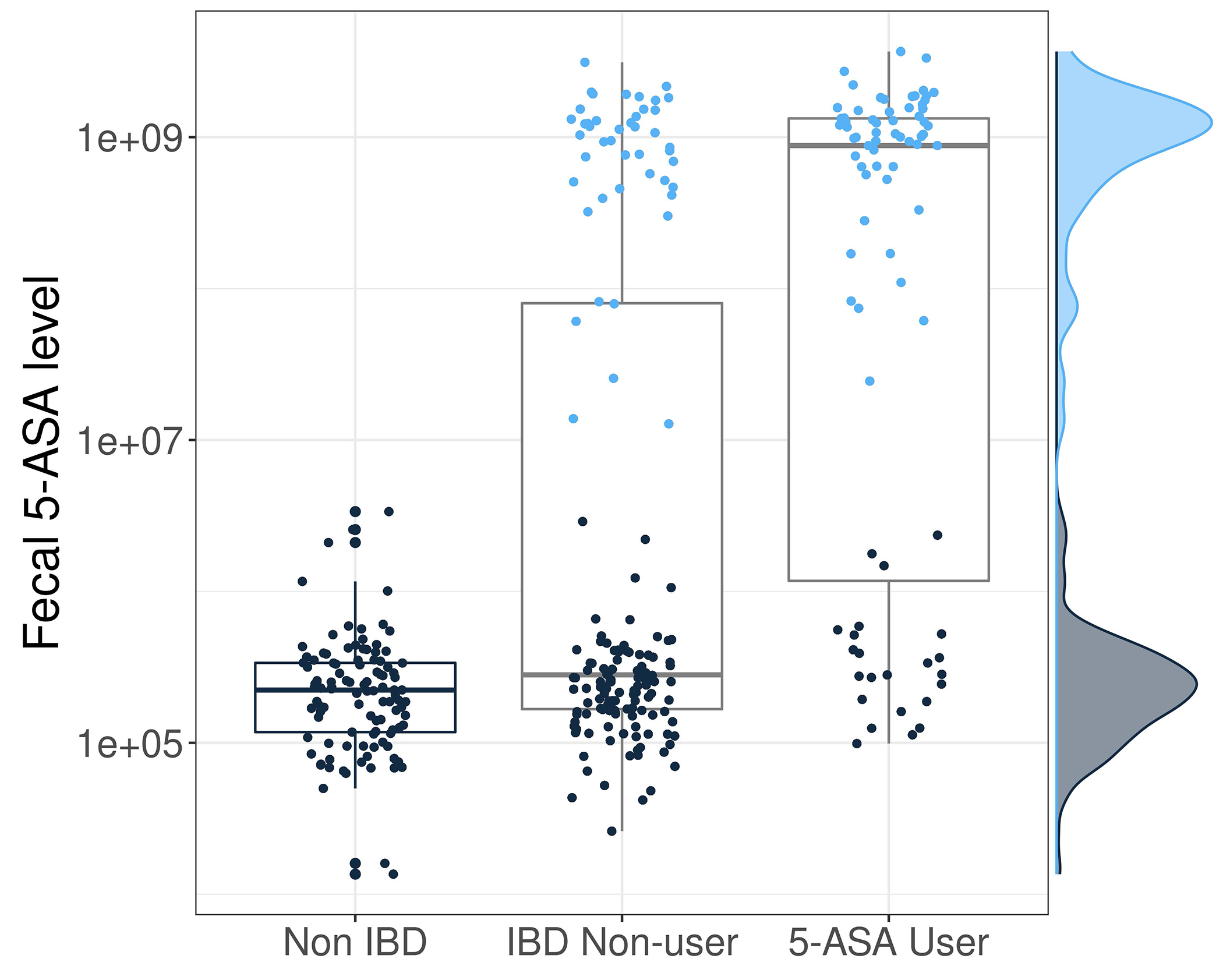
We determined 5-ASA use according to detection of drug levels in stool using LC-MS, defined as detection of fecal 5-ASA levels > 10e7. Concordance, determined according to statistical accuracy (Methods), between self-reported use of 5-ASA and detection of fecal 5-ASA was 80.3%. Users (light blue) had 5-ASA levels which were ~10,000x greater than non-users (navy blue). Each individual is represented by multiple points on this graph given that participants provided multiple samples across the year-long cohort. Boxplots show median and lower/upper quartiles; whiskers show inner fences.
Extended Figure 2. Representative indirect effects of 5-ASA on the fecal metabolome include shifts in Vitamin B3 and its metabolism towards glycine conjugation and formation of nicotinuric acid.
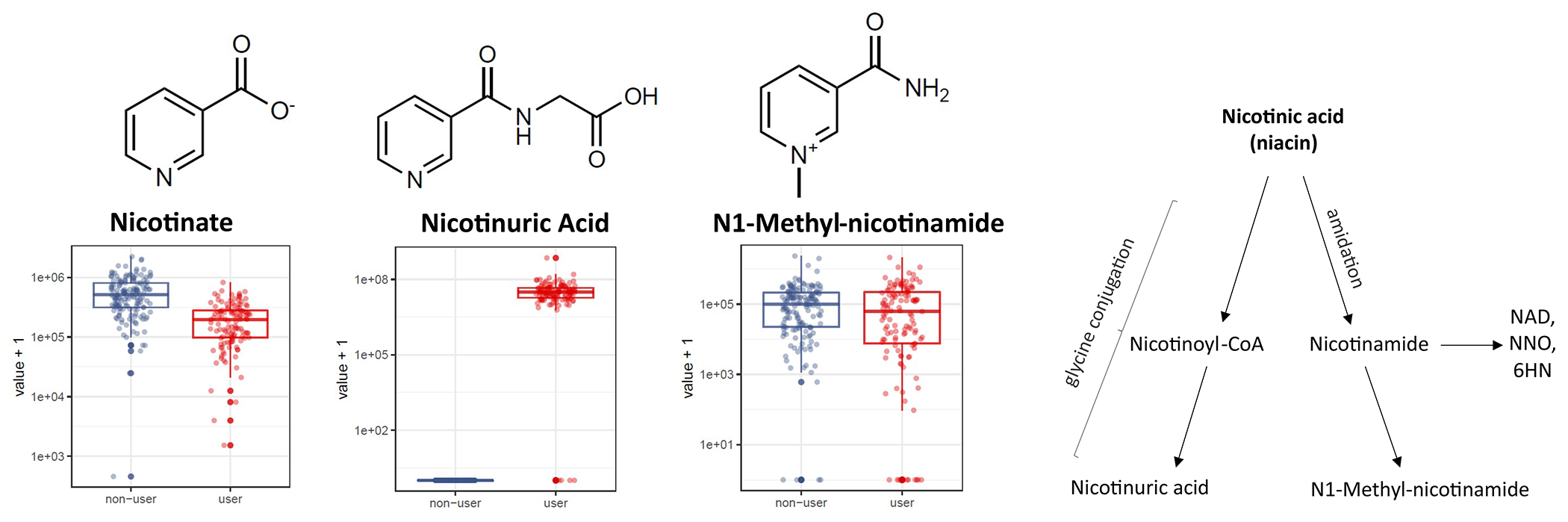
A) We considered that 5-ASA may lead to highly specific markers of medication use mediated through indirect microbial pathways, such as promotion of growth of certain bacteria which, in turn, generate compounds. Here, we find that the abundance of nicotinic acid (NA) dropped as nicotinuric acid (NUA) dramatically increased; there was no consistent change in the third niacin metabolite detected on our platform, N1-methyl-nicotinamide. While the precise role of 5-ASA in determining the fate of NA is unknown, for >70 years, anaerobic bacteria, including Clostridia species have been known to metabolize NA (24). Furthermore, medications, such as aspirin, are known to affect the balance of NA and NUA in the blood.79 Intriguingly, fecal NA levels have previously been detected at lower levels in IBD patients compared to healthy controls,80 but confounding by 5-ASA use was not explored. Given the role of anaerobic gut bacteria in metabolism of NA, it is conceivable that gut bacteria promoted by 5-ASA – or 5-ASA itself – shunts NA towards the glycine conjugation pathway without profound impact on amidation. This phenomenon was not seen in initiators of steroids or biological drugs in the cohort; NUA levels were undetectable in non-5-ASA users. B) These effects were highly specific to 5-ASA use and could not be attributed simply to suppression of inflammation: in a subset of 9 participants in the IBDMDB who started biologic drugs, there was no change in nicotinic acid levels, and nicotinuric acid levels were undetectable in non-users of 5-ASA. In both panels, boxplots show median and lower/upper quartiles; whiskers show inner fences.
Extended Figure 3. Impact of 5-ASA on the fecal metabolome is influenced by the gut microbiome.
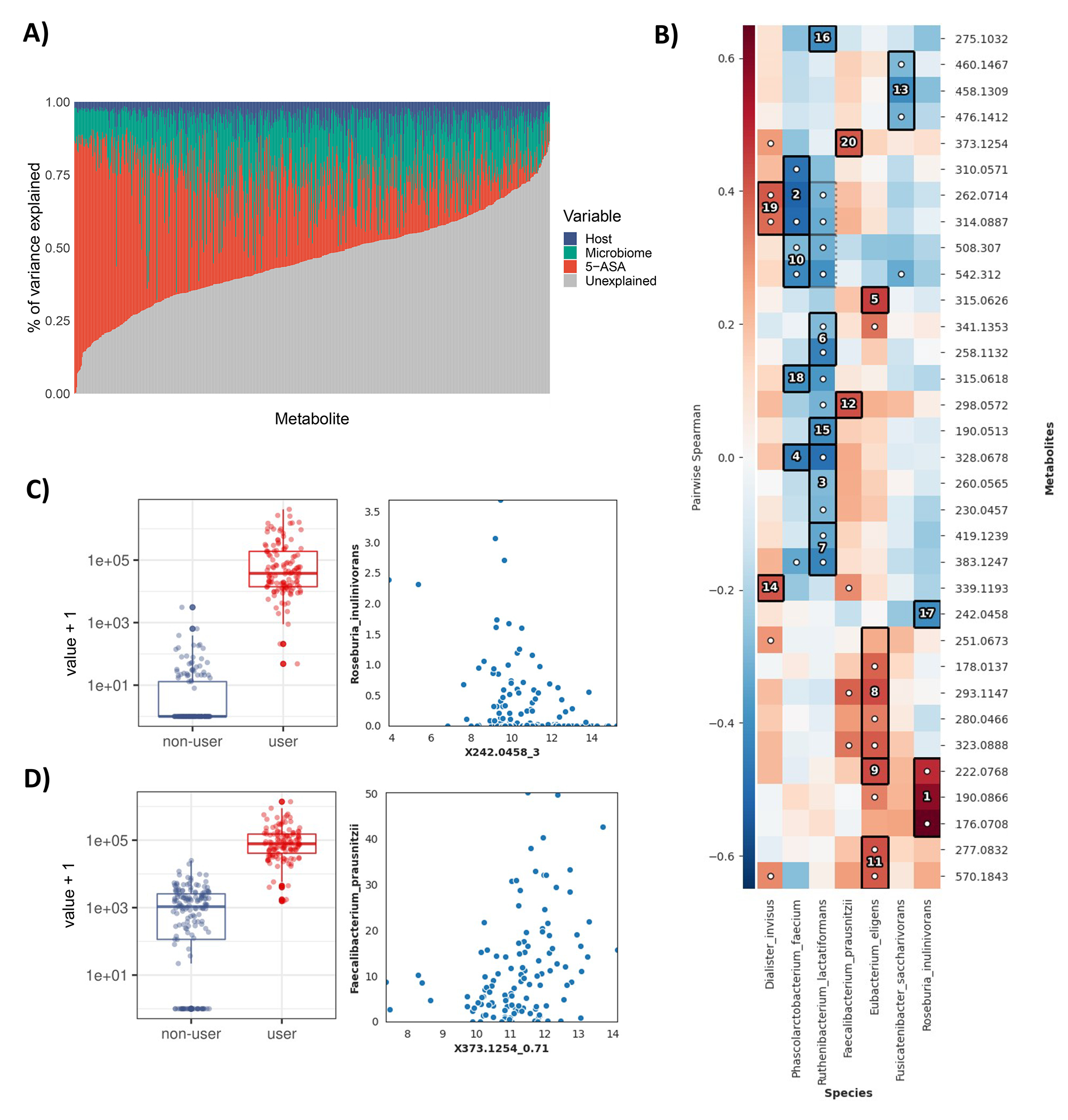
A) Drug levels, followed by gut microbiome taxonomic profiles, independently explain variation in most 5-ASA-derived metabolite levels. These were analyzed systematically by linear models constructed for each metabolite with independent variables being: fecal 5-ASA levels; microbiome data; host data, including diet, disease type, age, other medications, and sex; and other/unexplained. We quantified variance explained (EV) by each model and partitioned EV by each term for each 5-ASA-shifted metabolite (shown along the x-axis, as a stacked proportional bar plot). As an example, N-acetyl 5-ASA had a moderate correlation with 5-ASA levels in stool (Spearman rho 0.50, p=4e-9), with 35% of variance explained by drug levels, 15% explained by the microbiome, and 7% explained by other host features. B) Associations between differentially abundant (DA) metabolites and DA species using HAllA (Methods). Block associations are numbered in descending order of significance (max FDR<0.25), with each numbered block corresponding to a group of co-occurring metabolites with a species. A white dot indicates marginal significance of a particular pair of features (p<0.05). C) Abundance of R. inulinivorans was inversely correlated with the metabolite peak 242.0458 (represented by Block 17 in (B)), which was 37,661-fold greater on average among users than non-users (FDR 0.001). Boxplots show median and lower/upper quartiles; whiskers show inner fences. D) F. prausnitzii was positively associated with the peak 373.1254 (represented by Block 20 in (B)), which was 73-fold greater among users than non-users (FDR 0.001). Boxplots show median and lower/upper quartiles; whiskers show inner fences.
Extended Data Figure 4: Microbial species previously characterized to acetylate 5-ASA have low or absent representation in the gut microbiomes of patients with IBD.
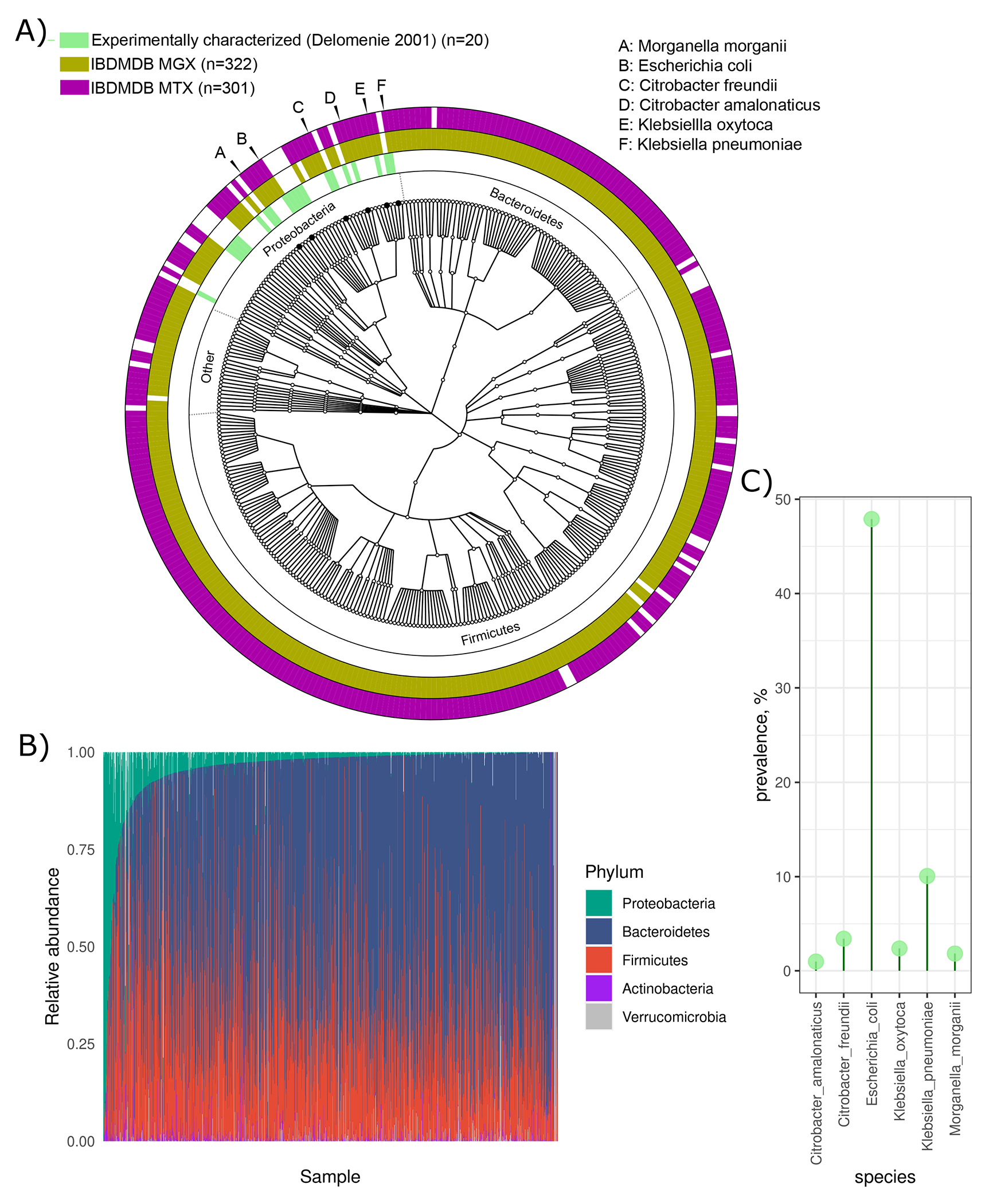
A) Phylogenetic analysis shows the taxonomic contributions to the MTX (purple, n=301 species) and MGX (gold, n=322 species) data in the IBDMDB. Also shown are bacteria (green, n=20 species) previously found to acetylate 5-ASA in vitro,18 all of which belong to the Proteobacteria phylum. Notably, only six of these had detectable MGX and MTX levels in the HMP2, which are labeled and listed. B) The abundance of Proteobacteria is low across the vast majority of participants, relative to Firmicutes and Bacteroidetes species. C) The prevalence of the six species with potential for 5-ASA acetylation activity is low, except E. coli. Most importantly, none of these six bacterial species have detectable arylamine N-acetyltransferase enzymes at the metatranscriptomic level in the IBDMDB.
Extended Figure 5. Human gut microbiome genes were prioritized for experimental characterization via a three-pronged multi-omics approach.
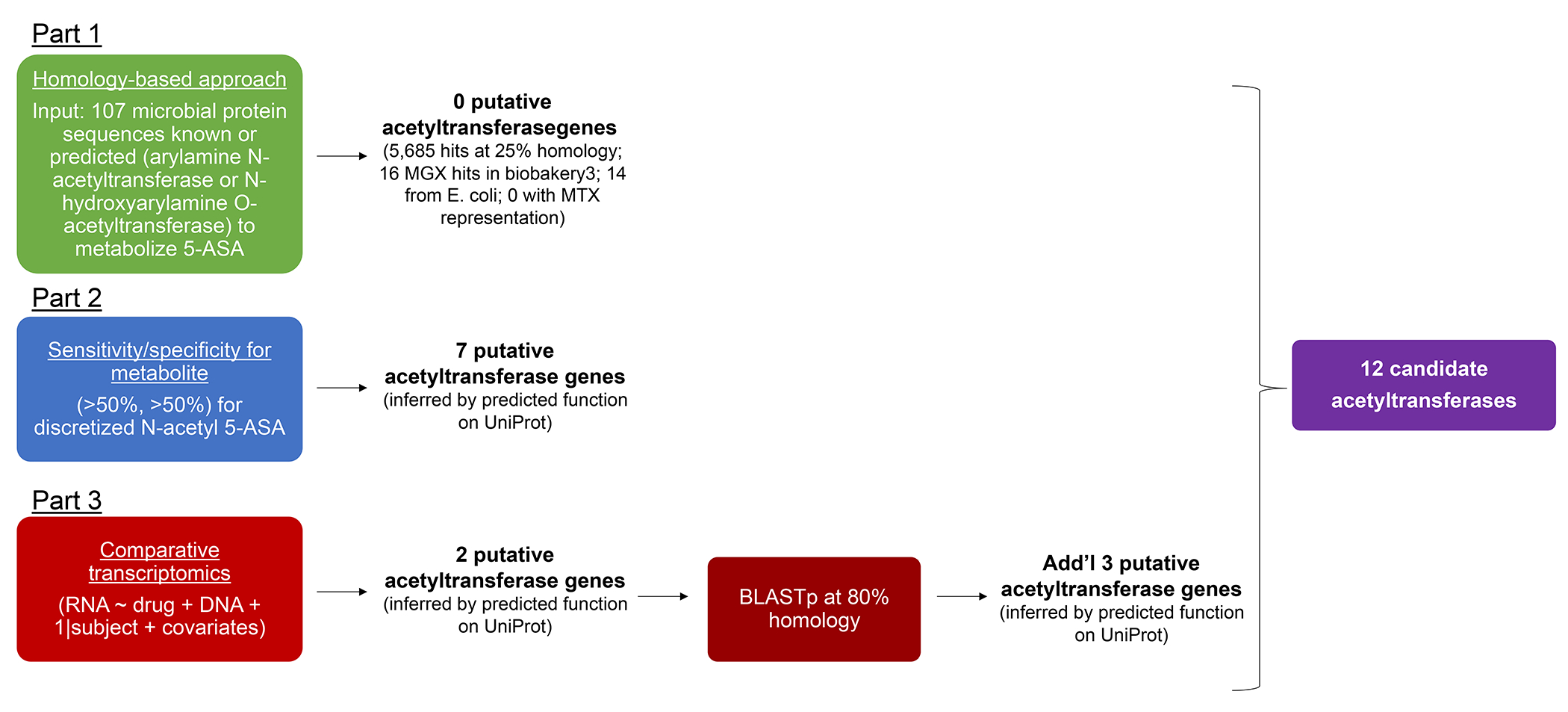
We took three-pronged approach to identifying 5-ASA metabolizing enzymes; each part was independent of the other. In Part 1, we used two arylamine N-acetyltransferase (NAT) sequences from Salmonella enterica serovar typhimurium LT2 (nhoA) (UniProt accession, Q00267)20 and Pseudomonas aeruginosa21 (UniProt accession, Q9HUY3), previously shown to metabolize 5-ASA, as well as 105 NAT or N-hydroxyarylamine O-acetyltransferase microbial protein sequences predicted to metabolize 5-ASA (Methods, Extended Table 4) as the query for a BLASTP search (Diamond v0.9.24.125) of the Human Microbiome Project (HMP) reference isolate genomes with an e-value of 10 down to the 25% identity. Given that there were no hits at the metatranscriptomic level, in Part 2, we then used differential abundance testing to identify significantly overexpressed acetyltransferases, fit with a per-feature linear mixed-effects model, which adjusted for DNA copy number, which allows for biological and technical zero values while also controlling for underlying DNA levels (q < 0.25). These two hits were expanded at 80% homology to find similar functional proteins. In Part 3 of our approach to identifying 5-ASA metabolizing enzymes, we compared metatranscriptomic abundance of individual gene families (present/absent) with the metabolomics readout of dichotomized N-acetyl 5-ASA (high/low) in each stool sample, and then derived measures of sensitivity (true positives / (true positives + false negatives)) and specificity (true negatives / (true negatives + false positives)) for each gene cluster to estimate how well a given cluster correlates with the drug metabolite. All of these were pooled to arrive at 12 candidates.
Extended Figure 6. Heterologous expression of thiolase and acyl-CoA N-acyltransferase enzymes in E. coli BL21.
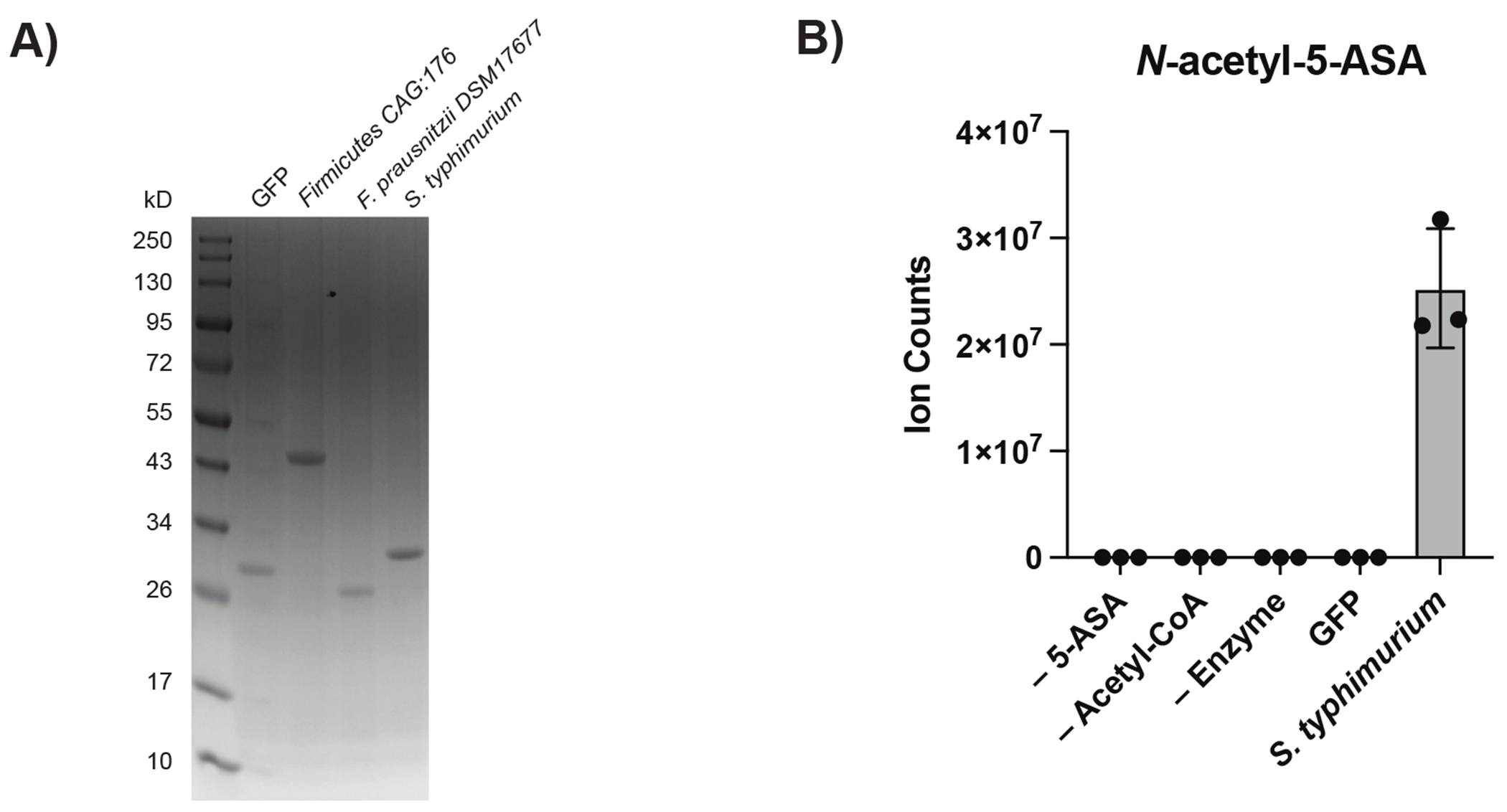
(A) Coomassie-stain gel shows purity of indicated enzymes after overexpression and cobalt purification. N=1 protein purification per enzyme; repeated independently n=2-4 for each enzyme. Abbreviations: FcTHL = a predicted thiolase (R6CZ24) from an uncultured Firmicutes; FpGNAT = a predicted acyl CoA N-acyltransferase (C7H1G6) from F. prausnitzii; StNAT = a known arylamine N-acetyltransferase from Salmonella enterica serovar typhimurium LT2. (B) Confirmation of in vitro acetylation of 5-ASA by known S. typhimurium enzyme using 1 mM of each substrate and 5 uM enzyme incubated for 1hr at RT. Data are presented as mean values +/− SEM.
Extended Fig. 7. Extended biochemical data for the 5-ASA metabolizing thiolase enzymes.
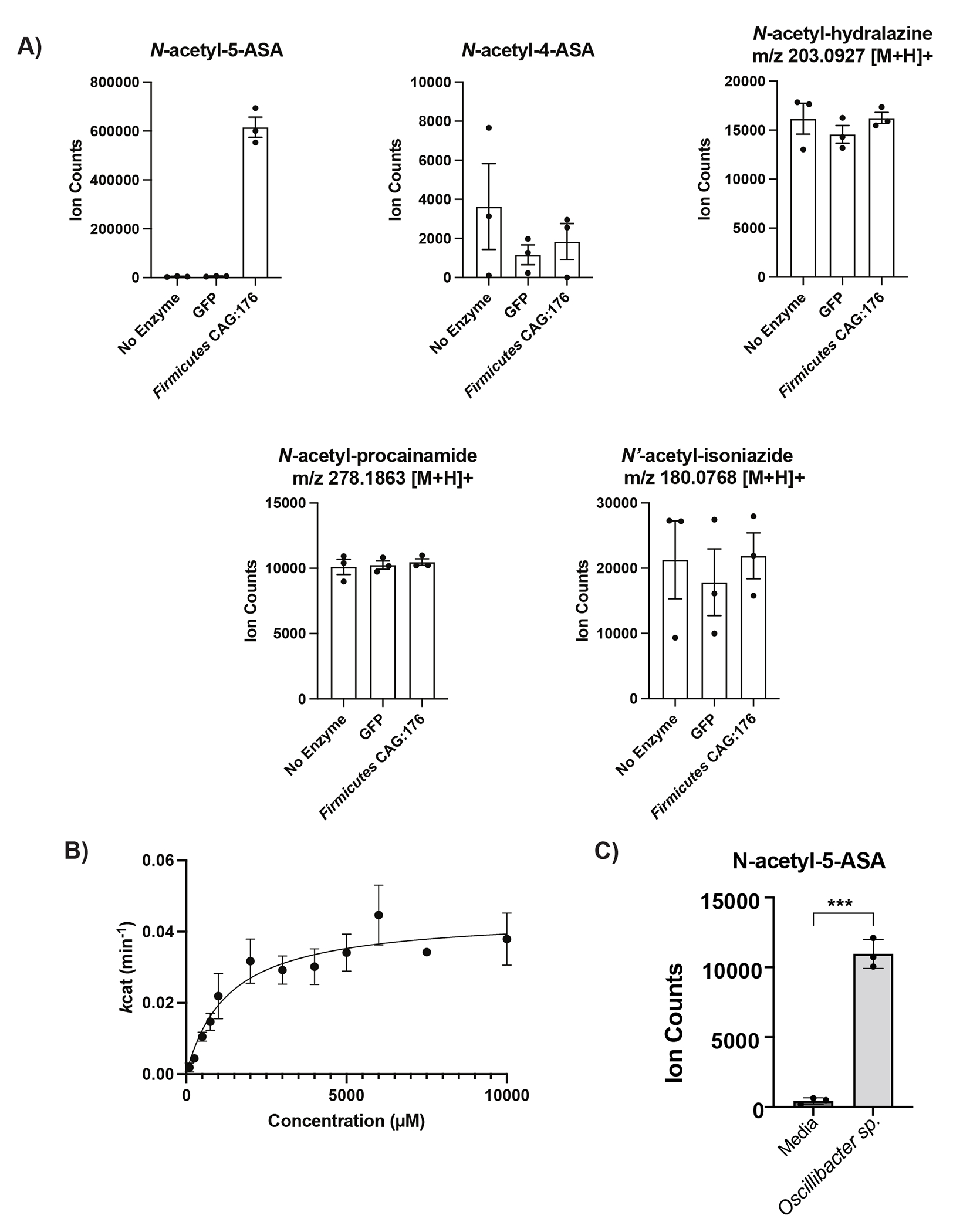
(A) In vitro competition assay with 1 mM of 5-ASA, 4-ASA, procainamide, hydralazine and isoniazide demonstrates relative specificity of the Firmicutes CAG:176 thiolase (FcTHL) for 5-ASA. N=3 biologically independent samples per enzyme/condition. (B) Shown is a representative Michaelis-Menten plot of n=1 thiolase enzyme preparation, conducted in technical triplicates at each concentration of 5-ASA; summary data is from n=5 biologically independent experiments each conducted in technical triplicate. (C) Live culture of Oscillibacter sp., strain KLE 1745 encoding another predicted thiolase gene (UniRef90 R6TIX3) was capable of acetylation of 5-ASA to N-acetyl 5-ASA. N=3 biologically independent samples per condition, representative data from n=2 independent experiments, unpaired, two-sided T-test, *** = p = 0.0015. Data are presented as mean values +/− SEM.
Extended Figure 8. Comparison of canonical acetyltransferase reaction mechanisms for thiolase and arylamine NAT enzymes, now both shown to acetylate 5-ASA.
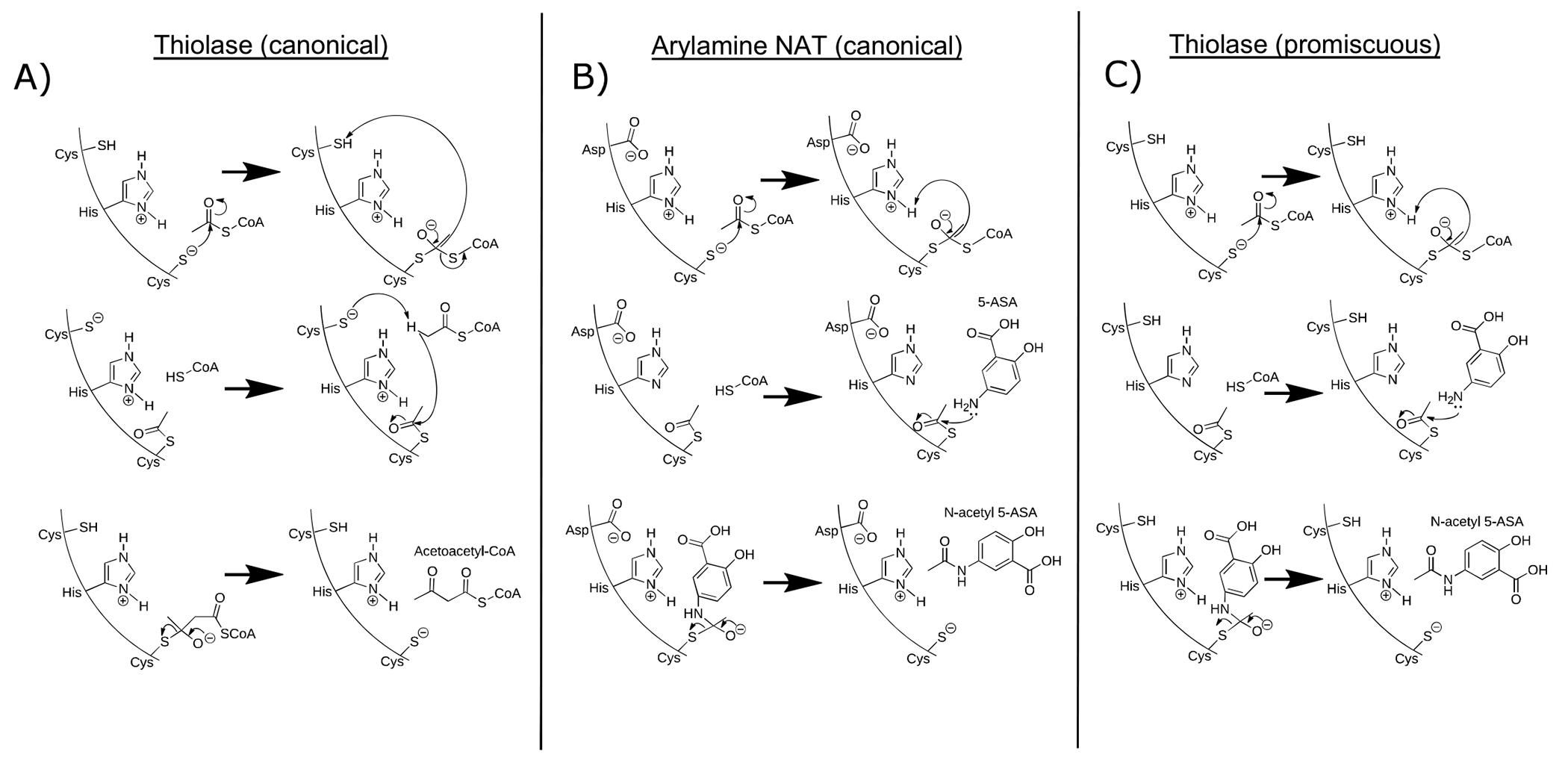
A) The thiolase two-step “ping pong” mechanism (Modis) begins after a cysteine is activated by a nearby histidine residue and then performs a nucleophilic attack on an acetyl CoA molecule to form a covalent acetyl-enzyme intermediate (shown in Fig 4d). In the second step, the substrate (classicaly a second acetyl CoA molecule) nucleophilically attacks the acetyl-enzyme intermediate to yield the final acetyoacetyl-CoA and enzyme. The second nucleophilic attack is activated by a second cysteine residue in the active site, which deprotonates the substrate. B) In a similar two-step “ping pong” mechanism in the arylamine NAT, a cysteine is also activated by a nearby histidine residue and then performs a nucleophilic attack on an acetyl CoA molecule to form a covalent acetyl-enzyme intermediate. In contrast, rather than abstracting a proton from another cysteine residue in the active site, the departing CoA molecule deprotonates the histidine residue, which allows an arylamine substrate to perform nucleophilic attack on the acetyl-enzyme intermediate to yield the final acetylated substrate and enzyme. C) We speculate that in some cases, an acetyl-thiolase enzyme intermediate is formed which may allow nucleophilic attack by an arylamine, such as 5-ASA.
Extended Figure 9. Overview of the Study of a Prospective Adult Research Cohort with IBD (SPARC IBD) study.
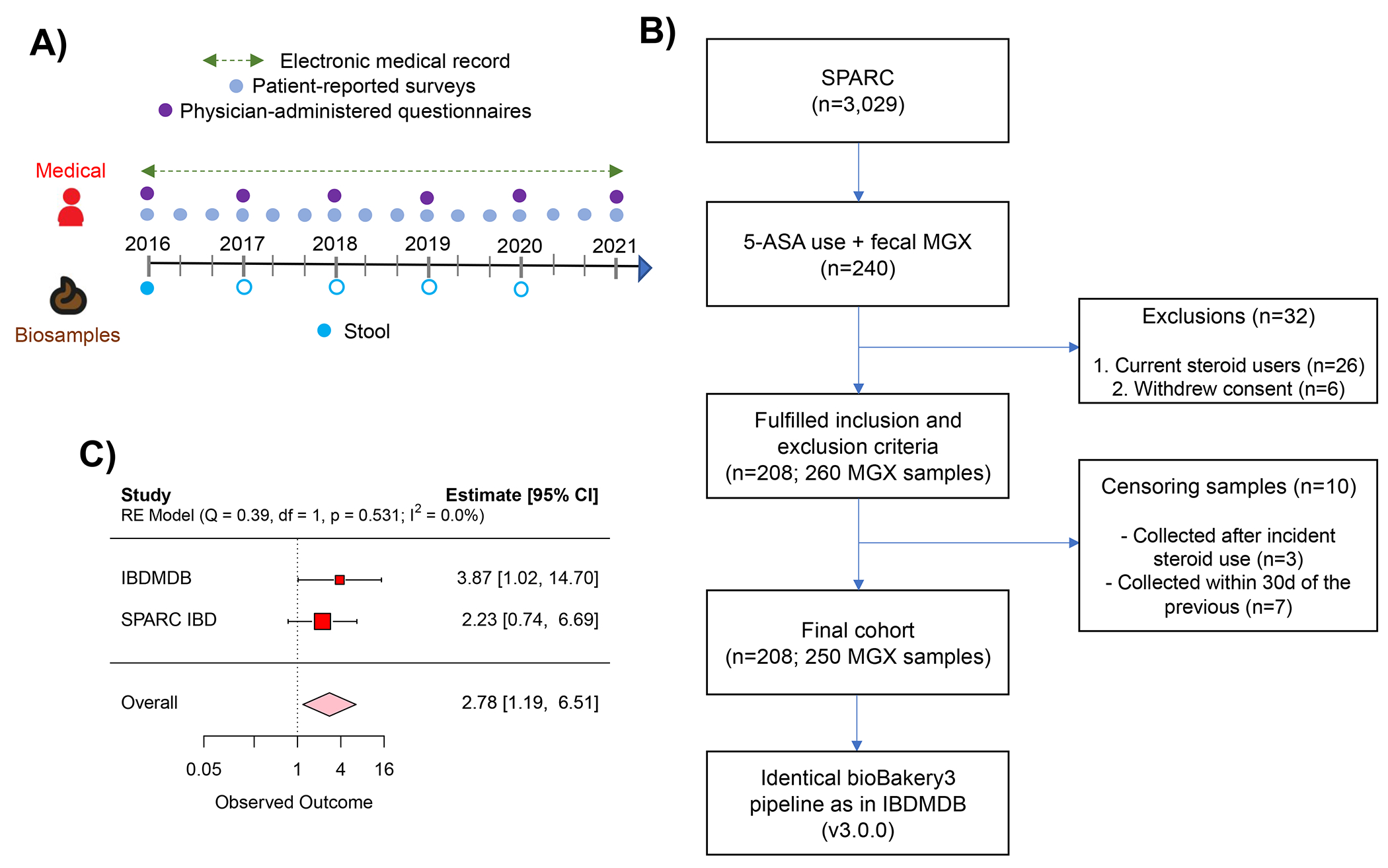
A) Timeline shows the stool sampling scheme in relation to clinical assessments throughout the cohort. At study entry, all participants provided a single stool sample. The median time to event (use of steroids) was 229 days. Among a minority of participants (n=33) who voluntarily provided additional stool samples throughout the cohort (denoted by empty blue circles, Methods), median interval sampling time was 133 days. B). Flow diagram illustrating inclusion and exclusion criteria for this analysis. C) In a sensitivity analysis, we limited our analysis to a single (baseline) stool sample per participant. With diminished power, the SPARC IBD-specific estimate was no longer significant (OR 2.22, 95% CI 0.74-6.69). Accordingly, age and CD were not significantly associated with risk of steroids despite being established risk factors for 5-ASA treatment failure.39 Nevertheless, the pooled meta-analysis was not meaningfully different from the primary analysis (OR 2.78, 95% CI 1.19-6.50).”
Extended Figure 10: Validity of 5-ASA and N-acetyl 5-ASA annotation by metabolomics methods.
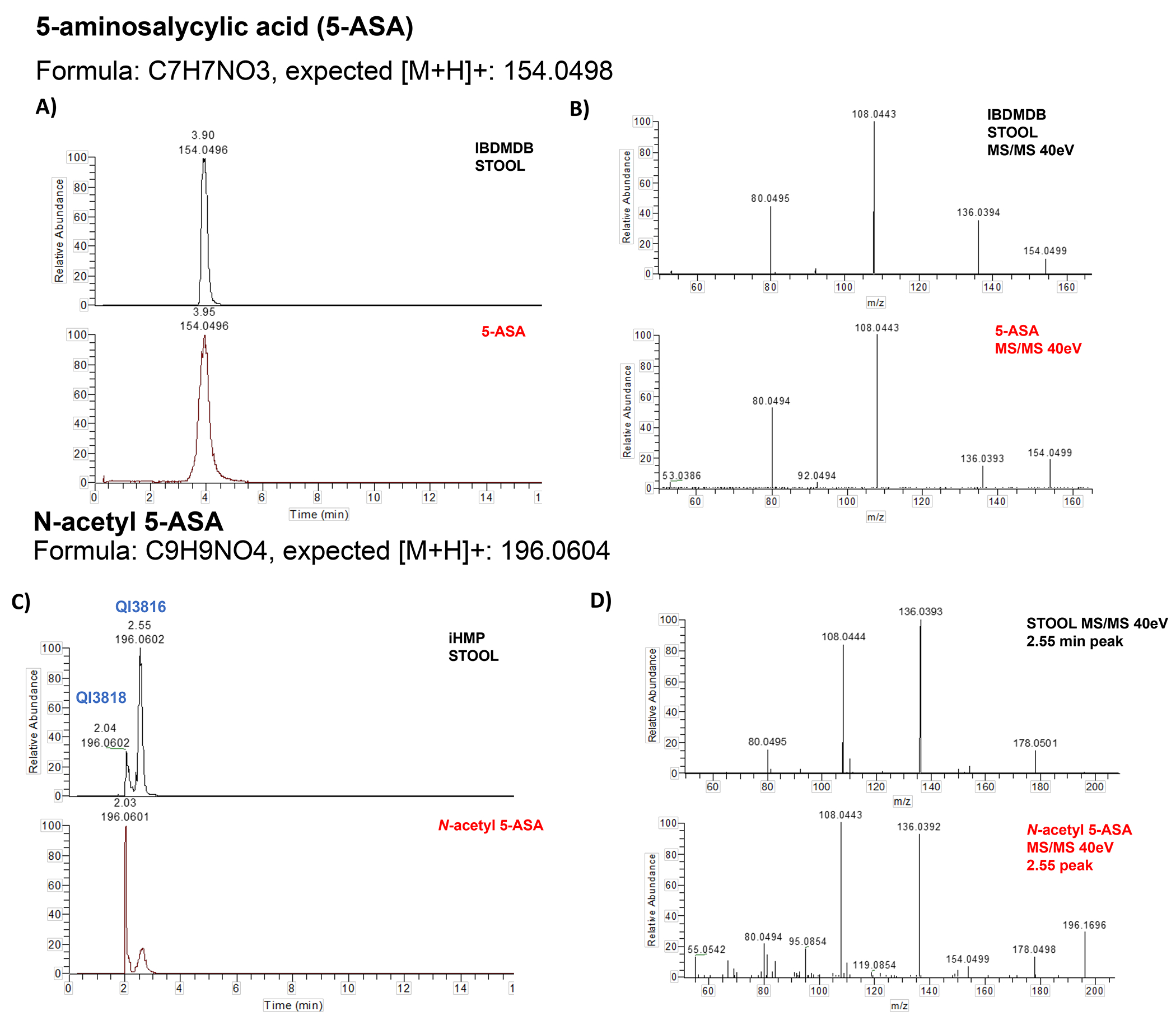
Two separate standards for 5-ASA (SIGMA catalog PHR1060 and 18858) confirmed the identity of the 5-ASA peak in the IBDMDB (m/z: 154.0502, RT: 3.83 min) through retention time (panel A) and spectral matching (panel B). A standard for N-acetyl 5-ASA (Cayman Catalogue 27618) produced two peaks which matched two peaks in the IBDMDB (QI3818 and QI3816) (panel C). The later eluting peak (QI3816), was more abundant in the IBDMBD stool, correlated better with 5-ASA (Pearson’s correlation r=0.89 vs r=0.60), and had excellent MS/MS spectral matching (panel D). Further still, levels of QI3816 perfectly discriminate 5-ASA users from non-users (c-statistic 0.99).
Extended Table 1. Characteristics of 5-ASA users vs non-users in the IBDMDB.
IBD users of 5-ASA were more likely to have ulcerative colitis. Overall, rates of hospitalization and history of bowel surgery were also lower for 5-ASA users compared to non-users.
| Non-users (n=34) | Users (n=45) | p-value | |
|---|---|---|---|
| Age at consent, mean (SD) | 27.7 (17.4) | 26.7 (17.1) | 0.81 |
| Age at diagnosis, mean (SD) | 22.1 (14.2) | 20.9 (11.0) | 0.68 |
| Male (%) | 17 (50) | 21 (47) | 0.95 |
| UC (%) | 6 (18) | 24 (53) | 0.003 |
| Race/ethnicity (%) | |||
| White | 30 (88) | 40 (89) | 0.13 |
| Other | 4 (12) | 5 (11) | |
| Prior bowel surgery (%) | 0.11 | ||
| Yes | 7 (21) | 4 (9) | |
| Antibiotic use (%) | 14 (41) | 25 (56) | 0.30 |
| Bonded 5-ASA use (%)a | 0 (0) | 4 (9) | 0.20 |
| Hospitalized during the study (%) | 13 (38) | 7 (15) | 0.04 |
| Dysbiosisb | 21 (62) | 19 (42) | 0.14 |
Bonded 5-ASA formulations include sulfasalazine, balsalazide, and olsalazine
As previously defined in the HMP2, Methods22
Extended data Table 2.
Extended Table 3. 5-ASA use is associated with large-scale differences across individual gut microbial species abundance in patients with IBD.
Significant associations (n=24, FDR q < 0.25) of 5-ASA use and microbial species. All models included each participant’s identifier as random effects and simultaneously adjusted for age, disease type, use of antibiotic, and dysbiosis status.
| Species | Beta | P | FDR |
|---|---|---|---|
| Gemmiger formicilis | −0.00518 | 0.000119 | 0.007644138 |
| Bifidobacterium longum | 0.024185 | 0.000221 | 0.009487238 |
| Dielma fastidiosa | 0.005456 | 0.000372 | 0.011991481 |
| Intestinimonas butyriciproducens | −0.0118 | 0.000763 | 0.015567799 |
| Lachnospira pectinoschiza | −0.02995 | 0.000915 | 0.015567799 |
| Faecalibacterium prausnitzii | 0.077469 | 0.000987 | 0.015567799 |
| Fusicatenibacter saccharivorans | −0.02626 | 0.001075 | 0.015567799 |
| Phascolarctobacterium faecium | −0.01404 | 0.001086 | 0.015567799 |
| Lactobacillus rogosae | −0.00461 | 0.002316 | 0.029877308 |
| Eubacterium eligens | 0.032305 | 0.003037 | 0.035614424 |
| Butyricimonas synergistica | −0.00486 | 0.003329 | 0.035790148 |
| Ruminococcus bromii | −0.02084 | 0.003658 | 0.036294803 |
| Paraprevotella xylaniphila | −0.00815 | 0.004008 | 0.03693114 |
| Turicimonas muris | −0.0055 | 0.006253 | 0.053774509 |
| Anaerostipes hadrus | 0.021856 | 0.011292 | 0.091043147 |
| Ruthenibacterium lactatiformans | −0.01123 | 0.013849 | 0.105092694 |
| Parasutterella excrementihominis | −0.0171 | 0.015598 | 0.11178451 |
| Anaerotignum lactatifermentans | −0.00775 | 0.018159 | 0.123290076 |
| Dialister invisus | −0.01012 | 0.024624 | 0.158826559 |
| Anaerotruncus colihominis | −0.00414 | 0.028712 | 0.173321541 |
| Bilophila wadsworthia | −0.00699 | 0.030248 | 0.173321541 |
| Akkermansia muciniphila | −0.0283 | 0.03201 | 0.173321541 |
| Roseburia inulinivorans | −0.0179 | 0.032246 | 0.173321541 |
| Proteobacteria bacterium CAG_139 | −0.01021 | 0.045073 | 0.232574975 |
Extended Table 4. M/z ratios and retention times for 5-ASA in the IBDMDB and PRISM cohorts.
Identity of N-acetyl 5-ASA in PRISM and for N-propionyl 5-ASA and N-butyryl 5-ASA in the IBDMDB and PRISM cohorts were inferred using mass and retention time matching coupled with known mass shifts from functional groups. As further confirmation of these two possible 5-ASA derivatives, we searched publicly available MS2 data from the Global Natural Product Social (GNPS) database (MassIVE MSV000084556) to generate molecular co-occurrence networks, inputting the known spectra of N-acetyl 5-ASA (46). In this analysis, we similarly found two unlabeled nodes linked with N-acetyl 5-ASA which matched the m/z of the compounds we identified, as well as a third node with an m/z of 238.11 which may correspond to yet another derivative of 5-ASA that has not been described, likely a 5-carbon acyl group plus 5-ASA given the additional mass shift (+12) from butyryl CoA.
| PRISM | IBDMDB | |||
|---|---|---|---|---|
| Compound | M/Z | Retention time | M/Z | Retention time |
| 5-ASA | 154.0493 | 3.98 | 154.0502 | 3.83 |
| N-butyryl 5-ASA | 224.0909 | 2.32 | 224.0922 | 2.24 |
| N-acetyl 5-ASA | 196.0599 | 2.97 | 196.0609 | 2.81 |
| N-propionyl 5-ASA | 210.0754 | 2.32 | 210.0764 | 2.52 |
Extended data Table 5.
Extended data Table 6.
Extended Table 7.
Data processing refinement statistics and quality of the FcTHL model.
| Protein Characteristics | |
|---|---|
| Wavelength | 0.9792 |
| Resolution range | 73.46 - 1.89 (1.958 - 1.89) |
| Space group | P 1 |
| Unit cell | 75.23 81.46 160.19 95.61 100.45 108.27 |
| Total reflections | 500387 (50866) |
| Unique reflections | 263887 (26397) |
| Multiplicity | 1.9 (1.9) |
| Completeness (%) | 94.02 (93.71) |
| Mean I/sigma(I) | 7.10 (0.81) |
| Wilson B-factor | 39.07 |
| R-merge | 0.0697 (1.012) |
| R-meas | 0.09856 (1.432) |
| R-pim | 0.0697 (1.012) |
| CC1/2 | 0.99 (0.391) |
| CC* | 0.997 (0.75) |
| Reflections used in refinement | 263708 (26331) |
| Reflections used for R-free | 13239 (1334) |
| R-work | 0.1758 (0.3703) |
| R-free | 0.2087 (0.3946) |
| CC(work) | 0.964 (0.695) |
| CC(free) | 0.952 (0.649) |
| Number of non-hydrogen atoms | 25714 |
| macromolecules | 24521 |
| ligands | 0 |
| solvent | 1193 |
| Protein residues | 3364 |
| RMS(bonds) | 0.013 |
| RMS(angles) | 1.18 |
| Ramachandran favored (%) | 96.87 |
| Ramachandran allowed (%) | 3.07 |
| Ramachandran outliers (%) | 0.06 |
| Rotamer outliers (%) | 1.00 |
| Clashscore | 4.15 |
| Average B-factor | 50.50 |
| macromolecules | 50.58 |
| solvent | 48.76 |
| Number of TLS groups | 34 |
Statistics for the highest-resolution shell are shown in parentheses.
Extended Table 8. Presence of 5-ASA metabolizing acetyltransferases are associated with steroid use in the HMP2.
Odds ratios with 95% CI are presented. Analyses were adjusted for age, sex, disease type (CD vs. UC), host acetylation phenotype, and smoking status.
| Feature | ||
|---|---|---|
| Genes | Absent | Present |
| C7H1G6 | 1 (referent) | 2.81 (1.68-4.68) |
| R5CY66 | 1 (referent) | 2.88 (1.66-5.00) |
| R6CZ24 | 1 (referent) | 2.58 (1.40-4.77) |
| T5S060 | 1 (referent) | 3.24 (1.63-6.42) |
| Host factors | ||
| Disease type | UC | CD |
| 1 (referent) | 3.11 (1.85-5.21) | |
| Age, y | 0.92 (0.89-0.94) |
Extended Table 9. Characteristics of the study population selected from the SPARC IBD cohort.
Compared to the IBDMDB, given that SPARC IBD is a cohort of adult patients, participants were older. They were also more likely to have UC and were less likely to be male. The prevalence of acetyltransferases that were linked with treatment failure was similar in both cohorts.
| Users (n=208) | |
|---|---|
| Age at consent, mean (SD) | 45.5 (15.1) |
| Male (%) | 88 (42.3%) |
| IBD subtype | |
| UC (%) | 140 (67.3) |
| CD (%) | 67 (32.2) |
| IBD-U a (%) | 1 (0.4) |
| Prevalence of acetyltransferases, % | |
| C7H1G6 | 44.0 |
| R5CY66 | 20.4 |
| R6CZ24 | 7.2 |
| T5S060 | 18.8 |
IBD-undifferentiated
Extended Table 10. Presence of 5-ASA inactivating acetyltransferases.
Gene prevalence (%) varies across participants with IBD, but does not vary significantly by 5-ASA status (GLMM p≥0.07), arguing for effect modification of the effects of 5-ASA on prevention of disease relapse. Among participants without IBD prevalence rates of these enzymes also appear similar.
| 5-ASA users | 5-ASA nonusers | Non-IBD | |
|---|---|---|---|
| UniRef90 ID | |||
| C7H1G6 | 47.8 | 52.3 | 67.0 |
| R5CY66 | 17.4 | 20.3 | 43.7 |
| R6CZ24 | 20.7 | 14.3 | 28.5 |
| T5S060 | 14.1 | 9.2 | 12.3 |
Extended data Table 11.
Extended data Table 12.
Supplementary Material
Acknowledgements & Funding:
We express our sincere thanks to the members of the Huttenhower lab for their editorial assistance. The computations in this paper were run on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University or on the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School. See https://it.hms.harvard.edu/our-services/research-computing for more information.
Funding:
National Institutes of Health grant R35 CA253185 (ATC)
National Institutes of Health grant R24 DK110499-01A1 (CH)
National Institutes of Health grant T32HL008633-36 (JRM)
National Institutes of Health grant 1K23DK125838 (LHN)
American College of Gastroenterology (RSM)
Crohn’s and Colitis Foundation (RSM, LHN)
American Gastroenterological Association (LHN, WM)
Pfizer Ulcerative Colitis Grant (ATC)
Stuart and Suzanne Steele MGH Research Scholarship (ATC)
Howard Hughes Medical Institute (EPB)
National Science Foundation Postdoctoral Research Fellowship in Biology grant 1907240 (NRG)
National Science Foundation Alan T Waterman Award 2038059 (JRM)
Linde Family Foundation (SDP)
Novartis Institute for Biomedical Research (SDP)
Doris Duke Charitable Foundation (SDP)
National Institutes of Health grants P30 GM124165 (SDP).
National Institutes of Health-ORIP HEI grant S10OD021527 (SDP).
Footnotes
Competing interests: CH is a consultant to Zoe Ltd (“Zoe”) and is on the Scientific Advisory Boards of Seres Therapeutics and Empress Therapeutics. ATC has been an investigator on a clinical study supported by Zoe. Other authors declare that they have no competing interests. R.S.M., J.R.M., E.P.B, A.T.C., and C.H. have filed a provisional patent application (XXXXX).
Code availability: All analysis code can be accessed at https://github.com/drraajmehta/hmp2_5asa for purposes of reproducibility.
Data availability:
All multi’omics and participant data from the HMP2 used in this analysis are available at http://IBDMDB.org. Access to PRISM data may be available after contact of rxavier@molbio.harvard.edu. The SPARC IBD data are available upon approved application to Crohn’s & Colitis Foundation IBD Plexus (https://www.crohnscolitisfoundation.org/ibd-plexus).
References
- 1.Plichta DR, Graham DB, Subramanian S & Xavier RJ Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell 178, 1041–1056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ham M & Moss AC Mesalamine in the treatment and maintenance of remission of ulcerative colitis. Expert Rev Clin Pharmacol 5, 113–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klag T, Stange EF & Wehkamp J Management of Crohn’s disease – are guidelines transferred to clinical practice? United European Gastroenterology Journal 3, 371–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford AC et al. Efficacy of 5-Aminosalicylates in Ulcerative Colitis: Systematic Review and Meta-Analysis. Official journal of the American College of Gastroenterology | ACG 106, 601–616 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Ford AC et al. Efficacy of 5-Aminosalicylates in Crohn’s Disease: Systematic Review and Meta-Analysis. Official journal of the American College of Gastroenterology | ACG 106, 617–629 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Javdan B et al. Personalized Mapping of Drug Metabolism by the Human Gut Microbiome. Cell 181, 1661–1679.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R & Goodman AL Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koppel N, Maini Rekdal V & Balskus EP Chemical transformation of xenobiotics by the human gut microbiota. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balaich J et al. The human microbiome encodes resistance to the antidiabetic drug acarbose. Nature 600, 110–115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haiser HJ et al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341, 295–298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rekdal VM, Bess EN, Bisanz JE, Turnbaugh PJ & Balskus EP Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haiser HJ, Seim KL, Balskus EP & Turnbaugh PJ Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 5, 233–238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JWJ et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe 29, 1294–1304.e4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forslund SK et al. Combinatorial, additive and dose-dependent drug–microbiome associations. Nature 600, 500–505 (2021). [DOI] [PubMed] [Google Scholar]
- 15.van Hogezand RA et al. Bacterial acetylation of 5-aminosalicylic acid in faecal suspensions cultured under aerobic and anaerobic conditions. Eur J Clin Pharmacol 43, 189–192 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Dull BJ, Salata K & Goldman P Role of the intestinal flora in the acetylation of sulfasalazine metabolites. Biochemical Pharmacology 36, 3772–3774 (1987). [DOI] [PubMed] [Google Scholar]
- 17.van Hogezand RA et al. Double-blind comparison of 5-aminosalicylic acid and acetyl-5-aminosalicylic acid suppositories in patients with idiopathic proctitis. Aliment Pharmacol Ther 2, 33–40 (1988). [DOI] [PubMed] [Google Scholar]
- 18.Sandborn WJ & Hanauer SB Systematic review: the pharmacokinetic profiles of oral mesalazine formulations and mesalazine pro-drugs used in the management of ulcerative colitis. Aliment Pharmacol Ther 17, 29–42 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Ireland A, Priddle JD & Jewell DP Comparison of 5-aminosalicylic acid and N-acetylaminosalicylic acid uptake by the isolated human colonic epithelial cell. Gut 33, 1343–1347 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deloménie C et al. Identification and functional characterization of arylamine N-acetyltransferases in eubacteria: evidence for highly selective acetylation of 5-aminosalicylic acid. J. Bacteriol 183, 3417–3427 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westwood IM et al. Expression, purification, characterization and structure of Pseudomonas aeruginosa arylamine N-acetyltransferase. Biochem J 385, 605–612 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beghini F et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafferty JD et al. Self-reported medication use validated through record linkage to national prescribing data. J Clin Epidemiol 94, 132–142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akobeng AK, Zhang D, Gordon M & MacDonald JK Oral 5-aminosalicylic acid for maintenance of medically-induced remission in Crohn’s disease. Cochrane Database of Systematic Reviews (2016) doi: 10.1002/14651858.CD003715.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neshich IA, Kiyota E & Arruda P Genome-wide analysis of lysine catabolism in bacteria reveals new connections with osmotic stress resistance. The ISME Journal 7, 2400–2410 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sell DR, Strauch CM, Shen W & Monnier VM 2-aminoadipic acid is a marker of protein carbonyl oxidation in the aging human skin: effects of diabetes, renal failure and sepsis. Biochem J 404, 269–277 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harary I Bacterial degradation of nicotinic acid. Nature 177, 328–329 (1956). [DOI] [PubMed] [Google Scholar]
- 29.Li J et al. Niacin ameliorates ulcerative colitis via prostaglandin D2-mediated D prostanoid receptor 1 activation. EMBO Mol Med 9, 571–588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franzosa EA et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nature Microbiology 4, 293 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Thompson KN, Huttenhower C & Franzosa EA Statistical approaches for differential expression analysis in metatranscriptomics. Bioinformatics 37, i34–i41 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devos D & Valencia A Practical limits of function prediction. Proteins: Structure, Function, and Bioinformatics 41, 98–107 (2000). [PubMed] [Google Scholar]
- 33.Fitzgerald CB et al. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics 19, 931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseaux C et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator–activated receptor-γ. J Exp Med 201, 1205–1215 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modis Y & Wierenga RK Crystallographic analysis of the reaction pathway of Zoogloea ramigera biosynthetic thiolase. J Mol Biol 297, 1171–1182 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Kim S et al. Redox-switch regulatory mechanism of thiolase from Clostridium acetobutylicum. Nat Commun 6, 8410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathieu M et al. The 1.8 Å crystal structure of the dimeric peroxisomal 3-ketoacyl-CoA thiolase of Saccharomyces cerevisiae: implications for substrate binding and reaction mechanism 11 Edited by R. Huber. Journal of Molecular Biology 273, 714–728 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Hyams JS et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): a multicentre inception cohort study. Lancet 393, 1708–1720 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ananthakrishnan AN Environmental Triggers for Inflammatory Bowel Disease. Curr Gastroenterol Rep 15, 302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ricart E et al. N-acetyltransferase 1 and 2 genotypes do not predict response or toxicity to treatment with mesalamine and sulfasalazine in patients with ulcerative colitis. Am. J. Gastroenterol 97, 1763–1768 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Yee J et al. The association between NAT2 acetylator status and adverse drug reactions of sulfasalazine: a systematic review and meta-analysis. Sci Rep 10, 3658 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lück H, Kinzig M, Jetter A, Fuhr U & Sörgel F Mesalazine pharmacokinetics and NAT2 phenotype. Eur J Clin Pharmacol 65, 47–54 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Ha CY, Newberry RD, Stone CD & Ciorba MA Patients with Late Adult Onset Ulcerative Colitis Have Better Outcomes than Those with Early Onset Disease. Clin Gastroenterol Hepatol 8, 682–687.e1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huberts DHEW & van der Klei IJ Moonlighting proteins: an intriguing mode of multitasking. Biochim Biophys Acta 1803, 520–525 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Hong J, Park W, Seo H, Kim I-K & Kim K-J Crystal structure of an acetyl-CoA acetyltransferase from PHB producing bacterium Bacillus cereus ATCC 14579. Biochemical and Biophysical Research Communications 533, 442–448 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Maier L et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallace BD et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 330, 831–835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Vos M et al. Concentrations of 5-ASA and Ac-5-ASA in human ileocolonic biopsy homogenates after oral 5-ASA preparations. Gut 33, 1338–1342 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references:
- 49.Lohman BK, Weber JN & Bolnick DI Evaluation of TagSeq, a reliable low-cost alternative for RNAseq. Molecular Ecology Resources 16, 1315–1321 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Suzek BE et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oksanen J et al. The vegan package. Community ecology package 10, 719 (2007). [Google Scholar]
- 52.Wickham H ggplot2: elegant graphics for data analysis. (springer, 2016). [Google Scholar]
- 53.Wang M et al. Mass spectrometry searches using MASST. Nature biotechnology 38, 23–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aron AT et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat Protoc 15, 1954–1991 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Mehta RS et al. Dietary Patterns and Risk of Colorectal Cancer: Analysis by Tumor Location and Molecular Subtypes. Gastroenterology 152, 1944–1953.e1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bar N et al. A reference map of potential determinants for the human serum metabolome. Nature 588, 135–140 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Groemping U & Matthias L Package ‘relaimpo’. Relative Importance of Regressors in Linear Models; R Fundation for Statstical Computing: Vienna, Austria: (2021). [Google Scholar]
- 58.Bustion A, Agrawal A, Turnbaugh PJ & Pollard KS A novel in silico method employs chemical and protein similarity algorithms to accurately identify chemical transformations in the human gut microbiome. 2022.08.02.502504 Preprint at 10.1101/2022.08.02.502504 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franzosa EA et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. U.S.A 111, E2329–2338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinheiro J et al. Package ‘nlme’. Linear and nonlinear mixed effects models, version 3, (2017). [Google Scholar]
- 61.Kenny DJ et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 28, 245–257.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Research 49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson JD, Higgins DG & Gibson TJ CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research 22, 4673–4680 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henikoff S & Henikoff JG Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences 89, 10915–10919 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hunter S et al. InterPro: the integrative protein signature database. Nucleic Acids Research 37, D211–D215 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitts PA et al. Assembly: a resource for assembled genomes at NCBI. Nucleic Acids Res 44, D73–80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Dawwas GK et al. Prevalence and Factors Associated With Fecal Urgency Among Patients With Ulcerative Colitis and Crohn’s Disease in the Study of a Prospective Adult Research Cohort With Inflammatory Bowel Disease. Crohn’s & Colitis 360 3, otab046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raffals LE et al. The Development and Initial Findings of A Study of a Prospective Adult Research Cohort with Inflammatory Bowel Disease (SPARC IBD). Inflammatory Bowel Diseases (2021) doi: 10.1093/ibd/izab071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carey VJ, and 4.4), T. S. L. (R port of versions 3 13, src/d*), C. M. (LINPACK routines in & updates), B. R. (R port of version 4 13 and. gee: Generalized Estimation Equation Solver. (2022).
- 71.Viechtbauer W Conducting Meta-Analyses in R with the metafor Package. Journal of Statistical Software 36, 1–48 (2010). [Google Scholar]
- 72.García-Closas M et al. NAT2 slow acetylation and GSTM1 null genotypes increase bladder cancer risk: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet 366, 649–659 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chan SL et al. Association and clinical utility of NAT2 in the prediction of isoniazid-induced liver injury in Singaporean patients. PLoS One 12, e0186200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selinski S et al. Genotyping NAT2 with only two SNPs (rs1041983 and rs1801280) outperforms the tagging SNP rs1495741 and is equivalent to the conventional 7-SNP NAT2 genotype. Pharmacogenetics and Genomics 21, 673–678 (2011). [DOI] [PubMed] [Google Scholar]
- 75.XDS Package. https://xds.mr.mpg.de/.
- 76.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coot. https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/.
- 78.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Ding RW et al. Pharmacokinetics of nicotinic acid – salicylic acid interaction. Clinical Pharmacology & Therapeutics 46, 642–647 (1989). [DOI] [PubMed] [Google Scholar]
- 80.Santoru ML et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep 7, 9523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All multi’omics and participant data from the HMP2 used in this analysis are available at http://IBDMDB.org. Access to PRISM data may be available after contact of rxavier@molbio.harvard.edu. The SPARC IBD data are available upon approved application to Crohn’s & Colitis Foundation IBD Plexus (https://www.crohnscolitisfoundation.org/ibd-plexus).