

Abstract
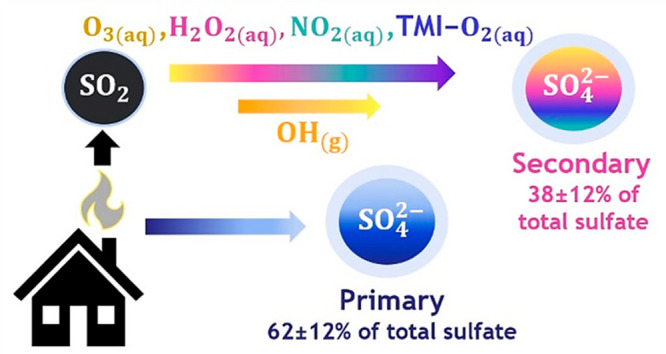
Within and surrounding high-latitude cities, poor air quality disturbs Arctic ecosystems, influences the climate, and harms human health. The Fairbanks North Star Borough has wintertime particulate matter (PM) concentrations that exceed the Environmental Protection Agency’s (EPA) threshold for public health. Particulate sulfate (SO42–) is the most abundant inorganic species and contributes approximately 20% of the total PM mass in Fairbanks, but air quality models underestimate observed sulfate concentrations. Here we quantify sulfate sources using size-resolved δ34S(SO42–), δ18O(SO42–), and Δ17O(SO42–) of particulate sulfate in Fairbanks from January 18th to February 25th, 2022 using a Bayesian isotope mixing model. Primary sulfate contributes 62 ± 12% of the total sulfate mass on average. Most primary sulfate is found in the size bin with a particle diameter < 0.7 μm, which contains 90 ±5% of total sulfate mass and poses the greatest risk to human health. Oxidation by all secondary formation pathways combined contributes 38 ± 12% of total sulfate mass on average, indicating that secondary sulfate formation is inefficient in this cold, dark environment. On average, the dominant secondary sulfate formation pathways are oxidation by H2O2 (13 ± 6%), O3 (8 ± 4%), and NO2 (8 ± 3%). These findings will inform mitigation strategies to improve air quality and public health in Fairbanks and possibly other high-latitude urban areas during winter.
Keywords: air quality, aerosols, stable isotopes, sulfate, particulate matter, sulfur oxidation
Short abstract
Sulfur and oxygen isotopes show that primary sulfur emissions from residential fuel oil can explain the majority of the particulate sulfate in Fairbanks, Alaska.
1. Introduction
Particulate matter smaller than or equal to 2.5 microns in diameter (PM2.5) causes cardiovascular and respiratory diseases and is responsible for over 4 million premature deaths per year globally.1−4 In urban environments, sulfate is a major contributor to particulate mass, accounting for 20% of PM2.5 on average.5,6 Air quality models often underestimate sulfate concentrations in polluted regions on the order of 2–6× lower than the observed ambient concentrations, suggesting unaccounted for primary sulfate emissions or secondary sulfate formation mechanisms in these environments.7−9
The Fairbanks North Star Borough (FNSB) is classified by the US Environmental Protection Agency (EPA) as a “serious” nonattainment area because the city exceeds the 24 h national standard of 35 μg m–3 every winter. The American Lung Association ranks Fairbanks in the top three worst cities for 24 h particle pollution, averaging 37 days where PM2.5 concentrations exceed 35.5 μg·m–1 per year between 2017–2021.10 Pollution events in Fairbanks often occur at temperatures less than −20 °C and are exacerbated by strong temperature inversions, low winds, and minimal vertical mixing.11,12 Most particulate matter (PM) mass in Fairbanks is organic PM from domestic woodburning (19–52%), gasoline exhaust (16–18%), and diesel (9–14%).13−16 After woodsmoke, sulfate is the second largest contributor to PM2.5 mass (15–33%).14−16 Community multiscale air quality (CMAQ) model simulations underestimate sulfate concentrations during winter in FNSB by 67%.17 This discrepancy makes it difficult to use the model to develop effective mitigation measures for reducing atmospheric sulfate.
Most studies conducted in Fairbanks have identified residential fuel oil as the dominant source of sulfur PM in Fairbanks.14−16,18 Wood-fired space heating is a minor source of atmospheric sulfur in Fairbanks. Emissions inventories in Fairbanks estimate that woodburning produces 4% as much SO2 as fuel oil in tons·day–1 for space heating sources.19 Coal is not considered an important sulfate source even though several coal-fired power plants are within the Fairbanks nonattainment area.19 These plants burn low-sulfur coal from the Usibelli mine (sulfur content < 0.20%).19 Most importantly, their smokestacks often emit above the stable and shallow inversion layer (<20 meters) during winter pollution events.11,12,19 This suggests that there is a minimal contribution of coal-derived sulfate on the highest pollution days in Fairbanks and that ground-level, residential fuel oil combustion is the major sulfur source.11,12,20
Primary sulfate is a sulfate that is emitted from a plume fully oxidized.21 It is parameterized in emissions inventories and air quality models using a bottom-up approach, where most sulfur is emitted in the form of gas-phase sulfur dioxide (SO2), and 1–5% is emitted directly as primary sulfate.1,22,23 Current CMAQ modeling in Fairbanks uses a primary sulfate emission factor of 0.5%. This value is calculated from speciation profiles of Fairbanks heating oil and the ratio of primary sulfate per gallon of fuel oil burned, where the latter is largely based on literature published between 1960–1980.19,23 The accuracy of this emission factor is limited by the paucity of both laboratory and ambient primary sulfate observations.
The main formation pathways of secondary sulfur PM in polluted environments are gas-phase oxidation of SO2 by OH and aqueous-phase oxidation of dissolved SO2 in cloud and aerosol particles by hydrogen peroxide (H2O2), ozone (O3), nitrogen dioxide (NO2), and oxygen (O2) (via transition metal ion catalysis (TMI-O2)) to form sulfate.7,24,25 Additionally, hydroxymethanesulfonate (HMS), an adduct of sulfite/bisulfite (SO32–/HSO3–) and formaldehyde (HCHO) that is present in PM, can be a major organosulfur species in wintertime haze events in HCHO-rich environments.26−29
Factors that influence PM sulfur formation in both cloud drops or aerosol particles include oxidant concentrations, liquid water content, pH, and ionic strength.3,7,18,25,30−32 Due to the short duration of daylight during winter in Fairbanks (4–6 h day–1), photochemically-produced oxidant abundances (OH and H2O2) may be low.33 Additionally, O3 is not abundant during ultrapolluted periods because it is titrated by NOx at the surface.11 Due to low oxidant abundances and cloud liquid water, it has been hypothesized that sulfur aerosol production in Fairbanks occurs via multiphase and heterogeneous oxidation of SO2 in aerosol liquid water (ALW) with high ionic strength.7,18,20,25,34−36 The high ionic strength of ALW and extremely low temperatures affect the solubility of gaseous SO2, the partitioning of S(IV) species (SO2 + HSO3– + SO32–), and the rate constants of aqueous oxidation.7,18,32 Finally, pH is important for O3, NO2, and TMI-O2 oxidation because it affects the partitioning of S(IV) species and metal solubility.34 The O3 oxidation pathway is only significant at pH > 5–6.37,38 NO2 oxidation can occur at lower pH but ultimately exhibits similar pH sensitivity as O3 since the reaction rate increases as pH and SO2 solubility increase.30 Conversely, the TMI-O2 pathway by both Fe(III) and Mn(II) requires acidic conditions since metal solubility increases as pH decreases.37 These reactions produce sulfate with a specific oxygen isotopic composition and fractionate sulfur isotopes, resulting in isotopic composition that reflects the sulfur source and sulfate formation pathways.
Oxygen isotopes reveal the prevalence of primary sulfate and characterize the dominant secondary oxidation pathways. Primary sulfate has the heaviest δ18O signature (δ18O(SO42– = +23.5 ± 0.3‰) because it is composed of molecular oxygen from combustion.39−41 Secondary sulfate is lighter than primary sulfate because SO2 exchanges its oxygen atoms with both water vapor and liquid water which has a relatively light oxygen isotopic composition (<0‰), with the most depleted δ18O values in the Northern Hemisphere at high latitudes (−20 to −30‰).42−45 Thus, the δ18O of emitted SO2 does not retain the oxygen composition of the sulfur source due to rapid isotopic exchange between SO2 and liquid and vapor H2O in the atmosphere.42,43 Δ17O(SO42–) refers to the enrichment of δ17O(SO42–) relative to δ18O(SO42) (eq 1) and has been used in many studies to estimate the importance of H2O2 (Δ17O(SO42–) = +0.8‰) and O3 (Δ17O(SO42–) = +9.8‰) oxidation in the atmosphere.25,34,46−50
| 1 |
δ34S(SO42–) measurements constrain the contribution of different secondary sulfate formation pathways because sulfur isotopes fractionate during the oxidation of SO2 to sulfate. Sulfur isotope fractionation factors are unique to a specific oxidation pathway and thus the extent of fractionation is sensitive to the oxidants involved.51 There are several factors that determine the sulfur isotopic signature: (1) the source signature of SO2 upon emission, (2) the ambient temperature during oxidation, (3) the oxidation pathway, and (4) the sulfur oxidation ratio (SOR) or degree of sulfate formation relative to its precursors.52 We present sulfur and oxygen isotope measurements of atmospheric sulfate collected during the Alaskan Layered Pollution and Chemical Analysis (ALPACA) field campaign in Fairbanks, AK in January and February 2022. We use these observations to quantify primary and secondary sulfate sources and show that primary sulfate is the dominant contributor to particulate sulfate.
2. Methods
2.1. Filter Sample Collection in Fairbanks and Gas-Phase Measurements of SO2 and O3
Quartz filters (TE-QMA and TE-230-QZ) were rinsed with 18 MΩ·cm water and pre-combusted at 500 °C for 8 h before being wrapped in aluminum foil in airtight polyethylene bags prior to the field campaign. A Volumetric Flow Controlled Particulate Sampling System (TE-5170) with a 4-stage cascade impactor (TE-230) was used to collect 24 h size-resolved aerosol samples at Fairbanks Community Technical College (CTC) (64.84064° N, 147.72677° W) between January 17th to February 25th, 2022. Size-resolved bins were determined by calculating the particle size cutoff (Dp,50) at 50% collection efficiency using the corrected flow rate in each sample (equation S1 in the Supporting Information). For each collection period, filters were combined to form three-size bins: particle diameters <0.7 μm (PM<0.7), 0.7–2.5 μm (PM0.7–2.5), and 2.5–10 μm (PM2.5–10). Both PM0.7 and PM0.7–2.5 fall within the EPA-regulated fine particle range deemed PM2.5, but they are analyzed in separate size bins here.
Filter samples were collected daily between 9:30 AM to 9:00 AM the following day, except for one exceptionally polluted period between January 31st to February 3rd when the filters were changed twice per day at approximately 9:30 AM and 5 PM local time (Table S2). Prior to each TE-5170 calibration, a 1 min blank with no flow through the air sampler was collected to yield four sets of blanks, for a total of 16 slotted filter blanks and 4 backup filter blanks. In-situ gas-phase SO2 (Thermo Scientific 43C) and O3 (Thermo Scientific 49C) were measured from an inlet at 3 meters above the valley floor at the CTC site at 1 min resolution, which was used to calculate the average SO2 and O3 concentrations during sample collection.
Ten snow samples were collected throughout the campaign to measure δ18O(H2Osnow). Snow samples were collected from the surface of undisturbed snowpack in 50 mL Nalgene bottles approximately 15 feet from the TE-5170 (Table S5).
Two filter samples were collected for 1 week each at the Poker Flat Research range (65.1256° N, 147.4919° W), a relatively clean site 46 km north of Fairbanks, to represent a two-week average of background sulfate. Atmospheric particles were collected using a high-volume sampler (Digitel, DH77, TSP inlet, 1 m3 min–1) on pre-combusted quartz filters (Whatman 150 nm diameter).
2.2. Ion Chromatography of SO42–, S(IV), and Hydroxymethanesulfonate (HMS)
Hydroxymethanesulfonate (HMS), non-HMS S(IV), and sulfate (SO42–) concentrations were measured in a Metrostep A Supp-5 ion chromatograph (IC) using a low concentration (1.0 mM NaHCO3 and 3.2 mM Na2CO3) isocratic elution method, as described by Campbell et al. (2022). With this IC configuration, non-HMS S(IV) (HSO3–, and SO32–) and HMS have identical retention times and were distinguished by running two aliquots with and without the addition of H2O2. Non-HMS S(IV) is oxidized by H2O2 to form sulfate, while HMS resists oxidation by H2O2 and remains intact in the aqueous phase.53 Ambient concentrations of SO42–, non-HMS S(IV), and HMS from the filter samples and mean SO2 concentration during each filter sampling period were used to calculate the sulfur oxidation ratio (SOR) for each filter sample. The SOR represents the number of moles of SO42– formed relative to the total moles of sulfur species and is an indicator of the degree of oxidation of SO2 to sulfate aerosol (eq 2):
| 2 |
where SO2 is in mols·m–3, S(IV) refers to total S(IV), including HMS in mols·m–3, and SO42– represents all S(VI) species (including H2SO4, HSO4–, and SO4=) in mols·m–3.
2.3. Isotope Measurements
For isotope analysis at the University of Washington, samples (including field blanks) were extracted into 18 MΩ·cm Millipore water and then filtered through a 0.2 μm poly(ether sulfone) (PES) syringe filter to remove insoluble species. Due to insufficient PM mass for isotopic analysis, PM0.7–2.5 and PM2.5–10 measurements combined several consecutive days of samples from 10 periods, which are detailed in Table S2 and indicated in Figure 1. Isotope samples were prepared for silver salt pyrolysis as described in Schauer et al. and Geng et al.54,55 Briefly, the filtrate was neutralized by converting anions to sodium form with an offline cation exchange resin (AG 50W-X8 Resin from Bio-Rad). This converts sulfuric acid (H2SO4) to sodium sulfate (Na2SO4), which prevents sample loss due to evaporation. This step is followed by the removal of soluble organics by adding 30% H2O2 and drying in a MiVAc Duo concentrator. Sulfate was separated from other ions in the sample matrix in a Dionex ICS-2000 before being converted to Ag2SO4 using Ag+-charged cation-exchange resin, as described in Geng et al.55
Figure 1.
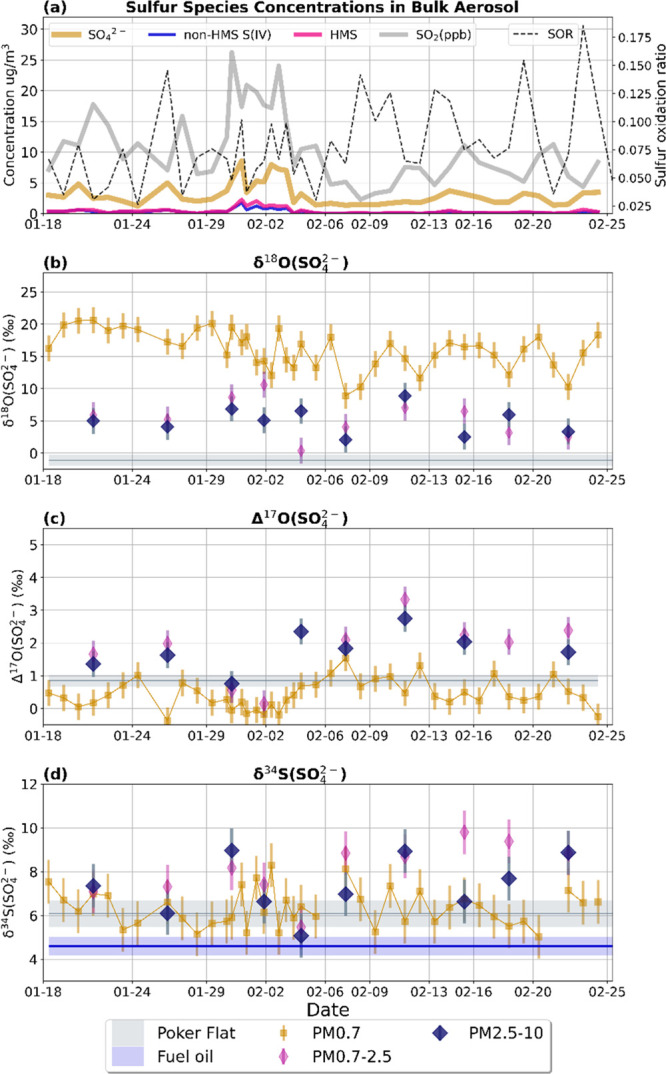
Time series of sulfur species concentrations (a), δ18O(b), δ18O (c), and δ18O (d) measurements. (a) Ambient concentrations of sulfur species including SO42– (gold), non-HMS S(IV) (blue), and HMS (magenta). The SOR for each sample is plotted with a black dashed line. Isotope observations in (b)–(d) are divided into three size bins: PM0.7 (gold squares), PM0.7–2.5 (narrow pink diamonds), and PM2.5–10 (wide blue diamonds). The error bars represent the propagated errors for each measurement. Daily PM0.7–2.5 and PM2.5–10 samples were combined into 10 periods as indicated by the vertical gridlines. A 2 week average of isotopic composition at Poker Flat is shown with gray shading in (b)–(d). The measured δ34S source signature for fuel oil is shown in blue in (d).
Oxygen isotope measurements were performed on a Finnegan MAT253 isotope ratio mass spectrometer using the same configuration as Geng et al.55 Oxygen isotope measurements were corrected for isotopic exchange with quartz and conversion of HMS and non-HMS S(IV) to sulfate during sample preparation (Supporting Information, section 1.4.1). Sulfur isotope composition was measured using a separate Finnegan MAT253 isotope ratio mass spectrometer with the same configuration as Jongebloed et al.56 δ34S values were normalized to the Vienna Canyon Diablo Troilite (VCDT) scale using four in-house reference materials that are regularly calibrated against the international reference materials IAEA-S-1, IAEA-S-3, and NBS-127. A sulfur isotope correction for sulfate formed from HMS and S(IV) during sample processing was estimated as described in Supporting Information, section 1.4.2. The δ34S composition of Fairbanks fuel oils #1 and #2 acquired during winter 2022 was measured by combusting 6 μL of fuel oil in a 50 μL tin capsule packed with tin powder. A source signature for δ34Semission was calculated using the δ34S measurements for fuel oil #1 (+3.7 ± 0.6‰) and fuel oil #2 (+4.9 ± 0.1‰) and weighing the values by sulfur content (896 and 2053 ppmv, respectively) and domestic use (33% and 67%, respectively; Table S4).19 This yielded a δ34Semission signature of +4.7 (±0.6)‰. The analytical error of the measurements (±0.8‰, ±0.2‰, and ±1.0‰ for δ18O, Δ17O, and δ34S, respectively) was estimated from duplicate sample analyses (performed on 30% of the Fairbanks samples) and replicate measurements of standards in quartz and silver capsules. The fully propagated errors, including isotopic corrections for the three measurements, are δ18O (±1.9‰), Δ17O (±0.4‰), and δ34S (±1.2‰).
2.4. Sulfate Source Apportionment Using a Bayesian Isotope Mixing Model
We developed an isotope mixing model to investigate the contributions of primary sulfate and five secondary sulfate formation pathways (H2O2, O3, TMI-O2, OH, and NO2). The model inputs are the δ18O(SO42–), Δ17O(SO42–), and δ34S(SO42–) observations. The δ18O(H2O) of Fairbanks snow (δ18O(H2O(precip)) was measured throughout the campaign with an average value of −24.8 ± 2.1‰ (Table S3). The relationship between measured δ18O(H2O(precip)) and ambient temperature was used to calculate the secondary δ18O(SO42–) source signatures for each sample (detailed in Supporting Information, sections 3.1 and 3.2). This represents a source of uncertainty, as the relationship between δ18O(H2O(precip)) and the temperature was not directly measured for each sulfate sample. For a given temperature, the error in δ18O(H2O(precip)) is ±0.35‰, which is the error in the intercept of Figure S14(b).
Table 1 summarizes the isotopic signatures used in the model (detailed in Supporting Information, section 3.3). Sulfate formed from the NO2 oxidation pathway has a light δ18O signature of −17.2 ± 1.6‰ because NO2SO3 decomposes and rapidly hydrolyzes to form SO42– and HONO.25,34,57 TMI-O2-derived sulfate is slightly heavier (−5.3 ± 1.2‰) as the fourth oxygen is from dissolved O2. The TMI-O2 path includes both the oxidation of inorganic S(IV) by Fe and Mn as well as by excited triplet states of brown carbon as these produce sulfate with the same O isotopic signature.24,58 Both H2O2 and O3 oxidation result in a heavier signature (δ18O(SO42–) = +10.0 ± 0.8 and +21.3 ± 1.2‰, respectively) because the oxidants themselves have relatively heavy δ18O values (+22 to +52‰ and +130‰, respectively).59,60 For H2O2, the oxidant supplies two of the four oxygen atoms of sulfate, leaving a smaller contribution from isotopically light water.50 The H2O2 path includes hydrogen peroxide formed in the particle phase and gas phase; however, we expect the gas-phase path to be minor during the most polluted periods because the very high NOx observed during the campaign may suppress formation of H2O2(g).61,62
Table 1. δ18O(SO42–), Δ17O(SO42–), and δ34S(SO42–) Isotopic Signatures Used in Mass Balance Equations.
| Pathway | δ18O(SO42–) (‰) avg ± 1σa | Δ17O(SO42–) (‰) | δ34S(SO42–) and ε34Sb (‰) avg ± 1σc |
|---|---|---|---|
| primary | +23.5 ± 0.3 | –0.34 | δ34Sprimary = +4.7 ± 0.6 |
| O3 | +21.3 ± 2.3 | +9.8 | ε34SO3 = +18.9 ± 0.6 |
| H2O2 | +9.8 ± 1.5 | +0.8 | ε34SH2O2 = +18.9 ± 0.6 |
| TMI-O2 | –5.6 ± 2.3 | –0.09 | ε34STMI-O2 = −1.2 ± 1.8 |
| NO2 | +17.5 ± 3.0 | 0.0 | ε34SNO2 = +1.0 ± 0.46565 |
| OH | –6.7 ± 2.1 | 0.0 | ε34SOH = +11.7 ± 0.03 |
Average ± 1σ reflects the range in δ18O(SO42–) signatures due to the temperature-dependence of water vapor and liquid water (see Supporting Information, section 1.3).
εoxidant = (α34oxidant – 1) × 1000 and α34oxidant = (34S/32S) products/(34S/32S) reactants.
For δ18O(SO42–) and Δ17O(SO42–), mass balance eqs 3 and 4 were used to represent the fractional contributions of the six sulfate formation pathways.
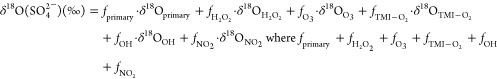 |
3 |
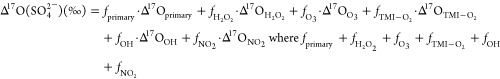 |
4 |
Equations 5–7 show the modeled sulfur isotope fractionation factors for SO2 oxidation by H2O2, O3, TMI-O2, and OH as a function of temperature51,50,51
| 5 |
| 6 |
| 7 |
The fractionation factor (εNO2) for SO2 oxidation by NO2 is +1.0‰.65 It should be noted that the average daily temperature in Fairbanks (−30 °C to 0.0 °C) was at times below the temperature ranges tested in laboratory measurements of δ34S fractionation factors (−25 °C for H2O2, O3, TMI-O2, OH,) and −7 °C for NO2.63,65 Yang et al. found that there was not a significant temperature difference for NO2 fractionation at temperatures <8 °C.65
Sulfur isotope fractionation in secondary sulfate formation is parametrized via Rayleigh distillation. eq 8 was used to calculate the δ34S isotope fractionation factors (ε) for secondary sulfate as a function of the isotopic primary source signature, average ambient temperature during sample collection (eqs 5–7), and the sulfur oxidation ratio (SOR). δ34Semission is assumed to be the same as primary sulfate (δ34Sprimary = +4.7(±0.6)‰) since sulfur isotope fractionation of fuel oil during high-temperature combustion is expected to be minimal.66 This is consistent with previous studies that assume primary sulfate from combustion retains the δ34S(SO42–) signature of the sulfur source.67−69 We assume that Fairbanks is a closed system, where long-range transport of biogenic and volcanic sulfur can be neglected. This assumption is further supported by prior literature showing that the pollution layer is often confined lower than 20 meters in Fairbanks with the highest PM2.5 concentrations below 3 meters.11,12,19,70 On-road mobile sampling performed by Robinson et al. (2023) found the lowest PM2.5 concentrations at the top of hills and asserted that residential neighborhoods were unequivocally the dominant PM source.
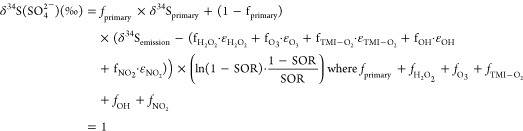 |
8 |
We used a Markov Chain Monte Carlo (MCMC) method to estimate the fractional contribution of primary sulfate and five secondary sulfate formation pathways in the isotope mixing model.71,72 We assumed that the observed δ18O(SO42–), Δ17O(SO42–), and δ34S(SO42–) follow a multivariate Gaussian distribution. We used a Dirichlet distribution as the prior for the fractional contributions such that each fraction is in the interval [0,1] and all fractions sum to 1. MCMC was used to calculate the fractional contributions of each sulfate formation pathway for each sample and estimate the uncertainty by providing a 95% confidence interval.
3. Results and Discussion
3.1. Ambient Sulfur Concentrations and Isotope Observations
Figure 1 shows the concentrations of sulfur species contributing to PM mass in Fairbanks between January 18th and February 25th, 2022. Sulfate concentrations range from 1 to 5 μg m–3 and contributes 85 (±9%) of total PM sulfur (sulfate + total S(IV), including HMS and non-HMS S(IV)) by mass throughout most of the campaign (Figure 1a). Non-HMS S(IV) and HMS on average contribute 6 (±3)% and 9 (±6)% of sulfur PM mass, respectively. Sulfur PM and SO2 concentrations were highest from January 29th and February 3rd, when temperatures averaged −25 °C and were as low as −30 °C. During this period, 23.5 h averaged sulfate concentrations were as high as 7.4 μg m–3 and averaged 5.6 μg m–3 (Figure 1a). The mass concentrations of S(IV) relative to sulfate were also highest during this period: combined, HMS + non-HMS S(IV) contributed 26% (±8%) sulfur PM mass. The average sulfur oxidation ratio is low during the ultrapolluted period (7 ± 2%) and was similar to the average SOR throughout the campaign (8 ± 4%; Figure 1a). Total sulfate concentrations are not correlated with SOR (r2 = 0.02, p-value > 0.1), but are positively associated with SO2 (r2 = 0.44, p-value < 0.01; Figure S13).
Figure 1 also shows the oxygen and sulfur isotopic measurements of sulfate in PM. The PM0.7 size bin has substantially higher δ18O and lower Δ17O (+16.2 ± 3.1‰ and +0.43 ± 0.42‰, respectively) compared to PM>0.7 (+5.2 ± 2.5‰ and +1.8 ± 1.2‰ for PM0.7–2.5 and PM2.5–10, respectively), suggesting a larger fraction of primary sulfate in the smaller size bin (Figures 1b,c). At times, PM0.7 δ18O and Δ17O observations approach the source signature of primary sulfate (+23.5‰ and −0.35‰, respectively; Table 1). δ34S(SO42–) of PM>0.7 (+7.7 ± 1.3‰) is more enriched than PM0.7 sulfate (+6.4 ± 1.1‰; Figure 1d). There is no relationship between surface temperature at 3 m and δ34S(SO42–) observations, suggesting that increased temperatures and enhanced vertical mixing does not have a detectable effect on sulfur isotope composition due to surface mixing of sulfur from coal-fired powerplants. This is consistent with findings in prior literature that suggest that source emissions from powerplants are likely minor contributors of atmospheric sulfate in Fairbanks.11,12,19 Bulk PM collected at Poker Flat has a δ34S = +6.1‰, which is similar to Fairbanks. Poker Flat has much lower δ18O values (−1.1‰) and enriched Δ17O (+0.85‰), suggesting that secondary sulfate is dominant at this clean location outside Fairbanks. HMS and non-HMS S(IV) concentrations at Poker Flat were below the detection limit.
Figure 2 shows relationships among observed δ18O, Δ17O, and δ34S in Fairbanks PM plotted adjacent to the isotopic source signatures of the six sulfate formation pathways. The Δ17O and δ18O measurements are negatively correlated (Figure 2a; r2 = 0.58, p-value < 0.01). The negative correlation is the opposite of the relationship observed in regions where secondary sulfate dominates due to enrichment in Δ17O and δ18O from O3 and H2O2 oxidation.40,73 A negative relationship between Δ17O and δ18O is expected when primary sulfate is significant, indicating a varying influence of primary (high δ18O, low Δ17O) and secondary (low δ18O, high Δ17O) sulfate. The positive correlation (r2 = 0.36, p-value < 0.01) between Δ17O and δ34S suggests enrichment in the sulfur isotopes composition with increasing secondary sulfate formation by H2O2 and O3 oxidation of S(IV) (Figure 2b), the only formation pathways leading to Δ17O > 0.0‰. This is consistent with the fact that these two sulfate formation pathways lead to the largest enrichment in δ34S (Table 1). In Figure 2c, the negative correlation between δ18O and δ34S (r2 = 0.38, p-value < 0.01) also suggests that secondary sulfate formation leads to lighter δ18O values and enriched δ34S.
Figure 2.

Regressions of (a) Δ17O vs δ18O, (b) Δ17O vs δ34S, and (c) δ18O vs δ34S, where the solid black line is the linear least-squares regression line. The three size bins are depicted by the shape of the marker, as defined in the legend. The color bar shows the sulfur oxidation ratio (SOR) for each sample. Poker Flat measurements are depicted with black triangles. The isotopic composition of fuel oil is shown by a blue line. The gray shaded region shows the full possible range of δ18O, Δ17O, and δ34S source signatures with the average source signature for each pathway plotted as a black star.
3.2. Sources and Formation of PM0.7 Sulfate
Figure 3 shows the mass concentrations and fractional contributions of the six sulfate formation pathways (primary, O3, H2O2, TMI-O2, OH, and NO2) calculated in the isotope mixing model for PM0.7 and PM>0.7 (see Supporting Information, section 1.4). Primary sulfate is the dominant source of sulfate for PM0.7 particles throughout the campaign contributing 2.1 ± 1.4 μg·m–3 (69 ± 15% of PM0.7 sulfate) on average. During the polluted period between January 30th and February 2nd, the fraction of primary sulfate is higher at nighttime (84 ± 9%) compared to daytime (65 ± 8%; p-value < 0.05). The daily-mean primary fraction during the polluted period is not significantly different from other periods in the campaign (p-value = 0.23), though the average mass concentration of primary sulfate was more than twice as high (4.0 ± 1.3 μg·m–3) compared to other periods (1.6 ± 1.0 μg·m–3; Figure 3a,b). Widespread use of residential heating oil creates a different pollution regime compared to pollution derived from coal-fired powerplants due the larger amount of direct fine mode primary sulfate emissions (<100 nm).41,74−77 This study’s top-down approach shows that CMAQ emissions inventories in Fairbanks likely underestimate residential sources of primary sulfate PM.
Figure 3.

Time series of the estimated contributions of primary sulfate (navy) and secondary sulfate formation via the NO2 (green), O3 (gold), OH (orange), TMI-O2 (light blue), and H2O2 (pink) pathways. Mass concentrations and average fractional contributions for PM0.7 sulfate are presented in (a) and (b), respectively. Likewise, (c) and (d) show mass concentrations and fractional contributions of PM0.7–2.5 and PM2.5–10 sulfate combined as PM>0.7 μm. The line graphs (a and c) show the estimated mass concentration for each sulfate formation pathway, and the shading represents the 95% confidence interval. The difference in scale for the y-axis for (a) and (c) should be noted. The bar charts (b and d) summarize the median fraction for each pathway and period during the campaign. “Day” and “Pol. Night” correspond to the daytime and nighttime samples collected during the ultrapolluted period between January 30th and February 2nd.
The dominant formation pathway for secondary sulfate in the PM0.7 size bin is H2O2, which contributes up to 2.6 ± 1.4 μg·m–3 (37 ± 6% of total PM0.7 sulfate) during the polluted period and 0.4 ± 0.3 μg·m–3 (14 ± 8%) on average throughout the campaign. The difference in the fraction of H2O2-derived sulfate in January (7 ± 4%) compared to February (14 ± 8%) is statistically significant (p-value < 0.01), likely due to increased photochemical activity and enhanced vertical mixing in February.
During the polluted period, HMS concentrations (1.1 ± 0.5 μg·m–3) are comparable to secondary sulfate (1.5 ± 1.0 μg·m–3), and at times exceed secondary sulfate concentrations (Figure S20). At night during the polluted period, average HMS concentrations (1.0 ± 0.2 μg·m–3) exceed secondary sulfate (0.79 ± 0.40 μg·m–3). During the day, secondary sulfate concentrations (2.1 ± 0.9 μg·m–3) are higher than HMS (1.3 ± 0.7 μg·m–3), largely due to H2O2-derived sulfate, which is responsible for 1.4 ± 0.8 μg·m–3 of secondary sulfate. Overall, the atmospheric formation of HMS during the most polluted period of the campaign rivals the abundance of secondary sulfate.
Ozone (O3) contributes 6 ± 4% of sulfate in the PM0.7 size bin on average, except during the polluted period where O3 concentrations are low (<1 ppb) and O3 contribution to particulate sulfate is less than 4% (Figure S22). O3 is likely the most important secondary oxidant during the January 24th “heat wave”, contributing 13 (±4)% of PM0.7, where the average ambient temperature and O3 concentrations were −0.14 °C and 18.3 ppb, respectively (Figure S22). The increased fractions of O3-derived sulfate on February 9th, 12th, and 17th correspond with relatively high O3 concentrations in Fairbanks (>20 ppb on average). The fraction of O3-derived sulfate in the PM0.7 size bin is moderately correlated with ambient O3 concentrations (r2 = 0.40, p-value < 0.01; Figure S22).
The fractional contributions of OH, NO2, and TMI-O2, shown in Figure 3, are more uncertain since their oxygen isotopic signatures are similar (Table 1). We used the δ34S observations and known sulfur isotope fractionation factors (eqns. 5−7, Table 1) in the model to help distinguish between these three pathways. On average, NO2 contribution to PM0.7 sulfate (6 ± 4%) is similar to that of the O3-derived sulfate and is higher in February (7 ± 4%) compared to January (3 ± 2%). NO2 may be the dominant secondary oxidant directly after the polluted period (February 3rd to February 5th) and for several days in mid to late February (February 9th and February 18th) based on the 95% confidence intervals (Figure 3a). NO2-oxidation contributes up to 18 (±16)% of sulfate in the PM0.7 size bin on February 22. The maximum OH contribution to sulfate 16 (±15)% was also observed February 22, signifying enhanced photochemical activity on that day.
On average, OH-derived sulfate contributes 6 ± 3% of PM0.7 sulfate, and, like H2O2 and NO2, it is higher in February (8 ± 3%) compared to January (4 ± 2%). The increase in OH, H2O2, and NO2 production of sulfate in February is likely due to increased solar intensity and longer days (8.5 h of daylight in February vs 6 h in January), leading to enhanced photolysis rates. NO2 and OH-derived sulfate are moderately correlated (r2 = 0.40, p-value < 0.01), likely due to the photochemical production of the oxidants themselves (Figure S21). NO2 oxidation is also correlated with O3-derived sulfate (r2 = 0.61, p-value < 0.01), as expected due to their similar pH dependencies (Figure S21). O3-derived sulfate is relatively constant throughout the campaign (6 ± 3% in January and 7 ± 4% in February), showing less sensitivity to differences in temperature and hours of daylight and more sensitivity to ambient O3 concentrations and aerosol pH.
The TMI-O2 pathway (1 ± 2%) is only a minor contributor to PM0.7 sulfate. This is consistent with the fact that average total water-soluble iron and manganese on the filters used for isotope analysis are <5 ng·m–3 combined and do not exceed 25 ng·m–3 combined (Figure S23). Though the model used in this study is a statistical model and not processed-based, the model results are consistent with the pH-dependence of NO2 and O3 oxidation of S(IV), the increase in photochemical oxidation in February, and the low solubility of transition metals in Fairbanks, showing that this methodology is useful in interpreting isotope observations with a statistically robust range of uncertainty.
3.3. Sources and Formation of PM>0.7 Sulfate
The combined PM0.7–2.5 and PM2.5–10 size bins, hereafter referred to as PM>0.7, only contribute approximately 0.3 ± 0.1 μg·m–3 (10 ± 5% of total sulfate mass), but show a noteworthy difference in isotopic composition compared to PM0.7 sulfate. Primary sulfate is a smaller portion of sulfate in the larger compared to smaller size bins, contributing 16 ± 11% of sulfate in PM>0.7. This is consistent with the size of primary sulfate from fuel oil combustion, which is typically <0.1 μm (Figure 3c,d).34 On average, the main secondary oxidants for PM>0.7 sulfate are H2O2 (12 ± 18%), O3 (22 ± 4%), and NO2 (25 ± 15%) (Figure 3c,d). During late January through early February, the combined O3 and NO2 pathways contribute 55 ± 11% of sulfate in PM>0.7 (Figure 3c,d). Depending on the abundance of ambient H2O2, these pathways are only dominant at pH > 4–5, suggesting aerosol pH within or above this range.
During the ultrapolluted period, primary sulfate is 23 ± 15% of sulfate in PM>0.7 during the day and 47 ± 13% at night. As with PM0.7 particles, H2O2 is the dominant secondary oxidant for PM>0.7 during this ultrapolluted period, contributing nearly as much as primary sulfate during the day at 56 ± 22% and 37 ± 24% at night. The combined NO2 and O3 pathways contribute 10 ± 8% of PM>0.7 sulfate during the daytime and 6 ± 4% at night. The decrease in the level of oxidation of O3 and NO2 during the polluted period is likely due to low ambient concentrations of O3 and may signify a lower pH that inhibits NO2 oxidation. TMI-O2 oxidation is insignificant (<1%), further suggesting limited metal solubility.
3.4. Atmospheric Implications
Sulfate is the most abundant PM sulfur species contributing 85 ± 9% of total PM sulfur (sulfate + total S(IV)) by mass throughout most of the campaign. During the ultra-polluted period, the fraction of HMS and non-HMS S(IV) relative to sulfate is the highest of the entire measurement period, contributing 26% (±8%) of PM sulfur mass. It is unclear whether the partially oxidized S(IV) in Fairbanks is primary or secondary in this study, though investigating the sources and speciation of unoxidized sulfur PM warrants further attention. For sulfur control measures, switching to an ultralow sulfur diesel (ULSD) heating oil (15 ppmv sulfur) would likely reduce emissions of primary sulfate and SO2, but more research is needed to understand how these measures may impact aerosol pH, secondary sulfate formation, and air quality overall. It is also important to acknowledge the economic impacts of increased fuel oil costs, since this financial burden may motivate residents to rely more heavily on woodburning and worsen air quality overall.
The dominance of primary sulfate demonstrated in this study may also highlight a bias in air quality modeling. Missing sulfate is often attributed to incomplete chemistry but could instead be due to dated emission factors or incomplete emission inventories due to the challenge of estimating residential and commercial space heating. These findings are applicable in regions with high rates of fuel oil use for space heating that have not mandated ULSD oil, places with nonexistent or rudimentary sulfur controls on their coal or oil-fired power plants, and over the ocean where high sulfur ship fuel is used. More work is also needed to quantify isotopic fractionation factors at ionic strengths representative of aerosol liquid water. This will improve assumptions in future sulfate isotope studies since literature values are based on bulk solutions meant to simulate cloud water.
Acknowledgments
A.M. and B.A. were supported by NOAA Grant NA20OAR4310295. We acknowledge James R. Campbell at UAF for his help with heating oil collection. We acknowledge Yuhan Yang for contributing water-soluble metal concentrations. U.J. acknowledges National Science Foundation Division of Polar Programs (PLR) Grants PLR 1904128 and PLR 629363. W.R.S. and M.C.-M. were supported by the NSF Navigating the New Arctic Program (Grant No. 1927750). S.A., S.B., and J.S. were supported by the Agence Nationale de la Recherche (ANR) via Contract ANR-21-CE01-0017 CASPA. R.J.W. and K.D. were supported by the NSF Atmospheric Geoscience Program (Grant No. AGS-2029730) and the NSF Navigating the New Arctic Program (Grant No. NNA-1927778). K.G. was supported by the Russian Science Foundation (Grant No. 21-17-00135). E.J.C. and K.A.P. were supported by the NSF Atmospheric Chemistry and Physical and Dynamic Meteorology (Grant No. AGS-2037091), NSF Navigating the New Arctic (Grant No. RISE-1927831), and the Univ. of Michigan, Dept. of Chemistry. L.M.D.H., M.O.S., and C.A. were supported by the National Science Foundation (Grant No. AGS-2109011). J.M. was supported by the NSF Atmospheric Geoscience Program (Grant No. AGS-2029747) and the NSF Navigating the New Arctic Program (Grant No. ICER-1927750).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsestair.3c00023.
Experimental methods for sample preparation, isotope analysis, and isotope corrections. It also includes assumptions, code, and performance analysis for the Bayesian isotope mixing model. Finally, there are additional model results based on the assumption that all H2O2 formation occurs in particles via the HOOHpm pathways (PDF)
Code for the Bayesian isotope mixing model used in this study, including source signature calculations (PDF)
Author Present Address
§ K.K.D.: Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, United States
Author Present Address
# L.T.: Department of Atmospheric and Oceanic Sciences, University of California, Los Angeles, CA 90095, United States.
Author Present Address
○ A.W.: Department of Climate and Space Sciences and Engineering, University of Michigan, Ann Arbor, MI 48109, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Beelen R.; Hoek G.; Raaschou-Nielsen O.; Stafoggia M.; Andersen Z. J.; Weinmayr G.; Hoffmann B.; Wolf K.; Samoli E.; Fischer P. H.; Nieuwenhuijsen M. J.; Xun W. W.; Katsouyanni K.; Dimakopoulou K.; Marcon A.; Vartiainen E.; Lanki T.; Yli-Tuomi T.; Oftedal B.; Schwarze P. E.; Nafstad P.; De Faire U.; Pedersen N. L.; Östenson C.-G.; Fratiglioni L.; Penell J.; Korek M.; Pershagen G.; Eriksen K. T.; Overvad K.; Sørensen M.; Eeftens M.; Peeters P. H.; Meliefste K.; Wang M.; Bueno-de-Mesquita H. B.; Sugiri D.; Krämer U.; Heinrich J.; de Hoogh K.; Key T.; Peters A.; Hampel R.; Concin H.; Nagel G.; Jaensch A.; Ineichen A.; Tsai M.-Y.; Schaffner E.; Probst-Hensch N. M.; Schindler C.; Ragettli M. S.; Vilier A.; Clavel-Chapelon F.; Declercq C.; Ricceri F.; Sacerdote C.; Galassi C.; Migliore E.; Ranzi A.; Cesaroni G.; Badaloni C.; Forastiere F.; Katsoulis M.; Trichopoulou A.; Keuken M.; Jedynska A.; Kooter I. M.; Kukkonen J.; Sokhi R. S.; Vineis P.; Brunekreef B. Natural-Cause Mortality and Long-Term Exposure to Particle Components: An Analysis of 19 European Cohorts within the Multi-Center ESCAPE Project. Environ. Health Perspect. 2015, 123 (6), 525–533. 10.1289/ehp.1408095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedoussi I. C.; Eastham S. D.; Monier E.; Barrett S. R. H. Premature Mortality Related to United States Cross-State Air Pollution. Nature 2020, 578 (7794), 261–265. 10.1038/s41586-020-1983-8. [DOI] [PubMed] [Google Scholar]
- Hoek G.; Krishnan R. M.; Beelen R.; Peters A.; Ostro B.; Brunekreef B.; Kaufman J. D. Long-Term Air Pollution Exposure and Cardio- Respiratory Mortality: A Review. Environ. Health 2013, 12 (1), 43. 10.1186/1476-069X-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waidyatillake N. T.; Campbell P. T.; Vicendese D.; Dharmage S. C.; Curto A.; Stevenson M. Particulate Matter and Premature Mortality: A Bayesian Meta-Analysis. Int. J. Environ. Res. Public. Health 2021, 18 (14), 7655. 10.3390/ijerph18147655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip S.; Martin R. V.; van Donkelaar A.; Lo J. W.-H.; Wang Y.; Chen D.; Zhang L.; Kasibhatla P. S.; Wang S.; Zhang Q.; Lu Z.; Streets D. G.; Bittman S.; Macdonald D. J. Global Chemical Composition of Ambient Fine Particulate Matter for Exposure Assessment. Environ. Sci. Technol. 2014, 48 (22), 13060–13068. 10.1021/es502965b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider G.; Weagle C. L.; Murdymootoo K. K.; Ring A.; Ritchie Y.; Stone E.; Walsh A.; Akoshile C.; Anh N. X.; Balasubramanian R.; Brook J.; Qonitan F. D.; Dong J.; Griffith D.; He K.; Holben B. N.; Kahn R.; Lagrosas N.; Lestari P.; Ma Z.; Misra A.; Norford L. K.; Quel E. J.; Salam A.; Schichtel B.; Segev L.; Tripathi S.; Wang C.; Yu C.; Zhang Q.; Zhang Y.; Brauer M.; Cohen A.; Gibson M. D.; Liu Y.; Martins J. V.; Rudich Y.; Martin R. V. Variation in Global Chemical Composition of PM2.5: Emerging Results from SPARTAN. Atmospheric Chem. Phys. 2016, 16 (15), 9629–9653. 10.5194/acp-16-9629-2016. [DOI] [Google Scholar]
- Liu T.; Chan A. W. H.; Abbatt J. P. D. Multiphase Oxidation of Sulfur Dioxide in Aerosol Particles: Implications for Sulfate Formation in Polluted Environments. Environ. Sci. Technol. 2021, 55 (8), 4227–4242. 10.1021/acs.est.0c06496. [DOI] [PubMed] [Google Scholar]
- Ye C.; Lu K.; Song H.; Mu Y.; Chen J.; Zhang Y. A Critical Review of Sulfate Aerosol Formation Mechanisms during Winter Polluted Periods. J. Environ. Sci. 2023, 123, 387–399. 10.1016/j.jes.2022.07.011. [DOI] [PubMed] [Google Scholar]
- Guo H.; Weber R. J.; Nenes A. High Levels of Ammonia Do Not Raise Fine Particle pH Sufficiently to Yield Nitrogen Oxide-Dominated Sulfate Production. Sci. Rep. 2017, 7 (1), 12109. 10.1038/s41598-017-11704-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Lung Association. https://www.lung.org/research/sota/city-rankings/msas/fairbanks-ak (accessed 2023-09-12).
- Cesler-Maloney M.; Simpson W. R.; Miles T.; Mao J.; Law K. S.; Roberts T. J. Differences in Ozone and Particulate Matter Between Ground Level and 20 m Aloft Are Frequent During Wintertime Surface-Based Temperature Inversions in Fairbanks, Alaska. J. Geophys. Res. Atmospheres 2022, 127 (10), 1–17. 10.1029/2021JD036215. [DOI] [Google Scholar]
- Tran H. N. Q.; Mölders N. Investigations on Meteorological Conditions for Elevated PM2.5 in Fairbanks, Alaska. Atmospheric Res. 2011, 99 (1), 39–49. 10.1016/j.atmosres.2010.08.028. [DOI] [Google Scholar]
- Kotchenruther R. A. Source Apportionment of PM2.5 at Multiple Northwest U.S. Sites: Assessing Regional Winter Wood Smoke Impacts from Residential Wood Combustion. Atmos. Environ. 2016, 142, 210–219. 10.1016/j.atmosenv.2016.07.048. [DOI] [Google Scholar]
- Wang Y.; Hopke P. K. Is Alaska Truly the Great Escape from Air Pollution? - Long Term Source Apportionment of Fine Particulate Matter in Fairbanks, Alaska. Aerosol Air Qual. Res. 2014, 14 (7), 1875–1882. 10.4209/aaqr.2014.03.0047. [DOI] [Google Scholar]
- Ward T.; Trost B.; Conner J.; Flanagan J.; Jayanty R. K. M. Source Apportionment of PM2.5 in a Subarctic Airshed - Fairbanks, Alaska. Aerosol Air Qual. Res. 2012, 12 (4), 536–543. 10.4209/aaqr.2011.11.0208. [DOI] [Google Scholar]
- Ye L.; Wang Y. Long-Term Air Quality Study in Fairbanks, Alaska: Air Pollutant Temporal Variations, Correlations, and PM2.5 Source Apportionment. Atmosphere 2020, 11 (11), 1203. 10.3390/atmos11111203. [DOI] [Google Scholar]
- Alaska Department of Environmental Conservation (DEC) Division of Air Quality Technical Analysis Modeling Report for Phase 1, 2 and 3; Alaska Department of Environmental Conservation, 2023.
- Campbell J. R.; Battaglia M.; Dingilian K.; Cesler-Maloney M.; St Clair J. M.; Hanisco T. F.; Robinson E.; DeCarlo P.; Simpson W.; Nenes A.; Weber R. J.; Mao J. Source and Chemistry of Hydroxymethanesulfonate (HMS) in Fairbanks, Alaska. Environ. Sci. Technol. 2022, 56 (12), 7657–7667. 10.1021/acs.est.2c00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaska Department of Environmental Conservation (DEC) State Air Quality Control Plan Emission Inventory Data; Alaska Department of Environmental Conservation, 2019; Vol. II: III.D.7.6.
- Shakya K. M.; Peltier R. E. Investigating Missing Sources of Sulfur at Fairbanks, Alaska. Environ. Sci. Technol. 2013, 47 (16), 9332–9338. 10.1021/es402020b. [DOI] [PubMed] [Google Scholar]
- Coykendall L. H. Formation and Control of Sulfur Oxides in Boilers. J. Air Pollut. Control Assoc. 1962, 12 (12), 567–591. 10.1080/00022470.1962.10468129. [DOI] [Google Scholar]
- Chin M.; Rood R. B.; Lin S.-J.; Müller J.-F.; Thompson A. M. Atmospheric Sulfur Cycle Simulated in the Global Model GOCART: Model Description and Global Properties. J. Geophys. Res. Atmospheres 2000, 105 (D20), 24671–24687. 10.1029/2000JD900384. [DOI] [Google Scholar]
- AP 42, Fifth ed., Vol. I Chapter 1: External Combustion Sources; United States Environmental Protection Agency, 1998.
- Alexander B.; Park R. J.; Jacob D. J.; Gong S. Transition Metal-Catalyzed Oxidation of Atmospheric Sulfur: Global Implications for the Sulfur Budget. J. Geophys. Res. 2009, 114 (D2), D02309. 10.1029/2008JD010486. [DOI] [Google Scholar]
- Shao J.; Chen Q.; Wang Y.; Lu X.; He P.; Sun Y.; Shah V.; Martin R. V.; Philip S.; Song S.; Zhao Y.; Xie Z.; Zhang L.; Alexander B. Heterogeneous Sulfate Aerosol Formation Mechanisms during Wintertime Chinese Haze Events: Air Quality Model Assessment Using Observations of Sulfate Oxygen Isotopes in Beijing. Atmospheric Chem. Phys. 2019, 19 (9), 6107–6123. 10.5194/acp-19-6107-2019. [DOI] [Google Scholar]
- Moch J. M.; Dovrou E.; Mickley L. J.; Keutsch F. N.; Liu Z.; Wang Y.; Dombek T. L.; Kuwata M.; Budisulistiorini S. H.; Yang L.; Decesari S.; Paglione M.; Alexander B.; Shao J.; Munger J. W.; Jacob D. J. Global Importance of Hydroxymethanesulfonate in Ambient Particulate Matter: Implications for Air Quality. J. Geophys. Res. Atmospheres 2020, 125 (18), 1–14. 10.1029/2020JD032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J. W.; Tiller C.; Hoffmann M. R. Identification of Hydroxymethanesulfonate in Fog Water. Science 1986, 231 (4735), 247–249. 10.1126/science.231.4735.247. [DOI] [PubMed] [Google Scholar]
- Olson T. M.; Hoffmann M. R. On the Kinetics of Formaldehyde-S(IV) Adduct Formation in Slightly Acidic Solution. Atmospheric Environ. 1967 1986, 20 (11), 2277–2278. 10.1016/0004-6981(86)90318-5. [DOI] [Google Scholar]
- Song S.; Ma T.; Zhang Y.; Shen L.; Liu P.; Li K.; Zhai S.; Zheng H.; Gao M.; Moch J. M.; Duan F.; He K.; McElroy M. B. Global Modeling of Heterogeneous Hydroxymethanesulfonate Chemistry. Atmospheric Chem. Phys. 2021, 21 (1), 457–481. 10.5194/acp-21-457-2021. [DOI] [Google Scholar]
- Cheng Y.; Zheng G.; Wei C.; Mu Q.; Zheng B.; Wang Z.; Gao M.; Zhang Q.; He K.; Carmichael G.; Pöschl U.; Su H. Reactive Nitrogen Chemistry in Aerosol Water as a Source of Sulfate during Haze Events in China. Sci. Adv. 2016, 2 (12), e1601530 10.1126/sciadv.1601530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T.; Furutani H.; Duan F.; Kimoto T.; Jiang J.; Zhang Q.; Xu X.; Wang Y.; Gao J.; Geng G.; Li M.; Song S.; Ma Y.; Che F.; Wang J.; Zhu L.; Huang T.; Toyoda M.; He K. Contribution of Hydroxymethanesulfonate (HMS) to Severe Winter Haze in the North China Plain. Atmospheric Chem. Phys. 2020, 20 (10), 5887–5897. 10.5194/acp-20-5887-2020. [DOI] [Google Scholar]
- Zhang H.; Xu Y.; Jia L. Hydroxymethanesulfonate Formation as a Significant Pathway of Transformation of SO2. Atmos. Environ. 2023, 294, 119474. 10.1016/j.atmosenv.2022.119474. [DOI] [Google Scholar]
- Hua W.; Chen Z. M.; Jie C. Y.; Kondo Y.; Hofzumahaus A.; Takegawa N.; Chang C. C.; Lu K. D.; Miyazaki Y.; Kita K.; Wang H. L.; Zhang Y. H.; Hu M. Atmospheric Hydrogen Peroxide and Organic Hydroperoxides during PRIDE-PRD’06, China: Their Concentration, Formation Mechanism and Contribution to Secondary Aerosols. Atmospheric Chem. Phys. 2008, 8 (22), 6755–6773. 10.5194/acp-8-6755-2008. [DOI] [Google Scholar]
- He P.; Alexander B.; Geng L.; Chi X.; Fan S.; Zhan H.; Kang H.; Zheng G.; Cheng Y.; Su H.; Liu C.; Xie Z. Isotopic Constraints on Heterogeneous Sulfate Production in Beijing Haze. Atmospheric Chem. Phys. 2018, 18 (8), 5515–5528. 10.5194/acp-18-5515-2018. [DOI] [Google Scholar]
- Shen X.; Lee T.; Guo J.; Wang X.; Li P.; Xu P.; Wang Y.; Ren Y.; Wang W.; Wang T.; Li Y.; Carn S. A.; Collett J. L. Aqueous Phase Sulfate Production in Clouds in Eastern China. Atmos. Environ. 2012, 62, 502–511. 10.1016/j.atmosenv.2012.07.079. [DOI] [Google Scholar]
- Schmale J.; Arnold S. R.; Law K. S.; Thorp T.; Anenberg S.; Simpson W. R.; Mao J.; Pratt K. A. Local Arctic Air Pollution: A Neglected but Serious Problem. Earths Future 2018, 6 (10), 1385–1412. 10.1029/2018EF000952. [DOI] [Google Scholar]
- Seinfeld J. H.; Pandis S. N.. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; J. Wiley: Hoboken, N.J, 2006. [Google Scholar]
- Tao W.; Su H.; Zheng G.; Wang J.; Wei C.; Liu L.; Ma N.; Li M.; Zhang Q.; Pöschl U.; Cheng Y. Aerosol pH and Chemical Regimes of Sulfate Formation in Aerosol Water during Winter Haze in the North China Plain. Atmospheric Chem. Phys. 2020, 20 (20), 11729–11746. 10.5194/acp-20-11729-2020. [DOI] [Google Scholar]
- Kroopnick P.; Craig H. Atmospheric Oxygen: Isotopic Composition and Solubility Fractionation. Science 1972, 175 (4017), 54–55. 10.1126/science.175.4017.54. [DOI] [PubMed] [Google Scholar]
- Dominguez G.; Jackson T.; Brothers L.; Barnett B.; Nguyen B.; Thiemens M. H. Discovery and Measurement of an Isotopically Distinct Source of Sulfate in Earth’s Atmosphere. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (35), 12769–12773. 10.1073/pnas.0805255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt B. D.; Kumar R. Oxygen-18 Study of High-Temperature Air Oxidation of SO2. Atmospheric Environ. 1967 1984, 18 (10), 2089–2094. 10.1016/0004-6981(84)90194-X. [DOI] [Google Scholar]
- Holt B. D.; Kumar R.; Cunningham P. T. Oxygen-18 Study of the Aqueous-Phase Oxidation of Sulfur Dioxide. Atmospheric Environ. 1967 1981, 15 (4), 557–566. 10.1016/0004-6981(81)90186-4. [DOI] [Google Scholar]
- Holt B. D.; Cunningham P. T.; Engelkemeir A. G.; Graczyk D. G.; Kumar R. Oxygen-18 Study of Nonaqueous-Phase Oxidation of Sulfur Dioxide. Atmospheric Environ. 1967 1983, 17 (3), 625–632. 10.1016/0004-6981(83)90136-1. [DOI] [Google Scholar]
- Aggarwal P. K.; Alduchov O. A.; Froehlich K. O.; Araguas-Araguas L. J.; Sturchio N. C.; Kurita N. Stable Isotopes in Global Precipitation: A Unified Interpretation Based on Atmospheric Moisture Residence Time. Geophys. Res. Lett. 2012, 39 (11), 1–6. 10.1029/2012GL051937. [DOI] [Google Scholar]
- Aggarwal P. K.; Alduchov O.; Araguás Araguás L.; Dogramaci S.; Katzlberger G.; Kriz K.; Kulkarni K. M.; Kurttas T.; Newman B. D.; Purcher A. New Capabilities for Studies Using Isotopes in the Water Cycle. Eos Trans. Am. Geophys. Union 2007, 88 (49), 537–538. 10.1029/2007EO490002. [DOI] [Google Scholar]
- Lee C. C.-W.; Thiemens M. H. The δ 17 O and δ 18 O Measurements of Atmospheric Sulfate from a Coastal and High Alpine Region: A Mass-Independent Isotopic Anomaly. J. Geophys. Res. Atmospheres 2001, 106 (D15), 17359–17373. 10.1029/2000JD900805. [DOI] [Google Scholar]
- Lyons J. R. Transfer of Mass-Independent Fractionation in Ozone to Other Oxygen-Containing Radicals in the Atmosphere. Geophys. Res. Lett. 2001, 28 (17), 3231–3234. 10.1029/2000GL012791. [DOI] [Google Scholar]
- Morin S.; Savarino J.; Bekki S.; Gong S.; Bottenheim J. W. Signature of Arctic Surface Ozone Depletion Events in the Isotope Anomaly (Δ<Sup>17</Sup>O) of Atmospheric Nitrate. Atmospheric Chem. Phys. 2007, 7 (5), 1451–1469. 10.5194/acp-7-1451-2007. [DOI] [Google Scholar]
- Sofen E. D.; Alexander B.; Kunasek S. A. The Impact of Anthropogenic Emissions on Atmospheric Sulfate Production Pathways, Oxidants, and Ice Core Δ17O(SO42–). Atmospheric Chem. Phys. 2011, 11 (7), 3565–3578. 10.5194/acp-11-3565-2011. [DOI] [Google Scholar]
- Savarino J.; Lee C. C. W.; Thiemens M. H. Laboratory Oxygen Isotopic Study of Sulfur (IV) Oxidation: Origin of the Mass-Independent Oxygen Isotopic Anomaly in Atmospheric Sulfates and Sulfate Mineral Deposits on Earth. J. Geophys. Res. Atmospheres 2000, 105 (D23), 29079–29088. 10.1029/2000JD900456. [DOI] [Google Scholar]
- Harris E.; Sinha B.; Hoppe P.; Crowley J. N.; Ono S.; Foley S. Sulfur Isotope Fractionation during Oxidation of Sulfur Dioxide: Gas-Phase Oxidation by OH Radicals and Aqueous Oxidation by H2O2, O3 and Iron Catalysis. Atmospheric Chem. Phys. 2012, 12 (1), 407–423. 10.5194/acp-12-407-2012. [DOI] [Google Scholar]
- Harris E.; Sinha B.; Hoppe P.; Foley S.; Borrmann S. Fractionation of Sulfur Isotopes during Heterogeneous Oxidation of SO2 on Sea Salt Aerosol: A New Tool to Investigate Non-Sea Salt Sulfate Production in the Marine Boundary Layer. Atmospheric Chem. Phys. 2012, 12 (10), 4619–4631. 10.5194/acp-12-4619-2012. [DOI] [Google Scholar]
- Kok G. L.; Gitlin S. N.; Lazrus A. L. Kinetics of the Formation and Decomposition of Hydroxymethanesulfonate. J. Geophys. Res. 1986, 91 (D2), 2801. 10.1029/JD091iD02p02801. [DOI] [Google Scholar]
- Schauer A. J.; Kunasek S. A.; Sofen E. D.; Erbland J.; Savarino J.; Johnson B. W.; Amos H. M.; Shaheen R.; Abaunza M.; Jackson T. L.; Thiemens M. H.; Alexander B. Oxygen Isotope Exchange with Quartz during Pyrolysis of Silver Sulfate and Silver Nitrate: Oxygen Isotope Exchange during Pyrolysis of Ag 2 SO 4 and AgNO 3. Rapid Commun. Mass Spectrom. 2012, 26 (18), 2151–2157. 10.1002/rcm.6332. [DOI] [PubMed] [Google Scholar]
- Geng L.; Schauer A. J.; Kunasek S. A.; Sofen E. D.; Erbland J.; Savarino J.; Allman D. J.; Sletten R. S.; Alexander B. Analysis of Oxygen-17 Excess of Nitrate and Sulfate at Sub-Micromole Levels Using the Pyrolysis Method: Analysis of Oxygen-17 Excess of Nitrate and Sulfate. Rapid Commun. Mass Spectrom. 2013, 27 (21), 2411–2419. 10.1002/rcm.6703. [DOI] [PubMed] [Google Scholar]
- Jongebloed U. A.; Schauer A. J.; Cole-Dai J.; Larrick C. G.; Wood R.; Fischer T. P.; Carn S. A.; Salimi S.; Edouard S. R.; Zhai S.; Geng L.; Alexander B. Underestimated Passive Volcanic Sulfur Degassing Implies Overestimated Anthropogenic Aerosol Forcing. Geophys. Res. Lett. 2023, 50 (1), 1–12. 10.1029/2022GL102061. [DOI] [Google Scholar]
- Liu T.; Abbatt J. P. D. Oxidation of Sulfur Dioxide by Nitrogen Dioxide Accelerated at the Interface of Deliquesced Aerosol Particles. Nat. Chem. 2021, 13 (12), 1173–1177. 10.1038/s41557-021-00777-0. [DOI] [PubMed] [Google Scholar]
- Wang X.; Gemayel R.; Hayeck N.; Perrier S.; Charbonnel N.; Xu C.; Chen H.; Zhu C.; Zhang L.; Wang L.; Nizkorodov S. A.; Wang X.; Wang Z.; Wang T.; Mellouki A.; Riva M.; Chen J.; George C. Atmospheric Photosensitization: A New Pathway for Sulfate Formation. Environ. Sci. Technol. 2020, 54 (6), 3114–3120. 10.1021/acs.est.9b06347. [DOI] [PubMed] [Google Scholar]
- Savarino J.; Thiemens M. H. Analytical Procedure to Determine Both δ18O and δ17O of H2O2 in Natural Water and First Measurements. Atmos. Environ. 1999, 33 (22), 3683–3690. 10.1016/S1352-2310(99)00122-3. [DOI] [Google Scholar]
- Vicars W. C.; Savarino J. Quantitative Constraints on the 17O-Excess (Δ17O) Signature of Surface Ozone: Ambient Measurements from 50°N to 50°S Using the Nitrite-Coated Filter Technique. Geochim. Cosmochim. Acta 2014, 135, 270–287. 10.1016/j.gca.2014.03.023. [DOI] [Google Scholar]
- Faust B. C.; Anastasio C.; Allen J. M.; Arakaki T. Aqueous-Phase Photochemical Formation of Peroxides in Authentic Cloud and Fog Waters. Science 1993, 260 (5104), 73–75. 10.1126/science.8465202. [DOI] [PubMed] [Google Scholar]
- Ye C.; Chen H.; Hoffmann E. H.; Mettke P.; Tilgner A.; He L.; Mutzel A.; Brüggemann M.; Poulain L.; Schaefer T.; Heinold B.; Ma Z.; Liu P.; Xue C.; Zhao X.; Zhang C.; Zhang F.; Sun H.; Li Q.; Wang L.; Yang X.; Wang J.; Liu C.; Xing C.; Mu Y.; Chen J.; Herrmann H. Particle-Phase Photoreactions of HULIS and TMIs Establish a Strong Source of H 2 O 2 and Particulate Sulfate in the Winter North China Plain. Environ. Sci. Technol. 2021, 55 (12), 7818–7830. 10.1021/acs.est.1c00561. [DOI] [PubMed] [Google Scholar]
- Harris E.; Sinha B.; Hoppe P.; Ono S. High-Precision Measurements of 33 S and 34 S Fractionation during SO 2 Oxidation Reveal Causes of Seasonality in SO 2 and Sulfate Isotopic Composition. Environ. Sci. Technol. 2013, 47 (21), 12174–12183. 10.1021/es402824c. [DOI] [PubMed] [Google Scholar]
- Au Yang D.; Bardoux G.; Assayag N.; Laskar C.; Widory D.; Cartigny P. Atmospheric SO2 Oxidation by NO2 Plays No Role in the Mass Independent Sulfur Isotope Fractionation of Urban Aerosols. Atmos. Environ. 2018, 193, 109–117. 10.1016/j.atmosenv.2018.09.007. [DOI] [Google Scholar]
- Urey H. C. The Thermodynamic Properties of Isotopic Substances. J. Chem. Soc. Resumed 1947, 562. 10.1039/jr9470000562. [DOI] [PubMed] [Google Scholar]
- Sinha B. W.; Hoppe P.; Huth J.; Foley S.; Andreae M. O. Sulfur Isotope Analyses of Individual Aerosol Particles in the Urban Aerosol at a Central European Site (Mainz, Germany). Atmospheric Chem. Phys. 2008, 8 (23), 7217–7238. 10.5194/acp-8-7217-2008. [DOI] [Google Scholar]
- Jongebloed U. A.; Schauer A. J.; Hattori S.; Cole-Dai J.; Larrick C. G.; Salimi S.; Edouard S. R.; Geng L.; Alexander B. Sulfur Isotopes Quantify the Impact of Anthropogenic Activities on Industrial-Era Arctic Sulfate in a Greenland Ice Core. Environ. Res. Lett. 2023, 18 (7), 074020. 10.1088/1748-9326/acdc3d. [DOI] [Google Scholar]
- Han X.; Guo Q.; Liu C.; Fu P.; Strauss H.; Yang J.; Hu J.; Wei L.; Ren H.; Peters M.; Wei R.; Tian L. Using Stable Isotopes to Trace Sources and Formation Processes of Sulfate Aerosols from Beijing, China. Sci. Rep. 2016, 6 (1), 29958. 10.1038/srep29958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson E. S.; Cesler-Maloney M.; Tan X.; Mao J.; Simpson W.; DeCarlo P. F. Wintertime Spatial Patterns of Particulate Matter in Fairbanks, AK during ALPACA 2022. Environ. Sci. Atmospheres 2023, 3 (3), 568–580. 10.1039/D2EA00140C. [DOI] [Google Scholar]
- Broecker W. S.; Oversby V. M.. Chemical Equilibria in the Earth; International Series in the Earth and Planetary Sciences; McGraw-Hill: New York, 1971. [Google Scholar]
- Salvatier J.; Wiecki T. V.; Fonnesbeck C. Probabilistic Programming in Python Using PyMC3. PeerJ. Comput. Sci. 2016, 2, e55 10.7717/peerj-cs.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.-W.; Savarino J. H.; Cachier H.; Thiemens M. H. Sulfur (32S, 33S, 34S, 36S) and Oxygen (16O,17O,18O) Isotopic Ratios of Primary Sulfate Produced from Combustion Processes. Tellus B 2002, 54 (3), 193–200. 10.1034/j.1600-0889.2002.01384.x. [DOI] [Google Scholar]
- Homolya J. B.; Lambert S. Characterization of Sulfate Emissions from Nonutility Boilers Firing Low-S Residual Oils in New York City. J. Air Pollut. Control Assoc. 1981, 31 (2), 139–143. 10.1080/00022470.1981.10465202. [DOI] [Google Scholar]
- Linak W. P.; Miller C. A.; Wendt J. O. L. Comparison of Particle Size Distributions and Elemental Partitioning from the Combustion of Pulverized Coal and Residual Fuel Oil. J. Air Waste Manag. Assoc. 2000, 50 (8), 1532–1544. 10.1080/10473289.2000.10464171. [DOI] [PubMed] [Google Scholar]
- Homolya J. B.; Fortune C. R. The Measurement of the Sulfuric Acid and Sulfate Content of Particulate Matter Resulting from the Combustion of Coal and Oil. Atmospheric Environ. 1967 1978, 12 (12), 2511–2514. 10.1016/0004-6981(78)90297-4. [DOI] [Google Scholar]
- Hays M. D.; Beck L.; Barfield P.; Lavrich R. J.; Dong Y.; Vander Wal R. L. Physical and Chemical Characterization of Residential Oil Boiler Emissions. Environ. Sci. Technol. 2008, 42 (7), 2496–2502. 10.1021/es071598e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.