

Abstract
Transcription start site (TSS) selection is a key step in gene expression and occurs at many promoter positions over a wide range of efficiencies. Here, we develop a massively parallel reporter assay to quantitatively dissect contributions of promoter sequence, NTP substrate levels, and RNA polymerase II (Pol II) activity to TSS selection by “promoter scanning” in Saccharomyces cerevisiae (Pol II MAssively Systematic Transcript End Readout, "Pol II MASTER"). Using Pol II MASTER, we measure the efficiency of Pol II initiation at 1,000,000 individual TSS sequences in a defined promoter context. Pol II MASTER confirms proposed critical qualities of S. cerevisiae TSS −8, −1, and +1 positions quantitatively in a controlled promoter context. Pol II MASTER extends quantitative analysis to surrounding sequences and determines that they tune initiation over a wide range of efficiencies. These results enabled the development of a predictive model for initiation efficiency based on sequence. We show that genetic perturbation of Pol II catalytic activity alters initiation efficiency mostly independently of TSS sequence, but selectively modulates preference for initiating nucleotide. Intriguingly, we find that Pol II initiation efficiency is directly sensitive to GTP levels at the first five transcript positions and to CTP and UTP levels at the second position genome wide. These results suggest individual NTP levels can have transcript-specific effects on initiation, representing a cryptic layer of potential regulation at the level of Pol II biochemical properties. The results establish Pol II MASTER as a method for quantitative dissection of transcription initiation in eukaryotes.
Introduction
In transcription initiation, RNA polymerase II (Pol II), assisted by General Transcription Factors (GTFs), binds promoter DNA through interactions with core promoter elements. Subsequently, a turn of promoter DNA is unwound, forming a Pol II-promoter open complex containing a single-stranded “transcription bubble”, and Pol II selects a promoter position to serve as the Transcription Start Site (TSS). At many Pol II promoters in eukaryotes, TSS selection occurs at multiple positions 1-11. Thus, the overall rate of gene expression at most Pol II promoters is determined by the efficiency of initiation from several distinct TSS positions. In addition, studies have suggested that alternative TSS selection can alter mRNA content, affecting translation activity and subsequent protein levels and functions 3,12,13, and is widespread in different cell types 6, developmental processes 4,12,14,15, growth conditions 16, responses to environmental changes, and cancers 17-19.
Pol II initiation in yeast proceeds by a promoter scanning mechanism, where the Pol II Pre-initiation Complex (PIC), comprising Pol II and GTFs, assembles upstream of an initiation region and then scans downstream to select TSSs 20-27. The efficiency of initiation at a given position depends on multiple features. Location relative to the core promoter region from which scanning will originate and proceed downstream, is critically important. DNA sequence in and around the TSS is critical for TSS specification. Here, the template base specifying the TSS position and the position immediately upstream of the TSS (positions +1 and −1, respectively) make the largest contributions. In particular, there is a strong preference for an R:Y base pair at position +1 and Y:R base pair at position −1 (reflected as a Y−1R+1 “initiator” sequence on the coding strand; Y=pYrimidine; R=puRine), and this preference is near universal for RNAPs 1,10,11,16,21,22,28-44. It has long been recognized that sequences beyond the −1/+1 also contribute to Pol II initiation efficiency. However, functions of these positions are difficult to determine from genomic usage alone.
An elegant study of promoter scanning from Kuehner and Brow established that TSS usage is determined by TSS priority during scanning 22. The study demonstrated that promoter sequences are examined by the transcription machinery in the order in which they are scanned with upstream TSSs having priority, independent of innate TSS strength. Kuehner and Brow introduced the concept of “TSS efficiency”, which accounts for how much Pol II reaches a particular TSS, allowing comparison of innate TSS sequence strengths (see Figure 1).
Figure 1. A high-throughput system for studying transcription TSS selection.

(A and B) Pol II initiation in yeast proceeds by promoter scanning. Yeast Pol II initiation usually occurs at multiple TSSs ~40 to 120 bp downstream of a core promoter comprising the PIC assembly position (e.g. a TATA element)). After PIC assembly upstream, scanning will proceed towards positions where TSS selection occurs (TSS region). Initiation across promoter positions is also controlled by multiple architectural features shown in A. These include the inhibition of initiation near a core promoter that diminishes downstream (“distance restriction”), biochemical restrictions on how far scanning can proceed downstream (“scanning processivity”), and “Pol II flux”, which represents the decrease in amount of scanning Pol II due to conversion of scanning Pol II initiating. (C) Construction of promoter libraries examining TSS sequence context. Top panel shows schematic of the SNR37 promoter and its TSS distribution based on TSS-seq 21. Bottom panel shows schematic of the Pol II MASTER libraries. A duplication of the SNR37 TSS region was inserted before native TSS region, and the −8 to +1 positions relative to native SNR37 +1 TSS (black box) were replaced by a 9 nt highly randomized region. The downstream SNR37 TSS region functions as a “Flux Detector” (FD) to capture Pol II that fails to initiate within the randomized region and allow measurement of initiation efficiency for upstream positions. A barcode (purple box) allows RNA products to be assigned to promoter variants. Other features (GAL1 UAS, GFP ORF, CYC1 terminator) support regulation and stabilization of RNAs. (D) TSS usage distributions at TSS and FD regions for promoter variant “AYR”, “BYR”, and “ARY” libraries are shown (left). TSS usages from designed +1 TSS and positions upstream are red/grey, respectively. TSS usage from the FD region is in brown. The definition of “TSS efficiency” and overall TSS efficiencies for aggregate +1 TSSs for different libraries are shown (middle). Example correlation plots of TSS efficiency calculations for +1 TSSs from individual promoter variants in Pol II MASTER libraries between representative biological replicates are shown (right) with Pearson r and N of variants.
Imbalanced promoter sequence distributions imposed by evolutionary processes also limit the ability to determine sequence-activity relationships for initiation. For example, it has been observed that yeast promoters have uneven base distributions across promoters being most obvious at highly expressed promoters for T on the transcribed strand upstream of the median TSS position (reflecting the middle of the TSS distribution), and A on the transcribed strand downstream of the median TSS. Furthermore, yeast promoters have a paucity of G/C in general 21,45-48. Therefore, the biased promoter distribution of bases leads to a biased distribution of TSS motifs. Preferred TSS motifs (those with a −8A) show enrichment downstream of promoter median TSS positions, and less-preferred motifs (those without a −8A) show enrichment upstream of the median TSS 21. We have found that hyperactive Pol II mutants (“gain-of-function” or GOF) and hypoactive Pol II mutants (“loss-of-function” or LOF) shift TSS usage upstream or downstream, respectively, while also showing differences in aggregate usage of TSS motifs. Disentangling apparent from actual differences in TSS efficiencies is difficult due to biased underlying sequence distributions. Moreover, other properties such as the biochemistry of transcription itself due to availability of NTP substrates, the biochemical properties of scanning processivity 20,25,27, promoter identity 49-51, or promoter chromatin could also contribute to initiation output 52.
To remove contextual differences among promoters we have developed a system to dissect determinants of initiation efficiency within a controlled promoter context. Here, we present “Pol II MASTER” based on bacterial MASTER (MAssively Systematic Transcript End Readout) 42,53-56, which couples initiation readout by sequencing with promoter identity. We apply Pol II MASTER to initiation by promoter scanning to investigate the initiation efficiency of ~80,000 promoter variants in Pol II WT and catalytic mutants and upon manipulation of NTP levels. We show that this system enables determination of the interface between initiation factor activity, transcription substrates, promoter sequence, and promoter output.
Results
High-throughput analysis of yeast Pol II initiation output
TSSs are specified within yeast promoters by scanning from upstream near the core-promoter to downstream (Figure 1A). While promoter melting occurs around +20 from the TATA box (if present), initiation is restricted adjacent to this region, with most TSS selection occurring ~40-150 nt downstream from the core promoter 57 (Figure 1B). Processivity of scanning, likely determined by TFIIH activity, will limit TSS usage downstream (Figure 1A, “unreachable TSS”). Once initiation happens (Figure 1A, B), Pol II “flux” – the amount of Pol II proceeding downstream – is reduced. Given variables in TSS context and the “first come-first served” basis of promoter scanning, innate TSS strengths can only be determined in a controlled context. We have established a massively parallel promoter variant assay “Pol II MASTER” to dissect how TSS sequence, Pol II activity, or NTP levels control initiation efficiency. We have embedded almost all possible sequences within a 9 bp randomized TSS region (Figure 1C) into promoter libraries for introduction into yeast. The sequence libraries are illustrated in Figure 1C and named by their base composition differences at positions −8, −1, and +1 on the transcribed strand. The “AYR” library has composition A−8NNNNNNY−1R+1 (N=A, C, G, or T, Y=C or T, R=A or G), with “BYR” having composition B−8NNNNNNY−1R+1 (B=C, G, or T), etc. Our libraries comprise 81,920 promoter variants. Because each promoter variant was evaluated for TSS initiation efficiency at up to 12 positions within or adjacent to the randomized region, we can analyze up to 983,040 distinct TSSs.
Our promoter context contains specific functionalities: inducibility (GAL1 UAS), a defined scanning region from an efficient promoter (SNR37), and RNA stabilization (GFP ORF and CYC1 terminator region) (Figure 1C). Critically, the native, highly efficient SNR37 TSS region was inserted downstream of the randomized TSS region as a “Flux Detector” (FD). Here, we employ the approach of Kuehner and Brow 22 that a highly efficient initiation region placed downstream of a TSS may capture polymerases that scan past the randomized TSS region. TSS efficiency is measured as in 22 as the usage of a TSS relative to usage at that TSS and downstream positions (Figure 1D). This metric allows upstream and downstream starts to be compared as it takes priority effects into account (upstream TSSs reduce the amount of polymerases scanning to downstream sites), with normalization of promoter usage using RNA only, allowing comparison across promoters and libraries. RNA products are assigned to promoter variants through a transcribed DNA barcode containing 20 randomized bases. Plasmid DNA and RNA products were extracted from yeast cells for DNA-seq and TSS-seq (Extended Data Figure 1A).
Several measures indicate high level of reproducibility and coverage (Figure 1D, Extended Data Figure 1). Base coverage in the randomized region was balanced (Extended Data Figure 1B). Correlation of DNA-seq variant counts indicates that transformation did not alter variant distribution (Extended Data Figure 1C, D). Bulk primer extension of libraries illustrated average library TSS selection and reproducibility (Extended Data Figure 1E). Only limited initiation was observed from the barcode region or downstream, validating flux detector function (Extended Data Figure 1E, F). Aggregate read distribution in our three libraries shows that as TSSs decreased in efficiency from the most efficient library (“AYR”) to the least (“ARY”) (Figure 1D, middle), reads shifted downstream from the designed +1 TSS (Figure 1D, left). The shift of TSS usage to position −1 in the ARY library was because the purine at the designed −1 position serves as a +1 for newly created TSSs. Biological replicates were merged given the high reproducibility (Figure 1D, right), keeping TSSs that contained at least five TSS-seq reads in each replicate and whose Coefficient of Variation (CV) in TSS-seq reads across replicates was less than 0.5 (as a proxy for reproducible behavior) (Extended Data Figure 1G). As a result, ~97% of TSS promoter variants were covered in each library (Supplementary Table 1). Finally, we analyzed potential interactions between TSS sequences and overall promoter expression (Extended Data Figure 1H). Normalization of individual promoter output (RNA levels) to promoter template number (DNA level) indicated that total promoter output based on TSS usage across each promoter was relatively unaffected by individual TSS strength.
Sequence-dependent control of S. cerevisiae TSS efficiency
To ask how our libraries recapitulated known TSS efficiencies, we first examined core sequences in our library matching the SNR14 TSS and its variants previously analyzed by Kuehner and Brow 22 (Figure 2A). Our randomized library contains the SNR14 TSS sequence embedded in our SNR37 context along with all single substitution variants. We found that Pol II MASTER recapitulated the single base effects on TSS efficiency previously observed while showing single base changes around a TSS can have large effects on TSS efficiency.
Figure 2. Wide range of initiation efficiency measured using MASTER.

(A) Comparison of single base mutation effects on TSS efficiency measured by Pol II MASTER and primer extension. (Left) Sequences of SNR14 and SNR37 TSS regions (in black boxes, including positions between −8 to +1 relative to TSS) and all possible single substitutions of SNR14 TSS region. Single substitutions included in both a prior SNR14 mutagenesis study 22 and Pol II MASTER libraries are in blue while those lacking in our study are in brown. Additional substitutions analyzed here are in gray. (Middle) High correlation of TSS efficiency measured by Pol II MASTER and primer extension. Mutation classes color coded as on left. (Right) Range of effects of single base substitutions on TSS efficiency for SNR14- and SNR37-related sequences. Mutation classes color coded as on left. Double substitutions of SNR14 and SNR37 TSS region included in Pol II MASTER “ARY” library are shown as orange inverted triangles and show almost no efficiency. (B) Pol II initiation shows a strong preference for A−8 and C−1A+1 containing variants. All promoter variants were divided into 20 groups defined by bases at positions −8, −1 and +1 relative to the designed +1 TSS, and their +1 TSS efficiencies were plotted as spots. Lines represent median efficiencies of each group. (C) Definition of “designed” +1 TSS (designated as +1) and positions relative to this TSS (blue TSS arrow and sequence). TSS usages generated from upstream or downstream of “designed” +1 TSS (green and brown TSS arrows and sequences, respectively) allow study of sequence preferences at positions −11 to −9 and +2 to +9 relative top designed +1.
Examining the designed +1 TSS variants, we first focused our analysis on positions −8, −1 and +1, which in vivo genome-wide data suggested are important determinants for TSS selection (Figure 2B, Extended Data Figure 2A). We divided +1 TSS variants into 64 groups defined by bases at positions −8, −1 and +1 relative to the TSS. The known importance of these three positions was recapitulated in our promoter context, but importantly, surrounding positions also had a considerable impact on TSS efficiency (Figure 2B). First, in our controlled context for TSSs at the designed +1 position, we found that Y−1R+1 was essential for initiation above a minimal background relative to R−1Y+1. Second, we demonstrate and quantify the very large effect of a −8A on TSS efficiency. A−8C−1A+1 motif-containing TSSs have the highest aggregate TSS usage from genomic promoters 21 and Pol II MASTER indicates they are also the most efficient. Third, among Y−1R+1 elements we found a clear hierarchy of efficiency – C−1A+1 > C−1G+1 > T−1A+1 ≈ T−1G+1 – that was not apparent from genome-wide usage, likely due to promoter sequence biases within genomic promoters.
To determine if −7 to −2 bases have similar effects regardless of −8, −1 and +1 identity, we rank ordered individual TSSs for each N−8N−1N+1 motif by the efficiency of their A−8C−1A+1 version (Extended Data Figure 2A). This rank ordering by A−8C−1A+1 efficiency was predictive of efficiency ranks for −7 to −2 sequences for different N−8N−1N+1 groups. We then set out to determine the contributions of individual bases at each position across our randomized region relative to the designed +1 TSS (Extended Data Figure 2B). Comparison of TSS efficiencies across base subgroups suggested individual base effects on TSS efficiency in aggregate for all examined positions. TSSs outside of the designed TSS +1 position allowed us to examine contribution of bases in our randomized region to the efficiency of nearly 1 million TSS sequences (Extended Data Figure 1E, F, Figure 2C, Extended Data Figure 2C). To visualize sequence preferences, we used the median initiation efficiencies of each base subgroup as indicators for preference. Centered median values were used to calculate “relative efficiency” and are shown in a sequence logo (Figure 3A). Datasets of designed +1 TSSs deriving from all libraries allowed us to nearly comprehensively study preference at positions −8 to +1 (Figure 3B, middle and bottom). Additionally, 10-25% of total TSS usage among libraries derived from the +4 position (Figure 2C, brown TSS arrow and sequences) allowing study of positions −11 to −9 relative to this TSS (Figure 3B, top). As noted above, positions −8, −1 and +1 were major determinants for TSS efficiency. Interestingly, position −9 showed a relatively strong effect in our defined promoter context, which was not obvious from genome-wide analyses. At positions −4 to −2, we observed modulation of initiation efficiency, where C/G were preferred while T was less preferred. The T effect is consistent mutation of Ts at positions −4 to −2 relative to −38 TSS of ADH1 to C significantly increased usage of that TSS 57. Preferences at positions within −9 to +1 were significant but −9 to −8 and −4 to +1 most contribute to TSS efficiency (Extended Data Figure 2B).
Figure 3. Sequence contributions to Pol II initiation efficiency from positions surrounding the TSS.

(A) Schematic illustrating how “relative efficiency” is calculated and visualized in B. At sequence positions relative to a TSS, all variants were divided into the four base subgroups at each position. Next, median values for each group were centered based on the mean of all median values. Centered median values were defined as “relative efficiencies”, representing preferences for bases at each position. Relative base efficiencies were visualized as sequence logos. Positive and negative values indicate relatively preferred or less preferred bases, respectively. (B) Pol II initiation shows distinct sequence preferences around TSSs. Preferences generated using designed +4 TSS deriving from “AYR”, “BYR” and “ARY” libraries (top). Preferences using datasets of designed +1 TSS deriving from “AYR” and “BYR” libraries (middle). Preferences using datasets of designed +1 TSS deriving from “AYR” and “ARY” libraries (bottom). Positions that contain fixed or not completely randomized bases are shown in grey. (C) Schematic illustrating how sequence interaction between positions is calculated and visualized as a heat map in D. Using −9/−8 positions as an example, relative efficiencies at position −8 were calculated when different bases were present at position −9. Next, relative efficiencies of each base were centered based on the mean of all relative efficiencies of a particular base. After centralization, negative and positive values indicate negative and positive interactions. Interaction scores for two sequence positions are read at the intersection of the x and y axes labeled by base and position. The centered relative efficiencies matrix was visualized as a heat map to represent the interaction between examined positions. (D) Sequence interactions were mainly observed at neighboring positions. Red and blue indicate positive and negative interactions, respectively. Missing values are shown in grey. Interactions related to positions −11 to −9 were calculated using datasets of designed +4 TSS deriving from “AYR”, “BYR” and “ARY” libraries. Other interactions were calculated using datasets of designed +1 TSS deriving from “AYR” and “BYR” libraries.
Experiments across species, including the description of the canonical initiator element (Inr), show contribution of downstream positions to TSS recognition or function 10,43,44,57-61. To determine downstream sequence contribution, we examined downstream preferences for the most efficient −8 TSS variants, whose positions +1 to +9 are located in the randomized region (Figure 2C, green TSS arrow and sequences). We found an A(/G)+2G(/C)+3G(/C)+4 enrichment for the top 10% most efficient TSSs, but not for the next 10% most efficient (Extended Data Figure 2D). These preferences were consistent with a TSS-specific study, where A(+2) to C/T, G(+3) to T, or C(+4) to T substitutions decreased TSS utilization, but A(+2) to G or T(+5) to C substitutions had minor effects 57.
Pairwise nucleotide-position dependencies have been observed for some processes, for example in 5′ splice sites 62-64. To investigate higher order sequence interactions i.e. positional coupling in TSS efficiency, we examined all pairwise interactions among positions −11 to +1 (Figure 3C, D, Extended Data Figure 2E, F). “Coupling” would entail a base at one position determining the contribution or effect of a base at another position. We found evidence for coupling at multiple positions with the strongest being between positions −9 and −8. Here, an A at one position suppresses the preference for A at the other. This observation is indicative of epistasis where an A at one position diminishes the impact of an A at the other (Extended Data Figure 2E). Using this −9/−8 interaction as an example, Figure 3C shows how coupling was detected and visualized. We mainly observed interactions at neighboring positions (Figure 3D). In addition to interaction with position −9 described above, position −8 was also showed interaction with position −7 (Extended Data Figure 2F).
Pol II mutants alter TSS efficiency in general
Pol II mutants affected total genomic usage of A−8 versus B−8 (non-A) TSSs in opposite directions depending on Pol II defect 21. This result could be a consequence of Pol II GOF or LOF mutants directionally shifting TSS usages coupled with the uneven distributions of bases and TSS motifs within promoters. Furthermore, TSS motifs with a −8A by definition have an increased probability of having a potential upstream TSS that can be used, e.g. a −8A could function as a +1R but might lack its own −8A leading to an apparent, but not real, alteration in TSS preference. To determine Pol II catalytic effects on initiation specificity, we measured TSS efficiencies for Pol II catalytic mutants (Figure 4, Extended Data Figure 3, Extended Data Figure 4). We introduced variant libraries into Pol II LOF mutants (rpb1 F1086S and H1085Q) and GOF mutants (rpb1 E1103G and G1097D). Data for all mutants showed high coverage of promoter libraries (Extended Data Figure 3A, Supplementary Table 1), were highly reproducible (Extended Data Figure 3B-E), and showed expected upstream or downstream shifts in TSS usage in aggregate across libraries (Extended Data Figure 3B) with broad effects on TSS efficiencies (Figure 4, Extended Data Figure 4). We found that LOF mutants decreased efficiencies across all sequences, and GOF mutants increased efficiencies across all sequences. Furthermore, mutants did not show strong effects on −8 in contrast to apparent effects on TSS usage from genomic promoters 21. Instead, we observed differences in Pol II mutants for efficiency of +1A TSS sequences relative to +1G TSSs (Figure 4A) across the range of TSS efficiencies (Figure 4B). These results are consistent with the potential for changes to the Pol II active site to alter activity for initiating NTPs.
Figure 4. Pol II mutants alter TSS efficiency for all possible TSS motifs while showing selective effects for base at +1.

(A) Pol II mutants alter TSS efficiencies across all motifs, corresponding to their direction of change to Pol II catalytic activity in vitro. TSS efficiency changes for each TSS variant were first determined by subtracting WT efficiency from Pol II mutant efficiency. The medians of efficiency changes for variant groups with indicated bases at each position relative to TSS were then calculated and illustrated in a heat map. Positive (red) values indicate Pol II mutants increased overall efficiency while negative (blue) values indicate decreased overall efficiency. (B) WT TSS efficiency for TSS variants divided into motif groups are plotted for mutant TSS efficiencies for the same TSS groups. The eight possible groups of TSSs for A/B−8C/T−1A/G+1 motifs were plotted and curve fit. Histograms show density of variants within each −8/−1/+1 subgroups. As to position −8, A−8 containing motifs show higher efficiency than B−8 containing motifs in both Pol II GOF (G1097D and E1103G) and LOF (F1086S and H1085Q) mutants (A−8 motifs: maroon and blue vs B−8 motifs: light coral and light blue). This is consistent with the proposed function of −8A to retain TSSs longer in the Pol II active site during scanning. This means that −8A may boost the positive effects of GOF mutants, therefore Pol II GOF mutants showed greater effects on A−8 motifs compared to B−8 motifs. In contrast, −8A compensates for active site defects of LOF mutants, therefore Pol II LOF mutants showed reduced effects on A−8 motifs compared to B−8 motifs. Both GOF and LOF mutants show reduced effects on G+1 motifs relative to A+1 motifs (G+1 motifs: light blue and blue vs A+1 motifs: light coral and maroon).
+1G TSSs are sensitive to GTP levels
Pol II active site alterations could result in altered substrate interactions, resulting in potential base-selective effects on TSS initiation efficiencies. Alternatively, NTP levels might be altered in Pol II mutants leading to indirect effects on initiation. To investigate if Pol II mutant effects for initiating NTPs could be due to altered NTPs, we measured NTP levels in Pol II WT, F1086S (LOF), and E1103G (GOF) strains (Figure 5A). WT and E1103G were similar while F1086S showed increased NTP levels. While both ATP and GTP were higher than WT in F1086S, we reasoned that initiation might be more sensitive to GTP levels as GTP levels are closer to the apparent KD of Pol II than are ATP levels. If so, the differential effect of F1086S on +1G versus +1A TSSs may relate to increased GTP levels buffering +1G TSSs from the F1086S effects to a greater extent than increased ATP levels would for +1A TSSs.
Figure 5. Pol II initiation is sensitive to NTP pools.

(A) NTP levels measured in WT, Pol II E1103G (GOF) and Pol II F1086S (LOF) mutants. For each genotype, N=6 biologically independent replicates were performed. Bars are mean +/− standard deviation. Statistical analyses by Ordinary one-way ANOVA with Dunnett’s multiple comparisons test for each NTP leave are shown. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. (B) NTP levels measured in WT, WT treated with 100% ethanol (the solvent for MPA) and WT treated with 20 ug/ml MPA. For each treatment, N=6 biologically independent replicates were performed. Bars are mean +/− standard deviation. Statistical analyses by RM one-way ANOVA with Dunnett’s multiple comparisons test for each NTP leave are shown. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. (C) MPA selectively decreases TSS efficiencies of +1G TSSs (right) but shows no effects on +1A TSSs (left) in Pol II MASTER libraries. The number (N) of TSSs examined and statistical analyses by Spearman’s rank correlation are shown. (D) MPA alters TSS efficiencies at genomic TSS positions. TSS efficiency change shown as difference between efficiency in EtOH and MPA treatment for all TSSs ≥ 2% efficiency in EtOH conditions within the 25%-75% of TSS distributions (see Methods). Lines on violin plots indicate medians computed based on density estimates. (E) Comparison of TSSs of any composition for first six nucleotides (NNNNNN) with those that lack G (HHHHHH, H=not G) or subsets of TSSs with A/C/G/T at positions +1 to +6. Lines on violin plots are as in D. All C/G/T datasets for all positions 1-5 are distinct from A as a baseline comparison (Wilcoxon rank sum test with continuity correction). At position 6 each base is not significantly different from A.
Reduction in GTP should result in selective reduction in initiation efficiency for +1G TSSs relative to +1A TSSs, if initiation was sensitive to GTP directly in vivo. Therefore, we examined how promoter variant libraries and genomic TSSs were affected by cell treatment with Mycophenolic Acid (MPA), which depletes cellular GTP through inhibition of inosine monophosphate dehydrogenase (IMPDH) activities encoded by IMD3 and IMD4 65,66. We treated our mixed AYR+BYR libraries with MPA or vehicle EtOH for 20 minutes prior to galactose induction. MPA decreased GTP level as expected while increasing ATP, CTP, and UTP levels (Figure 5B). MPA showed effects on promoter variant libraries in aggregate (Extended Data Figure 5A) with an overall downstream shift in TSS usage relative to control (Extended Data Figure 5A-B). When examining specific sequences, we observed a selective decrease in efficiency for essentially all +1G TSSs while +1A TSSs were essentially unaffected (Figure 5C). MPA was previously shown to depress efficiency of +1G TSSs at IMD2 67. Our results extend this observation to essentially all +1G TSSs in our MASTER library.
Genome-wide initiation changes in response to NTP changes
To determine if these effects were observed beyond our designed libraries, we performed TSS-seq on genome-derived RNAs from the same samples analyzed for MASTER. We found that MPA altered TSS efficiencies across the genome (Figure 5D, Extended Data Figure 5C). We observed efficiency reduction on average across all TSSs in MPA versus vehicle treatment. To explain global depression in TSS efficiency even for +1A TSSs, we speculated that there might be GTP effects at +2 or beyond, especially with initial bond formation requiring two substrates, and instability of initial transcribing complexes when RNAs are less than 5-6 nt. We found that TSSs specifying a G through position +5 show greater decreases in efficiency than non-G bases at those positions (Figure 5D, Extended Data Figure 5D). Conversely, we observed that presence of a +2C/U correlated with increased efficiency relative to +2A/G. These results are consistent with MPA-induced CTP and UTP increases promoting initiation efficiency for +2C/U TSSs. To examine TSSs more carefully for G effects, we examined TSSs lacking G entirely for the first 6 positions (Figure 5E). TSSs lacking a G in the first 6 positions were more efficient on average than all TSSs in the presence of MPA, explaining how MPA might globally affect TSS efficiency and demonstrating the effect of a single G in the initial transcript sequence.
Modeled TSS preferences predict genomic TSS efficiency
To ask how sequence determinants identified here relate to natural promoters, we compared our library-defined sequence efficiencies to TSS efficiencies observed at genomic promoters (Figure 6). To limit potentially confounding factors for genomic promoters, we focused on a single “median” TSS for each promoter in a defined set of promoter windows. The median TSS is defined as the TSS representing the position of 50th percentile of reads within each promoter window 21, i.e. the TSS representing the middle of the cumulative distribution function of TSS usage. We found that Pol II sequence preference at positions around median genomic TSSs was mostly consistent with MASTER data (Figure 3B, Figure 6A). Efficient genomic TSSs appear enriched for A at positions −7 to −5. The A-richness at positions between −10 to −3 and +5 to +10 has been noted in previous studies from our lab 21 (Figure 6B) and others 1,16,34. However, “A” bases at positions −7 to −5 were neutral in our promoter libraries (Figure 3B). The observed A-richness in the genome could reflect selection in vivo for additional promoter properties and not TSS efficiency per se. We confirmed the interaction between −9A and −8A discovered in our libraries is also reflected in genomic TSSs (Figure 6C). Presence of −9A decreased the enrichment of −8A relative to non-A at −9 (Figure 6C, left) for the top 20% expressed promoters. Moreover, when A was absent from position −8, a higher enrichment for −9A was observed (Figure 6C, right). These results suggest that −9A may function in similar fashion as −8A, but that −8A may be more effective and therefore has been evolutionarily favored (see Discussion).
Figure 6. Learned initiation preferences are predictive of TSS efficiencies at genomic promoters.

(A) Sequence preferences determined from TSSs representing the median of the distribution of usage from a set of 5979 yeast promoters (“median TSSs”) are congruent with library determined TSS preferences, except there is preference for A at positions −7 to −5 learned from genomic data. Sequence context and TSS efficiency of median TSSs were extracted from genomic TSS-seq data (GSE182792) 27. Calculation and visualization were as performed for promoter variant libraries. The number (N) of genomic median TSSs examined is shown. Statistical analyses by Spearman’s rank correlation test between relative efficiency at individual positions learned from promoter variant libraries and genomic median TSSs are shown beneath the sequence logo. (B) A-richness upstream and downstream of TSS is observed for highly expressed median TSSs. The number (N) of analyzed median TSSs is shown. Bars represent an approximate Bayesian 95% confidence interval. (C) Having an A at either position −9 or −8 reduces enrichment of A at the other position. Top 20% expressed median TSSs were divided into subgroups based on bases at position −9 or −8. Motif enrichment analysis was individually performed to subgroups. Numbers (N) of median TSSs within each subgroup are shown. Bars represent an approximate Bayesian 95% confidence interval.
Modeling identifies sequence features for TSS selection
We have found that DNA sequences around the TSS additively and interactively contribute to TSS efficiency. To quantitatively model sequence features for TSS efficiency, we compiled datasets deriving from all libraries and predicted TSS efficiency from sequence information by logistic regression (Figure 7A-E, Extended Data Figure 6A-C). We split compiled datasets generated from designed −8 to +2 and +4 TSSs deriving from “AYR”, “BYR”, and “ARY” libraries (Extended Data Figure 6A) into training (80%) and test (20%) sets. Sequences at positions −11 to +9 were extracted as potential predictors for TSS efficiency. We then used a forward stepwise strategy with 5-fold Cross-Validation (CV) to select robust features (predictors). By evaluating model performance with R2, sequences at nine positions (positions −9 to −7 and −4 to +2) and one interaction (−9/−8 interaction) were identified as robust features and selected for final modeling (Figure 7A).
Figure 7. Logistic regression model of DNA sequence contribution to TSS efficiency.
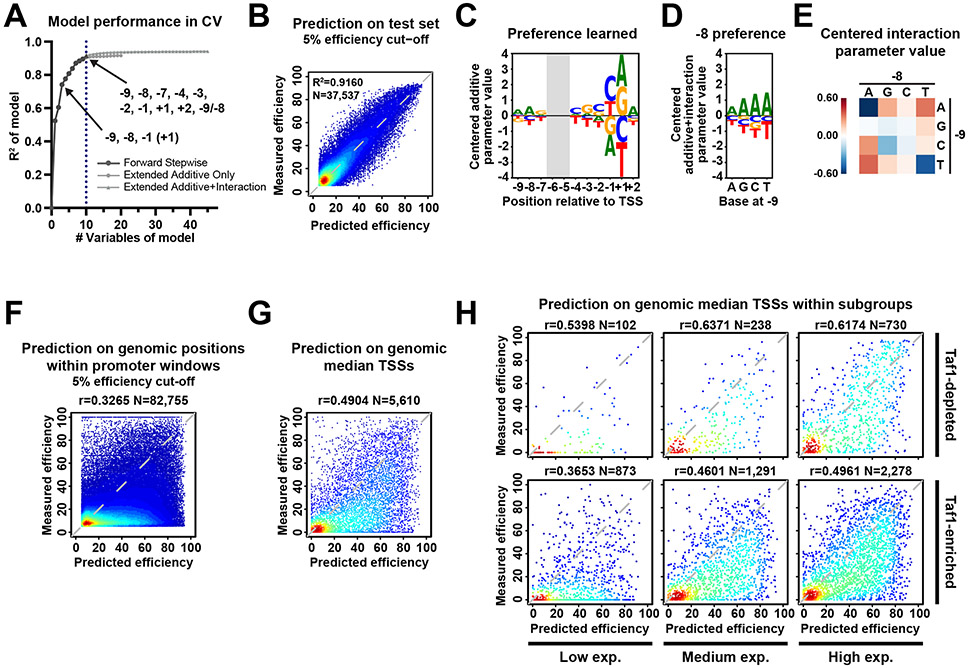
(A) Regression modeling identifies key DNA sequences and interactions contributing to TSS efficiency. Nine additive parameters plus one interaction were selected for the final model. Dots represents average R2 obtained in a 5-fold Cross-Validation (CV) strategy for logistic regression models using different numbers of features. The black line with SD error bars represents models with the best performance under a certain number of predictors. (B to E) Good performance of model including sequences at nine positions and the −9/−8 interaction indicates that TSS efficiency in our libraries is mainly regulated by the included features. (B) A scatterplot of measured and predicted efficiency of test sets, with a 5% efficiency cut-off. Model performance R2 on entire test and number (N) of data points are shown. (C) A sequence logo of centered additive parameters. The coefficients for bases were centered and visualized as a sequence logo. (D) A sequence logo showing learned preference at position −8 when different bases existing at position −9, with −9/−8 interaction included. The −9/−8 interaction parameters were added to corresponding additive coefficients for bases at position −8. The additive plus interaction parameters were then centered and visualized as sequence logos. (E) A heat map of centered parameters for −9/−8 interaction illustrating how bases at one position affect preference at another position. (F to G) Efficiency prediction for positions within known promoter windows in WT shows overall over-prediction. Scatterplots of comparison of measured and predicted TSS efficiencies of all positions (with a 5% efficiency cut-off) (F) or median TSSs (G) within 5979 known genomic promoter windows 21 with available measured efficiency. (H) Model shows better performance on Taf1-depleted promoters and promoters with medium to high expression. Scatterplots of comparison of measured and predicted TSS efficiency of median TSSs sub-grouped by promoter classes and expression levels. Genomic promoter expression was defined based on their total TSS-seq reads in the promoter window in the examined datasets: low, [0, 200); medium, [200, 1000); high, [1000, max). Pearson r and number (N) of compared variants are shown.
The final model containing the most predictive features explained 91.6% of the variance in TSS efficiency for WT test set while models with as few as three features could explain 74.1% of the variance (Figure 7A, B, Extended Data Figure 6B). Principal Component Analysis (PCA) on model variables trained with individual replicates across genotypes showed large differences between WT and Pol II mutants but very small differences between replicates (Extended Data Figure 6C), indicating that modeling captured features of different Pol II activity groups. We next asked whether the features learned by modeling were consistent with our sequence preference analysis using datasets with the most randomized bases (Figure 7C). As expected, positions −1 and +1 were major predictors, however the influence of −8 A was not as strong as in our simple preference analysis. After adding a −9/−8 interaction term, we observed emergence of the position −8 as an influential predictor (Figure 7D, E), emphasizing the contribution of the −9/−8 interaction. The +2A preference observed in previous motif enrichment analysis also emerged from modeling (Figure 7C, Extended Data Figure 2D). Furthermore, +2 preference appears to discriminate (Extended Data Figure 4D, Extended Data Figure 6C) between Pol II E1103G and G1097D and may indicate Pol II active site interactions with the +2 NTP.
Wider-context control of TSS efficiency during scanning
To evaluate the extent to which DNA sequence at TSSs contributes to TSS efficiency at genomic promoters, we compared the differences between observed and model-predicted efficiencies for all positions within genomic promoter windows or within specific subgroups of promoters (Figure 7F-H, Extended Data Figure 6D). As expected, we found most promoter positions showed low or no observed efficiency but were over-predicted by the sequence model (Figure 7F, Extended Data Figure 6D). This is explainable by TSSs needing to be specified by a core promoter followed by limited scanning. Therefore, features beyond TSS sequence, such as TSS distance from the site of PIC assembly, determine genomic initiation. We therefore extracted observed median TSSs as representatives of TSSs in contexts supporting efficient initiation to ask how our sequence-based predictor functioned on genomic TSSs (Figure 7G). We also separated median TSSs by promoter classes based on Taf1 enrichment 68, a proxy for the two main types of promoters in yeast, and by promoter expression levels (Figure 7H). We observed good prediction performance for many TSSs indicating that sequence determinants identified by MASTER contribute to TSS efficiency at genomic promoters. We observed increased performance for more highly expressed promoters and for Taf1-depleted promoters (Figure 7H). Conceivably, highly expressed or functionally promoters may be similarly sensitive to sequence effects as the MASTER promoter, explaining increased performance.
Discussion
Here we developed and employed Pol II MASTER to systematically investigate ~1 million TSS sequences in wildtype or Pol II mutant cells. This system allowed us to specifically and comprehensively study TSS efficiencies in initiation by promoter scanning by removing confounding effects from other architectural features, such as variability in core promoter-TSS distances, differences in promoter identities or chromatin configurations that may obfuscate analyses of genomic TSSs. We find that sequence variation at different positions around TSSs considerably tunes initiation efficiency in a predictable way and these contributions are important for initiation efficiency at genomic promoters.
Combining results from this study and others, we suggest how TSS selection during promoter scanning works through TSS sequence and Pol II activity (Figure 8). We find that two major sequence position groups contribute to TSS selection: bases around the TSS and bases around position −8. First, the TSS and adjacent bases interact with Pol II active site, recruiting the first and second NTPs, with early initiation in competition with continued scanning. Sensitivity of initiation efficiency to NTP levels for the first through fifth transcription positions in vivo supports this idea. This function of downstream positions in yeast are likely distinct from Inr or other elements as part of the TFIID or PIC binding site in other eukaryotes (see 69,70 and references therein). The universal initiating element, Y−1R+1, facilitates stable RNA polymerase binding to NTPs by base stacking between a template R−1 and initiating purine NTPs 43,44. Positions −4 to −2 upstream might contribute to this stacking or other template stabilization 44,71.
Figure 8. Model for TSS sequence preference regulated by multiple mechanisms.

Top panels show determined contribution of sequence at positions around TSS and proposed mechanisms. Two major groups of positions around TSS contribute to TSS selection: bases around TSS (actual initiating site) and bases around position −8. The TSS and adjacent bases interact with Pol II active site, the 1st NTP or each other to facilitate stable binding of 1st NTP thus stimulate RNA synthesis. −8 and −9 "T"s on the template strand with an additional interaction between −8 and −7 template strand positions may serve as an anchor point interacting with TFIIB B-reader domain allowing pausing of scanning and promotion of Pol II initiation at TSSs a fixed distance downstream, as proposed 45. These preferences are reflected as "A"s if the analysis is on the coding strand. Positions and interactions that were identified by regression modeling as robust features are labelled in bold. Bottom panel shows other architectural features involved in Pol II transcription initiation likely additionally contributing to TSS selection and initiation efficiency that will be accessible to Pol II MASTER analysis.
Selective effects of Pol II activity mutants on initiating NTP efficiency (Figure 4) supports that +1 NTP efficiency is directly sensitive to properties of the Pol II active site during initiation by promoter scanning. Meanwhile, the observation that Pol II initiation is sensitive to NTP pools (Figure 5C, Extended Data Figure 5A) reveals a mechanism for cellular state to regulate initiation via even modest alteration of NTP levels. For GOF Pol II mutants, we propose that differential preference for ATP versus GTP is a direct consequence of Pol II active site changes. Pol II has been observed to bypass cyclobutane pyrimidine dimers (CPD) in vitro through addition of an untemplated A across from CPD lesions and can be promoted by a GOF polymerase, consistent with increased selectivity for ATP in some situations 72. In contrast to GOF mutant effects, we propose that tested LOF mutants’ effects on +1A versus +1G TSS efficiency are due to indirect effects on cellular NTP levels. These defects might result from altered synthesis of nucleotide synthesis-related genes, some of which are themselves sensitive to Pol II activity 23,67,73-76. We observed the LOF mutant F1086S indeed alters all NTP levels (Figure 5A). Specifically, the LOF mutant increased GTP level, which may be due to the constitutive expression of IMD2 in Pol II LOF mutants 23,74, and the overall increase in expression for IMD genes at the mRNA level 23. Direct sensing and regulation of promoter activity by NTP levels has been proposed for the IMD2 promoter in yeast 67,77. Our results here argue that nucleotide sequence content at TSSs is sufficient to confer the observed regulation of IMD2 as we find that most +1G TSSs in the genome are sensitive to GTP levels.
Consistent with NTP effects described above, positions +2 to +4 downstream of TSS could establish initial RNA and NTP stability in the Pol II active site (Extended Data Figure 2D). We observed Pol II mutant effects on those preferences (Extended Data Figure 4D), suggesting these positions might function via directly interacting with the Pol II active site, as observed in other RNAP 43,44; for example, base-specific interactions of T7 RNAP 43 or base interactions of bacterial RNAP 44 with the +2 NTP. Alternatively, specific downstream bases might stabilize or promote Pol II translocation state as has been observed for bacterial RNAP 78. Our results detecting sensitivity in initiation efficiency based on levels of NTPs for positions downstream from +1 (Figure 5, Extended Data Figure 5) may relate to abortive initiation under conditions that slow phosphodiester bond formation early in transcript synthesis. Abortive initiation by yeast Pol II in vitro under NTP replete conditions is very low 79. However, these conditions bypassed initiation by a scanning PIC and were not tested at reduced NTP levels. Revisiting the biochemical properties of Pol II during bona fide initiation by promoter scanning could be valuable and our studies make specific predictions that can be tested.
The GTF TFIIB has been specifically implicated in the preference for bases near position −8, because the TFIIB B-reader domain has been observed to directly interact with a template T at −8 80. It is attractive to envision TFIIB functioning as an anchor point allowing pausing or slowing of the scanning process to promote Pol II initiation at a fixed distance downstream. In this model, it is plausible that sequences between −9 and −7 modulate or promote interaction with TFIIB during promoter scanning to engage initiation.
Whether or how DNA sequence surrounding TSSs is involved in other promoter properties is another question. “A” bases at positions −7 to −5 were observed to be neutral in our promoter libraries (Figure 3B), in contrast to the apparent A-enrichment observed for highly expressed and focused genomic TSSs (Figure 6A, B) 1,16,21,34,46. We speculate that observed A-richness around TSSs46,47 with T-richness upstream functions through other evolved promoter properties, for example facilitating template melting or lowering nucleosome occupancy 81 due to paucity G/C dinucleotides that promote it 82-85. TSS-proximal A-richness may remain from the evolution of promoter scanning. Examination of TSS usage in related yeasts suggests that A-richness upstream of TSSs was ancestral in promoter scanning, with subsequent refinement for −8A preference 45.
Our study highlights the strength of approaches that minimize contextual factors by isolating specific promoter attributes for study in high-throughput. Here we have applied Pol II MASTER to DNA sequence determinants of initiation efficiency during Pol II scanning. It will be valuable to apply this systematic analysis to other promoter architectural factors determining Pol II initiation, such as UAS identity, core promoter-TSS distance, and sequence composition within the scanning region. By applying Pol II MASTER across initiation mutants and promoter variants we may be able to determine initiation potential from DNA sequence and genome location alone.
Methods
Yeast strains, plasmids, oligonucleotides and media
Yeast strains, plasmids and oligonucleotide sequences are described in Supplementary Table 2-4. All oligonucleotides were obtained from IDT. Yeast strains used in this study were constructed as previously 21,23,73,86. Briefly, plasmids containing rpb1 mutants (G1097D, E1103G, F1086S, and H1085Q) were introduced by transformation into yeast strain CKY749 containing a chromosomal deletion of RPO21/RPB1 but with a wild type RPB1 URA3 plasmid, which was subsequently lost by plasmid shuffling. All plasmids and yeast strains are available upon request. Yeast media are following standard protocols 87. YPD solid medium is made of yeast extract (1% w/v; BD), peptone (2% w/v; BD, 211677), bacto-agar (2% w/v; BD, 214010) and dextrose (2% w/v; VWR, VWRBK876) supplemented with adenine (0.15 mM; Sigma-Aldrich, A9126) and L-tryptophan (0.4 mM; Sigma-Aldrich T0254). Minimal media plates are synthetic complete (“SC”) with amino-acids dropped out as appropriate as described in 87 with minor alterations as described in 23: per standard batch formulation, adenine hemisulfate (Sigma-Aldrich, A9126) was 2 g, L-Leucine (Sigma-Aldrich, L8000) was 4 g, myo-inositol was 0.1 g, para-aminobenzoic acid (PABA) was 0.2 g.
Construction and transformation of plasmid libraries
A 9 nt randomized TSS region and 20 nt randomized barcodes, with 4 fixed bases inserted between every 4 nt (NNNNANNNNCNNNNTNNNNGNNNN), were separately synthesized by IDT as oligo pools with specific randomized positions using “hand mixing” for N positions to ensure even randomization and avoid bias during machine mixing of precursors during oligo synthesis. Together with other components including the GAL1 UAS, SNR37 core promoter, SNR37 TSS region (“flux detector”), GFP ORF, and the CYC1 terminator, template libraries were constructed by PCR sewing and cloned into pRSII413 (a gift from Steven Haase, Addgene plasmid #35450; http://n2t.net/addgene:35450; RRID:Addgene_35450) 88 by ligation (Extended Data Figure 1A). Ligation products were transformed into Escherichia coli TOP10F’ cells (Invitrogen) and grown on LB plates supplemented with carbenicillin (100 μg/ml) at high density. 200,000-500,000 colonies were collected from each library to maximize variant representation. Plasmid libraries were isolated from cells pellets using ZymoPURE II Plasmid Maxiprep Kit (Zymo Research, D4203) per manufacturer’s instructions. Plasmid library pools were transformed into yeast strains with wildtype and mutated Pol II using chemical transformation and electroporation, respectively. For Pol II WT libraries, 500 ng plasmid pool per reaction was transformed following yeast transformation as described in 89. For Pol II mutant libraries, 2 μg plasmid pool per reaction was electroporated into Pol II mutant strains following yeast electroporation as described in 90, with 50 μg salmon sperm carrier DNA added. Transformants were grown on selective SC-His plates with 2% glucose as carbon source at high density. Three biological replicates were performed for each library and on average over two million colonies were collected apiece. Transformants scraped from densely plated transformation plates were inoculated into fresh SC-His medium with 2% raffinose (Amresco, J392) at 0.25 x 107 cells/ml and grown until 0.5-0.8 x 107 cells/ml. Subsequently, galactose (Amresco, 0637) was added for three hours (4% final concentration) to induce library expression. For MPA treated WT libraries, MPA (in 100% ethanol; Sigma-Aldrich, M3536) was added for 20 min (20 μg/ml final concentration) prior to the three-hour galactose induction, in parallel with the same volume of 100% ethanol as control. 50 ml and 5 ml culture aliquots, for RNA and DNA extraction, respectively, were harvested and then cell pellets were stored at −80 °C for further processing as described below.
Generation of DNA amplicons for DNA-seq
Plasmid DNA from yeast cell pellets was isolated using YeaStar Genomic DNA Kit (Zymo Research, D2002) per manufacturer’s instructions. Amplicon pools containing the TSS and barcode regions were generated using plasmid DNA from E.coli or yeast by Micellula DNA Emulsion & Purification (ePCR) Kit (EURx/CHIMERx, 3600) per manufacturer’s instructions. To minimize amplification bias, each sample was amplified in a 15-cycle ePCR reaction, purified and subject to an additional 10-cycle scale-up ePCR reaction. To create the necessary sequence template diversity for Illumina sequencing, 18-25 bp and 1-7 bp “stuffer” sequences were added to 5’- and 3’-ends, respectively, during amplicon preparation. Amplicon pools were subject to Illumina NovaSeq 6000 (150 PE) sequencing, and on average 20 M paired-end reads were obtained from each replicate, with high reproducibility and minimal perturbation of the variant distribution with each library (Supplementary Table 5).
Sample preparation for TSS-seq
Total RNA was extracted as in 91, followed by RNA purification (RNeasy Mini kit, QIAGEN, 74104) with on-column DNase digestion (RNase-Free DNase Set, QIAGEN, 79254) to remove DNA. TSS-seq was done using procedures described in 92. To prepare RNAs for cDNA library construction, samples were sequentially treated with Terminator 5’-Phosphate-Dependent Exonuclease (Lucigen), Quick CIP (calf-intestine alkaline phosphatase, NEB) and Cap-Clip™ Acid Pyrophosphatase (CellScript) to remove 5’ monophosphate RNA and convert 5’ triphosphate or capped RNAs to 5’ monophosphate RNAs. Next, RNA prepared with enzymatic treatments was ligated to the 5’-adapter (s1206-N15, 5’-GUUCAGAGUUCUACAGUCCGACGAUCNNNNNNNNNNNNNNN-3’) that contains Illumina adapter sequence and a 15 nt randomized 3’-end to reduce ligation bias and serve as a Unique Molecular Identifier (UMI). Next, cDNA was constructed by reverse transcription using RT primer CKO2191-s128A (5’-CCTTGGCACCCGAGAATTCCAAGTGAATAATTCTTCACCTTTA-3’) followed by emulsion PCR amplification for 20-22 cycles using Illumina PCR primers (RP1 and RPI3-30). Final DNA was gel size selected for 180-250 bp lengths and sequenced by Illumina NextSeq 500 (150 SE) or NovaSeq 6000 (200 SE) (Supplementary Table 6) using custom primer s1115 (5’-CTACACGTTCAGAGTTCTACAGTCCGACGATC-3’) to avoid potentially confounding effects of misannealing of the default pooled Illumina sequencing primers to the two randomized sequence regions.
Primer extension assay
Primer extension assays were performed on the same batch of total RNA extracted for TSS-seq as described in 93 with modifications described in 23. For each reaction, 30 μg total RNA was used. An RNA sample without library transformed was used as “no GFP” control. A sample containing same amount of nuclease-free water was used as “no RNA” control. A primer (CKO2191) complementary to the 6th to 27th bases of GFP ORF, which is the same annealing region for reverse transcription of TSS-seq sample preparation, was labeled with 32P γ-ATP (PerkinElmer, BLU502Z250UC) and T4 polynucleotide kinase (Thermo Scientific, EK0031). M-MuLV Reverse Transcriptase (NEB, M0253L), RNase inhibitor (NEB, M0307L), dNTPs (GE) and DTT were added to mix of RNA and labelled primer for reverse transcription. Before loading to sequencing gel, RNase A (Thermo Scientific, EN0531) was added to remove RNA. The products were analyzed by 8% acrylamide/bis-acrylamide (19:1 ratio, Bio-Rad, 1610145) gel containing 1x TBE and 7M Urea. Gels were visualized by a Molecular Imager PharosFX™ Plus System (Bio-Rad) and quantified by Image Lab 5.2 (Bio-Rad).
Determination of NTP levels
For each genotype or treatment, six biological replicates were performed. Cells from saturated overnight cultures were used to inoculate fresh SC medium containing 2% raffinose at 0.25 x 107 cells/ml and grown to a density of 0.5-0.8 x 107 cells/ml, as determined by cell counting. For each WT replicate, two 15 ml cultures were aliquoted and treated with 20 ug/ml MPA or 100% ethanol for 20 min, respectively. Subsequently, three-hour 4% galactose treatment was performed for all samples. About 1 x 108 cells were harvested, and then cell pellets were snap frozen using liquid nitrogen and immediately stored at −80 °C for further processing as described below.
Metabolic quenching and nucleotide phosphate metabolite pool extraction were performed by adding 1 ml ice-cold 2:2:1 acetonitrile:methanol:water with 0.1% formic acid with 1x MS-Stop phosphatase inhibitor (Sigma-Aldrich). Samples were spiked with 10 μl 100 mM mix of adenosine-15N5-5′-triphosphate, guanosine-15N5-5′-triphosphate, thymidine-13C10,15N2-5′-triphosphate, cytosine-2H14-5′-triphosphate, and uridine-15N2-5′-triphosphate (Sigma-Aldrich). Samples were homogenized via sonification at room temperature, and the supernatant was then cleared of protein by centrifugation at 16,000 xg. The protein pellet was resuspended in RIPA buffer for protein normalization. The cleared supernatant was dried down under nitrogen gas and resuspended in 100 μl 7.5 mM ammonium acetate / 0.05% ammonium hydroxide. 2μl of cleared supernatant was subjected to online LC-MS analysis. Purified adenosine-5′-triphosphate, guanosine-5′-triphosphate, thymidine-5′-triphosphate, cytosine-5′-triphosphate, and uridine-5′-triphosphate (Sigma-Aldrich) were serially diluted from 50 pmol/μl to 0.39 pmol/μl to generate calibration curves.
Analyses were performed by untargeted LC-HRMS. Briefly, samples were injected via a Thermo Vanquish UHPLC and separated over a reversed phase Phenomenex Kinetix-Polar C18 column (2.1 x 100 mm, 1.7 μm particle size) maintained at 55°C. For the 22.5-minute LC gradient, the mobile phase consisted of the following: solvent A (water/7.5 mM ammonium acetate/0.05% ammonium hydroxide) and solvent B (acetonitrile/0.05% ammonium hydroxide). The gradient was the following: 0-.1 min 2% B, increase to 70% B over 12 minutes, increase to 98% B over 0.1 min, hold at 98% B for 5 minutes, re-equilibrate at 2% B for five minutes. The Thermo IDX tribrid mass spectrometer was operated in positive ion mode, scanning in ddMS2 mode (2 μscans) from 200 to 800 m/z at 120,000 resolution with an AGC target of 2e5 for full scan, 2e4 for ms2 scans using HCD fragmentation at stepped 15,35,50 collision energies. Source ionization setting was 3.0 kV spray voltage for positive mode. Source gas parameters were 35 sheath gas, 12 auxiliary gas at 320°C, and 8 sweep gas. Calibration was performed prior to analysis using the Pierce™ FlexMix Ion Calibration Solutions (Thermo Fisher Scientific). Integrated peak areas were then extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). Calibration curves using purified standards were then used to convert peak area ratios to concentration.
DNA-seq analysis
High-throughput sequencing of template DNA amplicon products was used to assign each 9 nt randomized TSS sequence to a corresponding 24 nt barcode. First, paired-end reads were merged using PEAR (0.9.11) 94. Next, we considered only those reads that contained a perfect match to three sequence regions common to all variants: 27 nt sequence upstream of the TSS region, 24 nt sequence between TSS region and barcode, and 27 nt sequence downstream of barcode (5’-TTCAAATTTTTCTTTTGATTTTTTTTCNNNNNNNNNACATTTTCAAAAGGCTAACATCAGNNNNANNNNCNNNNTNNNNGNNNNATGTCTAAAGGTGAAGAATTATTCACT-3’, randomized TSS and barcode regions are underlined). From these reads, 9 nt TSS region and 24 nt barcode were extracted, followed by individual error correction using UMI-tools (1.0.0) 95. Next, for barcodes linked to multiple TSS variants, only barcodes for which >= 90% of the sequencing reads containing a specified barcode also contained a shared, exact 9 nt TSS region were kept. To generate a master pool of TSS-barcode linkages for all TSS-seq samples, for each library (“AYR”, “BYR”, “ARY”), TSS-barcode linkages that existed in at least two out of four samples (one E.coli sample plus three WT yeast replicates) and in which >= 5 reads existed were kept and pooled. Two types of processed data are available in GEO database, with accession numbers listed in (Supplementary Table 5): tables containing TSS-barcode linkages and corresponding DNA-seq read counts for each sample, tables of the master pool containing kept TSS-barcode linkages and corresponding DNA-seq read count in all related samples.
TSS-seq analysis for libraries
High-throughput sequencing of RNA samples was used to link RNA products to barcodes, therefore assigning TSS usage to corresponding DNA templates. For TSS identification and subsequent preference analysis, we considered only those reads that contained a perfect match to a 27 nt sequence region downstream of the barcode, as well as expected length of 5’-end: 5’-[15 nt 5’-UMI]-[>1 nt upstream of barcode region, designated as “RNA 5’-end”]-[24 nt barcode]-[the first 27 nt of GFP ORF, ATGTCTAAAGGTGAAGAATTATTCACT]-3’. Next, 15 nt 5’-UMIs, “RNA 5’-end” with varying length, and 24 nt barcode were extracted and individually corrected by UMI-tools (1.0.0). Deduplication was performed based on 5’-UMIs, meaning reads containing a shared UMI-“RNA 5’-end”-barcode linkage were counted as one deduplicated read for further analysis. Next, the identity of the 24 nt barcode was used to determine the template sequences of randomized TSS region. Only reads with “RNA 5’-end” sequence perfectly matched to corresponding template sequence were used for analysis of TSS efficiency, but all deduplicated reads, including reads with mismatch(es), were used for analysis of relative expression. Next, a TSS-seq count table containing TSS usage distribution of each TSS promoter variant was generated. In the count table, each row represents one TSS promoter variant, and each column represents one position between positions −68 to +25 relative to “designed” +1 TSS. The number in each cell represents TSS-seq reads generated from a particular position, with perfectly match to the DNA template. After investigating reproducibility (Extended Data Figure 3B-C, Extended Data Figure 5A-C), count tables generated from three biological replicates were merged into one by aggregating read counts at each position. Promoter variants with >= 5 TSS-seq reads in each replicate and whose Coefficient of Variation (CV) of TSS-seq reads is <= 0.5 were kept. Two types of processed data are available in GEO database, with accession numbers listed in Supplementary Table 6: tables containing “RNA 5’-end”-Barcode linkages and corresponding deduplicated TSS-seq read counts for individual sample, TSS-seq count tables for individual samples and after aggregating replicates. As an example, a TSS-seq count table including positions −10 to +25 relative to “designed” +1 TSS after aggregating replicates of WT libraries is shown as Supplementary Table 7.
TSS efficiency calculation
TSS efficiency for each position was calculated by dividing reads count at a particular position by the reads at or downstream of this TSS. TSS positions with >= 20% efficiency but with <= 5 reads left for this and following positions were filtered out, as well as their downstream positions.
Relative expression analysis for libraries
The relative expression from each library promoter variant was defined as the ratio of normalized TSS-seq reads generated from a particular promoter variant to normalized DNA-seq reads containing the variant. The normalized DNA-seq or RNA-seq reads were calculated by dividing reads per promoter variant by the total number of reads per sample, followed by averaging three biological replicates. Promoter variants with >= 10 DNA-seq reads in each replicate and whose Coefficient of Variation (CV) of normalized DNA-seq reads is <= 0.5 were kept.
Sequence preference analysis
For sequence preference at each position, all TSS variants were subgrouped based on the bases at a particular position. TSS efficiencies of TSS variants were visualized as scatter plots using GraphPad Prism 9. Kruskal-Wallis with Dunn’s test was performed to test sequence preference in GraphPad Prism 9. Next, TSS efficiency medians of each subgroup were calculated and centered to calculate “relative efficiency” at each position. The relative efficiencies were visualized as sequence logos using Logomaker (0.8) 96. In motif enrichment analysis, surrounding sequences relative to examined TSSs were extracted and visualized as sequence logos using WebLogo 3 97. Heatmaps, scatter plots and density plots for comparing Pol II WT and mutants were generated by Morpheus (https://software.broadinstitute.org/morpheus) or ggplot2 (3.3.3) R package.
Interaction analysis
The interaction between positions is defined as different bases existing at one position resulting in different sequence preferences at another position. For any two positions, all TSS variants were subgrouped based on bases at both positions. Median values of TSS efficiency distribution of each subgroup were calculated and centered twice to calculate “centered relative efficiency”. The centered relative efficiencies were visualized as heatmaps using Seaborn (0.11.0). Interactions related to positions between −11 to −9 were calculated using datasets of designed +4 TSS deriving from “AYR”, “BYR” and “ARY” libraries. Other interactions were calculated using datasets of designed +1 TSS deriving from “AYR” and “BYR” libraries.
TSS-seq analysis for genomic TSSs
Genomic TSS-seq datasets from our lab’s previous study were used for comparison of model to in vivo TSS usage 27. Quality control, read trimming and mapping were performed as described in 21,27 to generate a TSS count table that contains TSS-seq reads at each individual position within known promoter windows (“median” TSS, 250 nt upstream and 150 nt downstream from median TSS position). TSS efficiency calculation and subsequent sequence preference analyses were performed as that for Pol II MASTER libraries. EtOH and MPA treatment for genomic TSSs are described in Results and these libraries were analyzed as TSS-seq was above. TSSs within a promoter were analyzed by their positions within the cumulative distribution of usage from upstream to downstream, with 50% of the distribution representing the median TSS position. To limit potential effects of marginal TSSs, analyses of MPA treatment were limited to TSSs showing ≥2% efficiency in the EtOH condition within the 25%−75% of the TSS distribution.
Prediction of TSS efficiency
To prepare datasets for modeling, positions of designed −8 to +2 and +4 TSSs of each promoter variant that have valid TSS efficiency were compiled as sequence variants. For each TSS variant, sequences at −11 to +9 positions relative to TSS, together with corresponding TSS efficiency, were extracted. 80% of dataset were randomly partitioned as training set and the rest 20% as testing set. To select robust features, a forward stepwise strategy with a 5-fold Cross-Validation (CV) was employed in two major stages, for additive terms and for interactions. Starting with no variable in the model, logistic regression models with one additional variable (the sequence at a particular position) were trained to predict TSS efficiency on training set by train() of caret (6.0.86) R package 98, with a 5-fold CV. The R2, representing the proportion of variance explained, was calculated to indicate the performance of each model. The variable that provides the highest increased R2 for model was added into the model for next round of variable selection. This process was repeated until the increased R2 is less than 0.01. After identifying the most influential additive variables, same process was repeated for investigating robust interactions between selected additive variables. Next, a final model with selected robust features. including additive variables and interactions, was constructed on entire training set using glm() and investigated on testing set. Comparison between predicted and measured efficiencies was visualized as scatter plots using LSD (4.1.0) R package. Model parameters were extracted and used to further calculation. Visualizations were done in Logomaker (0.8) and Seaborn (0.11.0) in Python. Principal Component Analysis (PCA) was performed using prcomp() in R.
Extended Data
Extended Data Figure 1. High level of reproducibility and coverage depth of library variants.

(A) Schematic of experimental approach. Promoter libraries with almost all possible sequences within a 9 nt randomized region were constructed on plasmids. Libraries were designated “AYR”, “BYR”, and “ARY” based on randomized region composition. Plasmids were amplified in E. coli and transformed into yeast with wild type or mutated Pol II. DNA and RNA were extracted and prepared for DNA-seq and TSS-seq. (B) Base frequencies at positions within the randomized region of promoter variants demonstrate unbiased synthesis of randomized regions. Bars are mean +/− standard deviation of the mean for promoter variants in WT and four Pol II mutants. (C) Heatmap illustrating hierarchical clustering of Pearson correlation coefficients of reads per promoter variant E. coli libraries and three biological replicates of libraries transformed into yeast. (D) Example correlation plots of DNA reads count of promoter variants for E. coli and yeast WT biological replicates. Pearson r and number of compared variants are shown. (E) Bulk primer extension for RNA produced from promoter variant libraries transformed into WT yeast. “No GFP” control used RNA from an untransformed strain. “No RNA” control used a sample of nuclease-free water. Dots represent three biological replicates. Bars are mean +/− standard deviation of the mean. (F) TSS usage based TSS-seq read lengths from transformed libraries. Dots represent three biological replicates. Bars are mean +/− standard deviation of mean. Distributions are similar to the distributions in E. Note that primer extension will blur usage into adjacent upstream position due to some level of non-templated addition of C to RNA 5’ ends. (G) Heat scatter plots of Coefficient of Variation (CV, y axis) versus total RNA reads per promoter variant in each Pol II MASTER library. A cutoff of CV = 0.5 was used to filter higher variance variants. (H) Heat scatter plots of relative expression versus TSS efficiency of major TSSs per promoter variant, with contour lines indicating deciles of data. Number of promoter variants with [−1, +1] relative expression values (log2) and corresponding percentage of total promoter variants are shown.
Extended Data Figure 2. Surrounding sequence of TSSs modulates initiation efficiency.

(A) +1 TSS efficiency of all −7 to −2 sequences within each N−8N−1N+1 motif in WT, rank ordered by efficiency of A−8C−1A+1 version shown as a heat map. x-axis is ordered based on median efficiency for each N−8N−1N+1 motif group, as shown in Figure 2B. Spearman’s rank correlation tests between A−8C−1A+1 group and all groups are shown beneath the heat map. (B) Efficiencies of designed +1 TSSs grouped by base identities between −8 and +1 positions. Statistical analyses by Kruskal-Wallis with Dunn’s multiple comparisons test for base preference at individual positions relative to +1 TSS are shown beneath plots. Lines represent median values of subgroups. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05. (C) Histogram showing the distribution of measured efficiencies for all designed −8 to +4 TSSs of all promoter variants from “AYR”, “BYR” and “ARY” libraries in WT. Dashed lines mark the 5% efficiency cutoff. (D) A+2G+3G+4 motif enrichment is apparent for the top 10% most efficient designed −8 TSS. A(/G)+2G(/C)+3G(/C)+4 motif enrichment was observed for the top 10% most efficient −8 TSSs but not for the next 10% most efficient TSSs. A(/G)+1 enrichment observed for top 20% most efficient TSSs is consistent with the +1R preference of TSS. Numbers (N) of variants assessed are shown. Sequence logos were generated using WebLogo 3. Bars represent an approximate Bayesian 95% confidence interval. (E) An A at position −9 results in different sequence preferences at position −8. The dataset of designed +4 TSSs deriving from “AYR”, “BYR” and “ARY” libraries was used to detect the −9/−8 interaction. All variants were divided into 16 subgroups defined by bases at positions −9 and −8 relative to designed +4 TSS, and then their TSS efficiencies were plotted. Lines represent median values of subgroups. (F) An A at position −8 results in different sequence preferences at position −7. The dataset of designed +1 TSSs deriving from “AYR” and “BYR” libraries was used to detect −8/−7 interaction. Calculations same as −9/−8 interaction described in E.
Extended Data Figure 3. High level of reproducibility of library variants in Pol II mutants.

(A) Histograms showing the distribution of measured efficiencies for all designed −8 to +4 TSSs for MASTER libraries in Pol II mutants. Dashed lines mark the 5% efficiency cutoff with number of TSS variants shown. (B) TSS usage distributions at designed −10 to +25 TSSs for MASTER libraries in Pol II mutants. Dots represent three biological replicates. Bars are mean +/− standard deviation. (C) Hierarchical clustering of Pearson correlation coefficients of TSS efficiencies for major TSSs (designed +1 TSS for “AYR” and “BYR” libraries, +2 TSS for “ARY” library) for WT or Pol II mutants illustrated as a heat map for three biological replicates. (D) Example correlation plots of TSS efficiency of major TSSs between representative biological replicates. Pearson r and number of compared variants are shown. (E) Plots of CV versus total RNA reads (three biological replicates) for Pol II mutants. The red dashed lines mark the CV = 0.5 cutoff, an arbitrary cutoff for promoters with reasonable reproducibility across replicates. G1097D replicates contain outliers because this mutant is susceptible to genetic suppressors. A suppressor existing in one biological replicates generates a high CV allowing filtering.
Extended Data Figure 4. Pol II mutants alter TSS efficiency in general.

(A) TSS efficiency distributions of designed +1 TSSs of Pol II mutants for base subgroups at individual positions relative to +1. Identical analysis as in Extended Data Figure 2B for WT Pol II. (B) Pol II GOF G1097D showed greater increase in efficiency than GOF allele E1103G at upstream TSSs (designed −32 and −8 TSSs), while E1103G showed stronger effects at designed +1 TSS than G1097D. (C) Pol II initiation sequence preference in Pol II mutants. Identical analysis as in Figure 3B for WT Pol II. (D) Motif enrichment for top the 10% most efficient −8 TSSs for Pol II mutants. Identical motif enrichment analysis as in Extended Data Figure 2D top panel for WT Pol II. Numbers (N) of variants assessed are indicated. Bars represent an approximate Bayesian 95% confidence interval.
Extended Data Figure 5. High of reproducibility of TSS usage and efficiency upon MPA treatment.

(A) TSS usage distributions at designed −10 to +25 TSSs in WT “NYR” library (mixed AYR and BYR libraries) treated with 100% ethanol or with 20 ug/ml MPA. MPA treatment shifted TSS usage downstream relative to EtOH treatment. Dots represent three biological replicates. Bars are mean +/− standard deviation of the mean. (B) Hierarchical clustering of Pearson correlation coefficients of TSS efficiencies for designed +1 TSS for three biological replicates for MPA or EtOH treatment, illustrated as a heat map. (C) Hierarchical clustering of Pearson correlation coefficients of TSS efficiencies for all genome positions within defined promoter windows with >=3 reads in each replicate, illustrated as a heat map. (D) Correlation plots for combined biological replicates for TSS efficiency upon MPA treatment (y axes) versus EtOH treatment (x axes) for all TSSs ≥ 2% efficiency in the 25%-75% of the distribution for a curated set of 5979 yeast promoters (see Methods). TSSs are separated into groups depending on base identity at positions −3 (control) or positions +1 to +6.
Extended Data Figure 6. Modeling identifies sequence features for TSS selection in WT and Pol II mutants.

(A) Overview of TSS efficiency modeling. (1) TSS efficiencies including designed −8 to +2 and +4 TSSs deriving from “AYR”, “BYR” and “ARY” libraries were pooled for modeling. (2) Sequences from −11 to +9 relative to variant TSSs were extracted. (3) To identify robust features, a forward stepwise selection strategy coupled with a 5-fold cross-validation for logistic regression was used, with random splitting into training (80%) and test (20%) sets. Stepwise regression starting with a constant term only with stepwise variable addition, until a stopping criterion is met, was performed. Additive terms (sequences at positions −11 to +9) and interactions were tested in stages. Model performance was evaluated with R2. The stopping criterion for adding additional variables was an increase R2 < 0.01. (4) A logistic regression model containing selected robust features was trained using the training set and then evaluated with the test set. (B) Comparison of measured efficiencies and predicted efficiencies. Model performance R2 on entire test set and number of data points shown in plot are shown. (C) PCA analysis for parameters of models trained using individual replicates of WT and Pol II mutant. Close clustering of individual replicates indicates that models are not overfit. The top 15 contributing variables are shown. GOF and LOF mutants were separated from WT by the 1st principal component. GOF G1097D and E1103G were further distinguished by 2nd principal component by additional position +2 information, which is consistent with results in Extended Data Figure 4D, where G1097D and E1103G differentially altered +2 sequence enrichment. (D) A scatterplot of comparison of measured and predicted TSS efficiencies of all positions within 5979 known genomic promoter windows 21 with available measured efficiency. Pearson r and number (N) of compared variants are shown. Most promoter positions (82%, 1,678,406 out of 2,047,205) showed no observed efficiency, which is expected because TSSs need to be specified by a core promoter and scanning occurs over some distance downstream.
Supplementary Material
Acknowledgements
The authors thank Kaplan lab members for helpful comments on the manuscript. We are deeply grateful to Chenxi Qiu for discussions and comments on this project. We acknowledge Justin Kinney (Cold Spring Harbor Laboratory) and Shuoran Li (Statistical Consulting Center at University of Pittsburgh) for discussions on modeling. We thank Charles D. Johnson, Richard Metz (Texas A&M AgriLife Genomics and Bioinformatics Service), Andrew Hillhouse (Texas A&M Institute for Genome Sciences & Society), William A MacDonald, Rania Elbakri (the University of Pittsburgh Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh), Yinghong Pan (the UPMC Genome Center), Dibyendu Kumar (the Waksman Genomics Core Facility at Rutgers University), and Liz Freeman (Illumina) for discussions and advice regarding deep sequencing strategies. We thank Steven J. Mullett and Stacy Gelhaus Wendell (Metabolomics and Lipidomics Core, NIHS10OD023402) for performing NTP measurements. We acknowledge support from NIH grant R01GM097260 to C.D.K. for the early part of this work and NIH grants R01GM120450 and R35GM144116 to C.D.K. and R35GM118059 to B.E.N. This research was supported in part by the University of Pittsburgh Center for Research Computing, RRID:SCR_022735, through the resources provided. Specifically, this work used the HTC cluster, which is supported by NIH award number S10OD028483.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data availability
Raw sequencing data generated in this study are available in the NCBI BioProject, under the accession number PRJNA766624. Processed data are available in GEO, under the accession number GSE185290. Source data are provided with the manuscript.
Code availability
Code for analyses in this study is provided at https://github.com/Kaplan-Lab-Pitt/PolII_MASTER-TSS_sequence.
References
- 1.Zhang Z & Dietrich FS Mapping of transcription start sites in Saccharomyces cerevisiae using 5' SAGE. Nucleic Acids Res 33, 2838–51 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park D, Morris AR, Battenhouse A & Iyer VR Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res 42, 3736–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelechano V, Wei W & Steinmetz LM Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497, 127–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chia M et al. High-resolution analysis of cell-state transitions in yeast suggests widespread transcriptional tuning by alternative starts. Genome Biol 22, 34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nepal C et al. Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Res 23, 1938–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium F et al. A promoter-level mammalian expression atlas. Nature 507, 462–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamashita R et al. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res 21, 775–89 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carninci P et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–63 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Hoskins RA et al. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res 21, 182–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng H et al. Global identification of transcription start sites in the genome of Apis mellifera using 5'LongSAGE. J Exp Zool B Mol Dev Evol 316, 500–14 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Chen RA et al. The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Res 23, 1339–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng Z et al. Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923 e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojas-Duran MF & Gilbert WV Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18, 2299–305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batut P, Dobin A, Plessy C, Carninci P & Gingeras TR High-fidelity promoter profiling reveals widespread alternative promoter usage and transposon-driven developmental gene expression. Genome Res 23, 169–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P et al. Relatively frequent switching of transcription start sites during cerebellar development. BMC Genomics 18, 461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu Z & Lin Z Pervasive and dynamic transcription initiation in Saccharomyces cerevisiae. Genome Res 29, 1198–1210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demircioglu D et al. A Pan-cancer Transcriptome Analysis Reveals Pervasive Regulation through Alternative Promoters. Cell 178, 1465–1477 e17 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Thorsen K et al. Tumor-specific usage of alternative transcription start sites in colorectal cancer identified by genome-wide exon array analysis. BMC Genomics 12, 505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyd M et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nat Commun 9, 1661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lis C.G.a.J.T. DNA Melting on Yeast RNA Polymerase II Promoter. Science 261, 759–762 (1993). [DOI] [PubMed] [Google Scholar]
- 21.Qiu C et al. Universal promoter scanning by Pol II during transcription initiation in Saccharomyces cerevisiae. Genome Biology 21(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehner JN & Brow DA Quantitative analysis of in vivo initiator selection by yeast RNA polymerase II supports a scanning model. J Biol Chem 281, 14119–28 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Kaplan CD, Jin H, Zhang IL & Belyanin A Dissection of Pol II trigger loop function and Pol II activity-dependent control of start site selection in vivo. PLoS Genet 8, e1002627 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller G & Hahn S A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat Struct Mol Biol 13, 603–10 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fazal FM, Meng CA, Murakami K, Kornberg RD & Block SM Real-time observation of the initiation of RNA polymerase II transcription. Nature 525, 274–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hampsey M Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev 62, 465–503 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao T et al. Ssl2/TFIIH function in transcription start site scanning by RNA polymerase II in Saccharomyces cerevisiae. Elife 10(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahn S, H.E., Guarente L Each of three "TATA elements" specifies a subset of the transcription initiation sites at the CYC-1 promoter of Saccharomy cescerevisiae. Proceedings of the National Academy of Sciences 82, 8562–6 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortes T et al. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep 5, 1121–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bucher P Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J Mol Biol 212, 563–78 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Smale ST & Baltimore D The "initiator" as a transcription control element. Cell 57, 103–13 (1989). [DOI] [PubMed] [Google Scholar]
- 32.Corden J et al. Promoter sequences of eukaryotic protein-coding genes. Science 209, 1406–14 (1980). [DOI] [PubMed] [Google Scholar]
- 33.McNeil JB & Smith M Saccharomyces cerevisiae CYC1 mRNA 5'-end positioning: analysis by in vitro mutagenesis, using synthetic duplexes with random mismatch base pairs. Mol Cell Biol 5, 3545–51 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malabat C, Feuerbach F, Ma L, Saveanu C & Jacquier A Quality control of transcription start site selection by nonsense-mediated-mRNA decay. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Policastro RA, Raborn RT, Brendel VP & Zentner GE Simple and efficient profiling of transcription initiation and transcript levels with STRIPE-seq. Genome Res 30, 910–923 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Healy AM, Helser TL & Zitomer RS Sequences required for transcriptional initiation of the Saccharomyces cerevisiae CYC7 genes. Mol Cell Biol 7, 3785–91 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furter-Graves EM & Hall BD DNA sequence elements required for transcription initiation of the Schizosaccharomyces pombe ADH gene in Saccharomyces cerevisiae. Mol Gen Genet 223, 407–16 (1990). [DOI] [PubMed] [Google Scholar]
- 38.Carninci P et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38, 626–35 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Hashimoto S et al. 5'-end SAGE for the analysis of transcriptional start sites. Nat Biotechnol 22, 1146–9 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Suzuki Y et al. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep 2, 388–93 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D et al. Comparative Analysis of Regulatory Elements between Escherichia coli and Klebsiella pneumoniae by Genome-Wide Transcription Start Site Profiling. Plos Genetics 8(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vvedenskaya IO et al. Massively Systematic Transcript End Readout, "MASTER": Transcription Start Site Selection, Transcriptional Slippage, and Transcript Yields. Mol Cell 60, 953–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gleghorn ML, Davydova EK, Basu R, Rothman-Denes LB & Murakami KS X-ray crystal structures elucidate the nucleotidyl transfer reaction of transcript initiation using two nucleotides. Proceedings of the National Academy of Sciences of the United States of America 108, 3566–3571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basu RS et al. Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J Biol Chem 289, 24549–59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Z & Lin Z The origin and evolution of a distinct mechanism of transcription initiation in yeasts. Genome Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maicas E & Friesen JD A sequence pattern that occurs at the transcription initiation region of yeast RNA polymerase II promoters. Nucleic Acids Res 18, 3387–93 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lubliner S, Keren L & Segal E Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res 41, 5569–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dujon B The yeast genome project: what did we learn? Trends Genet 12, 263–70 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Lubliner S et al. Core promoter sequence in yeast is a major determinant of expression level. Genome Research 25, 1008–1017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blazeck J, Garg R, Reed B & Alper HS Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol Bioeng 109, 2884–95 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Dhillon N et al. Permutational analysis of Saccharomyces cerevisiae regulatory elements. Synth Biol (Oxf) 5, ysaa007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Schilbach S, Ninov M, Urlaub H & Cramer P Structures of transcription preinitiation complex engaged with the +1 nucleosome. Nat Struct Mol Biol (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vvedenskaya IO, Goldman SR & Nickels BE Analysis of Bacterial Transcription by "Massively Systematic Transcript End Readout," MASTER. Methods Enzymol 612, 269–302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vvedenskaya IO et al. Interactions between RNA polymerase and the core recognition element are a determinant of transcription start site selection. Proc Natl Acad Sci U S A 113, E2899–905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winkelman JT et al. Multiplexed protein-DNA cross-linking: Scrunching in transcription start site selection. Science 351, 1090–3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hochschild A Mastering Transcription: Multiplexed Analysis of Transcription Start Site Sequences. Mol Cell 60, 829–31 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Faitar SL, Brodie SA & Ponticelli AS Promoter-specific shifts in transcription initiation conferred by yeast TFIIB mutations are determined by the sequence in the immediate vicinity of the start sites. Molecular and Cellular Biology 21, 4427–4440 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deshpande AP & Patel SS Mechanism of transcription initiation by the yeast mitochondrial RNA polymerase. Biochim Biophys Acta 1819, 930–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Javahery R, Khachi A, Lo K, Zenzie-Gregory B & Smale ST DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol Cell Biol 14, 116–27 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arkhipova IR Promoter elements in Drosophila melanogaster revealed by sequence analysis. Genetics 139, 1359–69 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yarden G, Elfakess R, Gazit K & Dikstein R Characterization of sINR, a strict version of the Initiator core promoter element. Nucleic Acids Res 37, 4234–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong MS, Kinney JB & Krainer AR Quantitative Activity Profile and Context Dependence of All Human 5' Splice Sites. Mol Cell 71, 1012–1026 e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roca X et al. Features of 5'-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res 18, 77–87 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carmel I, Tal S, Vig I & Ast G Comparative analysis detects dependencies among the 5' splice-site positions. RNA 10, 828–40 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McPhillips CC, Hyle JW & Reines D Detection of the mycophenolate-inhibited form of IMP dehydrogenase in vivo. Proc Natl Acad Sci U S A 101, 12171–6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hyle JW, Shaw RJ & Reines D Functional distinctions between IMP dehydrogenase genes in providing mycophenolate resistance and guanine prototrophy to yeast. J Biol Chem 278, 28470–8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuehner JN & Brow DA Regulation of a eukaryotic gene by GTP-dependent start site selection and transcription attenuation. Mol Cell 31, 201–11 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Rhee HS & Pugh BF Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vo ngoc L, Huang CY, Cassidy CJ, Medrano C & Kadonaga JT Identification of the human DPR core promoter element using machine learning. Nature 585, 459–463 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luse DS, Parida M, Spector BM, Nilson KA & Price DH A unified view of the sequence and functional organization of the human RNA polymerase II promoter. Nucleic Acids Res 48, 7767–7785 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y et al. Structural basis of transcription initiation. Science 338, 1076–80 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walmacq C et al. Mechanism of translesion transcription by RNA polymerase II and its role in cellular resistance to DNA damage. Mol Cell 46, 18–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braberg H et al. From structure to systems: high-resolution, quantitative genetic analysis of RNA polymerase II. Cell 154, 775–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malik I, Qiu C, Snavely T & Kaplan CD Wide-ranging and unexpected consequences of altered Pol II catalytic activity in vivo. Nucleic Acids Res 45, 4431–4451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kwapisz M et al. Mutations of RNA polymerase II activate key genes of the nucleoside triphosphate biosynthetic pathways. EMBO J 27, 2411–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thiebaut M et al. Futile cycle of transcription initiation and termination modulates the response to nucleotide shortage in S. cerevisiae. Mol Cell 31, 671–82 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Steinmetz EJ et al. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell 24, 735–746 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Hein PP, Palangat M & Landick R RNA transcript 3'-proximal sequence affects translocation bias of RNA polymerase. Biochemistry 50, 7002–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cabart P, Jin H, Li L & Kaplan CD Activation and reactivation of the RNA polymerase II trigger loop for intrinsic RNA cleavage and catalysis. Transcription 5, e28869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sainsbury S, Niesser J & Cramer P Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature 493, 437–40 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Segal E & Widom J Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol 19, 65–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tillo D & Hughes TR G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 10, 442 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee W et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39, 1235–44 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Peckham HE et al. Nucleosome positioning signals in genomic DNA. Genome Res 17, 1170–7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Segal E et al. A genomic code for nucleosome positioning. Nature 442, 772–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 86.Jin H & Kaplan CD Relationships of RNA polymerase II genetic interactors to transcription start site usage defects and growth in Saccharomyces cerevisiae. G3 (Bethesda) 5, 21–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amberg DC, Burke D, Strathern JN, Burke D & Cold Spring Harbor Laboratory. Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual, xvii, 230 p. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 2005). [Google Scholar]
- 88.Chee MK & Haase SB New and Redesigned pRS Plasmid Shuttle Vectors for Genetic Manipulation of Saccharomycescerevisiae. G3 (Bethesda) 2, 515–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gietz RD & Schiestl RH High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2, 31–4 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Benatuil L, Perez JM, Belk J & Hsieh CM An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel 23, 155–9 (2010). [DOI] [PubMed] [Google Scholar]
- 91.Schmitt ME, Brown TA & Trumpower BL A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18, 3091–2 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vvedenskaya IO, Goldman SR & Nickels BE Preparation of cDNA libraries for high-throughput RNA sequencing analysis of RNA 5’ ends. Methods Mol Biol 1276, 211–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ranish JA & Hahn S The Yeast General Transcription Factor Tfiia Is Composed of 2 Polypeptide Subunits. Journal of Biological Chemistry 266, 19320–19327 (1991). [PubMed] [Google Scholar]
- 94.Zhang J, Kobert K, Flouri T & Stamatakis A PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tareen A & Kinney JB Logomaker: beautiful sequence logos in Python. Bioinformatics 36, 2272–2274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crooks GE, Hon G, Chandonia JM & Brenner SE WebLogo: a sequence logo generator. Genome Res 14, 1188–90 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuhn M Building Predictive Models in R Using the caret Package. Journal of Statistical Software 28, 1–26 (2008).27774042 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data generated in this study are available in the NCBI BioProject, under the accession number PRJNA766624. Processed data are available in GEO, under the accession number GSE185290. Source data are provided with the manuscript.
Code for analyses in this study is provided at https://github.com/Kaplan-Lab-Pitt/PolII_MASTER-TSS_sequence.