

Abstract
Defining monogenic drivers of autoinflammatory syndromes elucidates mechanisms of disease in patients with these inborn errors of immunity and can facilitate targeted therapeutic interventions. Here, we describe a cohort of patients with a Behçet’s- and inflammatory bowel disease (IBD)-like disorder termed ‘Deficiency in ELF4, X-linked’ (DEX) affecting males with loss-of-function variants in the ELF4 transcription factor gene located on the X chromosome. An international cohort of fourteen DEX patients was assessed to identify unifying clinical manifestations and diagnostic criteria as well as collate findings informing therapeutic responses. DEX patients exhibit a heterogeneous clinical phenotype including weight loss, oral and gastrointestinal aphthous ulcers, fevers, skin inflammation, gastrointestinal symptoms, arthritis, arthralgia, and myalgia, with findings of increased inflammatory markers, anemia, neutrophilic leukocytosis, thrombocytosis, intermittently low natural killer and class-switched memory B cells, and increased inflammatory cytokines in the serum. Patients have been predominantly treated with anti-inflammatory agents, with the majority of DEX patients treated with biologics targeting TNFα.
Introduction
Human monogenic autoinflammatory diseases (AIDs) comprise a subgroup of inborn errors of immunity (IEI) characterized by systemic inflammation not necessarily accompanied by increased infection susceptibility and without the features of classical autoimmune diseases such as high titers of autoantibodies and antigen-specific T cells [1,2]. These diseases are caused by single-gene defects, typically resulting in gain-of-function in innate immune mediators or loss-of-function in negative regulators of inflammation. Defined broadly, monogenic AIDs are largely driven by dysregulated and exuberant cytokine responses and can encompass inflammasomopathies, type I interferonopathies, and immune-driven very early onset IBD (VEO-IBD) syndromes, among others. Advances in genetic diagnostics have enabled discovery of a growing number of IEIs with predominant autoinflammatory features, leading simultaneously to breakthroughs in our mechanistic understanding of fundamental immune molecules and pathways and to further translation back to the clinic with targeted therapeutic interventions [1–3].
AID patients often present with periodic or recurrent fevers along with cutaneous, osteoarticular, serosal, and/or gastrointestinal manifestations [2–10]. Classic examples include Familial Mediterranean Fever (FMF), TNF receptor-associated periodic syndrome (TRAPS), Cryopyrin-associated periodic syndrome (CAPS), Deficiency of interleukin-1 receptor antagonist (DIRA), Haploinsufficiency of A20 (HA20), and Deficiency of adenosine deaminase 2 (DADA2), type I interferonopathies like Aicardi-Goutières syndrome, among many others [2]. Very early-onset inflammatory bowel disease (VEO-IBD) is a subgroup of IBD, often IBD-unclassified (IBD-U), with severe disease onset before 6 years old, and is often attributable to disrupted homeostasis of immune cells in the gastrointestinal tract caused by rare mutations in genes such as IL10, IL10RB, ADAM17, IL21, and XIAP [11–13]. However, it can be difficult for clinicians to confidently determine pathogenicity of rare variants found by next-generation sequencing and to find the information needed to manage treatment of these patients. Behçet’s syndrome (also known as Behçet’s disease) is characterized by recurrent, painful oral aphthous ulcers along with systemic manifestations that can include genital ulcers, gastrointestinal involvement, arthritis, various skin lesions, and ocular, vascular, and/or neurologic disease, and can often be difficult to distinguish from Crohn’s disease [14,15]. Here, we describe a cohort of patients with a recently described AID sharing features of Behçet’s syndrome and VEO-IBD that we termed ‘Deficiency in ELF4, X-linked’ (DEX).
DEX, also designated Autoinflammatory Syndrome, Familial, X-linked, Behçet-like 2 (AIFBL2; [MIM #301074]), is caused by hemizygous loss-of-function mutations in E74-like ETS transcription factor 4 (ELF4) located on the X chromosome (Xq26.1). DEX was initially described in three unrelated male patients presenting in childhood with recurrent fevers, oral ulcers, and inflammatory bowel disease (IBD)-like mucosal inflammation [16]. A fourth patient with a novel ELF4 mutation who presented with oral ulcers, constipation, arthritis, and recurrent bacterial and viral infections was later identified, demonstrating heterogenicity in the clinical phenotype [17]. The number of reported DEX patients increased to nine with the discovery of five more patients in a Chinese cohort with loss-of-function ELF4 variants who presented with symptoms similar to the previous patients [18].
ELF4 is a member of the family of transcription factors that share an evolutionarily conserved ‘E26 transformation specific’ (ETS) DNA-binding domain and are implicated in cellular growth, proliferation, apoptosis, senescence, development, differentiation, and angiogenesis [19]. Members of the ELF subfamily are specifically known to be involved in immune cell development as well as the regulation of immune responses [20,21]. In humans, ELF4 is most highly expressed in many hematopoietic cells, with the highest seen in natural killer (NK) cells, monocytes, and CD4+ T regulatory cells (Tregs) in the peripheral blood [22]. A number of mouse studies have attributed a variety of functions to ELF4 [23–34], including roles in NK cell development [35], CD8+ T cell proliferation and homing [36], and the regulation of antiviral immunity through type I interferon [37]. Notably, ELF4 has also been found in both mouse and humans (i.e., DEX patients) to restrain the differentiation of proliferating CD4+ T cells to T helper 17 (Th17) cells (Lee et al., 2014; Tyler et al., 2021) , a cell subset known to promote mucosal inflammation and neutrophil recruitment. Recent work with DEX patient samples and ELF4-deficient mice revealed that myeloid cells deficient in ELF4 are also hyperinflammatory [16].
We have now collated an international cohort of fourteen patients from thirteen unrelated families with loss-of-function mutations in ELF4 leading to DEX, nine of whom were reported previously [16–18]. Here, we report the immunological characteristics, clinical phenotypes, histopathological and laboratory findings, therapeutic interventions, and prognosis of all currently known DEX patients in order to highlight the unifying characteristics.
Results
DEX patients have pathogenic ELF4 mutations
De novo or maternally inherited ELF4 mutations in all DEX patients identified to date cause either an early stop codon/deletion that results in nonsense-mediated mRNA decay and loss of protein, or a missense variant that disrupts the ETS DNA binding domain in exon 7 of ELF4 (Table 1 and Figure 1a). All mutations except that of P2 have been confirmed by Sanger sequencing [16–18] (Supplemental Figure 1), and only two (W212C & R185X) are present in the gnomAD database of human exomes or genomes, both with minor allele frequencies of 5.46 × 10−6 [39]. Out of the fourteen patients, there are eleven distinct mutations, while W251S, R234X, and the deletion of exons 2–7 are shared by patients P6 and P7, patients P5 and P10, and identical twin patients P3 and P4, respectively. Most DEX patients (11/12) inherited their mutation from their mother, while one variant is confirmed to be de novo (P6), and the maternal genotype of two DEX patients is unknown (Table 1 and Figure 1b; pedigrees of P6–P14 previously published [16–18]).
Table 1:
ELF4 variants in patients with DEX
| Patient | Mutation | Inheritance | CADD score | Transcriptional Activity? | References | ||
|---|---|---|---|---|---|---|---|
| Location | cDNA | Protein | |||||
| P1 | Exon 5 | c.443delG | p.(G148Vfs*113) | Maternal | ------ | Loss of function | ------ |
| P2 | Exon 7 ETS domain | c.636G>T | p.(W212C) | Maternal | 32.0 | Loss of function | ------ |
| P3 | Exons 2–7 | Deletion exons 2–7 | N/A | Unknown | ------ | ------ | ------ |
| P4 | Exons 2–7 | Deletion exons 2–7 | N/A | Unknown | ------ | ------ | ------ |
| P5 | Exon 7 ETS domain | c.700C>T | p.(R234X) | Maternal | 35.0 | Loss of function | ------ |
| P6 | Exon 7 ETS domain | c.752G>C | p.(W251S) | de novo | 25.4 | Loss of function | Tyler et al. 2021 |
| P7 | Exon 7 ETS domain | c.752G>C | p.(W251S) | Maternal | 25.4 | Loss of function | Tyler et al. 2021 |
| P8 | Exon 8 | c.1015delG | p.(A339Pfs*32) | Maternal | ------ | Loss of function | Tyler et al. 2021 |
| P9 | Exon 7 ETS domain | c.691T>C | p.(W231 R) | Maternal | 27.8 | Loss of function | Sun et al. 2022 |
| P10 | Exon 7 ETS domain | c.700C>T | p.(R234X) | Maternal | 35.0 | Loss of function | Sun et al. 2023 |
| P11 | Exon 3 | c.115C>T | p.(Q39X) | Maternal | 36.0 | Loss of function | Sun et al. 2023 |
| P12 | Exon 5 | c.465delG | p.(H156Ifs*105) | Maternal | ------ | Loss of function | Sun et al. 2023 |
| P13 | Exon 7 ETS domain | c.743C>T | p.(S248F) | Maternal | 24.5 | Loss of function | Sun et al. 2023 |
| P14 | Exon 6 | c.553C>T | p.(R185X) | Maternal | 36.0 | Loss of function | Sun et al. 2023 |
Figure 1: Deleterious ELF4 mutations in patients with DEX.
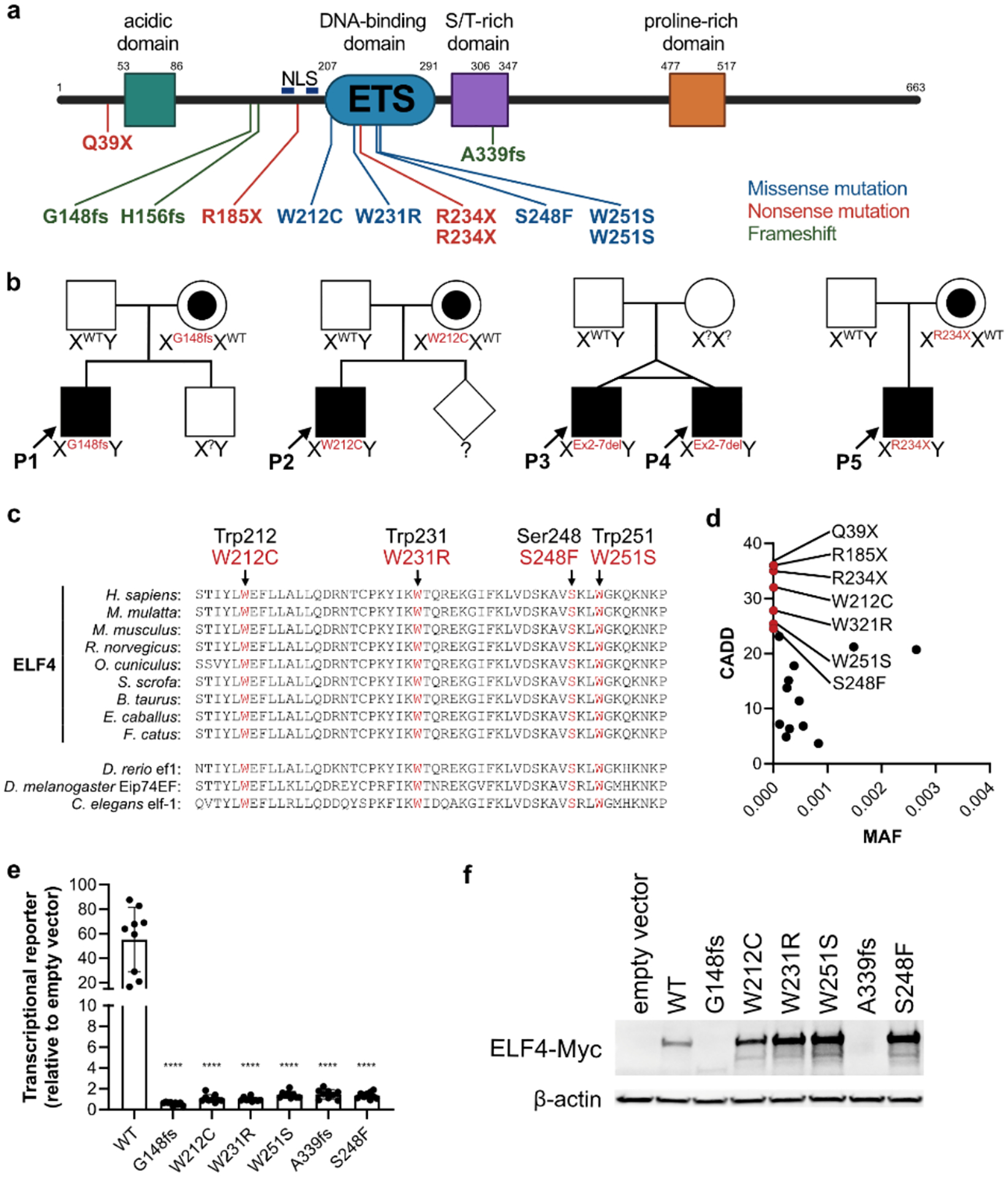
(a) Schematic diagram of ELF4 protein indicating the location of each known variant. Numbers indicate amino acid position. ETS, ETS DNA binding domain; NLS, nuclear localization sequence indicated by blue bars. (b) Pedigrees from four families with five newly reported patients with DEX (P1–P5). Square, males; circles, females; diamond, sex unknown; WT, wild-type; X, X chromosome; Y, Y chromosome; red, ELF4 variants with protein change indicated; ?, genotype unknown (c) Amino acid sequence alignment of ELF4 ETS domains across species and ETS domains from related proteins in other species (lower panel), with residues mutated in patients with DEX highlighted in red. Red text above sequences indicates DEX variant. (d) Combined annotation-dependent deletion (CADD) scores (GRCh37-v1.6) versus minor allele frequency (MAF) for ELF4 variants in DEX patients (highlighted in red, with protein change indicated) as compared with ELF4 variants present in males with a MAF cutoff of > 10−4 from the gnomAD database (v2.1.1). (e) Activity of ELF4 variants (protein change indicated) ectopically overexpressed in 293T cells co-transfected with a transcriptional luciferase reporter. WT, wild-type. Data shown are from 3 pooled independent experiments ± s.d. Statistical analyses were performed using two-tailed, unpaired Student’s t-test. ****, P<0.0001. (f) Immunoblot for myc-tagged ELF4 and β-actin in 293T cells overexpressing ELF4 variants used in luciferase reporter assay.
Alignment of the human ELF4 DNA-binding ETS domain in exon 7 with ETS domains of ELF4 and related proteins from other species highlights that the residues mutated in DEX patients within the ETS domain (Trp212, Trp231, Ser248, and Trp251) are all highly conserved amino acids (Figure 1c). Combined annotation-dependent deletion (CADD) scores, a measure of human variant deleteriousness, for all DEX mutations were >20 and these mutations are thus predicted to be pathogenic [40,41]. The variants are all rarer and have greater CADD scores than gnomAD variants in males with a minor allele frequency (MAF) cutoff of >10−4 (Figure 1d). Additionally, we have previously noted that missense variants are reduced within the conserved ETS domain of ELF4 [16], further emphasizing the predicted pathogenicity of variants within the ETS domain in DEX patients P2, P5, P6, P7, P9, P10, and P13. P1, P3, P4, P8, P11, P12, and P14 all have early stop codons and loss of ELF4 mRNA/protein [16,18]. Transfection of 293T cells with cDNA vectors expressing both a transcriptional luciferase reporter (using the mouse Ifnb1 promoter and multiple ISRE sites) and myc-tagged ELF4 wildtype (WT) or DEX patient-derived ELF4 variants to assess ELF4 transcriptional activity demonstrated variants for patients P1, P2, P6–P9, and P13 to be loss of function (Figure 1e & f). Sun et al. (2023) have also independently shown loss of function of the W231R (P9) and R234X (patients P5 and P10) variants by luciferase reporter assay, as well as verified loss of function for the variants of patients P11-P14. Except for the variants that result in a frameshift (A339Pfs*32, G148Vfs*113, and H156Ifs*105) or an early termination (R234X, Q39X, and R185X) all other ectopically overexpressed variants resulted in ELF4 protein with apparent molecular weight of full-length ELF4 (Figure 1f) [17,18].
Since DEX patients presented with signs of autoinflammation and recurrent fevers, many had more extensive genetic testing. P6 was tested for a number of genes associated with periodic fever syndromes (MEFV, MVK, LPIN2, TNFRSF1A, NLRP3, ELANE, and PSTPIP1) but no pathogenic variants were detected. Results from P8 were negative on a whole exome sequencing primary immunodeficiency (WES-PID) panel, and P9 also did not have any mutations in known causative genes of primary immunodeficiency, as assessed by whole exome sequencing [17]. P1 tested negative on a panel of >420 genes. However, testing in family members of P1 revealed that his mutation was inherited from his mother, who inherited the mutation from her own mother. To our knowledge, no DEX patients were tested for the presence of HLA-B51, which is associated with Behçet’s syndrome [42,43].
P3 and P4 are monozygotic twins who both were found by microarray to have an approximately 0.086 Mb deletion of chromosome 6q26, which includes PARK2, and an approximately 0.029 Mb deletion of chromosome Xq26.1, where ELF4 is located. Autosomal recessive pathogenic variants in PARK2 are associated with Parkinson disease, juvenile, type 2 (OMIM #600116), so it is unlikely that this mutation is leading to the symptoms seen in these patients, but they are likely carriers for this disorder. We confirmed the deletion in ELF4 by Sanger sequencing in both P3 and P4 and found it to be 29.046 kilobases in size (Supplemental Figure 1). Exons 2–7 are deleted, while coding exons 8 and 9 are present. Additionally, P3 was found on a sequencing panel and copy number analysis (NGS319) to have a heterozygous missense variant of uncertain significance in TNFRSF1A exon 10 (c.1328G>T; NM_001065; p.Gly443Val). This variant is listed in gnomAD with an allele frequency of 1.08 × 10−4 and CADD score of 15.69. However, it is believed that this variant is not related to the phenotype of P3 and that this patient does not have TRAPS. In addition to his DEX-related symptoms, P3 has a diagnosis of cerebral palsy, spastic quadriplegia, and dystonia for which he has had several surgeries related to muscle contractions. P14 was found on genetic testing to harbor a de novo large duplication that affects STAG2 and XIAP, which may contribute to his developmental delay and hydrocephalus [44].
DEX patients exhibit symptoms of autoinflammation
As DEX is an X-linked disorder, all fourteen patients are males, with a mean age of clinical presentation of 4.5 years old. Several patients presented within the first few years of life, with the latest age of onset being thirteen years. Clinical manifestations of the fourteen DEX patients are shown in Table 2. All patients have oral ulcers (canker sores), with the exception of P2, P5, and P13. However, oral ulcers did not develop in some patients (e.g., P3, P4, and P12) until a few years after the onset of other symptoms. These ulcers usually coincide with symptomatic flares that last for 2–5 days and consist of a recurrent constellation of symptoms including fevers (9/13), skin involvement (8/13), myalgias (3/12), arthritis (2/14), arthralgias (4/14), fatigue (6/14), abdominal pain (8/14), nausea/emesis (4/13), constipation (3/13) and/or diarrhea (7/14), and decreased appetite (12/13). Two patients had rectal bleeding (P2 and P6). Most patients (13/14) have also had poor weight gain or weight loss, often attributed to decreased intake secondary to pain or dysphagia due to oral ulcers. P3 had significant weight loss that required placement of a gastrostomy tube at age 12, though this was also likely related to his additional diagnosis of cerebral palsy. Two patients (P1 & P7) also have short stature. P11 presented first with oral ulcers and skin inflammation, developed fever four years later, and arthritis a year later [18]. P7 had episodes of lip swelling and oral ulcers, which initially led to suspicion of orofacial granulomatosis. P9 presented at 2 months old with infections (see below), developed constipation at 3 years old, followed by oral ulcers (4 years old), and then rash and arthritis (9 years old). His fevers were only ever associated with infections [17]. The location of arthralgias varies between patients, presenting as polyarthralgia in P5, while specifically the knees are affected in P1, P4, & P6. P9 was diagnosed with arthritis in the left hip and right ilium by MRI [17]. Some DEX patients have additional symptoms associated with their flares, including headache (P1), painful eyes with incidental conjunctival injection (P1; not evaluated by an ophthalmologist), periorbital swelling (P6), and irritability (P6). For some patients, the symptomatic flares were noted to have a periodicity, with P6 experiencing flares approximately once every 4 weeks and P1 approximately once every 3–8 weeks. In P3, fevers occurred periodically between 2 and 4.5 years of age, at which point the fevers improved and he started developing oral ulcers that were dependent on steroid dosing.
Table 2:
Clinical manifestations of patients with DEX.
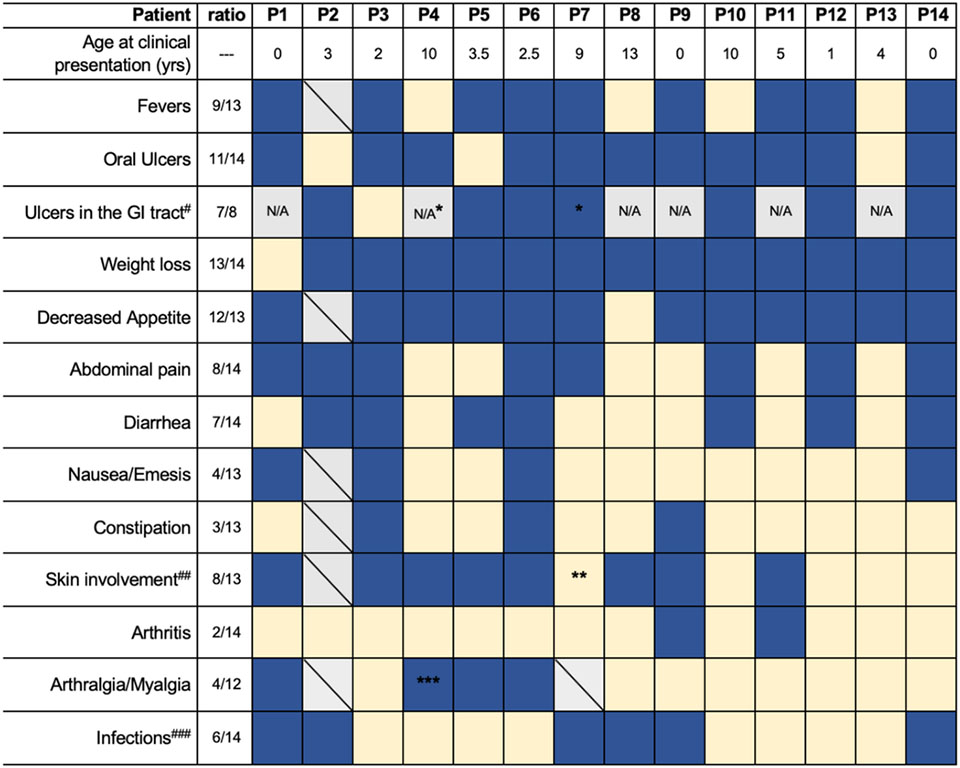
|
Dark blue, affected; light yellow, unaffected; boxes with a diagonal, unknown; N/A, not applicable as endoscopy was not performed.
patient had perianal ulcer (P7, also fissure);
eczema thought to be unrelated to DEX;
P4 only experiences arthralgia;
for more details on inflammatory involvement of the GI tract, refer to the text;
various skin lesions are described in the text;
for specific infections reported, refer to the text and Supplemental Table 1.
Data was not available for all patients.
Behçet’s syndrome-like aphthous ulcers in DEX patients
Aphthous lesions have been reported in several locations, including on the lips, hard palate, uvula, tonsils, tongue, posterior oropharynx, and buccal mucosa. The ulcers resemble those seen in recurrent aphthous stomatitis and Behçet’s syndrome, with an example shown in Sun et al. 2023 Figure 1B. A biopsy of an oral ulcer from P7 revealed acute ulceration with a prominent neutrophilic exudate in the epithelium and dermis as well as underlying granulation tissue with histiocytes and lymphocytes, but no areas of vasculitis. A biopsy of a lip ulcer from P3 demonstrated full thickness submucosal ulceration and marked submucosal acute and chronic inflammation with numerous plasma cells. In P4, an oral ulcer biopsy showed markedly inflamed fibroadipose tissue and focal marked acute and chronic inflammation with scattered eosinophils, and a biopsy of an ulcer in P10 also demonstrated inflammation. Due to his symptoms, P11 initially received a differential of either Behçet’s syndrome or juvenile idiopathic arthritis. Cultures of the oral ulcers did not reveal any abnormal growth in P3 or P6, while P8 was repeatedly positive for Candida albicans between the ages of 13 and 18. P8 was later evaluated several times while suffering from oral ulcers, but a pathogen was never identified. Specific emotional or physical triggers of ulcers were not identified in P1, P8, or P9–14. P3 does have worsening ulcers around times of slight physical trauma or emotional stress, though the causality is not clear. One patient (P7) reports that ulcers are worse when consuming certain foods such as pizza and carbonated beverages. In P9, oral ulcers went into remission while in lockdown during the SARS-CoV-2 pandemic lockdown but recurred immediately after returning to school [17]. In addition to ulcers in the gastrointestinal tract (discussed below), there have also been ulcerous lesions reported in other locations, including perianal (P7, persistent with fissures but without fistula; P10; and P12) and scrotal (P3). P12 initially presented with perianal ulcers and did not develop oral ulcers, fever, abdominal pain, or diarrhea until two years later [18].
Features of IBD-like gut inflammation in DEX
Signs of gut inflammation are common in DEX patients. Of these fourteen patients, seven have been evaluated with upper and/or lower endoscopies with histology, while the other six patients did not experience prominent GI symptoms (with the exception of P1). Seven of the eight (7/8) DEX patients who underwent endoscopy were found to have aphthous-like ulcers in their gastrointestinal tract, including the esophagus (P5 and P12), gastric sinus (P12 and P14), duodenum (P7), ileum (P14), terminal ileum (P7, and P10), ileocecal flap (P10), colon (P2, P5, P6, and P12), and rectum (P2 and P10) [18]. Several patients were also found to have gastritis (4/6; P5, P6, P7, and P2) and/or esophagitis (4/6; P2, P3, P5, and P7), with few eosinophils (5–7 per high powered field) seen in P7. P2 had continuous ulceration from the rectum through the cecum and terminal ileitis on colonoscopy with a single epithelioid granuloma in the right colon. He previously received a diagnosis of IBD-unclassified (IBD-U) that was later reclassified as Crohn’s disease. P3 was found to have esophagitis without ulcers and had a normal colonoscopy. P5 had aphthous lesions in the esophagus, foci of lymphocytic infiltrate in distal esophageal epithelium, gastritis with mild chronic inflammatory infiltrate (negative for Helicobacter pylori), terminal ileitis and pancolitis with erythematous and friable mucosa, as well as multiple aphthous lesions throughout the entire colon. Histopathology of the terminal ileum and colonic mucosa of P5 showed discontinuous inflammatory involvement with focal submucosal involvement, cryptitis, glandular distortion, neutrophilic infiltrate in the lamina propria with a discrete histiocytic component but no granulomas. P6 had scattered aphthous-like ulcerations throughout the colon, focally active colitis without granulomas on biopsies, and normal terminal ileum; more recent colonoscopy revealed pancolitis. P7 had mild chronic inflammation in the gastric body (with mononuclear cells), terminal ileum (with neutrophils), ileocecal valve, and cecum, without granulomas. Nodular lymphoid hyperplasia was also seen in the cecum and appendiceal orifice of P7. In addition to multiple ulcers throughout the GI tract, from esophagus to colon, P12 was found to have stenosis of the ascending colon and mild inflammation of the colonic mucosa with lymphoid hyperplasia [18]. Lymphocytic infiltration in terminal ileum was also seen in P14. These findings led to initial diagnoses of Crohn’s disease and IBD-U in P10 and P12, respectively [18].
Inflammatory skin lesions in DEX patients
The majority of DEX patients (8/13) experience some form of skin inflammation during symptomatic flares, with the locations and features varying between patients. P3 develops both scattered red tender papulonodular lesions with some overlaying scale that appear on his forehead, shin, arms, and feet (Figure 2a–c), with a biopsy showing lobular and septal panniculitis, as well as separate pustular lesions. He also develops lesions anywhere there is trauma such as a blood draw or vaccine injection, consistent with pathergy phenomenon described in Behçet’s syndrome. During symptomatic flares, P4 presents with lesions on his arms, legs, and buttocks that are initially pustular and then drain and ulcerate. P5 has skin inflammation described as bullous and prurigo-like lesions on his arms, legs, ears, and buttocks that resolve completely in a matter of days to weeks, leaving no scar or pigmentation (Figure 2d). P6 has experienced both a diffuse macular rash, as well as isolated scattered macules and petechiae on the legs and/or arms during fever episodes. P8 experienced a papulopustular rash with painful vesicles and redness shortly before presentation at age 13. The lesions started in the left inguinal region, expanded to the right thigh, and were not responsive to acyclovir. This patient no longer experiences such skin lesions. Around age 9, P9 experienced several episodes of a transient palmar rash [17]. During symptomatic flares, P1 sometimes develops widespread non-erythematous and non-pruritic micropapules covering his trunk that resemble piloerection. P7 has eczema, though this is thought to be unrelated to DEX, and it is unknown whether P2 experiences any form of skin inflammation. The location of the skin erythema in P11 was not reported [18].
Figure 2: Clinical symptoms and laboratory findings in DEX.
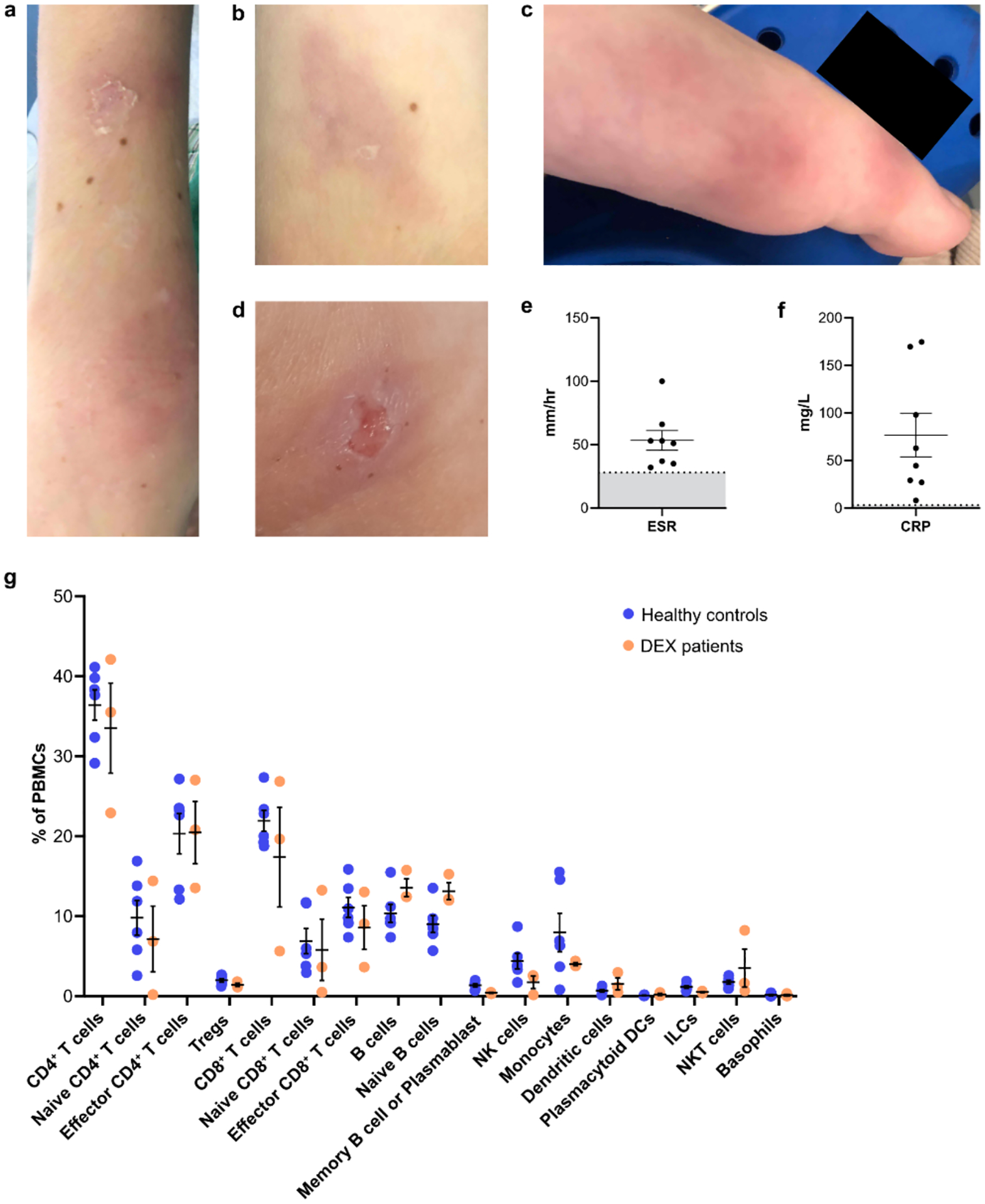
Representative images of skin inflammation on the arms (a & b) and foot (c) of P3, and the leg of P5 (d). Highest recorded (e) ESR and (f) CRP values reported for P1, P3, P4, P5, P6, P7, P8, and P9. Mean with SEM is shown, with shaded gray region in (e) indicating the most conservative normal range for ESR (< 28 mm/hr). Shaded gray region in (f) indicates a normal range of < 3mg/L for CRP. Exact reference ranges vary by age and laboratory. (g) Immunophenotyping of peripheral blood mononuclear cells (PBMCs) by mass cytometry (CyTOF) from DEX patients P1, P6, and P8 and sex-matched healthy pediatric controls. Cell subset frequencies are shown as a percentage of each patient’s total live PBMCs along with mean and SEM. Populations between HC (n = 6) and DEX (n =3) patients were not statistically significant. Treg, T regulatory cell; NK, natural killer cell; DC, dendritic cell; ILC, innate lymphoid cell.
Other clinical manifestations of DEX
Based on our cohort, most DEX patients do not present with notable or abnormal infectious phenotypes. However, 6/14 patients have had infectious components to their presentation. P1 had recurrent herpes labialis and an episode of giardiasis which could both be incidental. P2 had recurrent C. difficile colitis at presentation that was treated with a fecal transplant. Shortly after presentation, P8 was evaluated because of severe balanitis that was treated with acute circumcision complicated by granulomatous reaction. He was treated with antibiotics (without effect) and antifungals. Around the same time, P8 also suffered severe oral Candida albicans infection that led to weight loss from reduced oral intake secondary to mouth pain. He was treated with antifungals with good clinical response. Subsequently, he had recurrent oropharyngeal ulcers that were C. albicans-negative but responded to antifungals. His severe balanitis did improve gradually after antifungal treatment, but it is unclear whether the balanitis was also caused by fungal infection since cultures were negative for C. albicans. P7 is only reported to have been infected with Varicella (chickenpox) and subclinical intestinal spirochetosis, neither of which was considered concerning or unusual. After an initial infection with pneumonia at 2 months old that required a pediatric ICU admission and mechanical ventilation, P9 experienced frequent viral and bacterial pneumonia from the age of two years that often followed viral upper respiratory traction infections. He has required several hospitalizations and these infections have been controlled with intravenous antibiotics, intravenous immunoglobulins (IVIG), and supportive therapy [17]. At age five, P9 experienced desquamation of the skin of the tips of the thumb and finger as well as a dilated left coronary artery that was suspected to be “atypical Kawasaki’s disease” and resolved after several weeks of treatment [17]. He later presented at age 10 with abdominal pain and purpura on the lower legs felt to be typical of Henoch-Schönlein purpura and was treated with low dose glucocorticoids. While P1 has family members who have been diagnosed with Kawasaki’s disease but are not carriers of the ELF4 mutation, there have been no other reports in DEX patients. P14 has also experienced recurrent respiratory infections, but the causative organisms and whether hospitalization was required was not reported [18]. These infections in DEX patients are sporadic with different classes of microbes, suggesting the absence of specific immunodeficiency (Supplemental Table 1).
P13 initially presented with elevated transaminases and neutropenia. A diagnosis of systemic lupus erythematous was made based on multi-organ damage, multiple autoantibodies, and decreased C3 and C4. Renal biopsy showed mild mesangial proliferation with positive immunofluorescence for IgA, C3, and C1q, consistent with class II lupus nephritis according to the International Society of Nephrology/Renal Pathology Society classification [45]. Abnormal transaminases were attributed to lupus hepatitis. This patient also has coagulopathy, likely due to decreased activity of factor IX [18]. P14 also had persistent liver dysfunction with coagulopathy and decreased albumin, along with hydrocephalus, congenital hypospadias, inguinal and umbilical hernias, hepatosplenomegaly, and growth retardation due to growth hormone deficiency. Notably, this patient suffered from intrauterine growth retardation and later showed global developmental delay [18].
Laboratory findings and immunological markers in DEX
Diagnosis of DEX is facilitated by the overlap of abnormal clinical features and laboratory values (Table 3). Most DEX patients assessed (11/12) have elevated erythrocyte sedimentation rates (ESR), and C-reactive protein (CRP) is additionally elevated in all patients assessed (8/8) [18]. These values are generally elevated during symptomatic flares and normalize in between episodes or with treatment. The highest values recorded for ESR and CRP are shown in Figures 2e and 2f, respectively, for P1 and P3–P9. Eleven out of thirteen (11/13) DEX patients for whom we have data have anemia (Table 3). Nine out of thirteen (9/13) patients had microcytic anemia with low mean corpuscular volumes (MCV), while P1 had MCV values that were at the low range of normal during flares (76–81 fL). P5, P6, P7, P9, and P14 were also found to have low iron levels, supporting diagnoses of iron deficiency anemia; iron was not measured in the other patients [17,18]. P6 additionally had low ferritin levels (9 ng/mL), while P5 and P7 had ferritin within the normal range.
Table 3:
Main biological and immunological findings in DEX.
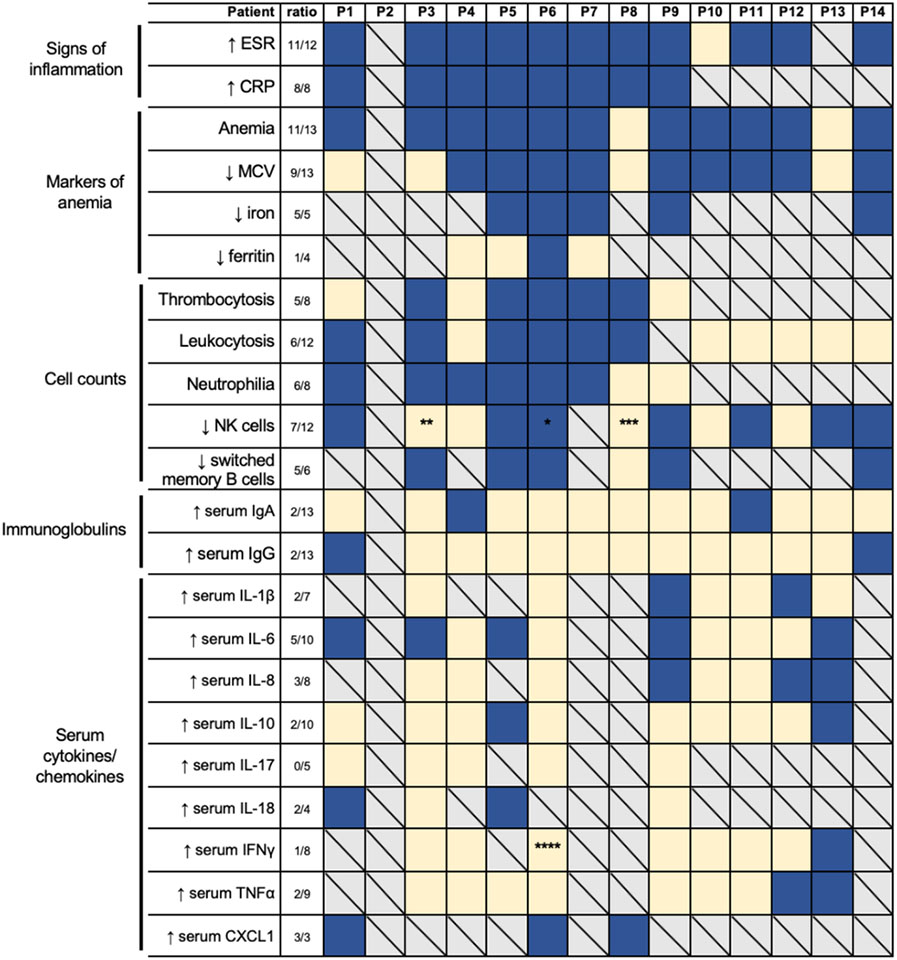
|
Laboratory findings, including signs of inflammation, markers of anemia, cell counts, immunoglobulins, and serum cytokines/chemokines. All relative values are reported based on the reference clinical range with the following exceptions: CXCL1 in P6 was measured in a research setting and is reported relative to values in 6 unrelated healthy controls and the patient’s mother and father; CXCL1 in P8 was measured in a research setting that is not approved as a diagnostic tool. P5 cytokine measurements were taken while on TNFa blockade therapy. Dark blue, affected; light yellow, unaffected; boxes with a diagonal, unknown or not evaluated. ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; MCV, mean corpuscular volume; Ig, immunoglobulin; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor. Switched memory B cells are CD19+CD27+IgD−. Natural Killer (NK) cells are CD56+CD3−.
patient intermittently had both increased and decreased frequencies of CD56+CD3− and CD56+CD3−CD16+ NK cells;
patient had increased frequencies and total numbers of CD56+CD3-CD16+ NK cells;
patient had increased total numbers of CD56+CD3−CD16+ NK cells once during active inflammation;
patient was on infliximab (anti-TNFα) at the time.
Data was not available for all patients.
Several patients analyzed have thrombocytosis (5/8 patients) and/or neutrophilia (6/8 patients) during symptomatic flares, with many additionally having leukocytosis (6/12 patients). Natural killer (NK) cell (CD56+CD3− and CD56+CD3−CD16+) numbers varied between and within DEX patients, with levels above, at, and below normal range. 7/12 patients displayed decreased NK cells at some point during the course of their disease. Percentages and absolute numbers of CD3+ T cells and CD3+CD4+ T cells were within normal range in all patients analyzed. With the exception of P9, who had an increased percentage of CD3+CD8− T cells [17], all other patients had normal frequencies. Most patients have normal levels of CD19+ B cells, but P6, P11, and P13 had transient episodes of decreased absolute numbers of B cells [18] and P7 had increased percentage and numbers of naïve (CD19+CD27−IgD+) B cells. Decreased class-switched memory B cells (CD19+CD27+IgD−) were observed in 5/6 patients in whom they were analyzed [17,18]. Multiparameter immunophenotyping by mass cytometry (CyTOF) of peripheral blood mononuclear cells (PBMCs) from P1, P6, and P8 did not reveal significantly different frequencies of any immune cell subsets compared to sex-matched healthy pediatric controls (HC) (Figure 2g and Supplemental Tables 2). Heterogeneity was noted in this small sample set, which may lead to differences in functional responses of these immune populations in DEX patients (Supplemental Figures 2 & 3).
Despite some patients having decreased frequencies of B cells, total antibody levels do not appear to be greatly impacted in most DEX patients. 2/13 patients have increased serum immunoglobulin A (IgA), while the other 11/13 patients have levels within the normal range. Serum IgG was only found to be increased in 2 patients out of the 13 assessed, and IgG2 was increased in only P6. P1 had increased total IgG, IgG1, and IgG4, but normal IgG2 and IgG3. DEX patients evaluated for serum IgM (6 patients; P1, P3, P4, P6, P8, and P9) or IgE (4 patients; P3, P4, P6, and P9) were all found to be within normal range [17]. P5 had no serological response to vaccination against hepatitis B (HBV), tetanus, and rubella. P6 was reported to have borderline immunoglobulin response to the diphtheria and tetanus vaccine and lack of response to the live, attenuated measles-mumps-rubella (MMR) vaccine and varicella zoster vaccine. P7 was also reported to have a suboptimal IgG response to the live, attenuated MMR vaccine. However, P1 and P4 reported normal immunoglobulin responses to the MMR vaccine. P9 also had normal responses to various other vaccines including Bacille Calmette-Guérin (BCG), HBV, poliovirus, and diphtheria-pertussis-tetanus (DPT) [17].
Several DEX patients have had serum levels of cytokines and chemokines assessed to characterize the nature of their inflammation more thoroughly. However, such testing was not available for all patients (P2, P7, P8, and P14), and of those tested, not all cytokines/chemokines were profiled. Several cytokines were found to be elevated in the serum: interleukin-1β (IL-1β) in 2/7 patients, IL-6 in 5/10 patients, IL-8 in 3/8 patients, IL-10 in 2/10 patients, IL-18 in 2/4 patients, interferon γ (IFNγ) in 1/8 patients, tumor necrosis factor α (TNFα) in 2/9 patients, and CXCL1 in 3/3 patients [16,17]. Serum IL-17 was within normal levels for the 5 patients evaluated. Additionally, macrophage inflammatory protein-1α (MIP-1α) and MIP-1β were elevated in P9, though within the range of healthy controls in P6. P6 and P9 had several other serum cytokines evaluated that were all found to be within reference ranges [16,17], P3 was found to have normal levels of CXCL9, and P5 had normal levels of CXCL5. P3 also had some intracellular cytokines assessed and was found to have decreased IFNγ in both CD8+ T cells and NK cells, and decreased TNFα in CD4+ T cells, CD8+ T cells, and NK cells. P4 had increased soluble IL-2 receptor.
Serum complement levels were reported in 5 DEX patients and showed no clear trend. Complement components C3 and C4 were decreased in P13 who has a diagnosis of SLE [18]. P6 and P7 had elevated levels of C3 and C4, and P12 had elevated level of C1q [18], indicative of an inflammatory state. P9 had normal C3 and C4 levels. Some DEX patients are positive for various autoantibodies, many of which improved with therapy, and these are shown in Supplemental Table 3.
Treatment considerations for DEX patients
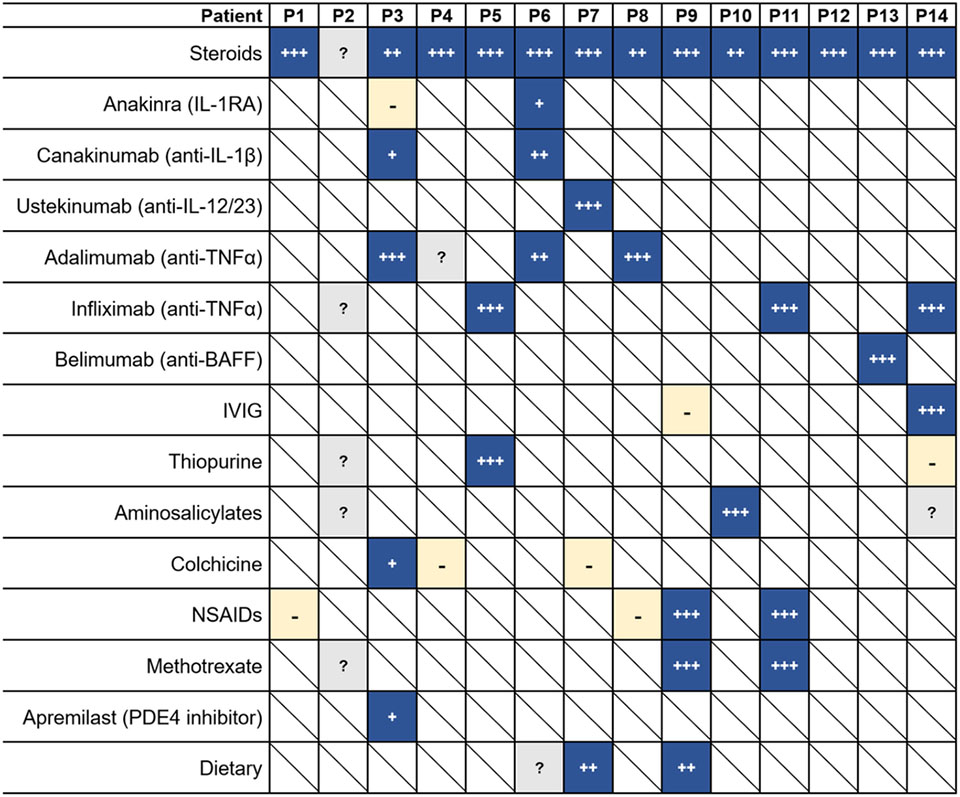
All fourteen identified DEX patients have received various treatments, with some successfully ameliorating their symptoms (Table 4 & Supplemental Table 4). All DEX patients have received steroids, usually as short courses, during periods of symptomatic flares to control their inflammation and provide quick relief of symptoms while avoiding the adverse effects of long-term glucocorticoid treatment in children. Every patient responded well to steroid therapy, and it is especially effective at resolving oral ulcers. Most patients (9/14) have also been successfully treated with some form of biologic therapy. These include monoclonal antibodies that target IL-1β (canakinumab), IL-12/23 p40 (ustekinumab), TNFα (adalimumab and infliximab), and B cell activating actor (BAFF; belimumab). TNFα-targeting agents have been the most widely used, with 6/8 patients achieving good clinical response, including resolution of oral ulcers, abolition of steroid-dependence, disappearance of inflammatory bowel lesions, alleviation of other symptoms, and even remission. For the other two patients treated with anti-TNFα, it is still too soon to assess response (P4) or the response is unknown (P2). However, canakinumab and ustekinumab have also been at least modestly effective at resolution of symptoms. Anakinra, a recombinant/modified version of human IL-1 receptor antagonist (IL-1RA), was effective in controlling fevers and abdominal symptoms and decreasing inflammatory markers for P6, but P3 found the injections difficult to tolerate. Both patients had better success after switching to canakinumab and then later to adalimumab. IVIG in replacement doses was associated with reduced frequency of infections in P9, and with symptom resolution in both P9 and P14, although it is difficult to assess its effect as it was used in combination with other immunomodulatory therapies [17,18].
Table 4:
DEX patient treatment regimens
|
Dark blue, patient responded to indicated treatment;
patient achieved remission or significant reduction of symptoms;
patient had good clinical response but did not lead to resolution of all symptoms;
patient had mild clinical response but relapsed or could not tolerate treatment;
light yellow with “-“, patient did not respond to indicated treatment;
gray boxes with “?”, patient was treated with indicated treatment but response is unknown;
white boxes with a diagonal, patient not treated with indicated treatment.
IL, interleukin; IL-1RA, interleukin-1 receptor antagonist protein; TNFα, tumor necrosis factor α; BAFF, B-cell activating factor; IVIG, intravenous immunoglobulin; NSAIDs, Non-steroidal anti-inflammatory drugs; PDE4, phosphodiesterase 4.
Other treatments used for the treatment of IBD and Behçet’s disease have also been used to treat DEX patients, with varying success. Antimetabolites azathioprine and mercaptopurine failed to induce long term remission in patients P2 and P14, respectively, but P5 is in clinical remission after a combination of infliximab and azathioprine therapy. P14 is currently being treated with aminosalicylate mesalazine along with thalidomide, and P10 achieved remission with a combination of mesalazine and steroid therapy [18]. The clinical response of P2 to sulfasalazine and mesalazine is unknown. Colchicine was trialed in P3, P4, and P7, but failed to prevent oral ulcers in any of these patients. Several DEX patients have been treated with either non-steroidal anti-inflammatory drugs (NSAIDs; 4/14 patients) or methotrexate (3/14 patients), usually for joint symptoms. The responses to these therapies were either minimal or unknown, except in P9 and P11, where the combination of NSAIDs and methotrexate alleviated joint symptoms [17]. P1 was initially trialed on naproxen, but it had little effect. Patient P3 was treated with apremilast (a phosphodiesterase 4 inhibitor) but had only a partial response. P14 failed to achieve long term remission with either tacrolimus or cyclosporine A (both calcineurin inhibitors) [18]. Some patients have found moderate success in modulating symptoms with dietary modifications, such as a benzoate-free or fiber-rich diet, but it can be difficult for children to maintain such diets. More details regarding treatment regimens and response are shown in Supplemental Table 4.
Discussion
This retrospective analysis comparing the clinical features of fourteen patients with DEX demonstrates a clinical phenotype with recurrent episodes of fevers of unknown origin, oral ulcers, ulcers in the gastrointestinal tract, and increased inflammatory markers (ESR and/or CRP) as the most common manifestations (Figure 3). Several patients also experience other symptoms such as skin inflammation, arthralgia/myalgia, arthritis, fatigue, decreased appetite, weight loss, and gastrointestinal symptoms such as abdominal pain, nausea/emesis, constipation, and diarrhea. With such a wide presentation of the disease, absence of any one of these clinical features should not rule out DEX. Due to the overlap in symptoms, DEX should be considered in the differential diagnosis of young male patients who are being evaluated for very early onset inflammatory bowel disease (VEO-IBD; before 6 years of age) [13,46], pediatric-onset IBD, periodic fever syndromes, Behçet’s syndrome, orofacial granulomatosis, or recurrent aphthous stomatitis (RAS). Not all DEX patients present with GI symptoms or have a family history typically seen in IBD, and most DEX patients identified to date lack clinical manifestations often seen in Behçet’s syndrome such as ocular, vascular, central nervous system, cardiac, renal, or pulmonary disease [14,15]. However, P3 exhibits pathergy as well as history of a scrotal ulcer, both findings often seen in Behçet’s syndrome. Additionally, P13 has systemic lupus erythematosus with biopsy-confirmed lupus nephritis class II and clinical diagnosis of lupus hepatitis. Despite the ELF4 variant of P13 (S248F) showing loss-of-function, this patient with SLE presents differently from other DEX patients; he lacks several of the common symptoms of oral ulcers, fevers, GI dysfunction, skin involvement, or arthralgias, and was not found to have anemia. Further investigation into the impacts of this ELF4 mutation is warranted.
Figure 3: Clinical and immunological findings of patients with Deficiency in ELF4, X-linked (DEX).
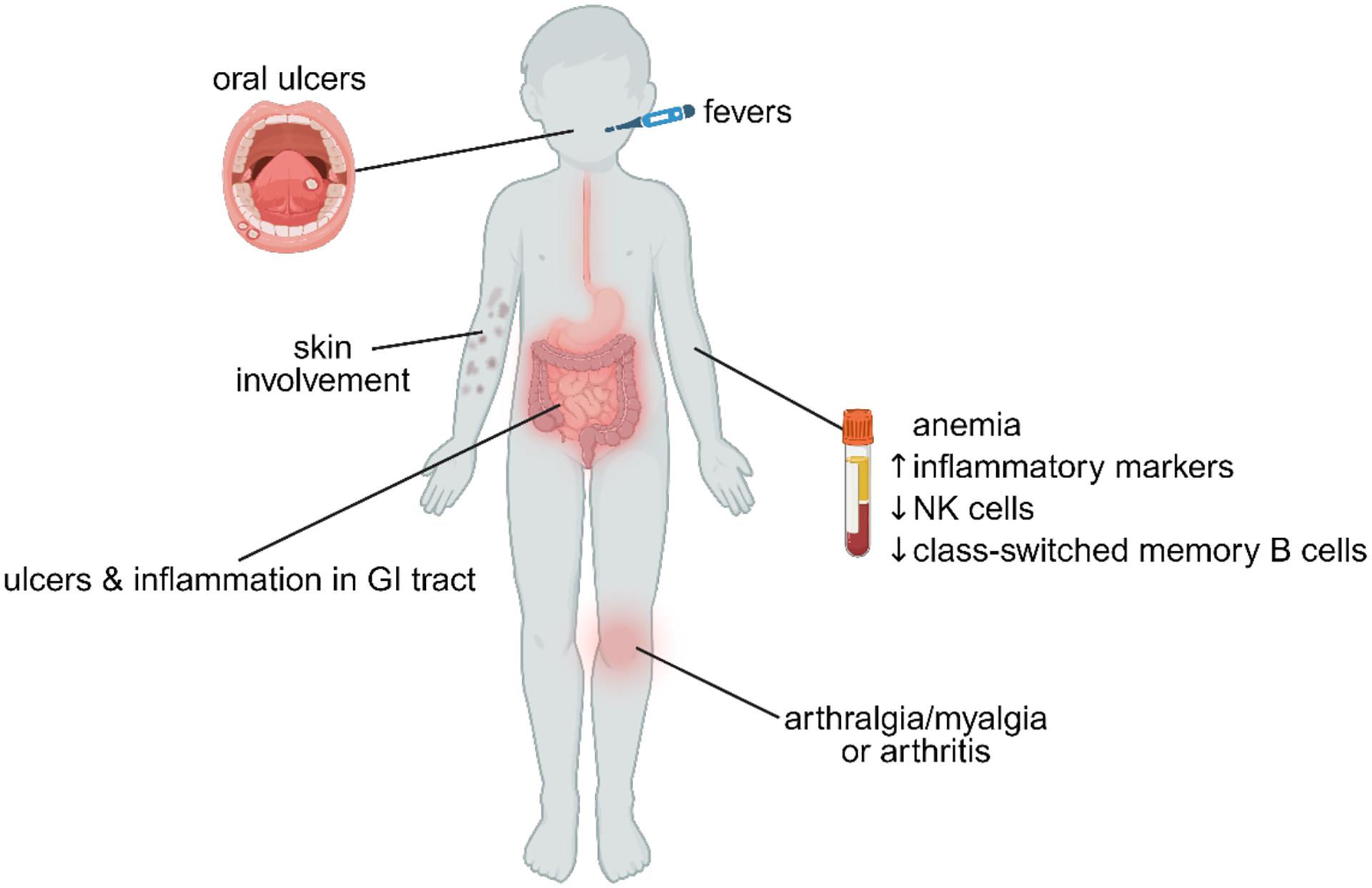
GI, gastrointestinal tract; NK, natural killer cells.
Definitive diagnosis of DEX is made by identification of mutations in ELF4 that are loss-of-function or lead to truncation/loss of protein. Missense mutations located within the ETS DNA binding domain in exon 7 of ELF4 or nonsense mutations/deletions that result in loss of ELF4 protein are good predictors of variant pathogenicity. The activity reporter assay with mouse Ifnb1 promoter used here to determine transcriptional activity of ELF4 provides “PS3_moderate” variant support as it has been validated with 12 benign gnomAD variants and is thus reliable at identifying loss-of-function variants of ELF4 [16,47]. Work is ongoing to identify the molecular and cellular mechanisms by which ELF4 variants lead to the observed phenotypes; deleterious mutations within the ETS DNA binding domain likely interfere with the ability of ELF4 to bind and initiate transcription at target genes, either directly or through interactions with other protein binding partners, as is common with ETS family members [48,49]. As most patients inherited mutations from their mothers, we recommend testing of family members upon genetic diagnosis of DEX. Females who are heterozygous for ELF4 mutations do not report symptoms and appear to be asymptomatic carriers, with the exception of the mother of P11, who has a diagnosis of Behçet’s syndrome and suffered from several ulcers on the mouth, face, and perineum along with episodes of fever, joint pain, and mild anemia [18]. Beyond rare cases of severe gene defects in ELF4, polymorphisms affecting ELF4 expression and function would be predicted to predispose to IBD more generally. It additionally does not escape our attention that somatic variants disrupting ELF4 function in a fraction of cells could be contributory toward inflammatory phenotypes overlapping in DEX that present later in life [50–52].
Based on the clinical courses of the fourteen DEX patients reported here, evaluation of several physical, immunological, and non-immunological factors within these patients will aid in monitoring disease course and guiding individualized treatment. The symptoms of several DEX patients have been found to evolve over time, so longitudinal monitoring for the appearance of new manifestations is needed. Ulcers within the GI tract were found in 7/8 DEX patients who were investigated, and in P5 and P7 this was in the absence of any abdominal pain or symptoms of constipation/diarrhea, respectively. Even in the absence of clear GI symptoms, initial evaluation by upper endoscopy as well as colonoscopy with ileoscopy may be informative to determine the extent and location of ulceration in the GI tract and to support therapeutic decisions. Magnetic resonance enterography (MRE) also has utility to evaluate for upper GI disease distal to the duodenum that is not accessible by endoscopy. Patients should also be evaluated for aphthous-like ulcers throughout the mouth, face, and perianal region, and signs of skin inflammation should be assessed. The oral ulcers and GI symptoms can cause great pain and discomfort, sometimes affecting oral intake and weight in DEX patients. Additionally, as several patients also have anemia, some due to iron deficiency, complete blood count (CBC), along with iron studies will help assess for iron deficiency. There have not been reports of other nutrient or mineral deficiencies in patients with DEX.
Mucosal inflammation paired with elevated pyrogens and inflammatory proteins seen in DEX fit with cytokinopathy being a major contributor to this autoinflammatory disease. In prior work, differentiation of CD4+ T cells of the T helper 17 (Th17) type was increased in both ELF4-deficient mice and DEX patients and associated with increased recruitment of neutrophils, a cell type implicated in the pathogenesis of IBD [16,38]. Consistently, in ascending colon biopsies of human DEX patients P6 and P7, there was also an increase in IL-17A and RORγT by immunofluorescence compared to human patients with IBD and healthy controls, and ELF4-mutant or total knockout mice displayed increased inflammatory responses in colitis models [16]. Additionally, ELF4-deficient myeloid cells have been shown to be hyperinflammatory and overproduce inflammatory cytokines in response to various stimuli in a mechanism partly dependent on elevated Trem1 and reduced IL-1Ra, the endogenous IL-1 receptor antagonist [16]. Sun et al. 2022 also demonstrated that DEX patient (P9) PBMCs or peripheral monocytes infected with virus produced more IL-6, IL-1β, and TNFα compared to healthy controls. The role of inflammatory cytokines in the pathology of DEX is underscored by the efficacy of biologics targeting IL-1, IL-12p40, and TNFα.
Discovery of DEX has illuminated an anti-inflammatory function of the ELF4 transcription factor in cells at the mucosal barrier [16,37]. As gastrointestinal and oral mucosa are sites of constant exposure to pattern recognition receptor (PRR) ligands from microbes, these sites being most affected in DEX patients fits with the function of ELF4 in promoting immune homeostasis at the mucosal barrier. However, a contribution of ELF4 in regulating the epithelial barrier is also plausible and requires further study [53,54]. Work is ongoing to elucidate the role of ELF4 as a transcriptional regulator of inflammation in immune cells and how variants in ELF4 lead to the inflammation seen in patients with DEX. Additionally, ELF4 could be playing a role in T regulatory cells, in which it is highly expressed [22], further contributing to immune dysregulation. Although ELF4 has been reported to be associated with other human immunological phenotypes, the associated variants (p.T187N [55] with a MAF of 1.15 × 10−4 and CADD score of 23.1 and p.S369P [56] with a MAF of 3.82 × 10−5 and a CADD score of 13.77) do not result in complete loss of ELF4 activity or cause disease consistent with DEX (Tyler et al., 2021 and unpublished).
During symptomatic flares, some patients have neutrophilic leukocytosis and thrombocytosis, as is well established in IBD flares and other acute inflammatory or reactive processes. Neutrophilic infiltration of inflamed mucosa was previously observed in colon and cheek biopsies from DEX patients, and the neutrophilia normalized in P6 upon treatment with anakinra and subsequently canakinumab [16]. It is unclear if intermittently decreased percentages of NK cells and class-switched memory B cells seen in some patients are due to cell-intrinsic or - extrinsic effects of ELF4 mutations in these immune cell types. Low peripheral NK cell numbers have been observed in pediatric patients with clinically active IBD [57], yet ELF4 has previously been shown in mouse models to be necessary for the development of NK cells and the expression of perforin, with ELF4-deficient mice showing both a reduction in the number of NK-T and NK cells and lower cytotoxicity [35]. These mice also displayed normal B cell development but a greater proliferative response of splenic B cells after lipopolysaccharide (LPS) treatment [35]. Despite intermittently decreased class-switched B cells seen in some DEX patients, only three DEX patients were reported to have suboptimal vaccine responses, and no patients have decreased immunoglobulin isotypes. However, we are lacking detailed information on class-switched B cells and vaccine responses for most DEX patients. Moreover, the infections seen in some DEX patients prior to the initiation of immunomodulatory therapy are from various classes of pathogens, do not indicate a clear immunodeficiency due to loss of ELF4, and may be related to other underlying factors/and or environmental context.
There is evidence of altered type 1 IFN responses in both ELF4-deficient mice and humans [16,17,37]. Mice lacking ELF4 display increased susceptibility to infection with West Nile Virus in vivo due to decreased induction of type 1 IFN responses, not through effects on NK, NK-T, or cytotoxic CD8+ T cells [37]. ELF4-mutant mouse macrophages as well as PBMCs and peripheral monocytes from DEX patient P9 have also displayed impaired viral clearance [17]. However, while low IFN-β was seen in the serum of LPS-challenged total ELF4 knockout and W250S mice (analogous mutation to P6 & P7) [16], virally-infected PBMCs and peripheral monocytes from P9 and macrophages from analogous point-mutant mice surprisingly produced more type 1 IFNs compared to healthy controls and wildtype mice [17]. The exact role that ELF4 plays in regulating interferons requires further investigation, and its role in viral susceptibility in particular may become clearer as more DEX patients are identified.
Clinical management of patients with DEX should be individualized based on the manifestations of each patient, with longitudinal evaluation of gut inflammation, oral ulcers, skin inflammation, and arthralgias. Short courses of steroids seem to be effective in the management of symptomatic flares, especially in resolving oral ulcers, but care should be taken in children to monitor adverse effects of prolonged glucocorticoid treatment. For longer-term maintenance, several DEX patients have been treated with therapies (aminosalicylates, thiopurines, methotrexate, apremilast, colchicine, thalidomide, and calcineurin inhibitors) often used in IBD and Behçet’s syndrome, with varying levels of response. Treatment with anti-IL-1β (canakinumab), anti-IL-12/23 p40 (ustekinumab), and anti-TNFα (adalimumab and infliximab), have proven effective in reducing inflammation, with some patients achieving symptomatic remission. Most patients (eight) have been treated with anti-TNFα biologics thus far, and they have been well-tolerated. For several DEX patients, ESR and/or CRP values appear to track with episodic symptomatic flares and also decrease upon treatment, making these helpful markers for monitoring disease severity. Stool calprotectin levels are correlated with disease severity in IBD and used to monitor response to treatment [58–61]. Stool calprotectin was found to be elevated in patients P5, P6, and P12 before substantial treatment. Patients P1, P3, and P7 had normal levels while undergoing treatment, while levels in P5 fluctuated greatly during treatment and are now trending downward. It is currently unclear whether stool calprotectin functions as an effective biomarker in evaluating the response of patients with DEX to treatment, as we only have longitudinal data for P5. Data are lacking on the effects of ELF4 variants as patients with DEX age, as all currently known patients are younger than 25 years of age. Disease progression should be monitored to identify manifestations that may present later in life.
Overall, compilation of clinical features from fourteen identified patients with DEX reveals that loss-of-function mutations in ELF4 lead to a disease with overlapping features of, though distinct from, IBD and Behçet’s syndrome. As more patients are identified, larger cohort studies can further characterize immune cell subsets and the extent/nature of inflammation in order to identify clinical or laboratory biomarkers that can reliably predict disease severity and follow DEX patients longitudinally. More effective targeted therapies may be identified as the mechanism by which ELF4 mutations lead to the symptoms seen in DEX are elucidated.
Supplementary Material
Acknowledgments:
The authors thank the patients and their families for participation in research and all clinical care staff for their contributions. We acknowledge the Yale Cancer Center for support. Figures 1A and 1B were created with BioRender.com.
Funding:
C.L.L. is funded by the Mathers Foundation, Rainin Foundation, NIAID/NIH, and Yale University. A.M.M. is funded by a Canada Research Chair (Tier 1) in Paediatric IBD, Canadian Institute of Health Research (CIHR) Foundation Grant, and NIDDK (RC2DK118640) grant and the Leona M. and Harry B. Helmsley Charitable Trust.
Conflicts of interest/Competing interests:
C.L.L. reports an advisory/consulting role for Pharming Healthcare Inc. and unrelated funding support from Ono Pharma. S.A.L. is part owner of Qiyas Higher Health, a startup company unrelated to this work. All other authors declare no competing interests.
Footnotes
Ethics approval: All human subjects in this study provided informed consent to use their samples for research and to publish de-identified data, in accordance with Helsinki principles for enrollment in research protocols that were approved by the Institutional Review Boards of Yale University, The Hospital for Sick Children, Erasmus University Medical Center, Radboud University Medical Center (regional Arnhem and Nijmegen Medical Ethics Committee), Hospital Universitario Virgen del Rocío, Children’s Hospital of Chongqing Medical University, the Children’s Hospital of Fudan University, Xiangya Hospital Central South University, West China Second University Hospital, and Shenzhen Children’s Hospital. Blood from healthy donors was also obtained under approved protocols. All relevant ethical regulations for work with human participants were followed.
Consent to participate: All human subjects in this study provided informed consent to participate.
Consent for publication: All human subjects in this study provided informed consent to publish de-identified data.
Availability of data and material:
May be available upon request.
References
- 1.Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nature Immunology 2017 18:8 [Internet]. 2017. [cited 2021 Dec 4];18:832–42. Available from: https://www.nature.com/articles/ni.3777 [DOI] [PubMed] [Google Scholar]
- 2.Rood JE, Behrens EM. Inherited Autoinflammatory Syndromes. https://doi.org/101146/annurev-pathmechdis-030121-041528 [Internet]. 2021. [cited 2021 Dec 4];17. Available from: https://www.annualreviews.org/doi/abs/10.1146/annurev-pathmechdis-030121-041528 [DOI] [PubMed] [Google Scholar]
- 3.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology 2022 42:7 [Internet]. 2022. [cited 2023 Feb 19];42:1473–507. Available from: https://link.springer.com/article/10.1007/s10875-022-01289-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum [Internet]. 2002. [cited 2021 Dec 4];46:3340–8. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/art.10688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An Autoinflammatory Disease with Deficiency of the Interleukin-1– Receptor Antagonist. New England Journal of Medicine [Internet]. 2009. [cited 2021 Dec 4];360:2426–37. Available from: https://www.nejm.org/doi/10.1056/NEJMoa0807865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity. 2011;34:755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF Receptor-Associated Periodic Syndrome (TRAPS): Current Perspectives. International Journal of Molecular Sciences 2020, Vol 21, Page 3263 [Internet]. 2020. [cited 2021 Dec 4];21:3263. Available from: https://www.mdpi.com/1422-0067/21/9/3263/htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konno H, Chinn IK, Hong D, Orange JS, Lupski JR, Mendoza A, et al. Pro-inflammation Associated with a Gain-of-Function Mutation (R284S) in the Innate Immune Sensor STING. Cell Rep. 2018;23:1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Q, Yang D, Ombrello AK, Zavialov A v., Toro C, Zavialov A v., et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. New England Journal of Medicine [Internet]. 2014. [cited 2021 Dec 4];370:911–20. Available from: https://www.nejm.org/doi/full/10.1056/nejmoa1307361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Q, Wang H, Schwartz DM, Stoffels M, Hwan Park Y, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nature Genetics 2015 48:1 [Internet]. 2015. [cited 2021 Dec 4];48:67–73. Available from: https://www.nature.com/articles/ng.3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pazmandi J, Kalinichenko A, Ardy RC, Boztug K. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol Rev [Internet]. 2019. [cited 2021 Dec 1];287:162–85. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/imr.12726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batura V, Muise AM. Very early onset IBD: Novel genetic aetiologies. Curr Opin Allergy Clin Immunol [Internet]. 2018. [cited 2021 Dec 4];18. Available from: https://journals.lww.com/co-allergy/Fulltext/2018/12000/Very_early_onset_IBD__novel_genetic_aetiologies.5.aspx [DOI] [PubMed] [Google Scholar]
- 13.Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology. 2014;147:990–1007.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yazici H, Seyahi E, Hatemi G, Yazici Y. Behçet syndrome: a contemporary view. Nature Reviews Rheumatology 2018 14:2 [Internet]. 2018. [cited 2023 May 2];14:107–19. Available from: https://www.nature.com/articles/nrrheum.2017.208 [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Yao X. A Contemporary Review of Behcet’s Syndrome. Clinical Reviews in Allergy & Immunology 2021 61:3 [Internet]. 2021. [cited 2023 May 2];61:363–76. Available from: https://link.springer.com/article/10.1007/s12016-021-08864-3 [DOI] [PubMed] [Google Scholar]
- 16.Tyler PM, Bucklin ML, Zhao M, Maher TJ, Rice AJ, Ji W, et al. Human autoinflammatory disease reveals ELF4 as a transcriptional regulator of inflammation. Nature Immunology 2021 22:9 [Internet]. 2021. [cited 2021 Oct 5];22:1118–26. Available from: https://www.nature.com/articles/s41590-021-00984-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun G, Qiu L, Yu L, An Y, Ding Y, Zhou L, et al. Loss of Function Mutation in ELF4 Causes Autoinflammatory and Immunodeficiency Disease in Human. J Clin Immunol [Internet]. 2022. [cited 2022 Mar 13];1:1–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/35266071 [DOI] [PubMed] [Google Scholar]
- 18.Sun G, Wu M, Lv Q, Yang X, Wu J, Tang W, et al. A Multicenter Cohort Study of Immune Dysregulation Disorders Caused by ELF4 Variants in China. Journal of Clinical Immunology 2023 [Internet]. 2023. [cited 2023 Feb 26];1:1–7. Available from: https://link.springer.com/article/10.1007/s10875-023-01453-3 [DOI] [PubMed] [Google Scholar]
- 19.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. [DOI] [PubMed] [Google Scholar]
- 20.Choi H-J, Geng Y, Cho H, Li S, Giri PK, Felio K, et al. Differential requirements for the Ets transcription factor Elf-1 in the development of NKT cells and NK cells. Blood [Internet]. 2011. [cited 2021 Oct 31];117:1880–7. Available from: http://ashpublications.org/blood/article-pdf/117/6/1880/1341410/zh800611001880.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wurster AL, Siu G, Leiden JM, Hedrick’ SM. Elf-1 binds to a critical element in a second CD4 enhancer. Mol Cell Biol [Internet]. 1994. [cited 2021 Dec 5];14:6452–63. Available from: https://journals.asm.org/doi/abs/10.1128/mcb.14.10.6452-6463.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 2018;175:1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sivina M, Yamada T, Park CS, Puppi M, Coskun S, Hirschi K, et al. The transcription factor E74-like factor controls quiescence of endothelial cells and their resistance to myeloablative treatments in bone marrow. Arterioscler Thromb Vasc Biol [Internet]. 2011. [cited 2021 Dec 5];31:1185–91. Available from: https://www.ahajournals.org/doi/abs/10.1161/ATVBAHA.111.224436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sashida G, Liu Y, Elf S, Miyata Y, Ohyashiki K, Izumi M, et al. ELF4/MEF Activates MDM2 Expression and Blocks Oncogene-Induced p16 Activation To Promote Transformation. Mol Cell Biol [Internet]. 2009. [cited 2021 Dec 1];29:3687. Available from: /pmc/articles/PMC2698769/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curina A, Termanini A, Barozzi I, Prosperini E, Simonatto M, Polletti S, et al. High constitutive activity of a broad panel of housekeeping and tissue-specific cis-regulatory elements depends on a subset of ETS proteins. Genes Dev [Internet]. 2017. [cited 2021 Oct 21];31:399. Available from: /pmc/articles/PMC5358759/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang Y, Wu T, He Y, He Y, Zhao D. Elf4 regulates lysosomal biogenesis and the mTOR pathway to promote clearance of Staphylococcus aureus in macrophages. FEBS Lett. 2021;595:881–91. [DOI] [PubMed] [Google Scholar]
- 27.Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–87. [DOI] [PubMed] [Google Scholar]
- 28.Lee JM, Libermann TA, Cho JY. The synergistic regulatory effect of Runx2 and MEF transcription factors on osteoblast differentiation markers. J Periodontal Implant Sci [Internet]. 2010. [cited 2021 Dec 1];40:39. Available from: /pmc/articles/PMC2872803/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sashida G, Bae N, Di Giandomenico S, Asai T, Gurvich N, Bazzoli E, et al. The Mef/Elf4 Transcription Factor Fine Tunes the DNA Damage Response. Cancer Res [Internet]. 2011. [cited 2021 Dec 1];71:4857–65. Available from: https://cancerres.aacrjournals.org/content/71/14/4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao JJ, Liu Y, Lacorazza HD, Soslow RA, Scandura JM, Nimer SD, et al. Tumor promoting properties of the ETS protein MEF in ovarian cancer. Oncogene 2007 26:27 [Internet]. 2007. [cited 2021 Dec 5];26:4032–7. Available from: https://www.nature.com/articles/1210170 [DOI] [PubMed] [Google Scholar]
- 31.Mao S, Frank RC, Zhang J, Miyazaki Y, Nimer SD. Functional and Physical Interactions between AML1 Proteins and an ETS Protein, MEF: Implications for the Pathogenesis of t(8;21)-Positive Leukemias. Mol Cell Biol [Internet]. 1999. [cited 2021 Oct 21];19:3635. Available from: /pmc/articles/PMC84165/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ando K, Tsushima H, Matsuo E, Horio K, Tominaga-Sato S, Imanishi D, et al. Mutations in the Nucleolar Phosphoprotein, Nucleophosmin, Promote the Expression of the Oncogenic Transcription Factor MEF/ELF4 in Leukemia Cells and Potentiates Transformation. J Biol Chem [Internet]. 2013. [cited 2021 Dec 1];288:9457. Available from: /pmc/articles/PMC3611015/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore SDP, Offor O, Ferry JA, Amrein PC, Morton CC, Dal Cin P. ELF4 is fused to ERG in a case of acute myeloid leukemia with a t(X;21)(q25–26;q22). Leuk Res. 2006;30:1037–42. [DOI] [PubMed] [Google Scholar]
- 34.Sashida G, Bazzoli E, Menendez S, Liu Y, Nimer SD. The oncogenic role of the ETS transcription factors MEF and ERG. Cell Cycle [Internet]. 2010. [cited 2021 Dec 1];9:3457. Available from: /pmc/articles/PMC3230474/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacorazza HD, Miyazaki Y, Di Cristofano A, Deblasio A, Hedvat C, Zhang J, et al. The ETS Protein MEF Plays a Critical Role in Perforin Gene Expression and the Development of Natural Killer and NK-T Cells. Immunity. 2002;17:437–49. [DOI] [PubMed] [Google Scholar]
- 36.Yamada T, Park CS, Mamonkin M, Lacorazza D. The transcription factor ELF4 controls proliferation and homing of CD8+ T cells via the Krüppel-like factors KLF4 and KLF2. Nat Immunol [Internet]. 2009. [cited 2021 Oct 21];10:618. Available from: /pmc/articles/PMC2774797/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You F, Wang P, Yang L, Yang G, Zhao YO, Qian F, et al. ELF4 is critical for induction of type i interferon and the host antiviral response. Nat Immunol [Internet]. 2013;14:1237–46. Available from: 10.1038/ni.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee P-H, Puppi M, Schluns KS, Yu-Lee L-Y, Dong C, Lacorazza HD. The Transcription Factor E74-like Factor 4 Suppresses Differentiation of Proliferating CD4 + T Cells to the Th17 Lineage. The Journal of Immunology. 2014;192:178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020 581:7809 [Internet]. 2020. [cited 2023 Apr 12];581:434–43. Available from: https://www.nature.com/articles/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med [Internet]. 2021. [cited 2023 Apr 12];13:1–12. Available from: https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 2014 46:3 [Internet]. 2014. [cited 2023 Apr 12];46:310–5. Available from: https://www.nature.com/articles/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Menthon M, LaValley MP, Maldini C, Guillevin L, Mahr A. HLA–B51/B5 and the Risk of Behçet’s Disease: A Systematic Review and Meta-Analysis of Case–Control Genetic Association Studies. Arthritis Rheum [Internet]. 2009. [cited 2023 May 22];61:1287–96. Available from: /pmc/articles/PMC3867978/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeno M The association of Behçet’s syndrome with HLA-B51 as understood in 2021. Curr Opin Rheumatol [Internet]. 2022. [cited 2023 May 22];34:4. Available from: /pmc/articles/PMC8635258/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johns Hopkins University BM. Online Mendelian Inheritance in Man, OMIM [Internet]. MIM Number: 300979. https://omim.org/; 2016. [cited 2023 May 12]. Available from: https://omim.org/entry/300979
- 45.Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The Classification of Glomerulonephritis in Systemic Lupus Erythematosus Revisited. Journal of the American Society of Nephrology [Internet]. 2004. [cited 2023 Jun 17];15:241–50. Available from: https://journals.lww.com/jasn/Fulltext/2004/02000/The_Classification_of_Glomerulonephritis_in.1.aspx [DOI] [PubMed] [Google Scholar]
- 46.Ouahed J, Spencer E, Kotlarz D, Shouval DS, Kowalik M, Peng K, et al. Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflamm Bowel Dis [Internet]. 2020. [cited 2023 May 2];26:820. Available from: /pmc/articles/PMC7216773/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med [Internet]. 2019. [cited 2023 Sep 17];12:1–12. Available from: https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li R, Pei H, Watson DK. Regulation of Ets function by protein–protein interactions. Oncogene 2000 19:55 [Internet]. 2001. [cited 2021 Oct 27];19:6514–23. Available from: https://www.nature.com/articles/1204035 [DOI] [PubMed] [Google Scholar]
- 49.Sharrocks AD. The ETS-domain transcription factor family. Nature Reviews Molecular Cell Biology 2001 2:11 [Internet]. 2001. [cited 2021 Oct 21];2:827–37. Available from: https://www.nature.com/articles/35099076 [DOI] [PubMed] [Google Scholar]
- 50.Sikora KA, Wells KV., Bolek EC, Jones AI, Grayson PC. Somatic mutations in rheumatological diseases: VEXAS syndrome and beyond. Rheumatology (Oxford) [Internet]. 2022. [cited 2023 May 1];61:3149. Available from: /pmc/articles/PMC9348615/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poulter JA, Collins JC, Cargo C, De Tute RM, Evans P, Ospina Cardona D, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood [Internet]. 2021. [cited 2023 May 1];137:3676–81. Available from: https://ashpublications.org/blood/article/137/26/3676/475476/Novel-somatic-mutations-in-UBA1-as-a-cause-of [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aluri J, Cooper MA. Genetic Mosaicism as a Cause of Inborn Errors of Immunity. J Clin Immunol [Internet]. 2021. [cited 2023 May 1];41:718–28. Available from: https://link.springer.com/article/10.1007/s10875-021-01037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kai H, Hisatsune A, Chihara T, Uto A, Kokusho A, Miyata T, et al. Myeloid ELF-1-like factor up-regulates lysozyme transcription in epithelial cells. Journal of Biological Chemistry [Internet]. 1999. [cited 2023 Sep 23];274:20098–102. Available from: http://www.jbc.org/article/S0021925819726210/fulltext [DOI] [PubMed] [Google Scholar]
- 54.Lu Z, Kim KA, Suico MA, Shuto T, Li JD, Kai H. MEF up-regulates human β-defensin 2 expression in epithelial cells. FEBS Lett [Internet]. 2004. [cited 2023 Sep 23];561:117–21. Available from: https://onlinelibrary.wiley.com/doi/full/10.1016/S0014-5793%2804%2900138-3 [DOI] [PubMed] [Google Scholar]
- 55.Salinas SA, Mace EM, Conte MI, Park CS, Li Y, Rosario-Sepulveda JI, et al. An ELF4 hypomorphic variant results in NK cell deficiency. JCI Insight [Internet]. 2022. [cited 2022 Dec 11];7. Available from: https://insight.jci.org/articles/view/155481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stewart DM, Tian L, Notarangelo LD, Nelson DL. X-linked hypogammaglobulinemia and isolated growth hormone deficiency: An update. Immunol Res [Internet]. 2008. [cited 2022 Jun 4];40:262–70. Available from: https://link.springer.com/article/10.1007/s12026-007-0028-9 [DOI] [PubMed] [Google Scholar]
- 57.Pappa A, Mührer J, Gast P, Hebbar Subramanyam S, Ohl K, Muschaweck M, et al. Pediatric IBD patients show medication and disease activity dependent changes in NK cell and CD4 memory T cell populations. Front Pediatr [Internet]. 2023. [cited 2023 Sep 24];11:1123873. Available from: /pmc/articles/PMC10345343/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sipponen T, Savilahti E, Kärkkäinen P, Kolho KL, Nuutinen H, Turunen U, et al. Fecal calprotectin, lactoferrin, and endoscopic disease activity in monitoring anti-TNF-alpha therapy for Crohn’s disease. Inflamm Bowel Dis [Internet]. 2008. [cited 2023 May 2];14:1392–8. Available from: https://pubmed.ncbi.nlm.nih.gov/18484671/ [DOI] [PubMed] [Google Scholar]
- 59.Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology [Internet]. 2011. [cited 2023 May 2];140:1817–1826.e2. Available from: https://pubmed.ncbi.nlm.nih.gov/21530748/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D’Haens G, Ferrante M, Vermeire S, Baert F, Noman M, Moortgat L, et al. Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm Bowel Dis [Internet]. 2012. [cited 2023 May 2];18:2218–24. Available from: https://pubmed.ncbi.nlm.nih.gov/22344983/ [DOI] [PubMed] [Google Scholar]
- 61.Aomatsu T, Yoden A, Matsumoto K, Kimura E, Inoue K, Andoh A, et al. Fecal calprotectin is a useful marker for disease activity in pediatric patients with inflammatory bowel disease. Dig Dis Sci [Internet]. 2011. [cited 2023 May 2];56:2372–7. Available from: https://link.springer.com/article/10.1007/s10620-011-1633-y [DOI] [PubMed] [Google Scholar]
- 62.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res [Internet]. 2010. [cited 2023 Jun 7];20:1297. Available from: /pmc/articles/PMC2928508/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res [Internet]. 2010. [cited 2023 Jun 7];38:e164. Available from: /pmc/articles/PMC2938201/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crowley E, Warner N, Pan J, Khalouei S, Elkadri A, Fiedler K, et al. Prevalence and Clinical Features of Inflammatory Bowel Diseases Associated With Monogenic Variants, Identified by Whole-Exome Sequencing in 1000 Children at a Single Center. Gastroenterology. 2020;158:2208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
May be available upon request.