

Abstract
目的
法尼酯X受体(farnesoid X receptor,FXR)是配体激活转录因子核受体超家族成员,属于胆汁酸受体。FXR在肾组织中表达,可通过调节糖脂代谢、抑制炎症反应、拮抗氧化应激及肾纤维化等机制减轻肾损伤。但FXR是否参与肾脏疾病中的自噬尚不明确。本研究旨在探讨FXR在顺铂所致急性肾损伤中的作用,并探讨其机制是否与调控自噬相关。
方法
选取12周雄性野生型(wild type,WT)或FXR基因敲除(knockout of FXR,FXR-KO)小鼠各12只,随机分为WT组、WT+顺铂组、FXR-KO组、FXR-KO+顺铂组,每组6只,WT+顺铂组和FXR-KO+顺铂组予腹腔注射顺铂(20 mg/kg),WT组和FXR-KO组予腹腔注射等体积的顺铂溶剂。72 h后处死小鼠,留取血液和肾组织标本。采用免疫比浊法检测血清BUN和SCr水平,HE染色后在光学显微镜下观察肾组织病理改变,采用蛋白质印迹法和免疫组织化学法检测LC3、p62的蛋白质表达水平,在电子显微镜下观察受损线粒体清除和溶酶体底物聚集的情况,采用TUNEL检测肾小管上皮细胞凋亡的情况。
结果
WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠SCr和BUN均明显升高(P<0.01或P<0.001),且FXR-KO+顺铂组小鼠SCr和BUN明显高于WT+顺铂组(均P<0.05)。在光学显微镜下可见WT组和FXR-KO组小鼠肾组织无明显病理改变,WT+顺铂组和FXR-KO+顺铂组小鼠均存在肾小管上皮细胞空泡样或颗粒样变性,细胞扁平,管腔扩张,刷状缘脱落,甚至出现基底膜裸露,管型形成。WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠肾小管损伤评分均明显升高(均P<0.001),且FXR-KO+顺铂组评分明显高于WT+顺铂组(P<0.05)。在透射电镜下可见WT+顺铂组和FXR-KO+顺铂组小鼠小管上皮细胞线粒体肿胀变圆、空泡化、嵴断裂或消失,溶酶体呈不均匀、高密度团块状,且以FXR-KO+顺铂组改变更为明显。蛋白质印迹法结果显示:WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠肾皮质LC3-II/LC3-I比值下降,p62表达增加(P<0.05或P<0.01);且相较于FXR-KO组,FXR-KO+顺铂组小鼠LC3-II/LC3-I比值下降,p62表达增加更为明显(均P<0.05)。免疫组织化学结果显示:WT+顺铂组和FXR-KO+顺铂组小鼠肾皮质总LC3和p62表达均明显增加,且以FXR-KO+顺铂组增加更为显著。TUNEL结果显示:WT组和FXR-KO组小鼠染色阴性或仅见数个小管上皮细胞凋亡,WT+顺铂组和FXR-KO+顺铂组凋亡细胞数量均增多;WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,肾小管上皮细胞凋亡率明显增加(均P<0.001),且FXR-KO+顺铂组凋亡率明显高于WT+顺铂组(P<0.05)。
结论
FXR基因敲除加重顺铂所致急性肾损伤,其机制可能与抑制自噬和促进凋亡有关。
Keywords: 法尼酯X受体, 基因敲除, 顺铂, 自噬, 凋亡, 急性肾损伤
Abstract
Objective
Farnesoid X receptor (FXR) is a member of the nuclear receptor superfamily of ligand activated transcription factors and belongs to bile acid receptor. Studies have shown that the expression of FXR in renal tissue can reduce renal injury via regulation of glucose and lipid metabolism, inhibition of inflammatory response, reduction of oxidative stress and renal fibrosis. However, it is unclear whether FXR is involved in autophagy in renal diseases. This study aims to investigate the role of FXR in cisplatin-induced acute renal injury and whether its mechanism is related to autophagy regulation.
Methods
Twelve male WT or FXR-KO mice at 12 weeks were randomly divided into a WT group, a WT+cisplatin group, a FXR-KO group, and a FXR-KO+cisplatin group, with 6 mice in each group. The WT+cisplatin group and the FXR-KO+cisplatin group were intraperitoneally injected with cisplatin (20 mg/kg), and the WT group and the FXR-KO group were intraperitoneally injected with equal volume of cisplatin solvent. Seventy-two hours later, the mice were killed and blood and renal tissue samples were collected. The levels of SCr and BUN were detected by immunoturbidimetry. After the staining, the pathological changes of renal tissue were observed under optical microscope. The protein levels of LC3 and p62 were detected by Western blotting and immunohistochemistry. The clearance of damaged mitochondria and the accumulation of lysosomal substrate were observed under electron microscope. The apoptosis of renal tubular epithelial cells was detected by TUNEL.
Results
Compared with the WT group or the FXR-KO group, both SCr and BUN levels in the WT+cisplatin group or the FXR-KO+cisplatin group were significantly increased (P<0.01 or P<0.001), and SCr and BUN levels in the FXR-KO+cisplatin group were significantly higher than those in the WT+cisplatin group (both P<0.05). Under the light microscope, there were no obvious pathological changes in the renal tissue of mice in the WT group and the FXR-KO group. Both the WT+cisplatin group and the FXR-KO+cisplatin group had vacuolar or granular degeneration of renal tubular epithelial cells, flat cells, lumen expansion, brush edge falling off, and even exposed basement membrane and tubular formation. The scores of renal tubular injury in the WT+cisplatin group and the FXR-KO+cisplatin group were significantly higher than those in the WT group and the FXR-KO group, respectively (both P<0.001), and the score in the FXR-KO+cisplatin group was significantly higher than that in the WT+cisplatin group (P<0.05). Under the transmission electron microscope, the mitochondria of mouse tubular epithelial cell in the WT+cisplatin group and the FXR-KO+cisplatin group was swollen, round, vacuolated, cristae broken or disappeared; the lysosome was uneven and high-density clumps, and the change was more obvious in the FXR-KO+cisplatin group. Western blotting showed that the ratio of LC3-II to LC3-I was decreased and the expression of p62 was increased in the WT+cisplatin group compared with the WT group and the FXR-KO+cisplatin group compared with FXR-KO group (P<0.05 or P<0.01); compared with the FXR-KO group, the ratio of LC3-II to LC3-I was decreased and the expression of p62 was increased significantly in the FXR-KO+cisplatin group (both P<0.05). Immunohistochemistry results showed that the expression of total LC3 and p62 in renal cortex of the WT+cisplatin group and the FXR-KO+cisplatin group was increased significantly, especially in the FXR-KO+cisplatin group. TUNEL results showed that the mice in the WT group and the FXR-KO group had negative staining or only a few apoptotic tubular epithelial cells, and the number of apoptotic cells in the WT+cisplatin group and the FXR-KO+cisplatin group were increased. The apoptosis rates of renal tubular epithelial cells in the WT+cisplatin group and the FXR-KO+cisplatin group were significantly higher than those in the WT group and the FXR-KO group, respectively (both P<0.001), and the apoptosis rate in the FXR-KO+cisplatin group was significantly higher than that in the WT+cisplatin group (P<0.05).
Conclusion
Knockout of FXR gene aggravates cisplatin induced acute renal injury, and its mechanism may be related to inhibiting autophagy and promoting apoptosis.
Keywords: farnesoid X receptor, gene knockout, cisplatin, autophagy, apoptosis, acute renal injury
急性肾损伤(acute kidney injury,AKI)是由肾缺血、脓毒症和肾毒性药物等因素引起的一类临床综合征,其特征点是肾脏结构或功能损伤引起的肾功能突然(48 h内)下降,代谢废物堆积,电解质和体液失衡。顺铂是一种广泛应用于治疗多种肿瘤的药物,主要经肾排泄,且易在肾皮质中蓄积。肾毒性被认为是是顺铂化学药物治疗的主要问题之一。顺铂诱导的AKI小鼠存在自噬缺陷,近端肾小管自噬相关基因7特异性敲除小鼠对顺铂更敏感,表现为肾功能丧失、组织损伤和细胞凋亡[1]。这表明自噬对顺铂所致AKI具有重要的保护作用。法尼酯X受体(farnesoid X receptor,FXR)是最早发现的胆汁酸受体,属于核受体超家族,在肝、胆囊、肾、心脏、肠道及胰岛中均有高表达。FXR参与包括AKI在内的多种肾脏疾病的发生和发展,并可通过调节糖脂代谢、抑制炎症反应和氧化应激、降低尿蛋白、拮抗肾纤维化等机制,减轻肾脏病变[2-3]。同时,野生型(wild type,WT)小鼠、自噬基因缺陷小鼠肝组织中的FXR对自噬通路具有重要的调节作用,与维持自噬稳态密切相关[4-5]。但FXR是否通过调控自噬在顺铂所致AKI中发挥保护作用尚不清楚。本研究建立顺铂诱导AKI小鼠模型,观察小鼠肾功能、肾组织病理及自噬相关指标的改变,研究FXR在顺铂所致AKI中的作用,并探讨其机制是否与自噬通路相关,以期为AKI发病机制及治疗药物的研究提供新的依据与思路。
1. 材料与方法
1.1. 实验动物与分组
选取12周同窝雄性FXR基因敲除(FXR-KO)小鼠和WT小鼠各12只,小鼠均由中南大学医学遗传学中心谭洁琼博士惠赠。实验分为WT组、WT+顺铂组、FXR-KO组、FXR-KO+顺铂组,每组6只,WT+顺铂组和FXR-KO+顺铂组小鼠予腹腔注射顺铂(20 mg/kg),WT组和FXR-KO组予腹腔注射等体积的顺铂溶剂。注射前及注射后72 h分别测量小鼠体重,计算体重变化值。注射后72 h处死小鼠,留取血液和肾组织标本。
1.2. 主要材料与试剂
BCA蛋白质浓度测定试剂盒购自美国Pierce公司,兔抗鼠p62抗体和兔二抗购自美国Sigma-Aldrich公司,兔抗鼠LC3抗体购自美国Cell Signaling Technology公司,DAB显色试剂盒购自北京中杉金桥生物技术有限公司,TUNEL细胞凋亡原位检测试剂盒购自瑞士Roche公司。
1.3. SCr和BUN的测定
将离心好的血清从-80 ℃冰箱中取出,恢复至室温,于全自动生化仪上检测SCr及BUN的水平。
1.4. 蛋白质印迹法
取适量肾皮质并充分研磨裂解组织,按BCA蛋白质浓度测定试剂盒说明书测定蛋白质浓度。行蛋白质上样、电泳、转膜后,取出PVDF膜放到封闭液中,置于摇床上、在室温下封闭1 h;加入一抗(兔抗鼠p62抗体 1꞉2 000,兔抗鼠LC3抗体1꞉1 000),置于摇床上,在4 ℃下孵育过夜;洗膜后分别加入相应兔二抗(1꞉10 000),置于摇床上,在室温下孵育1~2 h,用化学发光仪显影,使用Image J软件以目的条带与β-actin的吸光度值之比反映蛋白质水平。
1.5. 光学显微镜下观察
取已固定的小鼠肾脏,制作2 μm厚的石蜡切片,行HE染色后在光学显微镜下观察。光学显微镜下肾小管损伤的表现:管状扩张/扁平化,刷状缘丧失,细胞脱落到管状腔,管状铸型的形成,管状变性和空泡化。肾小管坏死评分[6]:无损伤为0分,受损肾小管占比<25%为1分,受损肾小管占比25%~50%为2分,受损肾小管占比51%~75%为3分,受损肾小管占比>75%为4分。
1.6. 免疫组织化学检测肾组织LC3、p62蛋白质的表达
将制作好的石蜡切片在65 ℃烘箱中烘60~ 120 min,脱蜡后进行加热以修复抗原,冷却后浸入蒸馏水洗涤3 min;加入3%的过氧化氢溶液后用蒸馏水清洗10 min;用5%山羊血清封闭1 h;在切片组织表面滴加稀释后的一抗(LC3或p62 1꞉100稀释),在4 ℃下过夜,次日用PBS冲洗5 min共3次;在切片组织表面滴加1꞉100稀释的二抗,于室温下孵育1 h;滴加DAB显色液后用蒸馏水冲洗以终止显色;用苏木精复染组织5 min,蒸馏水冲洗后PBS返蓝;用梯度乙醇脱水透明后放于二甲苯I、二甲苯II中各浸泡10 min;以中性树胶封片,在显微镜下扫描拍照,采用Image J软件分析结果。
1.7. 透射电镜下观察
将肾皮质组织制作成厚度约为80 nm的超薄切片,用饱和醋酸铀和柠檬酸铅进行染色,在日立HT7700透射电镜下观察小鼠肾组织线粒体损伤(表现为形态肿胀变圆、空泡化、嵴断裂及消失)和溶酶体底物聚集情况(表现为溶酶体内聚集的底物不能被降解,呈不均匀、高密度团块状)并拍片。
1.8. TUNEL分析
采用TUNEL细胞凋亡原位检测试剂盒检测小鼠肾组织的细胞凋亡情况,严格按照试剂盒说明书操作。在光学显微镜下观察细胞核:正常细胞核呈蓝色,细胞核染成棕色的细胞为TUNEL染色阳性的凋亡细胞。图片用Image J软件分析处理,每张图片随机选取互不重叠的5个视野并分别计数肾小管上皮细胞数和凋亡细胞数,然后计算凋亡率(细胞凋亡率=凋亡细胞数/细胞总数×100%)。
1.9. 统计学处理
每个实验至少重复3次,数据以均数±标准差 ( ±s)表示。用SPSS 22.0软件进行统计学分析,各组间差异的比较采用单因素方差分析(One-way ANOVA),两样本均数的多重比较采用独立样本t检验,P<0.05为差异具有统计学意义。
2. 结 果
2.1. 小鼠一般情况及体重变化
于腹腔注射顺铂或等体积的顺铂溶剂72 h后,WT+顺铂组和FXR-KO+顺铂组小鼠出现精神萎靡、嗜睡,进食及活动明显减少。WT组和FXR-KO组小鼠体重变化不明显,两组相比差异无统计学意义(P>0.05),WT+顺铂组和FXR-KO+顺铂组小鼠体重均较注射前下降,且以FXR-KO+顺铂组更为显著(P<0.05,图1)。
图1.
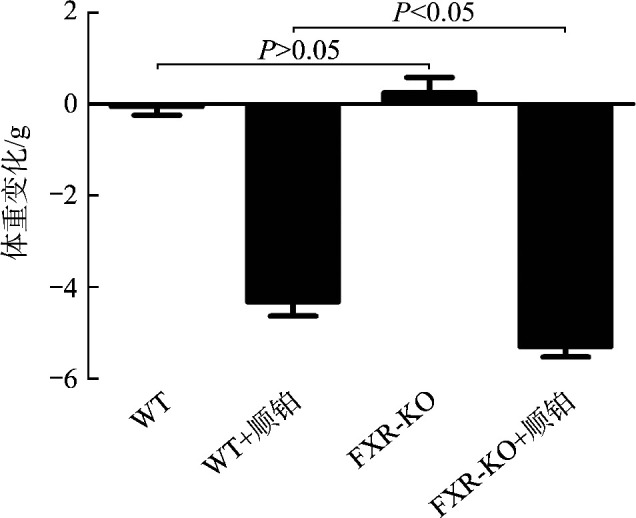
各组小鼠体重变化
Figure 1 Weight change of mice in each group
2.2. 小鼠肾功能变化
WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠SCr和BUN均明显升高(P<0.01或P<0.001),且FXR-KO+顺铂组小鼠SCr和BUN明显高于WT+顺铂组(均P<0.05,图2)。
图2.

各组小鼠SCr(A)和BUN(B)水平比较
Figure 2 Comparison of SCr (A) and BUN (B) level between groups *P<0.05, **P<0.01, ***P<0.001.
2.3. 肾组织学变化
在光学显微镜下可见WT组和FXR-KO组小鼠肾组织无明显病理改变,WT+顺铂组和FXR-KO+顺铂组小鼠均存在AKI改变,表现为肾小管上皮细胞空泡样或颗粒样变性,细胞扁平,管腔扩张,刷状缘脱落,甚至出现基底膜裸露,管型形成(图3A)。WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠肾小管损伤评分均明显升高(均P<0.001),且FXR-KO+顺铂组评分明显高于WT+顺铂组(P<0.05,图3B)。在透射电镜下可见WT+顺铂组和FXR-KO+顺铂组小鼠小管上皮细胞线粒体肿胀变圆、空泡化、脊断裂或消失,溶酶体呈不均匀、高密度团块状,且以FXR-KO+顺铂组改变更为明显(图4)。
图3.

各组小鼠肾组织病理改变(A,HE染色)和肾小管损伤评分(B)
Figure 3 Pathological changes of renal tissue (A, HE staining) and renal tubular injury score of mice in each group (B) *P<0.05, ***P<0.001.
图4.

透射电镜下观察各组小鼠肾小管上皮细胞线粒体清除和溶酶体底物聚集情况
Figure 4 Mitochondrial clearance and lysosomal substrate aggregation in renal tubular epithelial cells of mice in each group under transmission electron microscope The double arrow shows damaged mitochondria and the single arrow shows abnormal lysosome.
2.4. 肾皮质LC3、p62表达变化
蛋白质印迹法结果显示:与WT组相比,FXR-KO组小鼠肾皮质LC3-II/LC3-I比值升高,p62表达增加,但差异均无统计学意义(均P>0.05)。WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,小鼠肾皮质LC3-II/LC3-I比值下降,p62表达增加(P<0.05或P<0.01);且相较于WT+顺铂组,FXR-KO+顺铂组小鼠LC3-II/LC3-I比值下降,p62表达增加更为明显(均P<0.05,图5)。
图5.

蛋白质印迹法显示各组小鼠肾皮质LC3和p62的表达
Figure 5 Western blotting showing the expression of LC3 and p62 in renal cortex of mice in each group A: Protein electrophoretic diagram; B: Histogram of protein electrophoresis results. *P<0.05, **P<0.01.
免疫组织化学结果显示:WT+顺铂组和FXR-KO+顺铂组小鼠肾皮质总LC3和p62表达均明显增加,且以FXR-KO+顺铂组增加更为显著(图6)。
图6.

免疫组织化学显示各组小鼠肾皮质LC3(A)和p62(B)的表达
Figure 6 Immunohistochemistry showing the expressions of LC3 (A) and p62 (B) in renal cortex of mice in each group
2.5. 肾小管上皮细胞凋亡情况
TUNEL染色结果显示:WT组和FXR-KO组小鼠染色阴性或仅见数个小管上皮细胞凋亡,WT+顺铂组和FXR-KO+顺铂组凋亡细胞数量均增多;WT+顺铂组相较于WT组,FXR-KO+顺铂组相较于FXR-KO组,肾小管上皮细胞凋亡率明显增加(均P<0.001),且FXR-KO+顺铂组凋亡率明显高于WT+顺铂组(P<0.05,图7)。
图7.

各组小鼠肾小管上皮细胞凋亡情况
Figure 7 Apoptosis of renal tubular epithelial cells of mice in each group
*P<0.05, ***P<0.001.
3. 讨 论
顺铂用药后造成肾小管上皮细胞的损伤,诱发炎症反应、氧化应激以及肾脏血管损伤,这些改变共同导致AKI。本研究结果显示:无论是WT小鼠还是FXR-KO小鼠,顺铂处理后均出现肾功能损害并伴有典型急性肾小管坏死的病理改变,且FXR-KO+顺铂组小鼠的SCr、BUN水平及肾小管损伤评分更高,提示敲除FXR可加重顺铂诱导的AKI。FXR参与多种肾脏疾病的发生和发展,对肾具有潜在的保护作用。在喂食高脂食物或诱发糖尿病后,FXR-KO小鼠表现出明显的肝脂肪变性或肾脂质沉积[7]。FXR激动剂奥贝胆酸(obeticholic acid,OCA)可使单侧肾切除的小鼠免受肥胖诱导的肾损伤,减轻肾脂质沉积和脂质过氧化[7];6-乙基鹅去氧胆酸(6-ethyl-chenodeoxycholic acid,6-ECDCA)可减轻缺血再灌注诱导的AKI小鼠的肾小管坏死和细胞凋亡,改善小鼠的肾功能[8];FXR激动剂GW4064和鹅脱氧胆酸(chenodeoxycholic acid,CDCA)下调单侧输尿管梗阻小鼠Smad3的转录水平,抑制TGF-β1诱导的肾纤维化[9];GW4064或胆酸可以预防糖尿病小鼠出现蛋白尿并改善其肾功能[3]。FXR激动剂6-ECDCA在顺铂诱导AKI模型中发挥抗纤维化和拮抗炎症的作用[10]。过表达FXR下游靶基因SHP可抑制顺铂预处理HK2细胞中TGF-β1和磷酸化JNK(p-JNK)的表达,表明FXR保护肾脏的作用可能与SHP的抗纤维化、抗炎和抗细胞凋亡作用有关[10]。
对急性肝损伤[11]和肠缺血再灌注模型[12]的研究均提示FXR对维持自噬稳态发挥了重要作用。自噬又称为II型细胞死亡,能有效清除细胞中损伤、衰老、过量或异常的细胞器及生物大分子,促进肾小管的修复和再生[13],维持细胞稳态[1, 14],对顺铂诱导的AKI具有保护作用[14-15]。LC3是公认的自噬标志蛋白质,定位于前自噬泡和自噬泡膜表面。当细胞启动自噬时,细胞质中的LC3-I可与磷脂酰乙醇胺结合,形成膜结合状态的LC3-II,LC3-II/LC3-I比值常作为评估自噬水平的关键指标[16]。泛素结合蛋白p62也是一种选择性自噬底物,能与LC3结合靶向自噬体并促进泛素化蛋白的清除[17]。本研究显示:顺铂干预后,WT小鼠和FXR-KO小鼠肾皮质LC3-II/LC3-I比值均降低,p62蛋白质表达上调。提示在顺铂诱导AKI小鼠模型中自噬泡产生及底物降解减少,自噬流受阻,这与既往的研究[18-19]一致。其中FXR-KO+顺铂组小鼠肾皮质LC3-II/LC3-I比值降低、p62表达上调更为明显,提示FXR敲除阻碍了AKI小鼠的肾脏自噬体形成及清除过程。由于自噬流的形成是个动态过程,在没有使用自噬泡与溶酶体结合抑制剂Baf1时,难以在动物体内观察到自噬泡[20],因此,我们用透射电镜重点观察了自噬流损伤的后果,结果显示FXR-KO+顺铂组有更多受损的线粒体及聚集的溶酶体底物。Ceulemans等[12]发现:在肠缺血再灌注损伤大鼠模型中,自噬通量降低,LC3-I向LC3-II的转化受阻,FXR激动剂OCA预处理能减弱自噬抑制并下调p62 mRNA表达。启动FXR可保护小鼠免受LPS诱导的急性肝损伤,同时LC3-II/I比值升高,自噬活化[11]。此外,在63个自噬相关基因中,有13个可被FXR激动剂GW4064抑制[21],FXR还能与编码LC3-I和LC3-II基因的启动子结合[22]。因此,我们推测FXR可能通过直接调控自噬相关基因的表达,从而在AKI等肾脏疾病中发挥保护作用。
凋亡与自噬往往具有相互调节的作用,共同决定细胞的生存或死亡[23]。当自噬上调时,细胞凋亡途径可被启动[24],同时,凋亡相关蛋白的启动也可对自噬产生抑制作用,促进细胞死亡。研究表明:caspase-8对自噬死亡途径具有抑制作用[25],可以阻止自噬相关基因3和LC3的结合,或者切割自噬相关基因3阻止自噬的发生[26]。凋亡蛋白酶可以切割Beclin 1,从而破坏其自噬活性,由此产生的Beclin 1 C端片段可以促进线粒体介导的细胞凋亡[27]。本研究结果显示:WT+顺铂组和FXR-KO+顺铂组凋亡细胞数量明显增多,且以FXR-KO+顺铂组更为显著。Bae等[10]发现FXR配体6-ECDCA能降低顺铂干预的小鼠肾脏凋亡相关蛋白Bax/Bcl-2比值和TUNEL阳性小管上皮细胞数量,提示FXR在顺铂诱导的肾小管凋亡中发挥了保护作用。此外,FXR的缺乏导致肝细胞对凋亡的敏感性增加,过表达FXR能阻止死亡诱导信号复合物的形成和caspase 8的启动,抑制凋亡信号转导,FXR可能是一种内在的肝细胞凋亡抑制剂[28]。在本研究中,FXR缺陷导致自噬受阻,这可能与FXR直接调控自噬相关基因的表达有关。笔者推测FXR缺陷导致凋亡增加,肾小管细胞自噬受到抑制。有关凋亡相关蛋白对于自噬的影响仍有待进一步研究。
综上,本研究表明FXR基因敲除可加剧顺铂诱导的AKI,其机制与阻断自噬通路,促进细胞凋亡有关,FXR有望作为AKI新的有效药物靶点。但本研究仍属于动物研究,后续将收集人体组织标本进一步研究,并在细胞水平上探讨相关机制,评估FXR在脓毒血症、缺血再灌注等其他AKI模型中的保护作用,以期为临床上AKI的治疗提供更多的研究思路和实验依据。
基金资助
国家自然科学基金(82070759)。
This work was supported by the National Natural Science Foundation of China (82070759).
利益冲突声明
作者声称无任何利益冲突。
作者贡献
张礼君 实验操作,图片处理,论文撰写与修订;李爱梅 实验操作,实验器材等准备与提供;黄志军 协助实验,数据采集;王阳阳 数据采集,统计分析;易斌 实验设计者、负责人,提供实验指导及论文修改。
原文网址
http://xbyxb.csu.edu.cn/xbwk/fileup/PDF/202202174.pdf
参考文献
- 1. Jiang M, Wei Q, Dong G, et al. Autophagy in proximal tubules protects against acute kidney injury[J]. Kidney Int, 2012, 82(12): 1271-1283. 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c[J]. J Clin Invest, 2004, 113(10): 1408-1418. 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang T, Wang XX, Scherzer P, et al. Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy[J]. Diabetes, 2007, 56(10): 2485-2493. 10.2337/db06-1642. [DOI] [PubMed] [Google Scholar]
- 4. Seok S, Fu T, Choi SE, et al. Transcriptional regulation of autophagy by an FXR-CREB axis[J]. Nature, 2014, 516(7529): 108-111. 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khambu B, Li T, Yan S, et al. Hepatic autophagy deficiency compromises farnesoid X receptor functionality and causes cholestatic injury[J]. Hepatology, 2019, 69(5): 2196-2213. 10.1002/hep.30407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jablonski P, Howden BO, Rae DA, et al. An experimental model for assessment of renal recovery from warm ischemia[J]. Transplantation, 1983, 35(3): 198-204. 10.1097/00007890-198303000-00002. [DOI] [PubMed] [Google Scholar]
- 7. Gai Z, Gui T, Hiller C, et al. Farnesoid X receptor protects against kidney injury in uninephrectomized obese mice[J]. J Biol Chem, 2016, 291(5): 2397-2411. 10.1074/jbc.M115.694323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gai Z, Chu L, Xu Z, et al. Farnesoid X receptor activation protects the kidney from ischemia-reperfusion damage[J]. Sci Rep, 2017, 7(1): 9815. 1038/s41598-017-10168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao K, He J, Zhang Y, et al. Activation of FXR protects against renal fibrosis via suppressing Smad3 expression[J]. Sci Rep, 2016, 6: 37234. 10.1038/srep37234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bae EH, Choi HS, Joo SY, et al. Farnesoid X receptor ligand prevents cisplatin-induced kidney injury by enhancing small heterodimer partner[J/OL]. PLoS One, 2014, 9(1): e86553 (2014-01-27)[2020-03-17]. 10.1371/journal.pone.0086553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiong X, Ren Y, Cui Y, et al. Obeticholic acid protects mice against lipopolysaccharide-induced liver injury and inflammation[J]. Biomed Pharmacother, 2017, 96: 1292-1298. 10.1016/j.biopha.2017.11.08. [DOI] [PubMed] [Google Scholar]
- 12. Ceulemans LJ, Verbeke L, Decuypere JP, et al. Farnesoid X receptor activation attenuates intestinal ischemia reperfusion injury in rats[J/OL]. PLoS One, 2017, 12(1): e0169331 (2017-01-06)[2020-04-12]. 10.1371/journal.pone.0169331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair[J]. Nephron Clin Pract, 2014, 127(1/4): 56-60. 10.1159/000363677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levine B, Kroemer G. Autophagy in the pathogenesis of disease[J]. Cell, 2008, 132(1): 27-42. 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Livingston MJ, Dong Z. Autophagy in acute kidney injury[J]. Semin Nephrol, 2014, 34(1): 17-26. 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death[J]. Neurobiol Dis, 2008, 29(1): 132-141. 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 17. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death[J]. J Cell Biol, 2005, 171(4): 603-614. 10.1083/jcb.2005070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun CY, Nie J, Zheng ZL, et al. Renoprotective effect of scutellarin on cisplatin-induced renal injury in mice: Impact on inflammation, apoptosis, and autophagy[J]. Biomed Pharmacother, 2019, 112: 108647. 10.1016/j.biopha.2019.108647. [DOI] [PubMed] [Google Scholar]
- 19. Ning Y, Shi Y, Chen J, et al. Necrostatin-1 attenuates cisplatin-induced nephrotoxicity through suppression of apoptosis and oxidative stress and retains klotho expression[J]. Front Pharmacol, 2018, 9: 384. 10.3389/fphar.2018.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition)[J]. Autophagy. 2016, 12(1): 1-222. 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy[J]. Nature, 2014, 516(7529): 112-115. 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Preidis GA, Kim KH, Moore DD. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance[J]. J Clin Invest, 2017, 127(4): 1193-1201. 10.1172/JCI88893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song S, Tan J, Miao Y, et al. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress[J]. J Cell Physiol, 2017, 232(11): 2977-2984. 10.1002/jcp.25785. [DOI] [PubMed] [Google Scholar]
- 24. Chaudhari N, Talwar P, Parimisetty A, et al. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress[J]. Front Cell Neurosci, 2014, 8: 213. 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8[J]. Science, 2004, 304(5676): 1500-1502. 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 26. Mukhopadhyay S, Panda PK, Sinha N, et al. Autophagy and apoptosis: where do they meet? [J]. Apoptosis, 2014, 19(4): 555-566. 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- 27. Wirawan E, Vande WL, Kersse K, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria[J/OL]. Cell Death Dis, 2010, 1(1): e18 (2010-01-21)[2020-04-21]. 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H, Ge C, Zhou J, et al. Noncanonical farnesoid X receptor signaling inhibits apoptosis and impedes liver fibrosis[J]. EBioMedicine, 2018, 37: 322-333. 10.1016/j.ebiom.2018.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]