

Abstract
Autoimmune polyglandular syndromes (APS) are classified into four main categories, APS1–APS4. APS1 is caused by AIRE gene loss of function mutations, while the genetic background of the other APS remains to be clarified. Here, we investigated the potential association between AIRE gene promoter Single Nucleotide Polymorphisms (SNPs) and susceptibility to APS. We sequenced the AIRE gene promoter of 74 APS patients, also analyzing their clinical and autoantibody profile, and we further conducted molecular modeling studies on the identified SNPs. Overall, we found 6 SNPs (-230Y, -655R, -261M, -380S, -191M, -402S) of the AIRE promoter in patients’ DNA. Interestingly, folding free energy calculations highlighted that all identified SNPs, except for -261M, modify the stability of the nucleic acid structure. A rather similar percentage of APS3 and APS4 patients had polymorphisms in the AIRE promoter. Conversely, there was no association between APS2 and AIRE promoter polymorphisms. Further AIRE promoter SNPs were found in 4 out of 5 patients with APS1 clinical diagnosis that did not harbor AIRE loss of function mutations. We hypothesize that AIRE promoter polymorphisms could contribute to APS predisposition, although this should be validated through genetic screening in larger patient cohorts and in vitro and in vivo functional studies.
Keywords: AIRE, AIRE gene promoter, polymorphisms, autoimmune polyglandular syndrome, autoimmune etiopathogenesis, sequencing
1. Introduction
A book published to celebrate the 63rd anniversary of the discovery of autoimmune disorders (AIDs) edited by the “fathers” of autoimmunity describes more than a hundred of AIDs and estimated to affect about 7% of the general population [1,2,3]. In their natural history, it is generally observed progression from latent, to subclinical toward clinical disease with associated disease-related circulating autoantibodies (Abs) [4,5]. Subsequently, criteria for their frequent association called autoimmune polyglandular syndrome (APS) [6], multiple autoimmune (MAS) [4,7], or polyautoimmunity syndromes [8] were established. Indeed, the association was not limited to polyglandular diseases but it can also include multiple organ-specific autoimmune disorders, such as endocrine, gastrointestinal, skin, neurologic, and non-organ-specific rheumatologic conditions.
APS includes four main categories [4,5,6]. As regard APS1, also called autoimmune–polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (APECED, OMIM #240300, ORPHA 3453), is a rare monogenic recessive disorder caused by loss of function mutations in the AutoImmune REgulator (AIRE) gene, whose clinical diagnosis requires the presence of at least two of the following diseases: chronic mucocutaneous candidiasis (CMC), chronic hypoparathyroidism (HP) and primary adrenal insufficiency (Addison’s disease, AD) [9]. The combination of autoimmune thyroid disease (AITD), Type 1 diabetes mellitus, (T1DM), and AD is the autoimmune polyglandular syndrome Type 2 (APS2, Schmidt’s syndrome). This is a rare disease in humans, occurring in 1.4–4.5 per 100.000 inhabitants in Europe (OMIM #269200, ORPHA 3143). APS3 was defined as the association between AITD and one or more AIDs excluding AD. APS3 (ORPHA 227982) included four main subgroups based on the associated AITD: APS3A (AITD and autoimmune endocrine diseases), APS3B (AITD with autoimmune gastrointestinal, hepatic, or pancreatic diseases), APS3C (AITD with autoimmune skin, neurological and hematological diseases), APS3D (AITD with autoimmune rheumatological, cardiac, and vascular diseases). In the consideration that AITD is one of the most frequent autoimmune diseases and that about one-third of the patients can be associated with a non-thyroid AID during the entire lifespan, the APS3 can be considered the most prevalent APS worldwide [4,5,10,11]. APS4 (ORPHA 227990) is the last category including any other AID combination that cannot be assigned to APS1, APS2 or APS3 [4,5,6]. Overall, the incidence of APS2 to 4 is estimated between 1.4 and 4.5 per 100.000 inhabitants according to published studies [12].
Whereas the genetics of APS1 are clearly defined, APS2, APS3, and APS4 are genetically complex multifactorial syndromes [12]. The inheritance pattern seems to be autosomal-dominant with incomplete penetrance in some patients with several genetic loci being involved through interaction with environmental factors [13]. The cluster of several different organ-specific and non-organ-specific autoimmune diseases in patients can be due to shared common proinflammatory genetic background as well as a defect in immune regulation [14,15,16,17]. APS2, APS3, and APS4 are strongly associated with certain alleles of HLA genes of the major histocompatibility complex (MHC) located on chromosome 6. As regards the haplotypes, HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01 and HLA-DRB1*04:01-DQA1*03:01-DQB1*03:02 are significantly overrepresented and therefore predisposing, whereas the HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 haplotype is underrepresented and therefore protective of the development of these APS. The presence of the DRB1*04-DQ8 haplotype differentiates between APS1 and APS2. The DRB1*04:04-DQA1*03:01-DQB1*03:02 haplotype is associated with APS2, whereas the DRB1*04:01-DQA1*03:01-DQB1*03:02 haplotype confers susceptibility for APS3 [12].
Furthermore, several gene variations in non-HLA genes are present in APS. Among these, the PTPN22 (protein tyrosine phosphatase non-receptor type 22) C1858T variant encoding for the R620W Lyp (rs2476601) is frequently associated with T1DM, AITD, AD, and the APS2 syndrome [18]. Other gene polymorphisms associated with APS are detected in the CTLA4 gene [19] encoding for the cytotoxic T lymphocyte-associated antigen-4, the vitamin D receptor (VDR) gene [20], the IL2ra gene encoding IL2Ra (CD25) [21], the TNFα (tumor necrosis factor alpha) gene [22,23], the FOXP3 (forkhead box P3) gene which controls Treg development and function [24] and the MHC class I chain-related gene A (MICA) [25,26]. Further, susceptibility to T1DM is conferred by variable number of tandem repeats (VNTR) of the insulin gene [27,28]. It is generally recognized that genetic variability in the AIRE locus and the presence of heterozygous loss of function AIRE mutations can affect the presentation of self-antigens in the thymus and thus the development of certain organ-specific autoimmune disorders [29]. AIRE variants were detected in the DNA of patients affected by organ-specific autoimmune disorders [30]. AIRE gene monoallelic mutations located in the first plant homeodomain (PHD1) zinc finger with autosomal dominant inheritance were found associated with autoimmune disorders with later onset, milder phenotype, and reduced penetrance that did not satisfy the clinical diagnostic criteria for APECED [31]. In a recent paper by Oftedal et al. [32], 20 individuals from 11 kindreds with dominant AIRE mutations within the PHD1 e PHD2 domains were identified. These variants were shown to have dominant negative effects in vitro.
In the light of the foregoing, since the expression profile of peripheral tissue antigens in the thymus could not only be affected by AIRE deficiency, in the present study, we aimed to investigate whether the susceptibility to APS could be instead affected by SNPs of the AIRE gene promoter [33], potentially inducing alteration of the AIRE gene transcription.
2. Results
2.1. Clinical Phenotype and AIRE Gene Promoter Screening
As shown in Table 1, several AIRE gene promoter polymorphisms were identified in APS patients and conventionally numbered with respect to the AIRE start codon, which was assigned the value 0. These included the heterozygous -230Y (C/T) SNP (rs751032) (Table S1) and the homozygous -230T SNP, whose genotype frequencies were similar to those found in healthy controls (HD) (Table 2) [34]. Five additional heterozygous SNPs were rarely detected in five different APS patients: the -655R (G/A) (rs117557896), the -261M (C/A) (rs934375604), the -380S (C/G) (rs371261300), the -191M (C/A) (rs1048356976), and the -402S (C/G). The latter was not previously reported in genome databases (Table S1). Heterozygous polymorphisms -655R (G/A), -452Y (C/T) (rs547103905), -214M (A/C) (rs184978263), and -124M (C/A) (rs868650327) were detected in normal controls (Table 2B) (Table S1). Regarding the frequency of haplotypes -655G -230Y (C/T) and -655G -230T, there were no differences between patients and HD groups. Particularly, 19 patients and 18 HD presented haplotype -655G -230Y, while 2 patients and 3 HD showed haplotype -655G -230T (Table 2).
Table 1.
Clinical and genetic characteristics of the 74 APS patients.
| Pt n° |
Gender | Age (yrs) |
Age at Referral (yrs) | APS Type | Clinical Manifestations |
AIRE Promoter SNP | Therapy |
|---|---|---|---|---|---|---|---|
| 1 | F | 26.3 | 4.4 | 3A, 3C | T1DM, HT, vitiligo | Insulin aspart, insulin degludec | |
| 2 | F | 3.8 | 1.4 | 1 | CMC, HP, Vernal keratoconjunctivitis |
het -230Y (C/T) het -261M (C/A) |
Ketotifen fumarate eye drops, calcitriol |
| 3 | F | 33.2 | 12.7 | 3A | T1DM, HT | Insulin | |
| 4 | M | 28.6 | 8 | 3A | T1DM, HT | Insulin aspart, insulin degludec | |
| 5 | M | 40.3 | 14.2 | 3A | T1DM, HT | Insulin, bisoprolol fumarate, ramipril | |
| 6 | F | 17.5 | 4.4 | 3B | HT, CD | het -230Y (C/T) | None |
| 7 | F | 2.6 | 2.5 | 4 | AA, autoimmune piastrinopenia |
Topical therapy | |
| 8 | F | 11.9 | 6.2 | 3A, 3B | T1DM, HT, CD, AIH | Prednisone, AZA, VitD, insulin lispro | |
| 9 | F | 22.6 | 5.9 | 3B | HT, AIH | Prednisone, AZA, UDCA, LT4, VitD | |
| 10 | F | 18 | 3.9 | 3B, 3C | HT, suspected CD, AA, allergic rhinitis | het -230Y (C/T) | None |
| 11 | F | 19.3 | 6.2 | 3C, 3D | HT, AA, onychodystrophy, allergic rhinitis, arthralgia, Raynaud phenomenon, recurrent infections | LT4, VitD | |
| 12 | F | 12.1 | 5.2 | 3A, 3B | T1DM, HT, AIH | AZA, methimazole, VitD, insulin lispro |
|
| 13 | F | 20 | 11.7 | 3A, 3B, 3D | T1DM, HT, psoriasis, arthralgia, synovitis | LT4 | |
| 14 | F | 29.5 | 8.8 | 3A, 3B | T1DM, HT, CD | het -230Y (C/T) | Insulin |
| 15 | F | 12.7 | 8 | 2 | HT, AD, IGTT | LT4, hydrocortisone, acetate fludrocortisone, VitD, iron pidolate |
|
| 16 | F | NA | NA | 1 | AD, HP, secondary ovarian failure | het -230Y (C/T) | NA |
| 17 | M | 19.2 | 11.5 | 3C | Preclinical T1DM, HT, AA | None | |
| 18 | F | 38.7 | 14.8 | 3A, 3B | T1DM, HT, CD | LT4, insulin glulisine, insulin glargine |
|
| 19 | M | 25.1 | 2.8 | 3A, 3B | T1D, HT, CD | het -230Y (C/T) | Insulin |
| 20 | F | 33.2 | 9 | 3A, 3B | T1DM, Basedow’s disease, POF | Insulin aspart, VitD, candesartan cilexetil, methimazole, propranolol hydrochloride, folic acid |
|
| 21 | M | 35.3 | 3.9 | 3A | T1DM, HT | Insulin aspart | |
| 22 | F | 22.5 | 0.9 | 3A | T1DM, HT | Insulin aspart, Insulin glargine |
|
| 23 | M | 38.4 | 16.6 | 3A | T1DM, HT | het -230Y (C/T) | Human insulin, Insulin detemir, LT4 |
| 24 | M | 29.8 | 8.3 | 4 | T1DM, CD | het -230Y(C/T) | Human insulin, Insulin detemir, Insulin glulisine |
| 25 | F | 20 | NA | 2 | HT, AD, psoriasis | NA | |
| 26 | M | 14.2 | 12.8 | 3B | HT, AIH | het -655R (A/G) het -230Y (C/T) |
Cyclosporine, AZA, UDCA, LT4 |
| 27 | M | 29.8 | 13.5 | 3A | T1DM, HT | Insulin aspart, Insulin degludec |
|
| 28 | F | 25.1 | 3.8 | 4 | T1D, CD, vitiligo | het -230Y (C/T) | Insulin aspart |
| 29 | F | 32.1 | 5 | 3A | T1DM, HT | het -380S (C/G) het -230Y (C/T) |
Insulin aspart, LT4 |
| 30 | M | 39.9 | 19.6 | 4 | T1DM, CD | Insulin aspart | |
| 31 | F | 15.8 | 1.8 | 4 | T1DM, CD, AIH, IgA deficit, obesity | Metformin, insulin glargine, insulin aspart, insulin lispro |
|
| 32 | F | 22 | 8.1 | 2 | HT, CD, AD | Hydrocortisone, fludrocortisone, LT4 |
|
| 33 | F | 25.4 | 4.7 | 3A, 3B | T1DM, HT, suspected CD |
None | |
| 34 | M | 25.7 | 5.1 | 4 | T1DM, CD | het -230Y (C/T) | Human insulin, insulin glargine |
| 35 | M | NA | NA | 4 | T1DM, CD | NA | |
| 36 | F | 20.2 | NA | 3B | HT, CD, type 2 AIH | hom -230T | NA |
| 37 | M | 36.5 | 12.6 | 3A, 3B | T1DM, preclinical HT, CD | het -230Y (C/T) | Insulin aspart |
| 38 | M | 34.4 | 12.7 | 4 | T1DM, CD | Insulin glulisine, insulin glargine | |
| 39 | M | 30.6 | 9.5 | 3A | T1DM, HT | het -230Y (C/T) | Insulin aspart, insulin degludec |
| 40 | F | 29.2 | 5.4 | 3A | T1DM, HT | het -230Y (C/T) | Insulin aspart, insulin glargine |
| 41 | F | 30 | 2.2 | 3A, 3B | T1DM, HT, CD | LT4, insulin aspart, insulin glargine | |
| 42 | F | 35 | 15 | 3A | T1DM, HT | LT4, insulin aspart, insulin degludec |
|
| 43 | F | 23.8 | 2.8 | 4 | T1DM, CD | Insulin aspart, insulin degludec | |
| 44 | F | 29.7 | 9.2 | 3A, 3C | T1DM, HT, vitiligo | Insulin aspart | |
| 45 | M | 28.2 | 6.5 | 4 | T1DM, autoimmune hemolytic anemia | het -230Y (C/T) | Insulin aspart |
| 46 | M | 33 | 13.8 | 3A, 3B | T1DM, HT, AG | het -230Y (C/T) | LT4, Insulin aspart, insulin degludec |
| 47 | F | 33.7 | 10.5 | 3A, 3B, 3C | T1DM, HT, vitiligo, AG | LT4, Insulin aspart, insulin glargine | |
| 48 | F | 13.2 | NA | 2 | preclinical T1DM, AD | NA | |
| 49 | F | 32.2 | 6.8 | 3A, 3B, 3C | T1DM, HT, AG, iron deficiency anemia | Insulin aspart | |
| 50 | F | 28.6 | 11.2 | 4 | T1DM, CD | het -230Y (C/T) | None |
| 51 | M | 29.4 | 3.1 | 3A, 3B, 3C | T1DM, HT, CD, vitiligo, AG | het -191M (C/A) | Insulin aspart, insulin glargine, LT4, VitB12 |
| 52 | F | 33.4 | 8.2 | 3A | T1DM, HT | Insulin aspart, LT4 | |
| 53 | M | 22.2 | 7.8 | 3C | HT, vitiligo, inhalant allergy, selective IgA deficiency | Betacarotene | |
| 54 | M | NA | NA | 3C | HT, vitiligo | NA | |
| 55 | F | 15.2 | 1 | 3C | preclinical HT, AA | Folic acid | |
| 56 | M | 7.1 | 6.7 | 4 | AD, CMC, AA, arthritis, Perthes’ disease | Hydrocortisone, fludrocortisone, ibuprofen, omeprazole | |
| 57 | M | 8.8 | 4.7 | 3B, 3C | HT, CD, AA, atopy | Calcifediol, folic acid, LT4, betamethasone dipropionate, resorcin, levocystin |
|
| 58 | F | 13.9 | 5.9 | 3A, 3B, 3C, 3D | T1DM, HT, CD, AG, arthritis, vasculitis, algodystrophy, autoimmune encephalitis, ALPS | het -230Y (C/T) | Insulin, sirolimus, methotrexate, micronized PEA, 5-HTP, gabapentin |
| 59 | F | 13.3 | NA | 1 | T1DM, HT, CMC | het -402S (C/G) | NA |
| 60 | M | 16 | 1.9 | 4 | AD, Blackfan-Diamond anemia | het -230Y (C/T) | Fludrocortisone, hydrocortisone, deferasirox, folic acid |
| 61 | F | 16.4 | 1.1 | 4 | Preclinical CD, AD, MERS, MIS-C, hyperpigmentation, chronic asthenia | Omeprazole, hydrocortisone, fludrocortisone, topiramate |
|
| 62 | F | 17.2 | NA | 4 | CD, HP | NA | |
| 63 | F | 5.9 | NA | 1 | Vitiligo, CMC | NA | |
| 64 | M | 23.8 | 4.1 | 2 | T1DM, HT, preclinical AD, GH deficit, autoimmune leucopenia | Insulin aspart | |
| 65 | F | 30.9 | 3.2 | 3A | T1DM, preclinical HT | Insulin aspart, Insulin detemir |
|
| 66 | F | N.A | NA | 3A | T1DM, HT | NA | |
| 67 | F | NA | NA | 3A | T1DM, HT | hom -230T | NA |
| 68 | F | 26.4 | 4.7 | 3A, 3B | T1DM, preclinical HT, preclinical CD | Insulin aspart, Insulin degludec |
|
| 69 | F | 26.9 | 5.2 | 4 | T1DM, CD | NA | |
| 70 | F | NA | NA | 3A, 3C | T1DM, HT, vitiligo | NA | |
| 71 | F | 31.7 | 11.2 | 3B | HT, CD | LT4 | |
| 72 | F | 29.8 | 6 | 3A | T1DM, HT | LT4, insulin aspart, insulin glargine | |
| 73 | F | 30 | 0.6 | 4 | T1DM, CD | Insulin lispro, insulin degludec | |
| 74 | F | NA | NA | 1 | AD, CMC, AG, HP, POF | NA |
Pt, patients; Yrs, years; het, heterozygous; hom, homozygous; AD, Addison’s disease; AG, atrophic gastritis; AIH, autoimmune hepatitis; ALPS, autoimmune lymphoproliferative syndrome; CD, celiac disease; CMC, chronic mucocutaneous candidiasis; GH, growth hormone; HT, Hashimoto thyroiditis; HP, hypoparathyroidism; IGTT, impaired glucose tolerant test; MERS, mild encephalitis/encephalopathy with reversible splenial lesion; MIS-C, multisystem inflammatory syndrome in children; POF, primary ovarian failure; T1DM, type 1 diabetes. 5-HTP, L-5-hydroxytryptophan; AZA, azathioprine; LT4, levothyroxine; PEA, palmitoylethanolamide; UDCA, ursodeoxycholic acid; VitD, vitamin D; VitB12, vitamin B12. NA, not available.
Table 2.
Genotypes and alleles frequency of identified AIRE promoter variants in (A) 74 patients and (B) 81 controls of the present series.
| (A) | |||||
|---|---|---|---|---|---|
| GENOTYPES | N° | % | ALLELES | N° | % |
| -230Y (C/T) | 20 | 27.0 | -230C | 124 | 83.8 |
| -230T | 2 | 2.7 | -230T | 24 | 16.2 |
| -655R (G/A) | 1 | 1.4 | -655G | 147 | 99.3 |
| -655A | 0 | 0.0 | -655A | 1 | 0.7 |
| -655G -230Y | 19 | 25.7 | -261C | 147 | 99.3 |
| -655G -230T | 2 | 2.7 | -261A | 1 | 0.7 |
| -261M (C/A) | 1 | 1.4 | -380C | 147 | 99.3 |
| -261A | 0 | 0 | -380G | 1 | 0.7 |
| -380S (C/G) | 1 | 1.4 | -191C | 147 | 99.3 |
| -380G | 0 | 0.0 | -191A | 1 | 0.7 |
| -191M (C/A) | 1 | 1.4 | -402C | 147 | 99.3 |
| -191A | 0 | 0.0 | -402G | 1 | 0.7 |
| -402S (C/G) | 1 | 1.4 | |||
| -402G | 0 | 0 | |||
| (B) | |||||
| GENOTYPES | N° | % | ALLELES | N° | % |
| -230Y (C/T) | 19 | 23.5 | -230C | 137 | 84.6 |
| -230T | 3 | 3.7 | -230T | 25 | 15.4 |
| -655R (G/A) | 8 | 9.9 | -655G | 154 | 95.1 |
| -655A | 0 | 0.0 | -655A | 8 | 4.9 |
| -655G -230Y | 18 | 22.2 | -452C | 161 | 99.4 |
| -655G -230T | 3 | 3.7 | -452T | 1 | 0.6 |
| -452Y (C/T) | 1 | 1.2 | -214C | 161 | 99.4 |
| -452T | 0 | 0.0 | -214A | 1 | 0.6 |
| -214M (C/A) | 1 | 1.2 | -124C | 161 | 99.4 |
| -214A | 0 | 0.0 | -124A | 1 | 0.6 |
| -124M (C/A) | 1 | 1.2 | |||
| -124A | 0 | 0.0 | |||
No specific correlation was observed with peculiar serum autoantibody specificities and the presence of polymorphisms of AIRE gene promoter.
In Table S2, the AIRE gene pattern of each patient is also reported. AIRE gene polymorphisms IVS9+6 G>A (c.1095+6 G>A, rs1800525) and S278R (c.834 C>G, rs1800520), previously associated with APS [35], were identified in 17 and in 14 out of 74 patients, respectively. No significant association was found between the presence of these AIRE gene polymorphisms and the described AIRE gene promoter variants. As can be seen, among 17 patients harboring the intronic polymorphism, 3 had the -230Y SNP, one the -230Y SNP along with the -261M SNP, and one the -402S SNP. Among 14 patients harboring the S278R polymorphisms, 2 had the -230Y SNP, 1 the -230Y SNP along with the -261M SNP, 1 the -191M SNP, and 1 the -402S SNP (Table 1).
Furthermore, we analyzed the distribution of the identified AIRE gene promoter polymorphisms within the different APS subtypes in the present series of patients (Figure 1) (Table 3).
Figure 1.
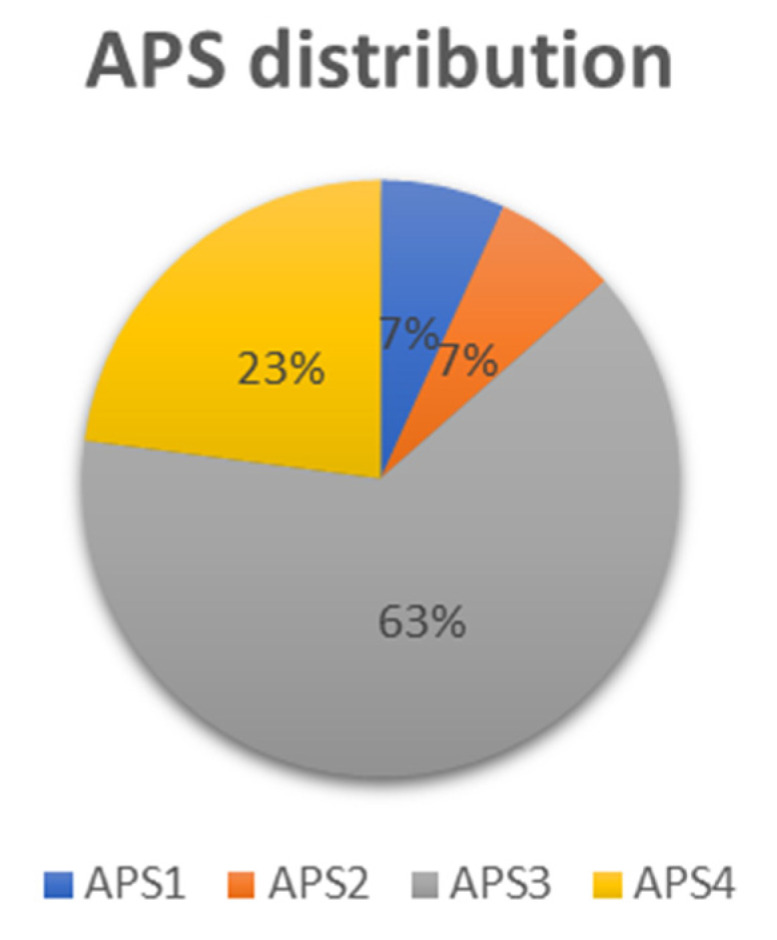
Frequency of APS subtypes in the present patient’s series. Total patients with APS1: 5 (blue); total patients with APS2: 5 (orange); total patients with APS3: 47 (gray); total patients with APS4: 17 (yellow).
Table 3.
Distribution of AIRE promoter SNPs according to APS subtypes.
| AIRE Promoter SNP | APS1 | APS2 | APS3 | APS4 |
|---|---|---|---|---|
| -230Y (C/T) | 2/5 | 0/5 | 12/47 | 6/17 |
| -230T | 0/5 | 0/5 | 2/47 | 0/17 |
| -655R (A/G) | 0/5 | 0/5 | 1/47 | 0/17 |
| -261M (C/A) | 1/5 | 0/5 | 0/47 | 0/17 |
| -380S (C/G) | 0/5 | 0/5 | 1/47 | 0/17 |
| -191M (C/A) | 0/5 | 0/5 | 1/47 | 0/17 |
| -402S (C/G) | 1/5 | 0/5 | 0/47 | 0/17 |
Of the 74 enrolled patients, 5 were affected by APS1, 5 by APS2, 47 by APS3, and 17 by APS4 (Figure 1). The heterozygous -230Y SNP was found in 2 out of 5 APS1 patients, one harboring the -261M SNP, the other the -402S SNP (Table 3). As regards the APS2-APS4 types, AIRE gene promoter SNPs were present in APS3 and APS4 patients but not in APS2 patients. Indeed, the heterozygous -230Y SNP was found in 12 out of 47 APS3 patients and in 6 out of 17 APS4 patients. The homozygous -230T SNP was found in two APS3 patients. Among APS3 patients, one had the -655R SNP, one the -380S SNP and another one the -191M SNP. Furthermore, we did not detect a clear association of the identified AIRE gene promoter polymorphisms with APS3 subtypes.
Notably, the five APS1 patients had received the clinical diagnosis based on their clinical manifestations although not genetically confirmed by AIRE gene screening (Table 1). In detail, patient No. 2 (Table 1) had a clinical APECED phenotype since affected by hypoparathyroidism and chronic mucocutaneous candidiasis. The patient was also affected by vernal keratoconjunctivitis caused by excessive allergic inflammation [36]. AIRE gene screening revealed the heterozygous loss of function mutation p.Arg203Ter (c.607 C>T, rs755490967) in exon 5 and the heterozygous polymorphism p.Ser278Arg (c.834 C>G, rs1800520) in exon 7 inherited from the mother; furthermore, the heterozygous intronic polymorphism IVS9+6 G>A [35], which could be inherited from both parents, was found. AIRE gene promoter analysis showed the presence of heterozygous -230Y and -261M SNPs, inherited from the father. Therefore, in this patient, one allele was affected by exons variants, the other allele by SNPs of the promoter; the final effect of this combination could lead to a reduced AIRE expression, leading to the pathological phenotype.
Remarkably, patient No. 16 (Table 1) was affected by hypoparathyroidism, AD, and secondary ovarian failure. Nevertheless, we did not detect variations through AIRE gene screening, but the heterozygous AIRE promoter -230Y (C/T) variant was present. The search for the C1858T polymorphism of the PTPN22 gene was negative. Although whole-exome sequencing studies could help to fully elucidate the contribution of additional genetic risk factors, the heterozygous AIRE promoter -230Y (C/T) variant could have contributed to the APECED phenotype of this patient.
APECED patient No. 74 (Table 1) had all three major APECED symptoms: AD, chronic hypoparathyroidism, and chronic mucocutaneous candidiasis. This female also suffered from atrophic gastritis and primary ovarian failure. AIRE gene sequencing revealed the heterozygous polymorphism p.Ser278Arg in exon 7 and two other heterozygous intronic polymorphisms: IVS5+14 C>T (c.652+14 C>T, rs41277546) and IVS13-55 A>G (c.1504-55 A>G, rs41277552). For the IVS5 SNP, there are conflicting interpretations of pathogenicity based on two submissions, as reported in the ClinVar database. Instead, the IVS13 SNP has a benign clinical significance based on one submission, as reported in the same database. Neither AIRE gene promoter SNPs nor C1858T polymorphism of the PTPN22 gene were present. Additional undiscovered new genetic risk factors could have contributed to the pathological phenotype in this patient.
2.2. Molecular Modeling of the SNPs of the AIRE Gene Promoter
Secondary structure prediction and lowest folding free energy calculations for the genomic sequence encompassing the sites of the nine variants indicate that compared to the wild type, all variants except for -261M (C/A) (rs934375604) either stabilize or destabilize the DNA structure (Figure 2). The changes in stability associated with the variants might impair the structural organization and any potential functional role presented by these regions.
Figure 2.

Scheme of AIRE gene and DNA secondary structure predictions. Shown is the genomic sequence scheme of AIRE showing the positions of exons/introns, the sites of the variants, and a representation of the lowest free energy DNA secondary structure (modelling was made across the nucleotide range indicated by lines). The lowest folding free energies for the wild type and the variants are displayed in the table (the arrows ↑ and ↓ flanking the variant energy values respectively indicate structural destabilization and stabilization with respect to the wild type AIRE).
We examined the conservation of the regions affected by the variants by aligning the genomic sequence of the human and other five mammalian species (Figure S1). We found that most of the variants fall within or near conserved blocks, suggesting that the affected regions may have possible functional roles. We also mapped on the same alignment the transcription factor binding sites, either predicted or confirmed, as reported by Lovewell et al. [34] and found that six out of the nine variants (i.e., -402S (C/G), rs371261300, rs934375604, rs751032, rs184978263, rs1048356976) fall within or near these functional sites (Figure S1). Indeed, owing to the possible changes induced on the three-dimensional DNA structure, the remaining three variants might also affect the transcription factor binding sites. Thus, we propose that impairments in the function of these sites might represent at least partially the pathological mechanism of the variants.
3. Discussion
The pathogenesis of complex autoimmunity phenotypes is contributed by SNPs of several susceptibility genes [12]. As can be seen, an altered AIRE gene expression causes a functional downstream effect on the transcription of peripheral tissue antigens at the thymus level in perinatal age, and thus, the escape of autoreactive T cells in the bloodstream leads to the occurrence of autoimmunity during postnatal lifetime [7,37,38,39]. In this investigation, we further unravel the possible influence of variations in the AIRE gene promoter that could potentially affect AIRE expression and entity of its transcriptional activity. We therefore investigated the potential presence of SNPs in the AIRE gene promoter in DNA samples from a cohort of 74 patients affected by different APS including APS1 to APS4 [4,5]. As control, a cohort of 81 sex-matched HD was analyzed.
We screened 751bp upstream from the AIRE start codon, including AIRE minimal promoter for SNPs. As shown in Table 2A, AIRE promoter gene polymorphisms identified in APS patients were: the -230Y (C/T), the -230T and the -655R (G/A), which also occurred in HD controls (Table 2B); the -261M (C/A), the -380S (C/G), the -191M (C/A), and the -402S (C/G), which were exclusive of four different patients. Notably, the -402S (C/G) was not previously reported in literature and genome databases, including ENSEMBL and dbSNP (Table S1).
Notably, molecular modeling studies revealed that all SNPs except for 261M (C/A) (rs934375604) were able to change the stability of nucleic acid structure confirming the possible functional effect of the identified AIRE promoter SNPs. As regards the AIRE -230Y polymorphism, it is located in a conserved region of the promoter and downstream of, but not within, a reporter ETS-1 (ETS Proto-Oncogene 1) transcription factor binding site and it is known to affect AIRE expression [33,34]. It has therefore been suggested that AIRE -230Y SNP has the potential to influence the promiscuous gene expression regulated by AIRE. In detail luciferase reporter assays demonstrated that the highest AIRE promoter activity is determined by the commonest haplotypes AIRE -655G AIRE -230C, while the lowest is associated with haplotype AIRE -655G AIRE -230T and detected in 10% of the controls [34]. By screening a cohort of 172 patients with alopecia areata associated with APECED, 4 patients were homozygous for this haplotype, suggesting that AIRE -655G AIRE -230T could be a susceptibility haplotype for alopecia areata outside APECED; nevertheless, it was pointed out by the authors that this hypothesis should be confirmed by the screening of a larger cohort [34,40]. In our investigation, the AIRE -655G AIRE -230T associated SNPs were more equally represented in the polyendocrine patients than in controls (Table 2 and Table S3). Furthermore, even for all the additional AIRE promoter SNPs identified in this preliminary investigation, especially for those affecting the fold of nuclei acids, no statistical significance of the frequency in patients versus controls was observed (Table S3). Therefore, their pathogenetic relevance and significance to autoimmune predisposition remain to be unraveled (vide infra).
Remarkably, within the 20 patients presenting the AIRE gene promoter -230Y SNP (Table 2A), 2 patients were affected by APS1, 12 by APS3, 6 by APS4 (Table 3). Of note, one patient with APS1 also presented the -261M heterozygous polymorphism (patient n° 2), one patient with APS3B the -655R heterozygous polymorphism (patient n° 26) and one patient with APS3A the -380S heterozygous polymorphism (patient n° 29). The -230T homozygous SNP was detected in one patient (n° 67) affected by APS3A and in one patient (n° 36) affected by APS3B (Table 1). Considering the other two identified AIRE gene promoter heterozygous polymorphisms, the -191M and the -402S, they were found in one patient affected by APS3A/3B/3C (n° 51) and in one patient affected by APS1 (n° 59), respectively. Based on these results, a rather similar percentage of APS3 and APS4 patients examined had polymorphisms in AIRE gene promoter (36.17% APS3 versus 35.29% APS4) (Table 3), suggesting that AIRE promoter SNPs could have a role in the pathogenesis of these autoimmune syndromes. Conversely, there was no association between APS2 and AIRE promoter polymorphisms (Table 3) although the analysis was carried out in a lower number of APS2 patients (Figure 1).
In a previous investigation carried out on 158 APECED patients in the Italian territory [41], 10 APS-1 patients had no detectable mutations in the AIRE gene in agreement with data obtained from other populations [42,43]. This suggests that not-yet-identified genes could be involved in the development of APS-1, including defects of AIRE partners or of other controllers of promiscuous gene expression. As can be seen, in the present study, we enrolled five patients with clinical APECED phenotypes, although they were not genetically confirmed since the disease is typically caused by AIRE gene loss of function mutations either in homozygosity or in compound heterozygosity. AIRE gene promoter SNPs were detected in these APS-1 patients (Table 3) suggesting their putative effect on the pathological phenotype.
Overall, based on the preliminary genetic screening results and the molecular modeling data obtained from this study, it is possible to hypothesize that AIRE gene promoter polymorphisms could contribute to autoimmune predisposition in APS patients as previously suggested for patients with alopecia areata [34]. However, future functional studies on cells in vitro and throughout animal models in vivo are necessary to validate this hypothesis and thus the actual contribution of AIRE gene promoter variants on AIRE gene expression. Finally, further extensive genetic screening of AIRE gene promoter polymorphisms should be undertaken in larger cohorts of APS patients to validate the effect of an altered AIRE gene transcription activity in addition to AIRE deficiency at the thymus level.
Based on the results of future screenings on an extended population of APS patients we could verify whether the distribution of the identified SNPs is selective in the different APS categories that present peculiar associations of autoimmune manifestations. We need to point out that, as reported in the Introduction, autoimmune polyglandular syndromes are complex for their clinical manifestations but also for their causative genetic background. SNPs of the AIRE gene promoter, at least those that in the molecular modeling analysis demonstrated being able to affect the structure of nucleic acids, could contribute to the pathogenesis in combination with SNPs of other discovered or not yet discovered susceptibility immune regulatory genes. As final remark, the results of an extended analysis could eventually allow to evaluate the presence of particular SNPs of the AIRE gene promoter and the response to the combined treatments that patients receive for the management of the clinical manifestations of each APS syndrome. These potential results could indeed have translational significance in clinical practice.
4. Materials and Methods
4.1. Study Population
In total, 74 patients affected by APS, i.e., variable association of organ and non-organ specific autoimmune disorders (24 males, 50 females with age ranges at presentation between 0.9 and 19.6 years old) were recruited from the University Department of Pediatrics (DPUO) at Bambino Gesù Children’s Hospital (OPBG) in Rome (Table 1). According to the current criteria for classification of APS [4,5,6,44,45], five patients were affected by clinical APS1 (patients No. 2, 16, 59, 63, 74) not confirmed by the detection of homozygous or compound heterozygous AIRE gene mutations. Additionally, 5 patients were affected by APS2 (patients n° 15, 25, 32, 48, 64), 47 patients were affected by APS3 (patients No. 1, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 17, 18, 19, 20, 21, 22, 23, 26, 27, 29, 33, 36, 37, 39, 40, 41, 42, 44, 46, 47, 49, 51, 52, 53, 54, 55, 57, 58, 65, 66, 67, 68, 70, 71, 72), and 17 patients were affected by APS4 (patients No. 7, 24, 28, 30, 31, 34, 35, 38, 43, 45, 50, 56, 60, 61, 62, 69, 73). Of the overall series, 35 patients could be classified as APS3A (patients No. 1, 3, 4, 5, 8, 12, 13, 14, 18, 19, 20, 21, 22, 23, 27, 29, 33, 37, 39, 40, 41, 42, 44, 46, 47, 49, 51, 52, 58, 65, 66, 67, 68, 70, 72), 23 patients could be classified as APS3B (patients No. 6, 8, 9, 10, 12, 13, 14, 18, 19, 20, 26, 33, 36, 37, 41, 46, 47, 49, 51, 57, 58, 68, 71), 14 patients could be classified by APS3C (patients No. 1, 10, 11, 17, 44, 47, 49, 51, 53, 54, 55, 57, 58, 70), and 3 could be classified as APS3D (patients No. 11, 13, 58).
Informed consent was obtained from all those who took part in the present study in accordance with the Declaration of Helsinki. The investigation was approved by the local Institutional Review Board (IRB) of Bambino Gesù Children’s Hospital (OPBG), which regulates human samples usage for experimental studies (study protocol No. 1385_OPBG_2017). A control group included 81 healthy blood donors (HD). Controls were recruited from the OPBG Blood Transfusion Centre; they had no history of autoimmunity or immunodeficiency, and no autoantibodies were detected in their serum.
4.2. Autoantibodies Screening
The patients’ sera were assayed for T1DM-related autoantibodies, i.e., islet cell antibodies (ICA) by immunofluorescence (IFL), glutamic acid decarboxylase isoform 65 Abs (GADAb), tyrosine phosphatase-related islet antigen 2 Abs (IA2Ab), insulin Abs (IAA), and zinc transporter 8 Abs (ZnT8Ab) by enzyme-linked immunosorbent assay (ELISA); for Addison’s disease, i.e., adrenal cortex Abs (ACA) and 21-hydroxylase Abs (21-OHAb) by ELISA; for AITD-related Abs, i.e., thyrotropin (TSH)-receptor Abs (TRAb) by immunoassay (Immulite TSI, Siemens Healthcare, Tarrytown, NY, USA), thyroglobulin Abs (TgAb), and thyroperoxidase Abs (TPOAb) via electrochemiluminescence immunoassay (ECLIA) (Siemens, Erlangen, Germany); for celiac-disease-related Abs by chemiluminescence (ADVIA Centaur analyzer, Siemens Healthcare, Germany), i.e., anti-transglutaminase-IgA Abs (TRGAb) and deaminated gliadin-IgG Abs (DGP-IgGAb) by EliA and endomysial Abs (EMA) by indirect immunofluorescence (IFL); for autoimmune hepatic diseases, i.e., anti-liver kidney microsomal Abs (LKMAb), smooth muscle Abs (SMA), liver cytosol type 1 Abs (LC1Ab), and soluble liver antigen Abs (SLAIgG); and for stomach-related Abs i.e parietal cells Abs (PCA) by IFL. Intrinsic factor Abs (IFIAb) were tested by ELISA. Non-organ-specific Abs, i.e., nuclear Abs (ANA), extractable nuclear antigen (ENA) (ELiA, Thermo Fisher, Waltham, MA, USA), neutrophil cytoplasmic Abs (ANCA), double-stranded DNA Abs (dsDNAAb), centromere Abs (CeAb), SCL-70 antigen Abs (SCL-70Ab), reticulin Abs (ARA), mitochondrial Abs (AMA), ribosomal Abs (RAb), phospholipid Abs or anti-cardiolipin Abs (CAb), beta2glycoprotein I-IgG or IgM Abs (β2GP-1IgGAb), anti-beta2glycoprotein I-IgM (β2GP-1IgMAb), dense fine speckled 70 Abs (DSF70Ab) (ELiA), and cyclic citrullinated peptide Abs (CCPAb) (EliA) were also tested.
4.3. Molecular Studies
To study the AIRE gene sequence, the AIRE promoter sequence, and the PTPN22 gene sequence, leukocyte genomic DNA was extracted from whole-blood samples of patients by QIAmp DNA blood mini kit (Qiagen, Hilden Germany) according to the manufacturer’s guidelines.
4.3.1. AIRE Gene Screening
All 14 exons and flanking exon-intron boundaries of the AIRE gene (GenBank ID: 326) were sequenced according to already described protocols (Genetic Analyzer 3500 Applied Biosystems HITACHI system, Thermo Fisher Scientific, Rodano, Italy) in the DNA of recruited patients [35]. AIRE gene promoter was screened by polymerase chain reaction (PCR) using the following primer sequences: forward 5′-GGAACCGAGGCTCAGAGAAGG-3′ and reverse 5′-CCTCAGAAGCCGGCGTAGC-3′ (annealing temperature 62 °C). These primers are positioned 751bp upstream and 33bp downstream relative to the AIRE start codon. The amplification lasted 35 cycles, generating PCR products of 787bp, which were purified using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Dueren, Germany) and sequenced with the Genetic Analyzer 3500 (Applied Biosystems HITACHI system).
4.3.2. Screening for the Presence of C1858T PTPN22
Detection of the C1858T variant in the PTPN22 gene was carried out by PCR with specific primers for exon 14 of the PTPN22 gene (protein tyrosine phosphatase N22, GenBank ID: 26191): forward 5′-GATAATGTTGCTTCAACGGAATTT-3′ and reverse 5′-CCTCAAACTCAAGGCTCACAC-3′ (annealing temperature 58.5 °C). The amplification lasted 35 cycles, generating PCR products of 318bp that were purified using NucleoSpin Gel and PCR Clean-up kit (Bioanalysis). PCR sequencing was carried out with the BigDye Terminator v.3.1 Cycle sequencing protocol (Life Technologies, Applied Biosystems, Paisley, UK), as reported [18,46].
4.4. Multiple Sequence Alignment and Modelling of DNA Secondary Structure
Multiple sequence alignment was performed with MAFFT (v7.490) [47]. Secondary structure predictions were carried out with RNAstructure (v6.4) [48] on the human sequence (ENST00000291582.6 for the wild type and for each variant by introducing the respective nucleotide change) in the range from 1kb upstream exon 1 to 144 bases downstream exon 2.
Acknowledgments
We acknowledge Anna Lo Russo for processing patients’ samples for subsequent molecular and serological analysis and Simone Piga of the Epidemiology Department OPBG for statistical analysis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25052656/s1.
Author Contributions
A.C. and C.N.: investigation, methodology. A.P.: investigation. E.B.: investigation, formal analysis. D.V.D.: writing—original draft. M.C.: resources, writing—original draft. C.B.: writing—original draft, supervision. A.F.: conceptualization, funding acquisition, project administration, supervision, validation, writing—original draft, writing—review and editing. All authors contributed to the article and approved the submitted version. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Bambino Gesù Children’s Hospital (Study protocol no. 1385_OPBG_2017).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study or parental consent in cases of children.
Data Availability Statement
The data presented in this study are available on request from the corresponding author and clinician Doctor Marco Cappa.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Funding Statement
This work was supported by the Italian Ministry of Health with “Current Research Funds”.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Roitt I.M., Doniach D., Campbell P.N., Hudson R.V. Auto-antibodies in Hashimoto’s disease (lymphadenoid goitre) Lancet. 1956;271:820–821. doi: 10.1016/S0140-6736(56)92249-8. [DOI] [PubMed] [Google Scholar]
- 2.Witebsky E., Rose N.R., Terplan K., Paine J.R., Egan R.W. Chronic thyroiditis and autoimmunization. J. Am. Med. Assoc. 1957;164:1439–1447. doi: 10.1001/jama.1957.02980130015004. [DOI] [PubMed] [Google Scholar]
- 3.Rose N., MacKay I.R. The Autoimmune Diseases. 6th ed. Academic Press; Dordrecht, The Netherlands: 2020. 895p [Google Scholar]
- 4.Betterle C., Sabbadin C., Scaroni C., Presotto F. Autoimmune Polyendocrine Syndromes (APS) or Multiple Autoimmune Syndromes (MAS) In: Colao A., Jaffrain-Rea M.L., Beckers A., editors. Polyendocrine Disorders and Endocrine Neoplastic Syndromes. Endocrinology. Springer; Cham, Switzerland: 2019. pp. 1–50. [DOI] [Google Scholar]
- 5.Betterle C., Furmaniak J., Sabbadin C., Scaroni C., Presotto F. Type 3 autoimmune polyglandular syndrome (APS-3) or type 3 multiple autoimmune syndrome (MAS-3): An expanding galaxy. J. Endocrinol. Investig. 2023;46:643–665. doi: 10.1007/s40618-022-01994-1. [DOI] [PubMed] [Google Scholar]
- 6.Neufeld M., Blizzard M.R. Polyglandular autoimmune disease. In: Pinchera A., Doniach D., Fenzi G.F., Baschieri. L., editors. Symposium on Autoimmune Aspects of Endocrine Disorders. Academic Press; New York, NY, USA: 1980. [Google Scholar]
- 7.Anaya J.M., Castiblanco J., Rojas-Villarraga A., Pineda-Tamayo R., Levy R.A., Gómez-Puerta J., Dias C., Mantilla R.D., Gallo J.E., Cervera R., et al. The multiple autoimmune syndromes. A clue for the autoimmune tautology. Clin. Rev. Allergy Immunol. 2012;43:256–264. doi: 10.1007/s12016-012-8317-z. [DOI] [PubMed] [Google Scholar]
- 8.Rojas-Villarraga A., Amaya-Amaya J., Rodriguez-Rodriguez A., Mantilla R.D., Anaya J.M. Introducing Polyautoimmunity: Secondary Autoimmune Diseases No Longer Exist. Autoimmune Dis. 2012;2012:254319. doi: 10.1155/2012/254319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferré E.M.N., Schmitt M.M., Lionakis M.S. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Front. Pediatr. 2021;1:723532. doi: 10.3389/fped.2021.723532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith T.J., Hegedüs L. Graves’ Disease. N. Engl. J. Med. 2016;375:1552–1565. doi: 10.1056/NEJMra1510030. [DOI] [PubMed] [Google Scholar]
- 11.Ralli M., Angeletti D., Fiore M., D’Aguanno V., Lambiase A., Artico M., de Vincentiis M., Greco A. Hashimoto’s thyroiditis: An update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential malignant transformation. Autoimmun. Rev. 2020;19:102649. doi: 10.1016/j.autrev.2020.102649. [DOI] [PubMed] [Google Scholar]
- 12.Frommer L., Kahaly G.J. Autoimmune Polyendocrinopathy. J. Clin. Endocrinol. Metab. 2019;104:4769–4782. doi: 10.1210/jc.2019-00602. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari S.M., Fallahi P., Antonelli A., Benvenga S. Environmental Issues in Thyroid Diseases. Front. Endocrinol. 2017;8:50. doi: 10.3389/fendo.2017.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flesch B.K., Matheis N., Alt T., Weinstock C., Bux J., Kahaly G.J. HLA class II haplotypes differentiate between the adult autoimmune polyglandular syndrome types II and III. J. Clin. Endocrinol. Metab. 2014;99:E177–E182. doi: 10.1210/jc.2013-2852. [DOI] [PubMed] [Google Scholar]
- 15.Barkia Beradhi S., Flesch B.K., Hansen M.P., Matheis N., Kahaly G.J. HLA Class II Differentiates Between Thyroid and Polyglandular Autoimmunity. Horm. Metab. Res. 2016;48:232–237. doi: 10.1055/s-0035-1559622. [DOI] [PubMed] [Google Scholar]
- 16.Dittmar M., Kahaly G.J. Genetics of the autoimmune polyglandular syndrome type 3 variant. Thyroid. Off. J. Am. Thyroid. Assoc. 2010;20:737–743. doi: 10.1089/thy.2010.1639. [DOI] [PubMed] [Google Scholar]
- 17.Dittmar M., Ide M., Wurm M., Kahaly G.J. Early onset of polyglandular failure is associated with HLA-DRB1*03. Eur. J. Endocrinol. 2008;159:55–60. doi: 10.1530/EJE-08-0082. [DOI] [PubMed] [Google Scholar]
- 18.Vang T., Congia M., Macis M.D., Musumeci L., Orrú V., Zavattari P., Nika K., Tautz L., Taskén K., Cucca F., et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 19.Houcken J., Degenhart C., Bender K., König J., Frommer L., Kahaly G.J. PTPN22 and CTLA-4 Polymorphisms Are Associated With Polyglandular Autoimmunity. J. Clin. Endocrinol. Metab. 2018;103:1977–1984. doi: 10.1210/jc.2017-02577. [DOI] [PubMed] [Google Scholar]
- 20.Sahin O.A., Goksen D., Ozpinar A., Serdar M., Onay H. Association of vitamin D receptor polymorphisms and type 1 diabetes susceptibility in children: A meta-analysis. Endocr. Connect. 2017;6:159–171. doi: 10.1530/EC-16-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Downes K., Marcovecchio M.L., Clarke P., Cooper J.D., Ferreira R.C., Howson J.M.M., Jolley J., Nutland S., Stevens H.E., Walker N.M., et al. Plasma concentrations of soluble IL-2 receptor α (CD25) are increased in type 1 diabetes and associated with reduced C-peptide levels in young patients. Diabetologia. 2014;57:366–372. doi: 10.1007/s00125-013-3113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan S., Mandal R.K., Jawed A., Dar S.A., Wahid M., Panda A.K., Areeshi M.Y., Khan M.E.A. TNF-α -308 G > A (rs1800629) Polymorphism is Associated with Celiac Disease: A Meta-analysis of 11 Case-Control Studies. Sci. Rep. 2016;6:32677. doi: 10.1038/srep32677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dittmar M., Kaczmarczyk A., Bischofs C., Kahaly G.J. The proinflammatory cytokine TNF-alpha -308 AA genotype is associated with polyglandular autoimmunity. Immunol. Investig. 2009;38:255–267. doi: 10.1080/08820130902766092. [DOI] [PubMed] [Google Scholar]
- 24.Hori S., Nomura T., Sakaguchi S. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3 Science 2003, 299, 1057–1061. J. Immunol. Baltim. 2017;198:981–985. [PubMed] [Google Scholar]
- 25.Bauer S., Groh V., Wu J., Steinle A., Phillips J.H., Lanier L.L., Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 26.Ide M., Dittmar M., Wurm M., Kanitz M., Kahaly G.J. Polymorphisms of MICA microsatellites in thyroidal autoimmunity. Med. Klin. Munich. 2007;102:11–15. doi: 10.1007/s00063-007-1001-z. [DOI] [PubMed] [Google Scholar]
- 27.Tomer Y., Menconi F. Type 1 diabetes and autoimmune thyroiditis: The genetic connection. Thyroid. Off. J. Am. Thyroid. Assoc. 2009;19:99–102. doi: 10.1089/thy.2008.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dittmar M., Kahaly G.J. Immunoregulatory and susceptibility genes in thyroid and polyglandular autoimmunity. Thyroid. Off. J. Am. Thyroid. Assoc. 2005;15:239–250. doi: 10.1089/thy.2005.15.239. [DOI] [PubMed] [Google Scholar]
- 29.Bruserud O., Oftedal B.E., Wolff A.B., Husebye E.S. AIRE-mutations and autoimmune disease. Curr. Opin. Immunol. 2016;43:8–15. doi: 10.1016/j.coi.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Eriksson D., Røyrvik E.C., Aranda-Guillén M., Berger A.H., Landegren N., Artaza H., Hallgren Å., Grytaas M.A., Ström S., Bratland E., et al. GWAS for autoimmune Addison’s disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat. Commun. 2021;12:959. doi: 10.1038/s41467-021-21015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oftedal B.E., Hellesen A., Erichsen M.M., Bratland E., Vardi A., Perheentupa J., Kemp E.H., Fiskerstrand T., Viken M.K., Weetman A.P., et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity. 2015;42:1185–1196. doi: 10.1016/j.immuni.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Oftedal B.E., Assing K., Baris S., Safgren S.L., Johansen I.S., Jakobsen M.A., Babovic-Vuksanovic D., Agre K., Klee E.W., Majcic E., et al. Dominant-negative heterozygous mutations in AIRE confer diverse autoimmune phenotypes. iScience. 2023;26:106818. doi: 10.1016/j.isci.2023.106818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murumägi A., Vähämurto P., Peterson P. Characterization of regulatory elements and methylation pattern of the autoimmune regulator (AIRE) promoter. J. Biol. Chem. 2003;278:19784–19790. doi: 10.1074/jbc.M210437200. [DOI] [PubMed] [Google Scholar]
- 34.Lovewell T.R.J., McDonagh A.J., Messenger A.G., Azzouz M., Tazi-Ahnini R. The AIRE -230Y Polymorphism Affects AIRE Transcriptional Activity: Potential Influence on AIRE Function in the Thymus. PLoS ONE. 2015;10:e0127476. doi: 10.1371/journal.pone.0127476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palma A., Gianchecchi E., Palombi M., Luciano R., Di Carlo P., Crinò A., Cappa M., Fierabracci A. Analysis of the autoimmune regulator gene in patients with autoimmune non-APECED polyendocrinopathies. Genomics. 2013;102:163–168. doi: 10.1016/j.ygeno.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Ghiglioni D.G., Zicari A.M., Parisi G.F., Marchese G., Indolfi C., Diaferio L., Brindisi G., Ciprandi G., Marseglia G.L., Del Giudice M.M. Vernal keratoconjunctivitis: An update. Eur. J. Ophthalmol. 2021;31:2828–2842. doi: 10.1177/11206721211022153. [DOI] [PubMed] [Google Scholar]
- 37.Liston A., Gray D.H.D., Lesage S., Fletcher A.L., Wilson J., Webster K.E., Scott H.S., Boyd R.L., Peltonen L., Goodnow C.C. Gene dosage--limiting role of Aire in thymic expression, clonal deletion, and organ-specific autoimmunity. J. Exp. Med. 2004;200:1015–1026. doi: 10.1084/jem.20040581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kont V., Laan M., Kisand K., Merits A., Scott H.S., Peterson P. Modulation of Aire regulates the expression of tissue-restricted antigens. Mol. Immunol. 2008;45:25–33. doi: 10.1016/j.molimm.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira E.H., Macedo C., Donate P.B., Almeida R.S., Pezzi N., Nguyen C., Rossi M.A., Sakamoto-Hojo E.T., Donadi E.A., Passos G.A. Expression profile of peripheral tissue antigen genes in medullary thymic epithelial cells (mTECs) is dependent on mRNA levels of autoimmune regulator (Aire) Immunobiology. 2013;218:96–104. doi: 10.1016/j.imbio.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Wengraf D.A., McDonagh A.J.G., Lovewell T.R.J., Vasilopoulos Y., Macdonald-Hull S.P., Cork M.J., Messenger A.G., Tazi-Ahnini R. Genetic analysis of autoimmune regulator haplotypes in alopecia areata. Tissue Antigens. 2008;71:206–212. doi: 10.1111/j.1399-0039.2007.00992.x. [DOI] [PubMed] [Google Scholar]
- 41.Garelli S., Dalla Costa M., Sabbadin C., Barollo S., Rubin B., Scarpa R., Masiero S., Fierabracci A., Bizzarri C., Crinò A., et al. Autoimmune polyendocrine syndrome type 1: An Italian survey on 158 patients. J. Endocrinol. Investig. 2021;44:2493–2510. doi: 10.1007/s40618-021-01585-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 2006;91:2843–2850. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 43.Ilmarinen T., Eskelin P., Halonen M., Rüppell T., Kilpikari R., Torres G.D., Kangas H., Ulmanen I. Functional analysis of SAND mutations in AIRE supports dominant inheritance of the G228W mutation. Hum. Mutat. 2005;26:322–331. doi: 10.1002/humu.20224. [DOI] [PubMed] [Google Scholar]
- 44.Ferre E.M.N., Rose S.R., Rosenzweig S.D., Burbelo P.D., Romito K.R., Niemela J.E., Rosen L.B., Break T.J., Gu W., Hunsberger S., et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. 2016;1:e88782. doi: 10.1172/jci.insight.88782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Husebye E.S., Anderson M.S., Kämpe O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018;378:1132–1141. doi: 10.1056/NEJMra1713301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arena A., Belcastro E., Ceccacci F., Petrini S., Conti L.A., Pagliarosi O., Giorda E., Sennato S., Schiaffini R., Wang P., et al. Improvement of Lipoplexes With a Sialic Acid Mimetic to Target the C1858T PTPN22 Variant for Immunotherapy in Endocrine Autoimmunity. Front. Immunol. 2022;13:838331. doi: 10.3389/fimmu.2022.838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reuter J.S., Mathews D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author and clinician Doctor Marco Cappa.