

Abstract
The existing T cell–centered immune checkpoint blockade therapies have been successful in treating some but not all patients with cancer. Immunosuppressive myeloid cells, including myeloid-derived suppressor cells (MDSCs), that inhibit antitumor immunity and support multiple steps of tumor development are recognized as one of the major obstacles in cancer treatment. Leukocyte Ig-like receptor subfamily B3 (LILRB3), an immune inhibitory receptor containing tyrosine-based inhibitory motifs (ITIMs), is expressed solely on myeloid cells. However, it is unknown whether LILRB3 is a critical checkpoint receptor in regulating the activity of immunosuppressive myeloid cells, and whether LILRB3 signaling can be blocked to activate the immune system to treat solid tumors. Here, we report that galectin-4 and galectin-7 induce activation of LILRB3 and that LILRB3 is functionally expressed on immunosuppressive myeloid cells. In some samples from patients with solid cancers, blockade of LILRB3 signaling by an antagonistic antibody inhibited the activity of immunosuppressive myeloid cells. Anti-LILRB3 also impeded tumor development in myeloid-specific LILRB3 transgenic mice through a T cell–dependent manner. LILRB3 blockade may prove to be a novel approach for immunotherapy of solid cancers.
Keywords: inhibitory receptor, ITIM, galectin, myeloid cells, cancer
Introduction
Immunosuppressive myeloid cells are a heterogeneous population of cells including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and certain groups of dendritic cells, neutrophils, and eosinophils (1,2). These cells are important elements in the tumor microenvironment (TME) where they contribute to the inhibition of antitumor immune responses and support tumor development, progression, and metastasis through a variety of mechanisms. Reprogramming these cells to stimulate proinflammatory properties and antigen presentation activities in the TME may overcome resistance to existing T cell–centered immune checkpoint blockade therapy and become an attractive novel anticancer immunotherapeutic strategy.
Leukocyte Ig-like receptor subfamily B (LILRB) proteins are a group of type I transmembrane, immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing receptors that are mainly expressed on hematopoietic cells (3-10). Activated LILRBs can recruit the tyrosine phosphatases SHP-1 and SHP-2 or the inositol-phosphatase SHIP. Prior studies indicate that LILRBs are immune checkpoint factors during cancer development (4,11-14). Work from our laboratory on these receptors has shown that LILRB2 is a receptor for the hormone Angptl2, and that several LILRBs and a related ITIM-containing receptor, LAIR1, support acute myeloid leukemia (AML) development (14-23). A number of studies have demonstrated that signaling blockade of certain LILRBs and related proteins through antibodies and recombinant proteins in normal or malignant human myeloid or lymphoid cells promotes antitumor immune activation (9,11,13,14,21,24-29).
LILRB3 is a member of the LILRB family that is solely expressed by myeloid cells, including monocytes, macrophages, dendritic cells, neutrophils, eosinophils, and basophils (as well as in vitro differentiated mast cells and osteoclasts) (4,30). Previous studies have reported that the cytoplasmic ITIMs of LILRB3 may contribute to negative regulation of immune responses (31,32) and that activating LILRB3 in human myeloid cells may lead to inhibition of immune activation (33,34). Furthermore, LILRB3 may be an inhibitor of allergic inflammation and autoimmunity (35,36). It also has been revealed that activation of LILRB3 with an agonist antibody in humanized mice induces tolerance and permits efficient engraftment of allogeneic cells (37). Recently, we demonstrated that in myeloid leukemia cells, LILRB3 exerts both activating and inhibitory functions (through the TRAF2/cFLIP/NFκB axis and recruiting SHP-1/SHP-2, respectively) (21).
It is noteworthy that LILRBs, including LILRB3, are expressed only in primates. The expression pattern and ligands of the mouse relative of LILRB3, PirB, differ from those of LILRB3 (4,6,9). Consequently, the use of PirB-deficient mice to investigate the biology of LILRB3 is likely to be of limited value. The limited knowledge of LILRB3 leaves important questions unanswered: what extracellular cues induce LILRB3 signaling, what are the unique functions of LILRB3, and can LILRB3 signaling be blocked to inhibit the tumor-supportive activity of myeloid cells in the TME.
Here, we identified galectin-4 and galectin-7 as extracellular proteins that activate LILRB3. We also found that LILRB3 was functionally expressed on immunosuppressive myeloid cells from patients with cancer. Moreover, LILRB3 blockade by a fully human anti-LILRB3 efficiently inhibited the immunosuppressive activity of human MDSCs and delayed cancer development in myeloid-specific LILRB3 transgenic mice. Our results suggest that LILRB3 signaling in immunosuppressive myeloid cells promotes cancer development, and that LILRB3 blockade may be an attractive option to treat certain patients with cancer.
Materials and Methods
Mice
Wild-type (WT) C57BL/6J mice were purchased from and maintained at the animal core facility of the University of Texas Southwestern Medical Center. Myeloid-specific (LysM-Cre) LILRB3 transgenic mice were produced by CRISPR and backcrossed with WT C57BL/6J mice for at least 6 generations, as previously described (21). Mice in each experiment were sex and age matched, and both male and female mice were utilized. All animal work in this study was approved by the UT Southwestern Institutional Animal Care and Use Committee.
Cell culture
293T, B16 mouse melanoma, and MC38 mouse colon cancer cell lines were maintained in a humidified atmosphere of 5% CO2 at 37°C in DMEM (ThermoFisher Scientific, Cat# MT10013CV) with 10% FBS (Gibco, Cat# A5209402) and 1% penicillin/streptomycin (ThermoFisher Scientific, Cat# 15140122) (this medium is referred to hereafter as D10). 293T cells were obtained from ATCC in 2020. B16 and MC38 cells were obtained as a gift from Dr. Yang-Xin Fu at University of Texas Southwestern Medical Center in 2020. LILRB reporter cells including the parental 2B4 cells were developed in 2012-2019 as previously described (14, 18, 21, 24) and were cultured in RPMI (Corning, Cat# 10-040-CV) with 10% FBS and 1% penicillin/streptomycin (this medium is referred to hereafter as R10). THP1 cells were obtained as a gift from Dr. Jian Xu at University of Texas Southwestern Medical Center in 2021 and maintained in R10. Primary human cells were isolated by autoMACS after purification of PBMCs from whole blood using Ficoll (Cytiva, Cat# 17144003) derived from healthy donors (Carter Bloodcare) or patients with cancer (UT Southwestern Tissue Management Shared Resource). Cell lines were cultured for a maximum of 20 passages before they were discarded. Cell lines were tested for mycoplasma at least every 10 passages using the Lookout Mycoplasma PCR Detection Kit (Sigma, Cat# MP0035-1KT). Cell lines were not re-authenticated within the past year.
Primary human samples
Primary whole blood samples from 76 patients with solid tumors were obtained during the patients’ visits to University of Texas Southwestern Medical Center from 2019-2023 in purple-top K2 EDTA tubes at 4°C and obtained by us through UT Southwestern Tissue Management Shared Resource. Informed written consent was obtained from all patients, and the study was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center (IRB STU 122013-023) and conducted in accordance with the Declaration of Helsinki. Samples were distributed to the laboratory in a de-identified fashion, and the proposed study was determined to not be human research.
Plasmids
LILRB3 (Accension# XM_006726314.4, AAI04994.1, XP_016885786) and human galectin-4 (Uniprot Entry# P56470) were cloned from synthesized cDNA (Genewiz) with a N-terminal FLAG tag after the signal peptide and inserted into the pSin lentiviral expression vector (obtained as a gift from Dr. Jiang Wu at University of Texas Southwestern Medical Center) under the regulation of an Ef1α promoter for signaling studies. LILRB extracellular domains (ECD) were inserted into the pSLAP expression vector (obtained as a gift from Dr. Hisashi Arase from Osaka University in 2012) with a mSLAF1 signal peptide and mPILRB transmembrane/intracellular domain. SHP1 (Uniprot Entry# Q9BZQ2), SHP2 (Uniprot Entry# Q06124), and Lyn (Uniprot Entry# P07948) were cloned from synthesized cDNA (Genewiz) and inserted into the pLVX expression vector (Takara, Cat# 631982) under the regulation of an Ef1a promoter. Plasmid sequencing was confirmed with whole plasmid sequencing (Eurofins Genomics).
Ligand Screening for LILRBs
20 μg/mL LILRB3-His (SinoBiological, Cat# 11978-H08H) was mixed with mesenchymal stem cells or a mixture of A549 and H157 cells in R10 for 24 hours at 37°C. The cells were fixed with 4% PFA (Sigma, Cat# 47608). The fixed cells were incubated and washed with anti-His magnetic beads (ThermoFisher Scientific, Cat# 10104D) per manufacturer’s protocol. The final eluted fraction was sent for mass spectrometry at the UT Southwestern Proteomics Core.
Virus production and infection
For lentivirus production, LILRB3 plasmids (see Plasmids) were mixed with psPAX2 (Addgene, Cat# 12260) and pMD2G (Addgene, Cat# 12259) at a ratio of 4:3:1 and transfected into 293T cells using Polyjet (SignaGen, Cat# SL100688). For retroviral production, LILRB3 plasmids were mixed with pCL-Eco (Addgene, Cat# 12371) at a ratio of 2:1 and transfected into 293T cells using Polyjet. 72 hours later, the viral supernatant was syringe-filtered through a 0.45um filter and centrifuged at 500g for 1 hour at 32°C. Lentiviral LILRB3 supernatant was centrifuged onto 293T and THP1 cells in the presence of 8 μg/mL polybrene (Sigma, Cat# H9268). Retroviral LILRB3 supernatant was centrifuged onto 2B4 cells in the presence of 4 μg/mL polybrene (Sigma, Cat# H9268). After 48 hours, virus supernatant was removed. 293T cells were selected in D10 with 5 μg/mL puromycin (Sigma, Cat# P7255). THP1 and 2B4 cells were selected in R10 with 5 μg/mL puromycin.
THP1-LILRB3 Signaling
THP1-LILRB3 cells were serum-starved (RPMI only) for 20 hours prior to signaling experiments. Cells were collected by centrifugation and resuspended with ice-cold PBS. For time-course experiments, cells were resuspended in ice-cold PBS containing LPS (Sigma, Cat# L2630, 200 ng/mL) and soluble human galectin-4 (Abnova, Cat# P3544, 20 μg/mL). Equivalent numbers of cells (8X106) were added to coated plates (non-treated flat-bottom plates with HLA-DR, Galectin-2, or Galectin-4). Cells were centrifuged onto the plate at 300g for 1 minute at 4°C. Plates were placed into a humidified 37°C tissue-culture incubator for indicated time points or 10 minutes (if not indicated). Then, ice-cold IP Lysis Buffer (1% NP-40, 0.025M Tris, 0.15M NaCl, 0.001M EDTA) containing protease inhibitor (Sigma, Cat# 11836153001) and phosphatase inhibitor (Sigma, Cat# 4906837001) was immediately added and the samples analyzed by Western blotting (see Western blotting and co-immunoprecipitation).
293T LILRB-Reconstituted Signaling
1X106 293T cells were plated into 6-well tissue-culture treated plates 24 hours before transfection. 3 μg of plasmid DNA (2 μg LILRB3-FLAG or LILRB3-FLAG ITIM mutants, 300 ng SHP1, 300 ng SHP2, 300 ng galectin-4, 100 ng Lyn) and 9 μL Polyjet was used to transfect 293T cells per recommended manufacturer’s instructions. 5 hours after transfection, media was replaced with DMEM containing 10% FBS and 20 μg/mL hIgG control or 20 μg/mL anti-LILRB3 (21) where indicated. 48 hours after transfection, cells were washed with PBS, and ice-cold IP Lysis Buffer containing protease inhibitor and phosphatase inhibitor was immediately added and the samples analyzed by Western blotting.
Flow cytometry
Cells from healthy patients, patients with cancer, tumor-bearing mice, and naïve mice were washed in FACS buffer (PBS containing 0.1% BSA, 2mM EDTA, 1% penicillin/streptomycin), blocked with human IgG (Sigma, Cat# I4506) for 15 minutes at 4°C, and stained with primary antibodies (Supplementary Table S1) for 30 minutes at 4°C. Flow data was collected using FACS Calibur, FACS Melody, or Cytek Northern Lights. Zombie yellow (Biolegend, Cat# 423104) staining before primary antibody staining or propidium iodide (Sigma, Cat# P4864) staining after primary antibody staining was used to exclude dead cells in analysis. Flow data was analyzed using Flowjo v10 or FCS Express 7.0 software. The anti-LILRB3 for flow cytometry was labeled using a Fc site-specific PE conjugation kit (ThermoFisher Scientific, Cat# S10467).
Biolayer interferometry (BLI)
BLI was conducted as previously described (22). Briefly, recombinant LILRB3-Fc proteins (R&D, Cat# 1806-T5-050) were loaded onto protein G biosensors. After soaking the sensors in kinetic buffer, the loaded biosensors were exposed to a series of recombinant Galectin-4-His (Abnova, Cat# P3544) concentrations (2-fold serial dilutions from 1000 nM to 1 nM) and background subtraction was used to correct for sensor drifting. Kd was calculated from the ratio koff/kon.
Coculture of human MDSCs and T cells
Coculture of human MDSCs and T cells was conducted as previously described (28). Phenotypically-enriched MDSCs (isolated with CD14 Microbeads (Miltenyi Biotec, Cat# 130-050-201)) and T cells (isolated from CD3 Microbeads (Miltenyi Biotec, Cat# 130-097-043)) were isolated from PBMCs from indicated patients with cancer. The cells were cocultured in R10 with CFSE-stained autologous T cells (T cells:MDSCs = 1:1) for 5 days, and treated with hIgG control or anti-LILRB3 (20 μg/mL). 6.25 μL/mL of ImmunoCult Human CD3/CD28 T Cell Activator (StemCell, Cat# 10971) was used to activate CD3+ T cells.
Human MDSC culture
MDSC culture was conducted as previously described (28). Briefly, phenotypically-enriched MDSCs from indicated patients with cancer were cultured in R10 for 5 days, and treated with hIgG control or anti-LILRB3 (20 μg/mL). Flow cytometry analysis of surface expression of CD86, CD206, and CD163 was then conducted.
Coculture of mouse MDSCs and T cells
Myeloid-specific LILRB3 transgenic mice and WT C57BL/6 mice were subcutaneously injected with tumor cells (5x105 B16 or MC38-hGal4). When tumor size reached the maximum-allowed size (2000 mm3), spleens were harvested and phenotypic MDSCs (Mac1+Gr1+) were isolated from the tumor-bearing LILRB3 transgenic mice using the EasySep Mouse MDSC Kit (StemCell, Cat# 19867) and T cells were isolated from the tumor-bearing WT C57BL/6 mice using the Mojosort Mouse CD3 T cell Isolation Kit (Biolegend, Cat# 480024), respectively. MDSCs from tumor-bearing LILRB3 transgenic mice were cocultured with CFSE-stained T cells from WT C57BL/6 mice (T cells:MDSCs = 1:4) in the presence of 5 μg/mL coated anti-CD3 (Biolegend Cat# 100202) and 5 μg/mL coated anti-CD28 (Biolegend Cat# 102115) or Dynabeads Mouse T-activator (ThermoFisher Scientific, 11456D) along with soluble hIgG control or soluble anti-LILRB3 (20 μg/mL). Cells were cocultured for 3-5 days and the percentage of proliferating T cells was measured using flow cytometry.
Coculture of naïve Mouse Mac1+Gr1+ cells and T cells
Mac1+Gr1+ cells and T cells were collected from the spleens of unchallenged LILRB3 transgenic mice and syngeneic WT C57BL/6J mice, respectively, using EasySep Mouse MDSC Kit (StemCell, Cat# 19867) and Mojosort Mouse CD3 T cell isolation Kit (Biolegend, Cat# 480024). Non-treated tissue culture plates were coated with 10 μg/mL human galectin-4 (Abnova, Cat# P3544) or BSA (GoldBio, Cat# A-421-100) overnight at 37°C. After the plates were blocked with 2% BSA/PBS for 30 minutes and washed with PBS, Mac1+Gr1+ cells and CFSE-stained T cells were incubated together (T cells:Mac1+Gr1+ = 1:1) in the presence of Mouse T cell Activator (ThermoFisher Scientific, Cat# 11456D), 10 ng/mL human IL7 (Peprotech, Cat# 200-07), 5 ng/mL human IL15 (Peprotech, Cat# 200-15) in Immunocult-XF T cell Expansion medium (Stemcell, Cat# 10981) along with soluble hIgG control or soluble anti-LILRB3 (20 μg/mL). Cells were cocultured for 3-4 days and the percentage of proliferating T cells was measured using flow cytometry.
Tumor experiments in LysM-Cre LILRB3 transgenic mice
5x105 B16, MC38, or MC38-hGal4 tumor cells were injected subcutaneously into each mouse (LysM-Cre+/+LILRB3+/+ in C57BL/6J background) at day 0 in their right flanks. Six days later, mice were randomized into two groups based on tumor size, followed by 200 μg intraperitoneal injection of hIgG control or anti-LILRB3 twice weekly. Mice were euthanized when tumors reached maximum allowed volume (2,000 mm3). Subcutaneous tumor was measured 2-3 times weekly using a digital caliper. Tumor volume was calculated as 0.52 X width2 X length. For indicated T-cell depletion experiments, 200 μg anti-CD4 (BioXCell, YTS177, Cat# BE-0003-3) and 200 μg anti-CD8 (BioXCell, YTS169.4, Cat# BE0117) or 400 μg isotype control (BioXCell, LTF-2, Cat# BE0090) were injected two days before tumor implantation and injected every 4 days thereafter.
Western blotting and co-immunoprecipitation
Cells were lysed with IP Lysis buffer containing protease inhibitor (Sigma, Cat# 11836153001) and phosphatase inhibitor (Sigma, Cat# 4906837001). The LILRB3 complex was immunoprecipitated with anti-FLAG L5 magnetic agarose (ThermoFisher Scientific, Cat# A36797) per manufacturer’s protocol. Samples were mixed with 4X LDS loading buffer (ThermoFisher Scientific, Cat# NP0007) containing 5% BME, boiled at 95°C for 10 min, and separated by reducing SDS-PAGE. After transfer to nitrocellulose membranes (Biorad Trans-Blot Transfer System), membranes were blocked with 5% dry milk diluted in TBST. Proteins were detected with specific primary antibodies (Supplementary Table S2) and HRP-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, Cat# 115-035-003) or HRP-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch, Cat# 111-035-003). When necessary, co-immunoprecipitated proteins were detected using Tidyblot (Biorad, Cat# STAR209PA, 1:200). Chemiluminescence was detected using chemiluminescent substrate (ThermoFisher Scientific, Cat# 34580) and chemiluminescent imaging (Biorad, Chemidoc).
Chimeric receptor reporter assay
LILRB3 chimeric receptor reporter cells were developed by us as previously described (14). Proteins (as shown in Supplementary Table S3) were coated onto 96-well plates by plating 50 μL of the desired protein concentration on 96-well plates at 37°C overnight. After removing the protein, 200 μL of 2% BSA diluted in PBS was added to each well for 30 minutes at 37°C. After washing with PBS, 5 x 104 LILRB reporter cells were seeded into each well. When soluble proteins were indicated, recombinant proteins were mixed with reporter cells prior to seeding. During competition assays used to screen in-house previously identified LILRB3 antagonist antibodies (21), 20 μg/ml hIgG control or anti-LILRB was added into culture media. After culture for 18 hours, the percentage of GFP+ reporter cells was measured by flow cytometry.
TCGA analyses
Patient data was obtained from TCGA GDC PANCAN, GDC KIRC, GDC GBM, GDC LGG, GDC THYM, and GDC LUSC (version: October 7, 2023) using the UCSC Xena Web Browser with inclusion of all patients in each cancer subset. The mRNA levels were determined by RNA-seq, and LILRB3 expression level groups (low and high) were determined from the median of each subtype. Overall survival was analyzed based on LILRB3 expression and corresponding patient survival data. Patients were grouped as “high LILRB3” if their LILRB3 expression was above the median LILRB3 level and grouped as “low LILRB3” if their LILRB3 expression was below the median LILRB3 level in each cancer subtype for overall survival analysis. Statistical significance for overall survival was calculated by Mann-Whitney’s log-rank test from UCSC Xena Web Browser.
Statistics
Statistical analysis was performed using Graphpad Prism 10.0. Statistical significance for comparisons was calculated by two-tailed Student’s t test. Statistical significance for overall survival was calculated by Mann-Whitney’s log-rank test. A p-value of 0.05 or less was considered significant and labeled with *. Values are reported as mean ± SEM.
Data availability statement
The data generated in this study are available within the article and its supplementary data files or from the corresponding author upon reasonable request.
Results
LILRB3 expression is associated with reduced survival in patients diagnosed with solid tumors.
Analysis of LILRB3 mRNA levels and survival of patients with cancer showed that the expression of LILRB3 negatively correlated with the overall survival of patients in the TCGA database, including pan-cancer and those with renal clear cell carcinoma, glioblastoma, lower grade glioma, thymoma, and lung squamous cell carcinoma (Fig. 1A).
Fig. 1∣. LILRB3 expression is associated with reduced survival in patients diagnosed with solid tumors.

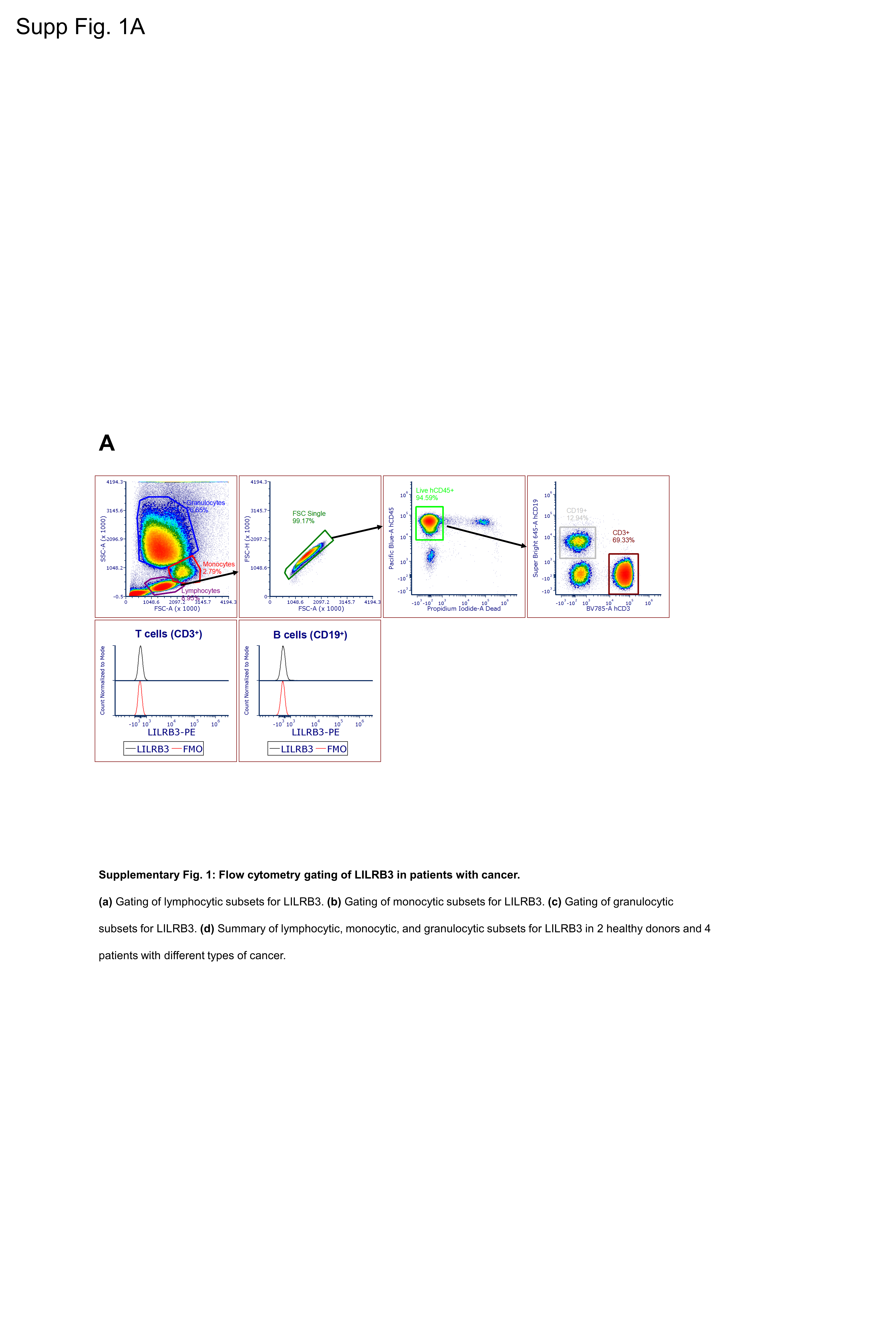
(a) Kaplan-Meier survival analysis of correlations between LILRB3 and overall survival of patients from the TCGA database. Patients were stratified with low levels of LILRB3 (blue) or high levels of LILRB3 (red) from TCGA subsets of pan-cancer, renal clear cell carcinoma (KIRC), glioblastoma (GBM), low-grade glioma (LGG), thymoma (THYM), and lung squamous cell carcinoma (LUSC). Statistical significance for overall survival was calculated by Mann-Whitney’s log-rank test. (b) Expression of LILRB3 on the cell surface of different subsets of lymphocytes, monocytes, and granulocytes in a representative human patient with melanoma by flow cytometry.
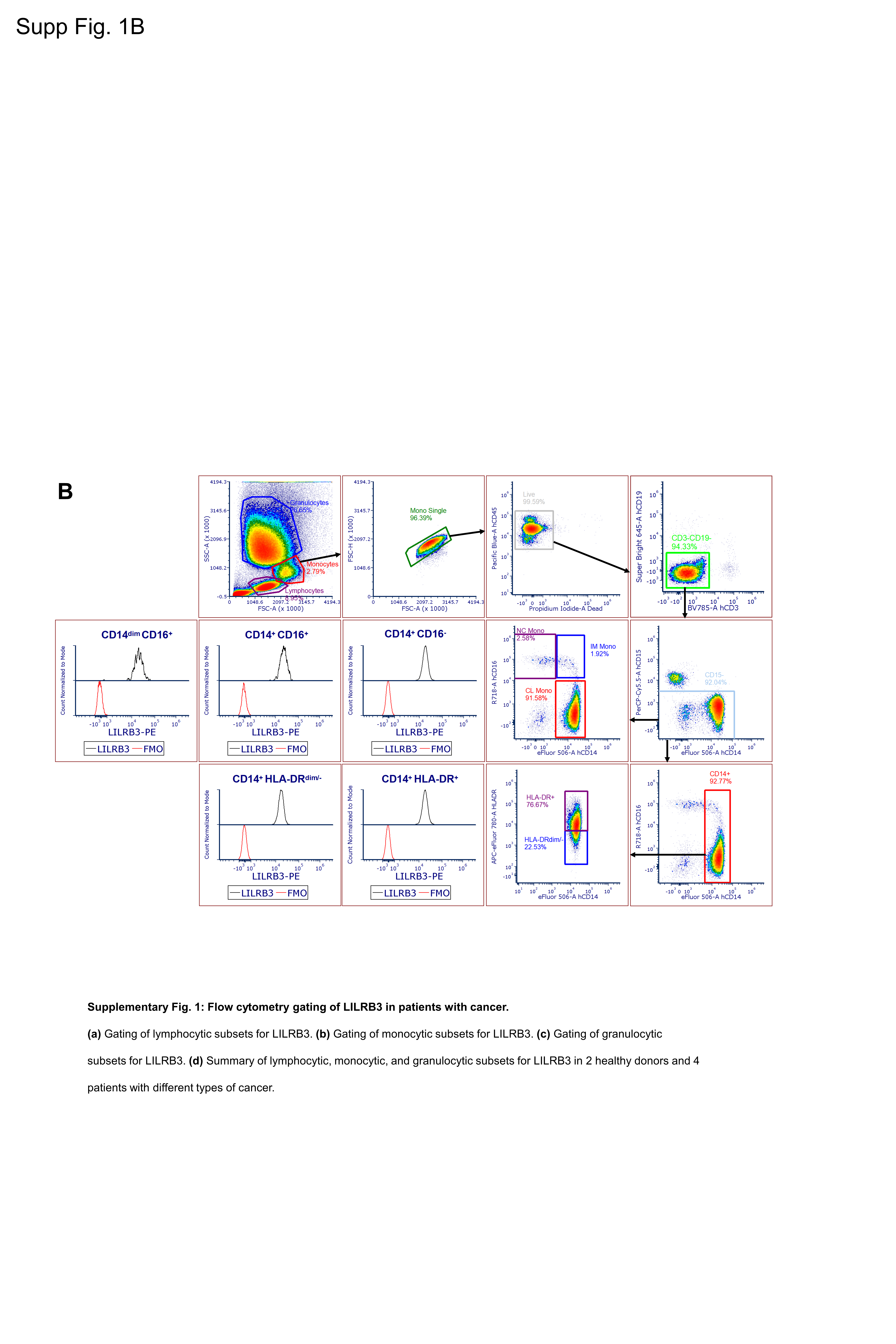
We analyzed peripheral blood samples from 2 healthy donors and 5 patients with solid cancers (including those with prostate cancer, melanoma, and renal cancer) to determine the expression pattern of LILRB3. To specifically stain LILRB3, we conjugated our anti-LILRB3 (21) with an Fc site-specific PE fluorophore via click chemistry. By flow cytometry, monocytes and granulocytes from both healthy donors and patients expressed LILRB3 with variation in expression levels while lymphocytes did not express LILRB3 (Fig. 1B, Supplementary Fig. S1A-1D). Further analysis showed that immunophenotypically-defined MDSCs in peripheral blood from patients with cancer (CD14+HLA-DRdim/−) (38) also expressed LILRB3.
Galectin-4 activates LILRB3 in a site-specific manner.
We developed chimeric receptor reporter cells (RC) for several LILRBs (14,16,18,22,24) as a sensitive system to detect the functional interaction between LILRBs and extracellular interacting proteins. MHC Class I (MHC-I) molecules (HLA-A, HLA-B, and HLA-G) and ApoE4 have been previously reported to bind LILRB3 (39,40). However, HLA-G (Fig. 2A) and all isoforms of ApoE (Fig. 2B), including ApoE4, did not activate LILRB3 reporter cells. In contrast, all ApoE isoforms induced the activation of LILRB4 reporter cells (Fig. 2C), consistent with prior observations (14,26).
Fig. 2∣. Galectin-4 activates LILRB3 in a site-specific manner.

(a) Percentages of indicated LILRB reporter cells activated by K562-Vec or K562-HLA-G cells. (b-c) Percentages of LILRB3 (b) or LILRB4 (c) reporter cells activated on recombinant coated ApoE or coated anti-LILRB3 (20 μg/mL). (d-h) Percentages of LILRB1 (d), LILRB2 (e), LILRB3 (f), LILRB4 (g), or LILRB5 (h) reporter cells activated on coated galectins (20 μg/mL). (i) Summary of LILRB reporter cells activated on coated galectins. (j) Percentages of LILRB3 reporter cells activated on coated or soluble galectin-4 or galectin-7 (20 μg/mL). (k) Percentages of LILRB31-443 or LILRB31-419 reporter cells activated on coated galectin-4 or anti-LILRB3 (20 μg/mL). Three technical replicates were performed for each condition and at least two independent experiments were performed for each condition. Error bars represent SEM and * indicates two-tailed student’s t-test p < 0.05.
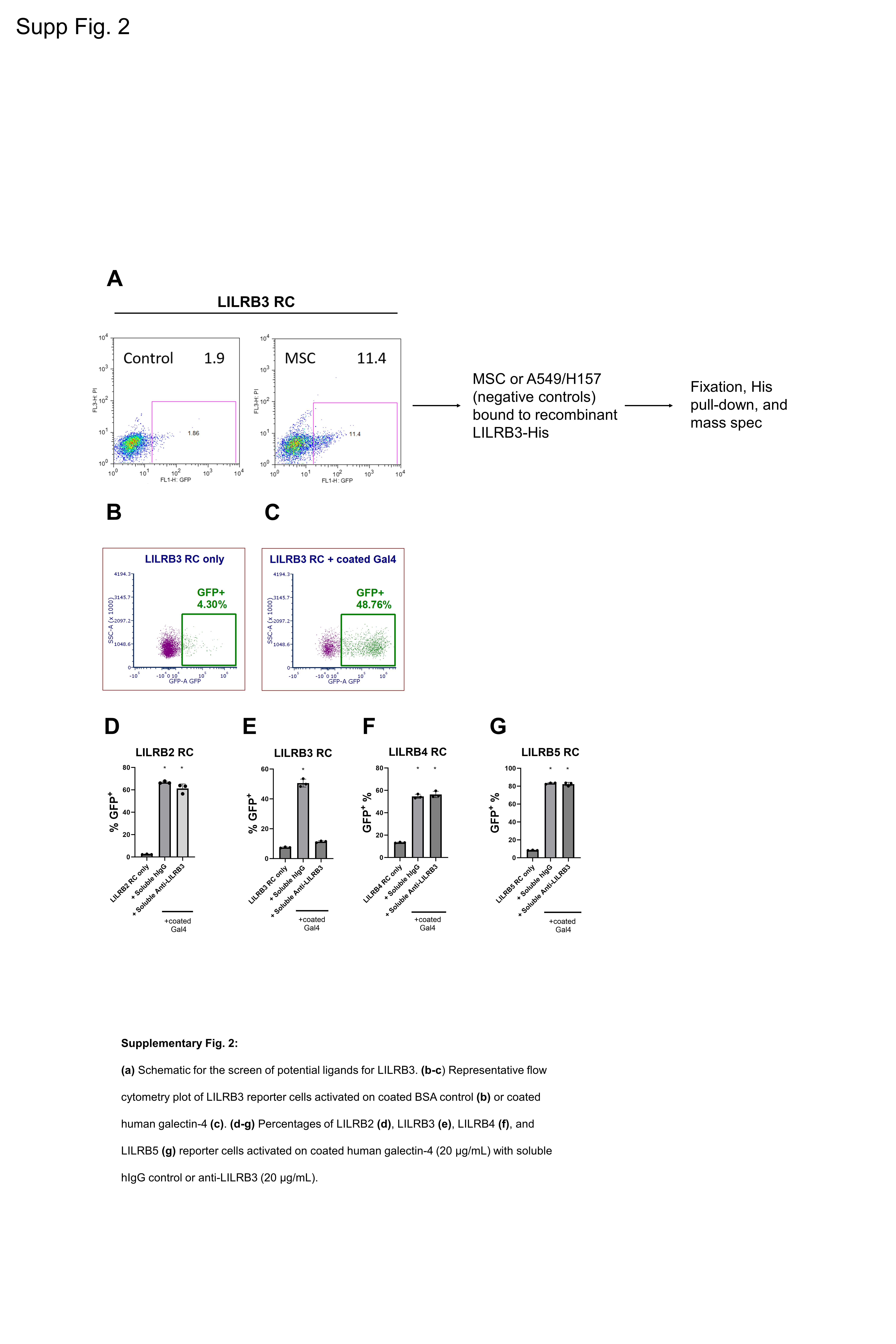
We found that mesenchymal stem cells (MSC) induced the activation of certain LILRB reporter cells (Supplementary Fig. S2A). This in turn, led us to discover that a number of galectins, which are galactoside-binding proteins (41), activate different LILRB reporter cells (Fig. 2D-I). Galectins have been reported to bind N-glycosylated asparagine residues or O-glycosylated serine and threonine residues (42). It has been observed that many cancer cells overexpress galectins, which can significantly increase their growth and metastasis. Galectins can be detected both intracellularly and extracellularly (41,43). Their presence in the extracellular space may be a consequence of secretion via the signal peptide–independent nonclassical pathway (43). In particular, we found that galectin-4 and galectin-7 activated LILRB3 reporter cells (Fig. 2F). Although both soluble and plates coated with human galectin-4 activated LILRB3, only plates coated with human galectin-7 and not soluble galectin-7 could effectively activate LILRB3 (Fig. 2J). Therefore, we focused on galectin-4 in our subsequent experiments.
To determine the binding region between galectin-4 and LILRB3, we generated mutant LILRB3 reporter cells with a truncated LILRB3 ECD. We found that plates coated with galectin-4 could not activate our LILRB3 reporter cells with a truncated membrane-proximal ECD. In contrast, the positive control, plates coated with anti-LILRB3, still activated those reporter cells (Fig. 2K). This suggests that the membrane-proximal segment of the ECD of LILRB3 is important for the interaction between galectin-4 and LILRB3.
Galectin-4 binds LILRB3 and induces downstream signaling in an ITIM-dependent manner.
We utilized several additional methods to confirm the binding between galectin-4 and LILRB3. Via co-immunoprecipitation, we found that LILRB3 interacts with human galectin-4 (Fig. 3A). Using an Octet Red Bio-Layer Interferometry (BLI) Assay, we determined the binding affinity of galectin-4 for LILRB3 to be 44 nM, which fits a 1:1 binding curve (Fig. 3B). Of note, the recombinant LILRB3 protein used in these binding assays is glycosylated due to its production from mammalian NS0 cells. Upon ligand binding, the tyrosines in the ITIMs of LILRB3 become phosphorylated (21). When using plates coated with HLA-DR to provide the initial signal for LILRB3 activation in THP1 cells overexpressing LILRB3 (44), additional plate coating with galectin-4, but not galectin-2, induced an increase in LILRB3-specific tyrosine phosphorylation (Fig. 3C). Furthermore, plates coated with galectin-4 induced tyrosine phosphorylation of LILRB3 in a time-dependent manner, peaking at 10 minutes (Fig. 3D). Using a system similar to the one that we previously described (21), we transfected SHP1, SHP2, Lyn, LILRB3-FLAG, and galectin-4 into 293T cells. When the ITIMs of LILRB3 were individually mutated into their inactive forms, we found that mutation of either the third or fourth ITIM of LILRB3 disrupted the ability of galectin-4 to elicit downstream SHP1 and pY signaling (Fig. 3E). Together, our results indicate that galectin-4 is an extracellular binding and activating protein of LILRB3.
Fig. 3∣. Galectin-4 binds LILRB3 and induces downstream signaling in an ITIM-dependent manner.

(a) Co-immunoprecipitation (Co-IP) demonstrates that recombinant Fc-tagged extracellular domain of LILRB3 (LILRB3-ECD) but not human IgG (hIgG) attached to Protein A Dynabeads interacts with recombinant human galectin-4 (hGal4). (b) Binding kinetics of His-tagged human galectin-4 to immobilized LILRB3-ECD were measured using biolayer interferometry and determined using a 1:1 binding curve. (c) Representative Co-IP of LILRB3-specific phosphotyrosine (pY) Western blot of THP1-LILRB3 lysates after 10 minutes on coated HLA-DR (20 μg/mL) and coated human galectin-2 (20 μg/mL) or human galectin-4 (20 μg/mL). (d) Representative Co-IP of LILRB3-specific pY Western blot of THP1-LILRB3 lysates after indicated time points with soluble LPS (200 ng/mL) and coated and soluble human galectin-4 (20 μg/mL). (e) Representative Co-IP of LILRB3-specific pY and SHP1 Western blot of 293T cells transfected with SHP1/SHP2/hGal4/Lyn and LILRB3-WT or LILRB3 ITIM-mutated plasmids after 48 hours. Three technical replicates were performed for each condition and at least two independent experiments were performed for each condition.
Anti-LILRB3 blocks Galectin-4 induced SHP1/SHP2 signaling through LILRB3.
The identification of galectins as extracellular activators of LILRB reporter cells provided us with a new tool to screen blocking antibodies against LILRBs. We screened a panel of fully human anti-LILRB3 that we previously generated (21) for their ability to potentiate or antagonize the activation of LILRB3 reporter cells by galectin-4 (Fig. 4A). Since anti-LILRB3 #1 could effectively antagonize the activation of LILRB3 reporter cells by galectin-4 (Fig. 4A), we used anti-LILRB3 #1 as “anti-LILRB3” in all subsequent assays. Consistent with our prior assays (21), anti-LILRB3 effectively antagonized the activation of LILRB3 reporter cells by galectin-7 (Figs. 4B). Therefore, we decided to use anti-LILRB3 for further experiments. This antibody was reengineered for this study and contains a mutated LALAPG-Fc fragment, thus lacking Fc effector functions (45). As a result, it is regarded as a pure LILRB3 blocking antibody. In a concentration-dependent manner, this LILRB3 blocking antibody abrogated LILRB3 reporter cell activation by galectin-4 with an IC50 of 1.55 nM (Fig. 4C). Using the reconstituted LILRB3 signaling pathway in 293T cells, we found that the anti-LILRB3 reduced galectin-4–induced tyrosine phosphorylation of LILRB3 and recruitment of SHP1 and SHP2 (Fig. 4D).
Fig. 4∣. Anti-LILRB3 antibody blocks Galectin-4 induced SHP1/SHP2 signaling through LILRB3.

(a) Percentages of LILRB3 reporter cells activated on coated human galectin-4 (20 μg/mL) with indicated soluble anti-LILRB3 antibodies (20 μg/mL). (b) Percentages of LILRB3 reporter cells activated on coated human galectin-7 (20 μg/mL) with soluble hIgG control or anti-LILRB3-1 (20 μg/mL). (c) Percentages of LILRB3 reporter cells activated on coated human Galectin-4 (20 μg/mL) with indicated concentrations of soluble anti-LILRB3-1. (d) Representative Co-IP of full-length LILRB3-specific pY/SHP1/SHP2 Western blot of 293T cells transfected with SHP1/SHP2/hGal4/LILRB3-WT/Lyn plasmids with soluble hIgG control or anti-LILRB3-1 antibody. Three technical replicates were performed for each condition and at least two independent experiments were performed for each condition. Error bars represent SEM and * indicates two-tailed student’s t-test p < 0.05.
Anti-LILRB3 inhibits human MDSC activity.
To investigate whether LILRB3 regulates the activity of immunosuppressive myeloid cells in patients with cancer, we collected immunophenotypically-defined MDSCs and autologous T cells from peripheral blood samples of patients with prostate cancer, melanoma, lung cancer, and breast cancer (Supplementary Table S4). Patient-derived enriched MDSCs effectively inhibited the proliferation of autologous T cells in a co-culture system consistent with previous literature (28) (Figs. 5A-F), suggesting that these are functional MDSCs. Anti-LILRB3 reversed the immunosuppressive activity of MDSCs and re-stimulated proliferation of autologous CD4+ and CD8+ T cells in the presence (Figs. 5C-D) or absence (Figs. 5E-F) of galectin-4 in a subset of patient samples. These results suggest that anti-LILRB3 could restore the proliferation of T cells in some patients with cancer (Table 1).
Fig. 5∣. Anti-LILRB3 antibody inhibits human MDSC activity.

(a-b) Representative histogram of 31 patients with cancer (total of 76 patients with cancer) shows that anti-LILRB3 attenuates MDSC suppressive functions on CD4+ (a) and CD8+ (b) T-cell proliferation in some patients with cancer as determined by flow cytometry. (c-d) Quantification of 4 individual patient samples from proliferating human CD4+ (c) and CD8+ (d) T cells cocultured with autologous MDSCs selected for patients with >10% proliferation with anti-LILRB3. Wells were coated with human galectin-4 (20 μg/mL) and cocultured cells were treated with hIgG control or anti-LILRB3 (20 μg/mL). (e-f) Quantification of 4 individual patient samples from proliferating human CD4+ (e) and CD8+ (f) T cells cultured with autologous MDSCs selected for patients with >10% proliferation with anti-LILRB3. Wells were not coated with human galectin-4 and cocultured cells were treated with hIgG control or anti-LILRB3 (20 μg/mL). (g-h) Median fluorescence intensity (MFI) change in CD86, CD163, and CD206 for isolated MDSCs treated with hIgG control or anti-LILRB3 (20 μg/mL) in patient samples with melanoma (g) or lung cancer (h). One to three replicates were performed for each condition and one experiment was performed for each condition. Error bars represent SEM and * indicates two-tailed student’s t-test p < 0.05.
Table 1:
Anti-LILRB3 inhibits the activity of MDSCs from some patients with cancer.
| Human cancer type | MDSC/T co-culture | MDSC culture | ||||
|---|---|---|---|---|---|---|
| No. of cases |
anti-LILRB3 efficacya |
% efficacy |
No. of cases |
anti-LILRB3 efficacyb |
% efficacy |
|
| Prostate cancer | 24 | 8 | 33 | N.A. | N.A. | N.A. |
| Melanoma | 41 | 18 | 44 | 30 | 4 | 13 |
| Lung cancer | 10 | 4 | 40 | 7 | 2 | 29 |
| Breast cancer | 1 | 1 | 100 | N.A. | N.A. | N.A. |
| Total Cases | 76 | 31 | 41 | 37 | 6 | 16 |
Increased T cell proliferation with anti-LILRB3 treatment.
Increased CD86 and decreased CD163 and CD206 with anti-LILRB3 treatment.
In parallel, we assessed the ability of anti-LILRB3 to inhibit the activity of immunosuppressive myeloid cells in culture with only MDSCs (28). LILRB3 blockade increased the expression of CD86, a marker of myeloid-cell activation, while it decreased the expression of immunosuppressive markers, CD163 and CD206, in a portion of patient samples with melanoma and lung cancer (13% and 29%, respectively) (Figs. 5G-H, Table 1). These findings indicate that LILRB3 is functionally expressed on MDSCs from patients with cancer, and that LILRB3 blockade can suppress the activity of immunosuppressive myeloid cells in a portion of patients with solid cancers.
Anti-LILRB3 ameliorates cancer development in vivo.
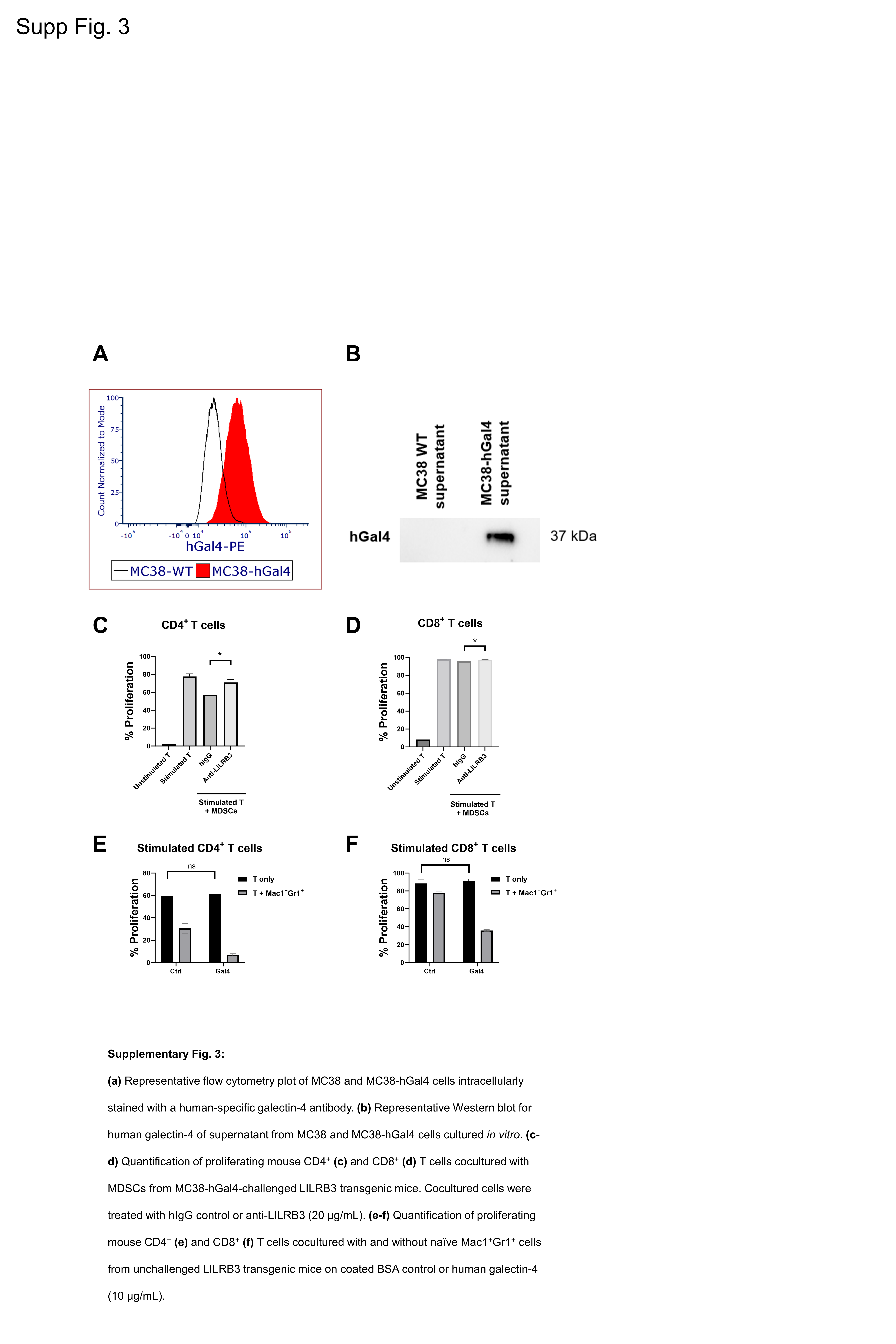
We previously developed myeloid-specific (LysM-Cre) LILRB3-transgenic C57BL/6 mice (21), which we used here to determine the efficacy of LILRB3 blockade in solid tumors. The LILRB3 blocking antibody effectively delayed tumor development following subcutaneous implantation of syngeneic B16 melanoma cells (Fig. 6A) but not that MC38 colon cancer cells (Fig. 6B). We found that although neither B16 nor MC38 cells expressed mouse galectin-7, B16 cells expressed mouse galectin-4 but MC38 cells did not (Fig. 6C). When we cultured LILRB3 reporter cells in plates coated with mouse galectin-4, we found that mouse galectin-4 activated LILRB3 reporter cells and anti-LILRB3 could blocked this activation (Fig. 6D). To test the functional significance of galectin-4 in LILRB3 activation, we stably expressed human galectin-4 on MC38 cells (MC38-hGal4) and implanted them into LILRB3 transgenic mice. We confirmed that MC38-hGal4 cells expressing human galectin-4 could secrete galectin-4 into the extracellular environment in vitro (Supplementary Fig. 3A-B). Similar to the situation with implanted B16 cells, anti-LILRB3 delayed MC38-hGal4 tumor development. We found that this effect was T-cell dependent since CD4 and CD8 T-cell depletion via anti-CD4 and anti-CD8 abrogated this effect (Fig. 6E).
Fig. 6∣. Anti-LILRB3 antibody ameliorates cancer development in vivo.

(a-b) Tumor progression of LILRB3 transgenic mice challenged with 5X105 B16 melanoma cells (a) or MC38 colon cancer cells (b) and treated with either hIgG control or anti-LILRB3. (c) Representative Western blot of B16 and MC38 whole cell lysates for mouse galectin-4, vinculin loading control, and mouse galectin-7. (d) Percentages of LILRB3 reporter cells activated on coated mouse galectin-4 (20 μg/mL) with hIgG control or anti-LILRB3 (20 μg/mL). (e) Tumor progression of LILRB3 transgenic mice challenged with 5X105 MC38-hGal4 and treated with hIgG control or anti-LILRB3, and a combination of anti-CD4 and anti-CD8 or isotype control. (f-g) Quantification of proliferating mouse CD4+ (f) or CD8+ (g) T cells cocultured with MDSCs from B16-challenged LILRB3 transgenic mice. Cocultured cells were treated with hIgG control or anti-LILRB3 (20 μg/mL). (h-i) Quantification of proliferating mouse CD4+ (h) or CD8+ (i) T cells cocultured with naïve Mac1+Gr1+ cells from unchallenged LILRB3 transgenic mice on coated human galectin-4 or BSA (10 μg/mL) and treated with hIgG control or anti-LILRB3 (20 μg/mL). Three technical replicates were performed for each condition and at least two independent experiments were performed for each condition. Error bars represent SEM and * indicates two-tailed student’s t-test p < 0.05. *** indicates two-tailed student’s t-test p < 0.0005.
To further explore the relationship between LILRB3 blockade and antitumor immune response, we established an ex vivo tumor model by coculturing MDSCs isolated from B16-challenged LILRB3 transgenic mice and syngeneic T cells from B16-challenged WT C57BL/6 mice. Similar to what we observed in certain samples from patients with cancer (Fig. 5), we found that LILRB3 blocking antibody partially rescued the proliferation of T cells inhibited by MDSCs (Fig. 6F-G, Supplementary Fig. 3C-D). Furthermore, we developed an in vitro assay where we cocultured naïve Mac1+Gr1+ cells from unchallenged LILRB3 transgenic mice with syngeneic CD3+ T cells from unchallenged WT C57BL/6 mice in the presence of BSA as a control or human galectin-4. Plates coated with galectin-4 protein induced naïve LILRB3+Mac1+Gr1+ cells to inhibit both CD4+ and CD8+ T-cell proliferation (Fig. 6H-6I, Supplementary Fig. 3E-F) and anti-LILRB3 ameliorated this effect. This result suggests that LILRB3 activation through galectin-4 induces immunosuppressive activity of myeloid cells that can be rescued by LILRB3 blockade. In summary, our results suggest that LILRB3-mediated signaling in immunosuppressive myeloid cells suppresses antitumor T-cell activity, and blocking LILRB3 signaling inhibits tumor development in a T cell–dependent manner.
Discussion
Although T-cell centered PD-1 and CTLA-4 immune checkpoint blockade has been successful in treating patients with cancer, most patients do not benefit from these monotherapies. Immunosuppressive myeloid cells are recognized as important elements in the TME that contribute to the inhibition of antitumor immune responses. LILRBs on these immunosuppressive myeloid cells have recently been recognized as critical myeloid checkpoint receptors, which may be targeted for cancer treatment. It was demonstrated that several LILRBs and LAIR1 may be attractive targets for cancer immunotherapy (9). Here, we report that galectin-4 is an extracellular binding partner that binds and activates LILRB3 signaling. In addition, we found that LILRB3 is functionally expressed on the immunosuppressive monocytic cells from some solid cancer patients. Importantly, we identified an LILRB3 blocking antibody that inhibits the activity of MDSCs and restores T-cell proliferation in some samples of patients with solid cancers in in vitro assays, and decreases tumor growth in our myeloid-specific LILRB3 transgenic mouse model in a T cell–dependent manner.
The ligands for LILRB3 are not clearly defined. Although it was previously reported that MHC-I molecules do not bind LILRB3 (46-48), a recent article suggested that MHC-I do bind LILRB3 (39). It was also shown that ApoE4 (but not ApoE2 or ApoE3) binds a variant of LILRB3 (Accension# XM_006726314.4) (40). Here, using LILRB3 chimeric reporter cells, we show that HLA-G and all isoforms of ApoE including ApoE4 do not activate LILRB3 (Accension# XM_006726314.4). By contrast, galectin-4 activates multiple variants of LILRB3 (Accension# XM_006726314.4, AAI04994.1, and XP_016885786). These results suggest that HLA-G and ApoE may bind LILRB3 with affinities that are unable to induce the functional conformational change required for LILRB3 activation in this system. Several galectins are considered to be cancer targets due to their aberrant expression in certain cancer types (49). Here, we demonstrate that some of these galectins can also activate LILRBs. Among these galectins, we show that galectin-4 and galectin-7 can activate LILRB3. Unlike the other inhibitory receptors in the LILRB family for which many ligands have been discovered, we believe that this is the first time that a potential binding partner for LILRB3 has been discovered that can activate downstream signaling events. Concurrently, we also discovered that galectin-4 and galectin-7 can bind and activate other LILRBs in our reporter cell systems, suggesting that galectins are not a specific binding partner solely for LILRB3. Since multiple LILRB family members are present on MDSCs, we hypothesize that aberrantly expressed galectins from cancer cells can potentially crosslink and activate multiple LILRBs to amplify the immunosuppressive effect of MDSCs.
Since LILRB3 signaling in human and transgenic mouse myeloid cells promotes their immunosuppressive activity, it is desirable to develop LILRB3-specific blocking antibodies that antagonize cancer growth by suppressing MDSC activity and enhancing antitumor T-cell functions. The outcomes of the cultures of MDSCs isolated from patients and those of MDSCs and autologous T cells together suggests that LILRB3 may be involved in the TME of some, but not all patients with cancer. Therefore, the immunosuppressive activity of LILRB3 signaling may be tumor-type and patient-specific. It is also interesting to note that in some patient samples, anti-LILRB3 were able to activate T cells without the presence of recombinant galectin-4. This suggests that endogenous activators for LILRB3, including galectin-4 and galectin-7, may already exist in the co-culture systems. In addition, the LILRB3 blocking antibody partially rescued the proliferation of autologous T cells in some of the MDSC/T-cell cocultures, suggesting that other LILRBs or LAIR-1, which is known to be expressed on MDSCs, may also be important in regulating MDSC activity. This is concordant with our recent report that LAIR1 blockade could suppress the activity of MDSCs and stimulate proliferation of T cells from patients with cancer (28).
The ability of anti-LILRB3 to reactivate T cells in some patient samples suggests that LILRB3 has a unique role in TME that is separate but also complementary to those of other LILRBs. Although the same myeloid cells can express multiple LILRBs, we hypothesize that each LILRB has a specific function that is influenced by a number of factors, including: 1) distinct sets of extracellular activators/ligands that crosslink various LILRBs in various contexts; 2) unique intracellular signaling domains of LILRBs (e.g., the distinct motif in LILRB3 for TRAF2/cFLIP signaling (21) and the functionally irreplaceable signaling domains of individual LILRBs (23)); and 3) a variety of extrinsic factors and transcription factors that regulate the expression of LILRBs that contribute to different expression dynamics (e.g., vitamin D3 upregulates LILRB4 but not LILRB3 (50)). Therefore, LILRB3 blockade may prove to be an effective new approach for immunotherapy of solid cancers.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Synopsis.
LILRB3 is an inhibitory receptor expressed by myeloid cells. The authors show galectin-4 and galectin-7 bind LILRB3, promoting the immunosuppressive function of myeloid cells. Blocking LILRB3 reduces immunosuppression and promotes antitumor immunity, suggesting a potential new immunotherapeutic approach.
Financial support:
This work was supported by the National Cancer Institute (R01CA248736, R01CA263079, 5P30CA142543, and Lung Cancer SPORE Development Research Program (DRP)), the Department of Defence (ME190050), the Cancer Prevention and Research Institute of Texas (RP220032, RP15150551, RP190561), the Welch Foundation (AU-0042-20030616), the Leukemia & Lymphoma Society (6629-21), and Immune-Onc Therapeutics, Inc. (Sponsored Research Grant #111077).
Footnotes
Conflict of interest statement: Authors R.H., X.L., J.K., M.D., H.C., G.W., X.G., J.X., Y.H., S.J., N.Z., Z.A., and C.C.Z. are listed as inventors in LILRB relevant patent applications that were exclusively licensed to Immune-Onc Therapeutic, Inc. by the Board of Regents of the University of Texas System. Authors Z.A., N.Z., and C.C.Z. hold equity in and have Sponsored Research Agreements with Immune-Onc Therapeutics, Inc. University of Texas has a financial interest in Immune-Onc in the form of equity and licensing.
References
- 1.Barry ST, Gabrilovich DI, Sansom OJ, Campbell AD, Morton JP. Therapeutic targeting of tumour myeloid cells. Nat Rev Cancer 2023;23(4):216–37 doi 10.1038/s41568-022-00546-2. [DOI] [PubMed] [Google Scholar]
- 2.Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front Oncol 2021;11:610303 doi 10.3389/fonc.2021.610303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samaridis J, Colonna M. Cloning of novel immunoglobulin superfamily receptors expressed on human myeloid and lymphoid cells: structural evidence for new stimulatory and inhibitory pathways. Eur J Immunol 1997;27(3):660–5 doi 10.1002/eji.1830270313. [DOI] [PubMed] [Google Scholar]
- 4.Kang X, Kim J, Deng M, John S, Chen H, Wu G, et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016;15(1):25–40 doi 10.1080/15384101.2015.1121324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirayasu K, Arase H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. Journal of human genetics 2015;60(11):703–8 doi 10.1038/jhg.2015.64. [DOI] [PubMed] [Google Scholar]
- 6.Deng M, Chen H, Liu X, Huang R, He Y, Yoo B, et al. Leukocyte immunoglobulin-like receptor subfamily B (LILRB): therapeutic targets in cancer Antibody Therapeutics 2021;4(1):16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother 2017;66(8):1079–87 doi 10.1007/s00262-017-2023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas R, Matthias T, Witte T. Leukocyte immunoglobulin-like receptors as new players in autoimmunity. Clin Rev Allergy Immunol 2010;38(2-3):159–62 doi 10.1007/s12016-009-8148-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhang CC. A perspective on LILRBs and LAIR1 as immune checkpoint targets for cancer treatment. Biochem Biophys Res Commun 2022;633:64–7 doi 10.1016/j.bbrc.2022.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Trowsdale J, Jones DC, Barrow AD, Traherne JA. Surveillance of cell and tissue perturbation by receptors in the LRC. Immunol Rev 2015;267(1):117–36 doi 10.1111/imr.12314. [DOI] [PubMed] [Google Scholar]
- 11.Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol 2018;19(1):76–84 doi 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suciu-Foca N, Feirt N, Zhang QY, Vlad G, Liu Z, Lin H, et al. Soluble Ig-like transcript 3 inhibits tumor allograft rejection in humanized SCID mice and T cell responses in cancer patients. J Immunol 2007;178(11):7432–41. [DOI] [PubMed] [Google Scholar]
- 13.Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest 2018;128(12):5647–62 doi 10.1172/jci97570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018;562(7728):605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng J, Umikawa M, Cui C, Li J, Chen X, Zhang C, et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 2012;485(7400):656–60 doi 10.1038/nature11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang X, Lu Z, Cui C, Deng M, Fan Y, Dong B, et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat Cell Biol 2015;17(5):665–77 doi 10.1038/ncb3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.John S, Chen H, Deng M, Gui X, Wu G, Chen W, et al. A Novel Anti-LILRB4 CAR-T Cell for the Treatment of Monocytic AML. Mol Ther 2018;26(10):2487–95 doi 10.1016/j.ymthe.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng M, Lu Z, Zheng J, Wan X, Chen X, Hirayasu K, et al. A motif in LILRB2 critical for Angptl2 binding and activation. Blood 2014;124(6):924–35 doi 10.1182/blood-2014-01-549162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 2019;19(10):568–86 doi 10.1038/s41568-019-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anami Y, Deng M, Gui X, Yamaguchi A, Yamazaki CM, Zhang N, et al. LILRB4-targeting Antibody-Drug Conjugates for the Treatment of Acute Myeloid Leukemia. Mol Cancer Ther 2020;19(11):2330–9 doi 10.1158/1535-7163.MCT-20-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu G, Xu Y, Schultz RD, Chen H, Xie J, Deng M, et al. LILRB3 supports acute myeloid leukemia development and regulates T-cell antitumor immune responses through the TRAF2-cFLIP-NF-kappaB signaling axis. Nat Cancer 2021;2(11):1170–84 doi 10.1038/s43018-021-00262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gui X, Deng M, Song H, Chen Y, Xie J, Li Z, et al. Disrupting LILRB4/APOE Interaction by an Efficacious Humanized Antibody Reverses T-cell Suppression and Blocks AML Development. Cancer Immunol Res 2019;7(8):1244–57 doi 10.1158/2326-6066.cir-19-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Deng M, Huang F, Jin C, Sun S, Chen H, et al. LILRB4 ITIMs mediate the T cell suppression and infiltration of acute myeloid leukemia cells. Cell Mol Immunol 2020;17(3):272–82 doi 10.1038/s41423-019-0321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Chen Y, Deng M, John S, Gui X, Kansagra A, et al. Antagonistic anti-LILRB1 monoclonal antibody regulates antitumor functions of natural killer cells. J Immunother Cancer 2020;8(2) doi 10.1136/jitc-2019-000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim A, Han CJ, Driver I, Olow A, Sewell AK, Zhang Z, et al. LILRB1 Blockade Enhances Bispecific T Cell Engager Antibody-Induced Tumor Cell Killing by Effector CD8(+) T Cells. J Immunol 2019;203(4):1076–87 doi 10.4049/jimmunol.1801472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paavola KJ, Roda JM, Lin VY, Chen P, O'Hollaren KP, Ventura R, et al. The Fibronectin-ILT3 Interaction Functions as a Stromal Checkpoint that Suppresses Myeloid Cells. Cancer Immunol Res 2021. doi 10.1158/2326-6066.cir-21-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos MIP, Tian L, de Ruiter EJ, Song C, Paucarmayta A, Singh A, et al. Cancer immunotherapy by NC410, a LAIR-2 Fc protein blocking human LAIR-collagen interaction. Elife 2021;10 doi 10.7554/eLife.62927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie J, Gui X, Deng M, Chen H, Chen Y, Liu X, et al. Blocking LAIR1 signaling in immune cells inhibits tumor development. Front Immunol 2022;13:996026 doi 10.3389/fimmu.2022.996026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh L, Muise ES, Bhattacharya A, Grein J, Javaid S, Stivers P, et al. ILT3 (LILRB4) Promotes the Immunosuppressive Function of Tumor-Educated Human Monocytic Myeloid-Derived Suppressor Cells. Mol Cancer Res 2021;19(4):702–16 doi 10.1158/1541-7786.MCR-20-0622. [DOI] [PubMed] [Google Scholar]
- 30.Tedla N, Bandeira-Melo C, Tassinari P, Sloane DE, Samplaski M, Cosman D, et al. Activation of human eosinophils through leukocyte immunoglobulin-like receptor 7. Proc Natl Acad Sci U S A 2003;100(3):1174–9 doi 10.1073/pnas.0337567100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coxon CH, Geer MJ, Senis YA. ITIM receptors: more than just inhibitors of platelet activation. Blood 2017;129(26):3407–18 doi 10.1182/blood-2016-12-720185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bashirova AA, Apps R, Vince N, Mochalova Y, Yu XG, Carrington M. Diversity of the human LILRB3/A6 locus encoding a myeloid inhibitory and activating receptor pair. Immunogenetics 2014;66(1):1–8 doi 10.1007/s00251-013-0730-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sloane DE, Tedla N, Awoniyi M, Macglashan DW, Jr., Borges L, Austen KF, et al. Leukocyte immunoglobulin-like receptors: novel innate receptors for human basophil activation and inhibition. Blood 2004;104(9):2832–9 doi 10.1182/blood-2004-01-0268. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, van Woudenbergh E, Zhu J, Heck AJR, van Kessel KPM, de Haas CJC, et al. The Orphan Immune Receptor LILRB3 Modulates Fc Receptor-Mediated Functions of Neutrophils. J Immunol 2020;204(4):954–66 doi 10.4049/jimmunol.1900852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Zheng Y, Ning M, Li T. KLRD1, FOSL2 and LILRB3 as potential biomarkers for plaques progression in acute myocardial infarction and stable coronary artery disease. BMC Cardiovasc Disord 2021;21(1):344 doi 10.1186/s12872-021-01997-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of Susceptibility Loci in IL6, RPS9/LILRB3, and an Intergenic Locus on Chromosome 21q22 in Takayasu Arteritis in a Genome-Wide Association Study. Arthritis Rheumatol 2015;67(5):1361–8 doi 10.1002/art.39035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeboah M, Papagregoriou C, Jones DC, Chan HTC, Hu G, McPartlan JS, et al. LILRB3 (ILT5) is a myeloid cell checkpoint that elicits profound immunomodulation. JCI Insight 2020;5(18) doi 10.1172/jci.insight.141593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150 doi 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayukawa S, Kamoshita N, Nakayama J, Teramoto R, Pishesha N, Ohba K, et al. Epithelial cells remove precancerous cells by cell competition via MHC class I-LILRB3 interaction. Nat Immunol 2021;22(11):1391–402 doi 10.1038/s41590-021-01045-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Wang Y, Huang G, Yang M, Zhu Y, Jin C, et al. LilrB3 is a putative cell surface receptor of APOE4. Cell Res 2023;33(2):116–30 doi 10.1038/s41422-022-00759-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao ZQ, Guo XL. The role of galectin-4 in physiology and diseases. Protein Cell 2016;7(5):314–24 doi 10.1007/s13238-016-0262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimitroff CJ. Galectin-Binding O-Glycosylations as Regulators of Malignancy. Cancer Res 2015;75(16):3195–202 doi 10.1158/0008-5472.CAN-15-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao US, Rao PS. Surface-bound galectin-4 regulates gene transcription and secretion of chemokines in human colorectal cancer cell lines. Tumour Biol 2017;39(3):1010428317691687 doi 10.1177/1010428317691687. [DOI] [PubMed] [Google Scholar]
- 44.Cella M, Dohring C, Samaridis J, Dessing M, Brockhaus M, Lanzavecchia A, et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J Exp Med 1997;185(10):1743–51 doi 10.1084/jem.185.10.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J Biol Chem 2017;292(9):3900–8 doi 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen RL, Raine T, Haude A, Trowsdale J, Wilson MJ. Leukocyte receptor complex-encoded immunomodulatory receptors show differing specificity for alternative HLA-B27 structures. J Immunol 2001;167(10):5543–7 doi 10.4049/jimmunol.167.10.5543. [DOI] [PubMed] [Google Scholar]
- 47.Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A, et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol 2011;186(5):2990–7 doi 10.4049/jimmunol.1003078. [DOI] [PubMed] [Google Scholar]
- 48.Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol 2003;4(9):913–9 doi 10.1038/ni961. [DOI] [PubMed] [Google Scholar]
- 49.Kapetanakis NI, Busson P. Galectins as pivotal components in oncogenesis and immune exclusion in human malignancies. Front Immunol 2023;14:1145268 doi 10.3389/fimmu.2023.1145268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rochat MK, Ege MJ, Plabst D, Steinle J, Bitter S, Braun-Fahrlander C, et al. Maternal vitamin D intake during pregnancy increases gene expression of ILT3 and ILT4 in cord blood. Clin Exp Allergy 2010;40(5):786–94 doi 10.1111/j.1365-2222.2009.03428.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files or from the corresponding author upon reasonable request.