

Abstract
Purpose:
We sought to delineate a multisystem disorder caused by recessive CRELD1 variants.
Methods:
The impact of CRELD1 variants was characterized through an international collaboration utilizing next generation DNA sequencing, gene knockdown and protein overexpression in Xenopus tropicalis, and in vitro analysis of patient immune cells.
Results:
Biallelic variants in CRELD1 were found in 18 participants from 14 families. Affected individuals displayed an array of phenotypes involving developmental delay, early-onset epilepsy, and hypotonia, with about half demonstrating cardiac arrhythmias and some experiencing recurrent infections. Most harbored a frameshift in trans with a missense allele, with one recurrent variant, p.(Cys192Tyr), identified in 10 families. X. tropicalis tadpoles with creld1 knockdown displayed developmental defects along with increased susceptibility to induced seizures compared to controls. Additionally, human CRELD1 harboring missense variants from affected individuals had reduced protein function, indicated by a diminished ability to induce craniofacial defects when overexpressed in X. tropicalis. Finally, baseline analyses of peripheral blood mononuclear cells showed similar proportions of immune cell subtypes in patients as compared to healthy donors.
Conclusion:
This patient cohort combined with experimental data provide evidence of a multisystem clinical syndrome mediated by recessive variants in CRELD1.
Keywords: CRELD1, developmental delay, epilepsy, hypotonia
INTRODUCTION
The Cysteine-Rich with Epidermal Growth Factor (EGF)-Like Domains 1 (CRELD1) gene (OMIM ID: 607170), located on the short arm of human chromosome 3, was first identified in 20021. Isoform 2 (NP_056328.3), the predominantly expressed form of CRELD1, is a highly conserved transmembrane protein containing a distinguishing tryptophan (W)- and glutamate (E)-rich WE domain, two EGF-like domains, two additional calcium-binding EGF-like (cbEGF-like) domains, and two carboxy-terminal Type III transmembrane domains1–3. The protein is localized primarily at the endoplasmic reticulum (ER) with evidence that it is found specifically at ER-mitochondria contact points, but it is also found at the cell membrane2–4. The EGF-like domains are particularly notable for their characteristic cysteine residues that form disulfide bonds that can contribute both to 3-dimensional protein structure as well as to protein redox activity5.
In 2003, CRELD1 was the first human gene to be identified as a susceptibility gene for atrioventricular (AV) septal defects, with heterozygous variants found in three unrelated individuals6. Since then, multiple heterozygous variants in CRELD1 have been linked to an increased risk of both nonsyndromic AV septal defects and to the occurrence of AV septal defects in Down syndrome, though penetrance of the phenotype has been incomplete with multiple unaffected individuals also carrying suspected susceptibility variants6–12. Recent work has also noted an association of rare CRELD1 variants with bicuspid aortic valves in Turner syndrome13. CRELD1 has been shown to be necessary for proper control of nuclear factor of activated T-cells (NFAT) and calcineurin signaling in the setting of endocardial and myocardial development3. CRELD1 also appears to interact with vascular endothelial growth factor-A (VEGFA) to modulate the morphogenesis of the embryonic AV canal10. Consistent with these findings, Creld1 knockout mouse embryos are grossly normal in appearance but die between E11.0-E11.5 with defective cardiac development3. Mice with conditional knockout of Creld1 specifically in the myocardium also display early postnatal lethality and have myocardial hypoplasia14.
CRELD1 has additionally been shown to have a role in the immune system2. Amongst its many physiological roles, the NFAT signaling pathway controls various aspects of the immune system, including T lymphocyte development and activation, IL-2 gene activation, and components of the allergy response15. CRELD1 has been shown to modulate NFAT and Beta-catenin pathways, impacting noncanonical and canonical Wnt signaling, as well as other pathways intersecting development and immunology such as PI3K/Akt and mTOR2. A population cohort of individuals with low CRELD1 expression was demonstrated to have low naïve CD4+ T cells counts, a finding reflective of the signatures from T cell-specific Creld1 conditional knockout mice, which had reduced T cell proliferation, increased T cell apoptosis, and reduced T cell numbers, suggesting a disruption in lymphocyte homeostasis2.
During development, CRELD1 displays prominent expression in the developing brain, heart, branchial arches, and limb buds, and in adult tissues there is high expression in the brain, heart, and skeletal muscle, suggesting other potentially important functional roles outside of the cardiac and immune systems1. Recent work showed that deletion of the only Creld gene found in Drosophila resulted in a locomotion defect in flies that was rescued by specific overexpression of wild type Creld in the nervous system4. The authors further demonstrated accumulation and elongation of mitochondria, impairment of mitochondrial activity, and disruption of reactive oxygen species homeostasis, which they linked to Creld localization to ER-mitochondrial contact points.
Despite these data, no highly penetrant monogenic phenotype has been established for CRELD1. Here we report 18 individuals from 14 unrelated families affected by deleterious biallelic recessive variants in CRELD1. Most presented with early-onset neurodevelopmental features, most notably hypotonia and epilepsy, with developmental plateauing and slowly progressive non-neurologic medical complexities in survivors, including cardiac rhythm disturbances and frequent infections. We provide both in vitro and in vivo functional evidence to support the pathogenicity of these patient variants, establishing CRELD1 variants as a cause of a novel multisystem genetic disorder in humans.
METHODS
PATIENT RECRUITMENT
Individuals were referred to this cohort from the United States, Canada, and the United Kingdom. An initial group of patients (n=10) was identified when they all underwent clinical DNA sequencing through the same laboratory, GeneDx. The remaining cohort was assembled via communication through Matchmaker Exchange16 and came from multiple clinical and undiagnosed genetic disease programs: individual providers with in the United Kingdom’s NHS (n=2); Deciphering Developmental Disorders (n=1), The Yale Pediatric Genomics Discovery Program (n=1), the NIH Neuromuscular and Neurogenetic Disorders of Childhood Section (n=1), The Undiagnosed Diseases Network (n=1), The 100,000 Genomes Project (n=1), and the Epilepsy Neurogenetics Initiative at the Children’s Hospital of Philadelphia (n=1). Clinical phenotypingwas done through review of medical records at each patient’s local site.
DNA SEQUENCING AND ANALYSIS
Clinical sequencing for probands in each family was performed through genome sequencing (GS) or exome sequencing (ES) using previously reported protocols from the sites noted above17–21. Variants in CRELD1 were identified through clinical diagnostic analysis or by using Institutional Review Board-approved research protocols (Supplementary Table S1). Sequencing of additional family members was done using GS, ES, or targeted single gene sequencing.
PHOTOGRAPHS, RADIOLOGY IMAGING AND HISTOLOGY
Patient photographs were provided by family members along with consent to publish identifying images. Selected frames from clinical magnetic resonance and ultrasound imaging studies were extracted and presented without additional processing. For microscopy, frozen tissue samples were obtained from the deltoid muscle during autopsy and processed in accordance with standard clinical pathology methodologies for hematoxylin and eosin staining, or for ATPase staining with pre-incubation at either pH 9.4 or pH 4.1 to distinguish between Type 1 and Type 2 muscle fibers, or for electron microscopy.
XENOPUS TROPICALIS KNOCKDOWN AND SEIZURE SCREENING
X. tropicalis were housed and cared for in our aquatics facility according to established protocols approved by the Yale IRB-Institutional Animal Care and Use Committee (IACUC). Embryos were produced by in vitro fertilization and raised to appropriate stages in 1/9X Modified Ringer’s media (MR). Creld1 was targeted for knockdown using morpholino as previously described22 with creld1-AUG-MO (AACATTCGGCGTGACATACCCATAG, GeneTools) injected at the 1-cell stage at 30ng/embryo, and standard control MO (CCTCTTACCTCAGTTACAATTTATA, GeneTools) injected at the same stage and dose as a control. Alternatively, 1-cell stage embryos were injected with sgRNAs directed against creld1. 400pg/embryo of sgRNAs targeting either TGACCGTCTATGGGAGCCTGTGG or GTGCTACGAAATACTCACAGAGG were injected along with 1.6ng of Cas9 protein (PNAbio). Embryos were genotyped for CRISPR efficiency using the following primers:
CRISPR1 F CTCAGCGTTCAGTGACCAGT
CRISPR1 R AAGGCAAACTTCCCCTTGAT
CRISPR2 F TGGCACACGGTCACTATCAT
CRISPR2 R TTTGGCTCCTGGGCTACTTA
Sanger trace files were obtained from PCR products and submitted to ICE (Synthego)23. Embryos were raised at 25 degrees Celsius in 1/9X MR and gross morphology was assessed using light microscopy at stage 22 and again at stage 42. For seizure screening, groups of two to three stage 42 tadpoles were transferred to individual wells in a 48 well plate. To model seizures we used pilocarpine, a well-established chemical convulsant for modeling epilepsy in animals24,25. A minimally phenotypic dose of pilocarpine (50mM) was added to media in individual wells in a 48 well plate, and tadpoles were monitored every 30 seconds over a 30-minute time period to determine seizure behavior as previously described18.
CRELD1 PATIENT VARIANT ASSAY USING X. TROPICALIS
Human CRELD1 mRNA was generated from the hORFeome-derived gateway pENTR plasmid HsCD00080555 (DNASU) after LR cloning. Primers to introduce variants were designed using NEBaseChanger, and variants were created using Q5 Site directed mutagenesis (NEB). Forward (F) and Reverse (R) primers utilized were as follows:
p.(Cys192Tyr): (F) GGTGAGGCCTATGGCCAGTGTG; (R) CCCGTAGCCGGCTTGGCA
p.(Cys262Arg): (F) TGACCAATTCCGCGTGAACACTGAG; (R) GCTCCACAGTTGGCTCCC
p.(Met369Val): (F) GCTGCAGCAGGTGTTCTTTGG; (R) ACCACCAACTCGTCTTCTG
p.(Asp386Asn): (F) TGCTAAGGGCAACTTGGTGTTCACCG; (R) GCCAGCGTGGCCAGTGCA
p.(Thr380Met): (F) GCACTGGCCAtGCTGGCTGCT; (R)
ACAGATGATGATGCCAAAGAACATCTGCTG
p.(Ala391Pro): (F) GGTGTTCACCcCCATCTTCATTGGG; (R) AAGTCGCCCTTAGCAGCC
Variant sequences were verified in pENTR before LR cloning (ThermoFisher) into pDEST. mRNA was produced by linearizing plasmids with KpnI and using the sp6 mMessage mMachine kit (ThermoFisher). mRNA was precipitated using LiCl and injected at the one-cell stage at 100pg/embryo along with mem-GFP (250pg/embryo). Embryos were sorted for fluorescence at 20 hours post fertilization and raised at 25 degrees Celsius until stage 42. Tadpoles were fixed in 4% PFA, washed in PBS, and imaged in an agarose mold using a Nikon dissecting microscope. Tadpole head area was measured in Fiji using the tracing tool, and the area in number of pixels was recorded. The straight line tool was used to measure the interocular distance (from center of left eye to center of right eye) and recorded in pixels. Data were graphed using Prism and unpaired Welch’s t tests were used to determine statistical significance.
ANALYSIS OF PATIENT-DERIVED LYMPHOCYTES
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood from by Ficoll-Paque PLUS (GE Healthcare) or Lymphoprep (STEMCELL Technologies) density gradient centrifugation. PBMCs were subsequently washed twice in phosphate buffered saline (PBS) and resuspended in complete RPMI 1640 (cRPMI) medium (Lonza) containing 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/ml each of penicillin and streptomycin (Invitrogen). PBMCs were then stored at 106 cells/ml in 10% dimethyl sulfoxide (DMSO) in FBS and stored in −80 °C overnight before further storage in liquid nitrogen. For controls we extracted PBMCs from sex and age-matched (+/− one year) healthy donors. Flow cytometric analyses were carried out on a MACSQuant10 Analyzer and data was analyzed using FlowJo and Prism 9 software programs, plotting mean and standard deviation. Flow antibodies (BioLegend) were anti-CD3, anti-CD4, anti-CD8, anti-CD19, anti-CD14, anti-CCR7, and anti-CD45RA (see Supplementary Figure 1 for antibody details and gating strategies).
RESULTS
CLINICAL PRESENTATIONS
Clinical information for all 18 individuals from 14 unrelated families in this study is summarized in Table 1 and comprehensive phenotyping is provided in Supplementary Table 1.
Table 1.
Genomic and clinical data for participants with recessive CRELD1 variants
| Family | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | F13 | F14 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||
| Individual | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | P13 | P14 | P15 | P16 | P17 | P18 |
|
| ||||||||||||||||||
| Ancestry, Race and
Ethnicity Reported by (when known) CRELD1 variants (hg19) NM_015513; NP_056328; GRCh37 |
Puerto Rican and Spanish (Canary Islands);
European (Irish, other) Self-reported |
Italian,Russian/Ukranian, Northern Irish,
Irish, German; Scottish, Irish, English,
Netherlands Self-reported |
Swedish and Mexican; German and other
European Self-reported |
German, mixed White European, American
Indian Self-reported |
German, Irish, Spanish; German, Prussian | NR | Irish; European Reported in medical record |
Ashkenazi Jewish; English,
Irish Self-reported |
White Reported in medical record |
White
British Self-reported |
White | French, English; Polish,
European. Self-reported |
White
British Self-reported |
White Self-reported |
||||
| c.196dup; p.(Asp66Glyfs*22); g.9976540dup |
c.254_257del; p.(Asp85Valfs*6); g.9976598_9976601del |
c.959del; p.(Gln320Argfs*25); g.9985110del |
c.1128_1129del; p.(Ala377Thrfs*7); g.9986128_9986129del |
c.959del; p.(Gln320Argfs*25); g.9985110del |
c.959del; p.(Gln320Argfs*25); g.9985110del |
c.959del; p.(Gln320Argfs*25); g.9985110del |
c.1128_1129del p.(Ala377Thrfs*7); g.9986128_9986129del |
c.959del; p.(Gln320Argfs*25); g.9985110del |
c.1128_1129del; p.(Ala377Thrfs*7); g.9986128_9986129del |
c.1171G>C; p.(Ala391Pro); g.9986171G>C |
c.959del; p.(Ala377Thrfs*7); g.9986128_9986129del |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.959del; p.(Gln320Argfs*25); g.9985110del |
|||||
| c.575G>A; p.(C192Y); g.9982648G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.1156G>A; p.(Asp386Asn); g.9986156G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.784T>C; p.(Cys262Arg); g.9984547T>C |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.1105A>G; p.(Met369Val); g.9986105A>G |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.1139C>T; p.(Thr380Met); g.9986139C>T |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
c.575G>A; p.(Cys192Tyr); g.9982648G>A |
|||||
| Sex/age (notation if deceased) | F/5 y | M/4 y | F/21 mo (d) | F/14 y | M/7 y | M/19 mo (d) | F/21 y | F/4 y | F/17 mo (d) | M/7 y | M/13 y | F/12 y (d) | M/25 mo | F/31 mo (d) | M/25 y | M/6 y | M/32 y | F/17 y (d) |
| Pregnancy/birth complications | –/– | –/– | –/C-section breech | NR/– | Oligohydramnios/28 day NICU stay | Oligohydramnios/2hrs of NICU care | –/repeat C-section | NR/– | NR/NR | GDM/nuchal cord, vacuum assisted delivery | Decreased fetal movement/C-section for breech | –/C-section for failure to progress | Polyhydramnios/- | –/– | -/vacuum assisted delivery | –/– | –/needed support only few mins | NR/NR |
| Neonatal complications | FTT | – | – | – | SGA, FTT, transfusion | FTT | Hypotonia | – | – | FTT, hypotonia | Poor PO feeding, hypotonia | FTT | – | FTT, constipation | Poor engagement, and feeding, no support required | Hypotonia | Hypotonia | NR |
| HEENT | Microcephaly. L Esotropia. Downturned corners of the mouth, open mouth, palpebral fissures | Difficulty tracking, nystagmus | Esotropia, low vision, nystagmus. L ear posteriorly rotated, mildly low set, prominent forehead | Macrocephaly, esotropia | PRS, cleft palate, Bilateral ptosis, strabismus, hyperopia, myopathic facies | PRS, cleft palate, large low-set ears, mild hyperopia, poor tracking | Microcephaly, tented vermillion of upper lip, tall chin, long palpebral fissures, bruxism, myopia, astigmatism, corneal abrasions, chalazions, myopathic facies | –/NR | Cleft palate, esotropia | Micro/brachycephaly, myopathic facies, open mouth, tented vermillion of upper lip pointed chin, ptosis, strabismus, long palpebral fissures | Borderline macrocephaly, high arched | Microcephaly, proptosis, subtle dysmorphic features, myopathic facies, roving eye movements | NR | Microcephaly, tented vermillion of upper lip, open mouth, tall chin, long palpebral fissures | Microcephaly, open mouth, tented vermillion of upper lip, pointed chin, deep set eyes. | Flattened nasal bridge, prominent supraorbital ridges, strabismus. | Normal OFC, prominent supraorbital ridges, coarse facies, sloping forehead, strabismus, mild hypermetropia | Microcephaly, exotropia, markedly downturned mouth with an overriding tented vermillion of upper lip and a small jaw, tapered, short fingers |
| Growth/feeding difficulties | +FTT, +G-tube | –/– | –/+G-tube | –/– | +FTT/+NG feeding | +FTT/+ | +/G-tube | NR/– | +/ G-tube | +FTT/G-tube | –/– | +/G-tube | +/– | –/+ (improved) | –/– | +FTT/+ | –/– | +/G-tube |
| Gross/fine motor delay | +/+ | +/+ | +/+ | –/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | –/+ | +/+ |
| Speech/cognitive delay | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ |
| Motor exam (tone/strength/reflexes) | Profound hypotonia, non-ambulatory, preserved DTRs | Hypotonia with head lag (3 mo), DTR3+, crossed adductors | Hypotonia, DTR 2+ | Hypotonia, normal muscle bulk and strength, preserved DTRs | Profound hypotonia, preserved
DTRs, CP-like features |
Profound hypotonia, preserved DTRs | Hypotonia, followed by CP-like features, non-ambulatory, DTRs 1+, 3–4 beats clonus | Axial & distal hypotonia, DTRs NR | Hypotonia, DTRs NR | Hypotonia, MRC 2/5, non-ambulatory, preserved but asymmetric DTRs | Improved tone. MRC 4/5 proximally, 5/5 distally, preserved DTRs | Hypotonia, weakness, spastic/dystonic quadriplegia, non-ambulatory, DTRs +/- | Hypotonia, non-ambulatory, DTRs NR | Hypotonia, non-ambulatory, DTRs NR | Truncal hypotonia, spasticity, DTRs + | Appendicular hypotonia, able to walk and self-feed, DTRs preserved | Hypotonic in infancy, now walks with orthotics, DTRs not available | Diffuse hypotonia, wheelchair dependent, non-ambulatory |
| Seizures, semiology (age of onset) | Intractable myoclonic seizures and GTC seizures (3 mo) | Intractable myoclonic jerks; recurrent status epilepticus (3 mo) | Early startle. Intractable myoclonic movements with eye deviation (3 mo) | Febrile seizures, staring with lip smacking, atonic, generalized, currently well controlled (2 y) | Infantile spasms (6m), myoclonic and intractable generalized seizures (8 mo) | Intractable generalized seizures (5 mo) | Myoclonic (16 m), now intractable of ‘various types’ | Myoclonic jerks, GTC seizures; status epilepticus (2 mo) | Myoclonic, GTC seizures; status epilepticus (5 mo) | Intractable, multiple types: GTC, atonic, myoclonic, focal (4 mo) | Arm shaking (9m). Possible complex partial and drop events (3 y). Sporadic. Normal EEG (10 y) | Infantile spasms (4 m), generalized tonic clonic seizures, atypical absence seizures, myoclonic jerks (8 mo); seizures are frequent, not intractable. | Febrile jerks, followed by intractable myoclonic jerks, eye twitching, less frequent GTCs (3 mo) | Pallor/floppy episodes (3 m); intractable limb jerks, twitching eyes/limbs, hypomotor, GTS (7 mo) | Frequent but not intractable seizures: febrile, focal, drop attacks, myoclonic epilepsy (7 mo) | Myoclonic astatic (6 mo), well controlled on keto diet | Febrile, recurrent status (10 mo). Currently intractable GTC, atonic, clonic, myoclonic and focal | Intractable generalized epilepsy (infancy) |
| Brain MRI abnormalities | + | + | + | – | + | NP | + | – | – | + | – | + | NP | + | + | – | – | + |
| Cardiac issues: Structural/arrhythmia | –/+ | –/– | –/+ | NP/– | –/+ | NP/- | –/– | +/+ | –/+ | –/– | –/– | –/+ | –/– | –/– | +/– | NP/– | NP/– | –/+ |
| Pulmonary involvement | - | Intubation with episodes of status epilepticus | - | OSA; CLD; frequent pneumonia | Mild OSA + central apnea | OSA and CLD requiring BiPAP | – | – | CLD, pulmonary hypoplasia, other issues | FVC 54% predicted (10 y); nocturnal BiPAP (11 y) | Frequent pneumonias and hypoventilation requiring BiPAP | – | Recurrent infection | Recurrent infections, asthma | – | – | – | |
| Immunologic findings/symptoms | + | – | – | – | + | – | + | – | – | + | – | + | + | + | + | – | – | + |
| Other | Hypercalcemia during keto, hypernatremia, medullary nephrocalcinosis. | Sudden fatal bradycardia leading to cardiac
arrest with status epilepticus (21 mo). Autopsy: brain increased gyral pattern with polygyria, delayed myelination. |
Ketogenic diet seemingly induced cardiac
arrhythmia. Frequent infections, history of milk protein allergy. |
Right inguinal hernia and undescended R
testicle Cause of death: prolonged seizure with intubation Autopsy: not performed |
Additional features: Short stature (<1% for age), alopecia, bilateral hip dysplasia, significant kyphoscoliosis (spinal fusion), hypoplastic patellae, small hands & feet, persistent thrombocytopenia neutropenia, macrocytosis, genital differences, hypothyroidism (12 y) | Chronic respiratory acidosis. Cystitis. Left
hemiparesis secondary to embolism from ECMO. Additional dysmorphic features: narrow forehead with bitemporal narrowing, malar flattening, prominent mandible, medial eyebrow flare, long and thin eyebrows, prominent eyes, full nasal ridge, high nasal root, wide nose, thin columella, smooth upper vermillion, small incisors, small mouth. |
Periods of regression and recovery. Oral
aversion, but PO feeds since infancy. Difficulty with eye contact and
social behavior. Additional dysmorphic features: Prominent metopic suture on forehead with wedging on the bilateral temporal areas. Distal joint laxity |
Short stature, episodes of hypothermia.
Bilateral frontal encephaloceles. Hypercalcemia. Episode of pancreatitis
due to Valproate. Regression and
deterioration. Additional dysmorphic features: Narrow forehead, mild mid-face flattening, subtle prognathia. |
Inflammatory markers elevated during seizures, responded well to NSAID and steroids. | Inflammatory markers elevated during seizures,
responded well to NSAID and steroids. Autopsy: NP; patient died suddenly overnight in sleep. |
Immune workup
normal. Additional dysmorphic features: Tapered fingers, small feet. Short philtrum, open mouth, tented vermillion of upper lip, micrognathia. History and exam or regression complicated by head injury age 14 years. |
Daily episodes of gait
disturbance/ataxia. Pectus excavatum. Single kidney stone. Splitting of R and L central renal sinus. Faded hemangioma behind left ear, left preauricular pit |
Mild biochemical
hypothyroidism. Small hands and feet. Regression after age 6 years, but some progress in development since. |
Dysautonomia. Intermittently low calcium
levels. History of thrombus. Autopsy: NP; patient died during bout of ventricular tachycardia (followed by bradycardia) and status epilepticus. |
||||
‘–’ denotes absent; ‘+’ denotes present; BiPAP, bilevel positive airway pressure; CLD, chronic lung disease; CP, cerebral palsy; d, days; DTRs, deep tendon reflexes; ECMO, extra-corporeal membrane oxygenation; FTT, failure to thrive; FVC, forced vital capacity; GDM, gestational diabetes mellitus; GTC, generalized tonic clonic; GTS, generalized tonic seizures; L, left; mo, months; MRC, Medical Research Council’s scale of muscle power; NG, nasogastric; NICU, neonatal intensive care unit; NP, not performed; NR, not reported; OFC, occipitofrontal circumference; OSA, obstructive sleep apnea; PO, per os (by mouth); PRS, pierre robin sequence; R, right; y, years.
This syndrome almost universally presented as a congenital developmental disorder with early-onset epileptic encephalopathy, though one patient with the mildest clinical course initially presented with hypotonia. While most individuals were living with the disorder, ranging in age from 2 to 32 years, six individuals were deceased from various causes at the time of this writing. While clear patterns of dysmorphic features were not readily apparent across the cohort, notable features (Figure 1) included microcephaly (n=7), tented vermillion border of upper lip (n=5), open mouth (n=4), myopathic facies (n=4), tall or pointed chin (n=4), and downturned mouth (n=2). Eye and periocular pathologies were identified in some patients including strabismus (n=9), long palpebral fissures (n=4), nystagmus (n=2), prominent supraorbital ridges (n=2), deep set eyes (n=1), and proptosis (n=1). Other comorbidities across the cohort included failure to thrive or other clinically significant growth issues (n=10), enteral tube feedings (n=8), recurrent infections (n=6), and need for chronic respiratory support (n=3).
Figure 1. Photographs of individuals with recessive CRELD1 variants.
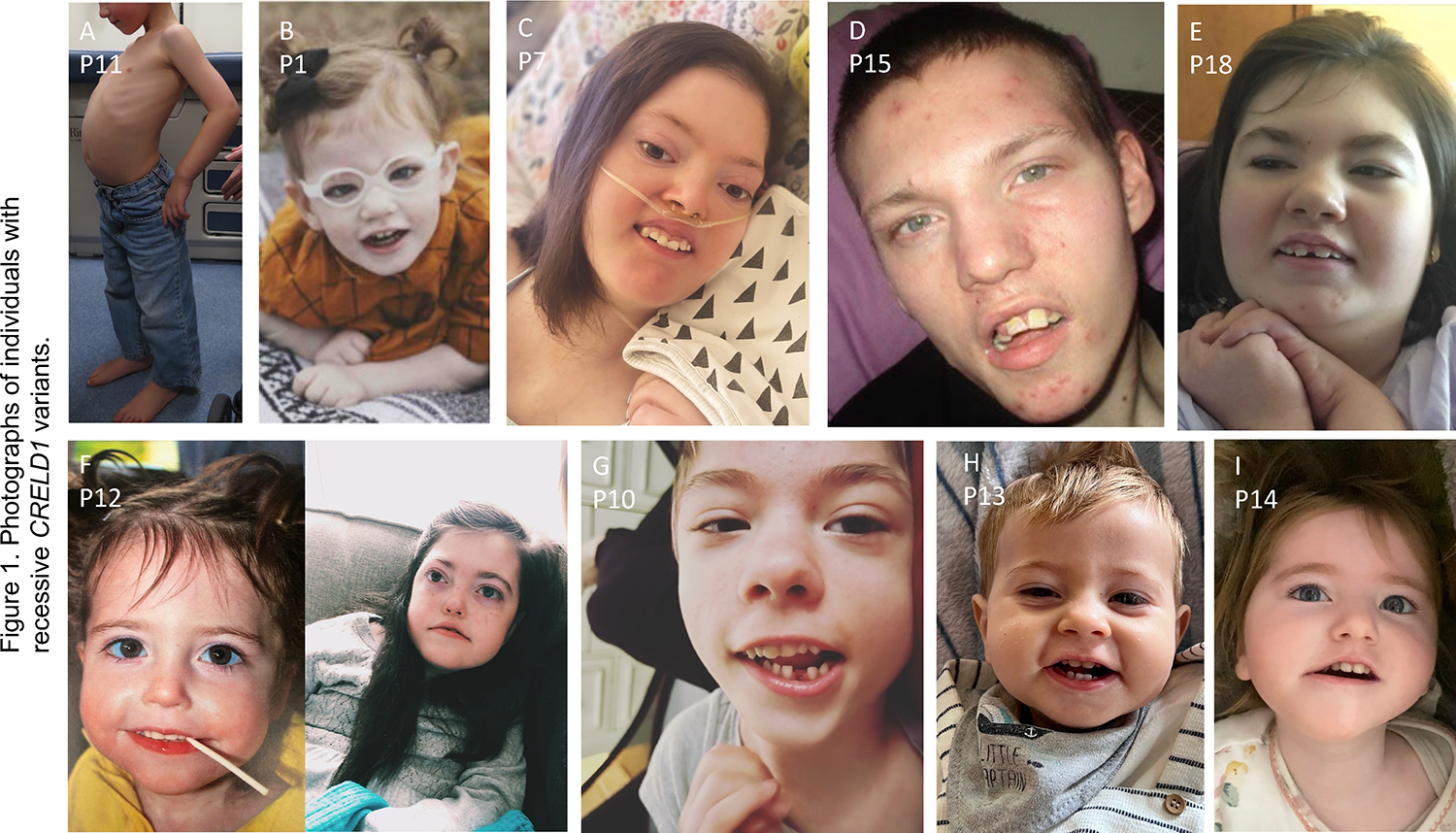
A. Lateral profile of P11 showing severe lordosis. For B to I, frequently seen features include open mouth, long palpebral fissures, tented vermillion border of upper lip, downturned corners of mouth, myopathic facies, narrow forehead with bitemporal narrowing, and tall or pointed chin. Figure F shows P12 at age 2 years (left) and 10 years (right).
All patients in this cohort had epilepsy that was often intractable with onset frequently in infancy (median age of 5 months). Seizures spanned a range of types including infantile and epileptic spasms, myoclonic seizures, atonic seizures, and generalized tonic-clonic seizures, and were frequently observed with elevated body temperature. Global developmental delays were universal and often severe, with eight individuals considered nonverbal. Abnormal findings were seen in 11 of the 16 patients who had undergone neuroimaging, and mainly consisted of variable degrees of cerebral volume loss and/or hypomyelination.
All individuals had some degree of hypotonia and most had motor delay with seven being nonambulatory. For most patients, low tone was attributed to the underlying central nervous system pathology or noted retrospectively after the assembly of this cohort. Three patients, however, had targeted neuromuscular evaluations. P7, an adult with severe neurodevelopmental symptoms, had a non-diagnostic muscle biopsy as part of an evaluation of possible mitochondrial disease. P11, a teenaged boy with moderate static developmental delays, was diagnosed with learning difficulties and an early-onset myopathy with respiratory insufficiency. He had a primary presentation of proximal more than distal muscular weakness with a positive Gower’s maneuver when arising from the floor and delayed motor milestones. Muscle ultrasound revealed patches of increased echogenicity throughout, indicative of fatty changes within the muscles, and similar findings were seen on magnetic resonance imaging (Figure 2A). Serum creatine kinase levels were normal. Muscle biopsy showed non-specific histologic features including variability of fiber size and fiber type grouping without angulated fibers. Electromyography (EMG) and repetitive nerve stimulations were attempted but not tolerated. P12 was a girl with chronic multisystem disease who had multiple hospitalizations during the final months of her life and underwent a neuromuscular evaluation during one such admission. Similar to P11, her muscle ultrasound showed increased echogenicity in the lower extremities, indicative of fatty infiltration (Figure 2B). However, rare fasciculations were also noted, suggestive of underlying neurogenic process. EMG evaluation did show possible mild axonal neuropathy with mild slowing of nerve conduction velocity, mild reduction in compound muscle action potential, and rare positive sharp waves; repetitive nerve stimulation was normal. She also had post-mortem muscle analysis that showed fiber size variation, a predominance of Type 1 fibers, and pleiomorphic mitochondria with abnormal cristae and paracrystalline-like inclusions (Figure 2C).
Figure 2. Muscle studies from patients.

A. Axial T1 MRI of mid-thigh (top panel) and lower leg (bottom panel) from P11 showing proximal more than distal muscle atrophy and fatty replacement. Mid-thigh image shows rectus femoris atrophy (black arrow) and streaky fatty infiltrate in the vastus lateralis and deep vastus medialis (red arrow). There is extensive fatty replacement of the adductor magnus and adductor longus (blue arrow), whereas other muscles are relatively spared. In the lower leg there is diffuse streaky fatty replacement in the soleus (yellow arrow) with sparing of surrounding muscles. B. Muscle ultrasound images from P12, demonstrating relative sparing of the triceps (top panel) compared to the gastrocnemius (bottom panel), which shows increased echogenicity, indicative of fatty infiltration. C. Muscle analysis performed on autopsy sample from P12. Hematoxylin and eosin stain (top panel) demonstrating variability in fiber size. ATPase staining (middle panels) showing predominance of Type 1 muscle fibers, which stain light at pH 9.4 (left) and dark at pH 4.1 (right). Electron microscopy (lower panel) showing enlarged, pleiomorphic mitochondria with abnormal cristae, paracrystalline-like inclusions, and osmophilic bodies.
Cardiac arrhythmias consisting of variable tachy- and bradyarrhythmias were identified in nearly half of the 18 individuals (n=8), and one patient required ICD placement. An additional patient whose epilepsy had been well-controlled and who did not have known dysrhythmia died suddenly in her sleep; this was classified as a sudden unexpected death in epilepsy (SUDEP). Two patients had structural heart defects: P8 had both an atrial septal defect (ASD) and a ventricular septal defect (VSD), and was also one of the patients with ventricular tachyarrhythmia; P15 had a VSD alone with no dysrhythmia. Their heterozygous parents had no signs of cardiac malformations, though they did not have formal cardiac echocardiography One patient, P10, had a transient cardiomyopathy with valvular insufficiency of unclear etiology that subsequently resolved; this patient did also have episodes of ventricular tachycardia, first degree AV block, and bradycardia.
IDENTIFICATION OF CRELD1 VARIANTS
Trio (proband and both biologic parents) ES or GS identified CRELD1 as a candidate gene in 12 affected individuals. Quad (two affected siblings and both biologic parents) ES identified CRELD1 as a candidate gene in an additional family, and duo (proband and mother, as father was unavailable) ES identified CRELD1 in one patient. Targeted Sanger sequencing confirmed compound heterozygous CRELD1 variants in the case of the remaining 3 affected children, who were siblings of probands identified through ES/GS. None of the 15 affected individuals analyzed with ES/GS were found to have variants in previously known disease-causing genes that could explain their clinical phenotypes. Three patients did have single nucleotide polymorphisms or copy number variants in genes/loci apart from CRELD1, but none of these were felt to be disease-causing based on American College of Medical Genetics (ACMG) criteria (Supplementary Table 1)26.
All 18 affected individuals harbored two CRELD1 (NM_015513.6) variants in trans, one inherited from each parent (Figure 3 and Table 1). In 16 cases, individuals harbored one frameshift variant with one missense variant; in the other two cases patients had compound heterozygous or homozygous missense variants. There were no patients with two predicted loss-of-function frameshift variants. Across the cohort of 18 patients, we identified 10 distinct variants – six missense and four frameshift – showing recurrence of multiple variants in unrelated families (Table 2). Most of the patients (n=14) carried p.(Cys192Tyr). Of these, 12 patients had p.(Cys192Tyr) in trans with a frameshift variant, one had p.(Cys192Tyr) in trans with a second missense variant, p.(Ala391Pro), and one was homozygous for p.(Cys192Tyr). Five of the variants – p.(Asp85Valfs*6), p.(Cys262Arg), p.(Met369Val), p.(Thr380Met), and p.(Asp386Asn) – were not found in gnomAD, with the others occurring with a maximum allele frequency of <0.05%. The two variants with the highest allele counts in gnomAD, p.(Cys192Tyr) and p.(Gln320Argfs*25), were both found predominantly in non-Finnish European population, suggestive of a founder effect. There were no homozygous individuals reported in gnomAD for any of the variants. Given that CRELD1 did not have a previously validated association with the patient phenotypes, all variants were classified as variants in a gene of uncertain significance as per ACMG criteria26.
Figure 3. Family Pedigrees.

Overview of pedigrees presented in this cohort showing heterozygous parents and affected individuals. The ten distinct CRELD1 variants are color-coded as indicated in the legend.
Table 2.
Genomic CRELD1 variants identified in this cohort
| GRCh37 (hg19) | Ref | Alt | Refseq (NM_015513.6) | Occurrence | Allele Frequency (gnomAD) | Consequence | CADD | REVEL | MetaSVM | SIFT | PolyPhen |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 3:9976540dup | - | G | c.196dup, p.(Asp66Glyfs*22) | 1 | 1.61 × 10−5 | frameshift | . | . | |||
| 3:9976598_9976601del | ACAG | - | c.254_257del, p.(Asp85Valfs*6) | 2 | 0 | frameshift | . | . | |||
| 3:9982648 | G | A | c.575G>A, p.(Cys192Tyr) | 15a | 1.84 × 10−4 | missense | 28.3 | 0.692 | D | D (0) | Pr D (1.0) |
| 3:9984547 | T | C | c.784T>C, p.(Cys262Arg) | 1 | 0 | missense | 25.5 | 0.968 | D | D (0) | Pr D (1.0) |
| 3:9985110del | A | - | c.959del, p.(Gln320Argfs*25) | 7 | 2.97 × 10−4 | frameshift | . | . | |||
| 3:9986105 | A | G | c.1105A>G, p.(Met369Val) | 1 | 3.18 × 10−5 | missense | 20.5 | 0.301 | T | D (0.01) | B (0.05) |
| 3:9986128_9986129del | TG | - | c.1128_1129del, p.(Ala377Thrfs*7) | 6 | 2.47 × 10−5 | frameshift | . | . | |||
| 3:9986139 | C | T | c.1139C>T, p.(Thr380Met) | 1 | 2.39 × 10−5 | missense | 17.14 | 0.348 | T | D (0.05) | B (0.14) |
| 3:9986156 | G | A | c.1156 G>A, p.(Asp386Asn) | 1 | 0 | missense | 18.47 | 0.18 | T | D (0) | Pos D (0.58) |
| 3:9986171 | G | C | c.1171G>C, p.(Ala391Pro) | 1 | 0 | missense | 16.13 | 0.405 | T | D (0.01) | Pos D (0.81) |
B, benign; D, damaging/deleterious; gnomAD, The Genome Aggregation Database; Pos, possibly; Pr, probably; T, tolerated.
This represents 14 patients, with one being homozygous for p.(Cys192Tyr).
We examined the location of the patient variants within the structural motifs of CRELD1 (Figure 4A). Cys192 was found to be in the first EGF-like domain and was predicted by ProSite27 to participate in a disulfide bond with Cys183. Cys262 was found to be in the first cbEGF-like domain, and ProSite was unable to predict disulfide bonding cysteine residues within this particular domain. The amino acid residues affected by missense variants were also noted to be completely conserved across a panel of 10 species (Figure 4B).
Figure 4. Gene variants in CRELD1.

A. Architecture of CRELD1 gene, based on the predominantly-expressed isoform 2 (NP_056328.3). Amino acid lengths of the 10 exons are shown at top. Locations of functional domains are also shown (colored squares; see text for descriptions of domains). Location of frameshift (orange text) and missense (blue text) variants from patients are indicated. B. Multiple species alignment showing reference sequence surrounding CRELD1 amino acid residues altered in missense variants found in this cohort (blue arrows). Note complete conservation across examined species at all residues that are altered by patient missense variants.
DEVELOPMENTAL DEFECTS IN XENOPUS TADPOLES WITH CRELD1 KNOCKDOWN
We sought to examine the in vivo effects of CRELD1 by utilizing two complementary methods for knockdown of creld1 in Xenopus tropicalis: start site morpholino (CRELD1-MO) to inhibit creld1 gene expression and CRISPR/Cas9-mediated editing (CRELD1-CRISPR) to directly mutate and disrupt the gene sequence. Roughly half of the CRELD1-CRISPR tadpoles had severe defects of either early gastrulation disruption or edema/late embryonic lethality, and some also had tail defects (Figure 5A), all three representing phenotypes that would render the tadpoles unscorable for seizure (see below). CRELD1-CRISPR tadpoles that did survive had a high incidence of morphological defects compared to controls, namely defects in heart development (looping defects) and craniofacial development (Table 3). In contrast, CRELD1-MO tadpoles were similar to uninjected controls in terms of morphology and survival to stage 42, though they did exhibit a mild reduction in normal tadpole movements, with reduced swimming and feeding behaviors compared to control MO and uninjected controls (data not shown).
Figure 5. Developmental defects and increased susceptibility to induced seizures in X. tropicalis tadpoles with creld1-knockdown.

A. X. tropicalis embryos were either uninjected controls (UiC) or injected with either morpholino targeting creld1 for knockdown (CRELD1-MO) or one of two sgRNAs plus CRISPR for direct creld1 gene knockout (CRELD1-CRISPR with sgRNA-1 or sgRNA-2). Tadpoles were then observed at set stages for phenotypes that would prevent scoring for seizures: at stage 22 for gastrulation defects or at stage 42 for edema/late lethality or tail defects. Results showed a high proportion of CRELD1-CRISPR embryos compared to UiC or creld1 MO with developmental defects preventing scoring for seizures. Data is shown from a representative experiment with n=indicating the number of embryos scored. B. X. tropicalis CRELD1-CRISPR embryos and UiC embryos observed for either spontaneous seizures or pilocarpine-induced seizures over time. All embryos showed minimal spontaneous seizure behaviors (<5%). By 30 minutes, approximately 70% of CRELD1-CRISPR embryos showed seizure behaviors in the presence of pilocarpine compared to approximately 40% of control embryos. Symbols indicate mean and error bars indicate standard deviation. Figure represents compilation of three biological replicates, each with >70 tadpoles per experimental condition.
Table 3.
Developmental defects seen in tadpoles with CRELD1 knockdown
| Observed Phenotypes | Uninjected Controls (n = 167) | CRELD1-MO (n = 170) | CRELD1-CRISPR1 (n = 137) | CRELD1-CRISPR2 (n = 141) |
|---|---|---|---|---|
|
| ||||
| Gastrulation defect at stage 22 | 8 (5%) | 11 (7%) | 44 (32%) | 48 (34%) |
| Edema/late lethality | 18 (11%) | 17 (10%) | 18 (13%) | 28 (20%) |
| Tail defects | 7 (4%) | 3 (2%) | 8 (6%) | 20 (14%) |
| Total unscorable embryos | 33 (20%) | 31 (18%) | 70 (51%) | 96 (68%) |
| Total scorable embryos | 134 (80%) | 139 (82%) | 67 (49%) | 45 (32%) |
| Scorable tadpoles with: | ||||
| heart looping defects | 7 (5%) | 6 (4%) | 8 (12%) | 7 (15%) |
| craniofacial defects | 12 (9%) | 8 (6%) | 18 (27%) | 23 (51%) |
CRISPR, clustered regularly interspaced short palindromic repeats; MO, morpholino antisense oligomer.
INDUCIBLE SEIZURES IN CRELD1-depleted TADPOLES
Given the prominence of seizures as a phenotype in our patient cohort, we assessed the relationship of CRELD1 to seizure propensity. We and others have previously described the use of Xenopus tadpoles as a seizure model using chemical convulsants or specific gene knockdowns18,25. Seizures are characterized by intermittent C-shaped body curvatures followed by inverted swimming and/or periods of immobility that can be quantified by their frequency. Spontaneous seizures were not observed in either CRELD1-CRISPR or CRELD1-MO tadpoles. However, when treated with a minimally-phenotypic dose of the chemical convulsant pilocarpine (50mM), CRELD1-MO tadpoles displayed more robust seizure behaviors within 30 minutes of treatment (mean 79%, n=78) compared to control MO (mean 33%, n=80) and uninjected control tadpoles (mean 41%, n=81) (Supplementary Figure 2). Similarly, although CRELD1-CRISPR tadpoles had a higher incidence of phenotypes rendering them unscorable for seizures, those that survived to stage 42 also displayed more seizures within 30 minutes of pilocarpine treatment (mean 76%, n=119) compared to uninjected control tadpoles (mean 40%, n=72) (Figure 5B). Representative analysis performed at 20 minutes indicates a statistically significant difference in seizures between CRELD1-depleted (both CRIPSPR and MO) and control tadpoles behavoir in response to pilocarpine treatment (Supplementary Figure 2).
REDUCED INDUCTION OF CRANIOFACIAL DEFECTS BY CRELD1 MISSENSE VARIANTS
To evaluate the effect of individual patient missense variants on CRELD1 function, we overexpressed human reference CRELD1 (as a control) and CRELD1 missense variants identified from patients in X. tropicalis embryos at the one cell stage to look at morphological changes during development. Given that this approach would result in high levels of CRELD1 protein as well as ectopic expression throughout development in cells CRELD1 may not normally be expressed, this was used specifically as an in vivo test of protein function and not as an assay of normal CRELD1 function in development. Overexpression of reference human CRELD1 mRNA resulted in stage 42 tadpoles that were similar in overall size to control tadpoles but had defects in craniofacial development (Figure 6A), indicating that ectopic overexpression of wild type CRELD1 at the one-cell stage induces defective craniofacial development. We quantified this by noting microcephaly (smaller head area of mean 120289 pixels (n=284) compared to control mean 154742 pixels (n=299)) and a reduction in interocular distance (IOD) (mean 223.1 pixels (n=283) compared to control mean 274.8 pixels (n=298)) (Figure 6B). In contrast, overexpression of individual patient variants led to a lesser degree of microcephaly and reduced interocular distance, consistent with a reduction in protein function of the patient variants. Of the variants tested, p.(Cys262Arg) (head area mean 147326 pixels (n=65) and IOD mean 265.5 pixels (n=65)), p.(Met369Val) (head area mean 149318 pixels (n=69) and IOD mean 267.9 pixels (n=69)), p.(Thr380Met) (head area mean 146508 pixels (n=77) and IOD mean 261.1 pixels (n=77)), and p.(Ala391Pro) (head area mean 146587 pixels (n=78) and IOD mean 264.1 pixels (n=78)) had the greatest reduction in protein function in this assay and looked more similar to uninjected control tadpoles. In contrast, p.(Cys192Tyr) (head area mean 134601 pixels (n=68) and IOD mean 242.4 pixels (n=68)) and p(Asp386Asn) (head area mean 130151 pixels (n=87) and IOD mean 238.3 pixels (n=87)) displayed function that was closer to, but still less than, the reference sequence, indicating that p.(Cys192Tyr) and p.(Asp386Asn) retained some residual function to induce microcephaly and reduced interocular distance when ectopically expressed at the one-cell stage, but not to the same extent as reference CRELD1 mRNA.
Figure 6. Diminished ability of missense variants of human CRELD1 to induce craniofacial defects in X. tropicalis tadpoles.

A. As an in vivo assay of CRELD1 protein function, X. tropicalis embryos were either uninjected controls (top panel) or injected with reference sequence human mRNA for CRELD1 (bottom panel). Tadpoles injected with reference CRELD1 were noted to have defective craniofacial development with microcephaly (head area indicated by dashed white lines) and shorter interocular distance (yellow line between eyes) as shown in these representative images. B. Violin plots showing quantification of findings illustrated in A, Head Area (left plot) and Interocular Distance (right plot) for reference sequence CRELD1 and patient variants as noted. All patient variants showed a reduced ability to induce craniofacial defects in tadpoles as compared to reference sequence CRELD1, and this difference was statistically significant. Figure represents compilation of eight experimental replicates for uninjected controls and reference sequence CRELD1, and three experimental replicates for each patient variant; each experimental replicate analyzed ~20 tadpoles. For statistical significance (unpaired Welch’s t tests), *p<10−2; **p<10−3; ***p<10−4; ****p<10−5.
COMPARISON OF PBMC POPULATIONS IN PATIENT AND HEALTHY DONORS
PBMCs were obtained for analysis from four patients; three of these were described as having frequent infections (P1, P5, and P12), while one was not (P11) (Supplementary Table 1). Given the prior description of populations with low CRELD1 expression having low naïve CD4+ T cells2, we characterized the PBMC populations of these patients and found no difference compared to healthy donors in percentage of B cells, T cells or monocytes (Figure 7A) or in percentage of naïve CD4+ or CD8+ T cells (Figure 7B). Notably, whereas the other patients had blood cell counts within the normal range, P12 had chronic leukopenia, anemia, and thrombocytopenia (Supplementary Table 2) and at time of her blood collection she had a white blood cell count of 3,500/μl (37% neutrophils, 44% lymphocytes, 15% monocytes), hemoglobin of 8.8 g/dL, and platelet count of 47,000/μl.
Figure 7. Baseline immune cell subsets.

A. Analysis of baseline peripheral blood mononuclear cells (PBMCs) gated on live singlets identified similar distribution of B cells (CD19+), T cells (CD3+), CD4+ T cells (CD4+), CD8+ T cells (CD8+), and monocytes (CD14+) in CRELD1 patients and healthy donors, who were matched by sex and age (+/− one year). CD4+ and CD8+ data is expressed as a percentage of total T cells. B. Delineation of various CD4+ and CD8+ T cell populations among CD3+ live singlets revealed similar subsets in CRELD1 patients and healthy donors. Anti-CCR7 and anti-CD45RA antibodies were used to define naïve (double positive), central memory (CM; CCR7+CD45RA−), effector memory (EM; double negative), or T effector memory cells that re-express CD45RA (TEMRA; CCR7-CD45RA+).
DISCUSSION
Here we present detailed clinical and genetic characterization of a cohort of patients with a recessive multisystem disease manifesting with prominent neurodevelopmental characteristics associated with biallelic variants in CRELD1. This study establishes CRELD1 as a monogenic cause of recessive disease in humans, provides delineation of its disease spectrum, and presents evidence for pathogenicity of patient variants. Unifying phenotypic characteristics across the cohort were early-onset hypotonia, early-onset refractory epilepsy, and neurodevelopmental delays of varying degrees. Other variable features included cardiac rhythm abnormalities, congenital heart defects, frequent infections, respiratory difficulties requiring positive pressure support, myopathic-appearing facies, and ocular and peri-ocular findings. One can note salient features with an acronym that correlates with the causative gene CRELD1: Cardiac abnormalities, Recurrent infections, Epileptic encephalopathy, Low tone, Developmental delay.
The cohort consisted of 18 individuals from 14 different unrelated families with six of the variants unique to single families: p.(Asp66Glyfs*22), p.(Cys262Arg), p.(Met369Val), p.(Thr380Met), p.(Asp386Asn) and p.(Ala391Pro). Of the remaining four variants, p.(Cys192Tyr) was identified in 10 families (with one homozygous individual), p.(Gln320Argfs) in 5 families, p.(Ala377Thrfs) in 2 families, and p.(Met369Val) also in 2 families. All affected individuals had compound heterozygous or homozygous variants, suggesting recessive inheritance, with 16 of the 18 patients having a missense variant in trans with a putative null allele and the other two having biallelic missense variants. Notably, despite the prevalence of frameshift variants in this cohort, no affected individual had two frameshift alleles. We suspect that these frameshifts lead to null alleles and that, similar to the prenatal lethality seen in mouse Creld1 knockouts, complete absence of CRELD1 protein is not compatible with life3. In contrast, our analysis of CRELD1 overexpression in X. tropicalis demonstrated that patient missense variants were less functional than the reference protein by their reduced ability to induce developmental defects, but some of them still appear to retain some residual effect. Additionally, as heterozygous parents harboring frameshift variants are healthy, simple haploinsufficiency does not appear to be the mechanism for disease. Therefore, it appears that there is an amount of CRELD1 function somewhere between 0 and 50% that is viable but functionally problematic. Additional work may help discern what that threshold may be, and if other factors might influence the severity of the phenotype.
The variant p.(Cys192Tyr) in the EGF-like domain comprised 15 of the 36 CRELD1 variant alleles in this cohort, and a second cysteine-altering variant p.(Cys262Arg) in the calcium-binding EGF-like domain was identified in one patient. Both of these cysteine residues are completely conserved across multiple species from human to zebrafish (Figure 3B). While cysteine is one of the least abundant amino acids, it is overrepresented in functionally important residues, such as those involved in catalysis, binding of cofactors, or other regulatory roles28. ProSite analysis predicts that Cys192 is involved in a disulfide bond with Cys183, suggesting that the p.(Cys192Tyr) variant disrupts protein structure and/or function by eliminating this disulfide bond. Although ProSite was unable to predict disulfide bonding in the calcium-binding EGF domain where Cys262 is located, it is possible that the p.(Cys262Arg) variant may also disrupt CRELD1 protein by eliminating a disulfide bond.
The precise effect of individual variants on the CRELD1 protein remains to be determined but may underlie variability in patient phenotypes. Two patients (P4 and P11) were less cognitively impacted than others in the cohort, had sporadic seizures rather than intractable epilepsy and macrocephaly instead of microcephaly. An additional patient (P16) with normocephaly similarly had moderate (not severe) developmental delays and seizures well-controlled on the ketogenic diet. These three probands (P4, P11, and P16) were the only ones in the cohort not to have at least one cysteine-substituting variant of CRELD1. The one patient (P17) with homozygous p.(Cys192Tyr) alleles interestingly had more developmental progress and longevity than those with p.(Cys192Tyr) in trans with a putative null allele, but still had infantile-onset seizures that were intractable. We also note that all eight patients who were reported to have cardiac dysrhythmias as well as all six patients who were deceased harbored p.(Cys192Tyr) alleles in trans with various frameshift variants, suggesting that this change in a cysteine residue may be more harmful than other missense variants. We also note, however, that the p.(Cys192Tyr) displayed greater protein function than some of the other variants tested in our overexpression assay. Other genotype-phenotype correlations appear to be less clear. For example, P9 had a cleft palate and a structurally normal heart while her sister with the same genotype, P8, had a normal palate but a ventricular septal defect. Further clinical data and functional experiments will be required to define genotype/phenotype relationships more clearly.
We have also demonstrated that creld1-deficient tadpoles are more sensitive to pilocarpine-induced seizures than wild-type tadpoles, supplying a connection between deficiency in CRELD1 and the universal presence of seizures in our patient cohort. Still, the precise mechanism of seizures in these patients remains unclear. Previous work with a nematode forward genetic screen identified crld1, the only CRELD family member gene found in C. elegans, as important for synaptic expression of the inotropic (nicotinic) acetylcholine receptor (AChR), and knockdown of CRELD1 in mouse muscle cells was shown to lead to a defect of inotropic AChR biogenesis and reduced cell surface expression29. Pilocarpine, the chemoconvulsant used in our studies, is a muscarinic AChR agonist. One may predict that if knockdown of creld1 in tadpoles results in a reduction in muscarinic as well as nicotinic AChR numbers, this could lead to a reduced sensitivity to pilocarpine-induced seizures; but our data showed the opposite effect. It is possible that CRELD1 does not impact the expression of muscarinic AChR or that modulation of AChR is variable in different tissues. Furthermore, pilocarpine appears to have additional effects beyond cholinergic stimulation that impact the development of seizures, including modulation of local inflammation and alterations in the blood-brain barrier30,31, that may be contributing to the increased seizures in both CRELD1-MO and CRELD1-CRISPR tadpoles.
Hypotonia was also present in all patients. A mechanistic link may be provided from Drosophila Creld mutants with locomotion defects, reduced neuronal function, and mitochondrial impairments, though further work will be required to clarify any connection4. Phenotypic characterization of our cohort did not reveal obvious manifestations of neuromuscular junction defects, which typically presents as fatigable weakness in early childhood. Interestingly, P11’s clinical presentation included gross motor delay, proximal more than distal muscle weakness and impaired respiratory function suggestive of a congenital myopathy. The family did report periods of developmental regression and recovery over a period of months; a similar history of progress and regression was reported in seven other patients. P12’s clinical, muscle ultrasound, and autopsy findings were also consistent with neuromuscular disease. Unfortunately, though, we have not been able to uniformly assess neuromuscular involvement in all individuals in this cohort, and it is possible that some of the individuals with profound delays who are nonverbal and non-ambulatory have neuromuscular disease that is not recognized due to the extent of CNS manifestations. At this time, it remains unclear whether the clinical spectrum of biallelic CRELD1 variants should expand to include myopathies, axonopathies, congenital myasthenic disease or an overlap syndrome.
There has been significant evidence for a role for heterozygous CRELD1 missense variants in AV septal defects, albeit with incomplete penetrance1,3,10,14. Only two patients in our cohort (P8 with a VSD and small ASD, and P15 with a VSD) had structural heart defects, and these were isolated defects, not true AV septal defects. The recent report of enrichment of CRELD1 variants in Turner syndrome patients with bicuspid aortic valve suggests that CRELD1 may play a broader role in cardiac development beyond the endocardial cushions. Interestingly, although mouse Creld1 knockouts are reported to have a highly penetrant cardiac developmental phenotype3, CRELD1-MO and CRELD1-CRISPR tadpoles had a lower incidence of structural cardiac defects, roughly 10% (Table 3), similar to what we observed in this human cohort. This suggests that the low penetration of structural heart disease in CRELD1 patients may be influenced by other modifying genetic factors, similar to those reported with heterozygous CRELD1 variants10,13. There was also a high prevalence of cardiac dysrhythmias in this cohort, some of them severe ventricular tachycardias, and all found in patients carrying the p.(Cys192Tyr) variant in combination with various frameshifts. The additional patient with p.(Cys192Tyr) who died suddenly in her sleep (P14) suggests the possibility of a fatal arrythmia; based on this, we recommend that CRELD1 patients, particularly those carrying the p.(Cys192)Tyr variant, should at least have a formal cardiology evaluation with a screening electrocardiogram. Multiple episodes of dysrhythmias across patients were associated with other factors such as intercurrent illnesses/fevers, seizure episodes, or use of ketogenic diet (Supplementary Table 1), hinting that CRELD1 variants may lower the arrhythmogenic threshold. The etiology of this remains to be determined, though possibilities based on prior work on CRELD1 include alterations in cholinergic signaling29 or disordered development or maintenance of the myocardium14.
While it has been recently established that CRELD1 is important for immune homeostasis, no variants in this gene have been linked directly to human immunodeficiency or inflammatory phenotypes. Clinically, recurrent infections were seen in this cohort (n = 6), with P12 having chronic leukopenia, frequent severe urinary tract infections (without indwelling catheters), and upper respiratory tract infections requiring critical care. Four probands with clinical concerns of auto-immune or inflammatory processes had clinically oriented immunology evaluations that were unrevealing. While patients with mobility issues and profound hypotonia certainly have higher inherent infection risks, these clinical histories suggest that patients with CRELD1 variants may be more prone to infections through a direct effect on immune function. We were able to obtain PBMCs from a limited number of patients who demonstrated similar immune cell subtypes as compared to age and sex-matched healthy donors. Ongoing work will examine T cell activation as well as Wnt signaling during lymphocyte activation to determine if there is an underlying molecular mechanism for the frequent infections seen in this cohort.
In summary, we provide detailed clinical and genetic characterization of 14 families with a novel syndrome with prominent neurodevelopmental phenotypes resulting from biallelic variants in CRELD1. We provide further evidence supporting this association by demonstration of reduced protein function of patient variants in an in vivo model and increased in vivo susceptibility to induced seizures with Creld1 knockout. Further research is needed to elucidate the mechanistic details of CRELD1 protein function to better understand the clinical manifestations of this syndrome and eventually to guide treatment.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the patients and their families for participating in our research study, with a particular note for Adam Clatworthy (https://www.creld1.com/). We also thank the multiple sources that have provided various forms of support for this work including funding, DNA sequencing services, and data analyses. We thank the Yale Center for Genome Analysis for DNA sequencing, the Yale DNA Diagnostic Lab for providing data access, Sara Pawlak at the Yale Electron Microscopy laboratory for assistance with micrographs, and Yale New Haven Hospital for internal funding. MKK was supported by NIH/NICHD R01 HD102186. We thank Christopher Mendoza, Gilberto ‘Mike’ Averion and Kia Brooks for their help in supporting the NINDS/NNDCS clinic, the NIH Intramural Sequencing staff for their help with the exome analysis, and NINDS/NIH/HHS for intramural funding. N.N.B. acknowledges funding from the Yale Center for Clinical Investigation and PCCTSDP/NICHD/NIH. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm. IH was supported by The Hartwell Foundation (Individual Biomedical Research Award), the National Institute for Neurological Disorders and Stroke (K02 NS112600, U24 NS120854, U54 NS108874), the Intellectual and Developmental Disabilities Research Center (IDDRC) at Children’s Hospital of Philadelphia and the University of Pennsylvania (U54 HD086984), and by the German Research Foundation (HE5415/3–1, HE5415/5–1, HE5415/6–1, HE5415/7–1). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR001878), by the Institute for Translational Medicine and Therapeutics’ (ITMAT) at the Perelman School of Medicine of the University of Pennsylvania, and by Children’s Hospital of Philadelphia through the Epilepsy NeuroGenetics Initiative (ENGIN). AJ and SB were supported by Solve-RD, which has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 779257; National Human Genome Research Institute (R01HG009141); Epilepsy Society, UK; 100,000 Genomes Project/National Institute for Health Research; National Health Service England; The Wellcome Trust; Cancer Research UK and the Medical Research Council; the Solve-RD project and the European Union's Horizon 2020 research and innovation program (grant agreement 779257); and the National Institute for Health and Care Research Biomedical Research Centres.
Footnotes
CONFLICT OF INTEREST
Two authors report part ownership of startup companies unrelated to this work: Qiyas Higher Health (SAL) and Victory Genomics (SAL and MKK). KM is an employee of GeneDx. KN is currently an employee of Cooper Surgical. BM is currently an employee of Genome Medical. No other authors have any disclosures to report.
ETHICS DECLARATION
All institutions involved in this research received approval from their local Institutional Review Board (IRB) or Research Ethics Committee (REC). Informed consent was obtained from all individuals or from their parents/legal guardians, through the IRB protocols at Yale University School of Medicine (main IRB), GeneDx, or through one of the other participating institutions. Individual data has been de-identified; for the presentation of identifiable patient images, express written consent has been obtained from the individuals or from their parents/legal guardians. Animal research was performed under an approved Institutional Animal Care and Use Committee Protocol at Yale University School of Medicine.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AVAILABILITY
De-identified data is available on request from authors.
REFERENCES
- 1.Rupp PA, Fouad GT, Egelston CA, et al. Identification, genomic organization and mRNA expression of CRELD1, the founding member of a unique family of matricellular proteins. Gene. 2002;293(1–2):47–57. [DOI] [PubMed] [Google Scholar]
- 2.Bonaguro L, Kohne M, Schmidleithner L, et al. CRELD1 modulates homeostasis of the immune system in mice and humans. Nat Immunol. 2020;21(12):1517–1527. [DOI] [PubMed] [Google Scholar]
- 3.Mass E, Wachten D, Aschenbrenner AC, Voelzmann A, Hoch M. Murine Creld1 controls cardiac development through activation of calcineurin/NFATc1 signaling. Dev Cell. 2014;28(6):711–726. [DOI] [PubMed] [Google Scholar]
- 4.Paradis M, Kucharowski N, Edwards Faret G, et al. The ER protein Creld regulates ER-mitochondria contact dynamics and respiratory complex 1 activity. Sci Adv. 2022;8(29):eabo0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tombling BJ, Wang CK, Craik DJ. EGF-like and Other Disulfide-rich Microdomains as Therapeutic Scaffolds. Angew Chem Int Ed Engl. 2020;59(28):11218–11232. [DOI] [PubMed] [Google Scholar]
- 6.Robinson SW, Morris CD, Goldmuntz E, et al. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet. 2003;72(4):1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo Y, Shen J, Yuan L, Li F, Wang J, Sun K. Novel CRELD1 gene mutations in patients with atrioventricular septal defect. World J Pediatr. 2010;6(4):348–352. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Cherry S, Klinedinst D, et al. Genetic modifiers predisposing to congenital heart disease in the sensitized Down syndrome population. Circ Cardiovasc Genet. 2012;5(3):301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maslen CL, Babcock D, Robinson SW, et al. CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am J Med Genet A. 2006;140(22):2501–2505. [DOI] [PubMed] [Google Scholar]
- 10.Redig JK, Fouad GT, Babcock D, et al. Allelic Interaction between CRELD1 and VEGFA in the Pathogenesis of Cardiac Atrioventricular Septal Defects. AIMS Genet. 2014;1(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zatyka M, Priestley M, Ladusans EJ, et al. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2). Clin Genet. 2005;67(6):526–528. [DOI] [PubMed] [Google Scholar]
- 12.Zhian S, Belmont J, Maslen CL. Specific association of missense mutations in CRELD1 with cardiac atrioventricular septal defects in heterotaxy syndrome. Am J Med Genet A. 2012;158A(8):2047–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinnaro CT, Beck CB, Major HJ, Darbro BW. CRELD1 variants are associated with bicuspid aortic valve in Turner syndrome. Hum Genet. 2023;142(4):523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckert V, Rassmann S, Kayvanjoo AH, et al. Creld1 regulates myocardial development and function. J Mol Cell Cardiol. 2021;156:45–56. [DOI] [PubMed] [Google Scholar]
- 15.Vaeth M, Feske S. NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res. 2018;7:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobreira NLM, Arachchi H, Buske OJ, et al. Matchmaker Exchange. Curr Protoc Hum Genet. 2017;95:9 31 31–39 31 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Ali S, Jeffries L, Faustino EVS, et al. A retrospective cohort analysis of the Yale pediatric genomics discovery program. Am J Med Genet A. 2022;188(10):2869–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sega AG, Mis EK, Lindstrom K, et al. De novo pathogenic variants in neuronal differentiation factor 2 (NEUROD2) cause a form of early infantile epileptic encephalopathy. J Med Genet. 2019;56(2):113–122. [DOI] [PubMed] [Google Scholar]
- 19.Vogtle FN, Brandl B, Larson A, et al. Mutations in PMPCB Encoding the Catalytic Subunit of the Mitochondrial Presequence Protease Cause Neurodegeneration in Early Childhood. Am J Hum Genet. 2018;102(4):557–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deciphering Developmental Disorders S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foley AR, Zou Y, Dunford JE, et al. GGPS1 Mutations Cause Muscular Dystrophy/Hearing Loss/Ovarian Insufficiency Syndrome. Ann Neurol. 2020;88(2):332–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khokha MK, Chung C, Bustamante EL, et al. Techniques and probes for the study of Xenopus tropicalis development. Developmental dynamics : an official publication of the American Association of Anatomists. 2002;225(4):499–510. [DOI] [PubMed] [Google Scholar]
- 23.Conant D, Hsiau T, Rossi N, et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022;5(1):123–130. [DOI] [PubMed] [Google Scholar]
- 24.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3(2):154–171. [DOI] [PubMed] [Google Scholar]
- 25.Hewapathirane DS, Dunfield D, Yen W, Chen S, Haas K. In vivo imaging of seizure activity in a novel developmental seizure model. Exp Neurol. 2008;211(2):480–488. [DOI] [PubMed] [Google Scholar]
- 26.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sigrist CJ, de Castro E, Cerutti L, et al. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41(Database issue):D344–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol. 2010;404(5):902–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Alessandro M, Richard M, Stigloher C, et al. CRELD1 is an evolutionarily-conserved maturational enhancer of ionotropic acetylcholine receptors. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchi N, Johnson AJ, Puvenna V, et al. Modulation of peripheral cytotoxic cells and ictogenesis in a model of seizures. Epilepsia. 2011;52(9):1627–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchi N, Oby E, Batra A, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48(10):1934–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
De-identified data is available on request from authors.