

Abstract
Site- and stereoselective C–H functionalization is highly challenging in the synthetic chemistry community. Although the chemistry of vinyl cations has been vigorously studied in C(sp3)–H functionalization reactions, the catalytic enantioselective C(sp3)–H functionalization based on vinyl cations, especially for an unactivated C(sp3)–H bond, has scarcely explored. Here, we report an asymmetric copper-catalyzed tandem diyne cyclization/unactivated C(sp3)–H insertion reaction via a kinetic resolution, affording both chiral polycyclic pyrroles and diynes with generally excellent enantioselectivities and excellent selectivity factors (up to 750). Importantly, this reaction demonstrates a metal-catalyzed enantioselective unactivated C(sp3)–H functionalization via vinyl cation and constitutes a kinetic resolution reaction based on diyne cyclization. Theoretical calculations further support the mechanism of vinyl cation-involved C(sp3)–H insertion reaction and elucidate the origin of enantioselectivity.
Subject terms: Synthetic chemistry methodology, Reaction mechanisms, Homogeneous catalysis
Site- and stereoselective C–H functionalization is challenging in the synthetic chemistry, and the catalytic enantioselective C(sp3)–H functionalization based on vinyl cations has scarcely explored. Here, the authors report an asymmetric copper-catalyzed tandem diyne cyclization/unactivated C(sp3)–H insertion reaction via a kinetic resolution.
Introduction
Carbon-hydrogen bond is one of the most common chemical bonds in organic frameworks. The enantioselective functionalization of C–H bonds is recognized as a very attractive strategy for synthesizing complex chiral molecules and has been widely used in organic synthesis and pharmaceutical chemistry1–6. In the past decades, several different approaches have been developed to modify C(sp3)–H bonds enantioselectively7,8, including transition metal-catalyzed C–H activation9–11, C–H insertion to a metal carbene or nitrene12–15, and biomimetic catalysis via radical reaction16–18. However, these C–H functionalizations generally involved a relatively activated C(sp3)–H bond next to a heteroatom or at a benzylic and allylic position. In contrast, unactivated C(sp3)–H functionalization is rarely reported, as these C–H bonds possess high bond energy and a similar chemical environment, resulting in site- and stereoselectivity challenges.
Catalytic kinetic resolution (KR) is a powerful and practical method to produce enantioenriched compounds19–21, and has been widely utilized in academia and industry in the past decades, especially for the catalytic asymmetric synthesis of chiral alcohols, amines, etc.22–27. However, the kinetic resolution based on alkynes has rarely been reported. In 2005, Fokin et al. reported the first enantioselective azide-alkyne cycloaddition reaction through kinetic resolution albeit with low selectivity factors28. After that, significant progress based on the azide-alkyne cycloaddition reaction was achieved by Zhou et al.29–33, which provided a series of chiral α-tertiary 1,2,3-triazoles (Fig. 1a). In addition to this strategy, a few examples of kinetic resolution reactions based on alkynes via cyclization have also been invoked34–40, but the nucleophiles are limited to alcohols34,35, alkenes36,37, and acyl groups38–40 (Fig. 1b). To the best of our knowledge, the kinetic resolution of alkynes via direct diyne cyclization41–44 has not yet been realized.
Fig. 1. Kinetic resolution reactions for asymmetric alkyne transformation.

a Kinetic resolution reactions based on azide-alkyne cycloaddition. b Kinetic resolution reactions based on alkynyl cyclization. c This work: enantioselective functionalization of unactivated C(sp3)–H bonds through copper-catalyzed diyne cyclization via kinetic resolution. KR kinetic resolution, FG functional group, PG protecting group.
Over the past decades, the chemistry of vinyl cations has attracted extensive attention due to their unique carbene-like reactivity45,46, and has been well explored in unactivated C(sp3)–H functionalization reactions47–53. However, the catalytic enantioselective C(sp3)–H functionalization based on vinyl cations is extremely difficult, likely due to the lack of catalytic methods for their generation and their high reactivity once formed. Recently, the first asymmetric version was achieved by Nelson et al.54, which was enabled by the imidodiphosphorimidate (IDPi) organocatalyst55 and led to the enantioselective synthesis of [4.3.0] and [3.2.1] bicycles. To our best knowledge, metal-catalyzed asymmetric C(sp3)–H insertion via vinyl cations has not been explored yet. Inspired by our recent studies on the chiral copper(I)-catalyzed diyne cyclization via vinyl cations56–62 and the application of ynamides in N-heterocycle synthesis63–70, we envisioned that copper-catalyzed cyclization of alkyl-substituted N-propargyl ynamides might generate the highly reactive vinyl cation intermediates, which could serve as the equivalent of donor-donor carbenes, and undergo an intramolecular [1,5]-H migration, eventually leading to the enantioenriched pyrrole derivatives and diynes through kinetic resolution. Here we describe the realization of such a chiral copper-catalyzed tandem diyne cyclization/unactivated C(sp3)–H insertion reaction (Fig. 1c). The reaction undergoes a kinetic resolution of the prochiral N-propargyl ynamides, and allows the efficient and practical synthesis of a range of polycyclic pyrroles in good to excellent yields with generally excellent enantioselectivities. Meanwhile, the remaining diyne substrates can be recovered with generally good to excellent yields and enantioselectivities. Significantly, this protocol not only demonstrates a kinetic resolution reaction based on diyne cyclization but also constitutes a metal-catalyzed enantioselective unactivated C(sp3)–H functionalization via vinyl cation. Theoretical calculations further support the reaction mechanism and the origin of enantioselectivity.
Results
To prohibit the background aromatic C–H insertion reaction56, 2,6-dimethylphenyl-substituted N-propargyl ynamide 1a was chosen as the model substrate under our previous related reaction conditions56–62, and selected results are listed in Table 1. First, some typical chiral biphosphine ligands and bisoxazoline (BOX) ligands were employed as chiral ligands in the presence of 10 mol % of Cu(MeCN)4PF6 as the catalyst and 12 mol % of NaBArF4 as the additive. To our delight, the use of various chiral ligands resulted in the kinetic resolution of ynamide 1a (Table 1, entries 1–6), and the use of BOX ligand L6 afforded the desired chiral product 2a with 88% ee (Table 1, entry 6). To further improve the enantioselectivity of this reaction, BOX ligands bearing different dibenzyl groups as the side-arm were investigated. These side-arm BOX (SaBOX) ligands71 significantly impacted the reactivity and enantioselectivity of the reaction. Screening of the SaBOX ligands L7–L10 (Table 1, entries 7–10) showed that the use of L10 led to the formation of 2a in 94% ee and 57.5 selectivity (s) factor (Table 1, entry 10). Typical solvents such as THF, toluene, PhCl, and mxylene were also screened (Table 1, entries 11–14). It was found that using mxylene as the solvent further increased the enantioselectivity of 2a to 95%, and the selectivity factor could be improved to 77.6 (Table 1, entry 14). Gratifyingly, an apparent temperature influence was observed. Decreasing the reaction temperature to 0 °C allowed the formation of the desired 2a in 95% ee with 145.7 selectivity factor, and the diyne substrate 1a was also recovered in 95% ee (Table 1, entry 15).
Table 1.
Optimization of reaction conditions for tandem diyne cyclization/C(sp3)–H insertion of N-propargyl ynamide 1a
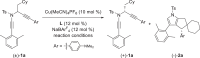 | ||||||
|---|---|---|---|---|---|---|
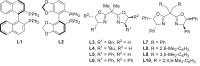 | ||||||
| Entry | L | Reaction conditions | Eesa | Eepa | C (%)b | sc |
| 1 | L1 | DCM, 10 °C, 1 h | 43 | 71 | 37.7 | 8.9 |
| 2 | L2 | DCM, 10 °C, 1 h | 91 | 44 | 67.4 | 7.4 |
| 3 | L3 | DCM, 10 °C, 1 h | 21 | 10 | 67.7 | 1.5 |
| 4 | L4 | DCM, 10 °C, 1 h | 15 | 75 | 16.7 | 8.1 |
| 5 | L5 | DCM, 10 °C, 1 h | <1 | <1 | / | / |
| 6 | L6 | DCM, 10 °C, 1 h | 19 | 88 | 17.8 | 18.9 |
| 7 | L7 | DCM, 10 °C, 3 h | 24 | 82 | 22.6 | 12.8 |
| 8 | L8 | DCM, 10 °C, 3 h | 18 | 95 | 15.9 | 46.5 |
| 9 | L9 | DCM, 10 °C, 3 h | 12 | 92 | 11.5 | 27.0 |
| 10 | L10 | DCM, 10 °C, 3 h | 57 | 94 | 37.7 | 57.5 |
| 11 | L10 | THF, 10 °C, 3 h | 98 | 70 | 58.3 | 24.9 |
| 12 | L10 | toluene, 10 °C, 3 h | 57 | 85 | 40.1 | 21.9 |
| 13 | L10 | PhCl, 10 °C, 3 h | 34 | 93 | 26.8 | 38.4 |
| 14 | L10 | mxylene, 10 °C, 3 h | 66 | 95 | 41.0 | 77.6 |
| 15 | L10 | mxylene, 0 °C, 14 h | 95 | 95 | 50.0 | 145.7 |
Reaction conditions: 1a (0.05 mmol), Cu(MeCN)4PF6 (0.005 mmol), L (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 ml), in Schlenk tubes.
Ts p-toluenesulfonyl, NaBArF4 sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate.
aEes are determined by HPLC analysis.
bConversion (C) = ees/(ees + eep).
cSelectivity factor (s) = ln[1 − C(1 + eep)]/ln[1 − C(1 − eep)].
Bold values indicate the optimal conditions.
With the optimal reaction conditions in hand (Table 1, entry 15), we started to investigate the scope of this tandem diyne cyclization/C(sp3)–H insertion via kinetic resolution under chiral copper catalysis. As depicted in Fig. 2, the reaction proceeded efficiently with a wide range of N-propargyl ynamides 1. First, ynamides bearing different N-protecting groups, such as Ts, Mbs, SO2Ph, Bs, and Ms, were well tolerated under the optimized conditions, affording the chiral polycyclic pyrroles (−)-2a–(−)-2e and recovered ynamides (+)-1a–(+)-1e in good yields with good to excellent enantioselectivities. Next, we turned our attention to the aryl groups at the propargylamide. Substrates with various heteroatom-containing electron-rich aryl groups, including Bn2N-, Et2N-, N-piperidinyl, and N-morpholinyl, were converted into the expected polycyclic pyrroles (−)-2f–(−)-2i in good yields with generally good to excellent enantioselectivities. Notably, the Me group at the ortho-position of the aryl ring had no impact on the enantioselectivity of the reaction, affording the recovered ynamide (+)-1j in 42% yield with 97% ee, and the polycyclic pyrrole (−)-2j in 44% yield with 95% ee. Then, a series of substituted 2,6-dimethylphenyl groups at the ynamide moiety were explored. Diynes bearing both electron-withdrawing and -donating groups on the aromatic ring, such as Me, F, Cl, Br, Ph, and alkynyl, were suitable substrates in this tandem reaction, furnishing the desired (−)-2k–(−)-2p in 39–42% yields with 95–97% ees, and chiral substrates (+)-1k–(+)-1p could also be recovered in 40–42% yields with 93–98% ees. 2,6-Dimethoxy-substituted substrate was also attempted, and the desired product (−)-2q could be obtained. However, the enantiocontrol was not satisfying. To our surprise, mono-substituted substrates, such as ortho methyl-, fluoro-, chloro-, and bromo-substituted diynes, could also deliver the target products (−)-2r–(−)-2u in good yields and enantioselectivities. Furthermore, the reaction also occurred smoothly with meta-and para-substituted substrates, and the corresponding sp3 C–H insertion products (−)-2v–(−)-2ab were obtained in moderate to good yields but with slightly decreased enantioselectivities. It is worth noting that these mono-substituted diynes failed to furnish the desired products in our previous diyne cyclization60 probably due to the competing aromatic C(sp2)–H insertion56. However, in our present mono-substituted cases, the corresponding C(sp2)–H insertion byproducts were almost not observed. Finally, it was found that the alkyl-substituted diyne substrate could also be converted into the desired product (−)-2ac in 49% yield and 90% ee, and (+)-1ac could be recovered in 46% yield and 97% ee. Our attempts to extend the reaction to the PMP- and TIPS-tethered substrates (2ad, 2ae) resulted in the formation of complicated mixtures. The molecular structure of 2b was confirmed by X-ray diffraction (Supplementary Fig. 9).
Fig. 2. Scope of tandem diyne cyclization/C(sp3)–H insertion of N-propargyl ynamides 1.

Reaction conditions: 1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), NaBArF4 (0.024 mmol), L10 (0.024 mmol), mxylene (4 ml), 0 °C, 2–48 h, in Schlenk tubes; yields are those for the isolated products; ees are determined by HPLC analysis. a30 °C. bL8 instead of L10. c15 °C. Mbs = 4-methoxybenzenesulfonyl, Bs = 4-bromobenzenesulfonyl, PMB = 4-methoxyphenyl.
Having established the reactivity of various substituted diyne moieties, we sought to investigate the reactivity with diynes bearing different C(sp3)–H bonds. As shown in Fig. 3, different ring sizes were first examined, including 5-, 7-, 8-, 12-, and 15-membered rings. These diynes underwent smooth tandem cyclization/C(sp3)–H insertion to furnish the desired enantioenriched products (−)-2af–(−)-2aj in 42–46% yields and 90–96% ees, and the corresponding chiral substrates (+)-1af–(+)-1aj were also recovered in good yields and good to excellent enantioselectivities. Next, diynes with different 6-membered rings were investigated, and the target products ((−)-2ak, (−)-2al) were obtained in good to excellent yields and enantioselectivities. Particularly, adamantane-substituted substrate was also applicable to this reaction, furnishing the corresponding polycyclic pyrrole (−)-2am in 45% yield and 98% ee with selectivity factor up to 750.0. In addition to the C(sp3)–H bonds of cyclic substrates, we also screened diynes containing the general C(sp3)–H bond of a tertiary carbon center, e.g., dimethyl, diethyl, and dipropyl substituted substrates. To our delight, the expected chiral products (−)-2an–(−)-2ap were delivered in 37–41% yields and 90–93% ees. Then, the diphenyl-substituted substrate with high reactivity was tried, and the target product (−)-2aq was formed in good yield but with significantly decreased enantioselectivity (71% ee). Moreover, an initial attempt to extend the reaction to substrate with an asymmetric 3 °C(sp3)–H bond resulted in the formation of the desired product (−)-2ar in good yield and enantioselectivity but only with poor diastereoselectivity (1:1.2). Besides the C(sp3)–H bond of a tertiary carbon center, the C(sp3)–H bond on a secondary carbon center was also studied. Interestingly, this substrate also afforded the target chiral product (−)-2as and recovered substrate (+)-1as in good yields and enantioselectivities but with only 1:1.3 diastereoselectivity. Furthermore, an unactivated secondary C(sp3)–H bond was also attempted, but failed to obtain the target product 2at. A likely reason for this is that the secondary C(sp3)–H bonds are electronically unfavoured compared with the tertiary C(sp3)–H bonds.
Fig. 3. Scope of tandem diyne cyclization/C(sp3)–H insertion of N-propargyl ynamides 1.

1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), NaBArF4 (0.024 mmol), L10 (0.024 mmol), mxylene (4 ml), 0 °C, 2–66 h, in Schlenk tubes; yields are those for the isolated products; ees are determined by HPLC analysis; drs are determined by crude 1H-NMR. a−20 °C.
Preparative-scale synthesis and the synthetic utility of this tandem diyne cyclization/C(sp3)–H insertion were then explored. As shown in Fig. 4, the kinetic resolution of the model substrate 1a (2.0 mmol, 1.11 g) under the standard conditions afforded the desired product (−)-2a in 41% yield with 94% ee, and (+)-1a was recovered in 42% yield with 96% ee (Fig. 4a). Meanwhile, 1f (1.0 mmol, 0.70 g) could undergo this kinetic resolution to deliver the target product (−)-2f in 39% yield with 95% ee and the recovered (+)-1f in 40% yield with 98% ee (Fig. 4a). Of note, the chiral diyne (+)-1a could be readily converted into the chiral product (+)-2a in 81% yield without employing the SaBOX ligand, and almost no erosion of the enantiopurity of the product was observed (Fig. 4b). Thus, both enantiomers of polycyclic pyrroles 2 could be synthesized by this strategy. Then, further transformations of the chiral products, such as (−)-2a and (−)-2f, were also conducted. First, the Ts group of (−)-2a was easily removed under basic conditions, affording the expected pyrrole product 3aa in 78% yield (Fig. 4c, (i)). Subsequently, the treatment of (−)-2a with NaBH3CN as the reductant led to the desired dihydropyrrole 3ab bearing two stereocenters in 95% yield with >20:1 dr (Fig. 4c, (ii)). Interestingly, a bridged polycyclic skeleton 3ac containing three stereocenters was also furnished in 83% yield with > 20:1 dr through a Diels-Alder (DA) reaction of the pyrrole ring of (−)-2a with dimethyl acetylenedicarboxylate (Fig. 4c, (iii)). In addition, the NMe2 group of pyrrole (−)-2a could be readily converted into the aryl group in 94% yield via a Pd-catalyzed cross-coupling with the aryl Grignard reagent (Fig. 4c, (iv)). Furthermore, the NBn2 group of pyrrole (−)-2f could also be transformed into the free aniline product 3fa through a debenzylation process in 90% yield (Fig. 4d, (v)), which could undergo further condensation with the corresponding acyl chloride to deliver the amide 3fb in 81% yield (Fig. 4d, (vi)). Similarly, the treatment of (−)-2f with dimethyl acetylenedicarboxylate allowed the formation of the expected 3fc in 87% yield (Fig. 4d, (iii)). Importantly, almost no erosion of the enantiopurity of these products was observed, and excellent diastereoselectivities (dr > 20:1) were achieved in all these cases. Notably, these bicyclo pyrrole skeletons can be found in a variety of bioactive molecules and may have some medicinal interest72,73. The absolute configuration of 3fb and the relative configuration of 3fc were confirmed by X-ray diffraction (Supplementary Figs. 10 and 11), and the former also determined the absolute configuration of the chiral polycyclic pyrroles 2.
Fig. 4. Scale-up reaction and product elaborations.

a Preparative-scale synthesis of products (−)-2a and (−)-2f. b Synthesis of the chiral product (+)-2a from chiral diyne (+)-1a. c Conversion of product (−)-2a into compounds 3aa–3ad. d Conversion of product (−)-2f into compounds 3fa–3fc. Reagents and conditions: (i) KOH (5 equiv), THF/MeOH = 1:1, 80 °C, 8 h. (ii) NaBH3CN (5 equiv), DCM/TFA = 10:1, 0 °C, 1 h. (iii) dimethyl acetylenedicarboxylate (10 equiv), toluene, 100 °C, 12 h. (iv) PhMgBr (1.2 equiv), Pd(PPh3)2Cl2 (5 mol %), THF, rt, 30 min. (v) Pd/C (10% w/w), H2 (1 atm), EtOH/EtOAc = 1:1, 80 °C, 5 h. (vi) p-BrC6H4COCl (1.5 equiv), Et3N (2 equiv), DCM, rt, 2 h.
To probe the reaction mechanism, several control experiments were next carried out. We performed a deuterium labeling experiment with the substrate (±)-[D]*-1a, and found that the deuterium atom was utterly retained in (−)-[D]*-2a under the standard conditions (Fig. 5a). Subsequently, the treatment of diyne (±)-1a with 10 equiv of D2O under the standard conditions revealed a significant deuterium incorporation into the pyrrole ring of (−)-[D]-2a (Fig. 5b). This observation was also found in the reaction of (±)-[D]-1a with 10 equiv of H2O as additive under the standard conditions (Fig. 5b). These results are consistent with our previous work58, in which water-assisted proton transfer was presumably involved in the formation of pyrrole moiety. Finally, the kinetic isotope effect (KIE) experiment was conducted with the mixture of diynes (±)-1a and (±)-[D]*-1a, and the result (kH/kD = 1.1) suggests that the cleavage of C(sp3)–H bond is not the rate-determining step (Fig. 5c).
Fig. 5. Control experiments.
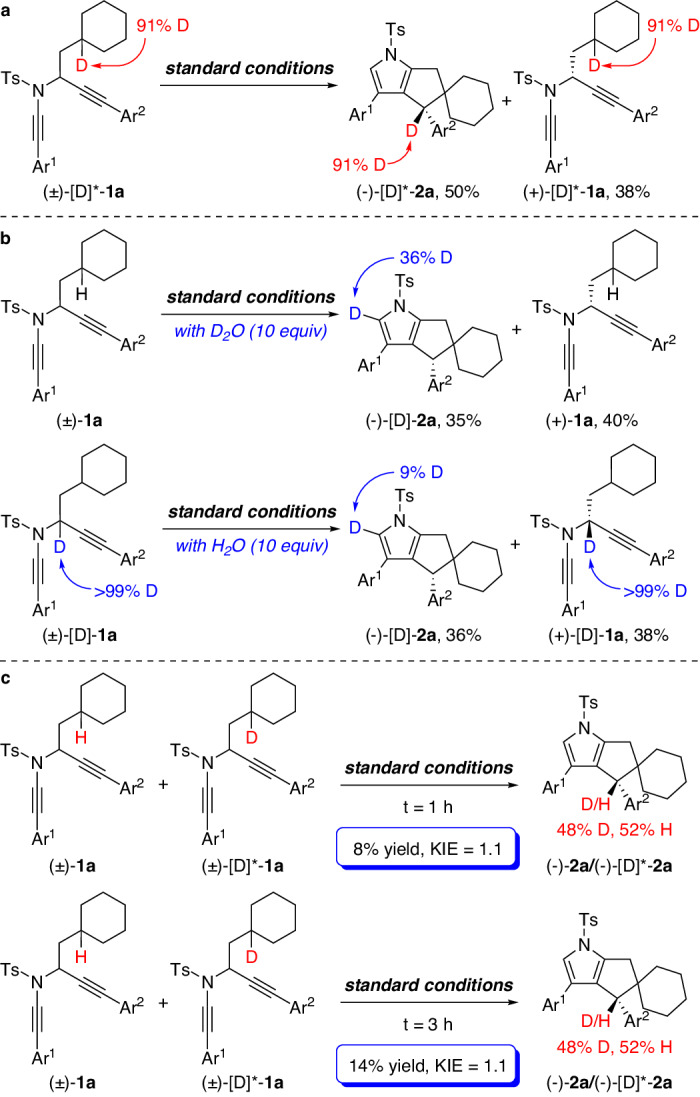
a The reaction of (±)-[D]*-1a under the standard conditions. b Hydrogen/deuterium exchange experiments of (±)-[D]-1a and (±)-1a. c KIE experiments. Ar1, 2,6-dimethylphenyl; Ar2, 4-NMe2phenyl.
Based on the above experimental results, density functional theory (DFT) calculations have been performed to gain more insights into the mechanism for the enantioselective synthesis of product 2a from 1a and elucidate the kinetic behavior of the reaction and the origin of enantioselectivity. As is exhibited in Fig. 6, similar to our previously reported studies56–62, at the beginning of the reaction the coordination of CuL10 copper species preferentially to the diyne (−)-1a leads to a much more stable [Cu]-bound intermediate (−)-A, which subsequently triggers an intramolecular cyclization to afford the vinyl cation intermediate (−)-B via a transition state (−)-TSA. Then, a C(sp3)–H insertion process gives the copper carbene intermediate (−)-C via a transition state (−)-TSB. Finally, as reported in our previous publications56–62, a H2O-assisted [1,4]-H migration and demetallation process occurs to afford the product (−)-2a, via a transition state (−)-TSC referring to the H-abstraction transition state with large barrier height (subsequent H-migration and demetallation step is omitted for slight barrier height). Afterwards, the recoordination of the desorbed catalyst CuL10 to diyne (+)-1a occurs and leads to the product (+)-2a, with much less reactivity due to a much larger RDS barrier height (28.2 kcal/mol) compared to the RDS barrier height leading to (−)-2a (24.9 kcal/mol), thus accounting for the kinetic behaviors of the reaction. The calculated energy profiles of the reaction with both enantiomers of 1a indicate that the irreversible enantiodetermining step is the intramolecular cyclization step. The optimized structures and barrier height of the enantioselective intramolecular cyclization transition states (−)-TSA and (+)-TSA are also exhibited in Fig. 6. Transition state (+)-TSA (from (+)-1a) is disfavored because of the H-H steric repulsion between the Ts group and the side arm of SaBOX ligand as a result of the more proximal orientation of the bulky Ts group. Such steric repulsions are not present in the favored transition state (−)-TSA (from (−)-1a), which leads to the difference in free energy between the two competing transition states.
Fig. 6. Plausible reaction mechanism.

Relative free energies (ΔG, in kcal/mol) of all the transition states and intermediates were computed at the SMD(solvent = mxylene)-PBE0-D3/Def2-TZVP//SMD(solvent = mxylene)-B3LYP-D3/6–31G(d,p) level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; brown = Cu.
Discussion
In summary, we have disclosed a chiral copper-catalyzed tandem diyne cyclization/C(sp3)–H insertion reaction via kinetic resolution, furnishing chiral polycyclic pyrroles and diynes in generally excellent enantioselectivities. Importantly, this reaction not only represents a kinetic resolution reaction based on diyne cyclization and a metal-catalyzed enantioselective unactivated C(sp3)–H functionalization via vinyl cation, but also constitutes an asymmetric unactivated C(sp3)–H insertion based on alkynes. In addition, theoretical calculations have been carried out to understand the reaction mechanism and the origin of enantioselectivity. We believe these findings will offer further perspectives and explorations in the field of kinetic resolution and enantioselective C(sp3)–H functionalization based on alkynes and vinyl cations.
Methods
General
For 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra of compounds in this manuscript and details of the synthetic procedures as well as more reaction condition screening, see Supplementary Information.
General procedure for the chiral copper-catalyzed kinetic resolution of diynes 1
The powered Cu(MeCN)4PF6 (0.02 mmol, 7.5 mg), L10 (0.024 mmol, 17.4 mg), and NaBArF4 (0.024 mmol, 21.3 mg) were introduced into an oven-dried Schlenk tube under argon atmosphere. After mxylene (2 ml) was injected into the Schlenk tube, the solution was stirred at rt under the argon atmosphere for 2 h. Then the reaction was cooled to 0 °C, and N-propargyl ynamide 1 (0.2 mmol) in mxylene (2 ml) was introduced into the system dropwise. The resulting mixture was stirred at indicating temperature and the progress of the reaction was monitored by TLC or HPLC. After concentration in vacuo, the residue was purified by flash chromatography on silica gel (eluent: hexanes/EA or hexanes/DCM) to give the final product (−)-2 and (+)-1.
Supplementary information
Source data
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22125108, 22331004, 22121001 and 92056104), the President Research Funds from Xiamen University (20720210002), and NFFTBS (J1310024).
Author contributions
Y.-B.C., Z.-Q.W., R.C., and B.Z. performed experiments. L.-G.L. and X.L. performed DFT calculations. L.-W.Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2270341 (2b), 2270342 (3fb) and 2270343 (3fc). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files or from the corresponding authors on request. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xin Lu, Email: xinlu@xmu.edu.cn.
Long-Wu Ye, Email: longwuye@xmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-46288-7.
References
- 1.Yu, J.-Q. & Shi, Z. C–H Activation (Topics in Current Chemistry 292) (Springer, 2010). [PubMed]
- 2.Hartwig JF, Larsen MA. Undirected, homogeneous C–H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi J, Yamaguchi AD, Itami K. C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 4.Davies HML, Du Bois J, Yu J-Q. C–H functionalization in organic synthesis. Chem. Soc. Rev. 2011;40:1855–1856. doi: 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]
- 5.McMurray L, O’Hara F, Gaunt MJ. Recent developments in natural product synthesis using metal-catalysed C–H bond functionalization. Chem. Soc. Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 6.Gutekunst WR, Baran PS. C–H functionalization logic in total synthesis. Chem. Soc. Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 7.Saint-Denis TG, Zhu RY, Chen G, Wu QF, Yu JQ. Enantioselective C(sp3)‒H bond activation by chiral transition metal catalysts. Science. 2018;359:eaao4798. doi: 10.1126/science.aao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park Y, Kim Y, Chang S. Transition metal-catalyzed C–H amination: scope, mechanism, and applications. Chem. Rev. 2017;117:9247–9301. doi: 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- 9.Lucas EL, et al. Palladium-catalyzed enantioselective β-C(sp3)−H activation reactions of aliphatic acids: a retrosynthetic surrogate for enolate alkylation and conjugate addition. Acc. Chem. Res. 2022;55:537–550. doi: 10.1021/acs.accounts.1c00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton CG, Wang SG, Oliveira CC, Cramer N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem. Rev. 2017;117:8908–8976. doi: 10.1021/acs.chemrev.6b00692. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, et al. Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science. 2016;353:1023–1027. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies HML, Liao K. Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization. Nat. Rev. Chem. 2019;3:347–360. doi: 10.1038/s41570-019-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu JCK, Rovis T. Complementary strategies for directed C(sp3)−H functionalization: a comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbene/nitrene transfer. Angew. Chem. Int. Ed. 2018;57:62–101. doi: 10.1002/anie.201703743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Díaz-Requejo MM, et al. Intermolecular copper-catalyzed carbon−hydrogen bond activation via carbene insertion. J. Am. Chem. Soc. 2002;124:896–897. doi: 10.1021/ja016798j. [DOI] [PubMed] [Google Scholar]
- 15.Caballero A, et al. Silver-catalyzed C–C bond formation between methane and ethyl diazoacetate in supercritical CO2. Science. 2011;332:835–838. doi: 10.1126/science.1204131. [DOI] [PubMed] [Google Scholar]
- 16.Golden DL, Suh SE, Stahl SS. Radical C(sp3)–H functionalization and cross-coupling reactions. Nat. Rev. Chem. 2022;6:405–427. doi: 10.1038/s41570-022-00388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Empel C, Jana S, Koenigs RM. C–H functionalization via iron-catalyzed carbene-transfer reactions. Molecules. 2020;25:880. doi: 10.3390/molecules25040880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang RK, et al. Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization. Nature. 2019;565:67–72. doi: 10.1038/s41586-018-0808-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng T, Li S, Yang D, Wang L. Recent advances in intramolecular kinetic resolution reactions. Org. Chem. Front. 2023;10:3401–3428. doi: 10.1039/D3QO00563A. [DOI] [Google Scholar]
- 20.Pellissier H. Catalytic non-enzymatic kinetic resolution. Adv. Synth. Catal. 2011;353:1613–1666. doi: 10.1002/adsc.201100111. [DOI] [Google Scholar]
- 21.Vedejs E, Jure M. Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed. 2005;44:3974–4001. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 22.Müller CE, Schreiner PR. Organocatalytic enantioselective acyl transfer onto racemic as well as meso alcohols, amines, and thiols. Angew. Chem. Int. Ed. 2011;50:6012–6042. doi: 10.1002/anie.201006128. [DOI] [PubMed] [Google Scholar]
- 23.Liu CX, et al. Kinetic resolution of planar chiral metallocenes using Rh-catalysed enantioselective C–H arylation. Nat. Synth. 2023;2:49–57. doi: 10.1038/s44160-022-00177-3. [DOI] [Google Scholar]
- 24.Mukherjee K, et al. Kinetic resolution of sulfur-stereogenic sulfoximines by Pd(II)-MPAA catalyzed C–H arylation and olefination. Chem. Sci. 2021;12:14863–14870. doi: 10.1039/D1SC04299H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma C, et al. Atroposelective access to oxindole-based axially chiral styrenes via the strategy of catalytic kinetic resolution. J. Am. Chem. Soc. 2020;142:15686–15696. doi: 10.1021/jacs.0c00208. [DOI] [PubMed] [Google Scholar]
- 26.Rajkumar S, He S, Yang X. Kinetic resolution of tertiary 2-alkoxycarboxamido-substituted allylic alcohols by chiral phosphoric acid catalyzed intramolecular transesterification. Angew. Chem. Int. Ed. 2019;58:10315–10319. doi: 10.1002/anie.201905034. [DOI] [PubMed] [Google Scholar]
- 27.Chu L, Xiao KJ, Yu JQ. Room-temperature enantioselective C–H iodination via kinetic resolution. Science. 2014;346:451–455. doi: 10.1126/science.1258538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng JC, Fokin VV, Finn MG. Kinetic resolution by copper-catalyzed azide–alkyne cycloaddition. Tetrahedron Lett. 2005;46:4543–4546. doi: 10.1016/j.tetlet.2005.05.019. [DOI] [Google Scholar]
- 29.Wang C, Zhou F, Zhou J. Recent advances in the enantioselective copper(I)-catalyzed azide-alkyne cycloaddition reaction. Chin. J. Org. Chem. 2020;40:3065–3077. doi: 10.6023/cjoc202005020. [DOI] [Google Scholar]
- 30.Liao K, et al. Highly enantioselective CuAAC of functional tertiary alcohols featuring an ethynyl group and their kinetic resolution. Angew. Chem. Int. Ed. 2021;60:8488–8493. doi: 10.1002/anie.202016286. [DOI] [PubMed] [Google Scholar]
- 31.Zhu RY, Chen L, Hu XS, Zhou F, Zhou J. Enantioselective synthesis of P-chiral tertiary phosphine oxides with an ethynyl group via Cu(i)-catalyzed azide–alkyne cycloaddition. Chem. Sci. 2020;11:97–106. doi: 10.1039/C9SC04938J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu EC, Topczewski JJ. Enantioselective copper catalyzed alkyne–azide cycloaddition by dynamic kinetic resolution. J. Am. Chem. Soc. 2019;141:5135–5138. doi: 10.1021/jacs.9b01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brittain WDG, Buckley BR, Fossey JS. Kinetic resolution of alkyne-substituted quaternary oxindoles via copper catalysed azide–alkyne cycloadditions. Chem. Commun. 2015;51:17217–17220. doi: 10.1039/C5CC04886A. [DOI] [PubMed] [Google Scholar]
- 34.Chen PF, Zhou B, Wu P, Wang B, Ye LW. Brønsted acid catalyzed dearomatization by intramolecular hydroalkoxylation/Claisen rearrangement: diastereo- and enantioselective synthesis of spirolactams. Angew. Chem. Int. Ed. 2021;60:27164–27170. doi: 10.1002/anie.202113464. [DOI] [PubMed] [Google Scholar]
- 35.Zhou B, et al. Stereoselective synthesis of medium lactams enabled by metal-free hydroalkoxylation/stereospecific [1,3]-rearrangement. Nat. Commun. 2019;10:3234. doi: 10.1038/s41467-019-11245-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohan PT, Toste FD. Well-defined chiral gold(III) complex catalyzed direct enantioconvergent kinetic resolution of 1,5-enynes. J. Am. Chem. Soc. 2017;139:11016–11019. doi: 10.1021/jacs.7b06025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He M, Lei A, Zhang X. Enantioselective syntheses of 3,4,5-trisubstituted γ-lactones: formal synthesis of (-)-blastmycinolactol. Tetrahedron Lett. 2005;46:1823–1826. doi: 10.1016/j.tetlet.2005.01.112. [DOI] [Google Scholar]
- 38.Deng L, et al. Kinetic resolution via Rh-catalyzed C–C activation of cyclobutanones at room temperature. J. Am. Chem. Soc. 2019;141:16260–16265. doi: 10.1021/jacs.9b09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka K, Fu GC. Parallel kinetic resolution of 4-alkynals catalyzed by Rh(I)/Tol-BINAP: synthesis of enantioenriched cyclobutanones and cyclopentenones. J. Am. Chem. Soc. 2003;125:8078–8079. doi: 10.1021/ja035489l. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka K, Fu GC. Enantioselective synthesis of cyclopentenones via Rhodium-catalyzed kinetic resolution and desymmetrization of 4-alkynals. J. Am. Chem. Soc. 2002;124:10296–10297. doi: 10.1021/ja0266161. [DOI] [PubMed] [Google Scholar]
- 41.Asiri AM, Hashmi ASK. Gold-catalysed reactions of diynes. Chem. Soc. Rev. 2016;45:4471–4503. doi: 10.1039/C6CS00023A. [DOI] [PubMed] [Google Scholar]
- 42.Dutta S, Mallick RK, Prasad R, Gandon V, Sahoo AK. Alkyne versus ynamide reactivity: regioselective radical cyclization of yne-ynamides. Angew. Chem. Int. Ed. 2019;58:2289–2294. doi: 10.1002/anie.201811947. [DOI] [PubMed] [Google Scholar]
- 43.Prabagar B, Mallick RK, Prasad R, Gandon V, Sahoo AK. Umpolung reactivity of ynamides: an unconventional [1,3]-sulfonyl and [1,5]-sulfinyl migration cascade. Angew. Chem. Int. Ed. 2019;58:2365–2370. doi: 10.1002/anie.201813143. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh N, Nayak S, Sahoo AK. Gold(I)-catalyzed 6-endo-dig hydrative cyclization of an alkyne-tethered ynamide: access to 1,6-dihydropyridin-2(3H)ones. Chem. Eur. J. 2013;19:9428–9433. doi: 10.1002/chem.201301599. [DOI] [PubMed] [Google Scholar]
- 45.Niggemann M, Gao S. Are vinyl cations finally coming of age? Angew. Chem. Int. Ed. 2018;57:16942–16944. doi: 10.1002/anie.201810701. [DOI] [PubMed] [Google Scholar]
- 46.Byrne PA, Kobayashi S, Würthwein EU, Ammer J, Mayr H. Why are vinyl cations sluggish electrophiles? J. Am. Chem. Soc. 2017;139:1499–1511. doi: 10.1021/jacs.6b10889. [DOI] [PubMed] [Google Scholar]
- 47.Liu XJ, Xu Y, Tang C, Qian PC, Ye LW. Unactivated C(sp3)–H functionalization via vinyl cations. Sci. China Chem. 2022;65:20–30. doi: 10.1007/s11426-021-1117-2. [DOI] [Google Scholar]
- 48.Wang Y, Cai PJ, Yu ZX. Mechanistic study on gold-catalyzed cycloisomerization of dienediynes involving aliphatic C–H functionalization and inspiration for developing a new strategy to access polycarbocycles. J. Am. Chem. Soc. 2020;142:2777–2786. doi: 10.1021/jacs.9b10362. [DOI] [PubMed] [Google Scholar]
- 49.Wigman B, et al. Vinyl carbocations generated under basic conditions and their intramolecular C–H insertion reactions. J. Am. Chem. Soc. 2019;141:9140–9144. doi: 10.1021/jacs.9b02110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikeuchi T, Inuki S, Oishi S, Ohno H. Gold(I)-catalyzed cascade cyclization reactions of allenynes for the synthesis of fused cyclopropanes and acenaphthenes. Angew. Chem. Int. Ed. 2019;58:7792–7796. doi: 10.1002/anie.201903384. [DOI] [PubMed] [Google Scholar]
- 51.Popov S, et al. Teaching an old carbocation new tricks: intermolecular C–H insertion reactions of vinyl cations. Science. 2018;361:381–387. doi: 10.1126/science.aat5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang F, et al. Cu-catalyzed cascades to carbocycles: union of diaryliodonium salts with alkenes or alkynes exploiting remote carbocations. J. Am. Chem. Soc. 2014;136:8851–8854. doi: 10.1021/ja504361y. [DOI] [PubMed] [Google Scholar]
- 53.Miguélez R, et al. C–H activation of unbiased C(sp3)–H bonds: gold(I)-catalyzed cycloisomerization of 1-bromoalkynes. Angew. Chem. Int. Ed. 2023;62:e202305296. doi: 10.1002/anie.202305296. [DOI] [PubMed] [Google Scholar]
- 54.Nistanaki SK, et al. Catalytic asymmetric C–H insertion reactions of vinyl carbocations. Science. 2022;378:1085–1091. doi: 10.1126/science.ade5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreyer L, Properzi R, List B. IDPi catalysis. Angew. Chem. Int. Ed. 2019;58:12761–12777. doi: 10.1002/anie.201900932. [DOI] [PubMed] [Google Scholar]
- 56.Hong FL, et al. Generation of donor/donor copper carbenes through copper-catalyzed diyne cyclization: enantioselective and divergent synthesis of chiral polycyclic pyrroles. J. Am. Chem. Soc. 2019;141:16961–16970. doi: 10.1021/jacs.9b09303. [DOI] [PubMed] [Google Scholar]
- 57.Hong FL, et al. Copper-catalyzed asymmetric reaction of alkenyl diynes with styrenes by formal [3+2] cycloaddition via Cu-containing all-carbon 1,3-dipoles: access to chiral pyrrole-fused bridged [2.2.1] skeletons. J. Am. Chem. Soc. 2020;142:7618–7626. doi: 10.1021/jacs.0c01918. [DOI] [PubMed] [Google Scholar]
- 58.Zhu XQ, et al. Copper-catalyzed asymmetric cyclization of alkenyl diynes: method development and new mechanistic insights. Chem. Sci. 2021;12:9466–9474. doi: 10.1039/D1SC02773E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong FL, et al. Copper-catalyzed asymmetric diyne cyclization via [1,2]-Stevens-type rearrangement for the synthesis of chiral chromeno[3,4-c]pyrroles. Angew. Chem. Int. Ed. 2022;61:e202115554. doi: 10.1002/anie.202115554. [DOI] [PubMed] [Google Scholar]
- 60.Qi LJ, et al. Enantioselective copper-catalyzed formal [2+1] and [4+1] annulations of diynes with ketones via carbonyl ylides. Angew. Chem. Int. Ed. 2022;61:e202210637. doi: 10.1002/anie.202210637. [DOI] [PubMed] [Google Scholar]
- 61.Chen YB, et al. Construction of axially chiral arylpyrroles via atroposelective diyne cyclization. Angew. Chem. Int. Ed. 2023;62:e202303670. doi: 10.1002/anie.202303670. [DOI] [PubMed] [Google Scholar]
- 62.Zhou JJ, et al. Copper-catalyzed enantioselective diyne cyclization via C(sp2)–O bond cleavage. Chem. Sci. 2023;14:3493–3500. doi: 10.1039/D2SC06152J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu YC, Zhao Y, Wan B, Chen QA. Reactivity of ynamides in catalytic intermolecular annulations. Chem. Soc. Rev. 2021;50:2582–2625. doi: 10.1039/D0CS00283F. [DOI] [PubMed] [Google Scholar]
- 64.Lynch CC, Sripada A, Wolf C. Asymmetric synthesis with ynamides: unique reaction control, chemical diversity and applications. Chem. Soc. Rev. 2020;49:8543–8583. doi: 10.1039/D0CS00769B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong FL, Ye LW. Transition-metal catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis. Acc. Chem. Res. 2020;53:2003–2019. doi: 10.1021/acs.accounts.0c00417. [DOI] [PubMed] [Google Scholar]
- 66.Chen YB, Qian P-C, Ye LW. Brønsted acid-mediated reactions of ynamides. Chem. Soc. Rev. 2020;49:8897–8909. doi: 10.1039/D0CS00474J. [DOI] [PubMed] [Google Scholar]
- 67.Luo J, et al. Exploiting remarkable reactivities of ynamides: opportunities in designing catalytic enantioselective reactions. ACS Catal. 2020;10:13978–13992. doi: 10.1021/acscatal.0c04180. [DOI] [Google Scholar]
- 68.Zhou B, Tan TD, Zhu XQ, Shang M, Ye LW. Reversal of regioselectivity in ynamide chemistry. ACS Catal. 2019;9:6393–6406. doi: 10.1021/acscatal.9b01851. [DOI] [Google Scholar]
- 69.Evano G, Theunissen C, Lecomte M. Ynamides: powerful and versatile reagents for chemical synthesis. Aldrichimica Acta. 2015;48:59–70. [Google Scholar]
- 70.Wang XN, et al. Ynamides in ring forming transformations. Acc. Chem. Res. 2014;47:560–578. doi: 10.1021/ar400193g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao S, Sun XL, Tang Y. Side arm strategy for catalyst design: modifying bisoxazolines for remote control of enantioselection and related. Acc. Chem. Res. 2014;47:2260–2272. doi: 10.1021/ar800104y. [DOI] [PubMed] [Google Scholar]
- 72.Bolli, M. et al. Preparation of 4-carbonyl substituted 1,1,2-trimethyl-1a,4,5,5a-tetrahydro-1H-4-aza-cyclopropa[a]pentalene derivatives as agonists for the G protein-coupled receptor S1P1/EDG1 and immunosuppressive agents. World patent 2005123677 (2005).
- 73.Yang, R. et al. Bicyclic compounds as HIF–2α protein inhibitors and their preparation, pharmaceutical compositions and use in the treatment of diseases. World patent 2022063115 (2022).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2270341 (2b), 2270342 (3fb) and 2270343 (3fc). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its Supplementary Information files or from the corresponding authors on request. Source data are provided with this paper.