

Key Points
-
•
Giroctocogene fitelparvovec is the first gene therapy for hemophilia A that uses a recombinant AAV serotype 6–based vector.
-
•
Giroctocogene fitelparvovec was generally well tolerated with appropriate clinical management and showed efficacy at the highest dose.
Visual Abstract
Abstract
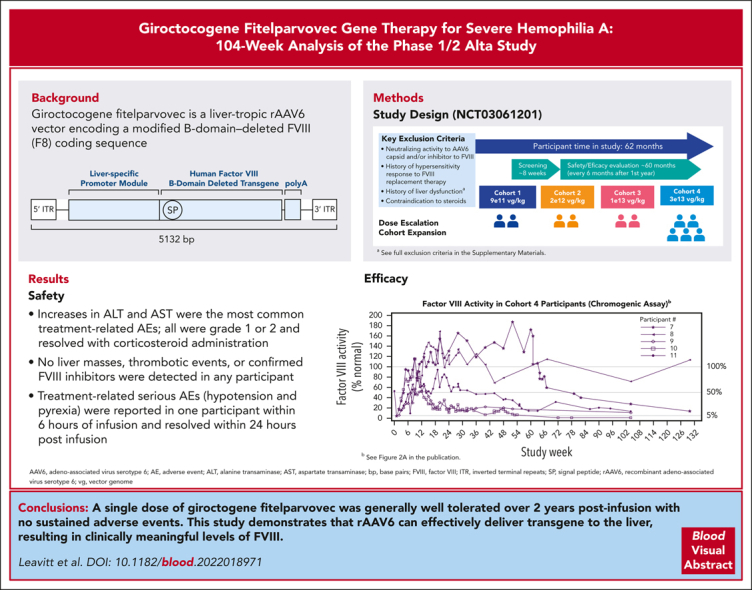
Patients with hemophilia A require exogenous factor VIII (FVIII) or nonfactor hemostatic agents to prevent spontaneous bleeding events. Adeno-associated virus (AAV) vector–based gene therapy is under clinical investigation to enable endogenous FVIII production. Giroctocogene fitelparvovec is a recombinant AAV serotype 6 vector containing the coding sequence for the B-domain–deleted human F8 gene. In the ongoing phase 1/2, dose-ranging Alta study, 4 sequential cohorts of male participants with severe hemophilia A received a single IV dose of giroctocogene fitelparvovec. The primary end points are safety and changes in circulating FVIII activity. Interim results up to 214 weeks after treatment for all participants are presented. Eleven participants were dosed. Increases in alanine and aspartate aminotransferases were the most common treatment-related adverse events (AEs), which resolved with corticosteroid administration. Two treatment-related serious AEs (hypotension and pyrexia) were reported in 1 participant within 6 hours of infusion and resolved within 24 hours after infusion. At the highest dose level (3 × 1013 vg/kg; n = 5), the mean circulating FVIII activity level at week 52 was 42.6% (range, 7.8%-122.3%), and at week 104 it was 25.4% (range, 0.9%-71.6%) based on a chromogenic assay. No liver masses, thrombotic events, or confirmed inhibitors were detected in any participant. These interim 104-week data suggest that giroctocogene fitelparvovec is generally well tolerated with appropriate clinical management and has the potential to provide clinically meaningful FVIII activity levels, as indicated by the low rate of bleeding events in the highest dose cohort. This trial was registered at www.clinicaltrials.gov as #NCT03061201.
Leavitt and colleagues report on the 2-year outcomes of gene therapy for hemophilia A with giroctocogene fitelparvovec, an adeno-associated virus vector carrying the coding sequence for the B-domain–deleted human factor VIII (FVIII) gene. At the highest dose, the mean circulating FVIII level at 1 year was 42.6%, with a range from 7.8% to 122.3%. Levels fell with time, with a mean level of 25.4% at 2 years. All of the patients at the highest dose level had meaningful clinical responses, with a low rate of bleeding events.
Introduction
Gene therapy for hemophilia A is potentially a 1-time treatment. Adeno-associated virus (AAV) vector–based gene therapies are under clinical investigation.1 Challenges to the development of AAV-based gene therapy include the presence of preexisting anti-AAV–neutralizing antibodies (NAbs), which can limit the pool of eligible patients, and posttreatment elevated transaminase levels, potentially due to immune responses stimulated by hepatocytes transiently displaying viral capsid antigens, which can diminish the treatment effect.2 Assessment of baseline NAb status and treatment with corticosteroids at the initial signs of a transaminase elevation may address these challenges.3
To our knowledge, giroctocogene fitelparvovec (PF-07055480, formerly SB-525; Pfizer Inc and Sangamo Therapeutics) is the first gene therapy for hemophilia A using a recombinant AAV (rAAV) serotype 6 (AAV6)–based vector. This serotype was chosen based on preclinical studies demonstrating a high degree of liver tropism and in vivo proof of concept suggesting clinical application.4, 5, 6, 7, 8, 9 Indeed, AAV6 was recently shown to target zinc-finger nucleases in the liver with a favorable safety profile, providing further clinical evidence supporting development of an AAV6-based vector for the treatment of hemophilia A.5 AAV6 antibody seroprevalence is comparable with other AAV serotypes, and the availability of multiple serotypes may increase the population of patients that can receive AAV-based gene therapy.10,11 Giroctocogene fitelparvovec carries a B-domain–deleted F8 complementary DNA encoding the same amino acid sequence as moroctocog α, followed by a polyA tail under the control of a liver-specific promoter using cis-acting regulatory modules to promote liver expression.12 The entire expression cassette is flanked by AAV2 inverted terminal repeats (supplemental Figure 1, available on the Blood website) and comprises a bioengineered hybrid liver promoter derived from the minimal transthyretin promoter. After culturing and expansion of the producer cell bank, rAAV production is initiated in the production bioreactor and continues until harvest, when the rAAVs are isolated using nuclease. The drug substance undergoes further purification before it is buffered and excipients are added to reach the desired concentration.
AAV-based gene therapy for the treatment of both hemophilia A and B has been met with encouraging results, yielding approved products in both the United States13,14 and the European Union.15,16 These studies have shown a favorable overall safety profile associated with marked clinical benefit with vectors leveraging different AAV serotypes. We present ≥2 years (104 weeks) of interim safety and efficacy data per participant with giroctocogene fitelparvovec, leveraging an AAV serotype that has not yet been tested clinically for hemophilia A.
Methods
Trial design
Alta is an ongoing, multicenter, dose-ranging, single-dose, 5-year US study of giroctocogene fitelparvovec (NCT03061201) that began in June 2017; this analysis includes data obtained through 1 October 2021. The study is being conducted in accordance with the Declaration of Helsinki, the Council for International Organizations of Medical Sciences International Ethical Guidelines, and the International Council for Harmonisation Good Clinical Practice guidelines. All participants provided written informed consent before participation.
This study used an adaptive design, with escalating doses starting at 9 × 1011 vg/kg to establish giroctocogene fitelparvovec dose levels sufficient to achieve factor VIII (FVIII) activity of 40% to 100% of normal. After a screening period of ∼8 weeks, the first participant in cohort 1 received a single IV dose (supplemental Figure 2). Once safety was deemed acceptable 6 weeks after dosing, a second participant received the same dose. Cumulative data, including FVIII expression and any dose-limiting toxicities, were then reviewed by an independent safety monitoring committee to assess the appropriateness of dose alteration, cohort expansion, or the addition of cohort(s) at higher dose levels.
On treatment administration day, participants were admitted to the infusion center, where they were observed for ∼24 hours after infusion. Participants on prophylactic FVIII replacement therapy before study treatment switched to on-demand therapy ∼2 weeks after infusion.
For 20 weeks after dosing, FVIII activity was monitored at least weekly, and hepatic enzymes were monitored at least twice weekly. For participants with alanine aminotransferase (ALT) levels > 1.5 × baseline, corticosteroid treatment was administered as described in the supplemental Materials.
Participants
The study enrolled males aged ≥18 years with severe hemophilia A (FVIII activity < 1% of normal) with ≥150 prior treatment or exposure days to FVIII concentrates or cryoprecipitate and who had ≥12 bleeding episodes over the 12 months preceding screening, if using on-demand treatment. Race/ethnicity information was self-reported by the patients. Candidates with preexisting anti-AAV6 NAbs, current or previous FVIII inhibitors, or chronic renal or liver disease were excluded. Full eligibility criteria are shown in the supplemental Materials.
End points
The primary end points were safety and efficacy. The primary end points for safety and tolerability included adverse events (AEs), serious AEs (SAEs), and clinical laboratory assessments. Detection of malignancy was included in long-term monitoring for SAEs, with assessments including magnetic resonance imaging of the liver or computerized tomography of the abdomen, alpha fetoprotein at weeks 12, 24, and 52 and annually thereafter, an abdominal examination for hepatomegaly, and routine liver panel assessments. The primary end point for efficacy was the change in circulating FVIII activity. The time course for FVIII expression was evaluated by a central laboratory using chromogenic (Coatest SP4 FVIII chromogenic assay kit [Chromogenix] on the BCSXP analyzer [Siemens Healthcare Diagnostics]) and 1-stage clotting (Actin FSL [Siemens Healthcare Diagnostics] on the BCSXP analyzer [Siemens Healthcare Diagnostics]) assays at screening, baseline, on the day of infusion, day 7 after infusion, weekly from weeks 2 through 20, then every 4 weeks beginning at week 24 through week 52, and every 6 months from week 52 to month 60 after infusion. FVIII activity data collected within 96 hours after administration of FVIII replacement therapy or after resumption of a prophylaxis regimen were excluded from analysis.
Secondary end points included change from baseline in use of FVIII replacement therapy, frequency and severity of bleeding episodes, FVIII inhibitor levels, and rAAV vector shedding in body fluids. Bleeding episodes (including date, time, location, and etiology) and FVIII concentrate usage (including date, time, reason, and dose) were self-reported by participants using handheld e-diaries. FVIII concentrate usage before baseline was collected retrospectively and via e-diary before giroctocogene fitelparvovec infusion. The presence of FVIII inhibitor was determined by a central laboratory using the Nijmegen-Bethesda assay. Vector shedding was assessed by polymerase chain reaction to detect and quantify vector DNA in plasma. Plasma samples were collected at 12 hours ± 1 hour after the start of infusion, and saliva, urine, stool, and semen samples were collected at weeks 1 and 2, then all samples were collected every 2 to 4 weeks until levels were undetectable in 3 consecutive specimens. Interferon-gamma enzyme-linked ImmunoSpot (ELISpot; Cellular Technology Limited, Shaker Heights, OH) was used to detect a proliferation of FVIII and capsid-specific T cells at baseline and week 6 (±3 days), with additional samples to be collected if a 50% drop in FVIII activity was detected and/or before the initiation of corticosteroid treatment.
Statistical analysis
Data analyses were descriptive in nature and were based on the safety population, defined as all participants who received any portion of study treatment. AEs were coded using the Medical Dictionary for Regulatory Activities version 24.0.
Plasma FVIII activity levels were plotted using units of % normal, defined as IU/mL × 100. Group-level plots were limited to protocol-defined study visits, whereas individual participant plots also included data points from unscheduled visits. Results with values below the limit of quantification were assigned a value of 0.009 IU/mL (0.9%) for analysis and plotting.
The pregene therapy annualized bleeding rate (ABR) was based on the total number of treated and untreated bleeding events reported during the 12-month prescreening. The post-gene therapy treated ABR was based on spontaneous and traumatic bleeding events (excluding bleed episodes associated with surgery or postsurgical rehabilitation), calculated as the number of episodes requiring exogenous FVIII administration during the participant’s observation period, divided by the number of days in the corresponding period, multiplied by 365.25. This observation period was defined as the period from day 22 after gene therapy to the first of (1) FVIII prophylaxis reinitiation (day before reinitiation), (2) the day of study completion or discontinuation, or (3) the planned data cutoff. Postinfusion total ABR (treated and untreated) events were calculated in a similar manner; however, all bleeding episodes were included, regardless of exogenous FVIII administration.
The pregene therapy annualized infusion rate (AIR) was calculated as the number of exogenous FVIII administrations between 30 days before screening visit and just before the giroctocogene fitelparvovec infusion, divided by the number of days in the corresponding period, multiplied by 365.25. Postinfusion AIR was calculated as the number of FVIII administrations on or after day 22 after infusion up to the data cutoff date or date of study completion or discontinuation (whichever occurred first), divided by the number of days in the corresponding observation period, multiplied by 365.25. Statistical analyses were performed using SAS system version 9.4.
Results
Participants
Eleven of 37 unique screened candidates were enrolled into 4 dose cohorts (supplemental Figure 3): cohort 1 (9 × 1011 vg/kg; participants 1 and 2), cohort 2 (2 × 1012 vg/kg; participants 3 and 4), cohort 3 (1 × 1013 vg/kg; participants 5 and 6), and cohort 4 (3 × 1013 vg/kg; participants 7, 8, 9, 10, and 11). Reasons for screening failure were detection of anti-AAV6 NAbs (n = 15), markers of hepatic inflammation or overt or occult cirrhosis (n = 3), or hematologic abnormalities (≥2 occurrences of any of the following: hemoglobin < 10 g/dL, platelets < 100 × 103/μL, or white blood cells < 4 × 103/μL; n = 1); individual candidates were excluded for other reasons, including infection, FVIII treatment less than 150 prior exposure days, or investigator decision. Additional information about screening failures is provided in the supplemental Materials. As on the analysis cutoff date, follow-up ranged from ∼114 weeks to 214 weeks. One participant in cohort 3 (participant 6) missed a visit at week 5, was lost to follow-up ∼35 weeks after dosing, and rejoined the study at ∼130 weeks.
Participants had a mean age of 30.3 years (range, 19-47; Table 1). In the 12 months before gene therapy infusion, participants had a mean (standard deviation [SD]) of 7.5 (8.8) total bleeding episodes (Table 2); 10 of 11 participants (91%) were receiving FVIII prophylaxis. A mean (SD) of 8.8 (8.3) bleeding events occurred in cohort 4 participants, who were all on prophylaxis (Table 2).
Table 1.
Participant demographics
| Characteristic | Cohort 1 (n = 2) | Cohort 2 (n = 2) | Cohort 3 (n = 2) | Cohort 4 (n = 5) | Total (N = 11) |
|---|---|---|---|---|---|
| Mean age, y (range) | 30.5 (24-37) | 35.5 (24-47) | 32.5 (32-33) | 27.2 (19-34) | 30.3 (19-47) |
| Sex, male, n (%) | 2 (100) | 2 (100) | 2 (100) | 5 (100) | 11 (100) |
| Race, n (%) | |||||
| White | 2 (100) | 1 (50) | 2 (100) | 4 (80) | 9 (82) |
| Asian | 0 | 1 (50) | 0 | 0 | 1 (9) |
| Other | 0 | 0 | 0 | 1 (20.0) | 1 (9) |
| Ethnicity, n (%) | |||||
| Hispanic or Latino | 0 | 0 | 0 | 2 (40) | 2 (18) |
| Not Hispanic or Latino | 2 (100) | 2 (100) | 2 (100) | 3 (60) | 9 (82) |
Table 2.
Frequency of bleeding episodes
| Parameter | Cohort 1 (n = 2) | Cohort 2 (n = 2) | Cohort 3 (n = 2) | Cohort 4 (n = 5) | Total (n = 11) |
|---|---|---|---|---|---|
| Experienced bleeds after study drug infusion, n (%)∗ | 2 (100) | 2 (100) | 1 (50) | 2 (40) | 7 (64) |
| Preinfusion total ABR†, mean (SD) | 3.5 (0.7) | 14.0 (17.0) | 1.5 (2.1) | 8.8 (8.3) | 7.5 (8.8) |
| Postinfusion total ABR before reinitiation of prophylaxis: all episodes‡, mean (SD) | 7.7 (3.0) | 2.3 (1.3) | 3.1 (4.4) | 0.7 (1.4) | 2.7 (3.3) |
n indicates number of participants with bleeding events.
Defined as the number of bleeding episodes during the 12 months before screening.
Postinfusion rate = (number of bleed episodes starting 3 weeks after study drug infusion and until the start of prophylactic dosing of FVIII [or date of data cut or conclusion date]) / (observation period for the participant, in years).
Safety
In total, 103 treatment-emergent AEs (TEAEs) occurred across all cohorts; 26 were considered related to study treatment, and all participants reported ≥1 TEAE (Table 3). The most common treatment-related TEAEs were increases in ALT levels (13 events in 5 [46%] participants) and aspartate aminotransferase levels (5 events in 3 [27%] participants). In cohort 4, 4 of 5 participants had elevations in ALT (defined as observed ALT value >1.5 times of baseline value) requiring tapering courses of corticosteroids, with 3 of the 4 participants needing ≥1 additional course for recurrent ALT elevation (Figure 1; supplemental Figure 5). In cohort 4, onset of initial ALT elevations ranged from week 2 to week 11. The duration of individual courses of corticosteroid treatment (regardless of reason for use) ranged from 7 to 187 days, with an average duration of 65.7 days. At investigator discretion, not all courses of steroids involved a full tapering regimen before discontinuing treatment. The minimum duration observed in those cases where steroids were tapered was 49 days. ALT elevations refractory to corticosteroid treatment were not observed.
Table 3.
Summary of TEAEs
| Event | Cohort 1 (n = 2) |
Cohort 2 (n = 2) |
Cohort 3 (n = 2) |
Cohort 4 (n = 5) |
Total (N = 11) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of participants | Number of events | Number of participants | Number of events | Number of participants | Number of events | Number of participants | Number of events | Number of participants (%) | Number of events | |
| Any AE∗ | 2 | 24 | 2 | 16 | 2 | 12 | 5 | 51 | 11 (100) | 103 |
| ALT increased | 2 | 3 | 2 | 3 | 1 | 1 | 4 | 12 | 9 (82) | 19 |
| AST increased | 2 | 3 | 1 | 2 | 1 | 1 | 3 | 4 | 7 (64) | 10 |
| Upper respiratory tract infection | 2 | 7 | 1 | 1 | 0 | 0 | 1 | 1 | 4 (36) | 9 |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 4 | 4 (36) | 4 |
| Otitis media | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4 | 2 (18) | 4 |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4 | 2 (18) | 4 |
| Arthralgia | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 2 (18) | 2 |
| Skin laceration | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 2 (18) | 2 |
| Fall | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 2 (18) | 2 |
| Oropharyngeal pain | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 2 (18) | 2 |
| Lymphopenia | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 2 (18) | 2 |
| Tachycardia | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 2 (18) | 2 |
| Hypotension | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 2 (18) | 2 |
| Any treatment-related AE† | 0 | 0 | 2 | 5 | 0 | 0 | 4 | 21 | 6 (55) | 26 |
| ALT increased | 0 | 0 | 2 | 3 | 0 | 0 | 3 | 10 | 5 (46) | 13 |
| AST increased | 0 | 0 | 1 | 2 | 0 | 0 | 2 | 3 | 3 (27) | 5 |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 3 | 3 (27) | 3 |
| Tachycardia | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 2 (18) | 2 |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 (9) | 1 |
| Myalgia | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 (9) | 1 |
| Hypotension | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 (9) | 1 |
AST, aspartate aminotransferase.
Number of participants with at least 1 TEAE reported in the study population.
For any AE, only the TEAEs reported by at least 2 participants are included in this table. Similarly for any treatment-related AE, only the treatment-related TEAEs reported by at least 1 participant are included.
Figure 1.

Summary of FVIII activity, ALT, AST, and corticosteroid dosing for individual participants in cohort 4 (giroctocogene fitelparvovec dose: 3 × 1013 vg/kg). Administration of corticosteroids (start and end) is shown for participants 7 (A), 8 (B), 9 (C), 10 (D), and 11 (E); participant 9 did not use glucocorticoids. Elevations in liver enzymes were managed with corticosteroids, with stabilization of transaminase levels and FVIII activity observed over time.
A total of 5 SAEs were reported in 3 participants. Two of these events were considered related to study treatment and occurred in 1 participant in cohort 4. This participant (no. 7) experienced grade 3 hypotension and grade 2 pyrexia 6 hours after completion of dosing, which resolved within 24 hours after treatment with electrolytes, norepinephrine, ondansetron, glucose, and paracetamol. Additional details about this episode are provided in the supplemental Materials additional safety findings. The participant was discharged per the protocol timeline (24 hours after infusion). The investigator considered both events related to study treatment based on the temporal association with study drug infusion. No hypotension events occurred in the 4 subsequent participants in cohort 4. One participant in cohort 2 (participant no. 3) experienced SAEs of grade 3 cellulitis of the buttock and perineum, and another (participant no. 4) experienced an SAE of grade 2 burns to the right anterior foot. Neither of these SAEs was considered related to study treatment. No confirmed FVIII inhibitors, thrombotic events, hypersensitivity, or malignancy were detected in any participant during the analysis period.
Efficacy
Changes in circulating FVIII activity
FVIII activity increased in a dose-dependent manner, with the highest levels achieved in cohort 4 (Figure 2A-B). One participant in cohort 2 (participant no. 4) had sustained FVIII activity levels of ≥1.4%, as assessed by a 1-stage assay beginning at week 3, but FVIII activity levels remained below the threshold of detection for the chromogenic assay (<3%).
Figure 2.

Changes in circulating FVIII activity. (A-B) Individual plots showing FVIII activity for individual participants in all cohorts over time based on the chromogenic (A) and 1-stage (B) assays. (C-D) Box and whisker plots showing the group mean (diamond) and median (horizontal line) with quartiles (blue boxes) and minimum to maximum (whiskers) of FVIII activity in cohort 4 at each assessment using the chromogenic (C) and 1-stage (D) assays. (A-D) For results reported as below the limit of quantitation, a value of 0.009 IU/mL (0.9%) was used for analysis and plotting.
All participants in cohort 4 achieved peak FVIII levels in the normal range (>50%) by week 9. Mean (SD; minimum-maximum) FVIII activity levels at weeks 52 and 104 in cohort 4 were 42.6% (53.5; 7.8-122.3; n = 4) and 25.4% (27.5; 0.9-71.6; n = 5) for the chromogenic assay (Figure 2C), representing a relative decrease of 40% in the mean FVIII activity level during year 2. For the 1-stage assay, values at weeks 52 and 104 were 66.4% (83.8; 12.0-191.3; n = 4) and 38.9% (36.7; 4.1-99.1; n = 5; Figure 2D). Mean FVIII activity results with the 1-stage assay were ∼1.5-fold higher than results with the chromogenic assay. Two participants in cohort 4 experienced transient chromogenic FVIII activity levels above the upper limit of normal (150%), with peak values of 169% and 187% at weeks 20 (participant no. 8) and 52 (participant no. 7), respectively (Figure 2A).
A scatterplot showing 1-stage assay results vs chromogenic assay results is shown in supplemental Figure 4.
Frequency of bleeding episodes
Treatment of bleeding episodes requiring the use of FVIII replacement therapy was reported in 2 of 2 participants each in cohorts 1 and 2, 1 of 2 participants in cohort 3, and 2 of 5 participants in cohort 4. In cohort 3, the mean (SD) total ABR was 3.1 (4.4) vs a mean (SD) total ABR of 1.5 (2.1) in the year before giroctocogene fitelparvovec infusion (Table 2). In cohort 3, 1 participant (no. 5) had 20 treated bleeding episodes, with the first bleeding episode (nontarget joint) occurring at week 12; 9 bleeding episodes were in a target joint (knee), with the first target joint episode occurring at week 18. The total ABRs for each participant in the year before and after infusion are shown in supplemental Table 1.
The cohort 4 mean (SD) total ABR was 0.7 (1.4) vs a mean (SD) total ABR of 8.8 (8.3) in the year before giroctocogene fitelparvovec infusion (Table 2). In cohort 4, 2 participants experienced bleeding events. One participant (no. 9) experienced a single treated bleeding event in a target joint (elbow) at week 67, with FVIII activity levels of 18.0% (week 52) and 10.9% (week 104) obtained via central chromogenic assay. Another participant (no. 10) had 6 treated bleeding episodes, the first occurring at week 67 after infusion. For this participant, 4 of 6 bleeding events were traumatic, with FVIII levels dropping below levels of quantification via central chromogenic assay as of week 72, but with FVIII values remaining detectable via 1-stage assay through week 104 (4.1% at week 104).
Changes in FVIII replacement therapy
At the time of data cutoff, relative to preinfusion, there was a mean decrease of 98.6% in the AIR of exogenous FVIII in cohort 4, and no participant in cohorts 3 or 4 resumed prophylaxis. In cohort 4, there were no infusions of FVIII products during the first year, and the postinfusion mean (SD) AIR of FVIII therapy was 1.8 (3.5) at data cutoff.
Vector shedding
Results for viral vector DNA shedding are summarized in Table 4. Overall, the highest concentration of vector DNA was detected in plasma, followed in descending order by saliva, semen, urine, and stool. The mean clearance rate was most rapid in urine (first negative in 7.0-28.0 days across the 4 cohorts), followed by semen, saliva, plasma, and stool. Positive vector DNA was not detected in urine in cohorts 1, 2, and 3 or stool in cohort 2. In cohort 4, mean (SD) times to the first of 3 consecutive negative results were 14.8 (8.2) days in urine, 42.2 (27.5) days in semen, 60.5 (17.6) days in saliva, 72.6 (62.1) days in stool, and 84.7 (47.9) days in plasma.
Table 4.
Vector shedding in body fluids
| Parameter | Cohort 1 (n = 2) | Cohort 2 (n = 2) | Cohort 3 (n = 2) | Cohort 4 (n = 5) | Total (N = 11) |
|---|---|---|---|---|---|
| Plasma | n = 2 | n = 2 | n = 2 | n = 5 | n = 11 |
| Peak value, vg/mL | 2.6 × 107 (2.7 × 107) | 2.9 × 108 (3.8 × 108) | 1.8 × 109 (1.4 × 109) | 3.4 × 109 (2.8 × 109) | 2.0 × 109 (2.4 × 109) |
| Days to peak value | 1.5 (0.7) | 1.0 (0.0) | 1.0 (0.0) | 1.4 (0.6) | 1.3 (0.5) |
| Days to first of 3 consecutive negative results | 22.0 (8.5) (n = 2) |
15.0 (0.0) (n = 2) |
56.5 (0.7) (n = 2) |
84.7 (47.9) (n = 3) |
49.0 (39.3) (n = 9) |
| Saliva | n = 2 | n = 2 | n = 2 | n = 5 | n = 11 |
| Peak value, vg/mL | 4.5 × 104 (2.2 × 104) | 1.6 × 105 (9.0 × 104) | 1.6 × 106 (4.2 × 105) | 7.0 × 106 (5.2 × 106) | 3.5 × 106 (4.7 × 106) |
| Days to peak value | 11.5 (5.0) | 7.5 (0.7) | 12.0 (4.2) | 11.8 (4.0) | 11.0 (3.7) |
| Days to first of 3 consecutive negative results | 29.0 (-) (n = 1) |
29.0 (0.0) (n = 2) |
57.0 (-) (n = 1) |
60.5 (17.6) (n = 4) |
48.3 (19.7) (n = 8) |
| Semen | n = 1 | n = 1 | n = 2 | n = 5 | n = 9 |
| Peak value, vg/mL | 1.5 × 104 (-) | 3.7 × 104 (-) | 1.5 × 105 (5.0 × 103) | 10.0 × 104 (1.5 × 105) | 9.3 × 104 (1.2 × 105) |
| Days to peak value | 8.0 (-) | 15.0 (-) | 11.0 (4.2) | 14.4 (9.0) | 13.0 (7.0) |
| Days to first negative result | 18.0 (14.1) (n = 2) |
15.0 (-) (n = 1) |
42.0 (18.4) (n = 2) |
42.2 (27.5) (n = 5) |
34.6 (23.3) (n = 10) |
| Stool | n = 2 | n = 0 | n = 2 | n = 5 | n = 9 |
| Peak value, vg/mL | 6.0 × 102 (4.1 × 102) | — | 2.5 × 103 (2.5 × 103) | 2.3 × 103 (2.8 × 103) | 2.0 × 103 (2.3 × 103) |
| Days to peak value | 8.0 (0.0) | — | 7.5 (0.7) | 17.0 (10.3) | 12.9 (8.8) |
| Days to first negative result | 15.0 (-) (n = 1) |
15.0 (-) (n = 1) |
42.0 (18.4) (n = 2) |
72.6 (62.1) (n = 5) |
53.0 (51.0) (n = 9) |
| Urine | n = 0 | n = 0 | n = 0 | n = 3 | n = 3 |
| Peak value, vg/mL | — | — | — | 9.7 × 103 (6.2 × 103) | 9.7 × 103 (6.2 × 103) |
| Days to peak value | — | — | — | 9.7 (3.8) | 9.7 (3.8) |
| Days to first negative result | 8.0 (0.0) (n = 2) |
7.5 (0.7) (n = 2) |
8.5 (0.7) (n = 2) |
14.8 (8.2) (n = 5) |
11.1 (6.3) (n = 11) |
Data are presented as mean (SD).
Immunogenicity
No confirmed FVIII inhibitors were detected during the study. ELISpot results specific to AAV6 and FVIII peptide pools are shown in supplemental Figure 5.
Discussion
Previous reports have described AAV-based or AAV-derived gene therapy in people with hemophilia A.17, 18, 19, 20 Here, we report results for giroctocogene fitelparvovec, a B-domain–deleted F8 gene therapy for the treatment of patients with severe hemophilia A using an AAV6 capsid. The data demonstrate that, with appropriate clinical management, giroctocogene fitelparvovec safely provided clinically relevant FVIII levels for effective treatment of patients with severe hemophilia A.
Alta is a phase 1/2 dose escalation study that included 4 dose cohorts. All 5 participants in the highest dose cohort (cohort 4) achieved FVIII activity levels in the normal range, suggesting some degree of consistency in initial treatment response and supporting that AAV6 is a viable hepatotropic serotype capable of delivering transgenes to produce clinically meaningful levels of FVIII. However, at the time of the data cutoff, only 1 participant (no. 8) remained in the normal range. In 4 of the 5 participants in cohort 4 (participants 7, 9, 10, and 11), FVIII expression showed a downward trend in activity over time. Although the precise etiology(s) for the decline from peak FVIII level is unknown, in 1 case, a temporal association with a rise in transaminase levels suggests a possible immune response and/or insult to hepatocytes harboring the transgene, whereas in other cases, rises in transaminase levels were not associated with a decline in FVIII levels, implicating alternative potential mechanisms. Some participants had FVIII activity level declines that stabilized over time (participants 9 and 10), whereas others exhibited a slow but continuous decline in FVIII activity (participants 7 and 11). The reasons for this difference are not known, and further follow-up is necessary to assess the trajectory of FVIII activity levels and whether there are potential patient characteristics that may predict the FVIII response over time. These findings are consistent with previous reports of hemophilia A gene therapy using an AAV5 vector that showed FVIII activity increases in the highest dose cohorts to 19% to 164% of normal in the 1-stage clotting assay,20 11% to 95% (median, 60%) of normal in the chromogenic assay,19 and a median of 23.9% (chromogenic assay)18 at 1 year after administration. As in our study, FVIII activity gradually declined, to chromogenic assay median values of 26%19 and 14.7%18 at 2 years, and levels of 4% to 100% (median, 20%) of normal (chromogenic assay) at 3 years.19 A previous report of AAV3-based gene therapy showed 15 of 18 enrolled participants had a FVIII activity level of 3.0% to 14.3% (mean, 6.9%) of normal (chromogenic assay) at 1 year after infusion without observable decreases to year 3; however, 2 other participants in the highest dose cohort lost expression of FVIII after an immune response to the rAAV3 vector.17
Transient asymptomatic liver transaminase elevations have been commonly observed after liver-directed AAV therapy for hemophilia.18,19,21 One potential etiology may be the activation of T cells that target hepatocytes displaying capsid peptides on their surface.22 Prior studies have shown that timely intervention with corticosteroids can quell potential immune responses, lower liver transaminase values, and stabilize transgene-derived factor activity levels.17,20,23, 24, 25 We also saw this with 4 of 5 cohort 4 participants requiring ≥1 course of corticosteroids for elevated liver transaminases. Notably, 3 participants received a second course of corticosteroids for a second rise in liver transaminase levels. This is not a unique finding and has been reported in another AAV-based gene therapy trial for the treatment of hemophilia A.20 The sampling schedule for ELISpot specimens in this study was too sparse to permit any meaningful conclusions regarding T-cell activation. Of note, 3 of the 4 participants (nos. 7, 8, and 11) showed maintenance of FVIII activity levels through corticosteroid treatment, and no ALT elevations refractory to corticosteroid treatment were observed. These results seem to suggest that use of the AAV6 vector may not reduce the incidence of liver transaminase elevations compared with other serotypes that have been used in other hemophilia A gene therapy studies.
The exact etiology for the secondary rise in liver transaminase levels in some participants is uncertain, as the initiation of second corticosteroid courses ranged from 8 to 43 weeks after gene therapy vector infusion, when capsid protein is unlikely to be present and prone to being targeted by activated T cells. There is mixed evidence that FVIII can induce a variable amount of endoplasmic reticulum stress in hepatocytes, contributing to the observed rise in transaminase levels.26, 27, 28 Regardless, the ALT elevations resolved after corticosteroid treatment, and all participants were able to maintain clinically efficacious levels of FVIII, as evidenced by low bleeding event rates. Further insights into the chronology and frequency of this finding will be determined as more patients are treated with giroctocogene fitelparvovec.
In our study, the 1-stage assay provided higher FVIII activity values than the chromogenic substrate assay, similar to findings in other studies.17,19 There is evidence that transgene-derived FVIII activity produces an initial burst in FXa and thrombin in the 1-stage assay that is not observed with the chromogenic assay; this may, in part, explain the increased values that are observed.29 Additional studies are planned to address this discrepancy.
In cohort 4, 2 participants (nos. 9 and 10) experienced bleeding events. One (participant 9) had a single bleeding event in a known target joint, whereas the other (participant 10) experienced 7 total (treated and untreated) bleeding events, with 5 noted to be traumatic. No bleeding events occurred in this cohort until 67 weeks after infusion. Both participants initially had FVIII levels in the normal range that subsequently declined to the mild range in 1 participant (no. 9) and to the moderate range in the other (participant 10). In this cohort, hemostasis is maintained over a broad range of FVIII activity levels, but with a tendency toward higher bleeding rates with FVIII activity in the low mild-to-moderate range. Most bleeding events for the participant (no. 10) with 7 bleeding episodes occurred once their FVIII level was in the moderate hemophilia range, as measured by the 1-stage assay, and below the level of quantification in the chromogenic assay (<3%), consistent with data from the mild hemophilia population in which the frequency of bleeding events increased with lower FVIII activity levels.30,31 Despite treated bleeding events, none of the participants in cohort 4 have elected to resume prophylaxis.
A consistent and acceptable feature of AAV-based gene therapy for the treatment of hemophilia, despite variability in AAV serotypes and transgenes used, is the overall safety profile.17, 18, 19,23 Giroctocogene fitelparvovec was well tolerated with appropriate clinical management in the 11 participants who received infusions at 4 different dose levels. The most notable findings were related to the SAEs of hypotension and pyrexia ∼6 hours after infusion, experienced by the first cohort 4 participant (no. 7) suggesting a hypersensitivity reaction, innate immune response, and/or hypovolemia as the participant had not received intravenous fluid supplementation in the prior 24 hours. The events were transient and responded to simple intervention. It is not known whether this hypersensitivity event was specifically related to the AAV6 serotype, the dose administered, and/or potentially patient predisposition; notably, the subsequent 4 participants, being well hydrated and treated with acetaminophen and diphenhydramine before infusion, did not experience similar hypotension. Consequently, with appropriate clinical management, giroctocogene fitelparvovec was well tolerated in the 11 participants who received infusions at 4 different dose levels.
The long-term effects of liver-directed gene therapy are not fully characterized. However, there have been no confirmed cases of the known gene therapy–mediated development of malignancy or liver dysfunction.17, 18, 19 Total follow-up time in this study ranged from 2 to 4 years. Over this interval, there was no discernible adverse impact on liver health. Overall, liver function assays (ALT, aspartate aminotransferase, alkaline phosphatase, bilirubin, direct bilirubin, and gamma-glutamyl transferase) were generally stable, and there were no significant findings on liver imaging and no elevations in alpha fetoprotein. Additionally, hepatocellular carcinoma was not reported. Longer follow-up and more participants are needed to gather additional data on the impact of giroctocogene fitelparvovec on long-term liver health. Furthermore, the use of corticosteroids and the duration of treatment did not appear to confer any long-term adverse consequences.
In this study, we present ≥2 years of follow-up data for patients treated with giroctocogene fitelparvovec, providing the first demonstration that an AAV6-based vector for the treatment of hemophilia A is generally well tolerated with appropriate clinical management and provides clinically relevant FVIII activity at the highest dose tested. FVIII activity levels were dose-dependent, with all 5 participants in cohort 4 reaching peak levels within or above the normal range. One participant (no. 7) in cohort 4 had a reaction shortly after completing the infusion; however, no other concerning safety events occurred during the study. Study limitations include the treatment of relatively few participants, potential variability in when participants decided to treat a bleeding event, and the relatively limited follow-up. Our current analysis did not provide information regarding the investigation of immune responses, such as T-cell responses, to the vector capsid because of limited data, precluding any robust assessment of ELISpot results and exploration of any correlation to transaminase elevation. In addition, liver biopsies were not performed to assess possible integration or other potential findings. Future studies with optional liver biopsies should provide these opportunities.
Overall, the emerging risk–benefit ratio of giroctocogene fitelparvovec remains generally favorable for the conduct of phase 3 investigations. Giroctocogene fitelparvovec was shown to have efficacy and safety comparable to other AAV gene therapy products while leveraging a serotype of AAV that was not used in prior studies. This has the potential to broaden the patient population eligible for gene therapy for the treatment of hemophilia A. A phase 3 study (NCT04370054) is ongoing, using the cohort 4 dose level.
Conflict-of-interest disclosure: A.D.L. has received research funding from BioMarin, Pfizer, and Sangamo; served on advisory boards for BioMarin, CSL, and Genentech; and owns stock in Pfizer. B.A.K. has received research funding from Pfizer, Spark Therapeutics, Takeda, and Uniqure and has served as a paid consultant for BioMarin, Octapharma, Pfizer, Regeneron, Spark Therapeutics, and Takeda. K.C.S. has served on an advisory board for BioMarin and ASC Therapeutics. N.V. has served on an advisory board for Biogen Idec. T.J.H. reports no conflicts to disclose. A.G. has served on advisory boards for BioMarin, Sanofi Genzyme, Pfizer, Hema Biologics, Genentech, and Uniqure; served on speakers bureau for BioMarin, Sanofi Genzyme; and received research funding from Pfizer, Spark Therapeutics, Uniqure, Genentech, Sangamo, and Freeline. S.A., A.F., F.P., A.Y., F.G., D.A., M.d.l.A.R., L.-J.T., G.D.R., and J.R. are employees of Pfizer Inc and may own stock/options in the company. B.M.C. and L.C. are employees of Sangamo Therapeutics and own stock/options in the company.
Acknowledgments
The authors thank the study participants, investigators, and study site personnel for their ongoing involvement with the Alta study.
Medical writing and editorial assistance were provided by Nate Connors and Michael Morren, of Peloton Advantage LLC, an OPEN Health company, and Margit Rezabek of Engage Scientific Solutions and funded by Pfizer. This study was sponsored by Pfizer.
Authorship
Contribution: K.C.S., G.D.R., and J.R. contributed to the study design. A.D.L., B.A.K., K.C.S., N.V., T.J.H., and A.G. participated as study investigators and provided patients or study materials. B.A.K., K.C.S., and L.C. participated in the collection and assembly of data. F.P., F.G., M.d.l.A.R., L.-J.T., D.A., A.Y., S.A., A.F., G.D.R., L.C., and J.R. contributed to data analysis; and all authors participated in data interpretation and critical review and revision of this manuscript and provided approval of the manuscript for submission.
Footnotes
Presented in abstract form at the 61st annual meeting and exposition of the American Society of Hematology, Orlando, FL, 7-10 December 2019; at the 63rd annual meeting and exposition of the American Society of Hematology, Atlanta, GA, 12 December 2021; and at the 15th annual congress of the European Association for Haemophilia and Allied Disorders, Virtual Meeting, 2-4 February 2022.
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Peyvandi F, Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia. 2019;25(5):738–746. doi: 10.1111/hae.13816. [DOI] [PubMed] [Google Scholar]
- 2.George LA. Hemophilia gene therapy comes of age. Blood Adv. 2017;1(26):2591–2599. doi: 10.1182/bloodadvances.2017009878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Favaro P, Finn JD, Siner JI, Wright JF, High KA, Arruda VR. Safety of liver gene transfer following peripheral intravascular delivery of adeno-associated virus (AAV)-5 and AAV-6 in a large animal model. Hum Gene Ther. 2011;22(7):843–852. doi: 10.1089/hum.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harmatz P, Prada CE, Burton BK, et al. First-in-human in vivo genome editing via AAV-zinc-finger nucleases for mucopolysaccharidosis I/II and hemophilia B. Mol Ther. 2022;30(12):3587–3600. doi: 10.1016/j.ymthe.2022.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang H, Lillicrap D, Patarroyo-White S, et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108(1):107–115. doi: 10.1182/blood-2005-12-5115. [DOI] [PubMed] [Google Scholar]
- 7.Nathwani AC, Gray JT, Ng CY, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107(7):2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone D, Liu Y, Li ZY, et al. Biodistribution and safety profile of recombinant adeno-associated virus serotype 6 vectors following intravenous delivery. J Virol. 2008;82(15):7711–7715. doi: 10.1128/JVI.00542-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 10.Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 11.Klamroth R, Hayes G, Andreeva T, et al. Global seroprevalence of pre-existing immunity against AAV5 and other AAV serotypes in people with hemophilia A. Hum Gene Ther. 2022;33(7-8):432–441. doi: 10.1089/hum.2021.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riley BE, Lee GK, inventors; Sangamo Therapeutics Inc, assignee . July 13, 2016. Liver-specific constructs, factor VIII expression cassettes and methods of use thereof. International patent W0/2017/074526. [Google Scholar]
- 13.Hemgenix (etranacogene dezaparvovec-drlb). Prescribing information; 2022. US Food and Drug Administration. https://www.fda.gov/vaccines-blood-biologics/vaccines/hemgenix
- 14.Roctavain (valoctocogene roxaparvovec-rvox). Prescribing information; 2023. US Food and Drug Administration. https://labeling.cslbehring.com/PI/US/Hemgenix/EN/Hemgenix-Prescribing-Information.pdf
- 15.European Medicines Agency Hemgenix (Etranacogene Dezaparvovec)-European Public Assessment Report (EPAR) https://www.ema.europa.eu/en/medicines/human/EPAR/hemgenix
- 16.European Medicines Agency Roctavian [Valoctocogene Roxaparvovec]-European Public Assessment Report (EPAR) https://www.ema.europa.eu/en/medicines/human/EPAR/roctavian-0
- 17.George LA, Monahan PE, Eyster ME, et al. Multiyear factor VIII expression after AAV gene transfer for hemophilia A. N Engl J Med. 2021;385(21):1961–1973. doi: 10.1056/NEJMoa2104205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N Engl J Med. 2022;386(11):1013–1025. doi: 10.1056/NEJMoa2113708. [DOI] [PubMed] [Google Scholar]
- 19.Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N Engl J Med. 2020;382(1):29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 20.Rangarajan S, Walsh L, Lester W, et al. AAV5-factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377(26):2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 21.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 22.George LA, Fogarty PF. Gene therapy for hemophilia: past, present and future. Semin Hematol. 2016;53(1):46–54. doi: 10.1053/j.seminhematol.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 23.George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377(23):2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.High KA, George LA, Eyster ME, et al. A phase 1/2 trial of investigational Spk-8011 in hemophilia A demonstrates durable expression and prevention of bleeds [abstract] Blood. 2018;132(suppl 1):487. [Google Scholar]
- 25.Nathwani AC, Tuddenham E, Chowdary P, et al. GO-8: preliminary results of a phase I/II dose escalation trial of gene therapy for haemophilia A using a novel human factor VIII variant [abstract] Blood. 2018;132(suppl 1):489. [Google Scholar]
- 26.Fong S, Handyside B, Sihn CR, et al. Induction of ER stress by an AAV5 BDD FVIII construct is dependent on the strength of the hepatic-specific promoter. Mol Ther Methods Clin Dev. 2020;18:620–630. doi: 10.1016/j.omtm.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zolotukhin I, Markusic DM, Palaschak B, Hoffman BE, Srikanthan MA, Herzog RW. Potential for cellular stress response to hepatic factor VIII expression from AAV vector. Mol Ther Methods Clin Dev. 2016;3 doi: 10.1038/mtm.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange AM, Altynova ES, Nguyen GN, Sabatino DE. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol Ther Methods Clin Dev. 2016;3 doi: 10.1038/mtm.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosen S, Tiefenbacher S, Robinson M, et al. Activity of transgene-produced B-domain-deleted factor VIII in human plasma following AAV5 gene therapy. Blood. 2020;136(22):2524–2534. doi: 10.1182/blood.2020005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 2011;17(6):849–853. doi: 10.1111/j.1365-2516.2011.02539.x. [DOI] [PubMed] [Google Scholar]
- 31.Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA, US Hemophilia Treatment Center Network The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2(16):2136–2144. doi: 10.1182/bloodadvances.2018020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.