

Abstract
The human papillomavirus (HPV) capsid is primarily composed of a structural protein denoted L1, which forms both pentameric capsomeres and capsids composed of 72 capsomeres. The L1 protein alone is capable of self-assembly in vivo into capsidlike structures referred to as viruslike particles (VLPs). We have determined conditions for the quantitative disassembly of purified HPV-11 L1 VLPs to the level of capsomeres, demonstrating that disulfide bonds alone are essential to maintaining long-term HPV-11 L1 VLP structure at physiological ionic strength. The ionic strength of the disassembly reaction was also important, as increased NaCl concentrations inhibited disassembly. Conversely, chelation of cations had no effect on disassembly. Quantitative reassembly to a homogeneous population of 55-nm, 150S VLPs was reliably achieved by the re-formation of disulfide linkages following removal of reducing agent at near-neutral pH and moderate NaCl concentration. HPV-11 L1 VLPs could also be dissociated by treatment with carbonate buffer at pH 9.6, but VLPs could not be regenerated following carbonate treatment. When probed with conformationally sensitive and/or neutralizing monoclonal antibodies, both capsomeres generated by disulfide reduction of purified VLPs and reassembled VLPs formed from capsomeres upon removal of reducing agents exhibited epitopes found on the surface of authentic HPV-11 virions. Antisera raised against either purified VLP starting material or reassembled VLPs similarly neutralized infectious HPV-11 virions. The ability to disassemble and reassemble VLPs in vitro and in bulk allows basic features of capsid assembly to be studied and also opens the possibility of packaging selected exogenous compounds within the reassembled VLPs.
Papillomaviruses are the causative agents of warts and have also been associated with certain cancers in a variety of animal hosts. They are members of the Papovaviridae family of nonenveloped, double-stranded DNA viruses (33), which in general are both species and tissue type specific. They are a large group of viruses, and more than 70 which infect humans only have been identified. Approximately one-third of the human papillomaviruses (HPVs) are targeted to genital mucosal epithelium. HPV types 6 and 11 (HPV-6 and -11) are the most common papillomaviruses associated with benign genital warts. Other HPV types, most significantly HPV-16 and -18, have been implicated as etiologic agents of cervical cancer (12, 28). The social and economic costs of these diseases are quite severe, and development of prophylactic or therapeutic measures to block or ameliorate the effects of HPV infection are obviously of great value.
The HPV capsid is composed of two structural proteins termed L1 and L2. L1 is an ∼55-kDa protein which is stable in two oligomeric configurations: capsomeres (pentamers of L1) and capsids composed of 72 capsomeres in a T=7 icosahedron (1, 2, 17). The L1 protein alone is capable of efficient self assembly in vivo into capsidlike structures referred to as viruslike particles (VLPs), as demonstrated by heterologous expression studies using recombinant baculovirus and vaccinia virus vectors and in recombinant yeast (18–21, 34, 38, 42, 44, 49). In most preparations isolated from eukaryotic cells, a variable population of VLPs approaching 55 nm in diameter, similar in appearance to virions, are observed. In a direct cryoelectron microscopic comparison, intact HPV-1 VLPs were indistinguishable from HPV-1 virions at a resolution of 3.5 nm (17). The assembly of VLPs is somewhat sensitive to cell type, however, as L1 expressed in Escherichia coli was observed to be largely in the form of capsomeres or smaller, with few or no capsids apparent either in the cell or upon purification (25), similar to polyomavirus VP1 protein expressed in E. coli (35). The L2 protein (∼72 kDa) comprises only a minor part of the total capsid protein, and its role appears to involve DNA binding and/or packaging (49, 50). Interestingly, HPV-1 VLPs composed of L1 and L2 appeared identical to VLPs composed of L1 alone when examined by cryoelectron microscopy at 3.5-nm resolution (17). VLPs composed of L1, alone or in combination with L2, are currently being tested as vaccine candidates by several groups.
HPV capsid assembly clearly requires correctly folded L1 capsomeres. However, additional factors which may be important for VLP formation and stability are less well defined. The importance of divalent cation binding, specifically calcium, in maintaining virion integrity has been demonstrated for polyomavirus (6) and rotavirus (14), and disulfide bonds also appear important in stabilizing polyomavirus (6, 45) and simian virus 40 (SV40) (8) virions. Additional factors such as pH and ionic strength also influence polyomavirus capsid stability, presumably by affecting electrostatic interactions (6, 35, 36). Finally, posttranslational modifications of viral capsid proteins, such as glycosylation, phosphorylation, and acetylation, may modify capsid stability and assembly (15, 46).
We describe here experiments designed to identify conditions for the maximal disassembly of purified HPV-11 VLPs in vitro, in a state conducive for subsequent reassembly. We find that prolonged incubation with relatively high concentrations of reducing agent at physiological ionic strength is both necessary and sufficient to generate homogeneous, soluble capsomeres from purified VLPs. Removal of reducing agent at higher ionic strength (0.5 M NaCl) efficiently yields a defined population of intact, appropriately sized VLPs which retain the ability to elicit virus-neutralizing antibodies.
MATERIALS AND METHODS
HPV-11 VLPs.
HPV-11 L1 proteins were heterologously expressed in Trichoplusia ni (High Five) cells infected with recombinant baculovirus encoding the complete L1 open reading frame downstream of the polyhedrin promoter as described previously (16). Cells were harvested approximately 72 h postinfection, pelleted by centrifugation, and frozen. For preparation of VLPs (all steps performed at 4°C), the cell paste was resuspended in homogenization buffer (20 mM NaH2PO4–150 mM NaCl [pH 7.4] containing leupeptin [10 μg/ml], aprotinin [1 μg/ml], and pepstatin A [1 μg/ml]) and lysed at ∼3,000 lb/in2 in a microfluidizer (Microfluidics model HC8000/3A). The homogenized lysate was then centrifuged at 100,000 × g for 90 min, and the supernatant was removed. The pellet, containing HPV-11 VLPs, was resuspended in phosphate-buffered saline (PBS) containing CsCl (405 g/liter) and centrifuged at 66,000 × g for 90 min to remove a contaminating buoyant layer. The clarified lysate was then centrifuged overnight at 83,000 × g in a vertical rotor, and the VLP band was collected (at a density of 1.28 g/cm3). The VLPs were diluted >2-fold in PBS–0.5 M NaCl, to reduce the density of the solution, and layered over a two-component step gradient composed of 30 and 63% (wt/wt) sucrose in PBS–0.5 M NaCl. The gradients were centrifuged at 167,000 × g for 3 h in a vertical rotor, and the purified VLP band was collected at the interface between the 30 and 63% sucrose solutions. The VLPs were then dialyzed into selected buffers (either PBS or PBS with NaCl added to a final concentration of 0.3 or 0.5 M) and stored at 4°C. Protein concentration was determined by the Bradford assay (5), using bovine serum albumin as the reference protein, and L1 content was determined as described previously (42). Starting with 25 to 30 g of wet cell paste, the above-described protocol yielded 15 to 25 mg of HPV-11 VLPs.
Sucrose gradient centrifugation.
Three types of sucrose gradients were used in these experiments. First, centrifugation on 30% sucrose cushions was used to identify conditions which favored the disassembly of VLPs into smaller, soluble components. Reaction mixtures of 100 to 200 μl containing VLPs (50 to 100 μg of total protein) plus or minus potential disrupting agents were layered atop 5-ml centrifuge tubes filled with 4.8 ml of 30% (wt/wt) sucrose in PBS–0.5 M NaCl and centrifuged at 197,000 × g for 2 h at 4°C in a swinging-bucket rotor. A 50-μl aliquot was taken from the very top of the tube and mixed with 2× Laemmli sample preparation buffer (24). The remainder of the 30% sucrose cushion was removed by pipette, and the pellet (typically none was visible) was resuspended in 100 μl of 1× Laemmli sample preparation buffer. The presence of HPV-11 L1 protein at the top or bottom of the 30% sucrose cushion was then determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the relative amount of L1 was quantified by analysis of digitized gels.
Second, the state of disassembled VLPs was determined by rate-zonal centrifugation through 5 to 20% linear sucrose gradients. Disassembled VLPs (100 to 200 μg of total protein per ml in 400 μl) were layered atop preformed 11.6-ml gradients composed of 5 to 20% (wt/vol) sucrose in PBS–0.5 M NaCl and centrifuged at 111,000 × g for 24 h at 4°C in a swinging-bucket rotor. Fractions (0.5 ml) were collected across the gradient, and the pellet (typically none was visible) was resuspended in 0.5 ml of PBS by Dounce homogenization. The position of HPV-11 L1 protein across the gradient was determined by immunoblotting. The gradients were calibrated by using standard proteins with established sedimentation coefficients (E. coli β-galactosidase, 19S; bovine liver catalase, 11.3S; bovine serum albumin, 4.3S), and the percentage of sucrose in the fractions was determined by refractometry.
Third, the state of initial, disassembled, and reassembled VLPs was determined by rate-zonal centrifugation through 10 to 65% linear sucrose gradients. HPV-11 L1 protein (100 to 200 μg of total protein per ml in 400 μl) in various states of assembly was layered atop preformed 11.6-ml gradients composed of 10 to 65% (wt/vol) sucrose in PBS–0.5 M NaCl and centrifuged at 188,000 × g for 2.5 h at 4°C in a swinging-bucket rotor. The gradients were collected (in 1.0-ml fractions), analyzed, and calibrated as described above, with parvovirus B19 (70S) and HPV-18 L1 (160S) VLPs used as additional calibration standards.
Gel electrophoresis. (i) SDS-PAGE.
SDS-PAGE was performed largely by the method of Laemmli (24). Samples were mixed with sample preparation buffer, boiled for 2 min, briefly spun in a minicentrifuge, and loaded onto 7.5% (Fig. 1) or 10% (Fig. 2 to 4) minigels with a 4% stacking gel. Gels were run for approximately 1 h at 20-mA constant current at room temperature, and protein was visualized by staining with Coomassie brilliant blue R250.
FIG. 1.
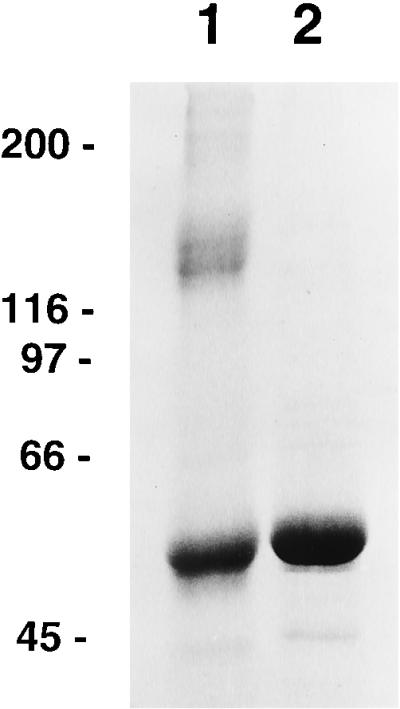
SDS-PAGE analysis of purified HPV-11 L1 protein. The protein was mixed with sample preparation buffer in the absence (lane 1) or presence (lane 2) of 2 mM DTT and boiled for 2 min prior to gel electrophoresis. Shown on the left are the positions at which molecular weight standards (in kilodaltons) migrated.
FIG. 2.

Thirty percent sucrose cushion analysis of HPV-11 VLP disassembly. HPV-11 preparations were treated at 4°C as described in the text, and samples were taken at the top (T) or bottom (B) of the sucrose cushion prior to gel electrophoresis. Group 1, untreated, purified HPV-11 VLP starting material in PBS; group 2, VLPs incubated with 5% βME for 16 h; group 3, VLPs incubated with 5% βME for 1 h; group 4, VLPs incubated with 2% βME for 16 h; group 5, VLPs incubated with 0.5% βME for 16 h; group 6, VLPs incubated with 10 mM DTT–5 mM EDTA for 16 h.
FIG. 4.

Ten to 65% linear sucrose gradient analysis of HPV-11 VLPs in various states of assembly. An aliquot of purified VLP starting material (a) was incubated with 5% βME for 16 h at 4°C (b). A portion of βME-treated VLPs were then reassembled by dialysis into PBS–0.5 NaCl to remove reducing agent (c). The samples were then centrifuged on 10 to 65% linear sucrose gradients as described in the text. Each gradient was collected in 12 fractions (1 ml), and the pellet (P) was resuspended in 1 ml of PBS. Shown are immunoblots demonstrating the positions at which the L1 protein migrated on the different gradients. Also indicated are the peak positions at which sedimentation standards migrated, as in Fig. 3.
(ii) Immunoblotting.
Electroblots of HPV-11 L1 from SDS-polyacrylamide gels were prepared largely by the method of Towbin et al. (43). The blots were blocked with 1% nonfat milk protein in PBS overnight at 4°C. The blots were probed with AU1 (Berkeley Antibody Co.), a mouse monoclonal antibody directed against a linear epitope on papillomavirus L1 proteins (27), for 90 min, washed with PBS–0.1% Triton X-100, and then reblocked for 30 min. The blots were then incubated with horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin G (Southern Biotechnology Associates, Inc.) for 40 min and washed as described above. The blots were then developed with ECL Western blotting reagent (Amersham) and exposed to X-ray film.
(iii) Analysis of gels.
The Mrs of monomeric and oligomeric L1 were determined from their Rf values on SDS–7.5% polyacrylamide gels in comparison to standard proteins (39). When indicated, gels were digitized on a Hewlett-Packard Scanjet Plus flatbed densitometer, and the relative intensity of bands was determined by using Scan Analysis software (version 2.2; Specom Research).
Electron microscopy.
Protein samples were allowed to settle on Formvar- and carbon-coated copper grids (Electron Microscopy Sciences), blotted dry, and stained with freshly filtered 2% phosphotungstic acid (pH 6.8). Grids were examined in a JEOL model 1005 transmission electron microscope at an accelerating voltage of 100 kV and photographed at nominal magnifications of ×15,000 to ×25,000.
Enzyme-linked immunosorbent assay (ELISA).
HPV-11 L1 VLPs (0.5 to 1.0 mg of L1 ml) in PBS–0.3 M NaCl were either stored without treatment at 4°C or incubated overnight at 4°C following addition of β-mercaptoethanol (βME) (to a final concentration of 5%) or 2.0 M carbonate buffer, pH 9.6 (to a final concentration of 200 mM carbonate). A portion of the treated samples was then dialyzed against 4 × 1 liter of PBS–0.5 M NaCl at 4°C for ≥24 h. All samples were diluted to a concentration of 0.8 μg of L1/ml and distributed into the wells of microtiter plates (80 ng of L1 per well). Untreated VLPs and dialyzed material were diluted into PBS. The sample treated with βME without subsequent dialysis was diluted into PBS containing 5% βME, and the undialyzed sample incubated in 200 mM carbonate was diluted into 200 mM carbonate, pH 9.6. Following incubation at 37°C for 1 h, the plates were washed with PBS–0.1% Tween 20 (PBS-Tw) and blocked with 5% nonfat milk protein in PBS. Monoclonal antibodies (AU1, or H11.F1 and H11.A3 purified from ascites; purchased from Pennsylvania State University [11]) were diluted in 1% nonfat milk in PBS and added to the wells. Following a 2-h incubation at room temperature, the plates were washed with PBS-Tw and HRP-labeled goat anti-mouse immunoglobulin G was added. After 1 h at room temperature, the plates were washed as described above and developed with HRP substrate (Kirkegaard & Perry Laboratories). Optical density measurements were made at 405 nm at the 15-min endpoint. Averages of duplicate wells were calculated as the final optical density values.
HPV-11 neutralization assay.
Antisera were generated in BALB/c mice against the initial purified HPV-11 VLPs and against HPV-11 VLPs which were reassembled from capsomeres upon removal of reducing agent. Mice were immunized by subcutaneous injection with 1 μg of VLPs adsorbed to 1 mg of aluminum hydroxide adjuvant (Alhydrogel; E. M. Sargent Pulp and Chemical Company) at weeks 0, 4, and 9. Sera collected 4 weeks after the last immunization were tested for the ability to neutralize authentic HPV-11 by the method of Smith et al. (40), with several modifications. Briefly, HPV-11Hershey virus stock (23) (from John Kreider) was incubated with serial dilutions of serum for 1 h at 37°C and then added to HaCat cells (4) (provided by Nobert Fusenig) which had been grown to confluency in 24-well plates. After 6 days, cells were harvested and total cellular RNA was prepared by using Tri Reagent (Molecular Research Center, Inc.). One microgram of total RNA was used for cDNA synthesis with a First Strand cDNA kit (Boehringer Mannheim) and an oligo(dT) primer. To amplify HPV-11 E1∧E4 cDNA, nested PCR was carried out with the primers described by Smith et al. (40). The first round of amplification utilized 12.5% of the cDNA from each reverse transcription (RT) reaction. Ten percent of the first-round PCR mixture was then used for the nested reactions. First-round and nested PCRs were set up with Hot Wax beads (1.5 mM) and pH 9.5 buffer (InVitrogen) with 200 μM deoxynucleoside triphosphates, 125 μg each of the forward and reverse primers, and 2.5 U of Taq polymerase (Perkin-Elmer) in a final volume of 50 μl and were carried out for 35 cycles. The temperature profile for both first-round and nested PCRs was 80°C for 5 min, 95°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 10 min.
As a control to demonstrate that the assay was able to detect mRNA extracted from HaCaT cells, all cDNA samples were used in separate PCRs with primers specific for spliced cellular β-actin mRNA as described previously (40).
All PCR products were separated by electrophoresis on a 2% agarose gel and visualized by ethidium bromide fluorescence.
RESULTS
Quantitative disassembly of HPV-11 VLPs.
Relatively large quantities of HPV-11 L1 VLPs were prepared as starting material for the study of VLP disassembly and reassembly. HPV-11 L1 VLPs were isolated from recombinant baculovirus-infected High Five cells by CsCl and sucrose gradient centrifugation. The calculated purity of these L1 preparations, based on densitometric analysis of SDS-PAGE, ranged between 70 and 90% (Fig. 1, lane 2). In addition, in linear sucrose gradients most of the protein migrated as expected for a mixture of individual and clumped VLPs (see Fig. 4a), and in the electron microscope a mixture of intermediate- and full-size (50- to 55-nm) particles was apparent (see Fig. 5a).
FIG. 5.

Electron micrographs of HPV-11 VLPs in various states of assembly. VLPs, treated as described below, were stained with 2% phosphotungstic acid, applied to grids, and photographed at magnifications of ×15,000 to ×25,000. (a) Purified VLP starting material; (b) VLPs disassembled to the level of capsomeres by incubation with 5% βME for 16 h at 4°C; (c) VLPs reassembled from disassembled VLPs by dialysis into PBS–0.5 M NaCl; (d) the central region of image c at greater magnification. Scale bars: a and c, 200 nm; b and d, 100 nm.
The covalent and noncovalent interactions which stabilize the assembled L1 VLPs are not entirely known, but earlier work on papillomavirus VLPs and related polyomavirus virions and VLPs suggested the importance of ionic strength, divalent cations (6, 35), and disulfide bonds (37, 44). In particular, Sapp and coworkers had demonstrated by immunoblotting that ∼50% of the L1 protein of HPV-33 VLPs was disulfide bonded into a range of larger oligomers with an apparent Mr consistent with trimers of L1 and that mild reducing conditions partially broke down HPV-33 VLPs to the level of capsomeres (37, 44). In our studies, in the absence of reducing agents, only a portion of the HPV-11 L1 protein migrated on SDS-PAGE with an apparent Mr of 55,000 (Fig. 1, lane 1). Approximately 40% (the percentage varied between different VLP preparations) of the L1 protein of HPV-11 VLPs was disulfide bonded into larger oligomers (Fig. 1, lane 1), with predicted Mr values of approximately 144,000 (possibly L1 trimer) and 210,000 (possibly L1 tetramer). The L1 oligomers did not migrate as a single band and appeared to be heterogeneous in size. The ∼200,000-Mr oligomer was also observed on immunoblots by Sapp and coworkers (37, 44) as part of a broad higher-molecular-weight band. These results indicate that a portion of the L1 proteins in HPV-11 VLPs are disulfide linked into higher oligomers.
To study the role of disulfide linkages and other interactions in VLP stability, a rapid screening assay for VLP disassembly was developed. Purified HPV-11 L1 VLPs, both before and after various treatments, were layered atop 30% sucrose cushions and centrifuged, and the distribution of L1 protein at the top and bottom of the 30% cushion was visualized by SDS-PAGE. Intact VLPs were expected to pellet through the 30% sucrose cushion; nonaggregated capsomeres and L1 monomer were expected to remain on the top of the cushion. An example of this assay is shown in Fig. 2. To quantitate the relative disposition of L1 protein, the gels were digitized, the total intensity of the L1 bands at the top and the bottom of the cushion was determined, and then the percentage of the L1 staining intensity found at either position was calculated. The results of a number of such determinations are given in Tables 1 and 2. As demonstrated in Fig. 2, the purified VLP starting material sedimented through the 30% sucrose, as predicted, with no L1 apparent at the top. However, upon incubation with a high concentration of the reducing agent βME, L1 protein was found largely at the top of the 30% sucrose cushion, indicating that the reducing agent had disassembled the HPV-11 VLPs to smaller, nonaggregated components. Interestingly, maximal disassembly of the VLPs typically required exposure to a very high concentration of reducing agent (in this instance 5%, or 713 mM, βME) for a relatively long duration (∼16 h at 4°C). Lower concentrations of reducing agent or shorter durations of reduction were not as reliably effective at VLP disassembly. Addition of a low concentration of a chelating agent did not enhance disassembly (Fig. 2 and Table 1).
TABLE 1.
Effects of reducing agents on disassembly of HPV-11 L1 VLPs
| Disassembly condition | Distribution (%)a
|
|||||
|---|---|---|---|---|---|---|
| 0.15 M NaCl
|
0.3 M NaCl
|
0.5 M NaCl
|
||||
| Top | Bottom | Top | Bottom | Top | Bottom | |
| Starting material | 3.8 ± 0.7 | 96.3 ± 0.8 | 3.2 ± 1.4 | 96.8 ± 1.4 | 4.2 ± 0.3 | 95.9 ± 0.6 |
| 5% βME, 16 h | 87.7 ± 3.2 | 12.4 ± 3.1 | 70.9 ± 12 | 29.1 ± 12 | 53.2 ± 6.8 | 46.8 ± 6.8 |
| 5% βME, 1 h | 68.1 ± 11 | 31.9 ± 11 | 68.0 ± 10 | 32 ± 10 | ||
| 2% βME, 16 h | 72.1 ± 2.7 | 27.9 ± 2.7 | 67.6 ± 12 | 32.3 ± 612 | ||
| 0.5% βME, 16 h | 45.8 ± 18 | 54.2 ± 18 | 28.8 ± 16 | 71.2 ± 16 | ||
| 10 mM DTT, 16 h | 44.5 ± 11 | 55.5 ± 11 | 43.8 ± 20 | 56.2 ± 20 | ||
| 10 mM DTT, 1 h | 9.5 ± 6.4 | 90.5 ± 6.4 | ||||
| 10 mM DTT, 5 mM EDTA, 16 h | 55.9 ± 6.2 | 44.1 ± 6.2 | ||||
VLPs (0.5 to 1.0 mg of protein per ml) were treated as indicated at 4°C, and the distribution of L1 across a 30% sucrose cushion was determined as described in Materials and Methods. Shown are the means of multiple determinations (n = 3 to 7) ± standard deviation.
TABLE 2.
Effects of chelators and buffers on disassembly of HPV-11 L1 VLPs
| Disassembly condition | Distribution (%)a
|
|
|---|---|---|
| Top | Bottom | |
| Starting material | 4 ± 2 | 96 ± 2 |
| 200 mM EDTA, pH 7.4 | 4 ± 3 | 96 ± 3 |
| 200 mM EDTA, 10 mM DTT | 10 ± 6 | 90 ± 6 |
| 200 mM EGTA, pH 7.4 | 13 ± 11 | 87 ± 11 |
| 200 mM EGTA, 10 mM DTT | 11 ± 6 | 89 ± 6 |
| 200 mM NaHCO3, pH 9.6 | 81 ± 2 | 19 ± 2 |
| 200 mM NaHCO3, 10 mM DTT | 74 ± 11 | 26 ± 11 |
| 200 mM glycine, pH 9.6 | 11 ± 1 | 89 ± 1 |
| 200 mM glycine, 10 mM DTT | 41 ± 12 | 59 ± 11 |
VLPs (0.5 to 1.0 mg of protein per ml) were treated as indicated for 16 h at 4°C, and the distribution of L1 across a 30% sucrose cushion was determined as described in Materials and Methods. Shown are the averages of duplicate determinations ± variation.
In addition to reductants, the other important variables for quantitative disassembly of VLPs were found to be the ionic strength during the disassembly reaction and the solubility of the VLP starting material. First, as observed earlier for polyomavirus virions, lower-ionic-strength conditions destabilize VLPs (6), although Sapp et al. (37) reported that generation of HPV-33 capsomeres from VLPs was insensitive to salt concentration between 0.15 and 0.6 M NaCl. For HPV-11 VLPs, maximum (∼90%) disassembly of VLPs exposed to 5% βME for 16 h was observed at physiological ionic strength (i.e., 0.15 M NaCl) but became correspondingly less effective as the ionic strength was increased (Table 1). The stabilizing effect of increased ionic strength could be partially overcome by incubating the VLPs with reducing agents for longer durations or at elevated temperatures. Thus, while incubating the VLPs with 5% βME for 120 h at 4°C or for 24 h at 24°C increased the extent of disassembly to 60 to 70% at 0.5 M NaCl, disassembly was still far from complete (data not shown). Second, for quantitative disassembly, the degree of aggregation of the VLP starting material was also important. In the experiments reported here, the VLP solutions were dialyzed into different ionic strength buffers and stored at 4°C until use in disassembly trials. After several days, particularly at 0.15 M NaCl, the solutions became slightly cloudy, indicating some degree of aggregation (although little or no precipitate was observed). Treatment of the clouded VLP solutions with reducing agents did not yield the same degree of disassembly as was observed with the initial soluble VLP solution, indicating that the aggregated VLPs were resistant to disassembly. However, upon removal of the aggregated material (which ranged from 10 to 50% of the total VLPs, depending on the age of the preparation) by filtration, the remaining soluble VLPs again could be disassembled to the same extent as the initial soluble VLP starting material.
Interestingly, even at high concentrations of chelators, chelation of cations did not significantly influence VLP disassembly. Dialysis of VLPs into 200 mM EDTA or EGTA buffer (PBS–0.3 M NaCl, pH 7.4) led to no apparent disassembly, and the addition of 10 mM dithiothreitol (DTT) to the dialysis buffers had little effect (Table 2). The inability of high concentrations of chelators to disassemble VLPs was confirmed by electron microscopic analysis, although EDTA (but not EGTA) appeared to swell the VLPs slightly (data not shown). Either these concentrations of chelator are insufficient to extract tightly bound, structurally important ions or cations are not essential to maintaining VLP structural integrity. Conversely, addition of a concentrated aliquot of NaHCO3 buffer (pH 9.6) to a solution of VLPs, to a final concentration of 200 mM carbonate (in PBS–0.3 M NaCl), caused significant breakdown of the VLPs (Table 2). Addition of DTT (to a final concentration of 10 mM) did not further enhance carbonate-induced breakdown. Incubation of VLPs with 200 mM carbonate–10 mM DTT is commonly used to denature HPV virions or VLPs in ELISAs (9, 10, 13). The effect of carbonate appears to be buffer specific, and not merely a function of pH, as incubation of HPV-11 VLPs with pH 9.6 glycine buffer (200 mM, final concentration) caused very little VLP breakdown, as measured by the 30% sucrose cushion assay (Table 2). Similarly, Brady et al. (6) observed that carbonate buffer at alkaline pH, but not alkaline pH alone, dissociated polyomavirus virions. However, the specific effect of carbonate at pH 9.6 does not appear to be due to carbonate’s potential chelating ability, as suggested by Brady et al. (6), as 200 mM EDTA at pH 9.6 (with or without 10 mM DTT) was completely ineffective at VLP disassembly (data not shown).
Characterization of disassembled VLPs.
Following long-term exposure to high concentrations of reducing agent, the purified VLPs appear to be broken down to the level of capsomeres. As shown in Fig. 3a, the disassembled VLPs generated by incubation in PBS with 5% βME for 16 h at 4°C migrated on 5 to 20% linear sucrose gradients with an average sedimentation coefficient of 11.3S ± 1.5S (n = 5), determined relative to sedimentation standards. Larger species, with a calculated sedimentation coefficient of 16S to 18S (perhaps dimeric capsomeres), and even pelleted material were occasionally observed. However, less than 10% of the L1 was detected at the top of the gradient (expected position for L1 monomer) or in the pellet (expected position for intact VLPs or aggregated capsomeres), suggesting that the purified VLP starting material was largely disassembled to the level of individual capsomeres upon prolonged reduction. This conclusion is supported by electron microscopic analysis of VLPs following prolonged incubation with 5% βME, which depicted a field of homogeneous capsomeres (see Fig. 5b) averaging 9.7 ± 1.2 nm (n = 15) in diameter, with occasionally a few larger aggregated structures apparent (monomeric L1 would not be detected with this technique). The estimated capsomere diameter is slightly smaller than that observed by cryoelectron microscopy (11 to 12 nm) (1, 2, 17), perhaps due to shrinkage during electron microscope grid preparation. The data demonstrated in Fig. 3a and 5b indicate that prolonged exposure to high concentrations of reductants quantitatively disassembles purified, soluble VLPs to a homogeneous population of capsomeres.
FIG. 3.

Five to 20% linear sucrose gradient analysis of disassembled HPV-11 VLPs. VLPs in PBS were incubated with 5% βME (a) or 200 mM NaHCO3 (pH 9.6) (b) for 16 h at 4°C and then centrifuged on a 5 to 20% linear sucrose gradient as described in the text. The gradient was collected in 25 fractions (0.5 ml), and the pellet (P) was resuspended in 0.5 ml of PBS. Shown is an immunoblot demonstrating positions of the L1 protein across the gradient. Also indicated are the peak positions at which sedimentation standards migrated when run on separate gradients.
Capsomeres generated from HPV-11 VLPs upon long-term exposure to high concentrations of reducing agent contain structural epitopes found on intact VLPs. A panel of HPV-11-specific monoclonal antibodies which react with intact HPV-11 L1 VLPs but not with denatured L1 has been described. These monoclonal antibodies include H11.F1, which has been demonstrated to recognize a dominant neutralizing epitope on HPV-11 virions, and H11.A3, a distinct nonneutralizing structure-dependent antibody (10, 11). As anticipated, H11.F1 and H11.A3 reacted strongly with the purified HPV-11 VLP starting material when analyzed by ELISA (see Fig. 6A). Interestingly, these antibodies also reacted with capsomeres generated from the VLP starting material by exposure to reducing agent (see Fig. 6B). Thus, capsomeres possess at least some of the structure-dependent epitopes found on the surface of intact VLPs and authentic virions, in agreement with studies performed by Li et al. (25) on HPV-11 capsomeres generated in E. coli. These results further demonstrate that monoclonal antibodies H11.F1 and H11.A3, while requiring a native-like conformation for binding, are not VLP dependent as has been previously described (29).
FIG. 6.

Reactions of intact and disassembled VLPs with HPV-11 structure-specific monoclonal antibodies. HPV-11 L1 VLP starting material (A), VLPs disassembled by treatment with 5% βME either without (B) or with (C) subsequent dialysis into PBS–0.5 M NaCl to remove reducing agent, and VLPs disassembled in the presence of 200 mM carbonate (pH 9.6) and then dialyzed into PBS–0.5 M NaCl (D) were attached to the wells of microtiter plates. HPV-11 structure-specific monoclonal antibodies H11.F1 (HPV-11 neutralizing; ▿) and H11.A3 (HPV-11 nonneutralizing; •) were tested for immunoreactivity to the bound antigens in an ELISA as described in Materials and Methods. Reactivity with monoclonal antibody AU1 (▪), which recognizes a linear epitope found on HPV-11 L1, was used as a control to demonstrate antigen attachment to the microtiter wells.
In contrast, monoclonal antibodies H11.F1 and H11.A3 failed to recognize HPV-11 VLPs dissociated by treatment with carbonate buffer at pH 9.6 (data not shown) (9). Carbonate treatment did not lead to a homogeneous solution of capsomeres but instead generated an indistinct mixture of small objects, partially aggregated, when examined by electron microscopy (data not shown). This view was partially confirmed by analysis of carbonate-treated VLPs on 5 to 20% linear sucrose gradients, in which the L1 protein largely migrated at ∼4S, although a small population at 9S to 11S was observed (Fig. 3b), in agreement with the effects of carbonate buffer (at pH 10.6, with 10 mM DTT) on bovine papillomavirus (BPV) virions (13). Finally, while treatment with glycine buffer at pH 9.6 did not dissociate VLPs to smaller, individual particles (Table 2), it did have some effect. VLPs treated with pH 9.6 glycine appeared in the electron microscope as a poorly defined mixture of intact and partially broken down and aggregated VLPs (data not shown).
Quantitative reassembly of VLPs.
VLP reassembly from HPV-11 capsomeres occurred upon removal of reducing agent, either by dialysis or by column chromatography. Starting with a homogeneous preparation of soluble capsomeres, prolonged dialysis in the absence of reducing agents consistently yielded a defined population of reassembled VLPs (Fig. 4c; Fig. 5c and d). The reassembled VLPs retained the structural epitopes recognized by monoclonal antibodies H11.F1 and H11.A3 (Fig. 6C).
For reassembly, capsomeres (1 to 5 ml at 0.5 to 1.0 mg of total protein per ml) were dialyzed versus 4 × 1 liter of PBS–0.5 M NaCl at 4°C for ≥24 h; the elevated salt concentration was designed to stabilize the VLPs. Whereas the addition of chelating agents did not appreciably enhance the ability of reducing agents to disassemble VLPs (Table 1), the presence of 2 mM EDTA moderately interfered with reassembly, yielding VLPs which migrated on a 10 to 65% linear sucrose gradient as a fairly discrete population of 150S particles but appeared flattened and partially opened up in the electron microscope (data not shown). Conversely, the addition of 2 mM Ca2+ during the reassembly reaction caused the VLPs to adhere to one another, as shown by 10 to 65% linear sucrose gradient analysis, in which VLPs reassembled in the presence of calcium migrated entirely in the pellet. However, the presence of Ca2+ did not otherwise appear to influence basic VLP morphology when examined in the electron microscope (data not shown). Finally, dialysis of carbonate-treated VLPs into PBS–0.5 M NaCl did not lead to the reassembly of VLPs. Instead, L1 protein remained as either small, soluble components or amorphous, aggregated precipitate, as evidenced by both electron microscopic and 10 to 65% linear sucrose gradient analysis (data not shown). Dialysis of carbonate-treated VLPs failed to restore reactivity with structure-specific monoclonal antibodies H11.F1 and H11.A3 (Fig. 6D).
Characterization of reassembled VLPs.
Following removal of the reducing agent, capsomeres quantitatively reassembled into VLPs. Surprisingly, the reassembled VLPs were much more homogeneous in particle size than the cesium- and sucrose-gradient purified VLP starting material. When the three stages of the disassembly/reassembly reaction were compared by 10 to 65% linear sucrose gradients, the purified VLP starting material was distributed across the gradient, with many particles migrating to the position expected for intact VLPs (150S to 160S) but with the majority of the protein further down the gradient and in the pellet (Fig. 4a). Similarly, when examined in the electron microscope (Fig. 5a), the VLP starting material was seen to be a mixture of different-sized particles, including full-size, 50- to 55-nm-diameter VLPs. It is possible that some disruption of VLPs occurred during extraction and purification, as linear sucrose gradient analysis of earlier stages of the purification process indicated a more homogeneous distribution of particle sizes (data not shown).
Upon long-term exposure to high concentrations of reducing agents, the VLPs were disassembled to capsomeres, as described above. Compared to the VLP starting material, the capsomeres migrated at the top of the 10 to 65% linear sucrose gradients (with little or no L1 detected in the pellet [Fig. 4b]) and in the electron microscope appeared as a lawn of capsomeres (Fig. 5b).
Reassembly of the capsomeres yielded a homogeneous population of spherical, full-sized VLPs. The reassembled VLPs banded in the middle of the 10 to 65% linear sucrose gradients, with a predicted sedimentation coefficient of 150.4S ± 4.6S (n = 7), with much less L1 detected either in the pellet or at the bottom of the gradient than was observed with the purified VLP starting material (Fig. 4c). The homogeneity of the reassembled VLPs was even more striking when examined in the electron microscope, as demonstrated in Fig. 5c and d. Predominantly particles in the range of full-size VLPs were detected, averaging 56.5 ± 7.0 nm (n = 15), with very few partially assembled VLPs or smaller complexes apparent. The yields of the reassembly process were also impressive (averaging 83% in terms of total L1 protein from starting material to reassembled VLPs under optimal disassembly conditions), as essentially all of the capsomeres appeared to reform soluble, filterable, full-size VLPs.
Finally, the ability of the reassembled VLPs to elicit virus-neutralizing antibodies when injected into experimental animals was tested. Polyclonal antisera to both the initial purified HPV-11 VLPs, and disassembled/reassembled HPV-11 VLPs, were generated in BALB/c mice as described in Materials and Methods. The two antisera were equally reactive against the corresponding immunogen when assayed in an ELISA format (data not shown). More importantly, when tested in the RT-PCR neutralization assay involving infectious HPV-11 virions (40), postimmune reassembled HPV-11 VLP-specific polyclonal antisera exhibited a neutralization titer of 10−5, equal to that obtained with the antisera generated against the initial, purified HPV-11 VLPs (Fig. 7). This result demonstrates that the reassembled HPV-11 VLPs retain the highly immunogenic, capsid-neutralizing antigenic domain of HPV-11 virions.
FIG. 7.

Comparison of the abilities of antisera raised against initial purified HPV-11 VLPs and against reassembled VLPs to neutralize HPV-11 virus. Serial log10 dilutions of anti-HPV-11 antiserum (10−3 to 10−7) raised against initial purified HPV-11 VLPs or reassembled VLPs were incubated with HPV-11 virions before addition to HaCaT cells. Control experiments without added virions (lane C) or virions added to cells without preincubation with serum (lane V) were also run. Six days postinfection, the cells were harvested and the presence of the E1∧E4 spliced message, diagnostic of HPV-11 infection, was determined by RT-PCR. PCR products were separated on 2% agarose gels, stained with ethidium bromide, and examined under UV light for the presence of the ∼0.6-kb E1∧E4 band (a). PCR amplification of β-actin was performed on all cDNA samples as an internal control (b). The expected size of the β-actin band is ∼0.6 kb. Lane S contains molecular size markers.
DISCUSSION
We report here, for the first time, conditions for the quantitative disassembly and subsequent reassembly of papillomavirus VLPs in vitro. Earlier attempts at papillomavirus VLP disassembly were to some extent influenced by work performed with polyomavirus, a structurally related papovavirus, where it was shown that both reduction of disulfides and chelation of calcium ions were essential for virion disassembly (6). However, the low levels of reducing agent (1 to 10 mM DTT) optimal for polyomavirus disassembly, in the presence of low levels of chelating agents (e.g., 0.5 to 10 mM EDTA), were found to be only slightly effective at disassembling papillomavirus VLPs (Table 1) (23), although partially trypsinized HPV-11 L1 VLPs were dissociated by the foregoing conditions (25). However, Sapp and coworkers demonstrated that capsomeres could be generated from HPV-33 VLPs by treatment with reducing agent alone (20 mM DTT), although the extent of VLP breakdown was not determined (37). In our studies, we found that when examining disassembly by gradient analysis, it was necessary to test for the presence of L1 protein in the pellet. In many cases, examination of fractions across the gradient would suggest that good breakdown had been achieved. However, examination of the pellet, even though none was visible, would indicate that a large percentage of the protein was still in the form of variably sized VLPs or otherwise aggregrated, as confirmed by electron microscopic analysis. The development of the 30% sucrose cushion assay allowed us to screen a number of disassembly conditions rapidly and identify those which consistently disassembled the VLPs to smaller, soluble components. We found that quantitative disassembly to a homogeneous solution of individual capsomeres could be consistently achieved by extended treatment of nonaggregated VLPs with high levels of reducing agent in moderate- to low-ionic-strength buffers.
The observation that chelation of cations did not materially affect VLP disassembly is in contrast to earlier studies with polyomavirus, which indicated that calcium chelation promoted virion disassembly and that added calcium could overcome the effect of chelators (6). Similarly, Montross et al. (31) observed that polyomavirus VLPs, which normally assemble only in the nucleus, could form in the cytoplasm following addition of a calcium ionophore, which presumably raised the cytoplasmic calcium concentration to the necessary level. However, calcium is apparently not important to HPV-11 L1 capsid stability, nor is it a general feature of other viruses. For example, even in virions where calcium binding sites have been described at atomic resolution, such as SV40 or rhinovirus, extraction of the bound calcium, or mutagenesis to remove the calcium binding site, did not significantly alter virus structure (26, 48). Conversely, treatment with carbonate buffer at alkaline pH did disassemble HPV-11 L1 VLPs, similar to results seen with polyomavirus virions (6). However, the dissociation of HPV-11 VLPs by carbonate treatment appears more complete than that caused by reducing agents, as in the electron microscope no regular structures were observed for carbonate-treated VLPs, nor could VLPs be regenerated by dialysis into PBS–0.5 M NaCl following carbonate treatment.
HPV-11 VLP disassembly by carbonate treatment resulted in L1 protein which failed to react with structure-dependent, HPV-11-specific monoclonal antibodies. In contrast, disassembly of HPV-11 L1 VLPs by prolonged reduction resulted in capsomeres which possessed structure-specific epitopes found on the surface of both intact HPV-11 L1 VLPs and HPV-11 virions. Previous studies by Li et al. (25) also indicated antigenic similarity between HPV-11 L1 capsomeres, VLPs, and authentic virions. Studies are currently in progress to determine whether, like HPV-11 VLPs, capsomeres generated by reduction of disulfides are capable of eliciting production of virus-neutralizing antibodies.
To reassemble full-size VLPs efficiently in vitro, our studies indicate that the structural integrity, solubility, and homogeneity of the capsomere starting material are crucial. Following generation of such a population of capsomeres by thiol reduction, reassembly occurs spontaneously upon removal of reducing agent. Whereas we have achieved some degree of reassembly by utilizing column chromatographic methods to remove reductant, dialysis against a large excess of buffer has reliably yielded uniform preparations of full-sized VLPs. In earlier studies of polyomavirus, Salunke et al. (36) observed that VLP assembly from capsomeres yielded multiple, polymorphic icosahedral assemblies as a function of the assembly conditions (pH, ionic strength, and calcium concentration). Interestingly, the most consistently formed structure was a 24-capsomere icosahedron, as well as a 12-capsomere icosahedron, in addition to the 72-capsomere icosahedron of the viral capsid. The authors noted that disulfide bond formation might aid in polyomavirus VLP assembly but that it was not essential, as at high ionic strength (2 M ammonium sulfate), variably sized capsids formed even in the presence of 15 mM βME. Similarly, Li et al. (25) have observed that during chromatographic purification of HPV-11 capsomeres expressed in E. coli, some capsid-like structures are formed in 1 M NaCl, again in the presence of 15 mM βME. However, while high-ionic-strength conditions apparently favor some degree of capsid formation, it is clear from our studies that at physiological ionic strength, disulfide bonds are necessary to hold HPV-11 L1 VLPs together.
The observation that VLPs form spontaneously from capsomeres at high-ionic-strength conditions but are thermodynamically unstable in a reducing environment at physiological salt concentrations suggests a possible model for viral disassembly during infection. Viral capsids form in the nucleus, where host and viral DNA may create a local, high-ionic-strength environment, as has been observed in studies of histone conformation and assembly (30). Disulfide bonds may then form in the nucleus or upon release of the virion from the cell. These disulfide bonds would then contribute to the stability of the virus as it traverses extracellular compartments. Upon infection, exposure of the capsid to the cytoplasm (a reducing environment containing 1 to 2 mM glutathione) could break some of the stabilizing disulfide bonds, leading to capsid disassembly at physiological ionic strength. Disulfide bond breakage might be accelerated by partial proteolysis of the capsid. Li et al. (25) have shown that proteolysis with trypsin leads to VLP disassembly under relatively mild reducing conditions. Similarly, we have observed that even mild proteolysis (removal of an ∼30-amino-acid peptide from the C terminus of the L1 protein) renders HPV-11 VLPs more susceptible to disassembly by low levels of reducing agent (data not shown). In sum, the ability of disulfide bonds to stabilize viral capsids while they endure the rigors of the external world combined with the susceptibility of disulfide bonds to the reducing environment of the cell suggests an important role for these bonds in the life cycle of the virus.
Even given that the disassembly reactions were typically performed at 4°C without agitation, it is interesting that maximal disassembly required prolonged exposure to very high levels of reducing agent. The most likely explanation is that the stabilizing disulfide bonds are buried and inaccessible and that exposure of these bonds to solvent by local structural fluctuations is very infrequent. Conversely, as summarized below, the most likely location of these disulfides is on the C-terminal tail of the L1 protein, a portion of the molecule which would be predicted to be relatively accessible to solvent. Whereas a comparatively small deletion (24 amino acids) of the C terminus of the BPV L1 protein did not interfere with VLP formation, a larger deletion (44 amino acids) did inhibit proper capsid formation (32). Similarly, an even more extensive C-terminal cleavage (86 amino acids) of the HPV-11 L1 protein yielded only capsomeres which did not appear capable of forming capsid-like structures (25). In the 3.8-Å structure of SV40, the C-terminal domain of the VP1 protein was demonstrated explicitly to form the intercapsomeric bonds which stabilize the capsid, by extending across the space between capsomeres and actually forming part of the extended β-sheet of the neighboring capsomeric L1 protein; disulfide bonds may also be important for this interaction but cannot be resolved in the crystal structure due to disorder in this portion of the molecule (26, 41). Fifteen-angstrom strands connecting capsomeres are also seen, at lower resolution, in the cryoelectron microscopic reconstruction of the BPV structure (1, 2) and in negatively stained HPV virions (47), suggesting that linker arms also stabilize Papovaviridae capsids. In combination, these studies support the notion that the C-terminal domain of the L1 protein is important for stabilizing capsomeric interactions in the assembled capsids and that the cysteine residue(s) involved in disulfide bond formation are localized in this domain. The conserved cysteine at amino acid position 424 in the HPV-11 L1 protein appears to be a good candidate.
The ability to reassemble full-sized VLPs in bulk opens a number of possibilities. The reassembled VLPs represent a homogeneous preparation of full-size VLPs (i.e., the size of infectious virus) capable of eliciting neutralizing antibodies. It is possible that the reassembled VLPs will be more potent immunogens than the initial, purified VLPs, which are heterogeneous in size. Whereas a number of different-sized and -shaped particles are observed in the nuclei of cells following infection in vivo (22), presumably only full-sized virus are productively infective. We are currently determining the potency of reassembled VLPs as immunogens by dosing mice with increasingly smaller amounts of initial and reassembled VLPs. We are also comparing the stability of initial and reassembled VLPs formed from the L1 proteins of a variety of HPV types. Additional aspects of the reassembly reaction which we are examining in greater detail are the effects of protein concentration, pH, ionic strength, and temperature, both to optimize reassembly under a greater range of starting conditions and to investigate the rules which dictate capsid assembly (3, 7). Finally, it appears possible to package exogenous compounds within VLPs by performing the reassembly reaction in the presence of a concentrated solution of the selected compound. This technology could be used to generate pseudovirions for use as surrogates for HPV types which are not currently available, or even as a possible delivery system for drugs or other targeted compounds.
ACKNOWLEDGMENTS
We thank Neil D. Christensen for providing HPV-11-specific monoclonal antibodies, John W. Kreider for providing HPV-11Hershey, Norbert Fusenig for providing HaCaT cells, Marco Cacciuttolo and Maria Patchan for culturing large amounts of baculovirus-infected T. ni cells, Steve Burke for helping to develop the 30% sucrose cushion assay, Kannaki Senthil for preparing purified HPV-11 L1 VLPs, and Eileen Rusnock and Hamp Edwards for help with electron microscopy.
REFERENCES
- 1.Baker T S, Newcombe W N, Olson B H, Cowsert L M, Olson C, Brown J C. Structure of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys J. 1991;60:1445–1456. doi: 10.1016/S0006-3495(91)82181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belnap D V, Olson N H, Cladel N M, Newcomb W N, Brown J C, Kreider J W, Christensen N D, Baker T S. Conserved features in papillomavirus and polyomavirus capsids. J Mol Biol. 1996;259:249–263. doi: 10.1006/jmbi.1996.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger B, Shor P W, Tucker-Kellogg L, King J. Local rule-based theory of virus shell assembly. Proc Natl Acad Sci USA. 1994;91:7732–7736. doi: 10.1073/pnas.91.16.7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boukamp P, Petrussekvka R T, Breifreutz D, Hornung J, Markham A, Fusenig N E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Brady J N, Winston V D, Consigli R A. Dissociation of polyoma virus by the chelation of calcium ions found associated with purified virions. J Virol. 1977;23:717–724. doi: 10.1128/jvi.23.3.717-724.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caspar D L D, Klug A. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp Quant Biol. 1962;27:1–24. doi: 10.1101/sqb.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- 8.Christensen N D, Höpfl R, DiAngelo S L, Cladel N M, Patrick S D, Welsh P A, Budgeon L R, Reed C A, Kreider J W. Assembled baculovirus-expressed human papillomavirus type 11 L1 capsid protein virus-like particles are recognized by neutralizing monoclonal antibodies and induce high titres of neutralizing antibodies. J Gen Virol. 1994;75:2271–2276. doi: 10.1099/0022-1317-75-9-2271. [DOI] [PubMed] [Google Scholar]
- 9.Christensen N D, Kreider J W. Antibody-mediated neutralization of infectious papillomaviruses. J Virol. 1990;64:3151–3156. doi: 10.1128/jvi.64.7.3151-3156.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen N D, Kreider J W, Cladel N M, Patrick S D, Welsh P A. Monoclonal antibody-mediated neutralization of infectious human papillomavirus type 11. J Virol. 1990;64:5678–5681. doi: 10.1128/jvi.64.11.5678-5681.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christiansen G, Landers T, Griffith J, Berg P. Characterization of components released by alkali disruption of simian virus 40. J Virol. 1977;21:1079–1084. doi: 10.1128/jvi.21.3.1079-1084.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Villiers E M. Heterogeneity of the human papillomavirus group. J Virol. 1989;63:4898–4903. doi: 10.1128/jvi.63.11.4898-4903.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favre M, Breitburd F, Croissant O, Orth G. Structural polypeptides of rabbit, bovine and human papillomaviruses. J Virol. 1975;15:1239–1247. doi: 10.1128/jvi.15.5.1239-1247.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gajardo R, Vende P, Poncet D, Cohen J. Two proline residues are essential in the calcium-binding activity of rotavirus VP7 outer capsid protein. J Virol. 1997;71:2211–2216. doi: 10.1128/jvi.71.3.2211-2216.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcea R L, Benjamin T L. Host range transforming gene of polyoma virus plays a role in virus assembly. Proc Natl Acad Sci USA. 1983;80:3613–3617. doi: 10.1073/pnas.80.12.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghim S, Schlegel R, Hines J F, Christensen N D, Kreider J, Jensen A B. Comparison of PV conformational epitopes expressed by L1 proteins in mammalian (cos) and insect (Sf9) cells. In: Stanley M A, editor. Immunology of human papillomaviruses. New York, N.Y: Plenum Press; 1994. pp. 147–153. [Google Scholar]
- 17.Hagensee M E, Olson N H, Baker T S, Galloway D A. Three-dimensional structure of vaccinia virus-produced human papillomavirus type 1 capsids. J Virol. 1994;68:4503–4505. doi: 10.1128/jvi.68.7.4503-4505.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagensee M E, Yaegashi N, Galloway D A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J Virol. 1993;67:315–322. doi: 10.1128/jvi.67.1.315-322.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann K J, Cook J C, Joyce J G, Brown D R, Schultz L D, George H A, Rosolowsky M, Fife K H, Jansen K U. Sequence determination of human papillomavirus type 6a and assembly of virus-like particles in Saccharomyces cerevisiae. Virology. 1995;209:506–518. doi: 10.1006/viro.1995.1283. [DOI] [PubMed] [Google Scholar]
- 20.Kirnbauer R, Booy F, Cheng N, Lowy D R, Schiller H R. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci USA. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirnbauer R, Taub J, Greenstone H, Roden R, Dürst M, Gissman L, Lowy D R, Schiller J T. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J Virol. 1993;67:6929–6936. doi: 10.1128/jvi.67.12.6929-6936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiselev N A, Klug A. The structure of viruses of papilloma-polyoma type. V. Tubular variants built of pentamers. J Mol Biol. 1969;40:155–171. doi: 10.1016/0022-2836(69)90465-3. [DOI] [PubMed] [Google Scholar]
- 23.Kreider J W, Howett M K, Leure-Dupree A E, Zaino R J, Weber J A. Laboratory production in vivo of infectious human papillomavirus type 11. J Virol. 1987;61:590–593. doi: 10.1128/jvi.61.2.590-593.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Li M, Cripe T P, Estes P A, Lyon M K, Rose R C, Garcea R L. Expression of the human papillomavirus type 11 L1 capsid protein in Escherichia coli: characterization of protein domains involved in DNA binding and capsid assembly. J Virol. 1997;71:2988–2995. doi: 10.1128/jvi.71.4.2988-2995.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liddington R C, Yan Y, Moulai J, Sahli R, Benjamin T L, Harrison S C. Structure of simian virus 40 at 3.8-Å resolution. Nature. 1991;354:278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- 27.Lim P S, Jenson A B, Cowsert L, Nakai Y, Lim L Y, Jin X W, Sundberg J P. Distribution and specific identification of papillomavirus major capsid protein epitopes by immunocytochemistry and epitope scanning of synthetic peptides. J Infect Dis. 1990;162:1263–1269. doi: 10.1093/infdis/162.6.1263. [DOI] [PubMed] [Google Scholar]
- 28.Lowy D R, Kirnbauer R, Schiller J T. Genital human papillomavirus infection. Proc Natl Acad Sci USA. 1994;91:2436–2440. doi: 10.1073/pnas.91.7.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ludmerer S W, Benicasa D, Mark G E, Christensen N D. A neutralizing epitope of human papillomavirus type 11 is principally described by a continuous set of residues which overlap a distinct linear, surface-exposed epitope. J Virol. 1997;71:3834–3839. doi: 10.1128/jvi.71.5.3834-3839.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy M P, Steffen P K, Allewell N M, Benedict R C, Moudrianikis E N, Ackers G K. Effects of ionic strength and state of assembly on kinetics of hydrogen exchange of calf thymus histones. Biochemistry. 1984;23:2227–2230. doi: 10.1021/bi00305a020. [DOI] [PubMed] [Google Scholar]
- 31.Montross L, Watkins S, Moreland R B, Mamon H, Caspar D L D, Garcea R L. Nuclear assembly of polyomavirus capsids in insect cells expressing the major capsid protein VP1. J Virol. 1991;65:4991–4998. doi: 10.1128/jvi.65.9.4991-4998.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paintsil J, Müller M, Picken M, Gissmann L, Zhou J. Carboxyl terminus of bovine papillomavirus type-1 L1 protein is not required for capsid formation. Virology. 1996;223:238–244. doi: 10.1006/viro.1996.0473. [DOI] [PubMed] [Google Scholar]
- 33.Pfister H. Papillomaviruses: general classification, taxonomy, and classification. In: Salzman N, Howley P N, editors. The Papovaviridae. New York, N.Y: Plenum Press; 1987. pp. 1–38. [Google Scholar]
- 34.Rose R C, Bonnez W, Reichamn R C, Garcea R L. Expression of human papillomavirus in type 11 L1 protein in insect cells: in vivo and in vitro assembly of viruslike particles. J Virol. 1993;67:1936–1944. doi: 10.1128/jvi.67.4.1936-1944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salunke D M, Caspar D L D, Garcea R L. Self-assembly of purified polyomavirus capsid proten VP1. Cell. 1986;46:895–904. doi: 10.1016/0092-8674(86)90071-1. [DOI] [PubMed] [Google Scholar]
- 36.Salunke D M, Caspar D L D, Garcea R L. Polymorphism in the assembly of polyomavirus capsid protein VP1. Biophys J. 1989;56:887–900. doi: 10.1016/S0006-3495(89)82735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapp M, Volpers C, Müller M, Streeck R E. Organization of the major and minor capsid proteins in human papillomavirus type 33 virus-like particles. J Gen Virol. 1995;76:2407–2412. doi: 10.1099/0022-1317-76-9-2407. [DOI] [PubMed] [Google Scholar]
- 38.Sasagawa T, Pushko P, Steers G, Gschmeissner S E, Hajibagheri M A N, Finch J, Crawford L, Tommasino M. Synthesis and assembly of virus-like particles of human papillomaviruses type 6 and type 16 in fission yeast Schizosaccharomyces pombe. Virology. 1995;206:126–135. doi: 10.1016/s0042-6822(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 39.See Y P, Jackowski G. Estimating molecular weights of polypeptides by SDS gel electrophoresis. In: Creighton T E, editor. Protein structure: a practical approach. New York, N.Y: IRL Press; 1989. pp. 1–21. [Google Scholar]
- 40.Smith L H, Foster C, Hitchcock M E, Leiserowitz G S, Hall K, Isseroff R, Christensen N D, Kreider J W. Titration of HPV-11 infectivity and antibody neutralization can be measured in vitro. J Invest Dermatol. 1995;105:1–7. doi: 10.1111/1523-1747.ep12321173. [DOI] [PubMed] [Google Scholar]
- 41.Stehle T, Gamblin S J, Yan Y, Harrison S C. The structure of simian virus 40 refined at 3.1 Å resolution. Structure. 1996;4:165–182. doi: 10.1016/s0969-2126(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 42.Suzich J A, Ghim S-J, Palmer-Hill F J, White W I, Tamura J K, Bell J A, Newsome J A, Jenson A B, Schliegel R. Systemic immunization with papillomavirus L1 protein completely prevents the development of viral mucosal papillomas. Proc Natl Acad Sci USA. 1995;92:11553–11557. doi: 10.1073/pnas.92.25.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volpers C, Schirmacher P, Streeck R E, Sapp M. Assembly of the major and minor capsid protein of human papillomavirus type 33 into virus-like particles and tubular structures in insect cells. Virology. 1994;200:504–512. doi: 10.1006/viro.1994.1213. [DOI] [PubMed] [Google Scholar]
- 45.Walter G, Deppert W. Intermolecular disulfide bonds: an important structural feature of the polyoma virus capsid. Cold Spring Harbor Symp Quant Biol. 1975;39:255–257. doi: 10.1101/sqb.1974.039.01.033. [DOI] [PubMed] [Google Scholar]
- 46.Xi S-Z, Banks L M. Baculovirus expression of the human papillomavirus type 16 capsid proteins: detection of L1-L2 protein complexes. J Gen Virol. 1991;72:2981–2988. doi: 10.1099/0022-1317-72-12-2981. [DOI] [PubMed] [Google Scholar]
- 47.Yabe Y, Sadakane H, Isono H. Connection between capsomeres in human papilloma virus. Virology. 1979;96:547–552. doi: 10.1016/0042-6822(79)90110-7. [DOI] [PubMed] [Google Scholar]
- 48.Zhao R, Hadfield A T, Kremer M J, Rossmann M G. Cations in human rhinoviruses. Virology. 1997;227:13–23. doi: 10.1006/viro.1996.8301. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Stenzel D J, Sun X-Y, Frazer I H. Synthesis and assembly of infectious bovine papillomavirus particles in vitro. J Gen Virol. 1993;74:763–768. doi: 10.1099/0022-1317-74-4-763. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Sun X-Y, Louis K, Frazer I H. Interaction of human papillomavirus (HPV) type 16 capsid protein with HPV DNA requires an intact L2 N-terminal sequence. J Virol. 1994;68:619–625. doi: 10.1128/jvi.68.2.619-625.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]