

Abstract
A novel strategy of not only stimulating the immune cycle but also modulating the immunosuppressive tumor microenvironment is of vital importance to efficient cancer immunotherapy. Here, a new type of spatiotemporal biomimetic “Gemini nanoimmunoregulators” was engineered to activate robust systemic photoimmunotherapy by integrating the triple-punch of amplified immunogenic cell death (ICD), tumor-associated macrophages (TAMs) phenotype reprogramming and programmed cell death ligand 1 (PD-L1) degradation. The “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 were constructed by taking the biocompatible mesoporous polydopamine (mPDA) as nanovectors to deliver metformin (Met) and toll-like receptor 7/8 agonist resiquimod (R848) to cancer cells and TAMs by specific biorecognition via wrapping of red blood cell membrane (RM) inlaid with T7 or M2 peptides. mPDA/Met@RM-T7 (abbreviated as PM@RM-T7) was constructed to elicit an amplified in situ ICD effect through the targeted PTT and effectively stimulated the anticancer immunity. Meanwhile, PD-L1 on the remaining cancer cells was degraded by the burst metformin to prevent immune evasion. Subsequently, mPDA/R848@RM-M2 (abbreviated as PR@RM-M2) specifically recognized TAMs and reset the phenotype from M2 to M1 state, thus disrupting the immunosuppressive microenvironment and further boosting the function of cytotoxic T lymphocytes. This pair of sister nanoimmunoregulators cooperatively orchestrated the comprehensive anticancer activity, which remarkably inhibited the growth of primary and distant 4T1 tumors and prevented malignant metastasis. This study highlights the spatiotemporal cooperative modalities using multiple nanomedicines and provides a new paradigm for efficient cancer immunotherapy against metastatic-prone tumors.
KEY WORDS: Biomimetic immunoregulator, Spatiotemporal delivery, Targeted photothermal therapy, Amplified immunogenic cell death, Immunosuppressive tumor microenvironment, PD-L1 degradation, TAMs phenotype reversion, Metastasis inhibition
Graphical abstract
A novel pair of biomimetic “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 were launched for the spatiotemporal cancer photoimmunotherapy via targeted hyperthermia-induced immunogenic cell death, immunosuppressive tumor-associated macrophages reeducation, and PD-L1 degradation.
1. Introduction
In the recent decade, immunotherapy has overwhelmed the conventional techniques in the area of cancer treatment due to its great power to harness the patient's immune system to eliminate the local and disseminated metastatic cells1,2. Great advances focused on immune checkpoint blockade3, chimeric antigen receptor T-cell immunotherapy4, and nanovaccines5,6 have benefited much to cancer patients. Prominently, photoimmunotherapy, which eliminates most cancer cells and induces immunogenic cell death (ICD) through in situ release of tumor-associated antigens and damaged-associated molecular patterns to invoke adaptive immune responses, represents a novel modality of anticancer immunotherapy, especially for the “immune cold” solid tumors7,8. Much effort has been dedicated to further improving the dendritic cells (DCs) maturation and the cytotoxic T lymphocytes (CTLs) activation by the codelivery nanosystem of photosensitive agents with immunoadjuvant based on the nanobiotechnology9. However, the tumor develops complicated and heterogeneous mechanisms to evade immune-mediated elimination10. Besides low antigenicity and immunogenicity, immunosuppressive tumor microenvironment (ITM) is the main culprit of the failure in the current immunotherapy, including photoimmunotherapy. Massive immune cells such as tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells in ITM contribute much to the limited infiltration and function exhaustion of CTLs, arousing the adaptive resistance11,12. Therefore, comprehensively modulating ITM from different aspects could profoundly elevate the efficacy of CTLs-mediated anticancer immunity and establish a long-term immune defense against tumors.
As a well-known immune checkpoint, programmed cell death ligand 1 (PD-L1)/programmed cell death 1 (PD-1) negative regulatory axile opens a new era of cancer immunotherapy, which was awarded the 2018 Nobel Prize in Medicine and Physiology. The overexpressed PD-L1 ligand on the surface of tumor cells binds to the PD-1 receptor on CTLs and induces the dysfunction and apoptosis of CTLs, thus protecting tumor cells from immune surveillance3. Antibodies of PD-1 or PD-L1 (e.g., OPDIVO® and KEYTRUDA®) block the interaction of PD-1/PD-L1 and rejuvenate exhausted CTLs. Their great success in several advanced tumors (the objective response rate was 46% in PD-L1 highly expressed tumors) brings exciting hope and opportunity for cancer patients13. However, the high costs of commercial anti-PD-1/PD-L1 antibodies carry a heavy financial burden on patients14. Moreover, systemic delivery of monoclonal antibodies is often accompanied by low targeting specificity and severe side effects (e.g., immune-mediated inflammation)3. Small molecule inhibitors provide an alternative avenue to solve the dilemma of antibody-based immunotherapy. Among them, metformin (Met), a small molecule drug prevalently used for type-II diabetes treatment, has been revealed to own diverse functions of metabolism regulation, tumor inhibition, and even immunomodulation15,16. Typically, it effectively blocks the PD-1/PD-L1 axile by activating the adenosine 5′-monophosphate-activated protein kinase to phosphorylate PD-L1 (at S195) and thus inducing the abnormal glycosylation and degradation of PD-L117. Repurposing of the old drug Met as a new immunoadjuvant remarkably reversed the immunosuppression and improved the anticancer immunity together with photodynamic therapy or chemotherapy18. Despite the advantages of effectiveness, safety, and low price, precisely and safely delivering the hydrophilic Met to the tumor site remains a formidable challenge.
On the other hand, TAMs, as the essential component of ITM (accounting for 30%–50% of the populations of innate immune cells), are closely associated with tumor progression, angiogenesis, metastasis, and even poor prognoses19. In oncology, TAMs usually feature an activated protumoral M2 phenotype and exhibit immunosuppressive effects by secreting many anti-inflammatory factors like interleukin-10 (IL-10), which further boost Tregs and directly inhibit T cell function20. Conversely, the tumoricidal M1 type macrophages not only own the ability to phagocytose tumor cells but also could secrete inflammatory cytokines (e.g., TNF-α, IFN-γ, and IL-6) to activate the antitumor immune response and remodel ITM. Therefore, TAMs have become an important target in cancer treatment, and reprogramming the phenotype of TAMs from M2 to M1 state was the most popular method to modulate anticancer activities of CTLs21,22. Resiquimod (R848) is a very effective TLR7/8 agonist to repolarize TAMs to produce the pro-inflammatory cytokines and ultimately activate the innate and adaptive immune responses, which was comparable to lipopolysaccharide (LPS)23. As an essential hydrophobic immune adjuvant, codelivery of R848 and photosensitizers in a nano platform significantly amplified the anticancer immunity after photoablation, thus improving the survival of tumor-bearing mice24. We propose that further combining R848 with PD-L1/PD-1 checkpoint inhibitors (e.g., Met) could fundamentally enhance CTLs-based immunotherapy. However, the all-in-one multicomponent nanoplatforms usually require cumbersome and multistep modifications25,26. Even it seems too idealistic to utilize a single system to deliver different drugs to different targets, including cancer cells and various immune cells (e.g., TAMs and/or PD-L1 checkpoint). It is highly desirable to design a multi-vector to spatiotemporally deliver different immune agents to the specific cell subtype for achieving synergistic anticancer immunity.
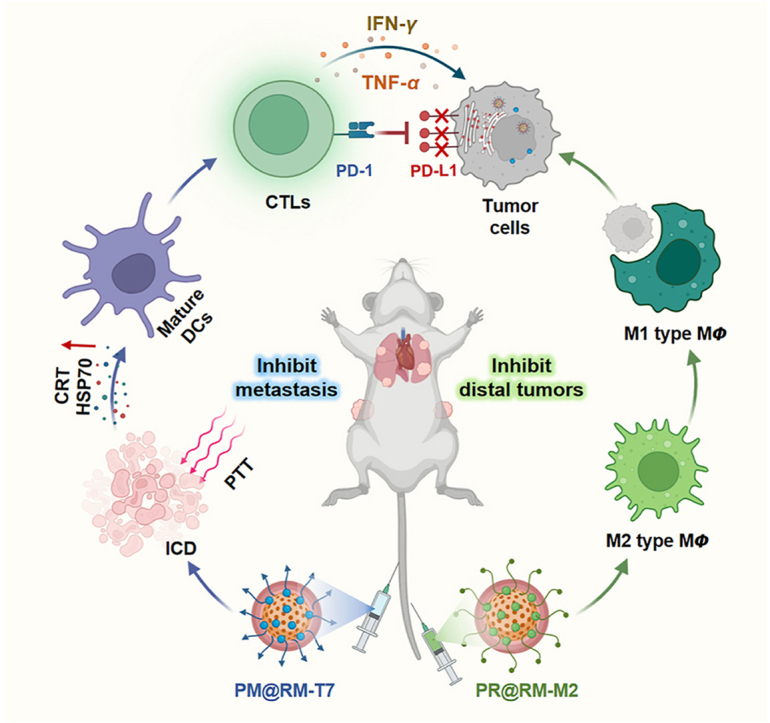
In light of the above considerations, we developed a new type of biomimetic “Gemini nanoimmunoregulators” for enhanced photoimmunotherapy via spatiotemporally targeting the tumor cells, PD-L1 checkpoint, and TAMs against the tumor growth and metastasis. As shown in Scheme 1A, the “Gemini nanoimmunoregulators” were constructed respectively by loading PD-L1 inhibitor Met and toll-like receptor 7/8 agonist R848 into the highly biocompatible mesoporous polydopamine (mPDA), which was an excellent photothermal nanoagent and efficient drug loading framework in the family of nanoscale biomaterials27. Coating mPDA/Met (abbreviated as PM) with T7 peptide-engineered red blood cell membrane (RM) yielded mPDA/Met@RM-T7 (abbreviated as PM@RM-T7) and rendered it high stability and recognition of tumor cells via transferrin receptors28. The sister nanoimmunoregulator mPDA/R848@RM-M2 (abbreviated as PR@RM-M2) was yielded by camouflaging mPDA/R848 (abbreviated as PR) with M2 peptide anchored RM, which enables it to bind to M2 type TAMs other than leukocytes selectively29. Through the cooperation of PM@RM-T7 and PR@RM-M2, enhanced CTLs activation was achieved through several paths (Scheme 1B): i) PM@RM-T7 precisely recognized the tumors and induced obvious ICD post photothermal therapy (PTT), increasing the immunogenicity of tumors and turning the “cold” tumor to “hot” for easy CTLs filtration. ii) PR@RM-M2 precisely repolarized TAMs to M1 phenotype, enhancing the phagocytosis of tumor cells and disarming immunosuppression of ITM. iii) The released Met from PM@RM-T7 further depleted PD-L1 of the remaining tumor cells and prevented immune escape. Thus, the triple-pronged strategy triggered profound anticancer immunity to control tumor proliferation and metastasis. Therefore, the “Gemini nanoimmunoregulators” provide a new paradigm of applying spatiotemporal nanomedicine to enhance the anticancer immune cycle from different aspects.
Scheme 1.

Schematic illustration of (A) the construction of the biomimetic “Gemini nanoimmunoregulators” and (B) the synergistic photoimmunotherapy via spatiotemporal targeted PTT, phenotype reversion of TAMs, and PD-L1 inhibition.
2. Materials and methods
2.1. Materials
Dopamine hydrochloride (98%) was purchased from Aladdin (Shanghai, China). Metformin and chlorine 6 (Ce6) were purchased from HEOWNS Biochemical Technology (Tianjin, China). Pluronic F127 was purchased from Sigma–Aldrich (St. Louis, MO, USA). Resiquimod (R848) was purchased from AdooQ Bioscience (Irvine, CA, USA). DSPE-PEG-MAL was purchased from Ruixi Biotechnology (Xi'an, China). SH-T7 peptide (Sequence: HAIYPRHC) was purchased from Taigu Biotechnology (Nanjing, China). SH-M2 peptide (Sequence: YEQDPWGVKWWYC) was purchased from Source peptide biology (Nanjing, China). Mouse IL-10, mouse IL-6, mouse tumor necrosis factor-α (TNF-α), and mouse interferon-γ (IFN-γ) enzyme-linked immunosorbent assay (ELISA) kits were provided by ABclonal Biotechnology (Wuhan, China). Nuclear dye 4′,6-diamidino-2-phenylindole (DAPI), the cell counting kit (CCK-8), and annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining kit, membrane dyes of 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1ʹ-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) were purchased from Beyotime Biotechnology (Shanghai, China).
2.2. Preparation and characterization of PM@RM-T7 and PR@RM-M2
PM@RM-T7 and PR@RM-M2 were constructed according to the following procedure. mPDA was respectively loaded with Met and R848 to obtain PM and PR. And the preparation details were given in the supporting information. RM was obtained through the method of hypotonic lysis of blood taken from the orbit of mice. After sonication by a cell ultrasonic disruptor for 5 min, RM were mixed with DSPE-PEG-T7 or DSPE-PEG-M2 at a ratio of 1/0.1 (m/m, RM/DSPE-PEG-T7, or RM/DSPE-PEG-M2), which was obtained by conjugating DSPE-PEG-MAL with SH-T7 or SH-M2 peptide. After incubation at 4 °C under stirring for 24 h, the obtained T7 and M2 peptide-embedded RM were bath-sonicated (180 W, 40 KHz) with PM or PR water solution for 10 min at the ratio of 1/1 (m/m, mPDA/RM) and then incubated overnight. Finally, they were continuously extruded with 200 nm polycarbonate porous membranes using an Avanti mini extruder (Avanti Polar and Lipids, Alabaster, USA). PM@RM-T7 or PR@RM-M2 was obtained after centrifugation (13,000 rpm, 10 min, 5804R, Eppendorf, Hamburg, German) and stored at 4 °C.
To verify the successful membrane modification, the protein concentrations of PM@RM-T7 and PR@RM-M2 were determined by bicinchoninic acid assay (BCA) and then treated with 12.5% sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 20 μg/well and stained with coomassie brilliant blue. After rinsing with the decolorizing solution three times, the gel was imaged by a digital camera. Furthermore, fluorescent indicator FITC was doped in mPDA to form PF@RM-T7 and then stained with membrane dye DiI for observation under fluorescent microscopy. Transmission electron microscope (TEM) images were acquired on JEM 2100F (JEOL, Tokyo, Japan). The absorption spectra of PM@RM-T7 and PR@RM-M2 were conducted on a UV–Visible spectrometer (UV2910, Hitachi, Tokyo, Japan). The pore size and specific surface area of mPDA were determined by the Brunauer-Emmett-Teller (BET) analyzer (JW-BK200C, Beijing, China). Zeta potential and dynamic light scattering (DLS) measurements were conducted on a Zetasizer Nanoseries (Malvern Instruments, Malvern, UK).
2.3. Photothermal effect and pH-responsive drug release
The solutions of PM@RM-T7 and PM (5 mL, at the equivalent concentration of mPDA = 1.0 mg/mL) containing 1.5 mg Met were transferred to a dialysis bag (MWCO = 3500 Da) and then stirred in 50 mL of phosphate buffer saline (PBS, pH 5.0 and 7.4) at 37 °C under stirring (500 rpm) (Eppendorf). PM@RM-T7 and PM solutions were laser irradiated (808 nm, 1.0 W/cm2, 10 min, BWT, Beijing, China) at the scheduled time. Then, 1.0 mL of PBS was collected, and an equal volume of fresh PBS was supplemented. The released Met was spectroscopically calculated by the standard curve. Likewise, the release of R848 from PR and PR@RM-M2 at different pH was measured with the same procedures as above.
2.4. Site-specific delivery of PM@RM-T7 and PR@RM-M2
To investigate the cellular uptake of PM@RM-T7 into 4T1 cancer cells, Met was replaced with the fluorescent molecule Ce6 to yield PC@RM-T7 as a surrogate. 4T1 cells seeded on six-well microplates (4 × 104 cells per well) were cultured overnight and then incubated with PC@RM and PC@RM-M2 (at the equivalent concentration of mPDA = 200 μg/mL) for 6 h. After being washed with PBS, the cells were digested by trypsin and collected for flow cytometry analysis (Agilent, Santa Clara, CA, USA). Furthermore, a laser scanning confocal microscope (CLSM, Zeiss, Oberkochen, German) was employed to evaluate the cellular uptake of PC@RM-T7 by 4T1 cells qualitatively.
To investigate the phagocytosis of PR@RM-M2 into M2 type RAW264.7 cells. RAW264.7 cells were polarized into M2 type by IL-4 (20 ng/mL) and then incubated with PC@RM, PC@RM-T7, and PC@RM-M2 (at the equivalent concentration of mPDA = 200 μg/mL) for 24 h. Flow cytometry (Agilent) and CSLM (Zeiss) were utilized to quantitatively and qualitatively analyze the cellular uptake.
2.5. Immunofluorescence assay of ICD
4T1 cancer cells (1 × 105) seeded in confocal petri dish were incubated with PM, PM@RM and PM@RM-T7 (at the equivalent mPDA = 200 μg/mL, Met = 60 μg/mL) for 6 h. Afterward, the cells were laser irradiated (808 nm, 1.0 W/cm2, 5 min) or not. The medium was changed to a fresh medium. After another incubation for 24 h, 4T1 cells were washed with PBS (pH 7.4) and incubated with 5% bovine serum albumin for 30 min to block unspecific binding of antibodies. Then, the cells were respectively incubated with anti-calreticulin (CRT) and anti-heat shock protein (HSP70) antibodies (Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C, followed by incubation with the corresponding secondary antibodies for 2 h at 37 °C in the dark. Finally, the cells were visualized under a CLSM (Zeiss) system. The procedures of immunofluorescence (IF) staining of PD-L1 (Beyotime) were the same as the above.
2.6. Western blot analysis of PD-L1
4T1 cancer cells were treated the same as in Section 2.5. The total protein of 4T1 cells was extracted to determine the protein concentration by BCA, and samples with equal protein amounts were separated by 12.5% SDS-PAGE gels. After being transferred to a 0.22 μm polyvinylidene fluoride membrane, the membrane was sealed with protein free rapid blocking buffer (Beyotime) at room temperature for 20 min. Subsequently, the membranes were respectively incubated with a rabbit anti-mouse PD-L1 antibody (Beyotime) and anti-GAPDH Rabbit pAb (Servicebio, Wuhan, China) at 4 °C overnight. Afterward, the membrane was incubated with horseradish peroxidase-labeled Goat anti-Mouse/Rabbit IgG (H + L) (Epizyme, Shanghai, China) for 2 h at room temperature. The protein expression was detected using a fully automatic chemiluminescence image analysis system (Tanon, Beijing, China) and quantified by Image J (NIH, MD, USA).
2.7. In vitro repolarization of M2 type RAW264.7 cells
RAW264.7 cells (2 × 105 cells/well) were cultured in six-well plates overnight before polarization. M1 type RAW264.7 cells were obtained by LPS stimulation (100 ng/mL) for 24 h as control, whereas M2 type RAW264.7 cells were obtained by IL-4 stimulation (20 ng/mL) for 24 h. For the in vitro repolarization experiments, M2 type RAW264.7 cells were treated with R848, PR@RM, and PR@RM-M2 (at the equivalent concentration of mPDA = 100 μg/mL, R848 = 3 μg/mL) for 24 h. Then, the cells were stained with FITC-anti-F4/80, PE-anti-CD206, and APC-anti-CD86 (BD, Franklin Lakes, NJ, USA) antibodies and were analyzed by flow cytometer (Agilent) to detect the expression of surface markers. Meanwhile, IF staining of CD86 (Abclonal) and CD206 (Proteintech, Wuhan, China) were performed, and phalloidin was used to label tubulin to observe the morphological changes of RAW264.7 cells by CSLM (Zeiss). Secreted cytokines (TNF-α, IL-6, IFN-γ, and IL-10) were detected by ELISA (Abclonal).
2.8. Macrophage phagocytosis of tumor cells
M2 type RAW264.7 cells (2 × 105 cells/well) seeded in the confocal petri dish were separately incubated with R848, PR@RM, and PR@RM-M2 for 24 h. Then, they were collected and labeled with DiO dye (Beyotime). In the meantime, 4T1 cancer cells were labeled with Hoechst and co-culture with the above DiO labeled RAW264.7 cells at the number ratio of 1:1. After 24 h, the phagocytosis of 4T1 cells by RAW264.7 cells was observed by CLSM (Zeiss).
2.9. Tumor model of mice
Female BALB/c mice aged 5 weeks were purchased from Sichuan Province People's Hospital (Chengdu, China). All the animal research was approved by the Institutional Animal Care and Ethics Committee of the University of Electronic Science and Technology. To construct the unilateral 4T1 tumor-bearing model. 1 × 106 4T1 cells were suspended in 100 μL of saline and then injected into the right flank of each mouse. A bilateral 4T1 tumor-bearing mouse model was established by subcutaneously injecting 1 × 105 4T1 cells (in 100 μL saline) on the left flank of the BALB/c mice 5 days after the primary tumor inoculated subcutaneously on the right side (1 × 106 4T1 cells in 100 μL saline).
2.10. In vivo evaluation of biodistribution
The unilateral 4T1 tumor-bearing model was constructed for the evaluation of the targeted tumor accumulation and biodistribution. Near-infrared fluorescent dye Cy5.5-loaded PCy@RM, PCy@RM-T7, and PCy@RM-M2 were intravenously injected into the mice at the dose of 500 μg/kg Cy5.5. After 2, 4, 8, 24, and 48 h, the mice were imaged by Lumina Series III imaging (PerkinElmer, Waltham, MA, USA). Then, the mice were sacrificed, and their major organs and tumors were isolated for ex vivo fluorescence imaging. Moreover, to observe the spatiotemporal targeting ability of PCy@RM-T7 and PCy@RM-M2, the tumor sections were stained with anti-CD206-FITC (Abclonal) and DAPI (Beyotime) and then were observed by fluorescent microscopy.
2.11. In vivo synergistic photo-immune antitumor effects
A bilateral 4T1 tumor-bearing mouse model was established, and these tumor-bearing mice were randomly divided into six groups, with ten mice in each group. G1: saline + L (the control), G2: P@RM-T7 + L, G3: PM@RM-T7, G4: PM@RM-T7 + L, and G5:PR@RM-M2 were administrated via intravenous injection and carried out every three days for three times (L was denoted as laser). G6: The “Gemini nanoimmunoregulators” treated group was i.v. injected with PM@RM-T7 on the first day, and then continued treatment of PR@RM-M2 was performed on the second day. The equivalent doses of mPDA, Met, and R848 were 10, 3, and 3 mg/kg, respectively. Laser irradiation (808 nm, 1.0 W/cm2, 10 min) was performed at the tumor site at 8 h after the first administration. The volumes of both primary and distal tumors and mice's body weights were monitored every three days during the whole period.
On Day 14, the remaining mice were sacrificed, and the main organs, primary and distal tumors were collected. Blood was collected for liver and kidney function testing. And primary and distal tumors were dissected and analyzed by hematoxylin and eosin (HE), TdT-mediated dUTP nick end labeling (TUNEL), and IF staining of Ki67, respectively.
2.12. Immune cascade and TAMs repolarization assay
2.12.1. In vivo ICD and PD-L1 degradation assay
On Day 10, after the beginning of treatment, the peripheral blood was collected for detection of TNF-α, IL-6, IFN-γ, and IL-10 levels in plasma using ELISA kits (Abclonal). The primary tumors were isolated from the treated mice and cut into sections for further examination. These sections were separately processed with primary antibodies against CRT, HSP 70, and PD-L1 (Beyotime) according to the manufacturer's protocols and then stained with secondary antibodies, followed by microscopic observation.
2.12.2. DCs maturation assay
To further detect DCs maturation, the tumor-draining lymph nodes and inguinal lymph nodes were collected and ground in a 2 cm-diameter dish by the rubber end of a syringe. The obtained cells were stained with flow antibodies CD11c, CD80, and CD86 (BD, NJ, USA) according to the procedure of the manufacturer. The frequency of DCs maturation in the lymph nodes was then investigated by flow cytometry (Agilent). All data analysis was performed using Flow Jo software (Biochemicals FlowJo, OR, USA).
2.12.3. Lymphocytes analysis
On Day 14, the remaining mice were sacrificed, and the spleen and primary tumors of the mice were processed into single-cell suspensions. Briefly, the spleen was placed in 1× PBS and gently minced with a syringe plunger. After that, the mixture was filtered through a 70 μm cell filter. The erythrocytes in the single-cell suspension were removed with 1× red blood cell lysis buffer. The single-cell suspension was obtained by centrifugation at 1300 rpm for 5 min (Eppendorf). The tumors were removed, cut into 3–4 mm pieces with scissors, and transferred to 15 mL centrifuge tubes supplemented with 1640 medium with collagenase type IV, hyaluronidase, and DNAase concentrations of 1.0 mg/mL, respectively, and placed in a 37 °C incubator for 1 h of digestion processing. The digested tissues were transferred to a sterile nylon mesh in a flat dish containing RPMI 1640 medium and well-ground with the tip of the inner plug of a 5 mL sterile syringe, and the nylon mesh was rinsed by continuously aspirating medium from the flat dish. The obtained cell liquid was filtered through 70 μm sterile nylon mesh collected in a centrifuge tube at 1300 rpm (Eppendorf) and centrifuged at 4 °C for 5 min. The supernatant was discarded, and the cells on the wall of the tube were retained and washed off three times with PBS. Then, the cells were further stained with the following fluorochrome-conjugated antibodies (BD) according to the manufacturer's protocol and the ratio of cytotoxic T cells (anti-CD3e, anti-CD8a, and anti-CD4) and Tregs (anti-Foxp3) was analyzed by flow cell cytometry (Agilent). Further, IF staining was also performed to analyze T cells (CD4+, CD8+, and Tregs) infiltration into the primary and distal tumors.
2.12.4. TAMs repolarization assay
The tumor sections were first stained with TAMs associated biomarkers, including anti-CD86 and anti-CD206, before observation by fluorescence microscope. For quantitative analysis, the tumor suspensions were prepared by homogenization and stained with anti-F4/80, anti-CD86, and anti-CD206 (BD), followed by flow cytometry (Agilent) analysis.
2.13. Inhibition of lung metastasis
A lung metastasis model was constructed, in which the primary tumors were constructed by subcutaneously injecting 4T1 (1 × 106) cells on the right side, and 5 days later, 4T1 cells (1 × 105) were injected into mice through the tail vein. It had been predicted that these tumor cells would enter the lung tissue of mice through blood circulation to form lung metastases. One week after inoculation, the mice were randomly divided into four groups (n = 3): (G1ʹ) Saline + L, (G2ʹ) PM@RM-T7 + L, (G3ʹ) PR@RM-M2, (G4ʹ) PM@RM-T7 + L and PR@RM-M2 (L was denoted as laser). Treatment was performed every three days and repeated three times. After 14 days, lung organs were extracted and counted for gross analysis of tumor nodules. The lung tissues were then examined with HE, immunohistochemistry of Ki67, and IF staining of CD4 and CD8.
2.14. Statistical analysis
The data in all experiments were expressed as mean ± standard deviation (SD). Statistical significance was analyzed by using analysis of variance (ANOVA) in the GraphPad prism (San Diego, CA, USA) for multiple group comparisons. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 were considered statistically significant.
3. Results and discussion
3.1. Construction of biomimetic “Gemini nanoimmunoregulators”
The biomimetic “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 were fabricated by respectively encapsulating Met and R848 into the inner cavity of mPDA and then camouflaging with RM inserted by T7 and M2 peptides for the specific recognition of tumor cells and TAMs (Scheme 1A). The spherical core of mPDA was about 100 nm with the typical mesoporous structure as shown in the TEM image (Fig. 1A), and its pore size was around 5 nm analyzed by BET (Supporting Information Fig. S1A). Met and R848 could easily be loaded into mPDA via the strong adhesive interaction, hydrogen interaction, and π−π stacking27. The representative absorption peak of the two drugs was observed in the UV–Vis absorption spectra of PM and PR (Fig. S1B). The maximum loading rate of Met and R848 on mPDA were 300 and 500 μg/mg, respectively, together with the high encapsulation efficiency (49.8% for Met and 85.9% for R848) (Fig. S1C). The drug loading did not change the morphology of mPDA (Fig. S1D and E). RM coating of the “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 were clearly observed from TEM images (Fig. 1B and C), which was prepared at the optimal ratio of 1/1 (m/m, mPDA/RM, Fig. S1F). The high overlapping of fluorescence signals of FITC (doped in mPDA) and DiI (embedded in RM) further verified the successful membrane modification around mPDA (Fig. 1D). Moreover, SDS-PAGE analysis of the “Gemini nanoimmunoregulators” showed good retention of the characteristic membrane protein (Fig. 1E). The successful anchoring of T7 and M2 peptides on the nanosystem was spectroscopically demonstrated and the conjugation rate could reach 10% and 5.6%, respectively (Fig. S1G and H). The biomimetic modification made the hydrodynamic size of PM@RM-T7 and PR@RM-M2 visibly increase by about 20 nm, together with slightly reduced zeta potentials in comparison with the naked counterparts (Fig. 1F and Fig. S1I). Also, the cargoes of Met and R848 still remained in mPDA, as proved by the UV–Vis absorption spectra (Fig. 1G). The “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 showed improved stability, and their hydrodynamic diameter did not fluctuate much in saline (Fig. 1H) or serum (Fig. S1J) during ten days. The hemolysis assay revealed that PM@RM-T7 and PR@RM-M2 showed excellent hemocompatibility with a shallow hemolysis rate of <3% (Fig. S1K and L).
Figure 1.

Characterization of biomimetic “Gemini nanoimmunoregulators”. TEM images of (A) mPDA, (B) PM@RM-T7, and (C) PR@RM-M2 (scale bar = 100 nm). (D) Fluorescence microscopic images of PF@RM-T7, FITC was doped into mPDA, and DiI was used to label RM (scale bar = 100 μm). (E) SDS-PAGE analysis of mPDA, RM, PM@RM-T7, and PR@RM-M2. (F) Z-average size and surface zeta potential of mPDA, PM, PR, PM@RM-T7, and PR@RM-M2. (G) UV–Vis absorption spectra of different formulations. (H) Hydrodynamic size of PM@RM-T7 and PR@RM-M2 in saline; (I) IR thermal images of mPDA and PM@RM-T7 (at the equivalent concentration of mPDA = 200 μg/mL) under laser irradiation (808 nm, 1.0 W/cm2, 10 min). (J) Photothermal curves of PM@RM-T7 in four cycles of laser on/off. (K) Cumulative release of Met at pH 7.4 and (L) pH 5.0 from PM and PM@RM-T7 with or without laser irradiation.
Furthermore, the photothermal performance of our “Gemini nanoimmunoregulators” was measured. PM, PM@RM, and PM@RM-T7 possessed nearly the same photothermal heating capacity as that of mPDA, and the temperature could rise to about 47 °C at the same concentration under laser irradiation (808 nm, 1.0 W/cm2, 10 min, Fig. 1I, and Supporting Information Fig. S2A). Also, the photothermal effect was concentration- and laser-power density-dependent (Fig. S2B and C). To note, PM@RM-T7 exhibited little attenuation on the temperature during four successive cycles of laser on/off (Fig. 1J), suggesting the stable photothermal conversion. Moreover, PM@RM-T7 exhibited outstanding photothermal conversion efficiency of around 31.7%, which was near to that of mPDA (about 34.3%, Fig. S2D and E). Therefore, PM@RM-T7 could be used as a promising PTT agent for cancer ablation.
The photothermal effect is an exogenous stimulus to control drug release, thus achieving an enhanced therapeutic efficacy. Subsequently, the releasing behavior of Met from PM@RM-T7 was explored at different pH with and without laser irradiation. The profiles showed that the release rate of Met from PM@RM-T7 at both pH 7.4 (Fig. 1K) and pH 5.0 (Fig. 1L) was slower than that from PM in the absence of irradiation, which was due to the shielding of outer RM. It was easy to see that much more Met was released from PM at pH 5.0 than at pH 7.4, e.g., a total of 55.2% (at pH 5.0) vs. 17.9% (at pH 7.4) within 48 h. This phenome could be attributed to Met, whose structure bears amino groups and is easily protonated under acidic conditions30. The acidity slightly affected Met release from PM@RM-T7, only 31.0% (at pH 5.0) vs. 13.0% (at pH 7.4) within 48 h, suggesting the relative stability in the physics environment. Upon irradiation (808 nm, 1.0 W/cm2, 10 min), the release rate of Met from PM@RM-T7 was dramatically enhanced, which was primarily due to the photothermal effect that accelerated the molecular diffusion of Met. To note, about 47.7% of Met burst from PM@RM-T7 after irradiation at pH 5.0 within 12 h, and then little increased in the later 36 h (Fig. 1L). PM@RM-T7 had a distinct on-demand drug release behavior triggered by the photothermal effect as well as the acidity. PR@RM-M2 showed similar drug release ability (Fig. S2F). The excellent photothermal effect and controlled drug release of our “Gemini nanoimmunoregulators” will be highly beneficial for the long circulation and intracellular rapid release for cancer photoimmunotherapy.
3.2. Targeted photothermal-immunol effect and downregulation of PD-L1 mediated by PM@RM-T7
The photoimmunol stimulation of PM@RM-T7 was evaluated against 4T1 murine mammary cells. Initially, the T7 peptide-mediated targeted cellular internalization of PM@RM-T7 was studied by using Ce6 encapsulated mPDA surrogate PC@RM-T7. Compared to the weak red fluorescence intensity in the groups treated by PC@RM and PC@RM-M2, the fluorescence intensity was significantly vital in the cytoplasm of 4T1 cells treated with PC@RM-T7 (Fig. 2A). This demonstrated that T7 peptide indeed aided more nanoimmunoregulators to be endocytosed by the cancer cells. Flow cytometry analysis also showed the targeted cellular uptake of PC@RM-T7 (Supporting Information Fig. S3A). Next, free Met and PM@RM-T7 (0–400 μg/mL) in the dark were found to have negligible cytotoxicity (Fig. 2B and Fig. S3B). However, upon irradiation (808 nm, 1.0 W/cm2, 10 min), PM@RM-T7 exhibited much higher photothermal killing efficiency than PM@RM and PM (Fig. 2C), confirming the targeted PTT effect. Consistently, the flow cytometry analysis using Annexin-V FITC/PI probe revealed that about 45.8% of 4T1 cells cultured with PM@RM-T7 + L went apoptosis, which was 11.9% higher than that of the cells treated by PM@RM + L (Fig. 2D and Fig. S3C). Clearly, PM@RM-T7 could selectively recognize and photothermally kill the cancer cells under the guidance of T7 peptide.
Figure 2.

Photoimmune activation and downregulation of PD-L1 by PM@RM-T7 in vitro. (A) Representative CLSM images of cellular uptake of PC@RM-T7 and relevant controls by 4T1 cells after 6 h incubation. Relative cell viability of 4T1 cells treated with PM@RM-T7 and the relevant controls (scale bar = 100 μm) (B) under dark and (C) laser irradiation. (D) Representative flow cytometry plots of 4T1 cells after different treatments. (E) CRT exposure and (F) CLSM images of 4T1 cells with IF staining of CRT (scale bar = 50 μm). Expression of PD-L1 in the 4T1 cells detected by (G) western blotting and (H) IF staining after treatment with PM@RM-T7 and relevant controls with or without laser irradiation (scale bar = 50 μm). Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗∗P < 0.0001 vs. indicated.
It has been reported that ICD usually occurs after photothermal ablation, which could trigger systemic anticancer immunity9. One characteristic marker of ICD is CRT, which acts as an “eat-me” signal to arouse the immune system identification and phagocytosis of cancer cells7. In our study, PM@RM-T7 maximally upregulated the exposure levels of CRT in 4T1 cells under laser irradiation compared with PM@RM and PM (Fig. 2E and F). Another marker of intracellular HSP70 also had the highest expression after PM@RM-T7 treatment (Fig. S3D and E), suggesting the amplified ICD effect induced by targeted PTT. Moreover, to identify whether PM@RM-T7 could effectively inhibit PD-L1 expression, western blot assay and IF staining were performed. The level of PD-L1 in 4T1 cells treated with laser or P@RM-T7 was almost identical to that of the control group. However, P@RM-T7 + L treatment caused the up-expression of PD-L1, which was probably due to the photothermal effect31. As expected, the amount of PD-L1 was significantly inhibited under the action of PM@RM-T7 and PM@RM-T7 + L, even lower than that of the control (Fig. 2G and Fig. S3F). The rationale behind this was that the photothermal heating accelerates Met released from PM@RM-T7 and thereby enhances the endogenous degradation of PD-L1. IF images also showed similar results (Fig. 2H and Fig. S3G). These data meant that PM@RM-T7 was potent in reducing the binding of PD-L1/PD-1 and then preventing the immune escape of tumors.
3.3. Selective recognition and phenotype reset of M2 type RAW264.7 cells by PR@RM-M2
To investigate the targeted repolarization ability of PR@RM-M2 on M2 type macrophages, the murine macrophage cell line RAW264.7 was polarized to M2 mode using IL-4. M1 type RAW264.7 cells, stimulated by LPS for 24 h, were used as the positive control (Supporting Information Fig. S4A). PR@RM-M2 showed negligible toxicity to M2 type RAW264.7 cells by the CCK-8 assay (Fig. S4B). The internalization of PR@RM-M2 by M2 type RAW264.7 cells was also explored by replacing R848 with a Ce6 fluorescence probe and observed by CLSM. As shown in Fig. 3A, the obvious red fluorescence intensity in CLSM images showed that PC@RM-M2 was significantly accumulated in M2 type RAW264.7 cells thanks to the active targeting of M2 peptide. However, PC@RM and PC@RM-T7 treated groups showed very dim red fluorescence signals in the cytoplasm under the same conditions (Fig. 3A). The flow cytometry analysis also proved that PC@RM-M2 was efficiently phagocytosed into M2 type RAW264.7 cells (Fig. S4C).
Figure 3.

Selective recognition and reprogramming ability of PR@RM-M2 on M2 type RAW264.7 cells. (A) Representative CLSM images of M2 type RAW264.7 cells incubated with PC@RM, PC@RM-M2, or PC@RM-T7 for 6 h (scale bar = 100 μm). (B) Representative flow cytometry plots of M1 type RAW264.7 cells (CD86high) and M2 type RAW264.7 cells (CD206high) gating on F4/80+ after different treatments and (C) the corresponding statistical analysis. (D) TNF-α, IL-6, IFN-γ, and IL-10 secretion by M2 type RAW264.7 cells after different treatments. (E) CLSM images of IF staining of CD86 (green) and CD206 (red) on the surfaces of M2 type RAW264.7 cells after various treatments (scale bar = 50 μm; zoom in: scale bar = 20 μm). (F) Representative CLSM images of the phagocytosis of 4T1 cells (labeled by Hoechst) by M2 type RAW264.7 cells (labeled by DiO) after treatment of R848, PR@RM, and PR@RM-M2 (scale bar = 100 μm; zoom in: scale bar = 20 μm). Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗∗P < 0.0001 vs. indicated. n.s., not significant.
Furthermore, the reprogramming ability of PR@RM-M2 towards M2 type RAW264.7 cells was investigated. As expected, compared with the nontargeted PR@RM, PR@RM-M2 significantly increased the expression of M1-related costimulatory molecules CD86 (51.6 ± 3.2% for PR@RM-M2 vs. 42.6 ± 3.9% for PR@RM) (Fig. 3B and C) and decreased the expression of CD206, typical markers for M2 type RAW264.7 cells (19.0 ± 11.4% for PR@RM-M2 vs. 25.4 ± 13.2% for PR@RM). Although this in vitro repolarization effect was weaker than that of R848, PR@RM-M2 still acted as an effective repolarizing agent to convert M2 type RAW264.7 cells to the M1 state. Cytokines secreted by macrophages also reflect the phenotype. As shown in Fig. 3D, R848, PR@RM, and PR@RM-M2 upregulated the secretion of pro-inflammatory cytokines (i.e., TNF-α, IL-6, and IFN-γ) and decreased the secretion of anti-inflammatory cytokines (i.e. IL-10) after incubated with M2 type RAW264.7 cells. In particular, PR@RM-M2 respectively resulted in 13.4-, 28.2- and 2.9-fold of TNF-α, IL-6, and IFN-γ (vs. control), which was significantly higher than that of the nontargeted PR@RM group (i.e. 9.7-, 22.5- and 2.1-fold of control). Furthermore, from the IF images of costimulatory molecules on the surface of M2 type RAW264.7 cells in Fig. 3E, the obvious green signals from anti-CD86-FITC but low red fluorescence signals from anti-CD206-Cy3 for PR@RM-M2 treated group proved the excellent repolarization capacity of PR@RM-M2 again. What's more, the changes in cell morphology have recently been identified as a reliable indicator for macrophage polarization, i.e., elongated projections for M2 type macrophages vs. the round and flattened morphology for M1 type counterparts32. In our study, the M2 type RAW264.7 cells stimulated by IL-4 as the control group showed an elongated morphology. However, after treatment with PR@RM-M2, the cell shape-shifted to a typical pancake-like form, indicating the phenotype being reprogrammed to M1 mode.
To further investigate whether PR@RM-M2 could increase the phagocytic capacity of macrophages, the Hoechst-labeled 4T1 tumor cells were co-incubated with DiO-labeled M2 type RAW264.7 cells after different treatments for 24 h. According to the colocalization of blue and green fluoresce signals in Fig. 3F, most 4T1 cancer cells were phagocytosed by M2 type RAW264.7 cells pretreated with PR@RM-M2, which was close to the observation in the R848 treated group. As a control, M2 type RAW264.7 cells pretreated with PBS were barely able to phagocytose 4T1 tumor cells. These phenomena were closely correlated with the phenotype reset of TAMs and enhanced phagocytic capacity due to the changes in central carbon metabolism33. All in all, these results demonstrated that PR@RM-M2 was a potent targeted immunoregulator to transform the phenotype of TAMs and remodel ITM.
3.4. In vivo spatiotemporal targeting behavior and biodistribution
Furthermore, the in vivo spatiotemporal targeting behavior and biodistribution of “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 were investigated by a whole animal fluorescence imaging system using Cy5.5 as a fluorescence probe. After intravenous injection, the biodistribution of PCy@RM, PCy@RM-T7, and PCy@RM-M2 in 4T1-bearing mice was examined at a series of predetermined time points. As shown in Fig. 4A, the fluorescent signal in the tumor area of each group gradually increased over 8 h but began to decrease after 24 h, indicating that 8 h was the appropriate time point for irradiation. During the whole process, the fluorescence signal of PCy@RM-T7 was significantly more potent than that of the PCy@RM treated group, which was owing to the guiding of T7 peptide. To note, the fluorescence signal intensities at the tumor site of the PCy@RM-M2 treated group were also relatively high but not the same as that of PCy@RM-T7. It was because PCy@RM-M2 targeted the population of M2 type TAMs in solid tumors, whose fraction was less than that of tumor cells19. Then, the dissected tumor and major organs were monitored by biofluorescence imaging at 48 h after injection (Fig. 4B and C). In comparison, the tumor fluorescence intensity in order of PCy@RM-T7 > PCy@RM-M2 > PCy@RM further confirmed the targeted delivery and accumulation in tumors for our “Gemini nanoimmunoregulators”.
Figure 4.

Spatiotemporal targeting behavior and biodistribution of “Gemini nanoimmunoregulators” in vivo. Biofluorescence images of (A) 4T1 tumor-bearing mice, (B) dissected tumors and organs, and (C) semiquantitative analysis of fluorescence signals after intravenous injection of PCy@RM, PCy@RM-M2 and PCy@RM-T7. (D) IF staining results of tumor sections (scale bar = 50 μm) and (E) quantitative analysis of the coincidence of fluorescence signals (Cy5.5 and anti-CD206-FITC) by Image J for groups treated by PCy@RM, PCy@RM-M2 and PCy@RM-T7. Data are presented as mean ± SD (n = 3). ∗P < 0.05 and ∗∗P < 0.01 vs. indicated.
To further analyze the site-specific delivery of “Gemini nanoimmunoregulators” in vivo, we performed IF analysis of tumor sections. DAPI was used to stain the nucleus of all cells, and anti-CD206-FITC was applied to label M2 type TAMs. As shown in Fig. 4D, the red fluorescent signal from PCy@RM-T7 was mainly accumulated in the DAPI-labeled tumor cells where the green signal of anti-CD206-FITC was weak, demonstrating that it could effectively enter tumor cells mediated by T7 peptide. However, the red fluorescent signal from PCy@RM-M2 overlapped with the green fluorescence of anti-CD206-FITC obviously, which indicated that PCy@RM-M2 was effectively phagocytosed by TAMs owing to the guiding of M2 peptide. In contrast, the PCy@RM treated group showed the weakest red fluorescence signal in the tumor, which was due to the absence of targeting peptides (Fig. 4E). The above results confirmed that the biomimetically engineered “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 could realize the site-specific delivery of their different cargoes to tumor cells and TAMs in vivo.
3.5. In vivo enhanced photo-immunol therapeutic effect
Inspired by the aforementioned results, the in vivo amplified photo-immunol therapeutic efficiency was evaluated in a bilateral 4T1 tumor-bearing mouse model (Fig. 5A). P@RM-T7 and PM@RM-T7 were also excellent photothermal agents, and the temperature at the tumor site raised to around 50 °C in 6 min upon laser irradiation (808 nm, 1.0 W/cm2, 10 min) (Supporting Information Fig. S5A and B). As displayed in Fig. 5B, the PTT group mediated by P@RM-T7 + L suppressed the tumor growth that was attributed to the photothermal ablation effect. However, the single PD-L1 interference group mediated by PM@RM-T7 or the single TAMs polarization group mediated by PR@RM-M2 moderately retarded the primary tumor growth in comparison with the rapid tumor growth curve of the control group. Though PTT combined with Met treatment (PM@RM-T7 + L group) significantly inhibited the growth of tumor, the treatment of “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 showed the most prominent tumor suppression effect during the whole process and even the tumors of some mice disappeared at a later stage, which may suggest the systemic anticancer immunity being activated. The weight and tumor photos of each group showed the same trend (Fig. 5C and D). Comparing the final tumor inhibition rate calculated by the tumor weight, “Gemini nanoimmunoregulators” PM@RM-T7 + L and PR@RM-M2 combined treatment achieved the highest final tumor inhibition rate up to about 89.4% vs. 42.5% for P@RM-T7 + L, 13.9% for PM@RM-T7, 67.6% for PM@RM-T7 + L and 28.9% for PR@RM-M2 (Fig. S5C). The results demonstrated that reprogramming TAMs in ITM by PR@RM-M2 further amplified the photothermal immunotherapeutic efficiency induced by PM@RM-T7, suggesting the superiority of “Gemini nanoimmunoregulators”. The tumors were then stained with HE to examine pathological changes (Fig. 5E). Compared to PM@RM-T7 + L and PR@RM-M2, “Gemini nanoimmunoregulators” caused the most damage to tumor cells being fragmented and lysed. Meanwhile, the TUNEL assay showed that most of tumor cells in the “Gemini nanoimmunoregulators” treatment group went apoptosis, which was significantly higher than that of the PM@RM-T7 + L and PR@RM-M2 single treatment group (Fig. 5E). Therefore, the “Gemini nanoimmunoregulators” cooperatively exerted the highest antitumor efficiency against the malignant primary tumor.
Figure 5.

Assessment of synergistic photoimmunotherapeutic effect against 4T1 primary tumors. (A) Therapeutic schedule for 4T1 bilateral tumor-bearing mice. (B) The volumes, (C) weight, and (D) photographs of primary tumors after various treatments. Microscopic images of primary tumor sections stained with (E) HE (scale bar = 200 μm), TUNEL (scale bar = 50 μm), and immunofluorescence staining of (F) CRT, HSP 70, and (G) PD-L1 (scale bar = 100 μm). Data are presented as mean ± SD (n = 3). ∗∗P < 0.01 and ∗∗∗∗P < 0.0001 vs. indicated. n.s., not significant.
To explore the mechanism behind the “Gemini nanoimmunoregulators”-mediated anticancer synergy, the ICD phenomenon was first analyzed by IF examination of the markers CRT and HSP 70 (Fig. 5F). Both CRT and HSP 70 were significantly upregulated in tumors treated with the irradiated groups including P@RM-T7 + L, PM@RM-T7 + L and “Gemini nanoimmunoregulators” group (Fig. S5D and E), consistent with the in vitro results. This PTT-induced ICD effect was expected to stimulate the adaptive immune system. However, the single PTT treatment mediated by P@RM-T7 + L dramatically boosted PD-L1 expression, which was most probably due to the IFN-γ secretion34. Fortunately, involvement of Met (for PM@RM-T7 + L and “Gemini nanoimmunoregulators” groups) remarkedly decreased the intratumoral PD-L1 level (Fig. 5G and Fig. S5F) resulting from the interference on PD-L1 biosynthesis. The PD-L1 expression of the PR@RM-M2 treated group was slightly decreased, which might be attributed to the inhibition and phagocytosis of tumor cells by TAMs after R848 repolarization. Prominently, our “Gemini nanoimmunoregulators” could effectively trigger ICD and blockade of PD-L1/PD-1 inhibitory axile in vivo.
3.6. Antitumor immunol cascade and TAMs polarization in vivo
Furthermore, the in vivo immune cascade triggered by our “Gemini nanoimmunoregulators” was evaluated. As the professional antigen-presenting cell, DCs in the tumor-draining lymph nodes and inguinal lymph nodes after treatment were initially detected to check the promotion of the anticancer immune cycle (Fig. 6A). The maturation rate of DCs in pure PTT group (mediated by P@RM + L), PTT combined with Met group (mediated by PM@RM-T7 + L) and the “Gemini nanoimmunoregulators” group gradually increased, i.e. 21.5 ± 3.4%, 32.2 ± 2.8% and 48.6 ± 3.9% respectively. These were approximately 2.7-, 4.1- and 6.2-fold of the control group (7.9 ± 2.5%) (Fig. 6B). Notably, our “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 tremendously activated the anticancer response. PM@RM-T7 treatment alone slightly caused the maturation of DCs (16.7 ± 1.4%), which would be owing to the multiple functions of Met to reduce the vascular compression and enhance DCs infiltration35. For the PR@RM-M2 treated group, the maturation rate was relatively high (42.3 ± 1.9%). On one hand, TAMs repolarized by R848 could secrete large amounts of pro-inflammatory cytokines, which in turn further promoted the maturation of DCs36. On the other hand, the released R848 could also facilitate DCs maturation as an excellent adjuvant37. In short, the above results suggested the remarkable induction of DCs maturation by “Gemini nanoimmunoregulators” through the combination of ICD effects and ITM modulation.
Figure 6.

TAMs reprogramming and immune activation by “Gemini nanoimmunoregulators”. (A) Representative flow cytometry plots and (B) corresponding percentages of matured DCs (gated by CD80+ CD86+ CD11c+) in lymph nodes. (C) Populations of the M1 type (gated by F4/80+ CD86+) and M2 type TAMs (gated by F4/80+ CD206+) in tumors. (D) The ratio of M1/M2 macrophages in tumors. (E) IF staining of CD86 and CD206 markers in the primary tumor section (scale bar = 100 μm). (F) Representative flow cytometry plots of the intratumor infiltration of CD4+ and CD8+ T cells (gated on CD3+ T cells) in the primary tumor after treatments. (G) Levels of TNF-α, IL-6, IFN-γ, and IL-10 in the serum of mice from each group were measured by ELISA. (H) Representative flow cytometry plots of the intratumoral infiltration of Tregs cells (CD4+ Foxp3+) in the primary tumor after treatments. Data are presented as mean ± SD (n = 3). ∗∗P < 0.01, ∗∗∗P < 001, and ∗∗∗∗P < 0.0001 vs. indicated. n.s., not significant.
Moreover, the effect of our “Gemini nanoimmunoregulators” on the phenotype of TAMs was examined. The representative flow cytometry plots (Fig. 6C) showed that PR@RM-M2 treatment caused the phenotype reversion from M2 to M1 type obviously as the percentage of CD86 positive TAMs increased from 21.8 ± 0.6% to 58.4 ± 8.5% while the percentage of CD206 positive TAMs decreased from 46.3 ± 1.0% to 21.3 ± 2.7%. This was because of the repolarizing rule of R848 via the TLR7/8 pathway23. Under the action of pure PTT (mediated by P@MT + L) and PTT combined with the Met group (mediated by PM@RM-T7 + L), the phenotype of TAMs was also mildly and similarly reversed from the M2 to M1 state. This was closely correlated with the photoablation effect, and it has been reported that IFN-γ induced by photothermal ablation could partially repolarize TAMs to M1 type38,39. Besides, the released Met could also repolarize TAMs (30.2 ± 3.6%), and this was evidenced by the reported data40. Excitedly, the “Gemini nanoimmunoregulators” treated group exhibited the best polarization effect on TAMs among all groups with the highest proportion of CD86 positive M1 type macrophages (62.8 ± 6.5%, 2.9-fold of the control group) and the lowest CD206 proportion of 13.0 ± 4.5% vs. 46.3 ± 0.9% for control. To note, compared to PR@RM-M2 (2.7) and PM@MT-T7 + L (1.6), the M1/M2 ratio of “Gemini nanoimmunoregulators” was dramatically increased to 4.9 (Fig. 6D). The IF images of tumor tissue were also consistent with the above results (Fig. 6E). Therefore, strong anticancer immunity would be expected via “Gemini nanoimmunoregulators” to reprogram TAMs in ITM maximally.
The activation of lymphocytes, including the helper T cells (CD3+ CD4+) and CTLs (CD3+ CD8+), is the critical event of adaptive immunity. The helper T cells could regulate the immunoactivity, while CTLs directly attack tumor cells via the secretion of cytotoxins such as perforin and granzymes41. The flow cytometry data (Fig. 6F and Supporting Information Fig. S6A) showed that the proportion of CD8+ T cells in the primary tumors of the two groups of P@RM-T7 + L (17.7 ± 2.5%) and PM@RM-T7 + L (18.3 ± 7.3%) were much higher than that of control (7.5 ± 0.6%), which was probably due to the photothermal stimulation of tumor changes from immune "cold" to immune "hot." For the PR@RM-M2 group, the amount of CD8+ T cells increased to 26.9 ± 5.5%, which could be due to the fact that the reprogrammed TAMs could engulf antigens and proteins from dying cancer cells, which then transported to draining lymph nodes to activate CD8+ T cells and CD4+ T cells20,42. Also, the inflammatory cytokines (e.g., IL-6) secreted by M1 type macrophages could enhance CTLs recruitment43. Excitingly, after “Gemini nanoimmunoregulators” treatment, the rate of CD8+ T cells increased to 36.5 ± 8.4% (4.9-fold of the control group), which was due to the synergistic effect of PM@RM-T7 and PR@RM-M2. Similarly, the percentage of CD4+ T cells in the primary tumors increased significantly after “Gemini nanoimmunoregulators” treatment. Thus, our “Gemini nanoimmunoregulators” maximized CTLs-based adaptive anticancer immunity via the integration of ICD, TAMs repolarization, and PD-L1 degradation. Besides, inflammatory cytokines are essential indicators of systemic immune activation44. As tested, the “Gemini nanoimmunoregulators” treatment aroused higher secretion of cytokines (i.e., TNF-α, IL-6, and IFN-γ) compared with other groups (Fig. 6G) accompanied by the low level of anti-inflammatory factor (i.e., IL-10), further indicating that the immune cascade was activated by “Gemini nanoimmunoregulators” in the tumor-bearing mice.
To note, Tregs (CD4+ Foxp3+) as the immunosuppressive subset of CD4+ T cells were only 2.7 ± 0.8% for the “Gemini nanoimmunoregulators” treated group, which was the lowest among all groups (Fig. 6H and Fig. S6B). The lymphocytes analyzed from spleen tissue were also consistent with the above trend (Fig. S6C and D), verifying that ITM was significantly modulated for tumor inhibition. Together, "Gemini nanoimmunoregulators" PM@RM-T7 and PR@RM-M2 ultimately evoked systemic anticancer immunity through the cooperation of amplified ICD, PD-L1 degradation, TAMs repolarization as well as Tregs inhibition.
3.7. Prominent inhibition of distal tumors and metastasis
Inspired by the above results, we subsequently investigated whether the established immune response primed by the “Gemini nanoimmunoregulators” was sufficient to suppress an untreated distal tumor. The photograph of the distal tumor after local treatment (Fig. 7A) and the tumor volumes (Fig. 7B and Supporting Information Fig. S7A) displayed that the “Gemini nanoimmunoregulators” treatment significantly inhibited the growth of the distal tumor. The tumor inhibition rate was as high as 86.3% (calculated by tumor weight), which was most probably a result of the abscopal effect of the induced systemic anticancer immunity (Fig. S7B). At the same time, the treatment by single nanoformulation PM@RM-T7 + L or PR@RM-M2 has limited inhibition of the distal tumors, with respective average tumor suppression rates of 28.2% and 55.2% for the two groups (Fig. S7B). By comparison, weak antitumor effects were also observed in the P@RM-T7 + L groups with a tumor suppression rate of about 25.1%, which was due to the limited anticancer immunity of pure PTT. The weight of the isolated distal tumor also confirmed the results (Fig. S7C). Moreover, the IF observations of proliferation-associated antigen Ki67 in distal tumors showed that the proliferation of tumor cells in the distal tumor was inhibited to the maximum extent after “Gemini nanoimmunoregulators” treatment, compared with the single group treated by PM@RM-T7 + L or PR@RM-M2 (Fig. 7C and D). Therefore, the “Gemini nanoimmunoregulators” could stimulate strong systemic anticancer immunity to effectively kill not only the primary tumor but also the distal tumor. Furthermore, the lymphocytes infiltrated into the distal tumor were assessed, and both CD8+ and CD4+ T cells had a more pronounced infiltration into the distal tumors after “Gemini nanoimmunoregulators” treatment. Meanwhile, the characteristic marker Foxp3 of immune-suppressive Tregs displayed the lowest expression in the “Gemini nanoimmunoregulators” treated group. The “Gemini nanoimmunoregulators” induced systemic anticancer immunity to cope with distal tumorigenesis and progression.
Figure 7.

Abscopal effect of “Gemini nanoimmunoregulators” and metastasis inhibition. (A) Photographs and (B) growth curves of distal tumors. (C) Microscopic images of distal tumor sections with IF staining of Ki67, CD4, CD8, and Foxp3 (scale bar = 100 μm), and (D) further quantified by Image J. (E) Therapeutic schedule of 4T1 lung metastatic model. (F) Representative photographs of lung tissues with tumor metastasis and HE staining images (scale bar = 2 mm). (G) Microscopic images of lung sections with immunohistochemistry analysis of Ki67 (scale bar = 100 μm) and IF staining of CD4 and CD8 (scale bar = 50 μm).
Encouraged by the “Gemini nanoimmunoregulators” mediated remarkable performance of inhibiting primary and distal tumors, we evaluated the potential inhibition effect on the tumor metastasis in a more aggressive model (Fig. 7E). After a period of 14 days, we collected lung tissues from each group to investigate if there were any metastatic cells. Interestingly, we did not observe any lung metastatic foci in the group treated with “Gemini nanoimmunoregulators”, but we did observe lung metastasis in the control group and in the groups that received single treatments of PM@RM-T7 or PR@RM-M2, the lung tissues of each group were obtained for metastatic investigation, except for the “Gemini nanoimmunoregulators” treated group, lung metastatic foci were observed in the groups of control and single treatment of PM@RM-T7 or PR@RM-M2 (Fig. 7F and Fig. S7D). The pathological observations from HE and immunohistochemistry of Ki67 illustrated that the “Gemini nanoimmunoregulators” displayed evident inhibition on the proliferation of metastasized 4T1 cells (Fig. 7G). Moreover, there was an abundance of CD8+ and CD4+ T cells infiltrated into the lungs of the mice treated with “Gemini nanoimmunoregulators”, compared with the two groups of PM@RM-T7 + L or PR@RM-M2 (Fig. 7G). There is no doubt that “Gemini nanoimmunoregulators”-mediated photoimmunotherapy owned distinguished antimetastatic potency.
At last, the biosafety of our “Gemini nanoimmunoregulators” was assessed in vivo. A slight increase in the body weight of mice was observed in all treatments during the whole period (Supporting Information Fig. S8A). HE staining of the major organs (heart, liver, spleen, lung, and kidney) showed no visible pathological changes after treatment with “Gemini nanoimmunoregulators” (Fig. S8B). Meanwhile, the analysis of blood samples taken from the treated mice showed that many indexes of alanine aminotransferase, aspartate aminotransferase, creatinine, total protein, urine acid, and lactate dehydrogenase were within a normal range (Fig. S8C). None of the main hematological parameters were abnormal (Fig. S8D). Therefore, our “Gemini nanoimmunoregulators” had favorable biocompatibility in vivo. Taken all together, “Gemini nanoimmunoregulators” PM@RM-T7 and PR@RM-M2 cooperatively induced the systemic immunotherapeutic effect not only against the primary but also the distal and metastatic tumors, which could be a promising and safe nanoplatforms for precise and efficient oncotherapy.
3.8. Discussion
Based on the mussel-inspired material polydopamine and RM, the biomimetic “Gemini nanoimmunoregulators” could spatiotemporally deliver the hydrophilic PD-L1 inhibitor Met and hydrophobic TLR7/8 agonist R848 into the cancer cells and TAMs precisely, achieving the amplified anticancer immunity in a three-pronged manner to combat tumor growth and metastasis. The whole system was biocompatible and safe. The endogenous RM camouflage minimized the immunogenicity of the delivery system and alleviated the rapid clearance by the reticuloendothelial system, which is clinically challenged for most synthetic delivery systems. The T7 and M2 peptides anchoring conferred the respective targeting ability to cancer cells and M2 type TAMs (Figure 2, Figure 3), which could also be changed to other ligands to target other immune cells (e.g., DCs). Different from the other “Gemini-like” nanoplatforms reported45,46, our system focused on not only the different stages of the immune process, i.e., tumor cells immunogenicity and CTLs dysfunction, but also the TAMs reprogramming in ITM to boost the robust anticancer immunity. The in vivo data demonstrated a dramatic increase in CD8+ T cell infiltration into the primary tumor of mice by approximately 4.9-fold after treatment with the “Gemini nanoimmunoregulators” (Fig. 6F). Also, the excellent inhibition on the abscopal tumor and lung metastasis had been achieved (Fig. 7). It seems that the photothermal effect may deplete TAMs since R848 was loaded in the photosensitive mPDA cores. However, the repolarization ability of TAMs had little been affected (Fig. 6C). The ratio of M1/M2 in the tumor could be adjusted to 4.9 (Fig. 6D). In addition, as the excellent TLR7/8 agonist, R848 could also improve DCs maturation23. Met has multiple functions, including interfering the production of PD-L1 and repolarizing TAMs15,40. So, utilizing other appropriate immunomodulators with less overlapping functions could be much better for avoiding the possible redundant dosages and side effects in practical usage. Finally, the construction procedures, as well as the storage and transportation techniques of our “Gemini nanoimmunoregulators”, should be optimized and standardized, which would guarantee clinical applications. Nevertheless, this study provides a promising spatiotemporal nanomedicine-based platform for precise and efficient combined cancer photoimmunotherapy.
4. Conclusions
In summary, a new type of spatiotemporal biomimetic “Gemini nanoimmunoregulators” was launched for robust cancer photoimmunotherapy through the combination of targeted PTT-induced ICD, PD-L1/PD-1 blockade, and immunosuppressive TAMs reeducation. PM@RM-T7 and PR@RM-M2 realized the on-demand delivery of small molecule inhibitors (Met) and immune adjuvant (R848) to tumor cells and TAMs. On the one hand, PM@RM-T7 spontaneously recognized tumor cells and triggered ICD to initiate immune activation. Simultaneously, the released Met effectively degraded the inhibitory PD-L1 on the remaining tumor cells and prevented its escape from CTLs. On the other hand, PR@RM-M2 specifically reprogrammed tumor-promoting M2 type TAMs toward M1 phenotype, thus shifting ITM to tumoricidal orientation. The cooperation of PM@RM-T7 and PR@RM-M2 finally primed potent anticancer immunity against both the primary and distal tumor as well as the metastasis through multidimensional regulating of the immune pathways. This study showed light on the ITM reconstitution with multi-nanomedicines to achieve the superior outcome of immunotherapy.
Acknowledgments
This research was supported, in part or whole, by the National Natural Science Foundation of China (Nos. 32171395, U19A2006, and 12132004), the Sichuan Science and Technology Program (Nos. 2021YJ0130, 2022NSFSC0048, and 2023NSFSC0715, China), and the Joint Funds of Center for Engineering Medicine (Nos. ZYGX2021YGLH010, ZYGX2021YGLH017, and ZYGX2021YGLH204, China).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.11.005.
Contributor Information
Yiyao Liu, Email: liuyiyao@uestc.edu.cn.
Chunhui Wu, Email: wuchunhui@uestc.edu.cn.
Author contributions
Honglin Huang: conceptualization, investigation, methodology, writing the original draft, revision, editing. Ningxi Li: investigation, methodology. Xiaodan Wei: software, revision. Qingzhi Li, and Junhan Guo: methodology. Geng Yang: software. Hong Yang and Lulu Cai: supervision. Yiyao Liu and Chunhui Wu: conceptualization, supervision, funding acquisition.
Conflict of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Mellman I., Coukos G., Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 3.Boutros C., Tarhini A., Routier E., Lambotte O., Ladurie F.L., Carbonnel F., et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–486. doi: 10.1038/nrclinonc.2016.58. [DOI] [PubMed] [Google Scholar]
- 4.Blache U., Popp G., Dunkel A., Koehl U., Fricke S. Potential solutions for the manufacture of CAR T cells in cancer immunotherapy. Nat Commun. 2022;13:5225. doi: 10.1038/s41467-022-32866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng C., Li Y.J., Ferdows B.E., Patel D.N., Ouyang J., Tang Z.M., et al. Emerging vaccine nanotechnology: from defense against infection to sniping cancer. Acta Pharm Sin B. 2022;12:2206–2223. doi: 10.1016/j.apsb.2021.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie X.X., Song T., Feng Y., Zhang H.X., Yang G., Wu C.H., et al. Nanotechnology-based multifunctional vaccines for cancer immunotherapy. Chem Eng J. 2022;437 [Google Scholar]
- 7.Krysko D.V., Garg A.D., Kaczmarek A., Krysko O., Agostinis P., Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 8.Wang S., Bai Y.P., Wang D.Y., Zhai Y., Qiao Y.Q., Zhao X.J., et al. Reversing tumor to “Hot”: a NIR light-triggered carrier-free nanoplatform for enhanced tumor penetration and photo-induced immunotherapy. Chem Eng J. 2022;442 [Google Scholar]
- 9.Yang Z., Gao D., Zhao J., Yang G.J., Guo M., Wang Y., et al. Thermal immuno-nanomedicine in cancer. Nat Rev Clin Oncol. 2023;20:116–134. doi: 10.1038/s41571-022-00717-y. [DOI] [PubMed] [Google Scholar]
- 10.Beatty G.L., Gladney W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura K., Smyth M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol. 2017;95:325–332. doi: 10.1038/icb.2016.126. [DOI] [PubMed] [Google Scholar]
- 12.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamid O., Robert C., Daud A., Hodi F.S., Hwu W.J., Kefford R., et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tartari F., Santoni M., Burattini L., Mazzanti P., Onofri A., Berardi R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: recent insights and future challenges. Cancer Treat Rev. 2016;48:20–24. doi: 10.1016/j.ctrv.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Hu C., He X.Q., Chen Y.X., Yang X.T., Qin L., Lei T., et al. Metformin mediated PD-L1 downregulation in combination with photodynamic-immunotherapy for treatment of breast cancer. Adv Funct Mater. 2021;31 [Google Scholar]
- 16.Wu Y.R., Yang Z.C., Cheng K., Bi H.C., Chen J.J. Small molecule-based immunomodulators for cancer therapy. Acta Pharm Sin B. 2022;12:4287–4308. doi: 10.1016/j.apsb.2022.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha J.H., Yang W.H., Xia W.Y., Wei Y.K., Chan L.C., Lim S.O., et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–620. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong W., Qi L., Jiang N., Zhao Q., Chen L.X., Jiang X., et al. Metformin liposome-mediated PD-L1 downregulation for amplifying the photodynamic immunotherapy efficacy. ACS Appl Mater Interfaces. 2021;13:8026–8041. doi: 10.1021/acsami.0c21743. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y., Lin Y.X., Qiao S.L., Wang J., Wang H. Progress in tumor-associated macrophages: from bench to bedside. Adv Biosyst. 2019;3 doi: 10.1002/adbi.201800232. [DOI] [PubMed] [Google Scholar]
- 20.Galli S.J., Borregaard N., Wynn T.A. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells, and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu M., Zhang J., Yu Y.L., Tu K., Yang T., Wang Y., et al. Injectable liquid crystal formation system for reshaping tumor immunosuppressive microenvironment to boost antitumor immunity: postoperative chemoimmunotherapy. Small. 2020;16 doi: 10.1002/smll.202004905. [DOI] [PubMed] [Google Scholar]
- 23.Kim S.Y., Kim S., Kim J.E., Lee S.N., Shin I.W., Shin H.S., et al. Lyophilizable and multifaceted toll-like receptor 7/8 agonist-loaded nanoemulsion for the reprogramming of tumor microenvironments and enhanced cancer immunotherapy. ACS Nano. 2019;13:12671–12686. doi: 10.1021/acsnano.9b04207. [DOI] [PubMed] [Google Scholar]
- 24.Jiang Y.Y., Huang J.G., Xu C., Pu K.Y. Activatable polymer nanoagonist for second near-infrared photothermal immunotherapy of cancer. Nat Commun. 2021;12:742. doi: 10.1038/s41467-021-21047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Y.Y., Chen Q., Guo Z.Y., Li M.W., Yang X.Y., Wan G.Y., et al. An intelligent biomimetic nanoplatform for holistic treatment of metastatic triple-negative breast cancer via photothermal ablation and immune remodeling. ACS Nano. 2020;14:15161–15181. doi: 10.1021/acsnano.0c05392. [DOI] [PubMed] [Google Scholar]
- 26.Liang S., Liu Y., Gao T., Liu X.Q., Zhang Z.P., Mu W.W., et al. An integrated nanoaircraft carrier modulating antitumor immunity to enhance immune checkpoint blockade therapy. Adv Funct Mater. 2021;31 [Google Scholar]
- 27.Cheng W., Zeng X.W., Chen H.Z., Li Z.M., Zeng W.F., Mei L., et al. Versatile polydopamine platforms: synthesis and promising applications for surface modification and advanced nanomedicine. ACS Nano. 2019;13:8537–8565. doi: 10.1021/acsnano.9b04436. [DOI] [PubMed] [Google Scholar]
- 28.Yu Y., Song M.Y., Chen C.L., Du Y.Y., Li C.G., Han Y., et al. Bortezomib-encapsulated CuS/Carbon dot nanocomposites for enhanced photothermal therapy via stabilization of polyubiquitinated substrates in the proteasomal degradation pathway. ACS Nano. 2020;14:10688–10703. doi: 10.1021/acsnano.0c05332. [DOI] [PubMed] [Google Scholar]
- 29.Cieslewicz M., Tang J.J., Yu J.L., Cao H., Zavaljevski M., Motoyama K., et al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci USA. 2013;110:15919–15924. doi: 10.1073/pnas.1312197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey C.J. Metformin: historical overview. Diabetologia. 2017;60:1566–1576. doi: 10.1007/s00125-017-4318-z. [DOI] [PubMed] [Google Scholar]
- 31.Peng J.R., Xiao Y., Li W.T., Yang Q., Tan L.W., Jia Y.P., et al. Photosensitizer micelles together with IDO inhibitor enhance cancer photothermal therapy and immunotherapy. Adv Sci. 2018;5 doi: 10.1002/advs.201700891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodell C.B., Arlauckas S.P., Cuccarese M.F., Garris C.S., Li R., Ahmed M.S., et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578–588. doi: 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M.G., O'Connor R.S., Trefely S., Graham K., Snyder N.W., Beatty G.L. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated ‘don’t-eat-me' signal. Nat Immunol. 2019;20:265–275. doi: 10.1038/s41590-018-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang L.P., Li Y.A., Du Y.N., Zhang Y.Y., Wang X.X., Ding Y., et al. Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat Commun. 2019;10:4871. doi: 10.1038/s41467-019-12771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J.C., Li G.Y., Wang B., Han S.X., Sun X., Jiang Y.N., et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J Exp Clin Cancer Res. 2019;38:235. doi: 10.1186/s13046-019-1211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Summers deLuca L., Gommerman J.L. Fine-tuning of dendritic cell biology by the TNF superfamily. Nat Rev Immunol. 2012;12:339–351. doi: 10.1038/nri3193. [DOI] [PubMed] [Google Scholar]
- 37.Wagner J., Gößl D., Ustyanovska N., Xiong M., Hauser D., Zhuzhgova O., et al. Mesoporous silica nanoparticles as pH-responsive carrier for the immune-activating drug resiquimod enhance the local immune response in mice. ACS Nano. 2021;15:4450–4466. doi: 10.1021/acsnano.0c08384. [DOI] [PubMed] [Google Scholar]
- 38.Rong L., Zhang Y., Li W.S., Su Z.G., Fadhil J.I., Zhang C. Iron chelated melanin-like nanoparticles for tumor-associated macrophage repolarization and cancer therapy. Biomaterials. 2019;225 doi: 10.1016/j.biomaterials.2019.119515. [DOI] [PubMed] [Google Scholar]
- 39.He L.Z., Nie T.Q., Xia X.J., Liu T., Huang Y.Y., Wang X.J., et al. Designing bioinspired 2D MoSe2 nanosheet for efficient photothermal-triggered cancer immunotherapy with reprogramming tumor-associated macrophages. Adv Funct Mater. 2019;29 [Google Scholar]
- 40.Wei Z.H., Zhang X.Q., Yong T.Y., Bie N.N., Zhan G.T., Li X., et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat Commun. 2021;12:440. doi: 10.1038/s41467-020-20723-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahjub R., Jatana S., Lee S.E., Qin Z., Pauli G., Soleimani M., et al. Recent advances in applying nanotechnologies for cancer immunotherapy. J Control Release. 2018;288:239–263. doi: 10.1016/j.jconrel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Jurk M., Heil F., Vollmer J., Schetter C., Krieg A.M., Wagner H., et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 43.Beyranvand Nejad E., Labrie C., van Elsas M.J., Kleinovink J.W., Mittrucker H.W., Franken K., et al. IL-6 signaling in macrophages is required for immunotherapy-driven regression of tumors. J Immunother Cancer. 2021;9 doi: 10.1136/jitc-2021-002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q.Q., Shi Z.Q., Zhang F., Zeng W.W., Zhu D., Mei L. Symphony of nanomaterials and immunotherapy based on the cancer–immunity cycle. Acta Pharm Sin B. 2022;12:107–134. doi: 10.1016/j.apsb.2021.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei B.C., Pan J.M., Yuan R.T., Shao B.F., Wang Y., Guo X., et al. Polarization of tumor-associated macrophages by nanoparticle-loaded Escherichia coli combined with immunogenic cell death for cancer immunotherapy. Nano Lett. 2021;21:4231–4240. doi: 10.1021/acs.nanolett.1c00209. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H.Y., Zhang J., Li Q., Song A.X., Tian H.L., Wang J.Q., et al. Site-specific MOF-based immunotherapeutic nanoplatforms via synergistic tumor cells-targeted treatment and dendritic cells-targeted immunomodulation. Biomaterials. 2020;245 doi: 10.1016/j.biomaterials.2020.119983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.