

Abstract
Liver fibrosis, characterized by scar tissue formation, can ultimately result in liver failure. It's a major cause of morbidity and mortality globally, often associated with chronic liver diseases like hepatitis or alcoholic and non-alcoholic fatty liver diseases. However, current treatment options are limited, highlighting the urgent need for the development of new therapies. As a reversible regulatory mechanism, epigenetic modification is implicated in many biological processes, including liver fibrosis. Exploring the epigenetic mechanisms involved in liver fibrosis could provide valuable insights into developing new treatments for chronic liver diseases, although the current evidence is still controversial. This review provides a comprehensive summary of the regulatory mechanisms and critical targets of epigenetic modifications, including DNA methylation, histone modification, and RNA modification, in liver fibrotic diseases. The potential cooperation of different epigenetic modifications in promoting fibrogenesis was also highlighted. Finally, available agonists or inhibitors regulating these epigenetic mechanisms and their potential application in preventing liver fibrosis were discussed. In summary, elucidating specific druggable epigenetic targets and developing more selective and specific candidate medicines may represent a promising approach with bright prospects for the treatment of chronic liver diseases.
Key words: Liver fibrosis, Epigenetics regulation, DNA methylation, Histone acetylation, Histone methylation, mRNA methylation, Non-coding RNA, Drug development
Graphical abstract
Developing specific candidate medicines targeting epigenetic modification, including DNA methylation, histone modification and RNA modification, may become a promising approach for the therapeutic strategy of liver fibrosis.
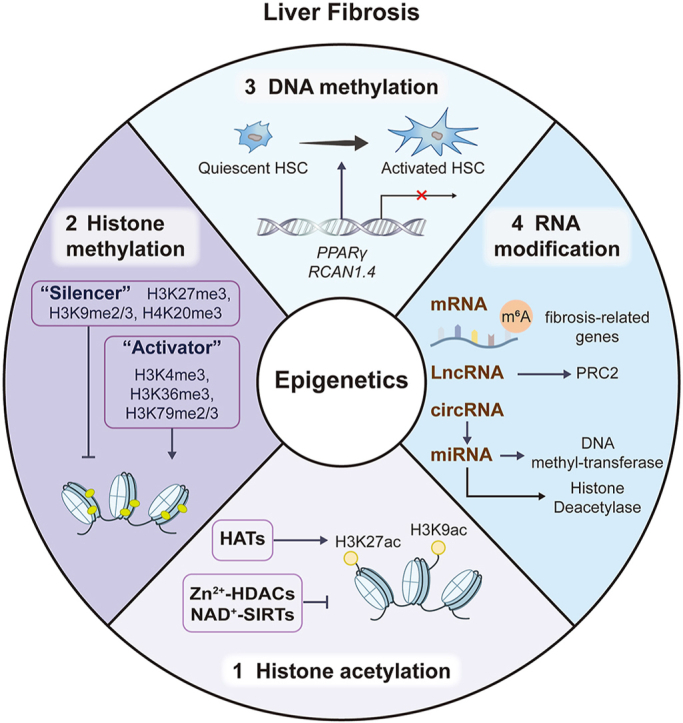
1. Epigenetics: A potential target regulating liver fibrosis
Epigenetics refers to the study of changes in gene expression or cellular phenotype that occur without alterations to the underlying DNA sequence. It encompasses different molecular mechanisms that can modify the activity of genome. These modifications can regulate the accessibility of DNA to transcriptional machinery and, therefore can have significant effects on gene expression1. The role and mechanisms of epigenetics in disease biology are currently understood to operate at multiple levels, including DNA methylation, histone modifications, chromatin remodeling, and regulation by non-coding RNA (ncRNA), leading to variable expression of critical pathogenesis-related gene sets (Fig. 1). Hence, epigenetics has significant implications for understanding human diseases and has been studied as a promising target for therapeutic interventions.
Figure 1.

The feature of epigenetic regulation. Epigenetic regulation involves DNA methylation, RNA modification, histone methylation and histone acetylation. DNMT family members mediate DNA methylation, which suppresses gene transcription by adding a methyl group to the 5′ position of cytosine to form 5-methylcytosine at cytosines within CpG dinucleotides. RNA modification, which includes mRNA and ncRNA, regulates gene expression by influencing the output and stability of genes in the nucleus. Histone methylation, catalyzed by HMTs and HDMTs, is involved in epigenetic remodeling in liver fibrosis by positively or negatively regulating transcriptional activation. Histone acetylation, regulated by HATs and HDACs, has also been found to be involved in liver diseases.
Liver fibrosis, characterized by the progressive deposition of extracellular matrix (ECM) and activation of hepatic stellate cells (HSCs). Given the increasing prevalence of liver diseases and the lack of treatment options, it remains a significant challenge to address liver fibrosis2. Liver fibrosis often occurs and starts the cicatrization process following environmental insults, but fundamentally, it is considered as an imbalance between the production and degradation of ECM3. Under the stimulation of external damage signals in the initial inflammation phase of fibrogenesis, hepatocytes undergo injury and subsequently trigger myofibroblasts to generate excess ECM. Meanwhile, myofibroblasts, epithelial and endothelial cells also produce matrix metalloproteinases (MMPs), which contribute to ECM degradation. In the subsequent remodeling phase, α-smooth muscle actin (α-SMA)-positive myofibroblasts continually release cytokines to achieve self-activation, leading to uncontrolled ECM deposition and scar formation4.
With genetic factors hardly explaining the pathogenesis of liver fibrosis and suffering the steady rise of cases over decades, increasing evidence has focused on the molecular mechanisms of extensive epigenetics remodeling in the profibrotic phenotype of various liver cells. Taking the above opinions into consideration, in the current review, we emphasize the interaction between epigenetic modification and liver fibrotic diseases and evaluate the potential of abnormal epigenetics as early diagnostic markers and therapeutic targets for liver fibrosis.
2. The modulation of DNA methylation in liver fibrosis
DNA methylation plays a crucial role in regulating gene expression by influencing the accessibility of DNA by transcription factors. It is catalyzed by DNA methyltransferases (DNMTs), which is responsible for 5-methylcytosine (5-mC) formation in the cytosine-guanosine dinucleotide (CpG) sites of the genome. Depending on the locations, DNA methylation manipulates gene silence and activation by recruiting certain proteins or regulating the binding of transcription factors to target genes5. Specifically, methylation in the transcription start point or promoters results in gene silence, while the gene-body methylation is associated with gene expression and alternative splicing. As major methylation inducer, DNMTs, including DNMT1, DNMT3a, DNMT3b and DNMT3L, are responsible for transferring methyl group from S-adenosyl-l-methionine (SAM) to the 5-position of DNA cytosine. Among DNMTs, DNMT1 mediated maintenance methylation while DNMT3a and DNMT3b are responsible for the de novo methylation6. DNMT3L does not have catalytic activity, but it enhances the DNA methylation activity of DNMT3A and DNMT3B by direct interaction with them. While a dynamic balance between methylation and demethylation was formed during the long-term transcriptional regulation. 5-mC formed in methylation can be oxidized by ten-eleven translocation (TET) proteins, including TET1/2/3. This results in the formation of 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), 5-carboxycytosine (5-caC) and 5-hydroxymethyluracil (5-hmU), initiating demethylation program that eliminates the methyl group from cytosine, ultimately reversing transcriptional suppression. Thus, the dynamic and reversible nature of DNA methylation also highlights the vital roles of DNA demethylation mediated by TET family. Emerging studies have identified that the DNA methylation pattern, including methylation and hydroxymethylation-mediated demethylation, is involved in HSC transdifferentiation during liver fibrotic diseases (Fig. 2). For instance, HSC transdifferentiation was accompanied by high densities of 5-mC and global reduction in 5-hmC, which was confirmed by comparing the numbers of 5-mC and 5hmC sites in activated-HSC and quiescent-HSC7. Meanwhile, when DNMT3a and DNMT3b were downregulated by siRNA, DNA methylation was reduced, and HSC activation was subsequently inhibited7,8.
Figure 2.

The modulation of DNA methylation in liver fibrosis. Advances have been made in the understanding of the roles of DNA methylation in liver fibrosis. It has been observed that a repressive chromatin structure in exons can lead to gene transcription suppression through the addition of a methyl group at the 5′ position of cytosine. This process is mediated by the DNMT and GNMT family. Several genes, including PPARγ, RCAN1.4, and PSTPIP2, and methylated binding protein Mecp2 have been found to experience DNA methylation, and their transcriptional suppression has been shown to be closely related to the transactivation of HSCs and liver fibrosis.
Growing evidence established that transcriptional regulation played a pivotal role in liver fibrosis9. Proliferator-activated receptor-γ (PPARγ) is one of the attractive targets and its transcriptional suppression was closely related the transactivation of HSCs and liver fibrosis10. Indeed, numerous attempts have been made to evaluate the clinical efficacies of PPARγ agonists, including troglitazone11, rosiglitazone12 and pioglitazone13, for treating NASH and liver fibrosis. Caldwell and his colleagues showed that short-term troglitazone therapy led to histological improvements and discontinued the progression of fibrosis in two NASH subjects. Mechanistically, quiescent HSCs contain lipid droplets responsible for hepatic vitamin A storage, maintaining liver homeostasis under physiological conditions. As a master negative-regulator of HSC activation, PPARγ binds to the receptor of retinoic acid, a vitamin A metabolite, and activates the expression of several lipid biosynthesis-related genes, including sterol regulatory element binding protein 1 (Srebp1), Seipin and Lpin, by recognizing DNA sequence element peroxisome proliferator response element (PPRE). Upon fibrogenesis, PPARγ undergoes transcriptional suppression, resulting in the impaired ability to induce certain genes. As a result, there is a loss of lipid droplets and transdifferentiation of myofibroblasts in activated HSCs, which are responsible for the accumulation of ECM, leading to fibrosis14. Several studies further demonstrated that PPARγ could directly regulate fibrotic classical signaling transforming growth factor-β (TGF-β)/Smad pathway to alleviate the generation of profibrotic myofibroblasts15. PPRE mutation could also promote HSC differentiation by abolishing the recruitment of PPARγ to the MAT2A promoter and subsequently increasing the expression cell prefoliation-related target methionine adenosyltransferase 2A (MAT2A)16. Indeed, plentiful CpG islands have been identified in the promoter, 5′UTR and gene body of PPARγ, suggesting a higher possibility of epigenetic modification. Thus, transcriptional suppression of PPARγ may be closely related to epigenetic regulation during fibrogenesis. Human NASH liver biopsies, animal studies and in vitro investigations have substantiated this hypothesis and provided evidence that the promoter region of PPARγ undergoes methylation remodeling, transforming to a hypermethylated pattern with increasing fibrosis severity. Zeybel and his colleagues17 identified CpG sites in human PPARγ promoter and further demonstrated that hypermethylation was positively correlated with liver fibrosis progression in male patients between the age of 46–65 with biopsy-proven NAFLD. Under myofibroblast phenotypic transformation, methyl CpG binding protein 2 (MeCP2), with high affinity to methylated DNA, maintained the hypermethylation (one with 109 CpG and the other with 21 CpG) of PPARγ promoter region and thus facilitated PPARγ suppression and liver fibrosis progression18. Recently, Moran-Salvador et al.19 established MeCP2 deletion and MeCP2 S80A mutated mouse model and further verified that the phosphorylation of MeCP2 at S80 site is required for HSC activation and collagen expression in the CCl4 mouse model. Additionally, MeCP2 upregulated myofibroblast DNA replication-related genes, including Cdc7, Has2 and Dna2, and thus enhanced active HSC proliferation and hepatic fibrogenesis. Despite multiple studies demonstrating a clear positive correlation between PPARγ methylation and liver fibrosis from bench to bed, research on PPARγ methylation is limited by the need for access to liver biopsy to measure methylation at the PPARγ promoter. Thus, several groups focused on identifying plasma PPARγ methylation markers to establish potential non-invasive biomarkers for stratification of liver fibrosis. Hlady et al.20 found hypermethylated PPARγ DNA in the plasma via genome-wide cell-free 5-mC landscape analysis and identified a set of differentially methylated CpGs (cg04645914, cg06215569, cg23663760, cg13781744 and cg07610777) in cell free PPARγ DNA by comparing plasma from cirrhosis patients and healthy controls. Notably, these high performing sites and differential CpG methylation events were also observed in different liver tissues, which distinguished cirrhosis and hepatocellular carcinoma patients from normal liver. Therefore, CpGs in cell free PPARγ DNA have the potential to differentiate non-cirrhosis from cirrhosis patients and may be used for the clinical non-invasive screen20. In addition to the diagnosis of liver diseases, differential plasma DNA methylation of PPARγ may be utilized as a non-invasive method to stratify the severity of liver fibrosis in NAFLD patients. Indeed, liver DNA methylation of PPARγ has been validated as a means to stratify patients in terms of fibrosis severity. A clinal study including 26 NAFLD patients provided further validation by demonstrating that sequence-specific quantification of methylation densities at PPARγ DNA in plasma correlated with the progression of fibrosis. This study also showed that hypermethylation degree at 2 positions, named CpG1 and CpG2, in PPARγ was similar between plasma and hepatocyte-rich liver tissue. Patients with mild live fibrosis exhibited lower levels of PPARγ methylation in plasma (CpG1:63%, CpG2:51%), while patients with severe fibrosis showed higher levels (CpG1:86%, CpG2:65%). Indeed, hepatocyte injury, which is a characteristic feature of NAFLD, may result in substantial leakage of cell-free DNA into the circulation. This suggests that liver contributes significantly to the pool of DNA present in plasma21. Taken together, DNA methylation patterns in plasma may potentially originate from injured hepatocytes and serve as a diagnostic marker for liver fibrosis. Paradoxically, another study reported that the promoter undergoes demethylation, leading to the accumulation of hepatic lipids in high-fat diet (HFD)-induced NAFLD mice, which in turn aggravates fibrosis. Specifically, this study showed that HFD increased PPARγ expression by decreasing cytosine methylation levels in the PPARγ promoter and thus activated its target genes, such as very low-density lipoprotein (VLDLR) and cluster differentiating 36 (CD36). While this study is inconsistent with mainstream understanding, it still highlights the importance of PPARγ methylation in the progression of liver diseases22.
In addition to PPARγ, another protein called Calcineurin (CaN), a calcium/calmodulin-activated serine/threonine phosphatase, has been shown to play a role in TGF-β-induced ECM accumulation. CaN can be activated by TGF-β in a time and dose-dependent manner23. The most studied substrates of CaN are the family of nuclear factors of activated T-cells (NFATs). CaN induces dephosphorylation and nuclear translocation of NFAT3 and subsequently leads to the overexpression of fibrosis-related genes, including Fibronectin (Fn) and vascular endothelial growth factor (Vegf). Interestingly, a member of the regulator of calcineurin (RCAN) family, RCAN1.4 has been shown to improve liver fibrosis by acting as an endogenous inhibitor of CaN24. Specifically, RCAN1.4 was significantly suppressed at transcription level in both CCl4-induced mouse model and TGF-β-activated HSCs due to the presence of two hypermethylation sites in its promoter region. In more detail, the hypermethylation of RCAN1.4 was mediated by DNMT1 and DNMT3, which was confirmed by DNMT inhibitor 5-aza-2′-deoxycytidine (5-AzaC) and DNMTs-siRNA in vitro. Furthermore, Pan and his colleagues25 developed a liver-specific RCAN1.4 overexpression mouse model by injecting RCAN1.4 plasmid specifically into to the liver tissue. In this study, the authors confirmed that the induction of RCAN1.4 had a beneficial effect on liver fibrosis by inhibiting CaN-NFAT3 signaling.
Persistence of chronic inflammation often caused by activated inflammatory immune cells such as macrophages is commonly associated with the progression of liver fibrosis. M2 macrophage-derived cytokines, including IL-4, IL-10 and IL-13, are related to epigenetic modifications that contribute to fibrotic processes. Proline-serine-threonine-interacting protein 2 (PSTPIP2), mainly expressed in Kupffer cells, has been found to be significantly decreased in the CCl4-induced mouse model of liver fibrosis and LPS-treated RAW264.7 cell line. Mechanically, DNMT3a and DNMT3b promote the hypermethylation of PSTPIP2 (chr18:77843840–77843968) in the 5′-UTR region and subsequently repress PSTPIP2 expression. This association was confirmed by representation bisulfite sequencing (RRBS) in primary macrophages derived from CCl4-induced mice. Further investigation revealed that liver-specific PSTPIP2 overexpression alleviated inflammation and liver fibrosis by regulating the expression and secretion of cytokines including IL-6, IL-1β and IL-10, which is relied on the suppression of signal transducer and activator of transcription 1 (STAT1) phosphorylation. On the other hand, accompanied with STAT6 phosphorylation, the overexpression of PSTPIP2 upregulated the mRNA levels of IL-10, IL-13 and CD16326. These findings highlight the complex role of PSTPIP2 in modulating cytokine expression and suggest its potential as a therapeutic target for liver inflammation and fibrosis.
In addition to DNMTs, another important enzyme involved in liver methylation processes is glycine N-methyltransferase (GNMT). GNMT is the most abundant SAM-dependent methyltransferase in the liver and hepatocytes. Interestingly, GNMT act epigenetic regulatory elements by competing with DNMT to remodel transmethylation flux. Recently, the inverse correlation between hepatic GNMT and miR-873-5p was identified in cirrhotic patients, BDL mice and primary mouse hepatocytes. Fernandez-Ramos et al.27 established that in BDL and the Mdr2-deficient mouse models, anti-miR-873-5p therapy ameliorates hepatocyte apoptosis, cholangiocyte proliferation and liver fibrosis by recovering GNMT expression and steering SAMe flux afar DNA.
3. Histone modifications in liver fibrosis
The transcriptional or replication machinery must arrive at the specific genomic region in order to function, which requires the DNA to be accessible. DNA is tightly organized into regulated structures by octameric protein complex, forming nucleosomes core particle. Each of these contains two of each core histones (H2A, H2B, H3 and H4) with 145–147 base pairs of DNAs. The N- and C-terminal histone tails are enriched with basic amino acid residues, such as lysine and arginine, which endow nucleosomes with unique properties and thus influence gene transcription and epigenetic states28. Histones modifications, including methylation and acetylation have the ability to influence the compaction of chromatin as well as the accessibility of DNA to transcription factors and other regulatory proteins29. The possible outcomes of different histone modifications in liver fibrosis are summarized in Table 1. Notably, histone lactylation and phosphorylation were both recently identified and are differentially involved in the mRNA expression of genes30,31. However, whether these novel histone modifications contribute to hepatic fibrogenesis is not elusive, yet worth to be investigated in the future.
Table 1.
Summary of histone modification in liver fibrosis.
| Histone modification | Consequence | Target | Cell type/animal model |
|---|---|---|---|
| Methylation | Inhibits transcription | ||
| H3K27me3 | TGF-β/IGFBP3 | HSC | |
| H3K9me2/3 | TLR4/NF-κB/PPARγ | HSC | |
| H4K20me3 | Bax | HSC | |
| Methylation | Promotes transcription | ||
| H3K4me1/3 | NLRP3/Bivalent domains in PPARγ | Hepatocyte/HSC | |
| H3K36me3 | Bivalent domains with H3K27me3 | HSC | |
| H3K79me3 | Cul4 with Clr4 | Hepatocyte | |
| Acetylation | Promotes transcription | ||
| H3K27ac | CCL2 | LSEC | |
| H3K9ac | Fibronectin and Serpine1 | Cholangiocytes | |
3.1. The modulation of histone methylation in liver fibrosis
Histone methylation is dynamically mediated by several histone methyl transferase (HMT) and histone demethylase (HDMT), which orchestrates gene expression in liver fibrosis by depositing and removing methylation marks, respectively (Fig. 3). The lysine residues of histones could be mono-, di- and trimethylated by adding one, two or three methyl groups respectively to act as active or repressive marks of gene expression32. Numerous studies have identified that methylation at different lysine sites play antipodal effects on transcription by influencing the binding of proteins to modified histones. The methylated histone 3 lysine 9 (H3K9), H3K27 and H4K20 function as “silencer” proteins and are associated with gene silencing, while methylated H3K4, H3K36, and H3K79 function as “activator” proteins to activate the transcription of implicated genes.
Figure 3.

The modulation of histone methylation in liver fibrosis. Mounting studies have shown correlations between histone methylation and liver fibrosis. Methylation at different lysine sites can have antipodal effects on transcription. As “Silencer” proteins, H3K9, H3K27, and H4K20 are involved in gene suppression, whereas H3K4, H3K36, and H3K79 function as “Activator” proteins that promote gene transcription. The “bivalent domains”, formed by H3K4me3, H3K36me3, and H3K79me2/3, as well as the silent markers H3K27me3 and H3K9me2/3, keep gene transcription at a low level, preparing for rapid gene expression with RNA polymerase II loaded. Since chromatin structure is created by DNA wrapping around histones, histone methylation works in conjunction with DNA methylation through EZH2, G9a, and DNMT1.
3.1.1. Histone methylation suppresses gene transcription as ‘Silencer’
Chromatin is organized as the euchromatin and heterochromatin in eukaryotes. Euchromatin is less condensed and allows transcription factors to access the DNA and initiate transcription. Euchromatin is characterized by a more open structure and is enriched in specific histone modifications associated with gene activation. However, heterochromatin is a tightly packed form of chromatin and is associated with gene silencing and a more repressive state of transcription. Methylation of H3K27, H3K9 and H4K20 is generally associated with packed heterochromatin and gene repression.
TGF-β, a master profibrogenic cytokine, triggers HSC activation and ECM deposition and thus plays a critical role in hepatic fibrogenesis. In these processes, TGF-β upregulated the expression of enhancer of zeste homolog 2 (EZH2) a polycomb repressive complex (PRC) component that specifically mediated the trimethylation of H3K27, and subsequently promoted H3K27me3 at the promoter of several downstream genes. Reversing H3K27me3 by EZH2 inhibitor GSK-503 or siRNA effectively attenuated TGF-β-stimulated HSC activation by inhibiting fibrotic gene transcription, including Fn, Col1a1 and Acta2 in CCl4-and BDL-mouse model33. Bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI) is regarded as the pseudo receptor of TGF-β due to the similar extracellular ligand binding domain structure as TGFβR, yet lacks an intercellular serine/threonine adaptor. BAMBI has been confirmed as a negative regulator of the TGF-β pathway and a potential anti-fibrosis target. EZH2 inhibitor 3-deazaneplanocin A (DZNep) promoted the expression of BAMBI, IL-10 and cell cycle regulators, including Cdkn1a, Gadd45a and Gadd45b, and thus led to TGF-β/Smads suppression and anti-inflammatory response by removing H3K27me3 signatures associated with these genes in TGF-β-induced HSCs34. Furthermore, by enhancing the transmembrane transport of TGF-β, the scaffold protein GIPC (also known as synectin) epigenetically upregulated Insulin-like growth factor binding protein 3 (IGFBP3) is predominantly expressed in HSCs, and promoted HSC activation and migration. Chromatin immunoprecipitation showed that GIPC significantly decreased H3K27me3 modification in Igfbp3 transcription start site region (chr7:45959379–45960659 and chr7: 45959718–45961110) and promoter region (chr7: 45961121–45962023). Usm and his colleagues further established a global Igfbp3 knockout mouse model, illustrating that Igfbp3 absent reversed HSC activation and migration and inhibited liver fibrosis. Thus, by employing EZH2 inhibitors, such as GSK-503 and DZNep or siRNAs, the studies confirmed that H3K27me3 is involved in epigenetic regulation of TGF-β-related targets, suggesting the therapeutic potential of targeting H3K27me3 modification in liver fibrosis35.
H3K9 methylations are also generally regarded as a crucial mark of heterochromatin formation and transcriptional silencing. G9a and Glp are required for H3K9 di-methylation and Suv39h enzymes, including Suv39h1 and Suv39h2, predominantly mediate H3K9 tri-methylation36. The transcriptional silencing of PPARγ during HSC activation is not only attributed to DNA methylation but can also be epigenetically repressed by H3K9me2 near the PPARγ gene promoter region. JMJD1A (Jumonji domain-containing protein 1A), a histone demethylase catalyzing the removal of H3K9me1 and H3K9me2, was abnormally decreased in TGF-β-treated HSCs and CCl4-treated mice. Meanwhile, JMJD1A overexpression significantly decreased fibrosis-associated gene expression and reversed PPARγ transcription by removing the two methyl groups from H3K937. Another histone demethylase catalyzing H3K9 di-, and tri-demethylation, lysine(K)-specific demethylase 4D (KDM4D) also gradually increased during HSC activation in CCl4-, BDL and TAA-treated mice and primary HSCs. KDM4D knockdown stimulated the enrichment of H3K9me2 and H3K9me3 in the Toll-like receptor 4 (Tlr4) promoter and therefore promoted fibrosis regression in vivo. In detail, Tlr4 was critically involved in pro-fibrogenic gene expression in HSCs and promoting liver fibrosis via triggering downstream nuclear factor kappa-B (NF-κB), a master regulator for the proinflammatory mediators38.
In addition to H3K27 and H3K9 methylation, H4K20 methylation was well-established as a repressive hallmark of histone modification contributing to fibrogenesis. Although the potential connection between H4K20 and liver fibrosis remains to be clarified, the figure of H4K20 mono- and trimethylation in pulmonary fibrosis and fibroblast senescence has attracted attention. In idiopathic pulmonary fibrosis myofibroblasts, H4K20me1 and its methyltransferase SET8 were higher than those in normal lung fibroblasts39. Fibroblast senescence, a state of irreversible growth arrest, is associated with apoptosis, including pro-apoptotic gene Bax and anti-apoptotic gene Bcl2. Due to the high-expression of Bcl2 and low level of Bax, senescent fibroblasts are more resistant to external injury. Chromatin immunoprecipitation indicated that H4K20me3 was enriched in the Bax promoter and decreased in the Bcl2 promoter to induce senescent phenotypes in fibroblasts, ultimately favoring the abrogation of fibrogenesis40.
3.1.2. Histone methylation promotes gene transcription as ‘Activators’
Except for inhibiting gene transcription, histone methylation could also be considered transcriptional-active marks of specific genes. The methylation at H3K4, H3K36 and H3K79 are known as active marks that are responsible for gene activation. H3K4me2 and H3K9me3 were demonstrated to be increased at the promoter regions of NOD-like receptor protein 3 (NLRP3) inflammasome, contributing stimulator of interferon genes (STING) overexpression-stimulated hepatocyte pyroptosis during liver fibrosis. Detailly, in response to STING agonist DMXAA plus TNF-α, the H3K4-specific histone methyltransferase WDR5 and H3K79 methyltransferase DOT1L formed a transcription activator complex with interferon regulatory transcription factor 3 (IRF3) thus recruited IRF3 to NLRP3 promoter and activated NLRP3 inflammasome pathway, which was significantly abrogated by WDR5 and DOT1L inhibitors ICR-9429 and EPZ004777, respectively41. In addition to the direct roles of these histone modifications, with the remission of liver fibrosis, the interaction between ‘Activator’ in ‘Silencer’ and the ‘bivalent domains’ formed by these active marks and recognized silence markers, including H3H27me3 and H3K9me were detected42. These ‘bivalent domains’ kept genes expressed at low levels while stimulated and poised for rapid activation if required. In detail, RNA polymerase II (poly II) is loaded in ‘bivalent domains’ formed by H3K4me3 and H3K27me3, preparing for rapid gene activation in embryonic fibroblast cells. Genes destined for activation lose the suppressive H3K27me3 and expand the active H3K4me3 to the gene body, establishing bimodal peaks of H3K4me3 through poly II elongation, and vice versa43. Meanwhile, bivalent H3K4me3 and H3K9me3 chromatin domains maintained adipogenic master regulatory genes, including PPARγ and Cebpα, low expression and stimulated poised for rapid activation when differentiation44. Meanwhile, H3K4me2 and H3K9me2 were reported to maintain a negative correlation. Depletion of LSD2, a specific H3K4me2 demethylase, increased H3K4me2 and thus decreased H3K9me245. While H3K36me2/3 and H3K79me3 rarely co-exist with H3K27me3 and H3K9me2 and the mutually exclusive effects were applied by influencing enzyme activity. Chromatin immunoprecipitation sequencing demonstrated that H3K36me3 suppressed H3K27me3 in the promoter region via inhibiting activity of H3K27 methyltransferase complex PRC246. H3K79me3 elimination, mediated by Cul4-mediated ubiquitination degradation, significantly increased methyltransferase Clr4-dependent H3K9me3 modification and turned off the expression of hepatocyte marker α-fetoprotein (AFP) and glypican-3 (GPC3) in the liver47. Thus, the methylation of H3K4, H3K36 and H3K79 may become a potential regulatory target in liver fibrosis by interacting with H3K9me3 and H3K27me3 and DNA methylation.
Additionally, H3K79me2/3, H3K36me3 and H3K4me3 have been illustrated to play vital roles in other organ fibrosis, including renal fibrosis, pulmonary fibrosis and cardiac fibrosis. For example, H3K79 methyltransferase DOT1L increased the enrichments of H3K79me3 on the promoter of Jagged gene and thus enhanced Jagged expression, then stimulating Notch signaling and fibrosis response in Bleomycin-stimulated pulmonary fibrosis48. Furthermore, Dot1L knockout rescued Ang II-induced and myocardial ischemia-induced cardiac fibrosis by decreasing H3K79me3 enrichment in forkhead box O3 (FOXO3) and thus inhibiting FOXO3 expression. Pharmacological targeting for Dot1L alleviated ECM deposition and inhibited cardiac fibroblast activation by epigenetically suppressing FoxO3a in cardiac fibrosis49. In renal tubular epithelial cells and myofibroblasts, treatment with EPZ5676 or Dot1L siRNA inhibited H3K79me3 enrichment in Snail, Twist and Notch1 promoter and thus inhibited their expression. Targeting Dot1L attenuated renal fibrosis via inhibiting renal fibroblasts and epithelial-to-mesenchymal transition (EMT)50. Additionally, Salvia miltiorrhiza and Carthamus tinctorius extract (SCE) treatment downregulated H3K4me3 and H3K36me3 at Smad3 promoter in cardiac fibroblasts and thus inhibited Smad3 transcription, which prevented myocardial fibrosis and adverse remodeling after myocardial infarction51. MM-102 or OICR-9429, both of which are MLL1/WDR5 protein–protein interaction inhibitors, could suppress the transcription of p16INK4a via decreasing H3K4me3, and attenuate kidney ischemia–reperfusion injury-related renal fibrosis and inflammation52. Meanwhile, sinefungin, a SET7/9 inhibitor were reported to reverse H3K4me1 modification and then suppressed TGF-β expression, which inhibited collagen deposition and ameliorated peritoneal fibrosis53. Therefore, the potential therapeutic effects of H3K79me2/3, H3K36me3, H3K4me1, and H3K4me3 on renal fibrosis, pulmonary fibrosis, cardiac fibrosis, and peritoneal fibrosis suggest a potential therapeutic avenue for “Activator” in treating liver fibrosis.
3.2. Histone acetylation in liver fibrosis
In the past decades, histone acetylation has been found to be involved in various diseases, including liver fibrosis (Fig. 4). The imbalance between acetylation and deacetylation of histone proteins, addition or removal of an acetyl group on histone lysine residue, is dominated by histone acetyltransferase (HAT) and histone deacetylases (HDACs)54. Additionally, bromodomain and extra-terminal (BET) proteins act as ‘readers’ and mediate histone acetylation by interacting with acetylated nucleosomes and transmitting acetylated lysine signals55. Histone acetylation has seemed as an active marker of gene transcription. By adding acetyl groups to lysine residues, the positive charge of histone was decreased and the binding between histone and negatively charged DNA was disturbed, leaving the underlying DNA exposed56. Chromatin is loosed into active transcription and the activities of transcription factors are upregulated after histone acetylation, which establishes a transcriptional connection between transcription factors and chromatin57. Several histone lysine residues could be acetylated, including H3K27, H3K9, H3K4 and H3K36 and so on. Hereafter, we mainly focus on well-investigated H3K27 and H3K9 acetylation and are committed to clarifying these potential modifications in liver fibrosis progression.
Figure 4.

Histone acetylation in liver fibrosis. This diagram represents the roles of histone acetylation in liver fibrosis. Acetylation of histone is mediated by a dynamic balance between HATs and HDACs. HATs, including P300, KAT2A and KAT8, regulate several fibrosis-related genes including Acta2, Ncf1 and Serpine1 by catalyzing H3K9ac and H3K27ac. HDACs are divided into 2 categories Zn2+-dependent HDACs and NAD+-dependent SIRTs. Several HDACs, including HDAC1, HDAC2, HDAC4, HDAC7, HDAC9 and SIRTs, including SIRT1, SIRT3, SIRT4 and SIRT6, are involved in fibrosis process via targeting TGF-β-Smads pathway, EMT, oxidative stress, cell senescence and HIF/β-catenin pathway.
3.2.1. Histone acetylation mediated by HATs
HATs, which catalyze histone acetylation, can be divided into three categories: p300/CBP, Gcn5-related N-acetyltransferases (GNATs) superfamily and MYST proteins58. P300/CBP proteins could stimulate histones acetylation at the enhancer and promoter regions of target genes and act as a transcription coactivator by cooperating with several transcription factors, including NF-κB, AP1 and STAT. GNAT superfamily includes PCAF, Gcn5, Elp3, Hpa2 and Hat1, and MYST superfamily is composed of Esa1, Sas2, Sas3, Tip60, MOF, MOZ, MORF and HBO1. These HATs are illustrated to be involved in multiple diseases, while, to be more specific, p300 (also called KAT3B), CBP (also known as KAT3A), Gcn5 (also called KAT2A), MOF (also called KAT8) and PCAF (also called KAT2B) were closely associated with the progression and development of fibrosis59.
P300, a transcriptional coactivator, initiated transcriptional regulation to promote the function of hepatic cells in liver fibrosis. As a direct target of TGF-β, p300 mediated transcriptional activation of fibrotic genes, including Acta2, Fn, and Col1a1, by interacting with Smad3 in fibroblasts60. Further study demonstrated that p300 interacted with Smad3 and thus enhanced TGF-β-induced EMT in hepatocytes61. The binding partners of p300 include transcription factor NF-κB and epigenetic reader protein bromodomain containing 4 (Brd4). The complex of p300-NF-κB-Brd4 is involved in pathological inflammatory in liver fibrosis. Indeed, Brd4 directly bonded with p300 and recruited NF-κB to the acetylated histone lysine, contributing to the formation and function of this acetyltransferase complex62. During fibrosis, p300 and its binding partners, NF-κB and Brd4, are required for the transcription of inflammatory factor C–C motif Chemokine 2 (Ccl2) in CCl4-stimulated liver sinusoidal endothelial cells (LSECs). Released from LSECs, Ccl2 subsequently recruited inflammatory cells, including CCR2+ monocyte/macrophage, into the sinusoids. These inflammatory cells activated HSCs by releasing factors such as TNF-α and TGF-β, which further promoted portal hypertension and liver fibrosis. Mechanically, chromatin immunoprecipitation showed that p300 upregulated Ccl2 expression by catalyzing H3K27ac at the Ccl2 promoter. Furthermore, Gao and his colleagues established LSEC-specific p300 deletion mice and further confirmed that p300 absence inhibited portal hypertension and hepatic fibrosis via reducing the hepatic accumulation of CCR2-positive monocyte/macrophage63. Previous studies also demonstrated the reliance of serum response factor (SRF), a transcription factor in myofibroblast transdifferentiation, on p300 for promoter binding. More importantly, SRF also interacted with KAT8 (known as MOF) and thereby recruited KAT8 to promoters of several genes, including NCF1 and NCF2. As members of NADPH oxidase (NOX), NCF1 and NCF2 promoted ROS production and HSC activation by organizing the NOX2 complex. Following HSC-specific SRF knockout, the reduction of H3K27ac and H4K16ac signatures was observed on the NCF1 and NCF2 promoters, leading to NCF1 and NCF2 transcriptional repression. Consistently, KAT8 knockdown by siRNA significantly downregulated pro-fibrogenic genes, including Acta2 and Col1a1, by decreasing Ncf1 and Ncf2 expression in primary mouse HSCs and LX-264.
H3K9ac was mediated by GNAT family members lysine acetyltransferase 2A (KAT2A) and KAT2B (also known as GCN5 and PCAF, respectively). As a master regulator of fibrosis, TGF-β directly activated HSCs and stimulated cholangiocyte transformation into reactive and secretory states, and therefore facilitating cholangiocyte-HSCs crosstalk. In TGF-β-stimulated cholangiocytes, the transcription of HSCs activators Fn and Serpine1 were promoted due to the enrichment of H3K9ac in promoters of these genes. Chromatin immunoprecipitation assay illustrated that KAT2A, which is predominantly expressed in cholangiopathies, was recruited to and acetylated H3K9 near the promoters of Fn and Serpine1 with the help of Smad3 and thereby promoted TGF-β-stimulated genes transcription in cholangiocytes. Subsequently, Fibronectin and SERPINE1 were released by cholangiocytes and swallowed by HSCs, leading to HSCs activation and biliary fibrosis in Mdr2−/− mice65.
3.2.2. Histone deacetylation mediated by HDACs
Opposite to HATs, histone deacetylation, catalyzed by HDACs, leads to gene repression via inducing chromatin compaction and subsequently preventing the binding of transcription factors66. To date, HDACs are divided into two categories and four families: Class I HDACs, including HDAC1, HDAC2, HDAC3, and HDAC8; Class II HDACs, including HDAC4, HDAC5, HDAC7, HDAC9, HDAC6, HDAC10; Class III HDACs, including SIRT1–7; and Class IV HDACs, including HDAC1167. Among these HDACs, Class I HDACs are predominantly expressed and located in nucleus, while Class II HDACs are located in cytoplasm and transferred to nucleus in response to cellular signals. Class III HDACs are different from other HDACs due to their requirement of cofactor nicotinamide adenine dinucleotide (NAD)+ in catalyzing histone deacetylation. Finally, HDAC11 is the sole member of Class IV localizing in the nucleus and uniquely shares sequence homology with the catalytic domains of both Class I and Class II. Numerous studies proposed the complex changes of HDACs in liver fibrosis. For example, HDAC1 and HDAC2 were significantly increased in early stages of HSC activation, and the upregulation of HDAC8 was found at later time points of HSC activation68. HDAC9 and HDAC10 were downregulated in HSC transdifferentiation, while HDAC4 and HDAC7 remained constantly expressed68. Additionally, recent studies illustrated that HDAC1, 2, 4, 5, 6, 8, 9 were increased during liver fibrosis, yet HDAC2, 6, 8 were decreased accompanied with increased HDAC11 during fibrosis resolution. Inconsistent understandings of the pathogenic roles of HDACs-mediated deacetylation hindered the discovery of novel therapeutic approaches targeting HDACs in liver fibrosis.
3.2.2.1. Zinc-dependent HDACs
The Zn2+-dependent HDACs were contained in Class I, Class II, and Class IV and involved in liver fibrosis progression and resolution. Many researchers have illustrated the regulation of histone deacetylation and further clarified the fundamental mechanism of Zn2+-dependent HDAC in the progression of liver fibrosis by integrating previous studies68. The hallmark of fibrotic lesions is constitutive TGF-β signaling and HDACs are involved in liver fibrosis by interfering with TGF-β-related genes. Accompanied with H3K9ac hyperexpression, miR-29a overexpression inhibited HDAC4 expression and its nuclear translocation and thereby improved TGF-β-stimulated HSC activation in LX-269. During HSC activation, HDAC4 was significantly enriched in the promoter regions of MMPs, enzymes responsible for collagen degradation, and inhibited their expression by repressing the recruitment of transcription factors, including c-JUN and NF-κB, resulting in abnormal collagen accumulation70. HDAC9 was also involved in the transcriptional suppression of TGF-β targets genes, including α-SMA and collagen, which was illustrated by introducing HDAC9 knockdown by siRNA in LX-271. Smad7 was a negative regulator of the TGF-β/Smads signaling pathway, and HDAC2 contributed to TGF-β-stimulated HSC activation via participating in Smad7 transcriptional repression72. Glioma-associated oncogene homolog 1 (Glil) are the profibrotic downstream of TGF-β. HDAC1 combined with lamina-associated polypeptide 2a (LAP2) and enriched in the promoter of Glil, leading to Glil hyperexpression and subsequent HSC activation73. Persistent inflammation almost always precedes liver fibrosis and TLR4 activates NF-κB and subsequently stimulates proinflammatory cytokines though adaptor protein MyD88. As mentioned above, BAMBI is a negative regulator in the TGF-β signaling pathway. HDAC1 interacted with NF-κB to form a transcriptional repression complex in the BAMBI promoter and thereby upregulated TGF-β signaling by decreasing BAMBI expression74. Additionally, hepatocyte growth factor (HGF) was regarded as an antifibrogenic mediator and significantly suppressed the TGF-β signaling pathway by inhibiting latent TGF-β1 activation. HDAC7 was enriched in promoter region of HGF and epigenetically repressed HGF expression and thus promoting NF-κB-stimulated expression of proinflammatory genes MCP-1 and IL-675.
3.2.2.2. NAD-dependent HDACs
Class III HDACs, also known as sirtuins (SIRTs), are localized to several subcellular compartments, including the nucleus (SIRT1, 2, 3, 6 and 7), cytoplasm (SIRT1 and 2), and mitochondria (SIRT3, 4 and 5), and SIRTs-mediated deacetylation can target both histones and non-histone proteins with the synergy of NAD. Owing to their dependence on NAD as the major substrate, SIRTs are linked with energy-sensing-related degenerative disorders. Indeed, SIRTs were reported to be involved in multiple organ fibrosis, especially liver fibrosis, through multiple mechanisms, including EMT, oxidative stress, cell senescence and HIF-β-catenin.
Increasing evidence demonstrated that SIRT1, SIRT3, and SIRT6 attenuated liver fibrosis by influencing the TGF-β pathway76,77. SIRT1 overexpression significantly blocked liver fibrosis by stimulating apoptosis and inhibiting the proliferation of activated HSCs, and SIRT1 knockdown enhanced CCl4-induced liver fibrosis76,78. Under TGF-β stimulation, Smad2/3 are phosphorylated and further bind to SMAD4 to form a complex, and the Smad2/3/4 complex triggers downstream genes transcription via relocating to the nucleus. SIRT1 decreased Smad3 expression by stimulating H3K9 deacetylation at Smad3 promoter and thereby inhibited the Smad2/3/4 complex formation and subsequent HSC activation79. Recent studies further demonstrated that SIRT1 is involved in the suppression of TGF-β-induced myofibroblast transdifferentiation by deacetylating EZH2 and thus reducing the stability of EZH2. In CCl4-and TGF-β-induced liver fibrosis, the variation in SIRT1 levels exhibited an opposite trend to the expression levels of EZH2. Mechanically, EZH2 acetylation at lysine 348 increased its stability and enhanced the PRC2 compound formation. SIRT1 disrupted EZH2 stability and played the hepatoprotective role through deacetylation. The SIRT1–EZH2 pathway may represent an attractive therapeutic target, suggesting the cooperative relationship between histone methylation and acetylation in liver fibrosis80. SIRT3 overexpression increased expression of glycogen synthase kinase 3β (GSK3β), a negative regulator of TGF-β1, and blocked TGF-β1 signaling, attenuating liver fibrosis in CCl4-induced mice. In detail, GSK3β was critically acetylated at residue K5, which negatively regulated GSK3β activity in TGF-β-stimulated HSC. SIRT3 is directly bound to and deacetylated GSK3β, thus reversing its activity and decreasing its downstream Smad3 expression81. SIRT6 was markedly decreased in patients as fibrosis progressed to cirrhosis, culture-activated and TGF-β-induced primary HSCs. SIRT6 stimulated H3K9 deacetylation at the Smad3 promoter and thereby inhibited Smad3 transcription. Meanwhile, SMAD3 acetylation was concurrently depended on and promoted by TGF-β-induced phosphorylation. SIRT6 also downregulated SMAD3 expression and thus alleviated liver fibrosis by directly deacetylating SMAD3 at key lysine residues K333 and K37882. Interestingly, recent studies have reported that SIRT6 may be an endogenous negative feedback regulator of TGF-β/Smad3. Lysine K333 and K378 of Smad3 were not necessary for SMAD3 to influence SIRT6, illustrating the coexistence of bidirectional regulation between SMAD3 and SIRRT6. SIRT6 limited TGF-β-stimulated fibrotic changes, while Smad3 formed a complex with SPTBN1 and bound to the promoter region of SIRT6, promoting SIRT6 transcription83. In addition to Smad3, recent studies illustrated that Smad2 acetylation plays a role in TGF-β-induced HSC activation. Lysine 54 in MH1 domain of Smad2 was the major position for phosphorylation, and acetylation of lysine 54 was required for phosphorylation and nuclear localization of Smad2. SIRT6 significantly inhibited Smad2 lysine 54 acetylation and subsequently alleviated TGF-β/Smads signaling pathway84. SIRT6 was also reported to interfere with Hippo pathway and acted as an anti-fibrosis factor. In cholestatic fibrosis, excess toxic bile acids may activate TGF-β/Smads and Yes-associated protein (YAP) signaling. YAP and transcriptional coactivator with PDZ-binding motif (TAZ) were implicated in Hippo pathway activation and liver fibrosis. SIRT6 downregulated the expression of YAP and TAZ by stimulating deacetylation, resulting in hepatic fibrosis-genes repression and liver fibrosis inhibition85. Furthermore, under pathological conditions, estrogen-related receptor γ (ERRγ) was increased and aggravated liver fibrosis by continually stimulating bile acid production. SIRT6 activation ameliorated BDL-induced cholestatic fibrosis by deacetylating orphan nuclear receptor ERRγ and inhibiting its transcriptional activity86.
Under oxidative stress, continuous ROS accumulation contributes to the activation of SIRTs, while SIRTs, including SIRT1, SIRT3 and SIRT6, are demonstrated to alleviate liver fibrosis via negatively regulating oxidative stress. In CCl4-stimulated rat fibrosis, oxidative stress induced the loss of fenestration in hepatic sinusoidal endothelial cells and promoted fibrogenesis. The overexpression of SIRT1 significantly reversed these processes and thus exerting therapeutic potential87. Furthermore, excessive oxidative stress is strongly associated with aging and exacerbates cellular dysfunction, tissue failure and fibrosis. SIRT1 was found downregulated in alcoholic-induced liver injury and fibrosis associated with aging and enhancing SIRT1 activity has yielded prominently prospects for new therapies88. HSCs isolated from middle-aged mice expressed lower levels of SIRT1 protein and were more susceptible to spontaneous activation in vitro89. EMT refers to a process wherein epithelial cells transform into mesenchymal cells, promoting liver fibrosis, which is also closely related to oxidative stress. Recently, EMT have been suggested to be the downstream event of SIRT1 regulation. Enforced expression of SIRT1 inhibited superoxide dismutase 3 (SOD3) deficiency-induced hepatocyte EMT and HSC activation, and depletion of SIRT1 counteracted the inhibitory effect of SOD3 in vitro90. Paradoxically, the activation of SIRT1/p53 signaling is required for TGF-β stimulated hepatocyte EMT, which contributed to liver fibrosis in rat91. In addition to liver fibrosis, SIRT1 could enhance SOD2-dependent anti-oxidative mechanism and inhibited high glucose-induced cardiac fibrosis by reducing ROS accumulation92. SIRT1 also increased eNOS level and therefore alleviated UUO-induced renal fibrosis93, providing the possibility for the therapeutic roles of SIRT1 in the liver.
SIRT3 blocked ROS-induced HSC transdifferentiation and ECM deposition by inhibiting oxidative stress. In detail, SIRT3 enhanced superoxide dismutase 2 (SOD2) activity through deacetylation and decreased NOX activity, which prevents the expression of fibrosis-related genes, including Acta2 and Fn94. Therefore, Withaferin A95 and γ-mangostin96 acted as SIRT3 agonists and thereby prevented CCl4-induced liver fibrosis via inhibiting oxidative stress. Meanwhile, SIRT6 overexpression also protected CCl4-induced liver fibrosis in vivo and SIRT6 knockout mice spontaneously developed liver injury, as characterized by remarkable increase of oxidative stress and inflammation97. Canthaxanthin has been illustrated to increase SIRT6 expression to alleviate CCl4-induced liver fibrosis98.
These SIRTs have also been illustrated to influence β-catenin-associated pathway, major pathways involved in fibrosis responses. Empagliflozin alleviated thioacetamide-induced hepatotoxicity and liver fibrosis via increasing SIRT1 and thereby downregulating HIF-1α/β-catenin axis99. SIRT1 deficiency promoted fibrosis and inflammation factors in glomerular mesangial cells via promoting HIF-1α and HIF-2α expression100,101. In addition to liver fibrosis, β-catenin target genes (Fsp1 and Axin2) are upregulated by TGF-β, and SIRT6 prevented β-catenin transcription and thus inhibited fibrosis-related genes via binding with Ctnnb1 promoter and thereby stimulating H3K56 deacetylation in tubular epithelial cells102. In Ang–II-induced cardiac fibrosis and TGF-β-stimulated renal fibrosis, SIRT1 and SIRT3 also alleviated fibrosis responses by catalyzing H3K56 deacetylation and epigenetically downregulating β-catenin expression103,104. Thus, further investigations on how SIRTs aggravate liver fibrosis by affecting β-catenin-associated pathways are urgently required.
However, unlike other members of SIRTs, SIRT2 is considered as a promoter in multi-organ fibrosis progression and inhibition of SIRT2 is demonstrated as an effective anti-fibrosis strategy. SIRT2 was abnormally increased in human fibrotic liver tissues and SIRT2 knockout downregulated fibrosis-related genes, including α-SMA and collagen105. SIRT2 activated the extracellular regulated protein kinases (ERK) pathway by stimulating ERK protein deacetylation. Thus, the degradation of c-MYC, the downstream target of ERK, was decreased, leading to c-MYC hyperexpression and fibrogenesis105. In idiopathic pulmonary fibrosis, SIRT2 inhibition, mediated by inhibitor AGK2 or siRNA, suppressed expression of Acta2 and Fn via downregulating TGF-β/Smad3106. While the underlying mechanism of SIRT2 deficiency in anti-fibrosis is still unclear, and the cause of its inconsistent effects with other SIRTs needs to be revealed.
4. RNA modification in liver fibrosis
As both targets and enactors of expression change, RNA, including mRNA, microRNA (miRNA), and long ncRNA (lncRNA), modulate gene expression through epigenetic mechanisms in liver fibrosis (Fig. 5). miRNA forms a RNA-induced silencing complex through interaction with argonaute, leading to the destabilization of the 3′ UTR in the target mRNA and subsequent inhibition of translation107. LncRNA regulates gene expression via recruiting histone-modifying enzymes to the chromatin, including HMT, HDMT, HAT and HDAC. Most recently, the field of mRNA epigenetic modifications has gained significant attention, with a particular focus on N6-methyladenosine (m6A) RNA modification. This modification is known to modulate gene expression by impacting mRNA nuclear export, translation, as well as the splicing and stability of ncRNAs108.
Figure 5.

Scheme diagram of RNA modification in fibrosis. Mounting evidence shows that RNA modification plays a vital role in the fibrosis process. Several miRNAs, including miR-152, miR-29b and miR-125b-5p, promote fibrosis by interacting with DNA methyltransferases and histone deacetylases. LncRNAs, including lncRNA-SCARNA10, lncRNA-HOTAIR, lncRNA-NEAT1, and lncRNA-GAS5, mainly regulate the PRC2 family. Several circRNAs, including hsa_circ_0007874, hsa_circ_0070963, circFBXW4, mmu_circ_34116, circ_PSD3 and circ_PWWP2A inhibited HSC activation via combining with miRNA and thus reversed miRNA-mediated transcription suppression.
4.1. mRNA methylation in liver fibrosis
m6A modification is a dynamic process regulated by methyltransferases, demethylases and m6A binding proteins, serving as a post-transcriptional mechanism in regulating mRNA stability109. The WT1-associated protein (WTAP), methyltransferase like 3 (METTL3), METTL4 and METTL14 all function as methyltransferases, while demethylases mainly include alkB homolog 5 (ALKBH5) and obesity-associated protein (FTO). Following methylation, modified mRNAs will be recognized and bound by m6A binding proteins, such as heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), YTH-domain-containing protein 1/2 (YTHDC1/2) and YTH-domain-family 1/2/3 (YTHDF1/2/3), and the cytosol transportation and translation of these mRNAs will be greatly regulated.
RNA m6A methylation has recently been reported to play a regulatory role in the activation of HSCs and then promote the progression of liver fibrosis. Yang and his colleagues110 established a spontaneous NASH, liver fibrosis, and HCC in type 2 diabetes mellitus (DM2) mouse model and confirmed that METTL3/14 overexpression promoted the production of triglyceride and cholesterol and ECM accumulation via increasing the protein levels of ATP citrate lyase (ACLY) and stearoyl-CoA desaturase1 (SCD1). m6A sequencing analysis confirmed that METTL3/14 overexpression bonded and stabilized mRNA of ACLY and SCD1 through m6A modification. Detailly, the coding sequence (CDS) of ACLY and 3′ UTR of SCD1 showed the relatively higher m6A enrichment, which was confirmed by luciferase reporter assay110. The upregulation of methyltransferases METTL3 and METTL14 and global RNA hypermethylation were also observed in HFD-stimulated NASH and LPS-activated KCs. In active-KCs, NF-κB p65 directly promoted the expression of METTL3 and METTL14 by binding to their promoter. Upregulated METTL3 and METTL14 then increased m6A modification in the TGF-β mRNA 5′UTR with the help of reader protein YTHDF3 and subsequently promoted cap-independent translation of TGF-β. This finding was further verified in METTL3/14 conditional knockout KCs111.
Recently, it is found that ferroptosis, a novel form of programmed cell death, functions as a two-edged sword in liver fibrosis. It has been reported that excessive hepatic iron deposition aggravates acetaminophen-induced liver fibrosis. While other studies indicated that ferroptosis could be considered as a new strategy to eliminate activated HSCs and regulate autophagy. Under the stimulus, different types of selective autophagy increased, including nuclear receptor coactivator 4 (NCOA4)-dependent ferritinophagy, RAB7A-dependent lipophagy, and p62-dependent clockophagy and chaperone-mediated autophagy, promoting ferroptosis via stimulating iron accumulation and lipid peroxidation112. Therefore, molecules triggered HSC ferroptosis might be able to alleviate liver fibrosis by regulating autophagy-related targets. Min and his colleagues identified that m6A reader YTHDF1 increased the mRNA stability of BECN1, an autophagy-related gene, by recognizing its coding region and thus triggering autophagy activation and ferroptosis in HSC. Recognized ferroptosis inducer has been demonstrated to promote ferroptosis-related elimination of activated-HSC by increasing YTHDF1 expression113.
4.2. ncRNA-mediated epigenetic modification in liver fibrosis
Recently, emerging evidence hint at a significant role for ncRNA in liver fibrosis and increasing emphasis is being placed on the role of ncRNA as epigenetic regulators114. A total of 32,461 differentially expressed ncRNAs have been identified by comparing fibrotic and normal liver samples in NASH patients, suggesting a strong link between ncRNA expression and fibrosis and a heightened risk of cirrhosis115. Consistently, more than 3600 differentially expressed ncRNAs were described during human HSC quiescence and activation115 and 381 ncRNAs were recognized in HSC under the conditions of TGF-β stimulation1. Indeed, gene expression is regulated not only by proteins but also by ncRNAs. Among them, miRNA and lncRNA are the most well-studied ncRNA. During fibrogenesis, the relationship between miRNA and lncRNA HSC activation has been widely disclosed (Table 2).
Table 2.
ncRNAs involved in the regulation of liver fibrosis.
| ncRNA | Target | Pathway or regulatory mechanism | Output | Ref. |
|---|---|---|---|---|
| miRNA | ||||
| miR-542-3p | BMP-7 | Decreases BMP-7 expression | Promotes liver fibrosis | 116 |
| miR-125b | RhoA | Activates RhoA pathway | Activates HSC | 117 |
| miR-199 | TGF-β | Activates TGF-β pathway | Promotes liver fibrosis | 118 |
| miR-200 | TGF-β | Activates TGF-β pathway | Promotes liver fibrosis | 118 |
| miR-21 | SMAD7 | Increases SMAD7 expression | Activates HSC | 119 |
| miR-129-5p | Collagen I | Reduces collagen I expression | Promotes liver fibrosis | 120 |
| miR-454 | Collagen I α-SMA |
Reduces collagen I and α-SMA expression | Promotes cirrhosis progression | 121 |
| miR-378-3p | Gli3 | Reduces Gli3 expression | Inhibits HSC activation | 122 |
| miR-222 | ICAM-1 | Decreases ICAM-1 expression | Promotes HSC proliferation and differentiation | 123 |
| miR-185 | SREBF1 | Increases SREBF1 expression | Promotes liver fibrosis | 124 |
| LncRNA | ||||
| H19 | Let-7 ZEB1 |
Increases expression of let-7 and ZEB1 | Promotes cholestatic liver fibrosis | 125,126 |
| MEG3 | SMO | Decreases SMO expression | Inhibits EMT in liver fibrosis | 127 |
| GAS5 | miR-222 | Competes with miR-222 and | Inhibits HSC activation and proliferation | 128 |
| Gm5091 | miR-27b/23b/24 | Increases TGF-β by sponging to miR-27b/23b/24 | Alleviates alcoholic hepatic fibrosis | 129 |
| Lnc-LFAR1 | Smadd2/3 phosphorylation | Promotes Smadd2/3 phosphorylation | Promotes liver fibrosis | 130 |
| NEAT1 | miR-122 miR-29b |
Increases miR-122 and miR-29b | Promotes liver fibrosis progression | 131 |
4.2.1. miRNA-directed epigenetic modification
miRNAs regulate target expression by affecting transcription and mRNA stability, thus modifying DNA methylation and histone modification in liver fibrosis. A recent study reported that some miRNAs stimulated DNA hypomethylation state and therefore decreased fibrogenic activities by downregulating methyltransferases, including DNMT1 and DNMT3b115. Increased miR-152-stimulated by salvianolic acid B directly decreased DNMT1 expression via binding to the 3′UTR of DNMT1. Therefore, PTCH1, a negative regulatory factor of the fibrosis-related Hedgehog pathway, underwent DNA hypomethylation and hyperexpression due to the absence of DNMT1. DNMT1 knockout and miR-152 inhibitor were consistently demonstrated to inhibit EMT in liver fibrosis132. Additionally, miR-29b was decreased in liver fibrosis associated with chronic HBV infection. While curcumin upregulated miR-29b and thus led to the suppression of activated HSCs. In detail, miR-29b directly downregulated DNMT3b expression, contributing to hypomethylation of phosphatase and tensin homolog deleted on chromosome (PTEN) DNA, as illustrated by luciferase activity assays133. Interestingly, epigenetic silencing of miRNA stimulated by DNA methylation also plays an important role in the miRNA-directed regulation of liver fibrosis. It has been observed that the CpG island in miR-125b-5p promoter was hypermethylated, and thus its expression was suppressed in NAFLD livers and hepatocytes. Cai et al.134 established the NAFLD mouse model and demonstrated that miR-125b-5p absence promoted liver fibrosis in NAFLD via activating RhoA and ITGA8. It is worth noting that some miRNAs act as the upstream factor of DNMTs and serve as the regulator of histone modification enzymes. miR-455-3p was reported to reduce the expression of profibrotic genes by binding to the 3′UTR of HDAC2 and upregulating the HDAC2 expression in activated HSCs72. Similarly, the interaction between miR-29b and HDAC4 in diabetic nephropathy was illustrated by luciferase assay. MiR-29b targeted 3′ untranslated region of HDAC4 and subsequently attenuated HDAC4 expression and renal fibrosis135. Overall, these studies indicate the involvement of miRNAs in epigenetic regulation in liver fibrosis, while the underlying mechanisms and specific targets have only been explored in a few studies. Further investigations on how varied miRNAs regulate liver fibrosis by affecting multifarious epigenetic modifications are still urgently required.
4.2.2. LncRNA-directed epigenetic modification
Accompanied by liver fibrogenesis, lncRNAs are involved in the promotion or suppression of liver fibrosis via mutual interaction with miRNAs and proteins. LncRNAs have been illustrated to interact with miRNAs as ‘sponges’ or competing endogenous RNA (ceRNA), thus altering the expression and function of miRNAs. Meanwhile, the majority of lncRNAs can interact with RNA-binding proteins (RBPs) to form ribonucleoprotein (RNP) complex, medicating mRNA stability, translation and post-translation. Notably, lncRNAs are involved in epigenetic regulation through interactions with epigenetic enzymes and its-associated miRNAs. It has been illustrated that lncRNA-ACTA2-AS1 contributed to ductular reaction and paracrine HSC activation by interacting with H3K27 acetyltransferase p300 and transcription factor Elk1. In detail, lncRNA-ACTA2-AS1 combined with Elk1 and subsequently guided p300 to the promoters of DR and fibrosis-related genes, including Platelet-derived growth factor β (Pdgfb), Acta2, Fn and Serpine1, which then catalyzed H3K27ac at binding sites for Elk1 (CCGGAA) and thus promoted the transcription of these genes in cholangiocytes136. As a typical lncRNA, homeobox transcript antisense RNA (HOTAIR) expression was upregulated in HSCs. Meanwhile, HOTAIR strikingly contributed to HSC activation and upregulated the expression of α-SMA and collagen I by promoting DNA methylation of PTEN and suppressing PTEN expression in vitro and in vivo. Detailly, HOTAIR, a ceRNA of miR-29b, downregulated miR-29b expression and attenuated its suppression on DNMT3b in HSCs. Restored DNMT3b subsequently catalyzed DNA methylation of PTEN and decreased its transcription, which was illustrated by HOTAIR knockdown mice137. Furthermore, HOTAIR was demonstrated to stimulate myofibroblast activation by interacting with EZH2 in systemic sclerosis characterized by vascular fibrosis. Wasson and his colleagues illustrated that upregulated HOTAIR inhibited miR-34a, functions as a Notch1 suppressor, and thus activated classical pro-fibrosis targets Notch signaling in fibroblasts. Mechanically, HOTAIR guided EZH2 to the promoter of miR-34a to catalyze H3K27me3 and thereby suppressed miR-34a transcription and increased downstream Notch1 expression, which was reversed by EZH2 inhibitor GSK126138. In addition to EZH2, other members of the PRC2 family, such as SUZ12 polycomb repressive complex 2 subunit (SUZ12), has been demonstrated to be involved in liver fibrosis through the interaction with various lncRNAs. LncRNA-SCARNA10 was increased in the serum and liver of patients with advanced hepatic fibrosis and functioned as a positive regulator of the TGF-β signaling pathway. Mechanically, LncRNA-SCARNA10 combined with SUZ12 and EZH2 and subsequently guided them away from the promoters of genes, including Tgfb, Smad2, Smad3 and Col1a1. Accompany with the unbinding of methylases, H3K27me3 was reversed and thus, the genes transcription was restarted, which promoted liver fibrosis progression139. In addition, epigenetic regulation mediated by lncRNAs through direct interaction with PRC2 family members were observed in fibrogenesis of other different organs, inspiring the investigation of these mechanisms in liver fibrosis. Similarly, lncRNA-NEAT1 promoted cardiac fibrosis by recruiting EZH2 to the promoter region of Smad7 and subsequently inhibiting Smad7 expression140. In diabetic nephropathy-related renal fibrosis, lncRNA-GAS5 recruited EZH2 to the Mmp9 promoter region and inhibited the Mmp9 expression, alleviating renal fibrosis progression141. Collectively, these results have verified that lncRNAs have great potential in guiding PRC2-mediated gene-specific epigenetic modification and targeting the complex of specific lncRNA and PRC2 may provide a promising therapeutic target against liver fibrosis, which is more selective than directly targeting the catalytic activities of PRC2.
4.2.3. Other ncRNA-directed epigenetic modification
Indeed, in addition to miRNA and lncRNA, circular RNA (circRNA) and piwi-interacting RNA (piRNA) may also play vital important roles in epigenetic modifications by affecting miRNA and lncRNA or directly affecting gene transcription. circRNAs are characterized by a covalently closed continuous loop and act through miRNA sponges or ceRNA mechanisms142. circRNA, named hsa_circ_0007874, increased PTEN transcription and thus inhibited HSC activation. Detailly, luciferase reporter assay and pull-down assays illustrated that hsa_circ_0007874 bound with miR-181b-5p in cytoplasm of HSC and further suppressed the inhibition of miR-181b-5p on PTEN143. As a miR-223-3p sponge, hsa_cir_0070963 bound with miR-223-3p and thus suppressed the inhibition effects of miR-223-3p on LEM domain containing 3 (LEMD3), which inhibited HSC activation in hepatic fibrosis144. Circ F-Box And WD Repeat Domain Containing 4 (CircFBXW4) was downregulated in liver fibrogenesis and was illustrated to bind to miR-18b-3p in primary mouse HSCs. Enforcing expression of circFBXW4 inhibited HSC activation by reducing the suppression of miR-18b-3p on FBXW7145. Similarly, mmu_circ_34116 inhibited HSC activation by binding with miR-22-3p and targeting miR-22-3p/BMP7 axis146. CircPSD3 bound with miR-92b-3p and thus suppressed its inhibition on Smad7, which alleviating hepatic fibrosis147. Meanwhile, circ-PWWP2A was significantly upregulated in TGF-β and LPS-stimulated HSC and was suggested to promoted HSC activation and proliferation by sponging miR-203 and miR-223 and increasing follistatin-like 1 (FSTL1) and TLR4148. Recently, a circRNA microarray identified that circRNA-0067835 promoted HSC activation by binding with miR-155 and reversing FOXO3 expression in thymosin beta 4 (Tβ4)-stimulated HSC149. Regarding piRNAs, Xue and his colleagues isolated primary HSCs from carbon tetrachloride and bile duct ligation-stimulated mice and demonstrated that piR-823 increased TGF-β expression and activated HSCs via binding with eukaryotic initiation factor 3B (EIF3B)150. Collectively, ncRNA network is more than just the interaction of cirRNAs and piRNAs with miRNA. Investigating the further connections among different ncRNAs could be a promising research direction in the future.
5. Promising therapeutic directions and challenges ahead
Based on all the evidence summarized above, it is not difficult to observe that many studies have clarified the epigenetic mechanisms involved in the pathogenesis of liver fibrosis. However, therapeutic strategies targeting epigenetic regulation to treat liver fibrosis have yet to be developed. Among epigenetic targets, specific inhibitors of EZH2, including DzNep, GSK126, GSK926, and GSK343 have been reported (Table 3). The first EZH2 inhibitor DzNep, an S-adenosyl-L-homocysteine (SAH) hydrolase inhibitor, inhibits EZH2 via repressing SAM-dependent histone methyltransferase activity151. Subsequently, highly selective SAM competitive inhibitors including GSK926 and GSK343 have been generated, which own a 2-pyridone core structure152,153. Among these inhibitors, GSK126 is demonstrated to be highly selective when compared with other inhibitors and its safety was evaluated by a phase 1 clinical trial.
Table 3.
Small molecules with EZH2-inhibiting activity.
| EZH2 inhibitor | Disease | Fibrosis-related output | Status | Ref./Identifier |
|---|---|---|---|---|
| DzNep | Tumor | Inhibits HSC cell cycle arrest | Drug discovery | 34 |
| GSK126 | Lymphomas, multiple myeloma | Inhibits hepatocyte autophagy and HSC activation | I clinical trial | 154 |
| GSK503 | Tumors | Inhibits TGF-β pathway | Drug discovery | 155 |
| EPZ-6438 | Lymphomas, advanced solid tumors | Induces HSC G2/M arrest and apoptosis | II clinical trial | 156 |
| MC4343 | Tumors | _ | I clinical trial | NCTO3854474 |
| MC4355 | Tumors | _ | II clinical trial | NCTO3213665 |
| CPI-1205 | B-cell lymphoma | _ | II clinical trial | NCT02395601 |
| CPI-360 | Tumors | _ | I clinical trial | NCTO2860286 |
| CPI-169 | Tumors | _ | Pre-clinical | WO2013120104A2 |
| EPZ005687 | Lymphoma, Solid tumors | _ | Pre-clinical | US20130317026A1 |
Indeed, histone methyltransferases play essential roles in the pathophysiology of cancer, thus, different types of specific inhibitors were initially developed for cancers. For example, the inhibitor of H3K36 methyltransferases SETD2, EZM0414, the inhibitors of H3K79 methyltransferases, including EPZ5676, SGC0946 and EPZ004777, and the inhibitors of KAT2A, YF-2, SR-18292 and CPTH2, have been demonstrated to be applied in antitumor treatments157. These inhibitors are gradually being evaluated in clinical trials and are promising to be used in further liver fibrosis therapeutics. Additionally, genetic approaches targeting methyltransferases were employed to alleviate liver fibrosis in basic studies, but no such candidate therapies have been demonstrated. For instance, G9a is responsible for the three methylation states of H3K9 and multiple studies confirmed that siRNA-G9a was the commonly used antifibrotic regimen158. While inhibitors of G9a have not yet been clearly characterized, suggesting that developing specific inhibitors of G9a may capture the windvane of further antifibrotic studies.
Notably, several pharmacological activators or inhibitors have been used to regulate histone acetylation, and HDAC enzyme inhibitors, including HNHA, Vorinostat and VPA, are a powerful group of chemotherapeutic in clinical (Table 4).
Table 4.
Small molecules with HDAC-inhibiting activity.
| HDAC inhibitor | Target | Disease | Fibrosis-related output | Status | Ref. |
|---|---|---|---|---|---|
| HNHA | Pan | Tumor | Improves hepatic function survival | FDA approval | 159 |
| Vorinostat | Pan | Cutaneous T-cell lymphoma, glioma | Inhibits TGF-β/SMAD signaling | FDA approve | 160 |
| VPA | Class I HDACs |
Epilepsies, Partial | Inhibits TGF-β and TNF-α signaling | FDA approve | 161 |
| Entinostat | Class I HDACs |
Breast cancer, lymphoma | Inhibits hepatocyte death and type 2 inflammation | III clinical trial | 162 |
| BRD4884 | Class I HDACs |
Memory disorders | Inhibits hepatocyte death and type 2 inflammation | Pre-clinical | 162 |
| NW21 | Class I HDACs |
Hematologic neoplasms, solid tumors | Inhibits hepatocyte death and type 2 inflammation | Drug discovery | 162 |
| Largazole | Pan | Tumor | Inhibits VEGF signaling | Drug discovery | 163 |
| Parthenolide | HDAC4 | Tumor | Downregulates TGF-β and upregulates CYP7A1 | Drug discovery | 164 |
| Valproate | Pan | Epilepsies | Inhibits HSC activation | Drug discovery | 165 |
| MC1568 | HDAC4/5/6 | Tumor | Inhibits HSC activation | Drug discovery | 166 |
It is not difficult to see the HDAC inhibitors are mainly broad-spectrum, targeting one class of HDACs or several HDACs, which hampers the development of specific anti-fibrosis drugs. Meanwhile, the roles of SIRTs, class III HDACs, are relatively consistent in diseases, and the SIRT activators and inhibitors have been identified (Table 5). At the same time, in anti-fibrosis studies, the plausible therapeutic or pathogenic roles of SIRTs are primarily verified by gene manipulation (overexpression or knockout). Thus, evaluating the antifibrotic activities of SIRTs regulators and developing specific SIRTs inhibitors instead of pan-HDACs inhibitors may become a feasible strategy.
Table 5.
Small molecules with SIRT-activating or SIRT-inhibiting activities.
| Compound name | Target | Disease | Fibrosis-related output | Status | Ref./identifier |
| SIRT activators | |||||
| Resveratrol | SIRT1, SIRT3, SIRT5 | Osteoarthritis | Increases IL-10 to reprogramme macrophages | III clinical trial | 167 |
| Sildenafil | SIRT1, SIRT3 | Huntington | Increases GSH and SOD and decreases TNF-α | II clinical trial | 168 |
| JP-022 | SIRT1, SIRT2, SIRT3 | Alzheimer's diseases | _ | Pre-clinical | WO2016028910A1 |
| SRT1460 | SIRT1 | Diabetes mellitus, type 2 | _ | Pre-clinical | CN103145738A |
| R-Cu | SIRT1 | Alzheimer diseases | _ | II clinical trial | CTRI/2019/07/020289 |
| SIRT inhibitors | |||||
| MDL-800 | SIRT6 | Hepatocellular carcinoma | Inhibits phosphorylation and nuclear localization of SMAD2 | Pre-clinical | 84 |
| EH-301 | SIRT5 | Acute kidney injury | Inhibits skin fibroblasts | II clinical trial | 169 |
| SP-624 | SIRT6 | Depressive disorder | _ | II clinical trial | NCT04510298, NCT04479852 |
| AGK2 | SIRT2 | Tumor | _ | Pre-clinical | 170 |
| Cambinol | Pan | Leukoencephalopathy, progressive multifocal | _ | Drug discovery | 171 |
| Ex-527 | SIRT1 | Tumor | _ | Drug discovery | 172 |
In recent decades, numerous epigenetic studies have demonstrated the potential of m6A demethylase, including ALKBH5 and FTO, as therapeutic targets and several inhibitors have been reported to regulate m6A in multiple diseases. Surprisingly, researchers have summarized the inhibitors of ALKBH5 and FTO and organized several detailed and comprehensive reviews173, 174, 175, 176. According to these reviews, available inhibitors of FTO and ALKBH5 are mainly 20G analogs or substrate-competitive inhibitors, and have been demonstrated to inhibit the proliferation of cancer cells, such as R-2HG, FB23-2, CS1, CS2 and NSC48890 in leukemia cells, MA2 in GSCs and MO-I-500 in breast cancer cells173. While only few inhibitors have been tested in fibrosis therapeutics. For example, ALKBH5 inhibitor IOX1 promoted m6A modifications of chemokine CCl28 mRNA and thus enhanced its stability. Therefore, IOX1 upregulated Treg recruitment and inhibited inflammatory cells via increasing CCl28 expression, which alleviated Ischemia–reperfusion injury-stimulated renal fibrosis mouse model177. It is noteworthy that radiation-induced ALKBH5 could inhibit toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) m6A methylation and thus activate NF-κB–Smad2 pathway to activate HSCs178. It may attract interest whether ALKBH5 inhibitors could disturb TIRAP–NF-κB–Smad2 pathway to suppress HSC activation and liver fibrosis for further studies. Collectively, further work is required to determine the anti-fibrosis effects of these antitumor m6A demethylase inhibitors in HSC activation and liver fibrosis mouse model.
Although molecular inhibitors of epigenetic-related enzymes are being evaluated and are expected to be used in liver diseases, it still faces considerable challenges. Currently, the molecular activators and inhibitors, including EZH2 inhibitors, HDAC inhibitors, SIRT activators and inhibitors are often used in cancer therapy, which has not been extended to liver fibrosis-related research. As aforementioned, the selectivity and specificity of these agonists and inhibitors need to be significantly improved. In addition, a more detailed understanding and analysis of epigenetic regulation mechanisms are required to ensure that the clinical use of these agonists and inhibitors reaches expected outcomes.
There may be therapeutic approaches by targeting the integrative effects between different epigenetic mechanisms, for instance, the interaction between DNA methylation and histone modification. Studies have demonstrated that combined effects of CpG binding protein Mecp2 and histone methyltransferase EZH2 are involved in PPARγ epigenetic repression and subsequent HSC activation. Mecp2 classically functions as a PPARγ transcriptional repressor by directly binding to the CpG island of PPARγ and thus mediating DNA methylation. Mann et al.179 established Mecp2–/y and EZH2 deletion mice and identified stimulation of EZH2 and subsequent H3K27me3 in the downstream exon of PPARγ gene as a second mechanism through which Mecp2 achieves transcriptional silencing. Yang et al.180 further reported polyphenolic rosmarinic acid reversed PPARγ expression and thus alleviated cholestatic fibrosis by reducing expression and recruitment of Mecp2 in PPARγ promoter and decreasing EZH2-mediated H3K27me3 at 3′exon, which suggested the possibility of Mecp2-EZH2 inhibition as effective anti-fibrosis strategies. In addition to the Mecp2-EZH2 targeting inhibitor, the simultaneous targeting of H3K9 methyltransferase G9a and DNA methyltransferase DNMT1 has been reported to alleviate liver fibrosis. During HSC activation, the serine–glycine metabolic pathway was indeed important for collagen synthesis, and TGF-β promoted HSC activation by decreasing gluconeogenic enzymes genes fructose-bisphosphatase 1 (FBP1) and phosphoenolpyruvate carboxykinase (PEPCK) and metabolic regulator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Mechanically, G9a and DNMT1 exited in a stable complex and colocalized at the promoter of several genes, including FBP1, PEPCK and PGC-1α. By forming a complex, G9a delivered transcriptional suppressor H3K9me2 signature and meanwhile, DNMT1 stimulated DNA methylation on these genes, which synergistically suppressed gene transcription. Thus, focused on the corporation between G9a and DNMT1, Marina and his colleagues observed that CM272, a novel/classic G9a/DNMT1 inhibitor, reversed the expression of FBP1, PEPCK and PGC-1α and thus inhibited the fibrotic phenotypes181. While several “Activator” histone methylation and active histone acetylation signatures have also been reported to interact with DNA methylation182,183. Overall, it remains unclear which factor—DNA methylation or histone modification—contributes more significantly to the outcome resulting from the interaction between the two. On the one hand, DNA methylation is the modification of gene sequence, which is more direct than histone modification in gene transcription regulation. On the other hand, histone modification is catalyzed by multiple enzymes and targets multiple sites of histones, providing more possibilities for regulating gene expression. The current understanding of these interaction effects is insufficient to support the development of therapeutic strategies. Further research is needed to deepen our knowledge in this area.
6. Conclusions
In summary, increasing evidence has comprehensively revealed the vital role of epigenetics in the progression of fibrosis, highlighting the complexity of underlying mechanisms and emphasizing the therapeutic potential of epigenetic modulation in the treatment of liver fibrosis. However, the findings are not fully consistent. Further characterization of the specificity of epigenetic modifications would offer novel perspectives for developing effective and highly selective epigenetic therapies and may represent a promising approach with bright prospects for the treatment of chronic liver diseases.
Author contributions
Xiaojiaoyang Li, Huiping Zhou, and Runping Liu conceived the original idea and supervised the study. Runping Liu and Yajing Li prepared the manuscript and figures. Qi Zheng and Mingning Ding revised figures. All data were generated in-house and no paper mill was used. All authors have approved the final manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
This study was supported by the National Key Research and Development Program on Modernization of Traditional Chinese Medicine (Grant No. 2022YFC3502104, China). This study also received supports from VA Merit Award 5 I01 BX005730 and National Institutes of Health Grant R01 R01DK-057543 to Huiping Zhou (USA), National High-Level Talents Special Support Program to Xiaojiaoyang Li (China), and Fundamental Research Funds for the Central Universities (Grant No. 2023-JYB-JBZD-046 to Xiaojiaoyang Li, China). Huiping Zhou is a VA Research Career Scientist (IK6BX004477, USA).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Huiping Zhou, Email: huiping.zhou@vcuheath.org.
Xiaojiaoyang Li, Email: xiaojiaoyang.li@bucm.edu.cn.
References
- 1.Xue T., Qiu X., Liu H., Gan C., Tan Z., Xie Y., et al. Epigenetic regulation in fibrosis progress. Pharmacol Res. 2021;173 doi: 10.1016/j.phrs.2021.105910. [DOI] [PubMed] [Google Scholar]
- 2.Li Y.J., Liu R.P., Ding M.N., Zheng Q., Wu J.Z., Xue X.Y., et al. Tetramethylpyrazine prevents liver fibrotic injury in mice by targeting hepatocyte-derived and mitochondrial DNA-enriched extracellular vesicles. Acta Pharmacol Sin. 2022;43:2026–2041. doi: 10.1038/s41401-021-00843-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Liu R., Wu J., Li X. Self-eating: friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics. 2020;10:7993–8017. doi: 10.7150/thno.47826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kisseleva T., Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151–166. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 5.Topper M.J., Vaz M., Marrone K.A., Brahmer J.R., Baylin S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17:75–90. doi: 10.1038/s41571-019-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- 7.Page A., Paoli P., Moran Salvador E., White S., French J., Mann J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J Hepatol. 2016;64:661–673. doi: 10.1016/j.jhep.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cullen S.M., Goodell M.A. Dynamic DNA methylation discovered during HSC differentiation. Cell Cycle. 2015;14:693–694. doi: 10.1080/15384101.2015.1006558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni X.X., Li X.Y., Wang Q., Hua J. Regulation of peroxisome proliferator-activated receptor-gamma activity affects the hepatic stellate cell activation and the progression of NASH via TGF-β1/Smad signaling pathway. J Physiol Biochem. 2021;77:35–45. doi: 10.1007/s13105-020-00777-7. [DOI] [PubMed] [Google Scholar]
- 10.Ratziu V., Harrison S.A., Francque S., Bedossa P., Lehert P., Serfaty L., et al. Elafibranor, an agonist of the peroxisome proliferator-activated Receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–1159. doi: 10.1053/j.gastro.2016.01.038. e5. [DOI] [PubMed] [Google Scholar]
- 11.Argo C.K., Iezzoni J.C., Al-Osaimi A.M., Caldwell S.H. Thiazolidinediones for the treatment in NASH: sustained benefit after drug discontinuation?. J Clin Gastroenterol. 2009;43:565–568. doi: 10.1097/MCG.0b013e31818f4fc2. [DOI] [PubMed] [Google Scholar]
- 12.Neuschwander-Tetri B.A., Brunt E.M., Wehmeier K.R., Sponseller C.A., Hampton K., Bacon B.R. Interim results of a pilot study demonstrating the early effects of the PPAR-gamma ligand rosiglitazone on insulin sensitivity, aminotransferases, hepatic steatosis and body weight in patients with non-alcoholic steatohepatitis. J Hepatol. 2003;38:434–440. doi: 10.1016/s0168-8278(03)00027-8. [DOI] [PubMed] [Google Scholar]
- 13.Promrat K., Lutchman G., Uwaifo G.I., Freedman R.J., Soza A., Heller T., et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 14.Panebianco C., Oben J.A., Vinciguerra M., Pazienza V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: a putative synergy between retinoic acid and PPAR-gamma signalings. Clin Exp Med. 2017;17:269–280. doi: 10.1007/s10238-016-0438-x. [DOI] [PubMed] [Google Scholar]
- 15.Choi J.H., Jin S.W., Choi C.Y., Kim H.G., Lee G.H., Kim Y.A., et al. Capsaicin inhibits dimethylnitrosamine-induced hepatic fibrosis by inhibiting the TGF-β1/smad pathway via peroxisome proliferator-activated receptor gamma activation. J Agric Food Chem. 2017;65:317–326. doi: 10.1021/acs.jafc.6b04805. [DOI] [PubMed] [Google Scholar]
- 16.Ramani K., Tomasi M.L. Transcriptional regulation of methionine adenosyltransferase 2A by peroxisome proliferator-activated receptors in rat hepatic stellate cells. Hepatology. 2012;55:1942–1953. doi: 10.1002/hep.25594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeybel M., Hardy T., Wong Y.K., Mathers J.C., Fox C.R., Gackowska A., et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18:1369–1377. doi: 10.1038/nm.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann J., Chu D.C., Maxwell A., Oakley F., Zhu N.L., Tsukamoto H., et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–714. doi: 10.1053/j.gastro.2009.10.002. 14 e1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moran-Salvador E., Garcia-Macia M., Sivaharan A., Sabater L., Zaki M.Y.W., Oakley F., et al. Fibrogenic activity of MECP2 is regulated by phosphorylation in hepatic stellate cells. Gastroenterology. 2019;157 doi: 10.1053/j.gastro.2019.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hlady R.A., Zhao X., Pan X., Yang J.D., Ahmed F., Antwi S.O., et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics. 2019;9:7239–7250. doi: 10.7150/thno.35573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardy T., Zeybel M., Day C.P., Dipper C., Masson S., McPherson S., et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut. 2017;66:1321–1328. doi: 10.1136/gutjnl-2016-311526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajri T., Zaiou M., Fungwe T.V., Ouguerram K., Besong S. Epigenetic regulation of peroxisome proliferator-activated receptor gamma mediates high-fat diet-induced non-alcoholic fatty liver disease. Cells. 2021;10:1355. doi: 10.3390/cells10061355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Creamer T.P. Calcineurin. Cell Commun Signal. 2020;18:137. doi: 10.1186/s12964-020-00636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ermak G., Davies K.J.A. Chronic high levels of the RCAN1-1 protein may promote neurodegeneration and Alzheimer disease. Free Radic Biol Med. 2013;62:47–51. doi: 10.1016/j.freeradbiomed.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan X.Y., You H.M., Wang L., Bi Y.H., Yang Y., Meng H.W., et al. Methylation of RCAN1.4 mediated by DNMT1 and DNMT3b enhances hepatic stellate cell activation and liver fibrogenesis through Calcineurin/NFAT3 signaling. Theranostics. 2019;9:4308–4323. doi: 10.7150/thno.32710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y., Wu X.Q., Li W.X., Huang H.M., Li H.D., Pan X.Y., et al. PSTPIP2 connects DNA methylation to macrophage polarization in CCl4-induced mouse model of hepatic fibrosis. Oncogene. 2018;37:6119–6135. doi: 10.1038/s41388-018-0383-0. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Ramos D., Fernandez-Tussy P., Lopitz-Otsoa F., Gutierrez-de-Juan V., Navasa N., Barbier-Torres L., et al. MiR-873-5p acts as an epigenetic regulator in early stages of liver fibrosis and cirrhosis. Cell Death Dis. 2018;9:958. doi: 10.1038/s41419-018-1014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling C., Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metabol. 2019;29:1028–1044. doi: 10.1016/j.cmet.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence M., Daujat S., Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 2016;32:42–56. doi: 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 30.James T.T., Aroor A.R., Lim R.W., Shukla S.D. Histone H3 phosphorylation (Ser10, Ser28) and phosphoacetylation (K9S10) are differentially associated with gene expression in liver of rats treated in vivo with acute ethanol. J Pharmacol Exp Therapeut. 2012;340:237–247. doi: 10.1124/jpet.111.186775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y., Hu H., Liu M., Zhou T., Cheng X., Huang W., et al. The role and mechanism of histone lactylation in health and diseases. Front Genet. 2022;13 doi: 10.3389/fgene.2022.949252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santos-Rosa H., Millán-Zambrano G., Han N., Leonardi T., Klimontova M., Nasiscionyte S., et al. Methylation of histone H3 at lysine 37 by Set1 and Set2 prevents spurious DNA replication. Mol Cell. 2021;81 doi: 10.1016/j.molcel.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin-Mateos R., De Assuncao T.M., Arab J.P., Jalan-Sakrikar N., Yaqoob U., Greuter T., et al. Enhancer of zeste homologue 2 inhibition attenuates TGF-β dependent hepatic stellate cell activation and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2019;7:197–209. doi: 10.1016/j.jcmgh.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y., Xiang C., Zhong F., Zhang Y., Wang L., Zhao Y., et al. Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics. 2021;11:361–378. doi: 10.7150/thno.46360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaqoob U., Luo F., Greuter T., Jalan Sakrikar N., Sehrawat T.S., Lu J., et al. GIPC-regulated IGFBP-3 promotes HSC migration in vitro and portal hypertension in vivo through a β1-integrin pathway. Cell Mol Gastroenterol Hepatol. 2020;10:545–559. doi: 10.1016/j.jcmgh.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montavon T., Shukeir N., Erikson G., Engist B., Onishi-Seebacher M., Ryan D., et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat Commun. 2021;12:4359. doi: 10.1038/s41467-021-24532-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Y., Wang S., Zhao Y., Lin C., Zhong F., Jin L., et al. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015;29:1830–1841. doi: 10.1096/fj.14-251751. [DOI] [PubMed] [Google Scholar]
- 38.Dong F., Jiang S., Li J., Wang Y., Zhu L., Huang Y., et al. The histone demethylase KDM4D promotes hepatic fibrogenesis by modulating Toll-like receptor 4 signaling pathway. EBioMedicine. 2019;39:472–483. doi: 10.1016/j.ebiom.2018.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ugai K., Matsuda S., Mikami H., Shimada A., Misawa T., Nakamura H., et al. Inhibition of the SET8 pathway ameliorates lung fibrosis even through fibroblast dedifferentiation. Front Mol Biosci. 2020;7:192. doi: 10.3389/fmolb.2020.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders Y.Y., Liu H., Zhang X., Hecker L., Bernard K., Desai L., et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013;1:8–16. doi: 10.1016/j.redox.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao Y., Zhao C., Tai Y., Li B., Lan T., Lai E., et al. STING mediates hepatocyte pyroptosis in liver fibrosis by Epigenetically activating the NLRP3 inflammasome. Redox Biol. 2023;62 doi: 10.1016/j.redox.2023.102691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeybel M., Luli S., Sabater L., Hardy T., Oakley F., Leslie J., et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: inhibition of liver fibrosis progression by 3-deazaneplanocin A. Mol Ther. 2017;25:218–231. doi: 10.1016/j.ymthe.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tee W.W., Reinberg D. Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development. 2014;141:2376–2390. doi: 10.1242/dev.096982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumura Y., Nakaki R., Inagaki T., Yoshida A., Kano Y., Kimura H., et al. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell. 2015;60:584–596. doi: 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Fang R., Barbera A.J., Xu Y., Rutenberg M., Leonor T., Bi Q., et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. 2010;39:222–233. doi: 10.1016/j.molcel.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li G., Ji T., Chen J., Fu Y., Hou L., Feng Y., et al. CRL4(DCAF8) ubiquitin ligase targets histone H3K79 and promotes H3K9 methylation in the liver. Cell Rep. 2017;18:1499–1511. doi: 10.1016/j.celrep.2017.01.039. [DOI] [PubMed] [Google Scholar]
- 48.Yang D., Xu P., Su H., Zhong W., Xu J., Su Z., et al. The histone methyltransferase DOT1L is a new epigenetic regulator of pulmonary fibrosis. Cell Death Dis. 2022;13:60. doi: 10.1038/s41419-021-04365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J., Wang J., Long F., Zhong W., Su H., Su Z., et al. Inhibition of the cardiac fibroblast-enriched histone methyltransferase Dot1L prevents cardiac fibrosis and cardiac dysfunction. Cell Biosci. 2022;12:134. doi: 10.1186/s13578-022-00877-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L., Zou J., Guan Y., Zhang Y., Zhang W., Zhou X., et al. Blocking the histone lysine 79 methyltransferase DOT1L alleviates renal fibrosis through inhibition of renal fibroblast activation and epithelial-mesenchymal transition. FASEB J. 2019;33:11941–11958. doi: 10.1096/fj.201801861R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J., Wang B., Li N., Zhou Q., Zhou W., Zhan Z. Salvia miltiorrhiza and Carthamus tinctorius extract prevents cardiac fibrosis and dysfunction after myocardial infarction by epigenetically inhibiting Smad3 expression. Evid Based Complement Alternat Med. 2019;2019 doi: 10.1155/2019/6479136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimoda H., Doi S., Nakashima A., Sasaki K., Doi T., Masaki T. Inhibition of the H3K4 methyltransferase MLL1/WDR5 complex attenuates renal senescence in ischemia reperfusion mice by reduction of p16(INK4a) Kidney Int. 2019;96:1162–1175. doi: 10.1016/j.kint.2019.06.021. [DOI] [PubMed] [Google Scholar]
- 53.Tamura R., Doi S., Nakashima A., Sasaki K., Maeda K., Ueno T., et al. Inhibition of the H3K4 methyltransferase SET7/9 ameliorates peritoneal fibrosis. PLoS One. 2018;13 doi: 10.1371/journal.pone.0196844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveira T., Hermann E., Lin D., Chowanadisai W., Hull E., Montgomery M. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cochran A.G., Conery A.R., Sims R.J., 3rd Bromodomains: a new target class for drug development. Nat Rev Drug Discov. 2019;18:609–628. doi: 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- 56.Papait R., Serio S., Condorelli G. Role of the epigenome in heart failure. Physiol Rev. 2020;100:1753–1777. doi: 10.1152/physrev.00037.2019. [DOI] [PubMed] [Google Scholar]
- 57.Dou C., Liu Z., Tu K., Zhang H., Chen C., Yaqoob U., et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. 2018;154:2209–2221. doi: 10.1053/j.gastro.2018.02.015. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen F., Zhuang S. Histone acetylation and modifiers in renal fibrosis. Front Pharmacol. 2022;13 doi: 10.3389/fphar.2022.760308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y., Liu H., Liu X., Zhang X., Wu J., Yuan L., et al. Histone acetylation plays an important role in MC-LR-induced apoptosis and cycle disorder in SD rat testicular cells. Chemosphere. 2020;241 doi: 10.1016/j.chemosphere.2019.125073. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh A.K., Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007;213:663–671. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- 61.Kaimori A., Potter J.J., Choti M., Ding Z., Mezey E., Koteish A.A. Histone deacetylase inhibition suppresses the transforming growth factor beta1-induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology. 2010;52:1033–1045. doi: 10.1002/hep.23765. [DOI] [PubMed] [Google Scholar]
- 62.Ma L., Gao Z., Wu J., Zhong B., Xie Y., Huang W., et al. Co-condensation between transcription factor and coactivator p300 modulates transcriptional bursting kinetics. Mol Cell. 2021;81:1682–1697. doi: 10.1016/j.molcel.2021.01.031. e7. [DOI] [PubMed] [Google Scholar]
- 63.Gao J., Wei B., Liu M., Hirsova P., Sehrawat T.S., Cao S., et al. Endothelial p300 promotes portal hypertension and hepatic fibrosis through C–C motif chemokine ligand 2-mediated angiocrine signaling. Hepatology. 2021;73:2468–2483. doi: 10.1002/hep.31617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kong M., Chen X., Lv F., Ren H., Fan Z., Qin H., et al. Serum response factor (SRF) promotes ROS generation and hepatic stellate cell activation by epigenetically stimulating NCF1/2 transcription. Redox Biol. 2019;26 doi: 10.1016/j.redox.2019.101302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aseem S.O., Jalan-Sakrikar N., Chi C., Navarro-Corcuera A., De Assuncao T.M., Hamdan F.H., et al. Epigenomic evaluation of cholangiocyte transforming growth factor-beta signaling identifies a selective role for histone 3 lysine 9 acetylation in biliary fibrosis. Gastroenterology. 2021;160:889–905.e10. doi: 10.1053/j.gastro.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Claveria-Cabello A., Colyn L., Arechederra M., Urman J.M., Berasain C., Avila M.A., et al. Epigenetics in liver fibrosis: could HDACs be a therapeutic target?. Cells. 2020;9:2321. doi: 10.3390/cells9102321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt O., Nehls N., Prexler C., von Heyking K., Groll T., Pardon K., et al. Class I histone deacetylases (HDAC) critically contribute to Ewing sarcoma pathogenesis. J Exp Clin Cancer Res. 2021;40:322. doi: 10.1186/s13046-021-02125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moran-Salvador E., Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2017;4:125–134. doi: 10.1016/j.jcmgh.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Y.H., Tiao M.M., Huang L.T., Chuang J.H., Kuo K.C., Yang Y.L., et al. Activation of mir-29a in activated hepatic stellate cells modulates its profibrogenic phenotype through inhibition of histone deacetylases 4. PLoS One. 2015;10 doi: 10.1371/journal.pone.0136453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang Z., Liu Y., Qin L., Wu P., Xia Z., Luo M., et al. Cathepsin H-mediated degradation of HDAC4 for matrix metalloproteinase expression in hepatic stellate cells: implications of epigenetic suppression of matrix metalloproteinases in fibrosis through stabilization of class IIa histone deacetylases. Am J Pathol. 2017;187:781–797. doi: 10.1016/j.ajpath.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yin S., Zhang Q., Yang J., Lin W., Li Y., Chen F., et al. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim Biophys Acta Mol Cell Res. 2017;1864:1207–1216. doi: 10.1016/j.bbamcr.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 72.You H., Wang L., Bu F., Meng H., Pan X., Li J., et al. The miR-455-3p/HDAC2 axis plays a pivotal role in the progression and reversal of liver fibrosis and is regulated by epigenetics. FASEB J. 2021;35 doi: 10.1096/fj.202002319RRR. [DOI] [PubMed] [Google Scholar]
- 73.Zhu X., Ye S., Yu D., Zhang Y., Li J., Zhang M., et al. Physalin B attenuates liver fibrosis via suppressing LAP2alpha-HDAC1-mediated deacetylation of the transcription factor GLI1 and hepatic stellate cell activation. Br J Pharmacol. 2021;178:3428–3447. doi: 10.1111/bph.15490. [DOI] [PubMed] [Google Scholar]
- 74.Liu C., Chen X., Yang L., Kisseleva T., Brenner D.A., Seki E. Transcriptional repression of the transforming growth factor beta (TGF-beta) pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by nuclear factor kappaB (NF-kappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J Biol Chem. 2014;289:7082–7091. doi: 10.1074/jbc.M113.543769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ko S., Russell J.O., Tian J., Gao C., Kobayashi M., Feng R., et al. Hdac1 regulates differentiation of bipotent liver progenitor cells during regeneration via Sox9b and Cdk8. Gastroenterology. 2019;156:187–202.e14. doi: 10.1053/j.gastro.2018.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Y., Liu X., Zhou Q., Huang C., Meng X., Xu F., et al. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol Appl Pharmacol. 2015;289:163–176. doi: 10.1016/j.taap.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 77.Yang Y., Bai T., Yao Y.L., Zhang D.Q., Wu Y.L., Lian L.H., et al. Upregulation of SIRT1–AMPK by thymoquinone in hepatic stellate cells ameliorates liver injury. Toxicol Lett. 2016;262:80–91. doi: 10.1016/j.toxlet.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 78.Li M., Hong W., Hao C., Li L., Xu H., Li P., et al. Hepatic stellate cell-specific deletion of SIRT1 exacerbates liver fibrosis in mice. Biochim Biophys Acta, Mol Basis Dis. 2017;1863:3202–3211. doi: 10.1016/j.bbadis.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 79.Jiang R., Zhou Y., Wang S., Pang N., Huang Y., Ye M., et al. Nicotinamide riboside protects against liver fibrosis induced by CCl4via regulating the acetylation of Smads signaling pathway. Life Sci. 2019;225:20–28. doi: 10.1016/j.lfs.2019.03.064. [DOI] [PubMed] [Google Scholar]
- 80.Zhao H., Wang Z., Tang F., Zhao Y., Feng D., Li Y., et al. Carnosol-mediated Sirtuin 1 activation inhibits enhancer of Zeste homolog 2 to attenuate liver fibrosis. Pharmacol Res. 2018;128:327–337. doi: 10.1016/j.phrs.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 81.Sundaresan N.R., Bindu S., Pillai V.B., Samant S., Pan Y., Huang J.Y., et al. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3beta. Mol Cell Biol. 2015;36:678–692. doi: 10.1128/MCB.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhong X., Huang M., Kim H.G., Zhang Y., Chowdhury K., Cai W., et al. SIRT6 protects against liver fibrosis by deacetylation and suppression of SMAD3 in hepatic stellate cells. Cell Mol Gastroenterol Hepatol. 2020;10:341–364. doi: 10.1016/j.jcmgh.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiang X., Ohshiro K., Zaidi S., Yang X., Bhowmick K., Vegesna A.K., et al. Impaired reciprocal regulation between SIRT6 and TGF-beta signaling in fatty liver. FASEB J. 2022;36 doi: 10.1096/fj.202101518R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang J., Li Y., Liu Q., Huang Y., Li R., Wu T., et al. Sirt6 alleviated liver fibrosis by deacetylating conserved lysine 54 on Smad2 in hepatic stellate cells. Hepatology. 2021;73:1140–1157. doi: 10.1002/hep.31418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chowdhury K., Huang M., Kim H.G., Dong X.C. Sirtuin 6 protects against hepatic fibrogenesis by suppressing the YAP and TAZ function. FASEB J. 2022;36 doi: 10.1096/fj.202200522R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hao L., Bang I.H., Wang J., Mao Y., Yang J.D., Na S.Y., et al. ERRgamma suppression by Sirt6 alleviates cholestatic liver injury and fibrosis. JCI Insight. 2020;5 doi: 10.1172/jci.insight.137566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo X., Bai Y., He S., Sun S., Jiang X., Yang Z., et al. Sirtuin 1 ameliorates defenestration in hepatic sinusoidal endothelial cells during liver fibrosis via inhibiting stress-induced premature senescence. Cell Prolif. 2021;54 doi: 10.1111/cpr.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Han X., Ding C., Sang X., Peng M., Yang Q., Ning Y., et al. Targeting Sirtuin1 to treat aging-related tissue fibrosis: from prevention to therapy. Pharmacol Ther. 2022;229 doi: 10.1016/j.pharmthera.2021.107983. [DOI] [PubMed] [Google Scholar]
- 89.Ramirez T., Li Y.M., Yin S., Xu M.J., Feng D., Zhou Z., et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol. 2017;66:601–609. doi: 10.1016/j.jhep.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun Y.L., Bai T., Zhou L., Zhu R.T., Wang W.J., Liang R.P., et al. SOD3 deficiency induces liver fibrosis by promoting hepatic stellate cell activation and epithelial–mesenchymal transition. J Cell Physiol. 2021;236:4313–4329. doi: 10.1002/jcp.30174. [DOI] [PubMed] [Google Scholar]
- 91.Song L., Chen T.Y., Zhao X.J., Xu Q., Jiao R.Q., Li J.M., et al. Pterostilbene prevents hepatocyte epithelial–mesenchymal transition in fructose-induced liver fibrosis through suppressing miR-34a/Sirt1/p53 and TGF-β1/Smads signalling. Br J Pharmacol. 2019;176:1619–1634. doi: 10.1111/bph.14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li K., Zhai M., Jiang L., Song F., Zhang B., Li J., et al. Tetrahydrocurcumin ameliorates diabetic cardiomyopathy by attenuating high glucose-induced oxidative stress and fibrosis via activating the SIRT1 pathway. Oxid Med Cell Longev. 2019;2019 doi: 10.1155/2019/6746907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen L., Wang Y., Li S., Zuo B., Zhang X., Wang F., et al. Exosomes derived from GDNF-modified human adipose mesenchymal stem cells ameliorate peritubular capillary loss in tubulointerstitial fibrosis by activating the SIRT1/eNOS signaling pathway. Theranostics. 2020;10:9425–9442. doi: 10.7150/thno.43315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou F., Wang A., Li D., Wang Y., Lin L. Pinocembrin from Penthorum chinense Pursh suppresses hepatic stellate cells activation through a unified SIRT3-TGF-beta-Smad signaling pathway. Toxicol Appl Pharmacol. 2018;341:38–50. doi: 10.1016/j.taap.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 95.Gu J., Chen C., Wang J., Chen T., Yao W., Yan T., et al. Withaferin A exerts preventive effect on liver fibrosis through oxidative stress inhibition in a Sirtuin 3-dependent manner. Oxid Med Cell Longev. 2020;2020 doi: 10.1155/2020/2452848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang A., Zhou F., Li D., Lu J.J., Wang Y., Lin L. γ-Mangostin alleviates liver fibrosis through Sirtuin 3-superoxide-high mobility group box 1 signaling axis. Toxicol Appl Pharmacol. 2019;363:142–153. doi: 10.1016/j.taap.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 97.Kim H.G., Huang M., Xin Y., Zhang Y., Zhang X., Wang G., et al. The epigenetic regulator SIRT6 protects the liver from alcohol-induced tissue injury by reducing oxidative stress in mice. J Hepatol. 2019;71:960–969. doi: 10.1016/j.jhep.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li J., Xian L., Zheng R., Wang Y., Wan X., Liu Y. Canthaxanthin shows anti-liver aging and anti-liver fibrosis effects by down-regulating inflammation and oxidative stress in vivo and in vitro. Int Immunopharm. 2022;110 doi: 10.1016/j.intimp.2022.108942. [DOI] [PubMed] [Google Scholar]
- 99.ElBaset M.A., Salem R.S., Ayman F., Ayman N., Shaban N., Afifi S.M., et al. Effect of empagliflozin on thioacetamide-induced liver injury in rats: role of AMPK/SIRT-1/HIF-1α pathway in halting liver fibrosis. Antioxidants. 2022;11:2152. doi: 10.3390/antiox11112152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li A., Peng R., Sun Y., Liu H., Peng H., Zhang Z. LincRNA 1700020I14Rik alleviates cell proliferation and fibrosis in diabetic nephropathy via miR-34a-5p/Sirt1/HIF-1alpha signaling. Cell Death Dis. 2018;9:461. doi: 10.1038/s41419-018-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lei J., Zhou M.H., Zhang F.C., Wu K., Liu S.W., Niu H.Q. Interferon regulatory factor transcript levels correlate with clinical outcomes in human glioma. Aging. 2021;13:12086–12098. doi: 10.18632/aging.202915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cai J., Liu Z., Huang X., Shu S., Hu X., Zheng M., et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking beta-catenin target gene expression. Kidney Int. 2020;97:106–118. doi: 10.1016/j.kint.2019.08.028. [DOI] [PubMed] [Google Scholar]
- 103.Simic P., Williams E.O., Bell E.L., Gong J.J., Bonkowski M., Guarente L. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 2013;3:1175–1186. doi: 10.1016/j.celrep.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guo X., Yan F., Shan X., Li J., Yang Y., Zhang J., et al. SIRT3 inhibits Ang II-induced transdifferentiation of cardiac fibroblasts through beta-catenin/PPAR-gamma signaling. Life Sci. 2017;186:111–117. doi: 10.1016/j.lfs.2017.07.030. [DOI] [PubMed] [Google Scholar]
- 105.Arteaga M., Shang N., Ding X., Yong S., Cotler S.J., Denning M.F., et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2016;310:G1155–G1168. doi: 10.1152/ajpgi.00271.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gong H., Zheng C., Lyu X., Dong L., Tan S., Zhang X. Inhibition of Sirt2 alleviates fibroblasts activation and pulmonary fibrosis via Smad2/3 pathway. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.756131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morselli M., Dieci G. Epigenetic regulation of human non-coding RNA gene transcription. Biochem Soc Trans. 2022;50:723–736. doi: 10.1042/BST20210860. [DOI] [PubMed] [Google Scholar]
- 108.Oerum S., Meynier V., Catala M., Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021;49:7239–7255. doi: 10.1093/nar/gkab378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sendinc E., Shi Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83:428–441. doi: 10.1016/j.molcel.2023.01.006. [DOI] [PubMed] [Google Scholar]
- 110.Yang Y., Cai J., Yang X., Wang K., Sun K., Yang Z., et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther. 2022;30:2342–2353. doi: 10.1016/j.ymthe.2022.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feng Y., Dong H., Sun B., Hu Y., Yang Y., Jia Y., et al. METTL3/METTL14 transactivation and m6A-dependent TGF-beta1 translation in activated Kupffer cells. Cell Mol Gastroenterol Hepatol. 2021;12:839–856. doi: 10.1016/j.jcmgh.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu J., Kuang F., Kroemer G., Klionsky D.J., Kang R., Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–435. doi: 10.1016/j.chembiol.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen M., Li Y., Wang Y., Shao J., Zhang F., Yin G., et al. N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He Z., Yang D., Fan X., Zhang M., Li Y., Gu X., et al. The roles and mechanisms of lncRNAs in liver fibrosis. Int J Mol Sci. 2020;21:1482. doi: 10.3390/ijms21041482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang J.J., Tao H., Deng Z.Y., Lu C., Li J. Non-coding RNA-mediated epigenetic regulation of liver fibrosis. Metabolism. 2015;64:1386–1394. doi: 10.1016/j.metabol.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 116.Ji F., Wang K., Zhang Y., Mao X.L., Huang Q., Wang J., et al. MiR-542-3p controls hepatic stellate cell activation and fibrosis via targeting BMP-7. J Cell Biochem. 2019;120:4573–4581. doi: 10.1002/jcb.27746. [DOI] [PubMed] [Google Scholar]
- 117.You K., Li S.Y., Gong J., Fang J.H., Zhang C., Zhang M., et al. MicroRNA-125b promotes hepatic stellate cell activation and liver fibrosis by activating RhoA signaling. Mol Ther Nucleic Acids. 2018;12:57–66. doi: 10.1016/j.omtn.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murakami Y., Toyoda H., Tanaka M., Kuroda M., Harada Y., Matsuda F., et al. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caviglia J.M., Yan J., Jang M.K., Gwak G.Y., Affo S., Yu L., et al. MicroRNA-21 and dicer are dispensable for hepatic stellate cell activation and the development of liver fibrosis. Hepatology. 2018;67:2414–2429. doi: 10.1002/hep.29627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen Y., Ou Y., Dong J., Yang G., Zeng Z., Liu Y., et al. Osteopontin promotes collagen I synthesis in hepatic stellate cells by miRNA-129-5p inhibition. Exp Cell Res. 2018;362:343–348. doi: 10.1016/j.yexcr.2017.11.035. [DOI] [PubMed] [Google Scholar]
- 121.Wang Y.Z., Zhang W., Wang Y.H., Fu X.L., Xue C.Q. Repression of liver cirrhosis achieved by inhibitory effect of miR-454 on hepatic stellate cells activation and proliferation via Wnt10a. J Biochem. 2019;165:361–367. doi: 10.1093/jb/mvy111. [DOI] [PubMed] [Google Scholar]
- 122.Hyun J., Wang S., Kim J., Rao K.M., Park S.Y., Chung I., et al. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat Commun. 2016;7 doi: 10.1038/ncomms10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Appourchaux K., Dokmak S., Resche-Rigon M., Treton X., Lapalus M., Gattolliat C.H., et al. MicroRNA-based diagnostic tools for advanced fibrosis and cirrhosis in patients with chronic hepatitis B and C. Sci Rep. 2016;6 doi: 10.1038/srep34935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li B.B., Li D.L., Chen C., Liu B.H., Xia C.Y., Wu H.J., et al. Potentials of the elevated circulating miR-185 level as a biomarker for early diagnosis of HBV-related liver fibrosis. Sci Rep. 2016;6 doi: 10.1038/srep34157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu R., Li X., Zhu W., Wang Y., Zhao D., Wang X., et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology. 2019;70:1317–1335. doi: 10.1002/hep.30662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song Y., Liu C., Liu X., Trottier J., Beaudoin M., Zhang L., et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule. Hepatology. 2017;66:1183–1196. doi: 10.1002/hep.29209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yu F., Geng W., Dong P., Huang Z., Zheng J. LncRNA-MEG3 inhibits activation of hepatic stellate cells through SMO protein and miR-212. Cell Death Dis. 2018;9:1014. doi: 10.1038/s41419-018-1068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dong Z., Li S., Wang X., Si L., Ma R., Bao L., et al. lncRNA GAS5 restrains CCl4-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2019;316:G539–G550. doi: 10.1152/ajpgi.00249.2018. [DOI] [PubMed] [Google Scholar]
- 129.Zhou B., Yuan W., Li X. LncRNA Gm5091 alleviates alcoholic hepatic fibrosis by sponging miR-27b/23b/24 in mice. Cell Biol Int. 2018;42:1330–1339. doi: 10.1002/cbin.11021. [DOI] [PubMed] [Google Scholar]
- 130.Zhang K., Han X., Zhang Z., Zheng L., Hu Z., Yao Q., et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat Commun. 2017;8:144. doi: 10.1038/s41467-017-00204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kong Y., Huang T., Zhang H., Zhang Q., Ren J., Guo X., et al. The lncRNA NEAT1/miR-29b/Atg9a axis regulates IGFBPrP1-induced autophagy and activation of mouse hepatic stellate cells. Life Sci. 2019;237 doi: 10.1016/j.lfs.2019.116902. [DOI] [PubMed] [Google Scholar]
- 132.Yu F., Lu Z., Chen B., Wu X., Dong P., Zheng J. Salvianolic acid B-induced microRNA-152 inhibits liver fibrosis by attenuating DNMT1-mediated Patched1 methylation. J Cell Mol Med. 2015;19:2617–2632. doi: 10.1111/jcmm.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y.L., Wang F.S., Li S.C., Tiao M.M., Huang Y.H. MicroRNA-29a alleviates bile duct ligation exacerbation of hepatic fibrosis in mice through epigenetic control of methyltransferases. Int J Mol Sci. 2017;18:192. doi: 10.3390/ijms18010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cai Q., Chen F., Xu F., Wang K., Zhang K., Li G., et al. Epigenetic silencing of microRNA-125b-5p promotes liver fibrosis in nonalcoholic fatty liver disease via integrin α8-mediated activation of RhoA signaling pathway. Metabolism. 2020;104 doi: 10.1016/j.metabol.2020.154140. [DOI] [PubMed] [Google Scholar]
- 135.Gondaliya P., Pd A., Jash K., Tekade R.K., Srivastava A., Kalia K. miR-29b attenuates histone deacetylase-4 mediated podocyte dysfunction and renal fibrosis in diabetic nephropathy. J Diabetes Metab Disord. 2020;19:13–27. doi: 10.1007/s40200-019-00469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Navarro-Corcuera A., Sehrawat T.S., Jalan-Sakrikar N., Gibbons H.R., Pirius N.E., Khanal S., et al. Long non-coding RNA ACTA2-AS1 promotes ductular reaction by interacting with the p300/ELK1 complex. J Hepatol. 2022;76:921–933. doi: 10.1016/j.jhep.2021.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu F., Chen B., Dong P., Zheng J. HOTAIR epigenetically modulates PTEN expression via microRNA-29b: a novel mechanism in regulation of liver fibrosis. Mol Ther. 2017;25:205–217. doi: 10.1016/j.ymthe.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wasson C.W., Abignano G., Hermes H., Malaab M., Ross R.L., Jimenez S.A., et al. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of NOTCH. Ann Rheum Dis. 2020;79:507–517. doi: 10.1136/annrheumdis-2019-216542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang K., Han Y., Hu Z., Zhang Z., Shao S., Yao Q., et al. SCARNA10, a nuclear-retained long non-coding RNA, promotes liver fibrosis and serves as a potential biomarker. Theranostics. 2019;9:3622–3638. doi: 10.7150/thno.32935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ge Z., Yin C., Li Y., Tian D., Xiang Y., Li Q., et al. Long noncoding RNA NEAT1 promotes cardiac fibrosis in heart failure through increased recruitment of EZH2 to the Smad7 promoter region. J Transl Med. 2022;20:7. doi: 10.1186/s12967-021-03211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang L., Zhao S., Zhu Y. Long noncoding RNA growth arrest-specific transcript 5 alleviates renal fibrosis in diabetic nephropathy by downregulating matrix metalloproteinase 9 through recruitment of enhancer of zeste homolog 2. FASEB J. 2020;34:2703–2714. doi: 10.1096/fj.201901380RR. [DOI] [PubMed] [Google Scholar]
- 142.Zeng X., Yuan X., Cai Q., Tang C., Gao J. Circular RNA as an epigenetic regulator in chronic liver diseases. Cells. 2021;10:1945. doi: 10.3390/cells10081945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jin H., Li C., Dong P., Huang J., Yu J., Zheng J. Circular RNA cMTO1 promotes PTEN expression through sponging miR-181b-5p in liver fibrosis. Front Cell Dev Biol. 2020;8:714. doi: 10.3389/fcell.2020.00714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ji D., Chen G.F., Wang J.C., Ji S.H., Wu X.W., Lu X.J., et al. Hsa_circ_0070963 inhibits liver fibrosis via regulation of miR-223-3p and LEMD3. Aging. 2020;12:1643–1655. doi: 10.18632/aging.102705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen X., Li H.D., Bu F.T., Li X.F., Chen Y., Zhu S., et al. Circular RNA circFBXW4 suppresses hepatic fibrosis via targeting the miR-18b-3p/FBXW7 axis. Theranostics. 2020;10:4851–4870. doi: 10.7150/thno.42423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou Y., Lv X., Qu H., Zhao K., Fu L., Zhu L., et al. Differential expression of circular RNAs in hepatic tissue in a model of liver fibrosis and functional analysis of their target genes. Hepatol Res. 2019;49:324–334. doi: 10.1111/hepr.13284. [DOI] [PubMed] [Google Scholar]
- 147.Bu F.T., Zhu Y., Chen X., Wang A., Zhang Y.F., You H.M., et al. Circular RNA circPSD3 alleviates hepatic fibrogenesis by regulating the miR-92b-3p/Smad7 axis. Mol Ther Nucleic Acids. 2021;23:847–862. doi: 10.1016/j.omtn.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu W., Feng R., Li X., Li D., Zhai W. TGF-β- and lipopolysaccharide-induced upregulation of circular RNA PWWP2A promotes hepatic fibrosis via sponging miR-203 and miR-223. Aging. 2019;11:9569–9580. doi: 10.18632/aging.102405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhu L., Ren T., Zhu Z., Cheng M., Mou Q., Mu M., et al. Thymosin-β4 mediates hepatic stellate cell activation by interfering with CircRNA-0067835/miR-155/FoxO3 signaling pathway. Cell Physiol Biochem. 2018;51:1389–1398. doi: 10.1159/000495556. [DOI] [PubMed] [Google Scholar]
- 150.Tang X., Xie X., Wang X., Wang Y., Jiang X., Jiang H. The combination of piR-823 and eukaryotic initiation factor 3 B (EIF3B) activates hepatic stellate cells via upregulating TGF-β1 in liver fibrogenesis. Med Sci Mon Int Med J Exp Clin Res. 2018;24:9151–9165. doi: 10.12659/MSM.914222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Miranda T.B., Cortez C.C., Yoo C.B., Liang G., Abe M., Kelly T.K., et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Therapeut. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Verma S.K., Tian X., LaFrance L.V., Duquenne C., Suarez D.P., Newlander K.A., et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fioravanti R., Stazi G., Zwergel C., Valente S., Mai A. Six years (2012–2018) of researches on catalytic EZH2 inhibitors: the boom of the 2-pyridone compounds. Chem Rec. 2018;18:1818–1832. doi: 10.1002/tcr.201800091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang Q., Jia R., Chen M., Wang J., Huang F., Shi M., et al. Antagonizing EZH2 combined with vitamin D3 exerts a synergistic role in anti-fibrosis through bidirectional effects on hepatocytes and hepatic stellate cells. J Gastroenterol Hepatol. 2023;38:441–450. doi: 10.1111/jgh.16126. [DOI] [PubMed] [Google Scholar]
- 155.Martin-Mateos R., De Assuncao T.M., Arab J.P., Jalan-Sakrikar N., Yaqoob U., Greuter T., et al. Enhancer of zeste homologue 2 inhibition attenuates TGF-beta dependent hepatic stellate cell activation and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2019;7:197–209. doi: 10.1016/j.jcmgh.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Du Z., Liu M., Wang Z., Lin Z., Feng Y., Tian D., et al. EZH2-mediated inhibition of KLF14 expression promotes HSCs activation and liver fibrosis by downregulating PPARgamma. Cell Prolif. 2021;54 doi: 10.1111/cpr.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hyun K., Jeon J., Park K., Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324. doi: 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kelly G.M., Al-Ejeh F., McCuaig R., Casciello F., Ahmad Kamal N., Ferguson B., et al. G9a inhibition enhances checkpoint inhibitor blockade response in melanoma. Clin Cancer Res. 2021;27:2624–2635. doi: 10.1158/1078-0432.CCR-20-3463. [DOI] [PubMed] [Google Scholar]
- 159.Park K.C., Park J.H., Jeon J.Y., Kim S.Y., Kim J.M., Lim C.Y., et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br J Pharmacol. 2014;171:4820–4830. doi: 10.1111/bph.12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang J., Qi H., Zhang X., Si W., Xu F., Hou T., et al. Saikosaponin D from Radix Bupleuri suppresses triple-negative breast cancer cell growth by targeting β-catenin signaling. Biomed Pharmacother. 2018;108:724–733. doi: 10.1016/j.biopha.2018.09.038. [DOI] [PubMed] [Google Scholar]
- 161.Elsakkar M.G., Eissa M.M., Hewedy W.A., Nassra R.M., Elatrebi S.F. Sodium valproate, a histone deacetylase inhibitor, with praziquantel ameliorates Schistosoma mansoni-induced liver fibrosis in mice. Life Sci. 2016;162:95–101. doi: 10.1016/j.lfs.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 162.Loh Z., Fitzsimmons R.L., Reid R.C., Ramnath D., Clouston A., Gupta P.K., et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br J Pharmacol. 2019;176:3775–3790. doi: 10.1111/bph.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu Y., Wang Z., Wang J., Lam W., Kwong S., Li F., et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-beta and vascular endothelial growth factor signalling. Liver Int. 2013;33:504–515. doi: 10.1111/liv.12034. [DOI] [PubMed] [Google Scholar]
- 164.Barbier-Torres L., Beraza N., Fernandez-Tussy P., Lopitz-Otsoa F., Fernandez-Ramos D., Zubiete-Franco I., et al. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology. 2015;62:1237–1248. doi: 10.1002/hep.27959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Aher J.S., Khan S., Jain S., Tikoo K., Jena G. Valproate ameliorates thioacetamide-induced fibrosis by hepatic stellate cell inactivation. Hum Exp Toxicol. 2015;34:44–55. doi: 10.1177/0960327114531992. [DOI] [PubMed] [Google Scholar]
- 166.Mannaerts I., Eysackers N., Onyema O.O., Van Beneden K., Valente S., Mai A., et al. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yu B., Qin S.Y., Hu B.L., Qin Q.Y., Jiang H.X., Luo W. Resveratrol improves CCL4-induced liver fibrosis in mouse by upregulating endogenous IL-10 to reprogramme macrophages phenotype from M(LPS) to M(IL-4) Biomed Pharmacother. 2019;117 doi: 10.1016/j.biopha.2019.109110. [DOI] [PubMed] [Google Scholar]
- 168.Abd El Motteleb D.M., Ibrahim I., Elshazly S.M. Sildenafil protects against bile duct ligation induced hepatic fibrosis in rats: potential role for silent information regulator 1 (SIRT1) Toxicol Appl Pharmacol. 2017;335:64–71. doi: 10.1016/j.taap.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 169.Fels J.A., Casalena G., Konrad C., Holmes H.E., Dellinger R.W., Manfredi G. Gene expression profiles in sporadic ALS fibroblasts define disease subtypes and the metabolic effects of the investigational drug EH301. Hum Mol Genet. 2022;31:3458–3477. doi: 10.1093/hmg/ddac118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Outeiro T.F., Kontopoulos E., Altmann S.M., Kufareva I., Strathearn K.E., Amore A.M., et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 171.Grozinger C.M., Chao E.D., Blackwell H.E., Moazed D., Schreiber S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 172.Gertz M., Fischer F., Nguyen G.T., Lakshminarasimhan M., Schutkowski M., Weyand M., et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci U S A. 2013;110:E2772–E2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.You Y., Fu Y., Huang M., Shen D., Zhao B., Liu H., et al. Recent advances of m6A demethylases inhibitors and their biological functions in human diseases. Int J Mol Sci. 2022;23:5815. doi: 10.3390/ijms23105815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Azzam S.K., Alsafar H., Sajini A.A. FTO m6A demethylase in obesity and cancer: implications and underlying molecular mechanisms. Int J Mol Sci. 2022;23:3800. doi: 10.3390/ijms23073800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Garbo S., Zwergel C., Battistelli C. m6A RNA methylation and beyond—the epigenetic machinery and potential treatment options. Drug Discov Today. 2021;26:2559–2574. doi: 10.1016/j.drudis.2021.06.004. [DOI] [PubMed] [Google Scholar]
- 176.Su R., Dong L., Li Y., Gao M., Han L., Wunderlich M., et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79–96. doi: 10.1016/j.ccell.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen J., Xu C., Yang K., Gao R., Cao Y., Liang L., et al. Inhibition of ALKBH5 attenuates I/R-induced renal injury in male mice by promoting Ccl28 m6A modification and increasing Treg recruitment. Nat Commun. 2023;14:1161. doi: 10.1038/s41467-023-36747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen Y., Zhou P., Deng Y., Cai X., Sun M., Sun Y., et al. ALKBH5-mediated m6 A demethylation of TIRAP mRNA promotes radiation-induced liver fibrosis and decreases radiosensitivity of hepatocellular carcinoma. Clin Transl Med. 2023;13:e1198. doi: 10.1002/ctm2.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mann J., Chu D.C., Maxwell A., Oakley F., Zhu N.L., Tsukamoto H., et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–714. doi: 10.1053/j.gastro.2009.10.002. 14.e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yang M.D., Chiang Y.M., Higashiyama R., Asahina K., Mann D.A., Mann J., et al. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor γ in hepatic stellate cells for their antifibrotic effect. Hepatology. 2012;55:1271–1281. doi: 10.1002/hep.24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Barcena-Varela M., Paish H., Alvarez L., Uriarte I., Latasa M.U., Santamaria E., et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut. 2021;70:388–400. doi: 10.1136/gutjnl-2019-320205. [DOI] [PubMed] [Google Scholar]
- 182.Li Y., Chen X., Lu C. The interplay between DNA and histone methylation: molecular mechanisms and disease implications. EMBO Rep. 2021;22 doi: 10.15252/embr.202051803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tang J., Zhuang S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin Sci (Lond) 2019;133:597–609. doi: 10.1042/CS20180465. [DOI] [PMC free article] [PubMed] [Google Scholar]