

Abstract
The vertebrate peripheral nervous system (PNS) is an intricate network that conveys sensory and motor information throughout the body. During development, extracellular cues direct the migration of axons and glia through peripheral tissues. Currently, the suite of molecules that govern PNS axon-glial patterning is incompletely understood. To elucidate factors that are critical for peripheral nerve development, we characterized the novel zebrafish mutant, stl159, that exhibits abnormalities in PNS patterning. In these mutants, motor and sensory nerves that develop adjacent to axial muscle fail to extend normally, and neuromasts in the posterior lateral line system, as well as neural crest-derived melanocytes, are incorrectly positioned. The stl159 genetic lesion lies in the basic helix-loop-helix (bHLH) transcription factor tcf15, which has been previously implicated in proper development of axial muscles. We find that targeted loss of tcf15 via CRISPR-Cas9 genome editing results in the PNS patterning abnormalities observed in stl159 mutants. Because tcf15 is expressed in developing muscle prior to nerve extension, rather than in neurons or glia, we predict that tcf15 non-cell-autonomously promotes peripheral nerve patterning in zebrafish through regulation of extracellular patterning cues. Our work underscores the importance of muscle-derived factors in PNS development.
Keywords: Lateral line nerve, Paraxial mesoderm, Schwann cells, Myelin, Zebrafish
1. Introduction
The patterning of the peripheral nervous system (PNS) is a precisely regulated developmental process that is essential for proper nervous system functioning. PNS axons must extend to specific sites of innervation so that sensory and motor cues can travel throughout the entirety of the body. Schwann cells, a class of myelinating glia that are essential for peripheral neuron survival and operation, undergo a distinct and complex developmental process to enwrap the axons of the PNS in myelin (Jessen and Mirsky, 2005; Muppirala et al., 2021). Early in development, Schwann cell precursors migrate from their neural crest origin in close proximity to the extending peripheral axons that they will eventually myelinate. Previous research has established the importance of a variety of intracellular and extracellular signaling molecules for proper patterning of peripheral axons and glia (Muppirala et al., 2021). However, the mechanisms that drive this process are incompletely understood.
Zebrafish have become a premier model to study the development of peripheral nerves. Previous work in zebrafish demonstrated that axons and Schwann cells have an interdependent signaling relationship to migrate into the periphery and stabilize myelinated axons (Gilmour et al., 2004; Lyons et al., 2005). Additionally, signaling molecules in the environment, including those expressed in muscle, are also crucial for directing axons and Schwann cell precursors to their final sites of innervation. For instance, Semaphorin/Neuropilin and Eph/Ephrin interactions are established cues for guiding axons through the surrounding environment (Bonanomi and Pfaff, 2010; Muñoz et al., 2005), and these critical migratory cues are also required by neural crest cells from which Schwann cells are derived (Banerjee et al., 2011; Gammill and Roffers-Agarwal, 2010). In zebrafish and mammals, the close proximity of muscle tissue to developing peripheral nerves means that specification of muscle cells and controlled expression of muscle-derived extracellular cues play an important role in nervous system patterning (Isaacman-Beck et al., 2015; Jing et al., 2009; Zhang et al., 2004). For instance, zebrafish larvae in which the muscle transcription factor foxc1 is disrupted exhibit defects in motor axon patterning; Foxc1 appears to regulate the expression of muscle-derived cues that are essential for motor axon migration (Banerjee et al., 2015). Similarly, zebrafish sensory axons rely upon local environmental cues for proper patterning of the posterior lateral line nerves, which co-develop with the lateral line primordia (Gallardo et al., 2010). Thus, PNS patterning is the result of complex interactions between numerous signaling molecules, both expressed in neural tissue (axons and glia) and the surrounding environment.
The transcription factor Tcf15 (sometimes referred to as paraxis) is a member of the basic helix-loop-helix (bHLH) transcription factor family and is expressed in a rostrocaudal gradient in the paraxial mesoderm prior to somite formation (Burgess et al., 1995, 1996; Lee et al., 2009; Topczewska et al., 2001). Tcf15 encodes a transcriptional activator that contains a binding domain for E-boxes in DNA as well as a ligand binding domain for dimerization (Wilson-Rawls et al., 2004). Tcf15 induces the expression of genes that promote the mesenchymal to epithelial transition that takes place during somitogenesis to form organized adhesive muscle blocks (Sánchez and Sánchez, 2015). Tcf15-negative mice exhibit altered expression of genes that are involved in cell-cell and cell-ECM adhesion, including Eph and Ephrin (Johnson et al., 2001; Rowton et al., 2013).
In this study, we identified tcf15 as a crucial regulator of PNS patterning via a forward genetic screen in zebrafish. The novel zebrafish mutant stl159 exhibits abnormal axon patterning and reduced glial development in the posterior lateral line nerve (PLLn) as well as its target sensory organs, neuromasts. Whole genome sequencing of the stl159 mutant reveals a premature stop codon in the tcf15 gene. Previous studies examining similar mutations in tcf15 have demonstrated that truncating protein synthesis prematurely compromises the DNA binding ability of the protein (Wilson-Rawls et al., 2004). It is therefore likely that gene expression normally regulated by tcf15 is disrupted in the muscle cells of stl159 mutant zebrafish, resulting in reduced expression of genes involved in cell-cell and cell-ECM adhesion and disorganized muscle ultrastructure. We hypothesize that a consequence of the tcf15stl159 mutation is reduced expression of muscle-derived cues that drive PNS patterning, resulting in the PNS hypomyelination and patterning abnormalities that characterize the stl159 mutant.
2. Methods
2.1. Zebrafish strains and husbandry
Zebrafish were maintained in accordance with Kenyon College IACUC-approved protocols and standards. The stl159 mutant was originally isolated in a large-scale genetic screen at Washington University in St. Louis (Sanchez et al., 2017). stl159 homozygous mutants are viable to adulthood when separated from wild-type siblings, but they are infertile; therefore, all experiments with stl159 mutants were performed by crossing heterozygous stl159 parents in pairs or harems to generate +/+ and +/stl159 siblings (“wild-type”) and homozygous stl159 mutants. Embryos were raised at 28.5 °C in egg water (Kimmel et al., 1995) in Petri dishes or, for time course data (Figs. 3–5), individually in 24-well plates with daily water changes. In addition to stl159, the following strains were used in this study: AB* as CRISPR/Cas9 control (Fig. S2), Tg(nbt:dsRed) (Peri and Nüsslein-Volhard, 2008), Tg(sox10:megfp) (Smith et al., 2014). stl159/+ mutants were crossed into transgenic backgrounds for analysis.
2.2. Identification and classification of stl159 phenotype
A traditional large-scale 3-generation forward genetic screen was used to identify mutants with defects in myelination (Sanchez et al., 2017). Myelin development was assessed using whole-mount in situ hybridization for myelin basic protein (mbp) using standard protocols and a previously described riboprobe (Cunningham and Monk, 2018; Lyons et al., 2005; Thisse and Thisse, 2008). mbp in situ hybridization of sibling larvae from stl159 carrier parents was scored along both the ALLn and PLLn as follows: First, presence of mbp along the nerve trajectory was categorized as “full” (mbp present along entire ALLn or along PLLn beyond yolk extension), “partial” (mbp beyond halfway but not full length of ALLn or between ganglion and yolk extension for PLLn), or “absent” (no mbp observed along nerve trajectory). Second, we qualitatively binned mbp intensity as “normal” (staining as intense as CNS mbp), “reduced” (less staining specifically in ALLn or PLLn relative to CNS), or “absent” (no mbp staining observed along nerve trajectory). stl159 was identified on the basis of both reduced mbp expression (either along nerve trajectory or staining intensity) and improper PLLn patterning, as well as aberrant pigment patterning in larvae and adults. Mutants were back-crossed to AB* ≥2 times before further analyses, and clutches with improper pigment patterning were raised to maintain the stl159 strain.
2.3. Identification of stl159 lesion and genotyping
Siblings from stl159/+ heterozygous in-crosses were sorted on the basis of pigment patterning, wild-type or stl159, and DNA was extracted from pooled fresh tissue for whole genome sequencing at the Genome Technology Access Center at Washington University in St. Louis. Sequenced DNA was aligned to Zv9 as the reference genome. Wild-type to mutant allele ratio was determined with a previously established bioinformatics pipeline (Sanchez et al., 2017) to identify linkage to chromosome 8. SNPs were filtered for non-synonymous mutations and tcf15 was pursued as the strongest lesion. To amplify the tcf15 locus from larvae and adult tissue for all subsequent analyses, the following primers were used: F 5′ GTGATGGCATTTGCCATGTT 3′ and R 5′ GCGCGGCTGTTTTCCTCCAC 3’ to yield a 450 bp product. Amplified DNA was purified and Sanger sequenced to identify the C-to-T mutation and identify carriers for future experiments.
2.4. CRISPR/Cas9 gene knockout
CRISPR sgRNA sequences were designed with a combination of analysis using CHOPCHOP (Labun et al., 2021) and the gRNA design tools from Integrated DNA Technologies (IDT). sgRNA sequences with high scores using both tools were selected with PAM sites in the 5’ UTR, adjacent to the tcf15 start site, and adjacent to the stl159 lesion. sgRNA and Cas9 protein were obtained from IDT (Alt-R® S.p. Cas9 Nuclease V3, Catalog #1081058, Lot Number: 0000434091). Embryos were injected at the 1–4 cell stage with 1 ng Cas9 protein and 50 pg each tcf15 sgRNA (100 pg total for combination). Uninjected and control-injected (no sgRNA) embryos were indistinguishable and thus pooled into a single “control” group. Lesions were identified with sequencing of individual injected larvae to compare control/uninjected larvae as described above.
2.5. Nerve imaging and analysis
High-resolution sox10:megfp PLLn imaging (Fig. 1) was obtained by embedding living 3 dpf larvae in low-melt agarose and imaging with a Zeiss Axio Observer with Apotome and ZEN software. For all other live imaging (nbt:dsRed, sox10:megfp, DASPEI, and brightfield), larvae were imaged in glass multiwell dishes with a Zeiss Discovery.V8 stereoscope and Axiocam 305 color camera. Measurements (counting, length, angle) were taken in ImageJ using static images. Immunostaining was performed as previously described (Cunningham and Monk, 2018) on clutches of tcf15stl159/+ in-cross progeny using 1:1000 mouse anti-acetylated tubulin (Sigma) and 1:2000 goat Alexa 488-conjugated anti-mouse IgG2b (Invitrogen).
Fig. 1. stl159 mutants have mispatterned neural crest cell derivatives.

(A–B) mbp expression in sibling (sib, A) and mutant (B) 5 dpf larvae stained via whole-mount in situ hybridization. Arrows indicate region of magnification for PLLn (A′-B′); arrowheads for ALLn (A″-B″). (C–D) Quantification of mbp phenotype among larval progeny of carrier in-cross. Bracket indicates stl159 PLLn-specific mutant phenotype in ~25% of larvae. *p < 0.05, ***p < 0.001, Fisher’s Exact test, normal (blue) vs. mutant (gray) phenotypes. Categorical definitions are reported in Methods. (C) Presence of mbp staining along ALLn and PLLn trajectory (full, partial, or absent) at 3 and 5 dpf. (D) Intensity of mbp staining along ALLn and PLLn trajectory (normal, reduced, or absent) at 3 and 5 dpf. (E–F) sox10:megfp labels neurons and glia including Schwann cells and precursors in the PLLn in sibling (E) and mutant (F) 3 dpf larvae. Arrows represent regions of magnification in the anterior (E′-F′) and more posterior, near the end of the yolk extension (E″-F″); scale bar, 20 μm. High-resolution imaging in E″-F″ shows individual glial cells on the PLLn marked with arrowheads. Dotted line in stl159 mutant (F″) indicates a non-PLLn GFP-labeled cell; asterisk marks a pigment cell obscuring PLLn fluorescence. Scale bar represents 100 μm in E-F, 20 μm in E′-F′ and E″-F″. (G) Quantification of sox10:megfp cells in the PLLn at 3 dpf along muscle segments 9–11. ****p < 0.0001, Student’s t-test. (H–I) Brightfield imaging showing melanocyte patterning in sibling (H) and mutant (I) 5 dpf larvae. Arrowheads indicate region of magnification just posterior to the ear; arrows for lateral body wall.
2.6. Neuromast labeling
DASPEI stock solution was prepared with 40 mg of DASPEI dissolved in 50 mL of distilled water and stored at 4 °C without light exposure until use (Cunningham et al., 2018). Larvae were anesthetized using standard protocols and incubated in 1:25 dilution of DASPEI stock:egg water for 20 min at room temperature prior to visualization. A GFP filter was used to visualize DASPEI-stained neuromasts. Neuromasts were counted and position was quantified by measuring the shortest distance, typically at a right angle. Neuromasts located anterior to the ganglia were not included in distance measurements. In mutants with no visible PLLn, only spinal cord measurements were taken.
2.7. Transmission electron microscopy (TEM)
TEM was performed according to standard techniques (Czopka and Lyons, 2011) on larval progeny of tcf15stl159/+ in-crosses at 5 dpf. Larvae were transected at muscle segment 5–6 and posterior halves processed for TEM. Samples were viewed with a Jeol JEM-1400 microscope (Jeol USA) and imaged with an AMT V601 digital camera (Advanced Microscopy Techniques Corp). PLLn were identified based on position and typical structure in sibling larvae. In mutants, landmarks were disorganized; thus both typical PLLn position and medial body wall muscle were scanned for presence of axons and glial cells.
2.8. Data handling and statistical analyses
For all experiments, data are pooled across at least two technical replicates with observer blind to genotype. Data were analyzed with GraphPad Prism. Fisher’s Exact test was used to compare percentages in the CRISPR enhancement experiment.
3. Results
3.1. The novel stl159 zebrafish mutant has developmental defects in neural crest derivatives
A recent large-scale forward genetic screen to identify regulators of myelination revealed the stl159 mutant, which has reduced mbp expression in the PLLn as visualized by whole-mount in situ hybridization. By 5 days post-fertilization (dpf), wild-type siblings show prominent mbp staining in the PLLn along the horizontal myoseptum and in the anterior lateral line nerve (ALLn) encircling the eye (Fig. 1A–A″). In contrast, stl159 mutants have reduced mbp expression along the PLLn, which is not restricted to the horizontal myoseptum (Fig. 1B–B′). We quantified mbp staining in clutches of larvae from stl159 carriers by scoring presence of mbp along nerve trajectory at both 3 and 5 dpf. Approximately one-quarter of larvae showed truncated mbp expression along typical PLLn length at both 3 dpf (7/29 larvae, 24.1%, p < 0.05) and 5 dpf (16/55, 29.1%, p < 0.001), with a subset having no mbp along the PLLn (3/29 at 3 dpf and 7/55 at 5 dpf, Fig. 1C). Furthermore, when mbp was present, the prominence of mbp staining was concomitantly reduced in these same larvae at both timepoints (6/29 larvae at 3 dpf, p < 0.05; 15/55 at 5 dpf, p < 0.001, Fig. 1D). Thus, stl159 mutants had mispositioned nerves with reduced mbp staining both spatially and in intensity. Interestingly, stl159 mutants have normal patterning and mbp expression in the ALLn compared to siblings (Fig. 1A″–B″, C–D). Taken together, these data suggest that stl159 is a recessive allele that results in a reduction of mbp(+) myelinating glia specifically along the PLLn.
Schwann cells are derived from sox10(+) neural crest lineage cells and express mbp upon terminal differentiation (Brosamle and Halpern, 2002; Jessen and Mirsky, 2005). To determine whether reduced mbp expression in stl159 is a consequence of earlier defects in Schwann cell precursor development, we examined sox10:megfp expression in stl159 mutants and siblings. sox10:megfp labels multiple cell types in the peripheral and central nervous systems (Smith et al., 2014), so we focused on the presumptive PLLn. Sibling larvae at 3 dpf have sox10:megfp(+) glia throughout the length of the PLLn (Fig. 1E–E″) in the expected position. We scored a defined region of the PLLn (dorsal to yolk extension along muscle segments 9–11) and found 7.7 ± 1.6 sox10:megfp(+) cells in wild-type siblings (Fig. 1G, n = 11 larvae). In contrast, the PLLn in stl159 mutants is incorrectly positioned (Fig. 1F–F″) and populated with fewer sox10:megfp(+) cells (3.4 ± 0.8, n = 10 larvae, p < 0.0001) (Fig. 1F–F″, 1G). Thus, the reduction of mbp in the PLLn of stl159 mutants could be a result of fewer myelinating glial cells along the nerve, rather than a specific disruption to the myelination program in PLLn Schwann cells.
Mispatterning of neural crest-derived glial precursors was accompanied by migration and morphology defects in melanocytes, which share an early developmental lineage with Schwann cells. In wild-type larvae, melanocytes migrate posteriorly along the horizontal myoseptum adjacent to the PLLn, where they adopt an elongated morphology on the anterior-posterior axis (Fig. 1H, Fig. S1A–D, I–J). stl159 melanocytes instead persistently cluster adjacent to the PLL ganglion posterior to the ear at larval stages, and those that migrate posteriorly are not restricted to the horizontal myoseptum (Fig. 1I, Fig. S1A–D, H–J). These early defects persist to adulthood, disrupting the characteristic stripe pattern (Figs. S1E–F). This phenotype is also fully penetrant and coincident with PLLn patterning defects, suggesting that stl159 disrupts a gene that is broadly necessary for neural crest-derived tissue patterning.
3.2. The muscle transcription factor tcf15 is truncated in stl159 mutants
To identify the lesion causative for the stl159 phenotype, we performed whole-genome sequencing on sibling and mutant stl159 larvae and compared single nucleotide polymorphisms (SNPs) to identify regions of linkage to the stl159 phenotype. Using a previously established pipeline (Sanchez et al., 2017), we identified candidate SNPs in annotated genes on chromosome 8 between 26 and 32 Mb (Fig. 2A–B). Most SNPs produced synonymous or weak non-synonymous amino acid changes; therefore, we focused our study on the nonsense allele in tcf15. Tcf15 encodes a bHLH transcription factor that cell-autonomously promotes muscle patterning in Xenopus and mouse (Burgess et al., 1996; Sánchez and Sánchez, 2015). stl159 contains a cytosine to thymine base pair substitution that produces an early stop codon. This mutation is predicted to truncate the wild-type 183 amino acid (aa) Tcf15 protein to 77 aa and presumably removes domains for dimerization and DNA binding (Fig. 2C–D). All assayed stl159 mutants with PLLn and melanocyte patterning defects possess this lesion, and all adults heterozygous for this lesion produce 25% stl159 phenotypic larvae when in-crossed. Together, these data suggest that the stl159 phenotype is a result of a recessive loss-of-function allele of tcf15.
Fig. 2. Mutation in tcf15 is causative for the stl159 phenotype.

(A) Whole genome sequencing reveals highest mutant/WT allele ratio on chromosome 8 (red box). Only SNP ratios >0.85 are shown on graph. (B) SNPs within region of strongest linkage are shown with wild-type in black and mutant SNP in red. Only tcf15 (bold) contains a nonsense allele. (C) Map of predicted unspliced tcf15 transcript is shown 5′ to 3′ with two introns marked in blue. The C to T substitution occurs at position 232 and produces a TAA stop codon that truncates the protein from 183 to 77 amino acids (aa). (D) Representative Sanger sequencing electropherograms of wild-type, stl159/+, and homozygous stl159 mutants in the tcf15 locus. Note transition of C (blue) in wild-type to T (red) in stl159. (E) Map of tcf15 transcript shown 5′ to 3′ with sgRNA targets sites marked in boxed A-C. sgRNA-A is in 5′ UTR 266 nucleotides upstream of the start site, while sgRNA-B and C both lie within the first intron (blue) at +21 and + 181. The stl159 C to T transition is marked for reference. (F) Quantification of tcf15 CRISPR enhancement of stl159 based on pigment phenotype. **p < 0.01, ****p < 0.0001, Fisher’s exact test wild-type vs. stl159 mutant pigment phenotype.
To test whether loss of tcf15 is sufficient to produce the stl159 phenotype, we generated multiple CRISPR sgRNA that target tcf15. Two sgRNA, labeled A and B, target the TATA box and start codon for tcf15, respectively. A third sgRNA, C, is adjacent but upstream to the stl159 lesion (Fig. 2E). We validated the efficacy of CRISPR sgRNA-C with Sanger sequencing of F0-injected wild-type larvae and observed mosaic disruption of tcf15 adjacent to the PAM target site (Fig. S2). Disruption is distinct from the lesion in stl159 mutants and was observed in 75% of sequenced injected larvae (12/16). Injection of each sgRNA with Cas9 protein produced a phenotype only rarely in homozygous wild-type larvae; therefore, we injected sgRNA-A and B in combination, or sgRNA-C alone, with Cas9 protein into clutches of stl159/+ in-cross progeny to test for enhancement of the stl159 phenotype in this sensitized background (Fig. 2F). As expected, we observed a Mendelian ratio (58/248, 23.4%) of stl159 phenotypic larvae in control conditions. Larvae injected with sgRNA-C alone, however, had a higher percentage of stl159 phenotype (140/332, 42.2%, p < 0.0001 vs. control). We tested a combination of sgRNA-A and sgRNA-B with Cas9 protein and found similar enhancement of heterozygous stl159/+ progeny (34/173 or 19.7% in control vs. 77/230 or 33.5% in multiple-injected larvae; p < 0.005). We conclude that disruption of tcf15 is causative for the mbp and pigment patterning defects observed in stl159 mutants and henceforth refer to this mutant as tcf15stl159.
3.3. tcf15 is required for sensory axon patterning
We next tested whether the mbp phenotype observed in tcf15stl159 is a consequence of mispatterned axons or strictly a glial defect. PLLn axons visualized at 5 dpf via immunostaining for acetylated tubulin (AcTub) have normal fasciculation and trajectory in siblings (Fig. 3A). In contrast, axon elongation and bundling fails in tcf15stl159 PLLn axons (Fig. 3B). We quantified the axon extension defects in tcf15stl159 with a transgenic neuronal fluorescent reporter, nbt:dsRed (Peri and Nüsslein-Volhard, 2008) and counted the number of muscle segments over which the nerve extends by 72 hours post-fertilization (hpf, Fig. 3C). Wild-type siblings have full PLLn extension by 72 hpf over 29.7 ± 1.3 muscle segments (n = 57 nerves). We binned tcf15stl159 PLLn into two phenotypic categories: those that extend posteriorly partway down the body (17/29, 58.6%) and those that fail to extend a PLLn (12/29, 41.4%). In tcf15stl159 mutants that extend a PLLn, nbt:dsRed + axons never extend the full body length, routing incorrectly through the embryo and stalling anteriorly (over 8.4 ± 3.5 muscle segments, n = 17, p < 0.0001). We conclude that tcf15stl159 inhibits proper PLLn axon elongation; furthermore, reduced PLLn sox10+ and mbp expression in tcf15stl159 is likely at least partially due to axon mispatterning and coincidently fewer myelinating Schwann cells along the PLLn.
Fig. 3. tcf15 is required for proper PLLn extension.

(A) Immunostaining for acetylated tubulin (AcTub) in sibling (sib, A) and tcf15stl159 (B) larvae at 5 dpf, immediately posterior to the PLL ganglia. Arrow indicates PLLn, which is bundled and extended in siblings and stalled in tcf15stl159; asterisk marks misplaced melanocyte in mutant. Scale bar, 100 μm. (C) Quantification of the farthest muscle segment position of the PLLn in siblings and tcf15stl159 larvae at 3 dpf. Tcf15stl159 larvae are divided into two categories, those in which a PLLn extended posteriorly from the ganglion (“extend”) and those in which a PLLn was not observed along the lateral body wall posterior to the ganglion (“stall/anterior”). All groups are significantly different from each other, p < 0.0001, 1-way ANOVA with Tukey test. (D–L) nbt:dsRed expression in the same PLLn of individual larvae at 36, 48, and 72 hpf. All images were taken at the same magnification immediately posterior to the PLL ganglion. Motor nerves are apparent but not quantified in these images. D-F represents a single sibling larva; G-I is a tcf15stl159 larva with stalled and misrouted extension, and J-L is a tcf15stl159 larva with no PLLn extension by 72 hpf. Arrows mark the PLLn at the anterior (left) and posterior-most position (right) in D-I; no PLLn is visible in J-L. Scale bar, 100 μm (M) Quantification of the farthest muscle segment position of the PLLn in individual sibling and tcf15stl159 larvae at 2 and 4 dpf. Each larva is represented in a different color and 2–4 dpf data points are connected with a line. (N) Representation of change in farthest PLLn position from 2 to 4 dpf for the sibling and tcf15stl159 larvae shown in M; there is no significant difference between groups (p > 0.05, Student’s t-test).
To test whether axonal defects observed at 3–5 dpf are due to failed axon extension or instability following outgrowth, we observed the PLLn outgrowth in individual larvae over time early in development. Sibling larvae have PLLn extension along the horizontal myoseptum over 30–36 hpf, and axons remain fasciculated through 72 hpf (Fig. 3D–F). PLLn of tcf15stl159 larvae, however, have initial outgrowth defects that persist over time. At 36 hpf, tcf15stl159 PLLn extension is delayed relative to siblings (Fig. 3G). Though axon growth may continue, the trajectory of the axon is misplaced by 48 hpf and the defect persists at 72 hpf (Fig. 3H–I). In tcf15stl159 mutants that fail to extend the PLLn by 36 hpf (Fig. 3J), no PLLn is observed later (Fig. 1K–L). We quantified these results by mapping PLLn extension in individual larvae at 2 and 4 dpf. While wild-type sibling larvae have stable PLLn outgrowth between 2 and 4 dpf (p > 0.1, 2 vs. 4 dpf extension, n = 12 larvae), tcf15stl159 PLLn defects present early and persist over time (p > 0.1, 2 vs. 4 dpf extension, n = 12 larvae, Fig. 3M–N). Neither wild-type nor tcf15stl159 mutants have significant changes in PLLn extension between 2 and 4 dpf (difference of 0.2 ± 0.6 segments in wild-type and 0.4 ± 2.2 segments in tcf15stl159, p > 0.05). Thus, tcf15 is required for stable PLLn axon outgrowth early in development.
3.4. Lateral line neuromasts are mispatterned in tcf15stl159 mutants
The PLLn contains sensory organs known as neuromasts at periodic intervals along the length of the PLLn, which are deposited by a migrating primordium during nervous system development. The primordium migrates in an anterior to posterior direction, directly preceding the migrating sensory axons and glial precursors. Neuromasts contain hair cells that detect vibrations in the surrounding water, and PLLn sensory axons innervate neuromasts to relay this information to the central nervous system (Ghysen and Dambly-Chaudière, 2004). Given the aberrant migration of PLLn axons, we questioned whether neuromasts were deposited correctly in tcf15stl159 mutants by visualizing neuromasts with DASPEI staining. By 4 dpf, wild-type larvae consistently have DASPEI-stained neuromasts, evenly spaced from anterior to posterior and in close apposition to the PLLn and horizontal myoseptum (16/16 wild-type siblings, Fig. 4A, G). In contrast, tcf15stl159 mutants display similar stalled depositions of neuromasts, with 9/16 or 56.2% larvae showing fewer neuromasts (Fig. 4B, G) and 7/16 or 43.8% showing no PLLn neuromasts posterior to the ganglia (Fig. 4C, G). Importantly, we found no difference in ALLn neuromast development (Fig. 4D–F). Together, these data suggest that tcf15 is required only for PLLn, and not ALLn, sensory organ deposition.
Fig. 4. tcf15 is required for neuromast development.
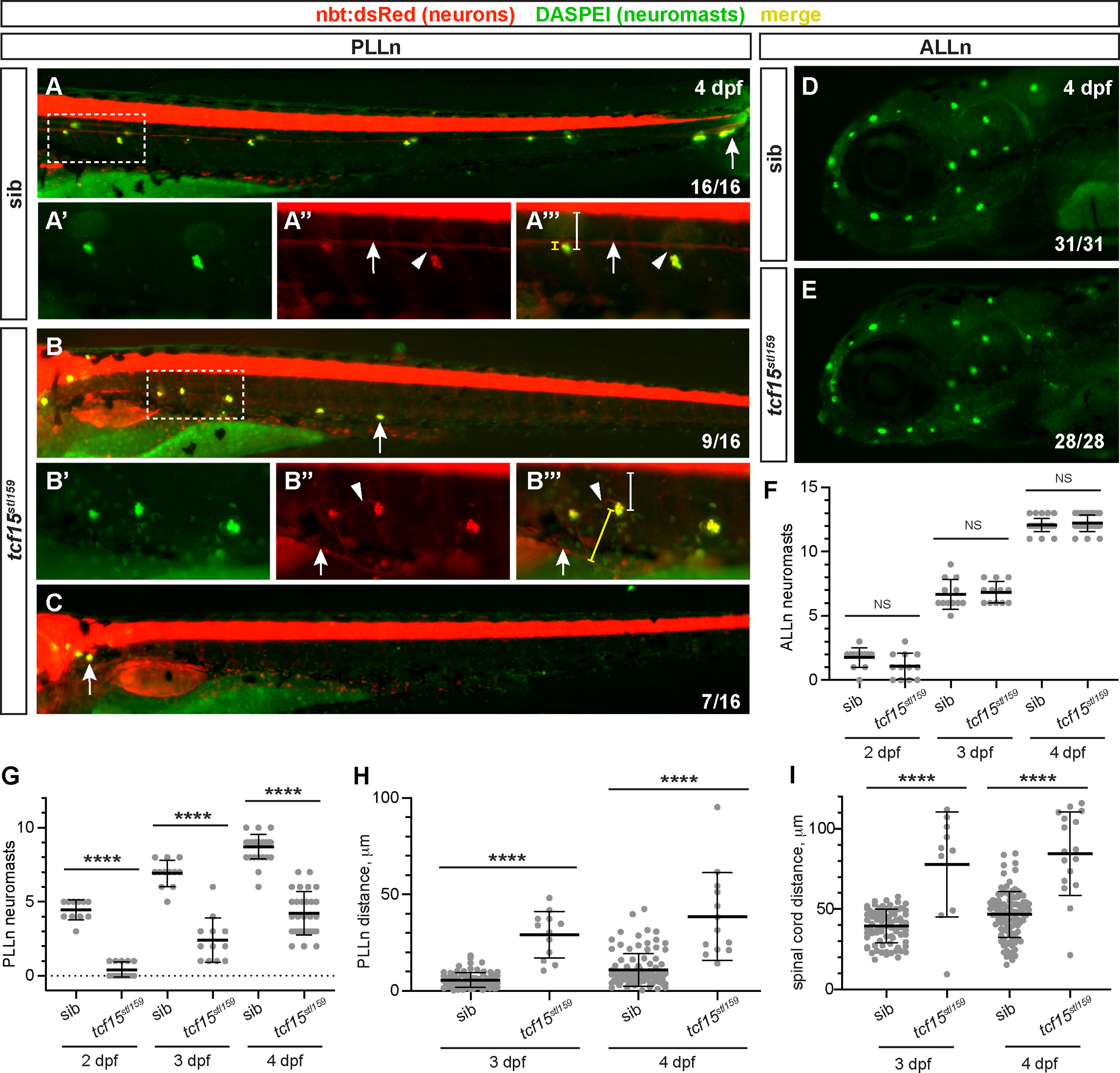
(A–C) DASPEI stain (green) marks neuromasts in nbt:dsRed-expressing (red) sibling (sib, A) and tcf15stl159 larvae at 4 dpf. Fraction in bottom right indicates number of larvae (of 16 siblings and 16 mutants) with each phenotype, normal (A), reduced extension (B), or stalled/failed to extend posteriorly (C). Arrows in A-C mark most posterior neuromast. Scale bar, 100 μm. Dotted boxes indicate high-magnification images, for DASPEI (A′-B′), PLLn marked with nbt:dsRed and arrow (A″-B″), and merge (A″′-B″′); scale bar, 100 μm. Arrowheads mark small PLLn processes that extend to the neuromast. Distance from neuromast to PLLn is bracketed in yellow; distance from neuromast to ventral spinal cord is bracketed in white. (D–E) DASPEI staining (green) of ALLn neuromasts shows no difference in sibling (sib, A) and tcf15stl159 larvae. (F) Quantification of ALLn neuromasts on the left side of individual larvae imaged at 2, 3, and 4 dpf. No significant differences are observed between genotypes at each timepoint (p > 0.05, 1-way ANOVA with Tukey test). (G) Quantification of PLLn neuromasts on the left side of individual larvae imaged at 2, 3, and 4 dpf. At each timepoint, fewer neuromasts have developed in tcf15stl159 larvae, ****p < 0.0001, 1-way ANOVA with Tukey test. (H–I) Quantification of shortest distance from individual neuromasts to main fascicle of PLLn (H) or ventral boundary of spinal cord (I) at 3 and 4 dpf. Neuromasts are more distant from the PLLn (****p < 0.0001, 1-way ANOVA with Tukey test) and more ventral and distant from the spinal cord (****p < 0.0001, 1-way ANOVA with Tukey test) in tcf15stl159 relative to siblings.
We next investigated whether neuromast deposition in tcf15stl159 mutants followed similar patterns to axon extension defects. To characterize neuromast deposition in tcf15stl159 mutants, we quantified neuromasts in individual larvae over time (n = 12 larvae per genotype) from 2 to 4 dpf. We found that ALLn neuromasts are deposited normally in both wild-type and tcf15stl159 siblings, increasing over time and at the same rate (1.8 ± 0.8 in wild-type vs. 1.1 ± 1.0 in tcf15stl159 at 2 dpf, p > 0.2; 6.7 ± 1.2 in wild-type vs. 6.8 ± 0.8 in tcf15stl159 at 3 dpf, p > 0.9, 12.1 ± 0.5 in wild-type vs. 12.2 ± 0.6 in tcf15stl159 at 4 dpf, p > 0.9, Fig. 4F). These findings are consistent with our original discovery that mbp is normal in tcf15stl159 ALLn at 5 dpf and that tcf15 is dispensable for ALLn development. However, tcf15stl159 mutant PLLn neuromasts are reduced in number at each timepoint and are deposited more slowly relative to wild-type siblings (4.5 ± 0.7 in wild-type vs. 0.4 ± 0.5 in tcf15stl159 at 2 dpf, p < 0.0001; 6.9 ± 0.9 in wild-type vs. 2.4 ± 1.5 in tcf15stl159 at 3 dpf, p < 0.0001, 8.7 ± 0.8 in wild-type vs. 4.2 ± 1.4 in tcf15stl159 at 4 dpf, p < 0.0001, Fig. 4G). Interestingly, neuromasts are deposited over time even in tcf15stl159 mutants, unlike PLLn axonal defects that appear relatively stable. This suggests that neuromast development may be less affected by loss of tcf15, as development partially continues in its absence.
In addition to fewer neuromasts in tcf15stl159 mutants, we measured neuromast position to demonstrate that developing neuromasts are misplaced over time in tcf15stl159 mutants relative to wild-type. At both 3 and 4 dpf, neuromasts are positioned farther from the main branch of the PLLn (5.7 ± 3.8 μm for wild-type neuromasts vs. 29.2 ± 12 μm in tcf15stl159 at 3 dpf, p < 0.0001; 11.0 ± 8.4 μm for wild-type neuromasts vs. 38.6 ± 22.8 μm in tcf15stl159 at 4 dpf, p < 0.0001; Fig. 4A″′–B″′, yellow bracketed line, 4H). However, we noted that while the PLLn was often rerouted to the ventral side of the body, the neuromasts tended to stay closer to their lateral position near the horizontal myoseptum. To characterize position within the body, we measured distance from neuromasts to spinal cord and found that they were positioned more ventrally in many, but not all, cases (39.5 ± 10.4 μm for wild-type neuromasts vs. 77.8 ± 32.6 μm in tcf15stl159 at 3 dpf, p < 0.0001; 46.7 ± 14.3 μm for wild-type neuromasts vs. 84.6 ± 26.0 μm in tcf15stl159 at 4 dpf, p < 0.0001; Fig. 4A″′–B″′, white bracketed line, Fig. 4I). The increased variation in distance from the PLLn and spinal cord at 4 dpf is likely because neuromasts deposited early in development migrate ventrally as the larvae continue to develop (Sapède et al., 2002). Altogether, tcf15stl159 mutants show severe patterning defects throughout the lateral line system, with misplaced sensory organs, reduced innervation based on axon position, and hypomyelination of PLLn axons based on mbp expression.
3.5. Motor nerves are directly mispatterned in tcf15stl159 mutants
PLLn axon and glial development has been previously shown to follow the migration of the primordium (Gilmour et al., 2002, 2004). Therefore, the PLLn patterning defects observed in tcf15stl159 larvae could be a secondary consequence of aberrant primordium migration, rather than a direct effect of environment on the nerve itself. We therefore investigated the development of motor nerves, which are comprised of axons emerging from the spinal cord and migrating along and innervating body wall muscle. nbt:dsRed labeled motor neurons were assayed in individual larvae at 36 and 72 hpf to characterize motor nerve development over time. In both wild-type and tcf15stl159 larvae, motor axons have exited the spinal cord and are migrating down muscle segments by 36 hpf (Fig. 5A–B). However, motor nerve length is shorter in tcf15stl159 larvae relative to wild-type siblings (82.0 ± 20.6 μm in wild-type vs. 60.4 ± 11.1 μm in tcf15stl159, p < 0.0001, Fig. 5A–B bracketed lines, 5F). By 72 hpf, both wild-type and tcf15stl159 siblings have elongated motor nerves (p < 0.0001 vs. 36 hpf timepoints), but motor nerves in tcf15stl159 remain shorter than wild-type overall (149.3 ± 12.7 μm in wild-type vs. 105.5 ± 10.2 μm in tcf15stl159, p < 0.0001, Fig. 5C–D bracketed lines, 5F). We also found slightly fewer motor nerves overall in tcf15stl159 larvae (Fig. 5E, 28.8 ± 1.2 motor nerves in n = 24 siblings vs. 26.4 ± 1.0 in n = 10 mutants, p < 0.0001). Because motor nerves interact directly with muscle during elongation, we suspect that tcf15 is generally required for axon elongation in the peripheral nervous system.
Fig. 5. tcf15 is required for motor nerve extension and fasciculation.

(A–D) Motor nerves in nbt:dsRed-expressing (red) at 36 (A–B) and 72 (C–D) hpf in sibling (sib) and tcf15stl159 larvae. Scale bar, 100 μm. Examples of motor nerve length measurement are indicated with a white bracketed line. PLLn are visible at 72 hpf and marked with gray arrows (C); arrowheads mark minor branches of motor nerves at 72 hpf (C–D). (E) Quantification of motor nerves on one side of 72 hpf larvae. There are slightly but significantly fewer motor nerves in tcf15stl159 relative to siblings, ****p < 0.0001, 1-way ANOVA with Tukey test. (F) Quantification of motor nerve length in sibling and tcf15stl159 larvae at 36 and 72 hpf. At both timepoints tcf15stl159 have shorter motor nerves, ****p < 0.0001, 1-way ANOVA with Tukey test. Dots represent individual motor nerves. (G) Schematic for counting motor nerve branches; both main and minor branches are counted equally. (H) Quantification of branches in motor nerves and tcf15stl159 larvae at 72 hpf. Tcf15stl159 have more branches motor nerves, ****p < 0.0001, 1-way ANOVA with Tukey test. Dots represent individual motor nerves.
In addition to extension defects, we observed that motor nerves in tcf15stl159 mutants were disorganized relative to wild-type siblings. By 72 hpf, wild-type motor nerves are neatly bundled and uniformly extended along the middle of the muscle segment, with few spurious processes (Fig. 5C, arrowheads). This patterning is disrupted in tcf15stl159 mutants, with multiple processes appearing to emerge from the spinal cord as well as processes extending off main branches. We quantified this defect by counting terminal branches, as our approach could not distinguish motor axons that were defasciculated vs. additional branches of a single motor axon (Fig. 5G). With this approach, we found excess terminal branches in tcf15stl159 mutants relative to wild-type siblings at 3 dpf (Fig. 5H, 2.3 ± 0.8 branches in wild-type vs. 3.3 ± 1.3 in tcf15stl159, p < 0.0001). This motor nerve phenotype is reminiscent of the mis-routed PLLn sensory nerve phenotype and suggests that tcf15 is broadly required both for proper nerve position as well as nerve elongation throughout the peripheral nervous system.
3.6. tcf15 is expressed in muscle cells prior to peripheral nerve patterning
Given the necessity of tcf15 across tissue types for proper development, we next questioned where tcf15 is expressed to have this broad role in development. Expression of tcf15 has been previously established across multiple systems as a critical regulator for muscle patterning (Burgess et al., 1995; Lee et al., 2009; Sánchez and Sánchez, 2015). We therefore examined the spatiotemporal expression of tcf15 using an established tcf15 probe (Topczewska et al., 2001) for in situ hybridization in wild-type larvae at 12 and 24 hpf, prior to nerve extension, and at 3 dpf when axons continue to elongate and premyelinating Schwann cells populate the PLLn. Consistent with previous reports, we found robust tcf15 expression in presomitic mesoderm at 12 hpf that becomes restricted to the posterior by 24 hpf (Fig. 6A–B), distal from the developing PLLn and motor nerves. Furthermore, tcf15 expression was not detected at 3 dpf or later (Fig. 6C), and notably was never detected in PLLn Schwann cells nor the ganglion from which PLLn axons extend. These data suggest that the role of tcf15 in nerve patterning is likely non-cell-autonomous and results from its transcriptional function in muscle that sets up the proper environment for nerve development, rather than functioning directly in neurons or glia.
Fig. 6. tcf15 is expressed in muscle early to set up proper architecture for nerve development.

(A–C) Whole mount in situ hybridization of tcf15 in wild-type embryos at 12 hpf (A-A′), 24 hpf (B), and 3 dpf (C). Note strong expression (arrowheads) at 12 hpf that moves to the posterior by 24 hpf. No tcf15 is observed in the PLLn (arrow, C) or surrounding muscle by 3 dpf. (D–E) Brightfield imaging showing normal gross muscle patterning in sibling (E) and tcf15stl159 mutant (F) 3 dpf larvae. Bracketed lines indicate one muscle segment; white curved arrow indicates measurement of muscle boundary angle from dorsal. (F) Quantification of number of muscle segments/larvae in sibling and tcf15stl159 mutant larvae at 3 and 5 dpf. No significant difference (NS) is observed between genotype or timepoints (p > 0.05, 1-way ANOVA). (G) Quantification of body length, measured from ear to tail tip (fin excluded), of sibling and tcf15stl159 mutant larvae at 3 and 5 dpf. No significant differences (NS) are observed between genotypes at each timepoint (p > 0.05, 1-way ANOVA). (H) Quantification of angle of muscle boundary from dorsal for five representative muscle segments (from segments 8–13) in sibling and tcf15stl159 mutant larvae at 3 and 5 dpf. Angle is slightly but significantly broader in tcf15stl159 mutants relative to age-matched siblings at both timepoints, ****p < 0.0001, 1-way ANOVA. (I) Schematic of larval cross-section at 5 dpf with major anatomical features noted. Regions of interest for panels J–K and L-M are indicated with dotted boxes. (J–K) TEM of sibling (J) and tcf15stl159 mutant (K) 5 dpf larvae. Horizontal myoseptum is prominent in sibling (J, arrow) and absent in mutant. Magnified regions of interest (J′-K′) are noted with a dotted box. In sibling larva (J′), there are numerous myelinated axons (pseudocolored green) with neighboring Schwann cells (sc) and muscle (m). Unmyelinated bundles of axons (a, white arrowhead) are present at this stage. The mutant larva (K′) has disorganized muscle (m) with unidentified adjacent cells, potentially Schwann cells (*). (L–M) Ultrastructure of medial body wall muscle (m) is organized in sibling (L), but has aberrant ultrastructure in mutants with small bundle of unmyelinated axons (a, arrowhead) in mutants (M).
We next examined body wall muscle development in tcf15stl159 mutants at both gross anatomical and ultrastructural levels. At 72 hpf, both wild-type and stl159 siblings have chevron-shaped muscle segments with distinct boundaries (Fig. 6D–E). We counted muscle segments and found no differences between wild-type and tcf15stl159 siblings in segment number (Fig. 6F, 29 ± 0.9 in wild-type vs. 28.5 ± 1.1 in tcf15stl159 at 72 hpf, p > 0.5; 29.4 ± 0.9 in wild-type vs. 29.5 ± 1.1 in tcf15stl159 at 5 dpf, p > 0.9). Furthermore, segments are the same size based on similarity of overall body length (Fig. 6G, 2.8 ± 0.1 μm in wild-type vs. 2.8 ± 0.1 μm in tcf15stl159 at 72 hpf, p > 0.99; 2.9 ± 0.1 μm in wild-type vs. 3.0 ± 0.1 μm in tcf15stl159 at 5 dpf, p > 0.1). The only slight difference observed was angle of apposition between muscle segments, with wild-type muscle boundaries forming a steeper angle compared to tcf15stl159 mutants at 72 hpf (Fig. 6H, 42 ± 4° in wild-type vs. 53 ± 6° in tcf15stl159, p < 0.0001). The difference is reduced, though still persists, at 5 dpf (45 ± 5° in wild-type vs. 50 ± 6° in tcf15stl159, p < 0.0001). We concluded that gross muscle organization is largely normal in tcf15stl159 mutants, with mild patterning defects relative to the severe disorganization of the peripheral nervous system.
Because muscle serves as the substrate for peripheral nerve development, we wanted to visualize the interface of PLLn development with body wall muscle. We therefore performed transmission electron microscopy (TEM) on cross-sections of stl159 sibling and mutant larvae at 5 dpf (Fig. 6I). In these cross sections, the PLLn can be located as a lateral superficial sensory nerve nestled between the skin and muscle at the junction of the horizontal myoseptum. PLLn development appeared normal in siblings (N = 3), with correctly positioned axons and normal myelinating Schwann cells (Fig. 6J–J′). Muscle patterning was also normal, with a prominent horizontal myoseptum (Fig. 6J) and dense skeletal muscle (Fig. 6J’, L). However, both nerve and muscle showed ultrastructural defects in tcf15stl159 mutants. PLLn axons were not visible in the expected lateral position in any tcf15stl159 mutants (N = 3), though some unidentified cells resembling migrating Schwann cells were located in a lateral position near skeletal muscle (Fig. 6K–K’). In addition, muscle ultrastructure was disorganized in tcf15stl159 mutants, with no apparent horizontal myoseptum and fewer intact skeletal muscle myocytes (Fig. 6K). A small bundle of small-calibre axons was found in a more medial position, adjacent to muscle but without myelin or presumptive Schwann cells (Fig. 6M). These defects were restricted to the periphery, as axons and myelin appear normal in the spinal cord of tcf15stl159 mutants (Fig. S3). Thus, while gross anatomy of muscle is normal in tcf15stl159 mutants, defects in the cellular substrate produce aberrant peripheral nerve migration and development. Taken together, our results suggest that early tcf15 expression in muscle is largely dispensable for gross muscle architecture, but is required for correct migration and differentiation of peripheral nerves and other neural crest-derived tissues.
4. Discussion
The bHLH transcription factor tcf15 has established roles in the development of paraxial mesoderm in vertebrates (Burgess et al., 1996; Sánchez and Sánchez, 2015; Topczewska et al., 2001). Formerly named paraxis, tcf15 expression is itself induced by surrounding tissues (Šošić et al., 1997) during development of the axial muscle. While tcf15 is not necessary for somite segmentation, in tcf15 mutants, somites fail to undergo mesenchymal-to-epithelial transition (Rowton et al., 2013; Takahashi et al., 2007) and as a result, lack normal patterning of surface markers including cell adhesion molecules and signaling cues on an epithelialized muscle surface. Furthermore, gene expression analysis in Paraxis−/− mice demonstrated loss of several cell adhesion molecules and classical guidance factors. Misexpression of these factors suggests that nerves, Schwann cells, and other developing neural crest-derived tissues would be disrupted in tcf15 mutant zebrafish.
Here, we identify a novel tcf15 mutant in zebrafish and demonstrate that tcf15 is necessary for the development of peripheral nerves and sensory organs. Though tcf15 is conserved and expressed in zebrafish muscle, mutant analysis had not been previously performed in zebrafish, and the role of tcf15 in development of myelinated peripheral nerves has not been explored via mutant analysis in any system. Using a combination of genomic and transgenic studies in zebrafish, we demonstrate that a novel tcf15 mutant has deficits in axon outgrowth and fasciculation alongside misplaced neural crest derivatives. We predict that these neural development defects are due to the incorrectly patterned muscle surface, resulting in a loss of appropriate guidance cues for peripheral nervous system patterning.
4.1. Axon outgrowth deviates from sensory and motor targets in the absence of tcf15
Previous work has demonstrated that axons of the PLLn typically follow the collectively migrating cells of lateral line primordia, with Schwann cell precursors migrating in close proximity. Genetic and transplantation experiments revealed that the lateral line primordium provides cues that are sufficient for axon extension (Gilmour et al., 2002, 2004). Primordium migration is itself dependent upon sdf-1 and cxcr4 as well as glypican4 that modulates Fgf/Wnt signaling (Dalle Nogare et al., 2014; Li et al., 2004; Venero Galanternik et al., 2016). Glypican4 and Sdf-1 are expressed by the developing muscle surrounding the lateral line. Additionally, the receptor tyrosine kinase Met and its ligand Hgf (hepatocyte growth factor) are necessary for proper muscle development as well as for proper neuromast deposition in a mechanism independent from sdf-1/cxcr4 (Haines et al., 2004). Altogether, this underscores that muscle-derived cues are necessary for proper development of the posterior lateral line (PLL) system. Importantly, in our study, we observed that neuromasts are not only mispatterned in tcf15 mutants, but innervating axons appear to extend further to innervate their neuromast targets, suggesting a loss of coordinated migration. Therefore, we predict that tcf15 is necessary to maintain the association of sensory axons with their peripheral targets.
Neuromasts in the PLL are misplaced in tcf15 mutants, which is consistent with prior observations suggesting the importance of muscle in patterning this system. However, we also observed inappropriate patterning of motor nerves, which use intrinsic and extrinsic cues that likely differ from lateral line axon development. Several muscle-derived factors have been reported for extension and branch patterns of zebrafish motor nerves during both development and regeneration; critically, the specification of the muscle is itself necessary for motor axon targeting (Banerjee et al., 2015; Bremer and Granato, 2016; Isaacman-Beck et al., 2015; Sainath and Granato, 2013; Walker et al., 2021). Given that we observed mispatterning of motor nerves concurrent with sensory nerve wandering and hypomyelination, we predict that tcf15 represents a critical global mediator of peripheral nerve patterning.
4.2. tcf15 is required for development of peripheral myelinating glia
In the PLLn, axons and glia co-develop as the nerve extends posteriorly from the ganglion. Axons are at least partially instructive for glial migration, as glial cells often follow wandering or stalling axons in mutants. This is likely due to Neuregulin-1 type III (Nrg1) expression on axons that activates ErbB2/3 receptors on Schwann cells (Lyons et al., 2005; Perlin et al., 2011). Though PLLn axons can extend normally in the absence of developing Schwann cells, the PLLn later defasciculates, suggesting that Schwann cells are necessary for maturation and stabilization of the PLLn (Gilmour et al., 2002; Raphael et al., 2010). In our tcf15 mutants, we observed fewer Schwann cell precursors, marked by sox10:gfp along the PLLn, as well as fewer mature myelinating Schwann cells as indicated by mbp expression and TEM. Altogether, this suggests that tcf15 is required non-cell-autonomously for the development of myelinated peripheral nerves, and more generally, that the external muscle environment can influence the intricate relationship between developing peripheral axons and their myelinating glia.
Our data demonstrating muscle influence on development of myelinated peripheral nerves are in accordance with earlier studies showing muscle influence on Schwann cell development. Axons of motor neurons emerge from ventral domains of the spinal cord as Schwann cell precursors delaminate from the neural crest and migrate along axonal trajectories (Eisen and Weston, 1993; Pike et al., 1992). Though axons are instructive for glial migration when mispatterned, glial cells are patterned correctly when motor axons are ablated, suggesting that axons are sufficient but not necessary for glial cell migration. Further investigation revealed that muscle-specific kinase (MuSK) serves as a cue for glial cell guidance, as it limits migration to the central muscle segment in the absence of axons (Banerjee et al., 2011). Previously, we demonstrated that expression of ectopic Laminin α2 expressed from adjacent muscle is sufficient to promote myelination in the PLLn of hypomyelinating zebrafish mutants (Petersen et al., 2015). In humans, mutations in lama2 can result in merosin-deficient congenital muscular dystrophy, and absence of Laminin α2 causes disorganized muscle fibers and decreased muscle tone in animal models and humans (Helbling-Leclerc et al., 1995; Miyagoe et al., 1997). Additionally, Laminin α2 interacts with integrins and dystrophin complexes expressed on both Schwann cells and muscle, and loss of integrin and dystrophin signaling in humans results in congenital neuromuscular disorders with coincident dysmyelination (Colognato et al., 1997; McKee et al., 2012; Pellegatta et al., 2013; Quattrini et al., 1996).
Taken together, the data suggest that glial cells and muscle have overlapping molecular factors mediating their development, and loss of these factors can affect glial and muscle development globally. Thus, the partial uncoupling of axon-glial development observed in mutant tcf15 PLLn presents two intriguing possibilities: first, that migrating Schwann cell precursors rely upon muscle-derived guidance cues in the absence of axonal cues, or second, that mispatterned muscle cues have the potential to supersede axonal cues for glial development. Future work can characterize this system and distinguish between these possibilities.
4.3. A transcriptional program in muscle for peripheral nerve patterning
While our study does not formally address cell autonomy, we predict that tcf15 exerts indirect effects on nervous system development through muscle expression. Though tcf15 is a well-established regulator of musculoskeletal development, bHLH in general, and tcf15 in particular, can be combined to induce neural reprogramming (Flitsch et al., 2020; Liu et al., 2018). This suggests that global loss of tcf15 could result in improper development of neurons and glia in a cell-autonomous fashion. However, we do not see expression of tcf15 via in situ hybridization in any peripheral axons or glia at any stage. Expression in larval zebrafish is restricted to developing muscle as previously described in other systems (Burgess et al., 1995; Carpio et al., 2004; Shanmugalingam and Wilson, 1998); we do not see expression in the neural tube as reported in Xenopus (Carpio et al., 2004). Tcf15 expression is also nearly undetected in mouse sciatic nerves based on the Sciatic Nerve Atlas (Gerber et al., 2021). Therefore, the most parsimonious model is that tcf15 directs muscle development to promote a proper scaffold for neurons and glial development, including directed co-migration of axons and glia, stabilization of axon fasciculation, and maturation of myelinating Schwann cells.
Our data suggest that, in zebrafish, muscle development appears largely normal with respect to size and number of somites, suggesting that tcf15 is dispensable for axis elongation and the somitogenic clock, but instead is necessary for patterning of the muscle as a microenvironment that directs nerve development. Previous work demonstrated that loss of the transcription factor foxc1a in zebrafish pectoral fin muscles results in misdirected axons, and identification of downstream targets of foxc1a in pectoral muscle revealed another transcription factor implicated in somitogenesis, foxd5, as well as classical guidance factors (Banerjee et al., 2015). Additionally, foxd5 is required for proper expression of paraxis/tcf15 and mediates zebrafish somitogenesis (Lee et al., 2009). Taken together, this predicts a transcriptional cascade within muscle necessary for peripheral nerve patterning. Furthermore, because we observe that tcf15 expression precedes posterior axon-glial outgrowth by ~12 h, we predict that tcf15 functions early in this cascade to mediate expression of factors for nerve development.
Given that tcf15 functions primarily as a transcriptional activator, numerous downstream targets could play a role in directing nerve development non-cell-autonomously. For example, eighteen genes encoding cell adhesion molecules, including vascular cell adhesion molecule 1 (Vcam1) and Neuroligin1, are downregulated in Paraxis−/− mice (Rowton et al., 2013), suggesting they might function in adhesion for epithelization during muscle development. Additionally, members of the A disintegrin and Metalloprotease (ADAM) family, Adam18 and Adamtsl3, were downregulated in the absence of tcf15, suggesting alteration of the extracellular environment (Rowton et al., 2013). Disruption of both intercellular and matrix interactions could prevent formation of muscle tissue with normal architecture, which is aligned with our findings of aberrant muscle ultrastructure in tcf15stl159 mutant zebrafish. This altered muscle environment could affect the trajectory of migrating axons and glia.
Guidance molecules implicated in PNS development are also reduced in Paraxis−/− mutant mice (Rowton et al., 2013), including Netrin 1 and Neurotrimin which can both direct neurite extension (Gil et al., 1998; Serafini et al., 1994). Eph/Ephrin signaling is critical for restricted migration of neural crest cells (Kuriyama and Mayor, 2008; Robinson et al., 1997) and axon targeting (Cramer and Miko, 2016; Muñoz et al., 2005). In Paraxis−/− mutant mice, the normal restriction of ephrinB2 to posterior halves of the somite is disrupted, and EphA3 and EphA7 receptor expression are reduced throughout somites as well (Johnson et al., 2001). Finally, although somites appear to form in Paraxis−/− mutants, Laminin expression is lost at intersomitic boundaries and diffuse along the muscle surface (Rowton et al., 2013). This is of particular importance given that Laminins direct multiple stages of Schwann cell development (Feltri and Wrabetz, 2005; Petersen et al., 2015). Thus, reduced Laminin levels in Paraxis−/− mutants could disrupt PNS patterning through the simultaneous disruption of Schwann cell development as well as muscle architecture.
Taken together, we predict that tcf15 controls a pattern of both restrictive and permissive or instructive molecules expressed on the muscle surface that promote peripheral nerve patterning across an intact skeletal muscle scaffold. Identification of these factors has the potential to uncover the complex relationships between neurons, glial cells, and their molecular environment, as well as understand the cause of inherited neuromuscular disorders.
Supplementary Material
Acknowledgements
We thank our colleagues at Kenyon College and members of the Petersen laboratory for critical feedback, thoughtful discussion, and generosity with equipment and reagents. We thank Rebecca Cunningham, Lila Solnica-Krezel, and members of the Monk and Solnica-Krezel laboratories at Washington University in St. Louis for reagents and the initial identification of the stl159 allele, as well as Sarah Kucenas and Cody Smith for additional zebrafish transgenic strains. We thank Becky Gallagher and the animal care staff at Kenyon College for excellent zebrafish care. This work was supported by NIH F32 NS087786 and NSF BIO-IOS #1941664 to S.C.P., NIH R01 HD080601 to K.R.M., and Kenyon College Summer Science Scholars fellowships to E.A.P., A.A., and K.P.
Footnotes
Declaration of competing interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ydbio.2022.07.001.
References
- Banerjee S, Gordon L, Donn TM, Berti C, Moens CB, Burden SJ, Granato M, 2011. A novel role for MuSK and non-canonical Wnt signaling during segmental neural crest cell migration. Development 138, 3287–3296. 10.1242/dev.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Hayer K, Hogenesch JB, Granato M, 2015. Zebrafish foxc1a drives appendage-specific neural circuit development. Development 142, 753–762. 10.1242/dev.115816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, Pfaff SL, 2010. Motor axon pathfinding. Cold Spring Harbor Perspect. Biol. 2, a001735. 10.1101/cshperspect.a001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J, Granato M, 2016. Myosin phosphatase fine-tunes zebrafish motoneuron position during axonogenesis. PLoS Genet. 12, e1006440. 10.1371/journal.pgen.1006440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosamle C, Halpern ME, 2002. Characterization of myelination in the developing zebrafish. Glia 39, 47–57. 10.1002/glia.10088. [DOI] [PubMed] [Google Scholar]
- Burgess R, Cserjesi P, Ligon KL, Olson EN, 1995. Paraxis: a basic helix-loop-helix protein expressed in paraxial mesoderm and developing somites. Dev. Biol. 168, 296–306. 10.1006/dbio.1995.1081. [DOI] [PubMed] [Google Scholar]
- Burgess R, Rawls A, Brown D, Bradley A, Olson EN, 1996. Requirement of the paraxis gene for somite formation and musculoskeletal patterning. Nature 384, 570–573. 10.1038/384570a0. [DOI] [PubMed] [Google Scholar]
- Carpio R, Honoré SM, Araya C, Mayor R, 2004. Xenopus paraxis homologue shows novel domains of expression. Dev. Dynam. 231, 609–613. 10.1002/dvdy.20147. [DOI] [PubMed] [Google Scholar]
- Colognato H, MacCarrick M, O’Rear JJ, Yurchenco PD, 1997. The laminin alpha2-chain short arm mediates cell adhesion through both the alpha1beta1 and alpha2beta1 integrins. J. Biol. Chem. 272, 29330–29336. 10.1074/jbc.272.46.29330. [DOI] [PubMed] [Google Scholar]
- Cramer KS, Miko IJ, 2016. Eph-ephrin signaling in nervous system development. F1000Res 5. 10.12688/f1000research.7417.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham RL, Monk KR, 2018. Whole mount in situ hybridization and immunohistochemistry for zebrafish larvae. Methods Mol. Biol. 1739, 371–384. 10.1007/978-1-4939-7649-2_25. [DOI] [PubMed] [Google Scholar]
- Cunningham RL, Herbert AL, Harty BL, Ackerman SD, Monk KR, 2018. Mutations in dock1 disrupt early Schwann cell development. Neural Dev. 13, 17. 10.1186/s13064-018-0114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czopka T, Lyons DA, 2011. Dissecting mechanisms of myelinated axon formation using zebrafish. Methods Cell Biol. 105, 25–62. 10.1016/b978-0-12-381320-6.00002-3. [DOI] [PubMed] [Google Scholar]
- Dalle Nogare D, Somers K, Rao S, Matsuda M, Reichman-Fried M, Raz E, Chitnis AB, 2014. Leading and trailing cells cooperate in collective migration of the zebrafish posterior lateral line primordium. Development 141, 3188–3196. 10.1242/dev.106690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JS, Weston JA, 1993. Development of the neural crest in the zebrafish. Dev. Biol. 159, 50–59. 10.1006/dbio.1993.1220. [DOI] [PubMed] [Google Scholar]
- Feltri ML, Wrabetz L, 2005. Laminins and their receptors in Schwann cells and hereditary neuropathies. J. Peripher. Nerv. Syst. : JPNS 10, 128–143. 10.1111/j.1085-9489.2005.0010204.x. [DOI] [PubMed] [Google Scholar]
- Flitsch LJ, Laupman KE, Brüstle O, 2020. Transcription factor-based fate specification and forward programming for neural regeneration. Front. Cell. Neurosci. 14, 121. 10.3389/fncel.2020.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo VE, Liang J, Behra M, Elkahloun A, Villablanca EJ, Russo V, Allende ML, Burgess SM, 2010. Molecular dissection of the migrating posterior lateral line primordium during early development in zebrafish. BMC Dev. Biol. 10, 120. 10.1186/1471-213X-10-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammill LS, Roffers-Agarwal J, 2010. Division of labor during trunk neural crest development. Dev. Biol. 344, 555–565. 10.1016/j.ydbio.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber D, Pereira JA, Gerber J, Tan G, Dimitrieva S, Yángüez E, Suter U, 2021. Transcriptional profiling of mouse peripheral nerves to the single-cell level to build a sciatic nerve ATlas (SNAT). Elife 10. 10.7554/eLife.58591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghysen A, Dambly-Chaudière C, 2004. Development of the zebrafish lateral line. Curr. Opin. Neurobiol. 14, 67–73. 10.1016/j.conb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Gil OD, Zanazzi G, Struyk AF, Salzer JL, 1998. Neurotrimin mediates bifunctional effects on neurite outgrowth via homophilic and heterophilic interactions. J. Neurosci. 18, 9312–9325. 10.1523/JNEUROSCI.18-22-09312.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DT, Maischein HM, Nüsslein-Volhard C, 2002. Migration and function of a glial subtype in the vertebrate peripheral nervous system. Neuron 34, 577–588. 10.1016/S0896-6273(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Gilmour D, Knaut H, Maischein HM, Nüsslein-Volhard C, 2004. Towing of sensory axons by their migrating target cells in vivo. Nat. Neurosci. 7, 491–492. 10.1038/nn1235. [DOI] [PubMed] [Google Scholar]
- Haines L, Neyt C, Gautier P, Keenan DG, Bryson-Richardson RJ, Hollway GE, Cole NJ, Currie PD, 2004. Met and Hgf signaling controls hypaxial muscle and lateral line development in the zebrafish. Development 131, 4857–4869. 10.1242/dev.01374. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. , 1995. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11, 216–218. 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Isaacman-Beck J, Schneider V, Franzini-Armstrong C, Granato M, 2015. The lh3 glycosyltransferase directs target-selective peripheral nerve regeneration. Neuron 88, 691–703. 10.1016/j.neuron.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R, 2005. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Jing L, Lefebvre JL, Gordon LR, Granato M, 2009. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron 61, 721–733. 10.1016/j.neuron.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Rhee J, Parsons SM, Brown D, Olson EN, Rawls A, 2001. The anterior/posterior polarity of somites is disrupted in paraxis-deficient mice. Dev. Biol. 229, 176–187. 10.1006/dbio.2000.9969. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF, 1995. Stages of Embryonic Development of the Zebrafish. Developmental Dynamics, vol. 203. an official publication of the American Association of Anatomists, pp. 253–310. 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Labun K, Krause M, Torres Cleuren Y, Valen E, 2021. CRISPR genome editing made easy through the CHOPCHOP website. Curr Protoc 1, e46. 10.1002/cpz1.46. [DOI] [PubMed] [Google Scholar]
- Lee HC, Tseng WA, Lo FY, Liu TM, Tsai HJ, 2009. FoxD5 mediates anterior-posterior polarity through upstream modulator Fgf signaling during zebrafish somitogenesis. Dev. Biol. 336, 232–245. 10.1016/j.ydbio.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Li Q, Shirabe K, Kuwada JY, 2004. Chemokine signaling regulates sensory cell migration in zebrafish. Dev. Biol. 269, 123–136. 10.1016/j.ydbio.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yu C, Daley TP, Wang F, Cao WS, Bhate S, Lin X, Still C, Liu H, Zhao D, et al. , 2018. CRISPR activation screens systematically identify factors that drive neuronal fate and reprogramming. Cell Stem Cell 23, 758–771. 10.1016/j.stem.2018.09.003e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DA, Pogoda HM, Voas MG, Woods IG, Diamond B, Nix R, Arana N, Jacobs J, Talbot WS, 2005. erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr. Biol. 15, 513–524. 10.1016/j.cub.2005.02.030. S0960-9822(05)00171-5 [pii]. [DOI] [PubMed] [Google Scholar]
- McKee KK, Yang DH, Patel R, Chen ZL, Strickland S, Takagi J, Sekiguchi K, Yurchenco PD, 2012. Schwann cell myelination requires integration of laminin activities. J. Cell Sci. 125, 4609–4619. 10.1242/jcs.107995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Takeda S, 1997. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415, 33–39. 10.1016/s0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- Muñoz LM, Zayachkivsky A, Kunz RB, Hunt JM, Wang G, Scott SA, 2005. Ephrin-A5 inhibits growth of embryonic sensory neurons. Dev. Biol. 283, 397–408. 10.1016/j.ydbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Muppirala AN, Limbach LE, Bradford EF, Petersen SC, 2021. Schwann cell development: from neural crest to myelin sheath. Wiley Interdiscip Rev Dev Biol 10, e398. 10.1002/wdev.398. [DOI] [PubMed] [Google Scholar]
- Pellegatta M, De Arcangelis A, D’Urso A, Nodari A, Zambroni D, Ghidinelli M, Matafora V, Williamson C, Georges-Labouesse E, Kreidberg J, et al. , 2013. alpha6beta1 and alpha7beta1 integrins are required in Schwann cells to sort axons. J. Neurosci. : the official journal of the Society for Neuroscience; 33, 17995–18007. 10.1523/jneurosci.3179-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri F, Nüsslein-Volhard C, 2008. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916–927. 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Perlin JR, Lush ME, Stephens WZ, Piotrowski T, Talbot WS, 2011. Neuronal Neuregulin 1 type III directs Schwann cell migration. Development 138, 4639–4648. 10.1242/dev.068072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SC, Luo R, Liebscher I, Giera S, Jeong SJ, Mogha A, Ghidinelli M, Feltri ML, Schoneberg T, Piao X, Monk KR, 2015. The adhesion GPCR GPR126 has distinct, domain-dependent functions in schwann cell development mediated by interaction with laminin-211. Neuron 85, 755–769. 10.1016/j.neuron.2014.12.057. S0896-6273(14)01166-0 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike SH, Melancon EF, Eisen JS, 1992. Pathfinding by zebrafish motoneurons in the absence of normal pioneer axons. Development 114, 825–831. 10.1242/dev.114.4.825. [DOI] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L, 1996. Beta 4 integrin and other Schwann cell markers in axonal neuropathy. Glia 17, 294–306. 10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Raphael AR, Perlin JR, Talbot WS, 2010. Schwann cells reposition a peripheral nerve to isolate it from postembryonic remodeling of its targets. Development 137, 3643–3649. 10.1242/dev.057521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowton M, Ramos P, Anderson DM, Rhee JM, Cunliffe HE, Rawls A, 2013. Regulation of mesenchymal-to-epithelial transition by PARAXIS during somitogenesis. Dev. Dynam. 242, 1332–1344. 10.1002/dvdy.24033. [DOI] [PubMed] [Google Scholar]
- Sainath R, Granato M, 2013. Plexin A3 and turnout regulate motor axonal branch morphogenesis in zebrafish. PLoS One 8, e54071. 10.1371/journal.pone.0054071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez RS, Sánchez SS, 2015. Paraxis is required for somite morphogenesis and differentiation in Xenopus laevis. Dev. Dynam. 244, 973–987. 10.1002/dvdy.24294. [DOI] [PubMed] [Google Scholar]
- Sanchez NE, Harty BL, O’Reilly-Pol T, Ackerman SD, Herbert AL, Holmgren M, Johnson SL, Gray RS, Monk KR, 2017. Whole genome sequencing-based mapping and candidate identification of mutations from fixed zebrafish tissue. G3 (Bethesda) 7, 3415–3425. 10.1534/g3.117.300212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapède D, Gompel N, Dambly-Chaudière C, Ghysen A, 2002. Cell migration in the postembryonic development of the fish lateral line. Development 129, 605–615. 10.1242/dev.129.3.605. [DOI] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M, 1994. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78, 409–424. 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- Shanmugalingam S, Wilson SW, 1998. Isolation, expression and regulation of a zebrafish paraxis homologue. Mech. Dev. 78, 85–89. 10.1016/s0925-4773(98)00150-6. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Morris AD, Welsh TG, Kucenas S, 2014. Contact-mediated inhibition between oligodendrocyte progenitor cells and motor exit point glia establishes the spinal cord transition zone. PLoS Biol. 12, e1001961. 10.1371/journal.pbio.1001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šošić D, Brand-Saberi B, Schmidt C, Christ B, Olson EN, 1997. Regulation of paraxis expression and somite formation by ectoderm- and neural tube-derived signals. Dev. Biol. 185, 229–243. 10.1006/dbio.1997.8561. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Takagi A, Hiraoka S, Koseki H, Kanno J, Rawls A, Saga Y, 2007. Transcription factors Mesp2 and Paraxis have critical roles in axial musculoskeletal formation. Dev. Dynam. 236, 1484–1494. 10.1002/dvdy.21178. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B, 2008. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69. 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Topczewska JM, Topczewski J, Shostak A, Kume T, Solnica-Krezel L, Hogan BL, 2001. The winged helix transcription factor Foxc1a is essential for somitogenesis in zebrafish. Genes Dev. 15, 2483–2493. 10.1101/gad.907401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venero Galanternik M, Lush ME, Piotrowski T, 2016. Glypican4 modulates lateral line collective cell migration non cell-autonomously. Dev. Biol. 419, 321–335. 10.1016/j.ydbio.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Walker LJ, Roque RA, Navarro MF, Granato M, 2021. Agrin/Lrp4 signal constrains MuSK-dependent neuromuscular synapse development in appendicular muscle. Development 148. 10.1242/dev.199790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Rawls J, Rhee JM, Rawls A, 2004. Paraxis is a basic helix-loop-helix protein that positively regulates transcription through binding to specific E-box elements. J. Biol. Chem. 279, 37685–37692. 10.1074/jbc.M401319200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lefebvre JL, Zhao S, Granato M, 2004. Zebrafish unplugged reveals a role for muscle-specific kinase homologs in axonal pathway choice. Nat. Neurosci. 7, 1303–1309. 10.1038/nn1350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.