

Abstract
Rationale.
Identifying genetic markers for heterogeneous complex diseases such as heart failure is challenging, and requires prohibitively large cohort sizes in genome-wide association studies (GWAS) in order to meet the stringent threshold of genome-wide statistical significance. On the other hand, chromatin quantitative trait loci (QTL), elucidated by direct epigenetic profiling of specific human tissues, may contribute towards prioritising sub-threshold variants for disease-association.
Objective.
Here, we captured non-coding genetic variants by performing epigenetic profiling for enhancer H3K27ac ChIP-seq in 70 human control and end-stage failing hearts.
Methods and Results.
We have mapped a comprehensive catalogue of 47,321 putative human heart enhancers and promoters. 3,897 differential acetylation peaks (FDR 5%) pointed to pathways altered in heart failure (HF). To identify cardiac histone acetylation QTLs (haQTLs), we regressed out confounding factors including HF disease status, and employed the G-SCI test1 to call out 1,680 haQTLs (FDR 10%). RNA-seq performed on the same heart samples proved a subset of haQTLs to have significant association also to gene expression (expression QTLs), either in cis (180), or through long range interactions (81), identified by Hi-C and HiChIP performed on a subset of hearts. Furthermore, a concordant relationship between the gain or disruption of transcription factor (TF) binding motifs, inferred from alternative alleles at the haQTLs, implied a surprising direct association between these specific TF and local histone acetylation in human hearts. Finally, 62 unique loci were identified by colocalisation of haQTLs with the sub-threshold loci of heart-related GWAS datasets.
Conclusions.
Disease and phenotype-association for 62 unique loci are now implicated. These loci may indeed mediated their effect through modification of enhancer H3K27-acetylation enrichment and their corresponding gene expression differences.
Keywords: Basic Science Research, Cardiomyopathy, Heart Failure, Mechanisms, Translational Studies, Histone acetylome, haQTL, GWAS, H3K27ac ChIP-seq, HiChIP, epigenetics, heart failure, genetic variation, chromatin
Graphical Abstract
INTRODUCTION
Genome-wide association studies (GWAS) have identified hundreds of genetic risk variants for some conditions such as atrial fibrillation2, 3, but for other conditions such as dilated cardiomyopathy4 and heart failure (HF)5, multiple HF GWAS6 have yielded only a limited number of significant hits meeting the genome-wide significance threshold of 5 × 10-8. Indeed many more “true” biological signals may be buried or hidden among the noise at sub-threshold p-values in these GWAS analyses, but for a variety of reasons including disease heterogeneity, underpowered sample sizes may explain why true signals do not emerge above the stringent genome-wide significance threshold6.
Chromatin quantitative trait loci (QTLs) on the other hand, have been discovered, in each case, using only a limited handful of tissue samples7–10. Moreover, instead of the analysis for expression QTL11, which is easily compromised by the instability of RNA molecules in difficult-to-obtain tissue such as human hearts, the relative stability of chromatin12 offers a distinct advantage with histone QTL analysis. In a wide range of different cell types, chromatin QTL studies have elucidated novel genes, and their contribution to phenotypic variation. To date however, chromatin QTL studies have only been performed on cell lines, with the exception of a few carried out on human brain10, and blood7. An increasing compendium of studies documents the association between non-coding genetic variants and epigenetic modifications such as histone methylation and acetylation, mapped out using the respective tissue or organ of disease relevance. We therefore hypothesized that an epigenomic profile of human left ventricles (LV), when integrated with well-designed LV-related GWAS datasets, may reveal new genetic variants at noncoding regulatory sequences that mediate their effect on LV traits and diseases through their influence on histone modifications, and downstream consequences of chromatin remodelling and gene expression9. Since many more functional genetic markers may be buried in the sub-threshold statistical significance limits of published GWAS datasets13, direct epigenetic profiling for chromatin QTLs in LV may prioritise variants and their corresponding genes for disease-association.
Hence, we performed epigenome profiling with H3K27-acetylation ChIP-seq to identify active cardiac enhancer loci, using 70 optimally-procured transmural explants of human LV from Caucasians, aged 36 to 76 years old (36 end-stage HF and 34 non-failing (NF) controls) (Online Table I). The preponderance of disease-associated non-coding SNPs lies in non-coding genomic enhancers, which can regulate multiple genes, some megabases away via chromatin folding. We therefore also integrated our H3K27ac ChIP-seq data with H3K27ac HiChIP to identify distal interacting genes and inferred target genes of the haQTLs. While there are examples of Hi-C and Capture Hi-C, such as performed on pluripotent stem cells-derived cardiomyocytes13–15, this is the first instance of enhancer HiChIP performed on human hearts. HiChIP generated focussed high-resolution contact maps16 and increased the yield of conformative-informative yield by over 10 fold16.
METHODS
Data availability.
All FASTQ files are deposited in EBI (EGA Study ID: EGAS00001003586). ChIP-seq read signal in the bigwig format has also been deposited onto UCSC Genome Browser (link: https://genome.ucsc.edu/s/wlwtan/EGAS00001003586). Raw data for RNA-seq have been deposited as part of the Myocardial Applied Genomics Network (MAGNet) study in NCBI GEO (Accession ID: GSE141910).
Ethical statement and human heart tissue.
The Myocardial Applied Genomics Network (MAGNet; www.med.upenn.edu/magnet), collects and banks human cardiac tissue for genomic research. All subjects or next of kin provided written informed consent for tissue donation, and analyses and all study protocols were approved by relevant institutional review boards. Left ventricular (LV) free-wall tissue was harvested at the time of cardiac surgery from subjects with heart failure undergoing transplantation and from unused donor hearts with apparently normal function. Hearts was perfused immediately with cold in-situ high-potassium cardioplegia prior to cardiectomy to arrest contraction and prevent ischemic damage, and tissue specimens were frozen in liquid nitrogen. LV sample metadata are provided in Online Table I.
Human cardiac H3K27ac ChIP-seq.
For each ChIP-seq library, 70 mg of snap frozen human heart left ventricle tissue was cut and pounded into fine powder in liquid nitrogen. Tissues were washed twice in cold PBS (Takara, 1x protease inhibitor), cross-linked with formaldehyde (1%) for 5 min at room temperature and subsequently quenched with glycine (125mM) for 5 min at room temperature. Cell pellets were rinsed twice with cold PBS. Cells were lysed in lysis buffer (50mM HEPES-KOH, pH 7.5, 150mM NaCl, 1mM EDTA, 1% Triton X, 0.1% Sodium deoxycholate, 0.1% SDS, Takara 1X protease inhibitors) using a glass douncer tight pestle ‘A’ with 10 to 15 strokes to release nuclei. Centrifuge was performed at 4000 rpm for 10 min at 4°C to collect the nuclei pellet. Nuclei were checked under microscopy for each preparation. Nuclei were lysed in nuclei lysis buffer (50mM HEPES-KOH, pH 7.5, 150mM NaCl, 1mM EDTA, 1% Triton X, 0.1% Sodium deoxycholate, 1% SDS, Takara, 1x protease inhibitor) and sonicated with Bioruptor sonicator to obtained chromatin fragments between 200 to 500 bp. Sheared chromatin was immunoprecipitated with 5 μg of H3K27ac antibody (catalogue #: ab4729, Abcam) with 50 μl of protein G beads (Invitrogen) overnight at 4°C. Beads were washed and eluted in 200 μl of elution buffer (50mM Tris-HCI, pH7.5 and 10mM EDTA), de-crosslink at 65°C overnight. Pulldown was phenol/chloroform treated and DNA purified by ethanol precipitation. Library preparation was performed using 2 ng ChIP DNA with NEB ultra II library preparation kit, according to manufacturer’s protocol. 10–12 PCR cycles were performed using indexed primers and libraries size between 300 to 500 bp were selected. The libraries underwent paired end sequencing at 2 × 100 bp read length on Illumina Hiseq 2500. Samples were processed in batches as enumerated in Online Table 1. For comparative analysis, we downloaded H3K27ac ChIP-seq data (Experiment Accession: ENCSR557DFM) and H3K4me1 ChIP-seq data (Experiment Accession: ENCFF072ZDB) from ENCODE. We also downloaded human cardiomyocyte H3K27ac and H3K4me1 ChIP-seq data from NCBI Bioproject PRJNA353755.
hES-CM CRISPR-targeted excision and gene knockdown.
Targeting sgRNAs were designed by identifying NGG PAM sites flanking the haQTL candidate at the distal enhancer of NOS1AP and C1orf226 (chr1:162033295–162034795). The bioinformatic tool at http://crispor.tefor.net/ was used. sgRNA constructs were cloned into the Esp3I-digested LentiCRISPRv2 plasmid backbone (Addgene #52961) using T4 DNA ligase (catalogue #: M0202, New England Biolabs, NEB), according to manufacturer’s instructions. Vectors were transformed into RapidTrans™ TAM1 Competent E. coli (catalogue #: 11096, Active Motif). Plasmid DNA was extracted by Monarch® Plasmid Miniprep Kit (NEB) and successful cloning confirmed by Sanger sequencing. Each vector DNA was packaged into individual lentivirus using the protocol described above. For all control groups, H1-hESC derived cardiomyocytes or endothelial cells were transduced with lentivirus carrying the non-targeting control (NTC) sgRNAs: pLentiCRISPRv2-Scramble1 (sgRNA sequence: 5’-AAAACAGGACGATGTGCGGC-3’) and/or pLentiCRISPRv2-Scramble2 (sgRNA sequence: 5’-AACGTGCTGACGATGCGGGC-3’). For the excision of NOS1AP/C1ORF226 distal enhancer (chr1:162033295–162034795), lentiviral pools of independent pairs of guides each were co-transduced at high multiplicity of infection (MOI). Two days post-infection, cells were selected with 3 μg/ml Puromycin (Sigma catalogue #: P9620) and sub-cultured for a further 10 days. Efficient CRISPR-targeted excision was verified by PCR amplification, gel electrophoresis and Sanger sequencing.
See Supplementary Material for full Methods and Statistical Analysis.
RESULTS
Differential H3K27-acetylation between non-failing control (NF) and failing (HF) hearts.
Figure 1A depicts the schematic workflow of our experimental design. We sequenced to an average of 117 ± 52 million unique paired-end reads per ChIP-seq library for each sample (Online Table II). Using the DFilter17 peak-caller, a consensus set of 47,321 H3K27ac peaks was obtained (Online Figure I; Online Table III). Peak calls were analysed carefully for the quality of enrichment scores, ChIP-seq fragment reads, and inter-sample replicability (Online Figure IA, Online Table II). As anticipated, our H3K27ac peaks also correlated consistently to ENCODE datasets for human heart18 (Online Figure IA, B).
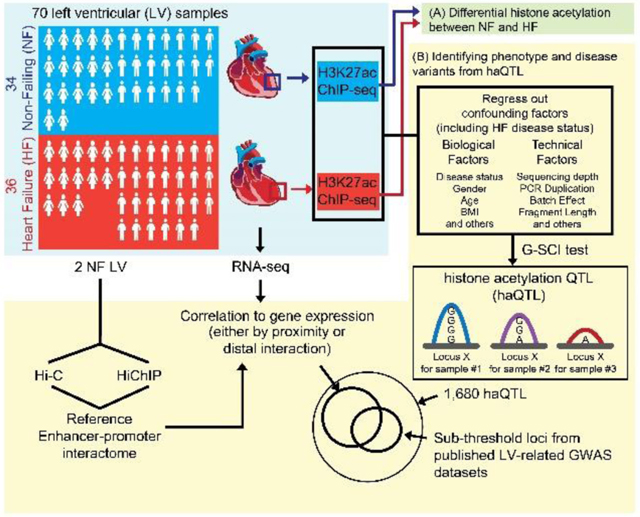
Figure 1. Schematic and differential H3K27-acetylation between non-failing control (NF) and failing (HF) hearts.

(A) Schematic for the experimental workflow. H3K27ac ChIP-seq was performed on 70 human left ventricles (LV). The first stage of the analysis involved (A), a direct analysis for differential histone acetylation between NF and HF. In the second stage (B), all ChIP-seq peaks were collected, and regressed for factors including HF disease status. The G-SCI test was performed to call out haQTLs, where genetic variants were associated with acetylation variation, independently of disease. This was followed by downstream analyses for correlation to gene expression and overlap with sub-threshold signals from published LV-related GWAS datasets. RNA-seq was performed so as to generate matched tissue transcriptomes. Hi-C and HiChIP were performed for 2 independent control (NF) LV to generate a reference cardiac chromatin interactome. (B) Heatmap showing 3,897 differential H3K27-acetylation loci (FDR<0.05, fold-change ≥ 1.3; Wilcoxon Signed-Rank test), and top 10 transcription factor (TF) binding motifs enriched from the loci of hypo- and hyperacetylated peaks, respectively.
To further confirm the functional validity of our acetylation peaks, we assessed for differential acetylation (DA) between control NF and diseased HF hearts. First, we normalized global peak heights for GC content (Online Figure IIA) and distributional skews. Next, we corrected for confounding effects by regressing out technical covariates, and biological covariates including age, sex, and proportion of cardiac myocytes for each sample (Online Figure IIB). Corrected peak heights were used to identify DA loci between NF and HF (Wilcoxon rank sum test; FDR < 0.05, fold change ≥ 1.3). 2,130 peaks showed significant decrease, while 1,767 peaks showed significant increase, for H3K27ac enrichment in HF (Online Table IV). Using the full set of 3,897 DA peaks, NF and HF hearts segregated well on unsupervised hierarchical clustering (Figure 1B). DA loci were subjected to the Genomic Regions Enrichment of Annotations Tool (GREAT)19, revealing gene ontology terms such as regulation of cardiomyocyte differentiation, DNA damage, concordant with well-known ongoing biological processes in heart disease, and familiar heart failure pathophysiological terms of cardioblast differentiation, skeletal muscle tissue development, wnt signalling pathways in upregulated loci, and DNA damage and sarcomere organization in downregulated loci11, 20 (Binomial FDR Q value < 0.05; Online Figure III, Online Table V).
As TFs are generally bound at regulatory loci of active enhancers as marked by H3K27ac 21, we also analysed DA loci for the enrichment of TF motifs using HOMER (Figure 1B). Key cardiac TF motifs for MEF2 and TEAD were enriched in upregulated peaks, consistent with previous evidence of their increased activity in HF22. AP1 and ATF3 motifs were enriched in downregulated peaks, also consistent with the reported reduced activity of ATF3 in worsening hypertensive LV modelling23. Hence, analysis of the loci for differential H3K27ac enrichment implicated established molecular pathways of heart failure disease pathophysiology and progression.
Human heart histone-acetylation Quantitative Trait Loci (haQTL).
Next, in order to curate for the genetic variants embedded in the cardiac H3K27ac enhancer loci, we performed variant-calling using GATK to identify SNPs within the H3K27ac peaks, arriving at a final high quality set of 249,732 SNPs (Figure 2A). Nearly all SNPs (99.46%) were as annotated before in gnomAD24 or dbSNP25. The remainder 1,367 novel SNPs were sufficiently well covered by sequencing read counts to give confidence of their validity (Online Figure IVA). Moreover, we also validated the accuracy of some SNPs using the orthogonal method of Sanger sequencing (Online Figure IVB, C). We then proceeded to use the G-SCI test1, to analyse for SNPs that correlated to their local cis acetylation peak heights (i.e. histone acetylation QTLs, or haQTLs). Importantly, in order to make sure that the general disease effect is removed from the effect of regulatory SNPs, we adjusted acetylation peak heights by regressing out HF disease status (Online Figure IIC). 1,680 H3K27ac peaks were finally identified to have significant association with at least one SNP at the same locus (10% FDR) (Figure 2B, Online Table VI). At each of these 1,680 acetylation peaks, a sentinel SNP was nominated to represent the haQTL, based on FDR ranking. Population MAF for the haQTLs are represented in Online Figure IVD. We validated a randomly selected list of these reference and alternative alleles for enhancer activity using Luciferase reporter assays (Online Figure V).
Figure 2. Histone-acetylation Quantitative Trait Loci (haQTL).

(A) Pie chart showing 249,732 SNPs underlying all 47,321 H3K27-acetylation loci. (B) Heatmap showing 1,680 haQTLs (FDR<10%). Each row displays the H3K27ac peak heights for all LV samples at a single haQTL, arranged in order of their reference/alternative allele read count at the haQTL. The alternative allele for each sentinel SNP of 1,680 haQTLs was associated with either increasing H3K27ac peak heights (higher in alt allele), or decreasing peak heights (lower in alt allele). (C) Quantile-quantile (Q-Q) plot of the linear regression P-values for the association between the expression of genes (±100 kb) and haQTL, based on haQTL-gene pairs (blue), compared to control groups of either random enhancer SNP-gene pairs (black), or scrambled haQTL-gene pairs where the labels between haQTL and genes have been scrambled (grey). 180 bona fide haQTLs showed gene expression associations (FDR<20%). (D) An example of a haQTL (sentinel: rs2080512, red spot; FDR=4.29 × 10−2) residing in an intergenic region ~13 kb upstream of LITAF. Black dots represent other SNPs in the enhancer locus. The alternative allele, T, is associated with reduced H3K27ac peak heights, and reduced gene expression (FDR=0.057; boxplots). This example, rs2080512, is also in linkage disequilibrium (LD) with two SNPs (rs8049607, r2=0.83; rs735951, r2=0.94; represented by +) that met genome-wide association significance for cardiac electrographic QT Interval. (E) An example of a haQTL (sentinel: rs17553284, red spot; FDR=1.25 × 10−4) residing in the promoter of DNAJC15. Black dots represent other SNPs at the enhancer locus. The alternative allele, T, is associated with reduced H3K27ac peak heights, and reduced gene expression (FDR=0.012; boxplots).
As haQTLs, localising to either gene promoters or intergenic distal enhancers, and correlating to enhancer peak heights, may correlate to proximal gene expression9, we overlapped the list of haQTLs with human LV eQTL mapping data from the GTEx project26. 167 haQTLs were in strong linkage disequilibrium (r2>0.8) with GTEx eQTL (Online Table VII). By random permutation analysis, we confirmed that haQTLs were more enriched for GTEx eQTL, compared to non-haQTL enhancer SNPs (20-fold enrichment, empirical P-value < 0.001; Online Figure VI). Concurrently, we also generated RNA-seq data from our set of left ventricles. A substantial number (180, 10.8%) of haQTLs also bore expression correlation to their proximal genes (±100 kb), using our RNA-seq data at the more inclusive FDR threshold of 20%, with 40% overlap to the set from GTEx eQTL (Online Table VII). A QQ plot (Figure 2C) showed haQTL to be more enriched for expression correlation at lower association P-value, compared to control groups of random enhancer SNPs, or to scrambled haQTL-gene pairs where haQTL labels were randomly swapped, strengthening the notion as before that haQTL significantly associates with proximal gene expression more than by random chance. For example, Figure 2D shows a haQTL located at the promoter region of LITAF (sentinel: rs2080512). The presence of alternative alleles correlated to reduced enhancer peak heights, and a corresponding decrease in LITAF gene expression. LITAF has been associated with cardiac electrocardiographic QT elongation with variants at the locus (LD r2>0.8) that met genome-wide significance in a previous GWAS27 (Figure 2D). LITAF regulates cardiac excitation by acting as a molecular adaptor, linking NEDD4–1 ubiquitin ligases to the cardiac L-type calcium channel, and LITAF knockdown prolonged action potential duration and increased cytosolic calcium28. Similarly, for another haQTL located in the DNAJC15 promoter (sentinel: rs17553284; Figure 2E), alternative alleles also correlated with decreased peak heights, and reduced DNAJC15 gene expression. While there is little information in the heart about DNAJC15, also known as Methylation-controlled J protein (MCJ), it has been described to play a role in the regulation of mitochondria function29.
Distal gene expression associated to haQTL are confined to within topologically associated domains (TADs).
Enhancers may also regulate distal gene expression (often more than just one gene) over long genomic distances by long-range interactions, resulting from chromatin folding 8. We therefore also examined the association between haQTLs and the expression of their putative distal interacting genes, after building up a cardiac chromatin interactome using in situ Hi-C that we performed in 2 control non-failing hearts (Online Table II). Hi-C libraries were sequenced to 540 million unique reads per sample, producing a chromatin contact matrix at 40 kb resolution. Topologically associated domains (TADs) are defined as self-interacting chromatin domains whose boundaries are enriched for insulator proteins such as CTCF and Cohesin, and whose composition have been shown to confine gene regulation via enhancer-promoter interactions to within TADs 30. As previously suspected, our 2 Hi-C libraries showed global similarity for TADs between hearts (Online Figure VIIA; Online Table VIII). Moreover, in a separate study, we have also found that, despite hallmark and characteristic gene expression changes in a mouse model of heart failure, the cardiac CTCF-based TAD chromatin architecture remains robust between healthy and failing hearts, in marked contrast to the alternative model of cardiac-specific CTCF knockout31. We also observed that our TAD boundaries were marked by characteristic epigenetic marks including CTCF30 (Online Figure VIIB-D) and were highly insulated 30 (Online Figure VIIE-F). As anticipated therefore, we observed that a correlation between haQTL and distal gene expression were more likely to occur for haQTL-gene pairs that resided in the same TAD, as opposed to haQTL-gene pairs spanning across TADs, or scrambled haQTL-gene pairs (Figure 3A, B). Some haQTLs had correlations in more than one haQTL-gene pair (examples in Figure 3B, C). Hence we found 65 associations (involving 57 haQTLs) for distal haQTL-gene pairs within the same TAD (at least 10 kb apart) that met the FDR threshold of 20% (Online Table VII). The example in Figure 3C shows the associations for sentinel SNP rs12143842, ~5 kb from the NOS1AP promoter. The alternative allele associated with increased enhancer peak height, and increased expression of the proximal NOS1AP gene and a distal interacting gene C1orf226, but not the closer OLFML2B gene which was in a neighbouring TAD. CRISPR-targeted excision of this enhancer (rs12143842; Online Figure VIIIA), validated a consistent downregulation of C1orf226 and NOS1AP, but not the other neighbouring genes outside the TAD (Online Figure VIIIB). While NOS1AP is a known regulator of neuronal nitric oxide synthase (NOS1), which is highly expressed in the mammalian heart and regulates cardiac excitation-contraction coupling and repolarization, the function of C1orf226 is unknown32.
Figure 3. Distal gene expression associated to haQTL are confined to within topologically associated domains (TADs).

(A) QQ plot of linear regression P-values for the association between gene expression and haQTLs, based on haQTL and distal gene pairs residing within the same TADs (black), compared to control groups of either haQTL-gene pairs spanning across neighbouring TADs (blue), or scrambled haQTL-gene pairs (as randomised in Figure 2C). 57 haQTLs with gene expression associations lying within the same TAD achieved FDR<20%. (B) Snapshot showing 4 haQTLs (orange vertical boxes), one residing in an upstream TAD (blue horizontal bar), and 3 residing in the downstream TAD (green bar). TADs are also delineated by yellow dotted lines in the Hi-C interaction matrix. Each haQTL is correlated to the expression of proximal and distal genes localised to only within the same TAD. For example, rs134620 is associated with the expression of KREMEN1, and distal interacting genes RHBDD3 and EWSR1, located 200 kb away but within the same TAD. rs713657 and rs2301402 are both correlated to the expression of a distal gene NIPSNAP1 235 kb away, again within the same TAD. Correlated genes are coloured in blue, distal interactions are represented by dotted black arrows. (C) An example of a haQTL with correlated distal gene expression. rs12143842 (red spot) located ~5 kb from the NOS1AP gene promoter, is correlated to its expression (FDR=0.14), and also to the expression of a distal interacting gene, C1orf226, located 500kb downstream (FDR=0.10). A chromatin loop (black circle) in the Hi-C interaction matrix highlights the interaction (dotted black arrow) connecting the 2 distal loci. See also Online Figure IXD for HiChIP validation of this distal interaction. Importantly, there was no expression correlation for this SNP to its upstream gene (OLFML2B) that was lacking Hi-C evidence for interaction, despite being in closer proximity. rs12143842 is also a significant GWAS hit for QT interval.
Since it would be of value to assign more haQTL to distal interacting genes, we also deepened our study further by constructing and sequencing H3K27ac HiChIP libraries from 2 control non-failing heart samples, enabling us to identify chromatin interactions based on H3K27ac enhancers at the anchors16. On average, 64 million unique valid read-pairs were sequenced per HiChIP library. Since both HiChIP libraries were again highly correlated (Online Figure IXA), we combined both replicates to attain the greater depth of 194 million unique di-tags (388 million reads). Using HICCUPS, 14,698 H3K27ac based chromatin loops were called (10% FDR; Online Table IX; Online Figure IXB). H3K27ac enrichment at loop anchors was confirmed, with 82% overlapping to H3K27ac ChIP-seq peaks, in at least 1 of the anchors (Online Figure IXC-E). 613 of these loops were anchored by a haQTL. The majority of haQTL-containing anchors (407/613, 60%) interacted with other H3K27ac-enriched loci ~30 kb away, and 45% (184/407) also contained expressed gene TSS. From these, we recognised 15 haQTLs (FDR 20%) that we had already noted to bear distal gene expression association (among those identified in Figure 3A). The analysis making use of the HiChIP connectome yielded a further 24 unique loci, where the distances between the haQTL and genes were greater than 100 kb apart (Online Table VII). Specific examples for more haQTLs with distal gene expression association are shown in Online Figure X.
HaQTLs inferred and validated to alter transcription factor (TF) binding.
of TF to genomic regulatory regions such as enhancers is important for transcriptional regulation21, and specificity of TF binding often depends on the recognition of unique DNA motif sequences21. Functional genetic variants located at enhancers may therefore be inferred to disrupt or create TF DNA motifs, thereby changing TF binding affinity, and in turn influencing histone acetylation, and corresponding gene expression differences 9, 33. We therefore analysed our haQTLs for putative disruption or gain of TF motifs using haploREG33 and motifbreakR34, based on the reference and alternative alleles at each locus (Online Table X). Keeping the relevance only to TF that are cardiac expressed based on our RNA-seq dataset, we analysed for TF binding motifs altered by alternative alleles (Figure 4). By comparing to 1,000 permutated SNP sets of non-haQTL, we first validated that haQTLs were significantly enriched for TF motif alteration (Online Figure XIA, B). Altogether, 963 loci were inferred to have either a de novo gain in TF motif, a loss in TF motif, or had SNP variants at the same locus that produced gains and losses for 2 or more different TF motifs. In analysing for the correlation between motif gain or disruption and acetylation peak heights, we then noticed a specific set of TFs that showed a remarkable converse relationship (Figure 4A, B). Gain of the MEF2 family motif (MEF2A and MEF2D), as well as that for ETV5, ETV3, SPI1 and others, was associated with increased acetylation peaks, whereas disruption of the same TF motifs was associated with reduced acetylation. As examples, Online Figure XIC, D show the gain and loss of MEF2 and ETV5 motifs at the KLF12 and DDX11 loci, respectively, associated with the increase or decrease of enhancer peak heights, and corresponding gene expression changes. In the example of Online Figure XIC, the haQTL (rs9573330) located in a KLF12 intronic locus, is in strong linkage disequilibrium with a GWAS hit rs728926 (LD r2=0.99) for QT interval, and associated with calcium signalling in myocardial polarization35. The gain in motif for a second set of TFs (Figure 4A, B): ZNF143, ZEB1, ID4 and TCF4, was associated with decreased acetylation peaks, and vice versa. Moreover, motif changes at haQTL with altered H3K27-acetylation also correlated concordantly and globally to gene expression changes (Online Figure XIE).
Figure 4. haQTLs inferred to alter transcription factor (TF) binding.

(A) Alternative variants for sentinel SNPs at 963 (out of 1,680) haQTLs are inferred to alter at least 1 TF binding motif for cardiac expressed TFs. Heatmaps show examples of 2 unique sets of TFs (in blue and red boxes) possessing a converse relationship where motif gain either correlated with increased or decreased acetylation peak height, and vice versa. (B) Boxplots showing the association between TF motif disrupted or gained with the direction of H3K27ac peak height change. The unique set of 28 TFs show significant opposite effect between TF motif alteration and peak height change (Wilcoxon Rank Sum Test, FDR<10%). 24 TFs show increased peak height (haQTL correlation coefficient, r>0) with de novo motif gain, and reduced peak height with motif disruption (r<0). 4 TFs show the opposite effect of increased peak height with motif disruption, and decreased peak height with motif gain. (C, D) Two examples of haQTL (sentinels: rs5758468 and rs11663468) both inferred to disrupt MEF2A binding motifs at H3K27ac loci, and associated with the reduction in H3K27ac peak heights (boxplots), respectively. (rs5758468: FDR=1.26 × 10−4; rs11663468: FDR=2.04 × 10−4). (E) Electrophoretic Mobility Shift Assay (EMSA) using MEF2A recombinant protein and labelled oligos. Positive gel-shift is seen for oligos containing the Reference variants, but not Alternative variants, represented by both loci from (C, D). Non-labelled oligos were used as controls for competitive binding.
Remarkably, to take 2 independent loci (rs5758468 and rs11663468) with inferred MEF2 binding motifs altered based on their respective SNP alleles, and with respective loss of the H3K27 acetylation associated with alternative alleles (Figure 4C, D), we performed validation by electrophoretic mobility shift assays using recombinant MEF2 protein and synthesised labelled oligo probes (Figure 4E). A corresponding gel-shift pattern in both cases for the reference variant, but not the alternative, was consistent with the predicted differential MEF2 binding affinity at these alleles in vitro. All together, these findings support the notion that altered TF binding affinity inferred from variants in haQTL likely leads directly to enhancer histone acetylation differences and changes in the expression of cardiac phenotype-driver genes.
HaQTLs enriched for GWAS sub-threshold SNPs.
Finally, in order to apply haQTLs to prioritising variants for disease- or phenotype-association, we sought to colocalise our 1,680 haQTL against published cardiac-related GWAS. For these, we chose the large meta-analysis performed for atrial fibrillation2, studies from the EchoGen consortium on cardiac structure and function36, CHARGE studies performed for incident heart failure5, and heart failure mortality37, and a three-staged GWA study performed on 4,100 dilated cardiomyopathy cases and 7,600 controls4. Using an inclusive P-value cutoff of 0.001 for sub-threshold GWAS SNPs13, we narrowed down further to those that lie in cardiac active regulatory loci as marked by H3K27-acetylation, and intersected them with our catalogue of haQTLs or SNPs in LD with our haQTLs (Figure 5; Figures S12, S13). For each GWAS dataset, we performed independent permutation analyses using random enhancer-localised SNPs that matched our haQTLs for MAF and distance to gene promoters (Figure 5B, E). Indeed, a 1.5 to 9 fold enrichment (Online Table XI) was observed for colocalized variants across the 8 GWAS datasets, confirming that our haQTLs were significantly enriched for heart disease-associated variants, compared to 1,000 sets of 1,000 randomly permutated enhancer-localised SNPs, and also in contrast to the non-significant enrichment with non-heart related GWAS datasets (Online Figure XIV, Online Table XI).
Figure 5. haQTLs enriched for GWAS sub-threshold SNPs.

(A) A Manhattan plot of adjusted P-values for all SNPs from the GWAS meta-analysis for atrial fibrillation2, in LD (r2>0.8) with SNPs in our 249,732 cardiac enhancers SNPs (black). Red represents colocalised SNPs in 22 loci that are either in LD or overlapping with haQTLs. (B) 1,000 permutation sets of 1,609 enhancer SNPs, matching for distribution to nearest TSS distance, and MAF, to simulate the expected number of overlap between GWAS and haQTLs. We observed 22 colocalisations between haQTL and atrial fibrillation GWAS SNPs, a 2-fold enrichment from the expected (empirical P-value<0.001). (C) An example of a haQTL (sentinel: rs17005892, red spot) where the alternative C allele is associated with reduced H3K27ac peak height (FDR=1.25 × 10−4). The distal enhancer interacted with the promoter of TMEM150C as evident on Hi-C and HiChIP (See also Online Table IX), and the alternative allele was associated with reduced gene expression of TMEM150C (FDR=0.08), but not the adjacent gene SCD5 (boxplots). Notably, rs17005892 is also a significant eQTL for TMEM150C in the GTEx project. LINC00575 is not expressed in the heart (FPKM~0). (D) A Manhattan plot of adjusted P-values for all SNPs from the GWAS for dilated cardiomyopathy5, in LD (r2>0.8) with our enhancer SNPs (black). Red represents colocalised SNPs in 9 loci that are either in LD or overlapping with haQTLs. (E) 1,000 permutation sets of 1,285 enhancer SNPs, matching for distribution to nearest TSS distance, and MAF, to simulate the expected number of overlap between GWAS and haQTLs. We observed 9 colocalisations between haQTL and dilated cardiomyopathy GWAS SNPs, a 9-fold enrichment from the expected (empirical P-value<0.001). (F) An example of haQTL (sentinel: rs7445264, red spot) residing in the promoter of DNAJC18. Samples with the alternative allele, T, show consistently reduced H3K27ac peak heights (FDR=5 × 10−7), and concordant reduced gene expression (FDR=0.0075), compared to GG homozygotes (boxplots).
Twenty-two loci were found in overlap between haQTLs and hits from the atrial fibrillation GWAS2, reflecting an LV basis for the pathophysiology of atrial fibrillation also noted previously2 (Figures 5A, B and S12A). Two of the top loci (sentinels: rs10821415 and rs1152591; Online Figure XIIA-C) showed correlation to acetylation peak heights at intragenic enhancers, and also correlation to cis gene expression of C9orf3 and SYNE2, respectively. SYNE2 (Nesprin-2) and other nesprin isoforms, are spectrin-repeat proteins, highly expressed in cardiac muscle, important for maintaining the integrity of the nuclear envelope by coupling the nucleoskeleton to the cytoskeleton38. SYNE2 mutations cause muscular dystrophy and dilated cardiomyopathy38. The function of C9orf3 is as yet unknown. At another subthreshold locus (Figure 5C, rs17005892), acetylation this time was correlated to the expression of a novel distal gene, TMEM150C, found to be interacting via Hi-C and HiChIP (Online Table IX), but not another adjacent gene SCD5. TMEM150C is a gene with no prior known function described before in the heart. In the overlap with GWAS for dilated cardiomyopathy4 (Figure 5D, E), a notable candidate was DNAJC18 with a haQTL located in its promoter (sentinel: rs7445264; Figure 5F), and whose alternative allele we had already validated by Luciferase assay to show reduced enhancer activity (Online Figure V). This corresponded to decreased acetylation peak heights and reduced DNAJC18 gene expression in our samples with alternative alleles (Figure 5F).
Validation of novel cardiac causal genes associated with haQTL.
Finally, Online Figure XIID, E shows another example SNP rs34866937, residing in an intergenic locus, also overlapping between haQTL and the GWAS for dilated cardiomyopathy4, as well as the EchoGen GWAS for cardiac structure and function36. Our Hi-C matrix revealed a chromatin loop connecting the intergenic enhancer locus to the MTSS1 gene promoter ~120 kb away, which we also confirmed by 3C-PCR (Online Figure XV). The alternative allele at this locus was correlated to reduced H3K27ac peak height, and reduced expression of MTSS1 (Online Figure XIIE). The Mtss1-knockout mouse indeed showed improved cardiac function, while luciferase and CRISPR edit assays proved the association of this enhancer locus to MTSS1 expression39. Altogether, we identified 62 loci (Figure 6, Online Table XII) as haQTL that intersected with the sub-threshold signals from at least one GWAS dataset. Since 3 of these were sub-threshold signals in at least one GWAS dataset, but also exceeded genome-wide significance in another GWAS (Online Table XII), overall, we have identified 59 unique new candidate loci for LV phenotype and disease based on this current analysis.
Figure 6. Circos plot of all H3K27acetylation peaks (blue, N=47,321), haQTLs (red, N=1,680) and 62 loci identified from the sub-threshold signals of 10 cardiac-related GWAS datasets.

SNP IDs are displayed with their corresponding candidate genes, based on cis-expression correlated to nearest gene, or correlation by distal Hi-C/HiChIP interaction. See also in Online Table XII.
DISCUSSION
The scale of millions of genetic variants and thousands of disease associated genes that has before appeared too big to tackle, stands to be rationalised in the next phase of human genetic discovery. Epigenetics offers such an approach, when applied to the corresponding disease-related organ7, 8, 10. This study provides the first comprehensive enhancer landscape profiling in a large cohort of human left ventricle explants, comprising of 70 donors. Regardless of the heterogeneous causes of end-stage heart failure, our differential acetylation analysis indicates that thousands of shared alterations in histone acetylation are consistently observed in dilated left ventricles (Figure 1). Histone acetylation changes, also observed in experimental models of heart disease31, and even in genetic mouse models with perturbed histone acetyl-modifications40, all correlate with hallmark differential gene expression changes, thus lending credence to the notion that underlying epigenomic changes regulate transcriptomic differences that drive the phenotype in heart diseases. Furthermore, motif analysis of differentially acetylated loci reveals the transcription factors (TFs) associated with both increased and decreased acetylation, many of which are known to be involved in cardiac pathology. The strong association between enhancer acetylation levels and gene expression changes is observed not only in disease conditions but also during differentiation41, underscoring the critical role of non-coding regulatory regions in modulating cell states. Furthermore, identifying differentially acetylated regions helps to uncover TF that are key regulators of cardiac disease akin to what is also seen in cardiac development41. The reproducibility of our findings underlie the robust basis of analysis, going beyond the prior database of human heart enhancers42, now revealing the integrated differences in human heart disease.
Beyond just describing the enhancer landscape in the heart, the main focus of this study investigated the genetic basis of the epigenetic variation in the cardiac enhancer landscape. In setting out to understand the association between genetics and epigenetics, we applied the G-SCI algorithm1, 10, and identified 1,680 enhancer loci whose H3K27-acetylation enrichment was associated to common genetic variants underlying each peak. A substantial number of them also bore correlation to gene expression abundance (Figure 2B-D), many involving distal interacting genes that are hundreds of kilobases away, mapped by concordant cardiac Hi-C and HiChIP, the first to be performed on human hearts. Luciferase reporter assays on a subset of these haQTLs corroborated the finding that the presence of a single nucleotide variation was sufficient to cause a change in luciferase expression (Online Figure V). Figure 3C offers an example of a haQTL at the cis-enhancer upstream of NOS1AP which is looped spatially to interact with a distal gene, C1orf226, with unknown function. The gene expression of C1orf226 bore correlation to the haQTL, and the regulatory role of the enhancer was confirmed by independent CRISPR-excision experiment in hESC-derived cardiomyocytes (Figure 5F). This proves the specificity of chromatin looping for regulating distal gene expression.
From our analysis, we have identified two interesting and unique sets of TFs where their inferred binding motifs are either positively or negatively correlated to the enrichment of histone acetylation (Figure 4A, B). As a proof of concept, we have validated in vitro binding using EMSA and recombinant MEF2 protein, for 2 different inferred MEF2 binding haQTL loci. The direct inverse relationship between histone acetylation and the 2 sets of TFs imply the unexpected notion that genetic variants creating or disrupting TF binding, at least for these 2 sets of TFs, are sufficient to alter histone acetylation enrichment. These TFs specifically, may have a primary functional role at haQTLs, promoting or repressing the active enhancer H3K27ac mark, respectively. For example, MEF2 TFs are important signal-responsive mediators in the transcriptional reprogramming of cardiac development and disease20, 40, known to bind to and recruit the histone acetyl-transferase p300 in cardiac cells43. MEF2, was found to be also enriched in upregulated loci, and is crucially implicated in promoting heart chamber dilation and contractile dysfunction in dilated cardiomyopathy in vivo44, 45. MEF2 was also enriched in the enhancers that were involved in modulating the gene expression that drive cardiac hypertrophy in a pressure overload model of cardiac disease46. Similarly, ETS TFs (ETV3, 5, 6, SPL1, ERG, ETS1, FL1) are important in cardiac development47, and also recruit the histone acetyl-transferase p30048. Conversely, TCF4 appears to be a negative regulator of gene expression, involving an interaction with histone deacetylases (HDACs)49. ZEB150 is a putative repressor of gene expression. Like TCF4, ZEB1 recruits HDACs at target gene promoters50. These latter TFs have no known cardiac function, and now merit further investigation. Moreover, population genetic variants at noncoding enhancer loci may thus have functional value for disease and phenotype if they perturb TF binding motifs, leading to chromatin and consequent gene expression difference9. It may be further extrapolated that for some such instances, differences are absent in the relevant tissue, until the onset of pathogenic stimuli when because of the noncoding genetic variant, a differential disease response ensues. Mechanistically, this therefore underpins the important inter-relationship between genetics and epigenetics for their relevance to disease and phenotypes.
Finally, to demonstrate the utility of the haQTL analysis in identifying phenotype and disease-relevant variants, we overlaid our 1,680 haQTL against the sub-threshold signals of 10 published datasets of cardiac-related GWAS, and identified 62 unique haQTL that intersected a sub-threshold signal from at least one GWAS. This supports the notion that valuable genetic signals may still be likely buried within sub-threshold GWAS results, not meeting stringent genome-wide statistical threshold perhaps due to the combination of prohibitive sample size challenges, disease heterogeneity and other confounding limitations. We have leveraged epigenetic profiling as a systematic means to curate for new hits, incorporating proposed corresponding mechanisms.
In summary, this haQTL dataset should now prove useful for discovering more genetic variants if integrated with further heart-related GWAS. The approach of chromatin QTL and 3D connectome analyses in disease-relevant tissue promises not just to resolve the identity of functional genetic variants, taking cardiac genomics to its next phase of discovery, but target genes with correlated expression changes can be accordingly implicated to represent important pathways for new disease therapy.
Supplementary Material
{kind=link}
NOVELTY AND SIGNIFICANCE.
What Is Known?
Genes are regulated by distal genomic enhancers that may be located, through 3-dimensional chromatin folding, at long and unpredictable linear distances away from the gene itself.
Millions of genetic variants exist in the noncoding portion of the whole human genome, but which of these are relevant for cardiac disease and/or phenotype relies on having a genomic map that, not only annotates the regulatory enhancers, but also connects them to their cognate genes.
What New Information Does This Article Contribute?
1680 enhancer loci that are histone acetylation quantitative trait loci (haQTL) were identified by chromatin profiling of a cohort of human hearts.
These haQTLs are inferred to alter important cardiac transcription factor binding.
This is the first instance of HiChIP performed on human hearts to link haQTL to their target genes.
62 haQTL intersected with heart-related GWA studies reveal putative novel causal variants/genes for cardiac disease and phenotype.
Novel disease-relevant variants can elucidate specific mechanism of action.
Novel causal genes associated with the haQTLs may be therapeutic targets for heart disease.
Identifying genetic markers for heterogeneous complex diseases such as heart failure is challenging, and requires prohibitively large cohort sizes in GWAS in order to meet the stringent threshold of genome-wide statistical significance. On the other hand, chromatin QTL, elucidated by direct epigenetic profiling of specific human tissues, may contribute towards prioritising sub-threshold variants for disease-association. Here, by using a series of human hearts, we identified 1680 histone acetylation QTL. This is the largest H3K27ac ChIP-seq performed in human left ventricles to date, and also the first instance of HiChIP performed on human hearts, linking haQTLs to their target genes. haQTLs were inferred to function through altered cardiac transcription factor binding. 62 haQTLs intersected with published GWA studies, revealing novel putative causal variants and genes for heart disease. This work combines epigenome profiling and 3D connectome analyses in disease-relevant tissue to resolve the identity of functional genetic variants and bring cardiac genomics to its next phase of discovery.
ACKNOWLEDGEMENT
We thank Arima Genomics for use of their Hi-C kits. We also thank Dr Wenjie Sun for her advice with haQTL methodology, and Mr Ken Bedi who assisted with human heart tissue procurement and processing.
SOURCES OF FUNDING
This work was funded by the Biomedical Research Council (BMRC), Agency for Science, Technology and Research (A*STaR) Special Positioning Fund (SPF2014/004), Individual Research Grants and a Clinician Scientist Award from National Medical Research Council (NMRC) of Singapore (R.S.Y.F.); and RO1-HL105993 (T.P.C. and K.B.M.).
Nonstandard Abbreviations and Acronyms:
- ChIP-seq
Chromatin immunoprecipitation followed by sequencing
- haQTL
Histone acetylation Quantitative Trait Loci
- H3K27ac
Acetylation of the Lysine Residue at the 27th position on histone H3 tail
- GWAS
Genome-wide Association Study
- SNP
Single Nucleotide Polymorphism
- HF
Heart Failure
- DCM
Dilated Cardiomyopathy
- TF
Transcription factor
- LV
Left Ventricle
Footnotes
DISCLOSURES
No conflict of interest.
Contributor Information
Collaborators:
Solmaz Assa, Patrick Ellinor, Janine Felix, Pim van der Harst, Benjamin Meder, Gustav J. Smith, Rewati Tappu, CHARGE-Heart Failure Working Group, and CHARGE-EchoGen Consortium
REFERENCES
- 1.del Rosario RC, Poschmann J, Rouam SL, Png E, Khor CC, Hibberd ML and Prabhakar S. Sensitive detection of chromatin-altering polymorphisms reveals autoimmune disease mechanisms. Nat Methods. 2015;12:458–64. [DOI] [PubMed] [Google Scholar]
- 2.Christophersen IE, Rienstra M, Roselli C, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meder B, Ruhle F, Weis T, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. 2014;35:1069–77. [DOI] [PubMed] [Google Scholar]
- 5.Smith NL, Felix JF, Morrison AC, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rau CD, Lusis AJ and Wang Y. Genetics of common forms of heart failure: challenges and potential solutions. Curr Opin Cardiol. 2015;30:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gate RE, Cheng CS, Aiden AP, et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat Genet. 2018;50:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grubert F, Zaugg JB, Kasowski M, et al. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell. 2015;162:1051–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kilpinen H, Waszak SM, Gschwind AR, et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science. 2013;342:744–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, Poschmann J, Cruz-Herrera Del Rosario R, et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell. 2016;167:1385–1397 e11. [DOI] [PubMed] [Google Scholar]
- 11.Heinig M, Adriaens ME, Schafer S, et al. Natural genetic variation of the cardiac transcriptome in non-diseased donors and patients with dilated cardiomyopathy. Genome Biol. 2017;18:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon JR, Gorkin DU and Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell. 2016;62:668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Tucker NR, Rizki G, et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertero A, Fields PA, Ramani V, Bonora G, Yardimci GG, Reinecke H, Pabon L, Noble WS, Shendure J and Murry CE. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat Commun. 2019;10:1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choy MK, Javierre BM, Williams SG, et al. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat Commun. 2018;9:2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mumbach MR, Satpathy AT, Boyle EA, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017;49:1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH and Prabhakar S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat Biotechnol. 2013;31:615–22. [DOI] [PubMed] [Google Scholar]
- 18.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM and Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kehat I and Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spitz F and Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–26. [DOI] [PubMed] [Google Scholar]
- 22.Kohli S, Ahuja S and Rani V. Transcription factors in heart: promising therapeutic targets in cardiac hypertrophy. Curr Cardiol Rev. 2011;7:262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C, Bond Lau W, Ma XL and Du J. Cardiac Fibroblast-Specific Activating Transcription Factor 3 Protects Against Heart Failure by Suppressing MAP2K3-p38 Signaling. Circulation. 2017;135:2041–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019.
- 25.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM and Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, Biospecimen Collection Source Site N, Biospecimen Collection Source Site R, Biospecimen Core Resource V, Brain Bank Repository-University of Miami Brain Endowment B, Leidos Biomedical-Project M, Study E, Genome Browser Data I, Visualization EBI, Genome Browser Data I, Visualization-Ucsc Genomics Institute UoCSC, Lead a, Laboratory DA, Coordinating C, management NIHp, Biospecimen c, Pathology, e QTLmwg, Battle A, Brown CD, Engelhardt BE and Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213.29022597 [Google Scholar]
- 27.Pfeufer A, Sanna S, Arking DE, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MOSHAL KS, Roder K, Werdich AA, et al. LITAF, A Novel Regulator of Cardiac Excitation. The FASEB Journal. 2017;31:686.3–686.3. [Google Scholar]
- 29.Hatle KM, Gummadidala P, Navasa N, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33:2302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS and Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee DP, Tan WLW, Anene-Nzelu CG, et al. Robust CTCF-Based Chromatin Architecture Underpins Epigenetic Changes in the Heart Failure Stress-Gene Response. Circulation. 2019;139:1937–1956. [DOI] [PubMed] [Google Scholar]
- 32.Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–51. [DOI] [PubMed] [Google Scholar]
- 33.Ward LD and Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coetzee SG, Coetzee GA and Hazelett DJ. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arking DE, Pulit SL, Crotti L, et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wild PS, Felix JF, Schillert A, et al. Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J Clin Invest. 2017;127:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JG, Felix JF, Morrison AC, et al. Discovery of Genetic Variation on Chromosome 5q22 Associated with Mortality in Heart Failure. PLoS Genet. 2016;12:e1006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q, Bethmann C, Worth NF, et al. Nesprin-1 and −2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–33. [DOI] [PubMed] [Google Scholar]
- 39.Morley MP, Wang X, Hu R, et al. Cardioprotective Effects of MTSS1 Enhancer Variants. Circulation. 2019;139:2073–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA and Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wamstad JA, Alexander JM, Truty RM, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickel DE, Barozzi I, Zhu Y, et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun. 2016;7:12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K, Chan JK, Zhu G and Wu Z. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol. 2005;25:3575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Oort RJ, van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, van der Nagel R, Doevendans PA, Schneider MD, van Echteld CJ and De Windt LJ. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. [DOI] [PubMed] [Google Scholar]
- 45.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH and Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem. 2006;281:9152–62. [DOI] [PubMed] [Google Scholar]
- 46.Papait R, Cattaneo P, Kunderfranco P, et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A. 2013;110:20164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lie-Venema H, Gittenberger-de Groot AC, van Empel LJ, Boot MJ, Kerkdijk H, de Kant E and DeRuiter MC. Ets-1 and Ets-2 transcription factors are essential for normal coronary and myocardial development in chicken embryos. Circ Res. 2003;92:749–56. [DOI] [PubMed] [Google Scholar]
- 48.Sun HJ, Xu X, Wang XL, Wei L, Li F, Lu J and Huang BQ. Transcription factors Ets2 and Sp1 act synergistically with histone acetyltransferase p300 in activating human interleukin-12 p40 promoter. Acta Biochim Biophys Sin (Shanghai). 2006;38:194–200. [DOI] [PubMed] [Google Scholar]
- 49.Wang H and Matise MP. Tcf7l2/Tcf4 Transcriptional Repressor Function Requires HDAC Activity in the Developing Vertebrate CNS. PLoS One. 2016;11:e0163267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–48. [DOI] [PubMed] [Google Scholar]
- 51.Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolstad BM, Irizarry RA, Astrand M and Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. [DOI] [PubMed] [Google Scholar]
- 53.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taliun D, Chothani SP, Schonherr S, Forer L, Boehnke M, Abecasis GR and Wang C. LASER server: ancestry tracing with genotypes or sequence reads. Bioinformatics. 2017;33:2056–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Law CW, Chen Y, Shi W and Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P and Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 2015;4:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J and Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J and Mirny LA. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J and Meyer BJ. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ and Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES and Aiden EL. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khan A, Fornes O, Stigliani A, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46:D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hagenaars SP, Hill WD, Harris SE, Ritchie SJ, Davies G, Liewald DC, Gale CR, Porteous DJ, Deary IJ and Marioni RE. Genetic prediction of male pattern baldness. PLoS Genet. 2017;13:e1006594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pankratz N, Beecham GW, DeStefano AL, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol. 2012;71:370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paternoster L, Standl M, Waage J, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47:1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sniekers S, Stringer S, Watanabe K, et al. Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence. Nat Genet. 2017;49:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ and Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haeussler M, Schonig K, Eckert H, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All FASTQ files are deposited in EBI (EGA Study ID: EGAS00001003586). ChIP-seq read signal in the bigwig format has also been deposited onto UCSC Genome Browser (link: https://genome.ucsc.edu/s/wlwtan/EGAS00001003586). Raw data for RNA-seq have been deposited as part of the Myocardial Applied Genomics Network (MAGNet) study in NCBI GEO (Accession ID: GSE141910).