

Summary
Thalassaemia is caused by genetic globin defects leading to anaemia, transfusion-dependence and comorbidities. Reduced survival and systemic organ disease affect transfusion-dependent thalassaemia major and thalassaemia intermedia. Recent improvements in clinical management have reduced thalassaemia mortality. The therapeutic landscape of thalassaemia may soon include gene therapies as functional cures. An analysis of the adult US thalassaemia population has not been performed since the Thalassemia Clinical Research Network cohort study from 2000 to 2006. The Centers for Disease Control and Prevention supported US thalassaemia treatment centres (TTCs) to compile longitudinal information on individuals with thalassaemia. This dataset provided an opportunity to evaluate iron balance, chelation, comorbidities and demographics of adults with thalassaemia receiving care at TTCs. Two adult cohorts were compared: those over 40 years old (n = 75) and younger adults ages 18–39 (n = 201). The older adult cohort was characterized by higher numbers of iron-related comorbidities and transfusion-related complications. By contrast, younger adults had excess hepatic and cardiac iron and were receiving combination chelation therapy. The ethnic composition of the younger cohort was predominantly of Asian origin, reflecting the demographics of immigration. These findings demonstrate that comprehensive care and periodic surveys are needed to ensure optimal health and access to emerging therapies.
Keywords: thalassaemia, iron overload, transfusions, chelation
Introduction
Thalassaemia syndromes encompass a wide clinical spectrum of inherited disorders of α- or β-globin production that result in chronic haemolytic anaemia. Thalassaemia can manifest in a transfusion-dependent disease that is associated with both high morbidity and premature mortality. Defective synthesis of β-globin production can result in β-thalassaemia major (TM), one of the most severe forms of thalassaemia.1 Individuals with thalassaemia develop significant systemic complications as they age. The accumulation of comorbidities is multifactorial and is a function of ineffective erythropoiesis, systemic iron accumulation and transfusion exposure. In 1973, the average age of a North American person with TM was 11·4 years, with <3% of this population surviving beyond the age of 25 years.2 Thirty years later, the average age increased to 21·1 years; however, the numbers of adults with thalassaemia over age 40 years remained quite small (<5%) and iron-related organ dysfunction was correlated with increasing age.3,4 The racial and ethnic demography of thalassaemia in the US also continues to change, with fewer new cases of thalassaemia among people of southern European descent and increased numbers of younger people with thalassaemia from the Middle East, Southeast Asia and the Asian Indian subcontinent. This trend, reported over two decades ago, has resulted in a new generation of young adults with thalassaemia in the US.4,5
The landscape of therapeutic options has expanded dramatically in the past 5–10 years. Oral dispersible as well as film-coated tablet formulations of deferasirox are now available and have shown improved compliance.6 A new twice daily formulation of deferiprone for transfusional iron overload offers more convenience.7 Luspatercept, an injectable transforming growth factor-beta (greek B) binding recombinant protein based on activin A, was approved in 2020 and has reduced transfusion requirements in thalassaemia intermedia (TI) and TM.8,9 Advanced ex vivo gene therapies using modified CD34+ haematopoietic stem cells now offer cures for many affected thalassaemia patients by restoring functional circulating red blood cell haemoglobin to near normal or normal levels. These cell-based therapies represent a true functional cure in the form of transfusion independence. Eliminating transfusion therapies may also alleviate much of the disease burden of thalassaemia including comorbidities, blood-borne infections and the need for lifelong iron chelation. However, eligibility for such curative therapy and gene-editing studies require adequate cardiac function, hepatic reserve and minimal excess iron. Therefore, good adherence to chelation is critical from childhood onwards for younger patients to tolerate these curative therapies for the coming decades.
For these reasons and many others, it is important to have a contemporary description of the US thalassaemia population. It has been recognized that people with thalassaemia face challenges when they transition out of paediatric care models and into the adult care system.10 This transition is a risk factor for poor treatment adherence and the development of comorbidities. Second, it is important to understand the populations that might benefit from promising new disease-modifying and curative therapies.
A consortium of thalassaemia treatment centres (TTCs) in the US was organized and supported by the Centers for Disease Control and Prevention (CDC). The goal of this collaboration was to describe the thalassaemia population and capture disease-specific outcomes that would guide improvements in US thalassaemia care. Here, we focus on the adult thalassaemia populations and provide a detailed description of relevant demographic and disease-focused characteristics. These insights will provide assistance to clinicians when preparing their thalassaemia populations for future therapies and clinical trial options.
Methods
Patient population
The CDC funded a cooperative agreement with seven TTCs in Boston, MA, Chicago, IL, Philadelphia, PA, New York, NY, Los Angeles, CA, Oakland, CA and Atlanta, GA. Written consent for data and blood specimen collection was obtained using a protocol that received local institutional review and was approval at each TTC. The study was conducted according to the Declaration of Helsinki, Good Clinical Practice guidelines and local regulations.
Individuals with thalassaemia were eligible if they received comprehensive care at one of these TTCs between 2004 and 2011. The entire cohort consisted of 501 participants of all ages.
Study variables
This analysis focused on the group who were aged 18 or older at time of enrolment (n = 276). Investigators completed web-based enrolment forms for each subject at time of study entry that were updated annually. Demographic information (age, sex, race, ethnicity and place of birth), diagnosis, transfusions, chelation use, iron levels, prior splenectomy and disease- or treatment-related complications were assembled from retrospective chart review based on the most recent clinical visits. Race and ethnicity were self-reported and defined in accordance with the CDC consensus definitions (https://www.cdc.gov/nchs/nhis/rhoi/rhoi_glossary.htm). Race is defined based on standard categories (White, African-American, etc.), while ethnicity is based on Hispanic or non-Hispanic culture or origin, regardless of race. Case definitions and clinical variables (development of red cell alloantibodies, requirement of treatment for hypothyroidism, hypoparathyroidism, growth hormone deficiency or gonadal failure, history of fractures, hepatitis C virus exposure (HCV), human immunodeficiency virus (HIV) exposure, thrombosis and diabetes mellitus) were defined by investigator expert consensus and agreed upon prior to study initiation. All data collection including comorbidities and mortality was abstracted by study personnel from the medical record to the case report form. Mortality data were included for those individuals who died during the study period. No additional patient-reported data were collected in this study.
Definitions
β-TM was defined as homozygous (or compound heterozygous) β-globin mutations in subjects requiring chronic transfusions. Chronic transfusion status was defined as ≥8 transfusions in the previous 12 months. Those who required fewer than eight transfusions annually were classified as β-TI. Any transfusion regimen in the last 12 months of lesser frequency was classified as intermittent. Other thalassaemia syndromes were defined by genotype and standard international classifications (haemoglobin E/β-thalassaemia, haemoglobin H disease, haemoglobin H-Constant Spring).11 Chelation medications included deferoxamine (DFO), deferiprone (DFP) and deferasirox (DFX).
Statistical analysis
Individuals were included in this analysis if they had a complete and unique medical record for abstraction. Abstractions that were deemed to be incomplete with respect to analysis variables or duplicate records were not included. The current analysis is based on sample data at enrolment. The prevalence of outcome variables between younger (18–39 years old) and older adult (≥40 years old) age groups was compared. Chi-square analysis was used to determine the statistical significance (P < 0·05) of categorical variables. For continuous variables (age, serum ferritin, liver iron content), a one-sample t-test was used to compare the differences of the sample means of each variable between the two age groups. All analyses were performed at the CDC data coordinating centre with computer software (SAS/STAT Software, Version 9.3; SAS Institute, Cary, NC, USA).
Results
The focus of this report was to describe the 276 individuals included in the adult cohort, defined as age 18 or older at time of enrolment. All eligible individuals who provided consent and had at least one data collection point were included in this study. Figure 1 depicts the age distribution of the adult cohort of 276 study participants.
Fig 1.
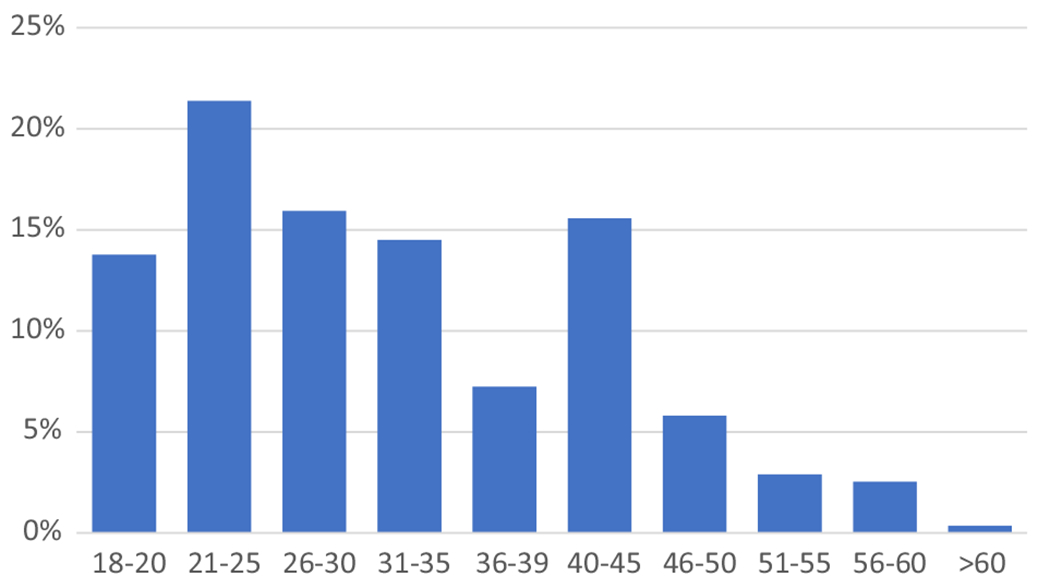
Age distribution of adult study participants with thalassaemia in the CDC Consortium.
Demographics
Table I provides demographic and clinical characteristics of the adult study participants. The younger adults (ages 18–39) consisted of 201 individuals with a mean age of 21·7 ± 6·1 years. The older adult group (≥40 years) was composed of 75 individuals with a mean age of 47 ± 6 years. Compared to younger adults, older adults were more likely to have diagnoses of β-TM or β-TI. Nearly all (99%) older adults carried a diagnosis of β-TM or β-TI, while 22% of younger adults had other thalassaemia syndromes such as haemoglobin E/β-thalassaemia, HbH-Constant Spring and HbH disease. Older adults were of European ancestry (73·4% were of Greek or Italian ancestry), while the majority of younger adults were of Asian ancestry as defined above. Most younger adults were diagnosed before the age of one year. The majority of older adults had undergone splenectomy (92%), compared to 55% in the group aged 18–39 (P < 0·001).
Table I.
Demographic and clinical characteristics of the CDC Thalassemia Treatment Center Consortium cohort.
| Total |
Age 18–39 |
Age ≥40 |
Chi-square P values 18–39 vs ≥40 | ||||
|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | ||
| n * | 501 | 100 | 201 | 100 | 75 | 100 | |
| Gender | 0·6606 | ||||||
| Female | 266 | 53·1 | 112 | 55·7 | 44 | 58·7 | |
| Male | 235 | 46·9 | 89 | 44·3 | 31 | 41·3 | |
| Race | <0·0001 | ||||||
| Other | 36 | 7·2 | 8 | 4 | 2 | 2·7 | |
| Asian | 256 | 51·1 | 105 | 52·2 | 9 | 12 | |
| White | 209 | 41·7 | 88 | 43·8 | 64 | 85·3 | |
| Ethnicity | 0·1681 | ||||||
| Hispanic or Latino | 20 | 4 | 5 | 2·5 | 0 | 0 | |
| Not Hispanic or Latino | 479 | 95·6 | 196 | 97·5 | 75 | 100 | |
| Place of birth | 0·1351 | ||||||
| Non-US | 145 | 28·9 | 67 | 33·3 | 18 | 24 | |
| US | 352 | 70·3 | 134 | 66·7 | 57 | 76 | |
| Diagnosis | <0·0001 | ||||||
| Beta thalassaemia intermedia | 48 | 9·6 | 15 | 7·5 | 20 | 26·7 | |
| Beta thalassaemia major | 330 | 65·9 | 142 | 70·6 | 54 | 72 | |
| HbE-beta thalassaemia | 44 | 8·8 | 20 | 10 | 0 | 0 | |
| HbH | 31 | 6·2 | 6 | 3 | 0 | 0 | |
| HbH-Constant Spring | 34 | 6·8 | 14 | 7 | 1 | 1·3 | |
| Other | 14 | 2·8 | 4 | 2 | 0 | 0 | |
| Age at diagnosis | 0·3888 | ||||||
| ≤6 months | 221 | 44·1 | 77 | 38·3 | 22 | 29·3 | |
| 7–12 months | 126 | 25·1 | 54 | 26·9 | 18 | 24 | |
| >1–5 years | 95 | 19 | 41 | 20·4 | 23 | 30·7 | |
| >5 years | 27 | 5·4 | 15 | 7·5 | 7 | 9·3 | |
| Ever had a splenectomy? | <0·0001 | ||||||
| No | 289 | 58 | 90 | 45 | 6 | 8 | |
| Yes | 212 | 42 | 111 | 55 | 69 | 92 | |
| All | 501 | 100 | 201 | 100 | 75 | 100 | |
CDC, Centers for Disease Control and Prevention.
Total may not add up to 100% because of missing or unknown values.
Table II describes the socioeconomic demographics of the adults with thalassaemia. Compared to younger adults, older adults were more likely to be married or partnered. The educational attainment and employment status were not significantly different, with approximately one third of patients having a bachelor’s degree or higher (27·9% vs 37·3%; P value not significant). While very few adults with thalassaemia reported being uninsured, a greater proportion (63%) of adults ≥40 years had commercial or private insurance (P = 0·0021).
Table II.
Socioeconomic demographics of younger and older adults with thalassaemia.
| Age 18–39 |
Age ≥40 |
Chi-square P values 18–39 vs ≥40 | |||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Education | 0·2401 | ||||
| Other | 13 | 6·5 | 5 | 6·7 | |
| ≤High school | 43 | 21·4 | 10 | 13·3 | |
| Some college | 68 | 33·8 | 22 | 29·3 | |
| Bachelor’s degree | 56 | 27·9 | 28 | 37·3 | |
| Master or doctoral | 21 | 10·4 | 10 | 13·3 | |
| Employment | 0·3311 | ||||
| Employed full-time | 72 | 35·8 | 35 | 46·7 | |
| Employed part-time | 31 | 15·4 | 11 | 14·7 | |
| Not employed | 89 | 44·3 | 28 | 37·3 | |
| Marital status | <0·0001 | ||||
| Single | 149 | 74·1 | 29 | 38·7 | |
| Married/partnered | 43 | 21·4 | 41 | 54·7 | |
| Separated/divorced/widowed | 0 | 0 | 4 | 5·3 | |
| Health insurance | 0·0021 | ||||
| Other | 46 | 22·9 | 12 | 16 | |
| Commercial | 99 | 49·3 | 47 | 62·7 | |
| Medicaid | 45 | 22·4 | 5 | 6·7 | |
| Medicare | 9 | 4·5 | 10 | 13·3 | |
| Uninsured | 2 | 1 | 1 | 1·3 | |
| All | 201 | 100 | 75 | 100 | |
Transfusion/chelation
To examine differences in disease burden, we compared transfusion status, iron management and clinical comorbidities between the two cohorts (Tables III and IV). Both age cohorts were predominantly transfusion-dependent (89%, ages 18–39 and 96%, age ≥40 years) and receiving iron chelation (79% and 89%; Table III). There was no significant difference in the types of chelation therapy being used, with the majority (42% and 55%) using DFX. However, younger adults were twice as likely to be prescribed combination iron chelation (26% vs 13%; P value 0·02).
Table III.
Transfusion and iron overload: transfusion, chelation, and iron assessment.
| Transfusion, chelation and iron assessment | Age 18·39 |
Age ≥40 |
Chi-square P values | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Transfusion status | 0·947 | ||||
| Chronically transfused | 163 | 81 | 64 | 85 | |
| Intermittently transfused | 3 | 1·5 | 1 | 1·3 | |
| Non-transfused | 26 | 13 | 9 | 12 | |
| Ever received regular chelation therapy | 0·06 | ||||
| No | 23 | 11 | 3 | 4 | |
| Yes | 178 | 89 | 72 | 96 | |
| Currently received regular chelation therapy | 0·04 | ||||
| No | 43 | 21 | 8 | 11 | |
| Yes | 158 | 79 | 67 | 89 | |
| Chelation therapy currently | 0·02 | ||||
| Combination | 52 | 26 | 10 | 13 | |
| Deferasirox | 85 | 42 | 41 | 55 | |
| Deferiprone | 4 | 2 | 4 | 5·3 | |
| Deferoxamine | 17 | 8·5 | 12 | 16 | |
| None | 43 | 21 | 8 | 11 | |
| Ever had a liver iron measurement | 0·16 | ||||
| No | 14 | 7 | 2 | 2·7 | |
| Unknown | 8 | 4 | 1 | 1·3 | |
| Yes | 179 | 89 | 72 | 96 | |
| Most recent method of liver iron measurement* | 0·58 | ||||
| Biopsy | 3 | 1·5 | 0 | 0 | |
| MRI | 126 | 63 | 46 | 61 | |
| SQUID† | 17 | 8·5 | 6 | 8 | |
| Total | 201 | 100 | 75 | 100 | |
MRI, magnetic resonance imaging; SQUID, Superconducting Quantum Interfering Device.
Liver iron content (mg iron per gram liver dry weight) measured by MRI (n = 126) and by liver biopsy (n = 3; one liver biopsy result not available for analysis) in n = 128 younger adults and by MRI in n = 46 older adults.
Liver iron content wet weight measured by SQUID in n = 7 younger adults and n = 6 older adults.
Table IV.
Transfusion and iron overload: cardiac, liver and serum iron values.
| Cardiac, liver and serum iron values | Age 18–39 |
Age ≥40 |
t-test P values | ||||
|---|---|---|---|---|---|---|---|
| n | Mean | SD | n | Mean | SD | ||
| Age, years | 201 | 27·2 | 6·1 | 75 | 47 | 6 | |
| Serum ferritin, last 12 months | 181 | 2434 | 2342 | 71 | 1251 | 1554 | <0·0001 |
| Serum ferritin by diagnosis | |||||||
| Beta thalassaemia major | 130 | 2395 | 2083 | 53 | 1415 | 1749 | 0·003 |
| Beta thalassaemia intermedia | 13 | 2171 | 2580 | 17 | 740·6 | 510·8 | 0·007 |
| HbH-Constant Spring | 13 | 2029 | 2992 | 1 | 1215 | 0 | |
| Cardiac iron, MRI T2 | 124 | 24·5 | 13·5 | 48 | 30·4 | 9·2 | 0·001 |
| Liver iron content | |||||||
| Liver dry weight (mg iron per gram liver weight); MRI or liver biopsy* | 128 | 10·6 | 10·5 | 46 | 4·5 | 5·7 | <0·0001 |
| Liver wet weight (mg iron per gram liver weight): SQUID† | 17 | 4·8 | 7 | 6 | 1·4 | 0·7 | 0·06 |
MRI, magnetic resonance imaging; SD, standard deviation; SQUID, Superconducting Quantum Interfering Device.
Liver iron content (mg iron per gram liver dry weight) measured by MRI (n = 126) and by liver biopsy (n = 3; one liver biopsy result not available for analysis) in n = 128 younger adults and by MRI in n = 46 older adults.
Liver iron content wet weight measured by SQUID in n = 7 younger adults and n = 6 older adults.
The degree of iron control was significantly different in the two age groups. Older adults had significantly lower mean serum ferritin levels at the most recent 12-month interval period compared to younger adults (1 251 ± 1 554 vs 2 434 ± 2 342 ng/ml; P < 0·0001) and older adults with thalassaemia had hepatic iron content closer to or within the therapeutic range compared to the younger adults by imaging (Table IV). In accordance with current standards of care, liver iron content was most commonly measured using magnetic resonance imaging. Similarly, the mean cardiac T2* values were significantly higher in older adults, indicating a lower level of cardiac siderosis compared to the younger adults, although the means for both subsets were above the threshold for significant cardiac overload (20 ms).
Comorbidities
Comorbidities by age cohort are described in Table V. Endocrine organs are known to be susceptible to iron toxicity and endocrinopathies are well characterized in adults receiving chronic transfusion support. Older adults were more likely to have diabetes mellitus compared to the younger adults. Gonadal failure, hypothyroidism and fractures were also more common in older adults.
Table V.
Complications of thalassaemia in younger and older adults.*
| Age 18–39 |
Age ≥40 |
Chi-square P values | |||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Ever developed an alloantibody | <0·001 | ||||
| Yes | 39 | 19·4 | 30 | 40 | |
| No | 149 | 74·1 | 43 | 57·3 | |
| Ever required treatment for hypothyroidism | 0·004 | ||||
| Yes | 22 | 10·9 | 19 | 25·3 | |
| No | 171 | 85·1 | 55 | 73·3 | |
| Ever required treatment for hypoparathyroidism | 0·254 | ||||
| Yes | 5 | 2·5 | 4 | 5·3 | |
| No | 188 | 93·5 | 70 | 93·3 | |
| Ever required treatment for growth hormone deficiency | 0·105 | ||||
| Yes | 24 | 11·9 | 15 | 20 | |
| No | 169 | 84·1 | 59 | 78·7 | |
| Ever required treatment for gonadal failure | <0·001 | ||||
| Yes | 57 | 28·4 | 39 | 52 | |
| No | 136 | 67·7 | 35 | 46·7 | |
| Ever experienced a fracture | <0·001 | ||||
| Yes | 50 | 24·9 | 46 | 61·3 | |
| No | 143 | 71·1 | 27 | 36 | |
| Ever had HCV | <0·001 | ||||
| Yes | 32 | 15·9 | 38 | 50·7 | |
| No | 161 | 80·1 | 36 | 48 | |
| Ever had HIV | 0·104 | ||||
| Yes | 2 | 1 | 3 | 4 | |
| No | 191 | 95 | 71 | 94·7 | |
| Ever had a thrombosis | 0·226 | ||||
| Yes | 23 | 11·4 | 13 | 17·3 | |
| No | 170 | 84·6 | 61 | 81·3 | |
| Ever had diabetes | <0·001 | ||||
| Yes | 25 | 12·4 | 23 | 30·7 | |
| No | 168 | 83·6 | 51 | 68 | |
| All | 201 | 100 | 75 | 100 | |
HCV, hepatitis C virus; HIV, human immunodeficiency virus.
Total may not add up to 100% because of missing or unknown values.
Hepatitis C, a blood-borne viral disease, has also been described in the thalassaemia population. The older adults in this report had a higher prevalence of prior hepatitis C exposure. A smaller proportion of younger adults had developed red cell alloantibodies. There were no differences in the proportions of HIV infection, thrombosis, hypoparathyroidism or growth hormone deficiencies between the two age cohorts.
During the study interval period, there were a total of nine adults who died (eight in TM, one in TI). Eight deaths occurred in the young adult cohort (three females and five males) and seven of the eight deaths were cardiac-related. The one case of death in TI was a younger male adult. The one death in the older cohort (one female) was non-cardiac (cancer; data not shown).
Discussion
Optimized transfusions, effective chelation and new modalities for clinical monitoring have resulted in improved survival of severe forms of thalassaemia over the past five decades.12,13 However, the burden of the disease with ageing may be underappreciated and the adult thalassaemia population in the US has not been described in detail since the Thalassemia Clinical Research Network (TCRN) cohort study ended in 2006. Therapeutic options continue to improve for thalassaemia, which is welcomed since the major contribution to morbidity and mortality is exposure to transfused packed red blood cells. Although age of diagnosis for specific comorbidities was not captured in the present study, these comorbidities have been well characterized in other adult and paediatric cohort studies and it remains clear that elimination of cumulative red blood cell exposure would substantially improve the health of the adult thalassaemia population.
Transfusion-sparing therapies such as luspatercept are now approved for use in transfusion-dependent thalassaemia and have demonstrated the ability to reduce transfusion requirements and iron burden.8,14,15 Stem cell transplant and ex vivo gene therapies may also represent a real option for long-term transfusion independence.16–19 However, as mentioned above, not all patients will be eligible based on comorbidities from transfusion exposure and chronic iron overload. Stem cell transplant and ex vivo CD34+-based gene therapies still carry a risk of mortality based on the need for myeloablation. The US thalassaemia population often has access to new therapeutic advances, but not all of them will be able to access these drugs without optimal care during childhood and early adulthood.
A substantial concern in this study was the iron control in younger adults, more deaths and the use of multiple chelation agents. Iron toxicity remains a risk factor for both morbidity and mortality in transfusion-dependent thalassaemia. In general, the older adult cohort was more likely to have better iron control, while the younger adult cohort had higher liver and cardiac iron, higher serum ferritins and a higher number reporting the use of combination chelation. This finding may imply that younger adults need more aggressive chelation if they are not compliant in childhood and that providers are prescribing more chelation in an attempt to attain negative iron balance. Practice guidelines have also evolved and younger adults may be prescribed chelation earlier and more aggressively than older generations. A predictor of survival to age 40 may therefore be related to chelation adherence in the teenage years or earlier. It is possible that the third decade of life is an inflection point where individuals with superior iron control begin to eclipse those with worse control in terms of future survival. Though not the focus of the current study, other reports have demonstrated that self-reported adherence to chelation was lowest in young adults aged 25–35 years and there is an opportunity to improve transition care in thalassaemia.20 In other diseases, transition models for young adults have demonstrated higher adherence with treatments and reduced risk of being lost to follow-up.21 In this study, historic or prospective compliance with chelation treatment could not be captured, although specific iron chelator prescribing is in line with previous studies, with DFX being the predominant chelator and the use of DFO and DFP is less common.22 There may also be a contribution due to survival bias in this report, as older individuals may have already achieved iron balance, accessed the healthcare system and thus did not succumb to cardiac disease earlier. It remains clear that adequate cardiac function and low iron burden are required for patients enrolling in ex vivo gene therapy trials for thalassaemia and that poor iron control may be a significant albeit mutable barrier for access. Thus targeting younger patients for close monitoring and enhanced compliance with chelation will remain critical to delivering functional cures.
Mortality in β-thalassaemia has been primarily related to cardiovascular disease, predominantly arrhythmias and iron-induced cardiomyopathy in most of the world.23–26 In addition, the thalassaemia population develops endocrine disorders, including diabetes mellitus, osteoporosis, fractures, hypogonadism, hypoparathyroidism and growth hormone deficiency at higher frequency than the general population. Finally, infections due to blood-borne pathogens such as hepatitis C, hepatitis B and HIV as well as red blood cell alloimmunization are associated with chronic transfusion and advancing age in North American thalassaemia populations.3,27
In this report, there was higher prevalence of comorbidities in adults ≥40 attributable to decades of exposure to transfusions (alloantibodies, hepatitis C exposure) and systematic iron exposure (e.g. endocrinopathies). These findings have been demonstrated in other geographies as well. In a recent Italian cohort study, the most significant complications were hypogonadism and osteoporosis. However, the percentage of the population with HCV was significantly higher in the Italian cohort compared to historic US controls (85% vs 35%),28 and in the current study, only 15% of the cohort under the age of 40 were positive for HCV. Blood-borne pathogen exposure is expected to decline as routine blood donor screening has been in place for decades (HIV in 1985, HCV in 1992). Similar results were reported in the 2004 TCRN study, where the prevalence of HCV as well as iron-related organ dysfunction and complications of iron chelation increased with age.29 The thalassaemia population enrolled in the CDC cohort included all thalassaemia subtypes, while the TCRN was limited to those with more severe phenotypes,30 though the largest thalassaemia subgroups in this report are also transfusion-dependent.
It has been previously reported that the ethnic background of the US thalassaemia population has changed in recent decades.4,5 In the present study, the younger adult cohort was composed of more patients of Asian ancestry, which also corresponded to enriching the cases of α-thalassaemia and haemoglobin E/β-thalassaemia syndromes in that group based on the known endemicity of these diseases.31,32 However, it is important to recognize that both adult cohorts were predominantly US-born, suggesting the difference in ethnic background in the younger adult cohort with thalassaemia is related to changes in immigration patterns from several decades earlier when their parents came to the US Other haemolytic anaemias are also prevalent in patients of Asian ancestry, such as G6PD33–35 and pyruvate kinase deficiency.36–38 Similarly, α-thalassaemia carrier rates are high and both of these disease-modifying factors should be considered in clinical evaluation of patients. Immigration status was not explored in depth in this study but is also a potential risk factor for poor chelation adherence because of health care access and should be an area for future evaluation. Furthermore, immigration status is a factor in health literacy and so targeting communications to address diverse populations from Asia is a key component of care.39
Cardiac disease remains the main cause of death in severe forms of thalassaemia, as has been previously reported in the TCRN cohort and others.22 The availability of cardiac imaging and its use to guide chelation has reduced cardiac death incidence in some thalassaemia populations.40 Female sex has also been identified as a predictor of survival alongside low ferritin and the absence of a cardiac disease diagnosis,41 and females have had significantly lower rates of heart failure in large studies. This study was not of sufficient duration to capture mortality events on a population level, though of the nine individuals with thalassaemia who died during follow-up, five were male and seven deaths out of nine were related to cardiac disease.
A recent cohort comparing TM and TI showed a narrowing gap in survival of the two phenotypes attributable to changes in practice patterns and chelation use.12 With new therapies and practice changes, it seems reasonable to expect that overall survival in thalassaemia will steadily improve in the US. Much of the improvement in survival will likely be related to early diagnosis, consistent evidence-informed transfusion practices and reduction in iron overload, enabled by improved iron measurements with non-invasive surveillance.28 Two new oral iron chelation drugs were also approved during this period (DFX in 2005 and DFP in 2011), so that patients and providers have access to multiple options if toxicities develop.
Improving provider knowledge of thalassaemia is also key to optimizing care. A recent CDC study reported that a large number of practitioners in California feel they are unfamiliar with thalassaemia guidelines and treatment and while 35·7% of paediatricians had some experience or expertise in thalassaemia, only 4·7% of internal medicine-trained haematologists reported having similar experience.42 Radke et al. also highlighted the need for this expertise, as 52·2% of respondent family physicians reported that they were solely or mainly responsible for the care of patients with thalassaemia. Fortunately, several professional societies have released comprehensive guidelines to manage patients with thalassaemia, most based on expert consensus.11,43–45 In a recent study from Italy, treatment at specialized care centres for thalassaemia using multidisciplinary clinical pathways has significantly improved iron chelation and survival.46 Therefore, transitional and interdisciplinary care can be seen as vital to the health of young adults with thalassaemia. Telemedicine and other eHealth options have been successful at improving clinical care in other rare diseases and may also be a viable option to bring expertise into areas across the US where thalassaemia practitioners are not present.47,48
Two major changes in thalassaemia clinical practice patterns are also evident in this study related to the age of diagnosis and splenectomy. First, older adults were more likely to be diagnosed between ages of two and five years, while the younger adults were often diagnosed at birth. This difference may be accounted for by the increased utlization of prenatal counselling and post implantation diagnosis in the younger group, which was not available prior to 1970. The α-thalassaemia syndromes in the younger cohort may have been recognized earlier by the clinician as well. Second, higher numbers of older adults had undergone splenectomy. This observation is in line with those of other multicentre studies, where younger cohorts had splenectomy rates of 22% compared to 57% in older cohorts and the progressing mean age of splenectomy in the cohort suggested that this practice may be declining.49 Although splenectomy has been a component of thalassaemia practice for decades, a recent Cochrane review concluded that high-quality studies of the effect of splenectomy in thalassaemia are lacking.50 Splenectomy has also been associated with increased risk of alloimmunization among younger patients.27 Current guidelines recommend splenectomy only in specific circumstances with appropriate consideration of technique and infection risk.11,45 It would be expected that, in the context of new therapies that reduce or eliminate transfusional iron overload and the known risks of splenectomy, the rate of this procedure will continue to decline in future US thalassaemia age cohorts.
Endocrinopathies are another known set of comorbidities in thalassaemia and are associated with iron overload and age of chelation initiation.51 In the present study, diabetes mellitus remained significantly more common in older adults compared to younger adults (30% vs 12%). Interestingly, the prevalence of diabetes in thalassaemia varies with geography. In an Italian cohort, diabetes mellitus was only diagnosed in 4%,12 while a recent UK cohort described diabetes at rates similar to this study.52 The prevalence of diabetes mellitus in thalassaemia is likely related to local dietary and environmental factors, similar to the general population.
Degenerative bone diseases and fractures are common in severe β-thalassaemia and contribute to morbidity.53 In this study, the older adult cohort were more likely to have had bone fractures. Higher rates of osteoporosis have been reported in β-thalassaemia and the risk factors appear to develop during childhood and adolescence.53–55 Current screening guidelines recommend regular evaluation of bone density.11,43
In terms of other demographic characteristics, rates of marriage by age cohort are consistent with broader trends in US Census data (https://www.census.gov/library/visualizations/2018/comm/percent-married.html), where only approximately one third of individuals ages 18–34 are married. It is also encouraging that most individuals in this study report having insurance coverage and are achieving education levels similar to those of the general US population, as has been previously reported.30
This study has several limitations. Systematic data on malignancies were not collected. Low numbers of malignancies are reported in most thalassaemia cohorts.12,22 However, recent reports suggest that haematologic malignancies may be higher in transfusion-dependent β-thalassaemia.56–58 It is now recognized as thalassaemia patients live longer and with consistent access to iron chelation they survive long enough to develop malignancies. Furthermore, the risk factors and natural history of some malignancies may be different in TM patients. Recent data from an Italian cohort indicated that hepatocellular carcinoma occurs at a younger age in TM patients compared to the general population and the additional risk factor of transfusional iron overload may play a role.59–61 Other contributors to differences in malignancy rates in thalassaemia may be related to immunomodulation by chronic alloantigen exposure via transfusion and iron overload induced oxidative stress.61,62 The need to understand long-term malignancy risk in transfusion-dependent β-thalassaemia should be a priority in future cross-sectional and longitudinal studies and to develop age-appropriate screening guidelines that are focused on this population.
Another important limitation is that this study reflects those individuals with thalassaemia who were receiving care in the funded TTCs and may not be representative of the experience elsewhere for the US population. The sample size collected here does not capture the entire thalassaemia cohort of the US However, it does capture a relevant and diverse group in the largest treatment centres. TTCs are reflective of the broader practice patterns in the US and act as referral centres for large regions of the country. The Federal Health Resources and Services Administration has also begun a National Coordinating Program initiative to ensure that thalassaemia patients across the US have access, including via telehealth, to consultants with thalassaemia to ensure that the latest consensus guidelines are being offered to patients at smaller centres. In addition, evidence-informed clinical practice guidelines from the Thalassemia International Federation reflect more global recommendations to improve outcomes.11,63 Despite published recommendations, transfusion practices and iron chelation may be more variable outside comprehensive thalassaemia centres where expertise and numbers of thalassaemia patients in a care setting may be far lower. Alternately, one could postulate that the population in this study treated by thalassaemia specialists may on par have more complications and health care needs than those with fewer complications who receive adequate care from non-specialists. This current study represents a large US cohort of adults where practice and expertise is likely to be the most consistent.
In conclusion, adults in the US with thalassaemia syndromes are composed of diverse and dynamic populations. This report provides a timely update from 2004 to 2011, as new therapeutic approaches have been or will be approved within the next five years. Clearly more of the population is reaching adulthood; however, older adults are still developing comorbidities related to iron overload and transfusion therapy and preventing these complications with chelation would ensure that they remain eligible for cures. Therapies to reduce or eliminate transfusions will continue to improve survival in future next generations and iron overload complications remain the top focus for those who cannot discontinue transfusions. Finally, focused attention to younger adults in clinical practice is warranted, as these patients remain at risk for complications. These measures will improve the likelihood that adults in the US of all ages will remain eligible for current and emerging curative therapies and address the health gaps in the thalassaemia population.
Acknowledgements
This study was supported by the Centers for Disease Control and Prevention (CDC; DD07-010). The findings and conclusions in this paper are those of the authors and do not necessarily represent the official position of the CDC. The investigators would also like to acknowledge the study coordinators, practitioners and research assistants who were instrumental in collecting study data.
Footnotes
Conflicts of interest
John Chapin is currently an employee and shareholder of Takeda Pharmaceuticals.
References
- 1.Taher AT, Musallam KM, Cappellini MD. β-thalassemias. N Engl J Med. 2021;384(8):727–43. [DOI] [PubMed] [Google Scholar]
- 2.Pearson HA, Cohen AR, Giardina PJ, Kazazian HH. The changing profile of homozygous beta-thalassemia: demography, ethnicity, and age distribution of current North American patients and changes in two decades. Pediatrics. 1996;97(3):352–6. [PubMed] [Google Scholar]
- 3.Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR. Complications of beta-thalassemia major in North America. Blood. 2004;104(1):34–9. [DOI] [PubMed] [Google Scholar]
- 4.Vichinsky EP, MacKlin EA, Waye JS, Lorey F, Olivieri NF. Changes in the epidemiology of thalassemia in North America: a new minority disease. Pediatrics. 2005;116(6):e818–25. [DOI] [PubMed] [Google Scholar]
- 5.Heer N, Choy J, Vichinsky EP. The social impact of migration on disease. Cooley’s anemia, thalassemia, and new Asian immigrants. Ann N Y Acad Sci. 1998;850:509–11. [DOI] [PubMed] [Google Scholar]
- 6.Tartaglione I, Origa R, Kattamis A, Pfeilstöcker M, Gunes S, Crowe S, et al. Two-year long safety and efficacy of deferasirox film-coated tablets in patients with thalassemia or lower/intermediate risk MDS: phase 3 results from a subset of patients previously treated with deferasirox in the ECLIPSE study. Exp Hematol Oncol. 2020;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FDA. Highlights of prescribing information FERRIPROX 2020. [cited 13 Oct 2021]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212266s000lbl.pdf
- 8.Cappellini MD, Viprakasit V, Taher AT, Georgiev P, Kuo KHM, Coates T, et al. A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2020;382(13):1219–31. [DOI] [PubMed] [Google Scholar]
- 9.Musallam KM, Rivella S, Taher AT. Management of non-transfusion-dependent β-thalassemia (NTDT): the next 5 years. Am J Hematol. 2021;96(3):E57–9. [DOI] [PubMed] [Google Scholar]
- 10.Levine L, Levine M. Health care transition in thalassemia: pediatric to adult-oriented care. Ann N Y Acad Sci. 2010;1202:244–7. [DOI] [PubMed] [Google Scholar]
- 11.Cappellini M, Cohen A, Porter J, Taher A & Viprakasit V Guidelines for the management of transfusion dependent thalassaemia (TDT). 3rd ed. Strovolos, Nicosia Cyprus: Thalaessemia International Federation; 2014. [PubMed] [Google Scholar]
- 12.Vitrano A, Calvaruso G, Lai E, Colletta G, Quota A, Gerardi C, et al. The era of comparable life expectancy between thalassaemia major and intermedia: is it time to revisit the major-intermedia dichotomy? Br J Haematol. 2017;176(1):124–30. [DOI] [PubMed] [Google Scholar]
- 13.Caocci G, Orofino MG, Vacca A, Piroddi A, Piras E, Addari MC, et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am J Hematol. 2017;92(12):1303–10. [DOI] [PubMed] [Google Scholar]
- 14.Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat Med. 2014;20(4):398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langdon JM, Barkataki S, Berger AE, Cheadle C, Xue Q-L, Sung V, et al. RAP-011, an activin receptor ligand trap, increases hemoglobin concentration in hepcidin transgenic mice. Am J Hematol. 2015;90(1):8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Negre O, Fusil F, Colomb C, Roth S, Gillet-Legrand B, Henri A, et al. Correction of murine β-thalassemia after minimal lentiviral gene transfer and homeostatic in vivo erythroid expansion. Blood. 2011;117(20):5321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Negre O, Bartholomae C, Beuzard Y, Cavazzana M, Christiansen L, Courne C, et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease. Curr Gene Ther. 2015;15(1):64–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2020;384:252–60. [DOI] [PubMed] [Google Scholar]
- 19.Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil J-A, Hongeng S, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479–93. [DOI] [PubMed] [Google Scholar]
- 20.Trachtenberg F, Vichinsky E, Haines D, Pakbaz Z, Mednick L, Sobota A, et al. Iron chelation adherence to deferoxamine and deferasirox in thalassemia. Am J Hematol. 2011;86(5):433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allemang B, Allan K, Johnson C, Cheong M, Cheung P, Odame I, et al. Impact of a transition program with navigator on loss to follow-up, medication adherence, and appointment attendance in hemoglobinopathies. Pediatr Blood Cancer. 2019;66(8):e27781. [DOI] [PubMed] [Google Scholar]
- 22.Kwiatkowski JL, Kim H-Y, Thompson AA, Quinn CT, Mueller BU, Odame I, et al. Chelation use and iron burden in North American and British thalassemia patients: a report from the Thalassemia Longitudinal Cohort. Blood. 2012;119(12):2746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107(9):3733–7. [DOI] [PubMed] [Google Scholar]
- 24.Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet. 2000;355(9220):2051–2. [DOI] [PubMed] [Google Scholar]
- 25.Telfer P, Coen PG, Christou S, Hadjigavriel M, Kolnakou A, Pangalou E, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980–2004. Haematologica. 2006;91(9):1187–92. [PubMed] [Google Scholar]
- 26.Ladis V, Chouliaras G, Berdousi H, Kanavakis E, Kattamis C. Longitudinal study of survival and causes of death in patients with thalassemia major in Greece. Ann N Y Acad Sci. 2005;1054:445–50. [DOI] [PubMed] [Google Scholar]
- 27.Thompson AA, Cunningham MJ, Singer ST, Neufeld EJ, Vichinsky E, Yamashita R, et al. Red cell alloimmunization in a diverse population of transfused patients with thalassaemia. Br J Haematol. 2011;153(1):121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgna-pignatti C, Cappellini MD, Stefano P, Vecchio GC, Forni GL, Gamberini MR, et al. Survival and complications in thalassemia. Ann N Y Acad Sci. 2005;1054:40–7. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR, Network TCR. Complications of beta-thalassemia major in North America. Blood. 2004;104(1):34–9. [DOI] [PubMed] [Google Scholar]
- 30.Pakbaz Z, Treadwell M, Kim H-Y, Trachtenberg F, Parmar N, Kwiatkowski JL, et al. Education and employment status of children and adults with thalassemia in North America. Pediatr Blood Cancer. 2010;55(4):678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134:522–31. [PMC free article] [PubMed] [Google Scholar]
- 32.Paulukonis S, Currier R, Coates TD, Vichinsky E, Feuchtbaum L. Impact of immigration and migration on thalassemia surveillance in California, 2004–2008. Blood. 2014;124(21):4855. [Google Scholar]
- 33.He Y, Zhang Y, Chen X, Wang Q, Ling L, Xu Y. Glucose-6-phosphate dehydrogenase deficiency in the Han Chinese population: molecular characterization and genotype-phenotype association throughout an activity distribution. Sci Rep. 2020;10(1):17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ninokata A, Kimura R, Samakkarn U, Settheetham-Ishida W, Ishida T. Coexistence of five G6PD variants indicates ethnic complexity of Phuket islanders, Southern Thailand. J Hum Genet. 2006;51(5):424–8. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee MB, Colah RB, Martin S, Ghosh K. Glucose-6-phosphate dehydrogenase (G6PD) deficiency among tribal populations of India - country scenario. Indian J Med Res. 2015;141(5):516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warang P, Kedar P, Ghosh K, Colah R. Molecular and clinical heterogeneity in pyruvate kinase deficiency in India. Blood Cells Mol Dis. 2013;51(3):133–7. [DOI] [PubMed] [Google Scholar]
- 37.Feng CS, Tsang SS, Mak YT. Prevalence of pyruvate kinase deficiency among the Chinese: determination by the quantitative assay. Am J Hematol. 1993;43(4):271–3. [DOI] [PubMed] [Google Scholar]
- 38.Secrest MH, Storm M, Carrington C, Casso D, Gilroy K, Pladson L, et al. Prevalence of pyruvate kinase deficiency: a systematic literature review. Eur J Haematol. 2020;105(2):173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HY, Rhee TG, Kim NK, Ahluwalia JS. Health literacy as a social determinant of health in Asian American immigrants: findings from a population-based surveyin California. J Gen Intern Med. 2015;30(8):1118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–93. [PubMed] [Google Scholar]
- 42.Radke T, Paulukonis S, Hulihan M, Feuchtbaum L. Providers’ perspectives on treating patients with thalassemia. J Pediatr Hematol Oncol. 2019;41(7):e421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tubman VN, Fung EB, Vogiatzi M, Thompson AA, Rogers ZR, Neufeld EJ, et al. Guidelines for the standard monitoring of patients with thalassemia: report of the thalassemia longitudinal cohort. J Pediatr Hematol Oncol. 2015;37(3):e162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet 2018;391(10116):155–67. [DOI] [PubMed] [Google Scholar]
- 45.Sleiman J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT. Non-transfusion-dependent thalassemia: an update on complications and management. Int J Mol Sci. 2018;19:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinto V, Poggi M, Russo R, Giusti A, Forni G. Management of the aging beta-thalassemia transfusion-dependent population - the Italian experience. Blood Rev. 2019;38:100594. [DOI] [PubMed] [Google Scholar]
- 47.Radtke HB, Klein-Tasman BP, Merker VL, Knight P, Ullrich NJ, Jordan JT, et al. The impact of the COVID-19 pandemic on neurofibromatosis clinical care and research. Orphanet J Rare Dis. 2021;16(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoogeveen IJ, Peeks F, de Boer F, Lubout CMA, de Koning TJ, te Boekhorst S, et al. A preliminary study of telemedicine for patients with hepatic glycogen storage disease and their healthcare providers: from bedside to home site monitoring. J Inherit Metab Dis. 2018;41(6):929–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piga A, Serra M, Longo F, Forni G, Quarta G, Cappellini MD, et al. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am J Hematol. 2011;86(9):808–10. [DOI] [PubMed] [Google Scholar]
- 50.Easow Mathew M, Sharma A, Aravindakshan R. Splenectomy for people with thalassaemia major or intermedia. Cochrane Database Syst Rev. 2016;(6):CD010517. [DOI] [PubMed] [Google Scholar]
- 51.De Sanctis V, Elsedfy H, Soliman AT, Elhakim IZ, Kattamis C, Soliman NA, et al. Clinical and biochemical data of adult thalassemia major patients (TM) with multiple endocrine complications (MEC) versus TM patients with normal endocrine functions: a long-term retrospective study (40 years) in a Tertiary Care Center in Italy. Mediterr J Hematol Infect Dis. 2016;8(1):e2016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jobanputra M, Paramore C, Laird S, McGahan M, Telfer P. Comorbidities and mortality associated with transfusion-dependent beta-thalassaemia in patients in England: a 10-year retrospective cohort analysis. Br J Haematol. 2020;191(5):897–905. [DOI] [PubMed] [Google Scholar]
- 53.Vogiatzi MG, Macklin EA, Fung EB, Cheung AM, Vichinsky E, Olivieri N, et al. Bone disease in thalassemia: a frequent and still unresolved problem. J Bone Miner Res. 2009;24(3):543–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogiatzi MG, Autio KA, Mait JE, Schneider R, Lesser M, Giardina PJ. Low bone mineral density in adolescents with beta-thalassemia. Ann N Y Acad Sci. 2005;1054:462–6. [DOI] [PubMed] [Google Scholar]
- 55.Vogiatzi MG, Macklin EA, Trachtenberg FL, Fung EB, Cheung AM, Vichinsky E, et al. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146(5):546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karimi M, Giti R, Haghpanah S, Azarkeivan A, Hoofar H, Eslami M. Malignancies in patients with beta-thalassemia major and beta-thalassemia intermedia: a multicenter study in Iran. Pediatr Blood Cancer. 2009;53(6):1064–7. [DOI] [PubMed] [Google Scholar]
- 57.Chung W-S, Lin C-L, Lin C-L, Kao C-H. Thalassaemia and risk of cancer: a population-based cohort study. J Epidemiol Community Health. 2015;69(11):1066–70. [DOI] [PubMed] [Google Scholar]
- 58.Halawi R, Beydoun H, Cappellini M, Ferla V, Taher A. Hematologic malignancies in thalassemia: adding new cases to the repertoire. Am J Hematol. 2017;92(5):e68–70. [DOI] [PubMed] [Google Scholar]
- 59.Borgna-Pignatti C, Garani MC, Forni GL, Cappellini MD, Cassinerio E, Fidone C, et al. Hepatocellular carcinoma in thalassaemia: an update of the Italian Registry. Br J Haematol. 2014;167(1):121–6. [DOI] [PubMed] [Google Scholar]
- 60.Mangia A, Bellini D, Cillo U, Laghi A, Pelle G, Valori VM, et al. Hepatocellular carcinoma in adult thalassemia patients: an expert opinion based on current evidence. BMC Gastroenterol. 2020;20(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moukhadder HM, Halawi R, Cappellini MD, Taher AT. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: a comprehensive review. Cancer. 2017;123(5):751–8. [DOI] [PubMed] [Google Scholar]
- 62.Leitch HA, Gattermann N. Hematologic improvement with iron chelation therapy in myelodysplastic syndromes: clinical data, potential mechanisms, and outstanding questions. Crit Rev Oncol Hematol. 2019;141:54–72. [DOI] [PubMed] [Google Scholar]
- 63.Farmakis D, Giakoumis A, Angastiniotis M, Eleftheriou A. The changing epidemiology of the ageing thalassaemia populations: a position statement of the Thalassaemia International Federation. Eur J Haematol. 2020;105(1):16–23. [DOI] [PubMed] [Google Scholar]