

ABSTRACT
MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) orchestrates diverse environmental signals to facilitate cell growth and is frequently activated in cancer. Translocation of MTORC1 from the cytosol to the lysosomal surface by the RRAG GTPases is the key step in MTORC1 activation. Here, we demonstrated that transcription factors MEF2A and MEF2D synergistically regulated MTORC1 activation via modulating its cyto-lysosome shutting. Mechanically, MEF2A and MEF2D controlled the transcription of FNIP1 and FNIP2, the components of the FLCN-FNIP1 or FNIP2 complex that acts as a RRAGC-RRAGD GTPase-activating element to promote the recruitment of MTORC1 to lysosome and its activation. Furthermore, we determined that the pro-oncogenic protein kinase SRC/c-Src directly phosphorylated MEF2D at three conserved tyrosine residues. The tyrosine phosphorylation enhanced MEF2D transcriptional activity and was indispensable for MTORC1 activation. Finally, both the protein and tyrosine phosphorylation levels of MEF2D are elevated in human pancreatic cancers, positively correlating with MTORC1 activity. Depletion of both MEF2A and MEF2D or expressing the unphosphorylatable MEF2D mutant suppressed tumor cell growth. Thus, our study revealed a transcriptional regulatory mechanism of MTORC1 that promoted cell anabolism and proliferation and uncovered its critical role in pancreatic cancer progression.
Abbreviation: ACTB: actin beta; ChIP: chromatin immunoprecipitation; EGF: epidermal growth factor; EIF4EBP1: eukaryotic translation initiation factor 4E binding protein 1; FLCN: folliculin; FNIP1: folliculin interacting protein 1; FNIP2: folliculin interacting protein 2; GAP: GTPase activator protein; GEF: guanine nucleotide exchange factors; GTPase: guanosine triphosphatase; LAMP2: lysosomal associated membrane protein 2; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MEF2: myocyte enhancer factor 2; MEF2A: myocyte enhancer factor 2A; MEF2D: myocyte enhancer factor 2D; MEF2D-3YF: Y131F, Y333F, Y337F mutant; MTOR: mechanistic target of rapamycin kinase; MTORC1: MTOR complex 1; NR4A1: nuclear receptor subfamily 4 group A member 1; RPTOR: regulatory associated protein of MTOR complex 1; RHEB: Ras homolog, mTORC1 binding; RPS6KB1: ribosomal protein S6 kinase B1; RRAG: Ras related GTP binding; RT-qPCR: real time-quantitative PCR; SRC: SRC proto-oncogene, non-receptor tyrosine kinase; TMEM192: transmembrane protein 192; WT: wild-type.
KEYWORDS: Autophagy, cell metabolism, oncogenesis, protein kinase, transcription factor
Introduction
Cancer cell growth is strictly dependent on aberrant signaling and reprogrammed cell metabolism, which sustain their adaptation to nutrient availability and energy-demanding in the tumor context. In particular, MTORC1 signaling is generally hyperactivated in most cancers and controls biomass biosynthetic processes to ensure sustained cancer cell proliferation [1–6]. Several MTORC1 inhibitors have been used for cancer treatment in cell and animal models but are ineffectual in clinical treatment of patients with cancers, indicating that there is redundant MTORC1 activation [2,7,8]. Therefore, advances in understanding the mechanisms underlying rewired MTORC1 activity in tumorigenesis may have potential benefits in developing drugs for cancer treatment.
To monitor and integrate multiple environmental cues such as growth factors and nutrients sufficiency, MTORC1 integrates these inputs by two types of small G proteins-RHEB (Ras homolog, mTORC1 binding) and the RRAG (Ras related GTP binding) guanosine triphosphatases [9,10]. RHEB preserves its active state on the surface of lysosome in response to growth factors [11–13]. Then MTORC1 interacts with and is activated by RHEB after its translocation to the lysosomal membrane by RRAG GTPases when nutrients, especially amino acids, are readily available [9,14]. Mammals have four RRAG proteins (RRAGA to RRAGD) that form heterodimers constituting RRAGA or RRAGB in association with RRAGC or RRAGD [14,15]. Nutrients induce RRAG heterodimers switching to an active form, in which RRAGA or RRAGB is GTP-bound forms and RRAGC or RRAGD is GDP-bound forms. The active heterodimeric RRAG GTPases interacts with RPTOR/raptor, a subunit of MTORC1, thus permitting MTORC1 to the lysosome surface [16–18]. Extensive previously studies have revealed the roles of several protein complexes which are called GTPase activator proteins (GAPs) and guanine nucleotide exchange factors (GEFs), in the regulation of RRAG GTPases activity by tightly controlling the nucleotide-binding states of the RRAG [19]. For example, heterotrimeric GATOR1 complex is the GAP for RRAGA-RRAGB and hence negatively regulates MTORC1 activity, and FLCN (folliculin) and its binding partner FNIP1 (folliculin interacting protein 1) or FNIP2 (folliculin interacting protein 2) have been identified as the GAP for RRAGC-RRAGD and positively regulates MTORC1 [19–23]. Apart from nutrient sufficiency, recent studies showed that growth factor signaling could also control RRAG GTPases activity, indicating crosstalk between growth factor and nutrients sensing machineries that deserve further investigation [24].
In many cancers, persistent RRAG GTPases activation contributes to uncontrolled MTORC1 signaling for cancer cell growth and proliferation by evasion of metabolic checks [6,7,25,26]. For example, mutations in GATOR1 complex subunits DEPDC5, NPRL3 and NPRL2 have been implicated in glioblastomas, and FLCN and RRAGC mutations have been reported in follicular lymphoma and Birt-Hogg-Dubé syndrome, respectively. Post-translational modifications (PTMs) including ubiquitination and phosphorylation of RRAG GTPases signaling have been recently reported and may have a pro-growth function in tumors. As an example, the E3 ubiquitin ligase KLHL22-mediated DEPDC5 degradation has been linked with breast cancer [27]. Moreover, deregulation of gene transcription is another critical mechanism underpinning cancer onset and progression. Yet the transcriptional regulatory mechanisms responsible for RRAG GTPases activation in both physical conditions and malignancies remain to be fully elucidated.
The transcription factors in the MEF2 (myocyte enhancer factor 2) family play crucial roles in determining cell fate and adaptive responses by controlling gene expression [28–30]. Mammals have four isoforms of transcription factor MEF2s (MEF2A to MEF2D) and each of these has a highly similar N-terminal sequence and binds the consensus sites in the proximal promoters of genes [30–33]. MEF2s have been known to play critical roles in multiple cell types such as neurons, muscle and hematopoietic cells. Still, they more recently have been implicated in triggering and maintaining the tumorigenic process [28,34–37]. A series of studies indicated that MEF2s acted as oncogenes in immature T-cell acute lymphoblastic leukemia, B-cell acute lymphoblastic leukemia and hepatocellular carcinoma by controlling various processes such as proliferation, apoptosis, or epithelial-mesenchymal transition [37–39]. However, the detailed mechanisms by which MEF2s promote tumor malignancy remained largely unknown. Interestingly, in drosophila, MEF2s promoted lipogenesis and glycogen storage in the drosophila fat body and profoundly influenced cell proliferation upon serum stimulation [29,40,41], suggesting that MEF2s positively regulate anabolic metabolism upon growth factor stimulus. This prompted us to ask whether MEF2s have a possible regulatory role in MTORC1 activation and hyper-activated MEF2s contribute to uncontrolled MTORC1 in cancers.
In this study, we reported that MEF2A and MEF2D controlled MTORC1 activation and subsequent cellular events including macroautophagy/autophagy and anabolic processes. Mechanistically, MEF2A and MEF2D directly regulated the expression of FNIP1 and FNIP2 to modulate the nucleotide-binding states of the RRAGC-RRAGD heterodimer and then promoted MTORC1 lysosomal localization. We further uncovered that MEF2D but not MEF2A was a direct substrate of the proto-oncogenic tyrosine kinase SRC upon pro-mitogenic signal. Phosphorylation of MEF2D on three highly conserved tyrosine residues increased its transcriptional activity and was essential for MTORC1 activation by growth factors. Finally, we proved that the SRC-MEF2D-MTORC1 pathway regulated pancreatic cancer cell growth and was aberrantly activated in human pancreatic cancer. Thus, our study established a transcriptional regulatory mechanism of MTORC1 by MEF2A and MEF2D and uncovered an SRC-MEF2D-MTORC1 pathway for growth factor-mediated control of the RRAG GTPase machinery and its role in pancreatic cancer malignancy.
Results
MEF2A and MEF2D positively regulate MTORC1 activity
By analyzing The Cancer Genome Atlas (TCGA) pan-cancer public datasets, we demonstrated that the expression level of transcription factor MEF2s was upregulated across multiple cancer cohorts compared to normal samples and positively correlated with the MTOR pathway in pancreatic cancer (Figure S1A and S1B). RNA-sequencing data from HeLa cells with constitutively active MEF2 (MEF2-VP16) overexpressed also showed a positive correlation between MEF2s transcriptional activity and genes set in MTOR signaling (Figure 1A). To further investigate whether MEF2s play a role in MTORC1 activation, HeLa cells were transfected with either trans-dominant negative MEF2 (dnMEF2) or MEF2-VP16 plasmids and then treated with the amino acid fasting-refeeding paradigm for MTORC1 activation. The results showed that dnMEF2, which abrogates the function of the endogenous MEF2s attenuated MTORC1 activation upon amino acid stimulation, as evidenced by the decreased phosphorylation levels of RPS6KB1/S6K1 and EIF4EBP1/4E-BP1, two well-characterized MTORC1 substrates (Figure 1B and Figure S1C) [42]. By contrast, MEF2-VP16 amplified MTORC1 activation in HeLa cells upon amino acid stimulation (Figure 1C). These results suggested that MEF2s indeed promoted MTORC1 activity. Mammals have four isoforms MEF2s which are usually functionally redundant except for the most distant family member MEF2B by controlling the transcription of overlapping sets of genes. To identify the specific MEF2 isoform that participated in MTORC1 regulation, HeLa cells were depleted of MEF2A, MEF2C or MEF2D expression with small interfering RNA (siRNA) and then deprived of and restimulated with amino acids. Notably, the results exhibited that depletion of MEF2A and MEF2D, but not MEF2C, inhibited MTORC1 activation (Figure 1D and Figure S1D). Moreover, simultaneous knockdown of MEF2A and MEF2D with either transient siRNAs or stable short hairpin RNA (shRNA) transfection synergistically decreased MTORC1 activation (Figure 1D and Figure S1E), indicated that MEF2A and MEF2D acted coordinately in controlling MTORC1 activity. Similar to a complete set of amino acids, depletion of MEF2A and MEF2D resulted in decreased MTORC1 activation upon stimulation with solely leucine, the key amino acid that activates MTORC1 (Figure 1E). Since previous studies [43] showed that MEF2A positively regulated PI3K-AKT signaling which was an efficient MTORC1 activator, we tested whether MEF2A or MEF2D accommodated the activity of AKT. The results displayed that neither overexpression nor depletion of MEF2A and MEF2D altered AKT activity in HeLa cells, as evidenced by the phosphorylation level of Ser473 in AKT protein (Figure 1(B,C), and Figure S1C and S1E), suggested that MEF2A and MEF2D modulated MTORC1 through other mechanisms. A rescue experiment was performed by re-introducing MEF2A with or without MEF2D in MEF2A and MEF2D double-knockdown cells. Only coincident MEF2A and MEF2D reconstitution maximally restored MTORC1 activation upon amino acid stimulation, compared with individual MEF2A or MEF2D overexpression (Figure 1F). Taken together, these results indicated that MEF2A and MEF2D positively regulated MTORC1 activity.
Figure 1.
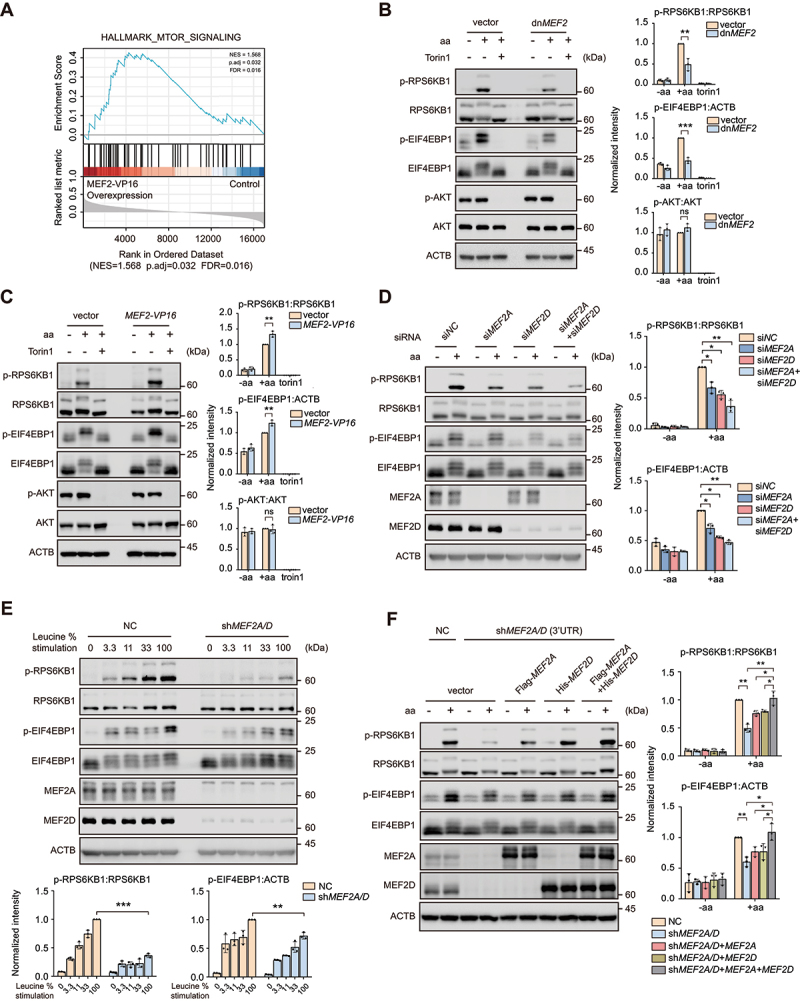
Transcription factors MEF2A and MEF2D regulate MTORC1 activity upon amino acid stimulation. (A) Gene set enrichment analysis of the MTOR pathway gene list in HeLa cells with overexpression of MEF2-VP16 versus controls by using RNA-seq data. (B) HeLa cells transfected with either dnMEF2 or vector as control were treated with amino acid (aa) starvation and amino acid restimulation with or without troin1 (250 nM). Western blot analysis of phosphorylation of RPS6KB1, EIF4EBP1 and AKT is shown. Right plots show phosphorylated p-RPS6KB1:RPS6KB1 (top), p-EIF4EBP1:ACTB (middle) and p-AKT:AKT (bottom) ratios. (C) HeLa cells transfected with MEF2-VP16 or vector were treated and then analyzed by immunoblotting similar to (B). (D) HeLa cells transfected for 48 h with indicated siRNA were starved of amino acids and restimulated before immunoblotting analysis. Right plots show the p-RPS6KB1:RPS6KB1 (top), p-EIF4EBP1:ACTB (bottom) ratios. (E) HeLa cells that stably knocked-down both MEF2A and MEF2D (shMEF2A/D) or control (NC) were starved for amino acids and restimulated with increasing levels (expressed as % of the concentration in DMEM medium) of leucine. Lysates were analyzed for MTORC1 activity as in (D). Bottom plots show the p-RPS6KB1:RPS6KB1 (right), p-EIF4EBP1:ACTB (left) ratios. (F) MEF2A and MEF2D double-knockdown HeLa cells transfected with MEF2A or/and MEF2D and then subjected to amino acid administration. MTORC1 activity was confirmed by immunoblotting. Plots on the right show p-RPS6KB1:RPS6KB1 (top) and p-EIF4EBP1:ACTB (bottom) ratios. Data are presented as the mean ± S.E.M. (n = 3 independent experiments, two-sided Student’s t-test for B, C and E, one-way analysis of variance [ANOVA] for D, two-way ANOVA post hoc test for F, *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant compared with indicated group).
MEF2A and MEF2D are essential for MTORC1-dependent anabolic processes and cell growth
MTORC1 promotes protein and lipid biosynthesis and inhibits catabolic processes such as autophagy [1,42,44–46]. We determined whether MEF2A and MEF2D affected these cellular functions known to be downstream of MTORC1 signaling. HeLa cells depletion of both MEF2A and MEF2D were subjected to amino acid starvation and replenishment in the presence or absence of puromycin, which is known as the surface sensing of translation (SUnSET) experiment utilized to monitor and quantify global protein synthesis in mammalian cells [47]. Detection of puromycin incorporation by immunoblot assay revealed that depletion of both MEF2A and MEF2D impaired protein synthesis (Figure 2A). Furthermore, we stained the neutral lipids by using oil red O assay to detect the intracellular lipid level in stably MEF2A and MEF2D knockdown HeLa cells. As expected, loss of MEF2A and MEF2D dramatically reduced the accumulation of lipids in HeLa cells. Similar results were obtained in TSC2 (TSC complex subunit 2)-depleted cells which accumulate high levels of lipids as MTORC1 is hyperactivated (Figure 2B). Moreover, in MEF2A and MEF2D double-knockdown HeLa cells with or without TSC2 depletion, reconstitution with MEF2A and MEF2D largely recovered the content of intracellular lipid (Figure 2B). MTORC1 is a well-established negative regulator of autophagy. To investigate the impacts of MEF2A and MEF2D depletion on cellular autophagic activity, we monitored the dynamic change of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) lipidation level by measuring LC3B-II:GAPDH ratio. The results showed that, in control cells, amino acid replenishment significantly induced ULK1 (unc-51 like autophagy activating kinase 1) phosphorylation at Ser757 targeted by MTORC1 and suppressed LC3B lipidation. By contrast, depletion of MEF2A and MEF2D largely inhibited ULK1 Ser757 phosphorylation and reversed the suppressive effects of amino acids on LC3B lipidation (Figure 2C). Similarly, time-lapse imaging of GFP-LC3 puncta formation that reflecting autophagy induction in stable HeLa cell lines revealed that amino acid restimulation led to a reduction in GFP-LC3 puncta, which was inversed by MEF2A and MEF2D knockdown (Figure 2D). Moreover, compared with control cells, depletion of MEF2A and MEF2D sustained the autophagic flux upon amino acid restimulation, as measured by a decreased ratio of yellow:red puncta using the tandem GFP-RFP-LC3 construct (Figure 2E). Depletion of MEF2A and MEF2D also reduced cell size (Figure 2F), a phenotypic characteristic of MTORC1 inactivation [48]. These results suggested that MEF2A and MEF2D were required for MTORC1-mediated anabolic events and autophagy.
Figure 2.
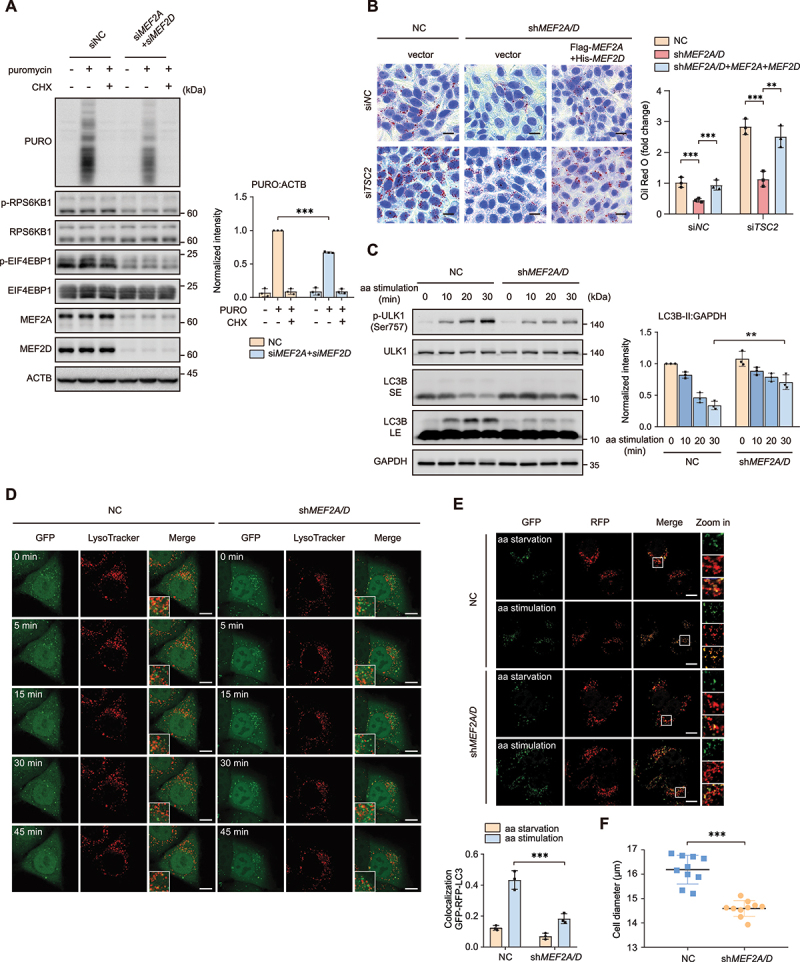
MEF2A and MEF2D are required for MTORC1 mediated protein, lipid synthesis and inhibit autophagy. (A) HeLa cells depletion of both MEF2A and MEF2D by indicated siRNA were treated with amino acid administration in the presence or absence of 10 μg/ml puromycin (PURO) and cycloheximide (CHX). Cell lysates were analyzed by western blot with an antibody to puromycin (17H1). Right plots show the ratio of PURO:ACTB. (B) MEF2A and MEF2D double-knockdown HeLa cells with or without transiently re-expressing MEF2A and MEF2D after utilized TSC2-targeting (siTSC2) or control (siNC) siRNA for 48 h under normal culture condition. Oil red O staining was performed to quantify intracellular lipid levels. Scale bar: 20 μm. (C) control or shRNA-mediated depletion of MEF2A and MEF2D HeLa cells were treated with the starvation of amino acids for 1 h and then restimulated with amino acids for indicated time. Phospho-ULK1 (Ser757), ULK1, LC3B-II levels were analyzed by western blotting. Right plot shows the quantification of LC3B-II:GAPDH. Short exposure (SE), long exposure (LE). (D) MEF2A and MEF2D knockdown and control HeLa cells that stably express GFP-LC3 were starved of amino acids for 1 h and then restimulated with amino acids for indicate duration and stained with LysoTracker. Time-lapse images were taken. Scale bar: 5 μm. (E) MEF2A and MEF2D double-knockdown and control HeLa cells were transfected for 24 h with GFP-RFP-LC3 adenovirus. After that, cells were subjected to amino acid starvation for 4 h and restimulation for 30 min. Autophagy flux was tested by confocal. Plot on bottom shows the ratios of colocalization of GFP and RFP. Scale bar: 10 μm. (F) statistical analysis of the cell diameters of control HeLa cells and the cells that depletion of MEF2A and MEF2D by using a cell counter. Data are presented as the mean ± S.E.M. (n = 3 independent experiments for A, B and C, n = 3 independent fields per condition for E, n = 10 per group for F. two-sided Student’s t-test for A, C, D and E, one-way ANOVA for B and E, *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant compared with indicated group).
MEF2A and MEF2D promoted the lysosomal localization of MTORC1 through modulating RRAG GTPase activity
Translocation of MTORC1 from the cytosol to the lysosomal membranes is a key step for amino acid induced MTORC1 activation. Since MEF2A and MEF2D regulated MTORC1 activity in an amino acid sensitive manner (Figure 1), we hypothesized that MEF2A and MEF2D might regulate MTORC1 lysosomal translocation. To support this idea, lysosome immunoprecipitation (Lyso-IP) was performed in control and MEF2A and MEF2D double-knockdown HeLa cells expressing Flag-tagged TMEM192 (transmembrane protein 192). We found that in MEF2A and MEF2D knockdown HeLa cells, MTORC1 could not obviously translocate on lysosomes upon amino acid restimulation (Figure 3A). Consistently, depletion of MEF2A and MEF2D decreased the colocalization of MTOR and lysosomal protein LAMP2 in HeLa cells when amino acids were supplemented (Figure 3B). Thus, we concluded that MEF2A and MEF2D controlled MTORC1 lysosomal translocation in response to amino acids. RRAG GTPases are considered as critical regulators of the MTORC1 cytosol-lysosomal shutting process through anchoring to lysosome surface via Ragulator complex and physically interacting with RPTOR/raptor. Immunoprecipitation assay revealed that MEF2A and MEF2D depletion attenuated the interaction between RRAGC and MTORC1 when amino acids were supplemented (Figure 3C). We reasoned that MEF2A and MEF2D might transcriptionally regulate the expression of RRAG GTPases components. Yet mRNA levels of RRAG GTPases transcripts remained unchanged in MEF2A and MEF2D knockdown cells (Figure S2A). To further investigate the mechanistic details of how MEF2A and MEF2D regulated MTORC1 lysosome translocation, we expressed constitutively active RRAG GTPases (RRAG[CA]: Flag-tagged RRAGAQ66L and Flag-tagged RRAGCS75N) [15] in MEF2A and MEF2D double-knockdown cells, and found that RRAG(CA) completely rescued MTOR and LAMP2 colocalization in cells (Figure 3D). Consistently, RRAG(CA) also fully restored MTORC1 activation by amino acids (Figure 3E), indicating that MEF2A and MEF2D acted upstream of RRAG GTPases to modulate its activity. Collectively, these data demonstrated that MEF2A and MEF2D controlled MTORC1 lysosomal translocation via regulating the activity rather than the protein level of RRAG GTPases.
Figure 3.
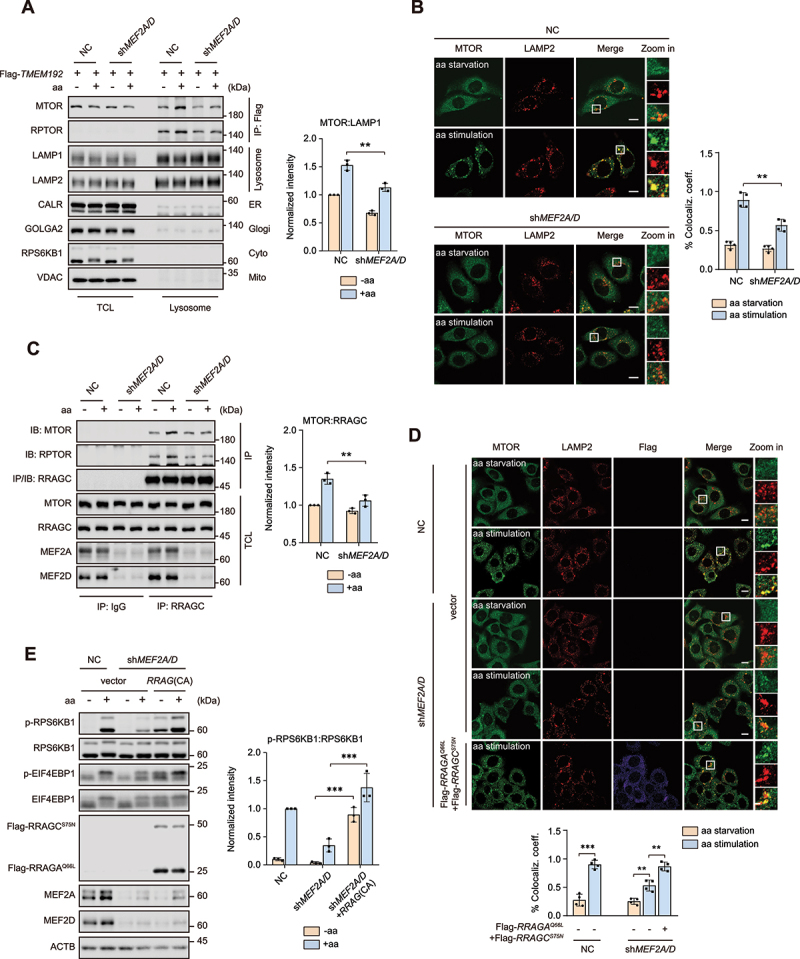
MEF2A and MEF2D positive regulate MTORC1 translocation to lysosomes. (A) MEF2A and MEF2D double-knockdown and control HeLa cells transfected with Flag-tagged TMEM192 for 24 h were subjected to amino acid starvation and restimulation. Purified lysosomes via lyso-IP assay (methods). Protein levels of MTOR and RPTOR (MTORC1 components), LAMP1 and LAMP2 (lysosome), CALR (endoplasmic reticulum), VDAC (mitochondria), GOLGA2 (Golgi) and RPS6KB1 (cytosol) were confirmed by immunoblotting. Right graph shows the quantification result of MTOR after normalization for LAMP1. (B) cells as in (A) were starved of and restimulated with amino acids for the indicated times before being analyzed by immunofluorescence and quantified to calculate the percentage of colocalization coefficient of MTOR (green) and lysosomal protein LAMP2 (red). Scale bar: 10 μm. (C) MEF2A and MEF2D double-knockdown HeLa cells were subjected to amino acid starvation and restimulation paradigm. Cell lysates were immunoprecipitated with anti-RRAGC antibody. Immunoprecipitates or total cell lysate (TCL) samples were probed for MTOR, RPTOR and RRAGC. Right graph shows the quantification result of MTOR after normalization for RRAGC. (D) MEF2A and MEF2D double-knockdown and control HeLa cells that either transfected with constitutively active RRAG GTPases (Flag-tagged RRAGAQ66L and RRAGCS75N) or vector coimmunostained for lysosomal marker LAMP2 (green), MTOR (red) and Flag-tag (violet). Cells were starved of amino acid and restimulated with amino acids before processing and imaging. The graph on the bottom shows the colocalization coefficient. Scale bar: 10 μm. (E) control (lanes 1–2) and knockdown of both MEF2A and MEF2D (lanes 3–6) HeLa cells that transfected with indicated RRAG GTPases were then treated with amino acids administration. Phosphorylation and protein levels of RPS6KB1 and EIF4EBP1 were determined by immunoblotting. Right graph shows the RPS6KB1:RPS6KB1 ratios. Data are presented as the mean ± S.E.M. (n = 3 independent experiments for A, C and E, n = 4 independent fields per condition for B, D. two-sided Student’s t-test for A, B, C and E, one-way ANOVA for D, **P < 0.01, ***P < 0.001).
MEF2A and MEF2D controlled RRAG GTPases activity via directly transcribing FNIP1 and FNIP2
Since MEF2A and MEF2D exerted a transcriptional activity-dependent effect on MTORC1 signaling (Figures 1 and 2), we speculated that certain downstream genes were targeted by MEF2A and MEF2D to control RRAG GTPases activity and subsequent MTORC1 recruitment. To identify the potential MEF2s regulated genes, we applied CUT&Tag assay [49] in HeLa cells to identify MEF2-regulated loci in transcriptionally active euchromatic regions with high resolution and sensitivity. We observed that MEF2D-bound signals were more prominent at promoter regions compared with IgG-bound signals and had a strong association with MEF2D in our motif analysis (Figure 4A and Figure S2B). We also revealed nominally significant differentially expressed genes (DEGs) in our previous transcriptome data and identified 678 genes were downregulated, and 801 genes were upregulated in MEF2-VP16 overexpressed HeLa cells (Figure 4B). KEGG analysis of the DEGs found that the upregulated genes were associated with the MTOR signaling pathway and cancer (Figure S2C). Among the DEGs, 175 downregulated and 289 upregulated genes overlapped with MEF2D bound genes disclosed by the CUT&Tag assay (Figure 4C). Notably, FNIP2, a component of the FLCN-FNIP2 complex which is a GAP for RRAGC-RRAGD was included in the upregulated overlapping genes cluster Figure 4(B, C). We also searched for MEF2s consensus DNA binding sites (5′-CC[A/t][t/a]AAATAG-3′) in the promoters of human genes known to regulate RRAG GTPases activity besides FNIP2. The results claimed that multiple genes including FNIP1, FNIP2, FLCN and RRAGD are potential MEF2s targets. Further studies revealed that MEF2A and MEF2D depletion clearly decreased both the mRNA and protein level of FNIP1 and FNIP2, while the levels of FLCN and RRAGD transcript showed no significant difference between MEF2A and MEF2D double-knockdown and control cells (Figure S2A, S2D and S2F). These data strongly indicated FNIP1 and FNIP2 as putative MEF2D target genes. To confirm this finding, we further performed chromatin immunoprecipitation-polymerase chain reaction (ChIP-PCR) and luciferase assays which showed that FNIP1 and FNIP2 were indeed direct transcriptional targets of MEF2D (Figure 4D, Figure S2E and S2G). Because the FLCN-FNIP complex functions as a GAP toward RAAGC-RAAGD, we speculate that MEF2A and MEF2D may affect RAAGC-RAAGD GTPase activity through transcription of FNIP1 and FNIP2. To test this, we detected the activity of RRAGC by isolating RRAG heterodimers from cells ectopically expressing wild-type RRAGC coupled with a RRAGBQ99L construct lacking GTPase activity, considering that the RRAGs function as obligate heterodimers as previously described [50,51]. The results showed that compared with control cells, knockdown of MEF2A and MEF2D led to a reduction in RRAGC activity, whereas FNIP1 and FNIP2 overexpression significantly rescued RRAGC activity in MEF2A and MEF2D-depleted cells (Figure S2H), indicating that MEF2A and MEF2D modulates RRAG GTPases activity via controlling FNIP1 and FNIP2 expression level. In line with these results, overexpression of either FNIP1 or FNIP2 in MEF2A and MEF2D-depleted HeLa cells rescued MTORC1 lysosomal localization and its downstream substrates phosphorylation upon amino acid restimulation Figure 4(E,F). In summary, these findings suggested that MEF2A and MEF2D were transcription factors for the FNIP1 and FNIP2 gene, which underlies its regulation of MTORC1 signaling.
Figure 4.
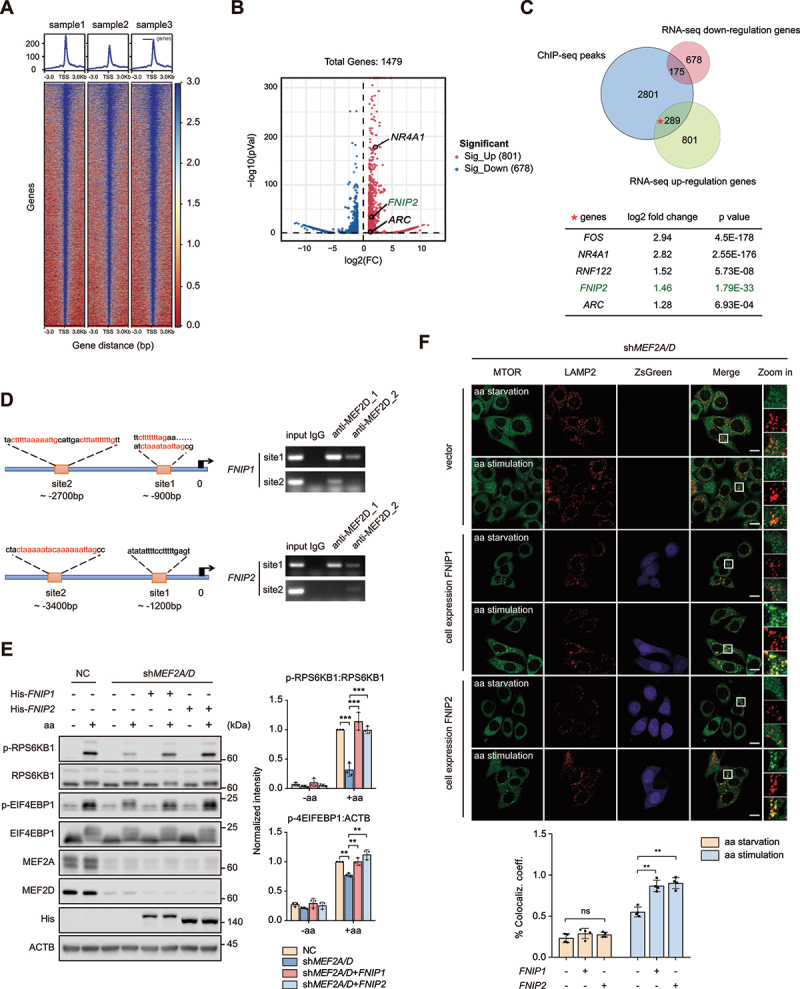
MEF2A and MEF2D control MTORC1 lysosome localization and activation via transcriptional regulation of FNIP1 and FNIP2. (A) heatmap of the binding sites of MEF2s at the positions − 3.0 kb upstream to + 3.0 kb downstream relative to the transcription start site (TSS). (B) volcano plot showing DEGs in control and MEF2-VP16 overexpression HeLa cells. The significant changed upregulated genes were labeled with gene names, NR4A1 and ARC are well-characterized substrates of MEF2s. (C) venn diagrams displaying the overlap of downregulated and upregulated genes from the transcriptome with MEF2 bound genes. Bottom table shows FNIP2 and other genes which are well known as MEF2s substrates including Fos/c-fos, NR4A1, ZNF122 and ARC in the subset of overlap of upregulated genes with MEF2 bound genes. (D) left-schematic shows the predicted MEF2 binding sites by conserved MEF2 consensus at the positions upstream to FNIP1 and FNIP2 genes transcription start site (TSS). Right plots show the PCR results revealed by chromatin immunoprecipitation assay. MEF2D_1,2 indicate the ChIP-PCR results by different antibodies (1-BD biosciences, 610774; 2-CST, 77986). (E) control (lanes 1–2) and MEF2A and MEF2D double-knockdown (lanes 3–8) HeLa cells that either transfected with indicated plasmids were starved of and restimulated with amino acids. Phosphorylation and protein levels of RPS6KB1 and EIF4EBP1 were determined by immunoblotting, as indicated. Plots on the right show p-RPS6KB1:RPS6KB1 (top) and p-EIF4EBP1:ATCB (bottom) ratios. (F) MEF2A and MEF2D double-knockdown HeLa cells stably overexpressing FNIP1 or FNIP2 by pLVX-IRES-ZsGreen constructs were starved of and restimulated with amino acids before coimmunostained of LAMP2 and MTOR and analyzed by confocal. MTOR (green), LAMP2 (red), ZsGreen-FNIP1 (violet) and ZsGreen-FNIP2 (violet). The plot on the bottom shows the colocalization coefficient. Scale bar: 10 μm. Data are presented as the mean ± S.E.M. (n = 3 independent experiments for E. n = 4 independent fields per condition for F. one-way ANOVA for F, two-way ANOVA for E **P < 0.01, ***P < 0.001).
SRC directly interacted with and phosphorylated MEF2D
SRC proto-oncogene is a protein tyrosine kinase that involved in cells proliferation, division and migration signaling pathways [52]. Aberrantly activated SRC has been known as a potent driver during oncogenesis [36,53,54]. Posttranslational modifications of MEF2s, such as phosphorylation, have been established as critical manners for modulating its transcriptional activity [28,55–57]. We then tried to delineate whether SRC could regulate MEF2s function in cancer cells. Co-immunoprecipitation (Co-IP) assay revealed that endogenous MEF2D, but not MEF2A, interacted with SRC (Figure S3A). In consistent, the transfected exogenous SRC and MEF2D were also bound with each other (Figure 5A and Figure S3B and S3C). GST affinity isolation experiment showed a direct interaction between MEF2D and SRC (Figure 5B). Intriguingly, the SRC-MEF2D interaction was markedly enhanced by EGF stimulation (Figure 5C), indicated that an active construction of SRC kinase appeared to be critical for its binding with MEF2D. We then asked whether SRC directly phosphorylates MEF2s. MEF2A or MEF2D together with SRC constructs were transfected into HeLa cells, and the phosphorylation level of immunoprecipitated MEF2A or MEF2D was determined by a pan phospho-tyrosine antibody. The data clearly showed that only MEF2D, but not MEF2A was phosphorylated by SRC (Figure S3D). In vitro kinase assay corroborated that SRC could directly phosphorylate MEF2D, which was abolished by SU6656 and Dasatinib, two specific SRC inhibitors (Figure 5D and Figure S3E). We also found that the level of tyrosine-phosphorylated MEF2D positively correlated with the transfected cDNA doses of SRC (Figure 5E). In addition, compared to wild-type SRC, the kinase-dead SRC failed to phosphorylate MEF2D (Figure 5F). We next examined whether growth factors increased SRC-mediated MEF2D tyrosine phosphorylation. The results displayed that serum and EGF stimulation significantly increased MEF2D tyrosine phosphorylation level, concurring with SRC activation evidenced by its elevated phospho-Tyr416 level (Figure 5G). By contrast, EGF lost its ability to increase the level of phospho-tyrosine of MEF2D in SRC-depleted cells (Figure 5H). Likewise, treatment of cells with SU6656 or Dasatinib also abrogated EGF-mediated MEF2D phosphorylation (Figure 5I). Together, these results illustrated that SRC proto-oncogene kinase interacted with and phosphorylated MEF2D upon mitogenic stimulation.
Figure 5.
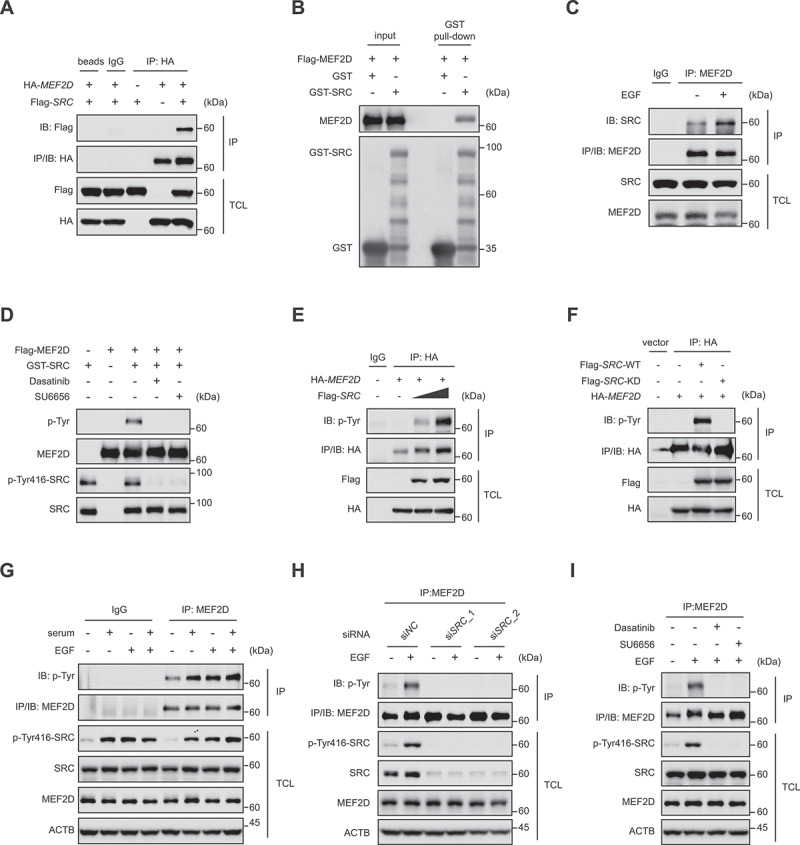
SRC directly interacts with and phosphorylates MEF2D upon mitogenic stimulation. (A) lysate derived from human embryonic kidney 293T (HEK293T) cells transfected as indicated were immunoprecipitated with IgG or anti-hemagglutinin (HA) antibody. Immunoprecipitation (IP) and TCL were probed for indicated antibodies. (B) GST affinity isolation assay was performed using HEK293T cells purified Flag-tagged MEF2D protein and bacterially purified GST or GST-tagged SRC (method), followed by immunoblotting with indicated antibodies. (C) HeLa cells were cultured in a serum free medium for 4 h, then treated with or without EGF for 30 min. Cell lysates were immunoprecipitated with anti-MEF2D antibody and blotted with anti-SRC antibody (top panel). TCL was probed for indicated antibodies. (D) HEK293T purified Flag-tagged MEF2D protein was pre-treated with lambda PP and then incubated with commercial active GST-tagged SRC kinase in a kinase assay buffer, followed by immunoblotting with indicated antibodies. (E) HEK293T cells transfected as indicated were immunoprecipitated with anti-HA antibody and blotted with pan p-Tyr antibody. TCL was analyzed by immunoblotting for indicated antibodies. (F) HEK293T cells transfected HA-tagged MEF2D with either Flag-tagged SRC-WT (wild-type) or Flag-tagged SRC-KD (SRCK298M, a kinase-dead form of SRC). Cell lysates were immunoprecipitated with anti-HA antibody. IP and TCL samples were probed for indicated antibodies. (G) HeLa cells were maintained in a serum free medium for 4 h, and then treated with or without serum, EGF or serum plus EGF for 30 min. Total cell lysates and endogenous MEF2D immunoprecipitated were analyzed by immunoblotting with indicated antibodies. Actin as a loading control. (H) HeLa cells transfected with indicated siRnas against SRC (in two RNAi sequences) or control (siNC) for 48 h were subjected to serum free culture for 4 h, followed by stimulation with or without EGF for 30 min. Cells were lysed and subjected to immunoprecipitation with anti-MEF2D antibody, followed by immunoblotting with indicated antibodies. (I) HeLa cells were subjected to serum free medium for 4 h, and then either untreated or restimulation with EGF in the presence or absence of SRC kinase inhibitor Dasatinib or SU6656. Endogenous MEF2D was immunoprecipitated, followed by immunoblotting with indicated antibodies.
Phosphorylation of MEF2D by SRC was required for MTORC1 activation by growth factors
An online phosphorylation sites prediction tool NetPhos-3.1 (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1) was performed to identify the potential sites in MEF2D protein sequence. To verify if these predicted residues in MEF2D are indeed SRC targeted phosphorylation sites, we mutated each of these tyrosine residues to phenylalanine and tested their phosphorylation ability by SRC. The evidence showed that phosphorylation levels of MEF2DY131F, MEF2DY333F and MEF2DY337F constructs decreased compared with wild-type MEF2D (Figure 6A). Combined mutation of Tyr131, Tyr333 and Tyr337 (3YF) in MEF2D nearly abolished SRC-mediated tyrosine phosphorylation of MEF2D (Figure 6B). Consistent results were obtained in the in vitro kinase assay (Figure 6C). Sequence alignment indicated that all three tyrosine residues in MEF2D are highly conserved in multiple species (Figure 6D and Figure S3F). Moreover, compared to wild-type MEF2D, the unphosphorylable MEF2D-3YF mutation exhibited a much less level of phospho-tyrosine even after EGF treatment (Figure 6E). To sum up, these data demonstrated that MEF2D was phosphorylated by SRC on multi-tyrosine residues. As MEF2D functions as a transcriptional factor and shuttles between nucleus and cytoplasm [58], we then tested whether SRC-mediated MEF2D phosphorylation impacted its cellular localization and transcriptional activity. We found that ectopic expression of SRC induced the nuclear translocation of MEF2D (Figure S3G). Moreover, the results also showed that SRC caused an increased reporter activity in wild-type but not 3YF mutated MEF2D-reconstituted HeLa cells with endogenous MEF2A and MEF2D depletion (Figure 6F), indicating an elicited transactivation potential of MEF2D by its tyrosine phosphorylation. Quantitative reverse transcription PCR (RT-qPCR) revealed that wild-type MEF2D increased the mRNA levels of FNIP1 and FNIP2 whereas the 3YF mutation had little effect on their levels. It was the same case for NR4A1, ZMAT4 and DAAM1, which are the well-characterized substrate of MEF2D (Figure 6G). To confirm whether SRC-MEF2D phosphorylation was involved in MTORC1 activation by growth factors, we determined MTORC1 activation upon growth factor stimulation in control and knockdown of MEF2A and MEF2D HeLa cells. The results showed that EGF treatment led to a more pronounced upregulation of p-RPS6KB1 and p-EIF4EBP1 in control cells, indicating that EGF boosted MTORC1 activity in a MEF2A and MEF2D-dependent manner (Figure 6H). Then either wild-type MEF2D or the 3YF mutation together with MEF2A was reintroduced into MEF2A and MEF2D double-knockdown cells, and the data showed that wild-type MEF2D fully restored MTORC1 activity upon EGF stimulation while the 3YF mutation failed to do so (Figure 6I). Collectively, these results proved that phosphorylation of tyrosine residues in MEF2D by SRC enhanced its transcriptional activity, representing an important mechanism linking growth factor stimulus to MTORC1 activation.
Figure 6.
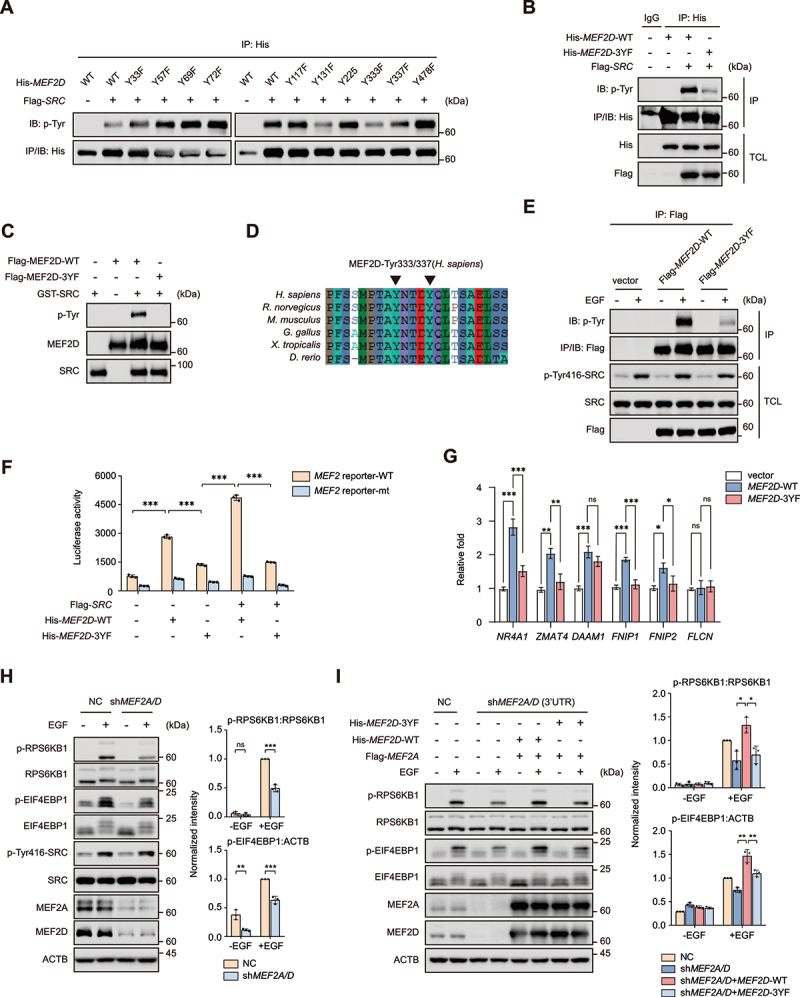
SRC-mediated tyrosine phosphorylation of MEF2D is required for transcriptional activity of MEF2D and in regulation of MTORC1 activity. (A) His-tagged MEF2D-WT or MEF2D mutant constructs (mutation of Tyr33, 57, 69, 72, 117, 131, 225, 333, 337 and 478 residues) were co-transfected with or without Flag-tagged SRC in HEK293T cells for 24 h. Cells were lysed and subjected to immunoprecipitation against His-tag, followed by immunoblotting with p-tyr antibody. (B) HEK293T cells transfected His-tagged MEF2D-WT or MEF2D-3YF mutants with or without Flag-tagged SRC were lysed and subjected to immunoprecipitation against His-tag, followed by immunoblotting with indicated antibodies. (C) Flag-tagged MEF2D-WT or its 3YF mutant protein purified from HEK293T cells was incubated with commercial active GST-tagged SRC kinase in a kinase assay buffer, followed by immunoblotting with p-tyr antibody. (D) sequence alignment of the residues flanking across different species. Black arrowheads point to the tyrosine residues corresponding to the Tyr333 and tyr 337 residues in human MEF2D. (E) HeLa cells that transfected with Flag-tagged MEF2D-WT or MEF2D-3YF were maintained in a serum free medium for 4 h, followed with or without EGF treatment. Cell lysates were prepared and immunoprecipitation were analyzed by immunoblotting. (F) luciferase assay was performed in depletion of both MEF2A and MEF2D HeLa cells after co-transfection of indicated expression plasmids and wild-type (MEF2 reporter-WT) or mutated (MEF2 reporter-mt) luciferase reporter plasmids for 24 h. (G) qRT-PCR analysis was performed in MEF2A and MEF2D double-knockdown HeLa cells that reconstructed with MEF2D-WT or MEF2D-3YF. The mRNA levels of FNIP1, FNIP2, FLCN, NR4A1, ZMAT4 and DAAM1 were shown. (H) MEF2A and MEF2D double-knockdown HeLa cells were starved with serum for 4 h and then treated with EGF for 3 h before immunoblotting analysis of the activation of MTORC1 with indicated antibodies. Right plots show phosphorylated p-RPS6KB1:RPS6KB1 (top), p-EIF4EBP1:ACTB (bottom) ratios. (I) MEF2A and MEF2D double-knockdown HeLa cells that transfected with indicated plasmids were subjected to serum starvation for 4 h and restimulated with EGF for 3 h. MTORC1 activity was analyzed similarly to (H). Right plots show phosphorylated p-RPS6KB1:RPS6KB1 (top), p-EIF4EBP1:ACTB (bottom) ratios. Data are presented as the mean ± S.E.M. (n = 3 independent experiments. two-sided Student’s t-test for H, one-way ANOVA for I, *P < 0.05, **P < 0.01, ***P < 0.001).
SRC-MEF2D-MTORC1 axis regulates pancreatic cancer cell growth and predicts clinical outcomes
Aberrant MTORC1 and SRC activity has been linked to cancer development and progression [2,53,54]. Analysis of the TCGA database revealed that several types of cancer including pancreatic cancer exhibited elevated MEF2s expression level (Figure S1A), which positively correlated with the activation of MTOR pathway in pancreatic cancer as shown by GSVA analysis (Figure S1B). These data suggested that SRC-MEF2D-MTORC1 axis may play a role in pancreatic cancer. Consistently, depletion of MEF2A and MEF2D significantly impeded pancreatic cancer cells proliferation, as displayed by the cell proliferation and colony formation assay using AsPC-1 and PANC-1 pancreatic cancer cell lines Figure 7(A,B). Notably, the decreased cell proliferation rate implicated by MEF2A and MEF2D depletion could be almost entirely retrieved by re-expression of wild-type MEF2D but not the 3YF mutation (Figure 7A). Knockdown of MEF2A and MEF2D also significantly reduced tumor growth of the xenografts Figure 7(C,D). These results suggested that MEF2A and MEF2D were required for sustaining the proliferation of pancreatic cancer cells. Analysis of the TCGA dataset also showed that mRNA level of MEF2D is upregulated in human pancreatic cancer samples and is the predictive factor for pancreatic cancer with poor clinic outcome (Figure 7E, Figure S4A, S4B and S4C). Moreover, a strong positive correlation between the mRNA levels of MEF2D and EGFR, SRC, FNIP1, and FNIP2 was revealed (Figure S4D). The immunohistochemistry data consistent with immunoblotting proved that protein levels of MEF2D were markedly increased in human pancreatic cancer and positively correlated with that of FNIP1, FNIP2 or p-EIF4EBP1 Figure 7(F,G). Finally, we identified that tyrosine phosphorylation of endogenous MEF2D was increased significantly in human pancreatic cancers, correlating with hyperactivated SRC (Figure 7H). These results demonstrated that MEF2A and MEF2D are crucial drivers of pancreatic cancer cell growth and the SRC-MEF2D-MTORC1 axis may be involved in this process.
Figure 7.
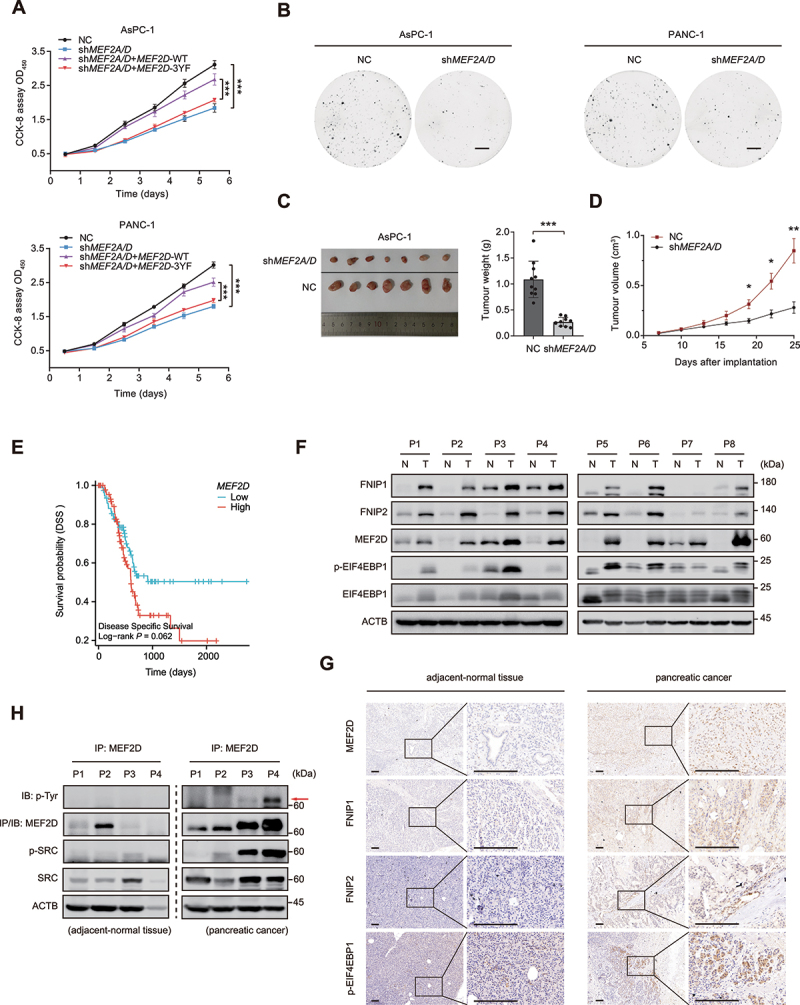
MEF2D promotes pancreatic cancer cell proliferation and correlates with clinical and pathological parameters. (A) pancreatic cancer cell line AsPC-1 (top) and PANC-1 (bottom) that depletion of MEF2A and MEF2D, transfected with either Flag-tagged MEF2D-WT or MEF2D-3YF, respectively and subjected to the cell proliferation assay (CCK8) to determine viable cell number. NC and knockdown of both MEF2A and MEF2D group co-transfected with empty vector. (B) the colony formation assay was performed with MEF2A and MEF2D double-knockdown AsPC-1 cells (left) or PANC-1 cells (right). The cells were maintained in complete medium containing 10% FBS for two weeks before staining and imaging. Scale bars: 5 mm. (C) Representative images (left) and weights (right) of xenograft tumors from nude mice implanted with MEF2A and MEF2D double-knockdown AsPC-1 cells versus control cells. n = 10 mice per group. (D) volumes of tumor burden in nude mice implanted with MEF2A and MEF2D double-knockdown or control AsPC-1 cells were measured after implantation. n = 10 mice per group. (E) TCGA cohort of pancreatic cancer patients were divided into two groups according to the median level of MEF2D mRNA expression. Overall survival was compared between these two groups, as shown in Kaplan-Meier curves. Log-rank P values are indicated. (F) immunoblot analysis of MEF2D, FNIP1, FNIP2, EIF4EBP1 and p-EIF4EBP1 were performed in pancreatic tumors and paired adjacent normal tissues. ACTB was probed for as a loading control. Tumor-adjacent normal tissue (N), tumor (T). (G) randomly selected pancreatic cancer samples and their paired adjacent normal tissues were lysed and subjected to IP against MEF2D, followed by immunoblotting with the pan p-Tyr antibody. (H) Representative immunohistochemistry of pancreatic cancer and paired adjacent normal tissue sections were stained for MEF2D, FNIP1, FNIP2 and p-EIF4EBP1 respectively. Scale bars: 200 μm. Data are presented as the mean ± S.E.M. (n = 3 independent experiments for a and B, n = 8 biologically independent samples for F, n = 4 biologically independent samples per group for G and H. two-sided Student’s t-test for a and D, *P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
MTORC1 pathway is usually aberrantly activated in human cancers and deregulated cell anabolic processes for sustaining their survival and impetus for growth [44,48,59]. While numerous studies have indicated that MTORC1 functions as a downstream effector for many frequently mutated oncogenes, resulting in its hyperactivation, the role of transcription factors in regulation of MTORC1 activation is still poorly understood. It has been reported that MiT/TFE transcription factors controlled MTORC1 activity by directly transcribing RRAGD, and this modulation was critical for metabolic adaptation to nutrient availability and multiple types of cancer cell proliferation, demonstrating a crucial role of transcription factors in MTORC1 signaling [60]. Thus, it would be interesting to test if other transcription factors may also contribute to modulating MTORC1 activation in cancers. Here, we uncovered a novel transcriptional regulatory mechanism of MTORC1 activation supporting cancer progression, involving the transcriptional factors MEF2s. Our results confirmed that MEF2A and MEF2D acted as positive regulators of MTORC1 by directly transcribing FNIP1 and FNIP2 to promote MTORC1 recruitment to lysosomes upon amino acid stimulation. Importantly, MTORC1 activation mediated by MEF2s was particularly relevant to pancreatic cancer, as shown by the analysis of TCGA datasets and our experimental evidence obtained from human pancreatic cancer samples, which all demonstrated a strong positive correlation among expression levels of MEF2D, FNIP1, FNIP2 and MTORC1 activity. Therefore, our finding proved that MEF2A and MEF2D were essentially required for MTORC1 activation and broadened the molecular regulatory mechanisms of aberrant MTORC1 activation in cancer. Interestingly, although functional redundancy of MEF2 family members due to the overlapping DNA binding sites, our data indicated that both MEF2A and MEF2D were indispensable for full responsiveness of MTORC1 to amino acids, suggesting that MEF2A and MEF2D regulated MTORC1 activity in a non-redundant manner. The mechanistic details of how MEF2A collaborated with MEF2D in controlling MTORC1 activation remained to be deciphered. One possibility is that MEF2A forms heterodimers with MEF2D and then positively regulates MEF2-dependent gene expression, which has been well elucidated in previous studies [61,62].
Post-translation modifications such as phosphorylation represent a unique integrated code to diversify MEF2s function. For example, MAPK14/p38 (mitogen-activated protein kinase 14) and PKC (protein kinase C) have been shown to phosphorylate MEF2A and MEF2C [28,56,63], enhancing their transcriptional activity. While CDK5 (cyclin-dependent kinase 5) phosphorylated MEF2A and MEF2D in the C-terminal trans-activation domain and impaired their transcriptional potential [55,64]. Here, we determined that MEF2D was a novel substrate of proto-oncogene protein kinase SRC which is commonly activated in human cancers, and mapped Y131, Y333 and Y337 residues in the trans-activation domain of MEF2D as SRC targeting sites. Phosphorylation at these sites enhanced MEF2D transcriptional activity. Moreover, our data indicated that phosphorylation and activation of MEF2D by SRC were essentially required for growth factor-stimulated MTORC1 signaling. Extensive studies illustrated two distinctive pathways, the growth factor stimulatory pathway governed via RHEB and the amino acid sensing pathway mainly consisting of the RRAG GTPases and their regulators, cooperatively controlled MTORC1 activation in a parallel manner. Yet recent studies showed that these two pathways were interactive rather than mutually repelled. For instance, investigations proved that TSC2, the critical molecular responsible for sensing growth factor, was also recruited to lysosome upon removal of amino acids, interacting with and allosterically inhibiting RHEB [11]. Analogically, growth factors could modulate the phosphorylation level of RRAGC to regulate both amino acid and growth factor mediated MTORC1 activation [24]. Our study demonstrated that MEF2A and MEF2D involved in amino acid sensing by transcriptional regulation of FNIP1 and FNIP2 could also readily detect growth factor stimulus, which was attained via the phosphorylation of MEF2D by SRC, a well-established growth factor-activated kinase. Therefore, similar to the previous research, our study implied another mechanism mediating crosstalk between amino acid and growth factor monitoring machinery during the activation of MTORC1. Notably, our data indicated that unlike MEF2D, MEF2A was not a substrate for SRC, which may be due to its failure to interact with SRC, suggesting that SRC-mediated phosphorylation of MEF2D was a specific regulatory process. The fact that SRC only phosphorylated MEF2D but both MEF2A and MEF2D were required during growth factor evoked MTORC1 activation was not contradictory. A previous study showed that phosphorylation of MEF2A by MAPK14 in the MEF2A-MEF2D heterodimer enhances its trans-activation activity [61]. We speculated that it might work in a similar manner in our research, that is, phosphorylation of MEF2D by SRC under mitogen stimulation also amplified MEF2A-MEF2D heterodimer transcriptional potential.
Aberrantly activated SRC has been known to be a potent oncogenic protein and is actively involved in a variety of human cancers hallmarks, including cell survival, invasion, metastasis and angiogenesis [52,53,65,66]. While multiple studies have shown that SRC gained its activity via either direct gene mutations or non-genetic events including increased signaling from receptor tyrosine kinases (RTKs) upon growth factor stimulation like EGF, the mechanisms relating SRC to oncogenesis remained to be completely deciphered. Recently, several studies have specified the role of SRC in cell proliferation with unexpected mechanisms. For example, in human breast cancer, SRC promoted tumor malignancy by boosting glycerolipid synthesis via an SRC-LPIN1/lipin1 axis [54]. In addition, SRC was involved in the activation of MTORC1 by facilitating the dissociation of GATOR1 from the RRAGs in many cancer types [67]. In this study, we also demonstrated that SRC regulated growth factor-mediated MTORC1 activation but added another layer of regulation involving the transcriptional modulation of the amino acid sensing machine, shedding new light on the mechanistic details underlying the tumor-promotion effects of SRC.
According to previous studies, MEF2s are well-characterized oncogenes for multiple cancers [39,68]. Yet the specific roles of MEF2s in tumorigenesis are largely unknown, especially in solid tumor. Our discoveries supported that one possible mechanism underlying the tumor-promoting effects of MEF2A and MEF2D may be that they controlled the activation of MTORC1. Supporting this notion, analysis of the TCGA dataset showed that MEF2A and MEF2D were significantly upregulated and were predictive factors for pancreatic cancer, and a positive correlation between MEF2D expression and genes directly regulated by MTOR in pancreatic cancer was further evidenced by GSVA analysis. Moreover, our data deriving from in vitro cell proliferation assay, in vivo animal studies and examination of human tumor tissues experimentally illustrated the critical role of SRC-MEF2D-MTORC1 axis in pancreatic cancer. We showed that SRC activity and the MEF2D tyrosine phosphorylation level were markedly increased in human pancreatic cancers. We also identified that upregulation of MEF2D was positively associated with FNIP1, FNIP2 and high MTORC1 activity in pancreatic cancers. Both MEF2A and MEF2D expression and phosphorylation of MEF2D were required for sustained pancreatic cancer cell proliferation. Yet our study was limited as we failed to generate specific antibodies recognizing the three tyrosine residues in MEF2D targeted by SRC, which should be utilized to accurately determine the phosphorylation level of MEF2D in pancreatic cancer samples. Also, considering the critical role of MTORC1 signaling in multiple human cancer types, further investigations are needed to check the role of SRC-MEF2D-MTORC1 pathway in other malignancy diseases apart from pancreatic cancer.
Materials and methods
Cell culture and treatments
All cells were maintained at 37°C, 5% CO2 and cultured in the following media: HeLa cells (American Type Culture Collection, CCL-2), HEK293T cells (American Type Culture Collection, CRL-3216), PANC-1 human pancreatic cancer cells (American Type Culture Collection, CRL-1469) and their derivatives were held in high glucose Dulbecco’s modified Eagle’s medium (DMEM, Corning, 10–013-CV); AsPC-1 human pancreatic cancer cells (American Type Culture Collection, CRL-1682) and its derivative were grown in RPMI-1640 (Corning, 10–040-CV). All media were supplemented with 10% fetal bovine serum (AusGeneX, FBS500-S), penicillin and streptomycin (Hyclone, SV30010). All cell lines were authenticated by STR profiling and validated to be free of mycoplasma contamination.
For amino acid starvation experiments, culture cells were rinsed twice with PBS (Corning, 21–040-CMR) and then maintained in amino acid-free DMEM (USBiological, MBS6120661) for 60 min. For amino acid restimulation, cells were treated with a handmade medium by directly adding commercial essential (Gibco, 11130077) and non-essential (Gibco, 11140076) amino acids to 1× in amino acid-free DMEM supplemented with glutamine (Gibco, 25030081). For leucine restimulation, cells were restimulated with handmade leucine solutions by suspending leucine powder (Sigma-Aldrich, L8192) in amino acid-free DMEM. All media in amino acid starvation or restimulation experiments were supplemented with insulin (Tocris, 3435/10) at 200 ng/ml instead of regular FBS. To remove the serum, cells were washed twice with PBS and then cultured at serum-free DMEM for the indicated time.
Antibodies and chemicals
The antibodies and other reagents used in this study were from the following sources: phospho-RPS6KB1/p-S6K1 (9234; 1:1,000 WB), RPS6KB1/S6K1 (9202; 1:1,000 WB), phospho-EIF4EBP1/p-4E-BP1 (9451; 1:3,000 WB), EIF4EBP1/4E-BP1 (9644; 1:1,000 WB), phospho-AKT (4060; 1:6,000 WB), AKT (4691; 1:6,000 WB), MTOR (2972; 1:1,000 WB; 1:200 immunofluorescence [IF]), RPTOR/raptor (2280; 1:1,000 WB), RRAGC (3360; 1:1,000 WB), LC3B (3868; 1:1,000 WB), FNIP2 (57612; 1:1,000 WB; 1:300 immunohistochemistry [IHC]), FLCN (3697; 1:1,000 WB), MEF2D (77986; 1:1,000 WB; 1:400 IHC; 1:50 ChIP), HA (3724; 1:2,000 WB), His (2365; 1:2,000 WB), VDAC (4661; 1:1,000 WB), GOLGA2/GM130 (12480; 1:1,000 WB), Flag (8146; 1:1,000 WB), CALR/calreticulin (12238; 1:1,000 WB), phospho-tyrosine/p-Tyr (9411; 1:3,000 WB), SRC (2109; 1:1,000 WB), phospho-SRC (59548; 1:1,000 WB) and ACTB/β-actin (3700; 1:10,000) were purchased from Cell Signaling Technology/CST. LAMP2 (ab25631; 1:500 WB; 1:120 IF), LAMP1 (ab24170; 1:1,000 WB), FNIP1 (ab25631; 1:1,000 WB; 1:300 IHC), phospho-EIF4EBP1 (ab278686; 1:200 IHC), MEF2A (ab264329; 1:1,000 WB) and MEF2C (ab231859; 1:1,000 WB) were obtained from Abcam; MEF2D (610774; 1:50 ChIP) from BD biosciences; puromycin (MABE341; 1:1,000 WB) was purchased from Sigma-Aldrich. Flag tag (NB-600-344; 1:500 IF) was purchased from Novus Biologicals. Flag tag (20,543–1-AP; 1:150 IP), HA tag (66006–2-lg; 1: 200 IP), His tag (66005–1-lg; 1:200 IP) were purchased from Proteintech; HRP-conjugated goat anti-mouse IgG (AS003), HRP-conjugated goat anti-rat IgG (AS028), HRP-conjugated mouse anti-rabbit IgG Light Chain (AS061), HRP-conjugated goat anti-rabbit IgG Heavy Chain (AS063) and HRP-conjugated goat anti-rabbit IgG (AS014) were obtained from ABclonal; Alexa Fluor 488-adsorbed donkey anti-rabbit IgG (A-21206), Alexa Fluor 594-adsorbed donkey anti-mouse IgG (A-21203), Alexa Fluor 647-adsorbed donkey anti-rabbit IgG (A-31573), Alexa Fluor 647-adsorbed donkey anti-goat IgG (A-21447) and LysoTracker™ Red DND-99 (L7528) were purchased from Invitrogen.
The chemicals used were torin1 (S2827), SU6656 (AS061) and Dasatinib (S1021) from Selleckchem; cycloheximide (CHX; HY-12320) from MedChemExpress; puromycin (60210ES25), hygromycin (60225ES03) and G418 (60220ES03) from Yeasen; Recombinant human EGF (236-EG) from R&D Systems; protease inhibitor cocktail (539134) and phosphatase inhibitor cocktail (524625) from EMD Millipore.
Plasmids and small interfering (si) RNAs
The Flag-tagged MEF2A, Flag-tagged MEF2D, Flag-tagged dnMEF2, MEF2-VP16 and MEF2 reporter plasmids were the gifts generously provided by Dr. Zixu Mao (Departments of Pharmacology and Neurology, School of Medicine, Emory University). The cDNA for human MEF2D (NM_005920.4), FNIP1 (NM_133372.3) and FNIP2 (NM_020840.3) were amplified according to their mRNA by using high-fidelity DNA polymerase (Vazyme, P515–01) and cloned into BamHI and EcoRV sites of pcDNA™3.1/myc-His (Invitrogen, V80020) through in-fusion cloning assay (In-Fusion Snap Assembly Kit; Clontech, 638974). HA or Flag tag sequence was synthesized and inserted into HindIII and KpnI sites of pcDNA™3.1/myc-His vector to re-construct the vector. The cDNA encoding TMEM192 (NM_001100389.2), human SRC (NM_005417.5) and MEF2D (NM_005920.4) were amplified and cloned into BamHI and EcoRV sites of HA/Flag-pcDNA™3.1 vector similarly. Kinase dead mutation SRCK298M was generated from wild-type Flag-tagged SRC using the site-directed mutagenesis kit (Agilent, 210513). Y33F, Y57F, Y69F, Y72F, Y117F, Y131F, Y225F, Y333F, Y337F and Y478F mutants MEF2D were generated from wild-type His-tagged MEF2D. The primers used were listed as follows: K298M F: 5ʹ-GGCTTCAGGGTCATGATGGCCACCCTGGTGGTAC-3ʹ and R: 5ʹ- GTACCACCAGGGTGGCCATCATGACCCTGAAGCC-3ʹ;
Y33F F: 5”-GCACGCTCAGCTCAAACGCCTTCTTCATCAG-3‘ and R: 5’-CTGATGAAGAAGGCGTTTGAGCTGAGCGTGC-3”;
Y57F F: 5”-GGTGCTGGCGAACTGGAACAGCTTGTTGGA-3‘ and R: 5’-TCCAACAAGCTGTTCCAGTTCGCCAGCACC-3”;
Y69F F: 5”-GCTCATTGTACTCCGTGAACTTGAGCAGCACCTTG-3‘ and R: 5’- GCTCATTGTACTCCGTGAACTTGAGCAGCACCTTG-3”;
Y72F F: 5”-CTCGTGTGGCTCATTGAACTCCGTGTACTTGAG-3‘ and R: 5’-CTCAAGTACACGGAGTTCAATGAGCCACACGAG-3”;
Y117F F: 5”- GCGCGTCGGAACTTGTCCTCCAGCAGG-3‘ and R: 5’- CCTGCTGGAGGACAAGTTCCGACGCGC-3”;
Y131F F: 5”-CAGTTGACCCAAAGCGCCGGAAGAGCCC-3‘ and R: 5’- GGGCTCTTCCGGCGCTTTGGGTCAACTG-3”;
Y225F F: 5”-CGAGCACTGACGAAGCCATTCCCAACAGGG-3‘ and R: 5’- CCCTGTTGGGAATGGCTTCGTCAGTGCTCG-3”;
Y333F F: 5”-CTGGTAATCTGTGTTGAAGGCAGTGGGCATGGA-3‘ and R: 5’- TCCATGCCCACTGCCTTCAACACAGATTACCAG-3”;
Y337F F: 5”-CTGGTCAACTGGAAATCTGTGTTGTAGGCAGTGGGC-3‘ and R: 5’- GCCCACTGCCTACAACACAGATTTCCAGTTGACCAG-3”;
Y478F F: 5”- CTCCCGTCTCAAAGGATCCCCCGGC-3‘ and R: 5’- GCCGGGGGATCCTTTGAGACGGGAG-3”;
For siRNA-mediated protein knockdown experiments, cells were transfected using Lipofectamine 2000 Transfection Reagent (Invitrogen, 11668019) according to the manufacturer’s instructions with the indicated siRNA and analyzed after 48 h (unless stated otherwise). Sequences of siRNAs were listed as follows: siTSC2 5ʹ-GCUGUUACCUCGACGAGUA-3ʹ; siSRC-776 5ʹ-GCGGCUCCAGAUUGUCAACAA-3ʹ; siSRC-1395 5ʹ-GUCAUGAAGAAGCUGAGGCAU-3ʹ; siMEF2A 5ʹ-CUGGCAGCAAGAACACAAU-3ʹ; siMEF2C 5ʹ-GAUAAUGGAUGAGCGUAAC-3ʹ; siMEF2D 5ʹ-GUAGCUCUCUGGUCACUCC-3ʹ.
Lentiviral production and stable cell lines generation
HeLa, AsPC-1, and PANC-1 stably knockdown MEF2A and MEF2D cell lines were established by a lentiviral system. The following lentiviral constructs were used: pLKO.1-puro (Addgene, 8453; deposited by Bob Weinberg) and pLKO.1-neo (Addgene, 13425; deposited by Sheila Stewart). The sequence of shRNA targeting MEF2A and MEF2D mRNA or 3’UTR was obtained from Sigma-Aldrich (TRCN0000015897, TRCN0000274112, TRCN0000005134 and TRCN0000005132). To generate a stable expression of FNIP1 or FNIP2 in stable knockdown of both MEF2A and MEF2D HeLa cells, FNIP1 and FNIP2 fragments were cloned into pLVX-IRES-ZsGreen1 (Clontech, 632187) for stable cell line generation. GFP-LC3 was cloned into pLVX-IRES-hygro (Clontech, 632185) to generate a stably expressing HeLa cell line. All constructs used were verified by DNA sequencing (Tsingke Biotechnology). The RFP-GFP-LC3II adenovirus were purchased from HANBIO Biotechnology (AP21122705).
Lentiviruses were generated by transfection of HEK293T cells in 60 mm plate with pLKO.1-neo-shMEF2A, pLKO.1-puro-shMEF2D or pLVX-IRES-ZsGreen-FNIP1, pLVX-IRES-ZsGreen-FNIP2, PLVX-IRES-Hygro-GFP-LC3 constructs and control (indicated lentiviral backbone) in combination with the psPAX2 and pMD2.G packaging plasmids using Lipofectamine 2000 transfection reagent. After 12 h, medium was switching with fresh DMEM supplemented with 10% FBS to remove the transfection reagent. After an additional 24 h, supernatants medium with viral particles were harvested and a 0.45 μm filter was used to eliminate cell debris. For stable cell line generation, target cells at about 70% confluent were added viral particles supplemented with 8 mg/ml polybrene (Sigma Aldrich, TR-1003) and cultured at 37°C, 5% CO2 for another 24 h. Cells were selected by puromycin, G418 or hygromycin after transfected for 72 h.
Cell lysis and western blotting
Cells were harvested and lysed in pre-cold RIPA lysis buffer (Millipore, 20–188) supplemented with protease and phosphatase inhibitors on ice for 20 min. Total lysates were gently sonicated and then centrifuged in a microcentrifuge at 16,000 g for 20 min at 4°C. The supernatant was transmitted to a new microcentrifuge tube as total protein sample. The protein concentration of the sample was measured by Bradford assay kit (Thermo Fisher Scientific, 23225). Samples were added loading buffer and boiled at 95°C for 5 min before loaded on 8%~15% SDS-PAGE gel, then transferred to the 0.22 μm PVDF membrane (Roche, 03010040001) and incubated membrane with the indicated antibodies overnight at 4°C. Secondary HRP-coupled antibodies were incubated for 2 h at room temperature (RT). The blots were then visualized with the chemiluminescent detection system (Bio-Rad). Immunoblotting was quantified by calculating the intensity of bans by densitometry analysis using the Fiji software.
Immunoprecipitation and transfection
For immunoprecipitation experiment, cells were lysed by ice-cold TX-100 lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM DTT, 1% Triton X-100 [MP bio, 9002-93-1]) supplemented with protease and phosphatase inhibitors. Gently sonication was used to completely lyse cells and shear DNA. For co-immunoprecipitation, cells were prepared in pre-cold Pierce™ IP buffer (Thermo Fisher Scientific, 87787) supplemented with protease inhibitors. The cell lysate was gently rotated at 4°C for 4 h (HulaMixer, Invitrogen) and then centrifuged at 16,000 g for 15 min at 4°C. The indicated antibodies were incubated with the supernatants of cell lysates and gently rotated overnight at 4°C, subsequently mixed with pre-washed protein A/G-plus agarose beads (Santa Cruz Biotechnology, sc-2003) at 4°C for 4 h. Immunoprecipitates were isolation by centrifuging samples at 1,000 g for 3 min and washed 5 times with the lysis buffer before being separated by SDS-PAGE and immunoblotted with indicated antibodies.
For transient transfection experiments, HEK293T and HeLa cells were seeded in culture dishes and incubated for approximately 24 h. Cells were then transfected with indicated pcDNA3.1-based expression vectors by Lipofectamine 2000 transfection reagent for another 24 h.
RNA extraction and real-time quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, 15596026) according to the manufacturer’s protocol. RNA input (1 μg) was reverse transcribed by PrimeScript™ RT reagent Kit (Takara, RR047A) to synthesize cDNA. Quantitative PCR analysis on cDNAs was carried out using the iQ5™ PCR system (Bio-Rad, CA, USA) with TB Green® Premix Ex Taq™ II (Takara, RR820A). Fold change values were calculated with the ΔΔCt method. The following primers were used in qPCR: RRAGA F: 5’-CATGAGGTCGATAATCTTCGCC-3’ and R: 5’-GAGTGTTCCACGTCAATGGTG-3’; RRAGB F: 5’-GGTGGGCAAGACACCTTCAT-3’ and R: 5’-GCGGCTCTCCACATCAAAGA-3’; RRAGC F: 5’-AGGGCCAATGATGACCTTGC-3’ and R: 5’-GTGGAATGAGTTTCTGCACCA-3’; RRAGD F: 5’- CTAGCGGACTACGGAGACG-3’ and R: 5’-ATGAGCAGGATTCTCGGCTTC-3’; NR4A1/Nur77 F: 5’-TCTGCTCAGGCCTGGTGCTAC-3’ and R: 5’-GGCACCAAGTCCTCCAGCTTG-3’; ZMAT4 F: 5’- TACGAGAGTCGAAAACATGCAA-3’ and R: 5’-CCACCGCTGAAGTGAATGACA-3’; DAAM1 F: 5’-AGTATGCCAGCGAAAGGACC-3’ and R: 5’-TTCATCTCGATACCGCCCAGT-3’; FNIP1 F: 5’-GGTTCTCGGTGCTCTTCTGAT-3’ and R: 5’-GCTGTGGAGGGGAACGAAT-3’; FNIP2 F: 5’-TCGTACTGGAAGTAACCTAGCA-3’ and R: 5’-AGGCTGATCGAGCAATGCC-3’; FLCN F: 5’-TCTTCAGCATTGTCCGCCAG-3’ and R: 5’-AGTTGATGAGGTAGATCCGGTC-3’; GAPDH F: 5’-TGCTGGTGCTGAGTATGTCG-3’ and R: 5’-CATGTCAGATCCACAACGG-3’.
Luciferase reporter assay
MEF2 Luciferase Reporter experiment was performed as described previously [69]. Briefly, cells were co-transfected with indicated constructs with MEF2 luciferase reporter plasmid (WT, reporter with wild-type MEF2 DNA binding sites; mt, reporter with the MEF2 DNA binding sites mutated) using Lipofectamine 2000. The β-galactosidase expression plasmid (Promega, E1081) was used to determine the efficiency in each transfection. The total amount of DNA for each transfection was kept constant using empty vectors. Cell lysates were analyzed for luciferase and normalized for transfection efficiency by β-galactosidase activity according to the manufacturer’s instructions (Promega, E6110 and E4720). For FNIP1 and FNIP2 Luciferase Reporter assay, the predicted MEF2 DNA binding sites on FNIP1 and FNIP2 promoter regions were amplified by PCR from the HeLa cell genome and cloned into KpnI and XhoI sites of pGL3-basic luciferase reporter vector (Promega, E1751). HEK293T cells were transfected with indicated FNIP1 or FNIP2 reporter plasmids with an increasing amount of MEF2-VP16 and subsequently processed as described above. The primers used were listed as following: FNIP1-900 F: 5”-TACGCGTGCTAGCCCGGGAGACTGTGAGGTCACATGTGAG-3‘ and R: 5’-CAGTACCGGAATGCCTCAGGAAGCCTAAAGACATATATG-3‘; FNIP1-2700 F: 5’-TACGCGTGCTAGCCCGGGTGACTGATAGGATCTACACTTCTG-3‘ and R: 5’-CAGTACCGGAATGCCTCCTCTGGCATCAAATGCTGAA-3‘; FNIP2-1200 F: 5’-TACGCGTGCTAGCCCGGGCACAGCTAGTTCCCAACCAAG-3‘ and R: 5’- CAGTACCGGAATGCCAGGAGGACTCTTGGAGACTCA-3‘; FNIP2-3400 F: 5’-TACGCGTGCTAGCCCGGGTGGATCACTTGAGGTCAGGAGTTCAAGACC-3‘ and R: 5’-CAGTACCGGAATGCCAGAGTAGCTGGGACTACAGATACCCACC-3”.
Lysosome immunoprecipitation (Lyso-IP)
Lysosome immunoprecipitation assay was conducted as a previously described protocol [70]. Briefly, Control and knockdown of MEF2A and MEF2D HeLa cells in a 15 cm dish were transfected for 48 h with Flag-tagged TMEM192 then performed amino acid starvation for 60 min and restimulation for 20 min. Cells were quickly rinsed twice and scraped in ice-cold KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25 was adjusted with KOH). Cells were collected under centrifuged at 1,000 g for 2 min at 4°C and then resuspended in 950 μl fractionation buffer (50 mM KCl, 90 mM K-gluconate [Sigma-Aldrich, 1550001], 1 mM EGTA, 5 mM MgCl2, 50 mM sucrose [Sigma-Aldrich, 573113], 5 mM glucose, 20 mM HEPES, pH 7.4, 2.5 mM ATP [Sigma-Aldrich, A1852], as previously described [27]) supplement with protease inhibitor. 50 μl content was reserved for further use as total cell lysis. Cells were physically broken using a 2 ml homogenizer (Thermo Fisher Scientific, K8853000002) and then centrifuged at 1,000 g for 2 min at 4°C. The supernatant containing the cellular organelles was mixed with 150 μL anti-Flag magnetic beads (Sigma-Aldrich, M8823) on a gentle rotator for 10 min at 4°C to enrich lysosomes. Immunoprecipitates with 1× SDS loading buffer were denatured for 5 min at 95°C and then analyzed by immunoblotting.
Protein purification and in vitro GST affinity-isolation assay
Full-length cDNA of SRC was cloned into pET-41b(+) expression vector (Novagen, 70557). Protein expression was performed in the engineered BL21 E. coli bacteria. The transformed E. coli cells were cultured in 100 ml LB medium at 37°C in an orbital shaker for 12 h and then incubated for 6 h with 1 mM isopropyl-D-thiogalactopyranoside (IPTG; Sigma Aldrich, I6758) to induce the expression of GST-tagged SRC and GST (as control). Glutathione sepharose beads (MCLAB, GAB-200) were used to purify GST-tagged SRC. Recombinant human Flag-tagged MEF2D was immunopurified from HEK293T cells. Cells were transfected with indicated expression plasmid for 48 h and then lysed and immunoprecipitated as before. Protein-beads complex was washed twice by high salt lysis buffer (20 mM Tris-HCl, pH 7.4, 0.5 M NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100) to remove the nonspecific binding proteins. Competitive elution with free Flag peptide (Sigma Aldrich, F3290) was used to purify the Flag tag fusion protein from immunoprecipitates.
In the GST affinity-isolation assay, the glutathione beads-bound GST or GST-SRC proteins were incubated with purified Flag-tagged MEF2D protein in GST affinity-isolation buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 0.5% Nonidet P-40 [Thermo Fisher Scientific, 85124], 1 mM PMSF [Beyotime, ST506]) at 4°C for 1.5 h. Precipitates were washed 3 times with affinity-isolation buffer, then loading sample on SDS-PAGE gel and blotting with anti-MEF2D and anti-GST antibodies to detect Flag-tagged MEF2D, GST or GST-SRC.
In vitro kinase assay
In vitro kinase assay was performed based on a previously described protocol [54,71]. In brief, HEK293T purified Flag-tagged MEF2D-WT or MEF2D-3YF proteins were pre-treated with lambda protein phosphatase (Lambda PP; NEB, P0753S) according to the manufacturer’s protocol to release phosphate groups from phosphorylated serine, threonine and tyrosine residues in proteins. Active GST-tagged SRC kinase (Abcam, ab60884) was incubated with fully dephosphorylated Flag-tagged MEF2D-WT or MEF2D-3YF in a kinase buffer containing 25 mM HEPES (pH 7.4), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.5 mg/ml BSA (Sangon Biotech, A600903), 1 mM Na3VO4, 30 μM cold ATP for 30 min at 30°C. Reactions were stopped by addition of SDS loading buffer, followed by SDS-PAGE, transferring to PVDF membrane and immunoblotting with phospho-tyrosine antibody.
CUT&Tag and chromatin immunoprecipitations (ChIP)
CUT&Tag assay was performed according to the previously described protocol [49] and the manufacturer’s instructions (Novoprotein, N259-YH01). Briefly, 50000 HeLa cells were harvested and incubated with concanavalin A-coated magnetic beads (Novoprotein, N251-01A) for 15 min. The beads binding cells were permeabilized with 0.05% digitonin (Novoprotein, N253-YH01) and then incubated with primary MEF2D antibody (BD biosciences, 610774) or mouse IgG1 (CST, 5415) as a control on a rotating platform overnight at 4°C. An appropriate secondary antibody (Abcam, ab6708) was incubated with a primary antibody-cell-bead complex to increase the number of protein A binding sites. After removing unbound antibodies by twice washing, the complex was incubated with pA-Tn5 (Novoprotein, M059-YH01) and subjected to a transposition reaction. Next, cells were resuspended in fragmentation buffer and incubated at 37°C for 1 h. Then the DNA fragments were extracted, purified and subjected to library construction using the reagents provided by the kit. All the generated libraries were sequenced by Illumina sequencer (DIATRE Biotech Co., Ltd).
ChIP assay was performed from 2 × 107 fixed HeLa cells using a ChIP Assay Kit (Millipore, 17–295) according to the manufacturer’s protocol. Purified sheared DNA was subjected to PCR using Phanta Max Master Mix (Vazyme, P515–01) for fragment amplification. The primers used in ChIP-PCR were listed as following: FNIP1-900 F: 5”-AGACTGTGAGGTCACATGTG-3‘ and R: 5’- ACCAACGAAGTCAGGAAGCCT-3‘; FNIP1-2700 F: 5’-TGCCAGTCATGGACAGCCA-3‘ and R: 5’-AGTGGAAACAGGCCAAAGTC-3‘; FNIP2-1200 F: 5’-CACAGCTAGTTCCCAACCAAG-3‘ and R: 5’-AGGAGGACTCTTGGAGACTCA-3‘; FNIP2-3400 F: 5’-TGGATCACTTGAGGTCAGGAG-3‘ and R: 5’-GCCTAACCATCCAGAGTAGCT-3‘; RRAGD-500 F: 5’-TGGTGGGAAGCACTTTGAAC-3‘ and R: 5’-TCCTGCTTACAGTCCTGAGCT-3”. PCR products were separated by agarose gel electrophoresis and visualized with ethidium bromide under ultraviolet.
Immunofluorescence assays
For the MTOR lysosomal localization experiment, HeLa cells were cultured on coverglass bottom dishes (NEST, 801001) for 24 h. Cell plates were washed twice with PBS, starved in amino acid-free DMEM supplemented with insulin for 60 min, and then left untreated or stimulated with amino acids for 20 min. After that, cells were aspirated medium, washed with PBS twice, and fixed for 10 min with 4% paraformaldehyde in PBS at RT. Cell dishes were then rinsed 3 times with PBS and permeabilized with 0.1% Triton X-100 for 20 min at RT. Cells were subsequently incubated with blocking buffer (0.01% Triton X-100 plus 10% donkey serum [Jackson ImmunoResearch, 017-000-121] and 2% BSA in PBS) for 1 h at RT and then incubated with primary antibodies (LAMP2 and MTOR) in 5% donkey serum at 4°C overnight. Cells were then rinsed five times with PBS and incubated with secondary antibodies in 5% donkey serum for 2 h at RT. After washing, cells were incubated with DAPI (Roche, 28718-90-3) for 10 min. Images were detected on Nikon A1 confocal microscopes using a 60×oil immersion objective. Colocalization analysis was performed using Fiji 1.0 software.
Immunohistochemistry assay
Human pancreatic cancer and paired adjacent samples were post-fixed with 4% paraformaldehyde for 24 h at 4°C. They were dehydrated in a graded series of ethanol, cleared with xylene (Lecia, TP1020), and infiltrated with paraffin (Leica, EG1150H). Paraffin-embedded blocks were cut on a microtome in 6 μm sections (Leica, HistoCore BIOCUT). Immunohistochemistry was performed following the manufacturer’s instructions (ZSGB-BIO, SAP-9100). Images were taken using an automated high throughput slide scanner (Pannoramic MIDI, 3DHIESTECH).
Oil red O staining
Oil Red O staining was performed according to the manufacturer’s instructions (ZHONGHUIHECAI, CD011). For statistical analysis of Oil Red O content levels, isopropanol was added to each sample shaken for 5 min at RT, and each sample was assessed spectrophotometrically at 510 nm (Molecular Devices, SpectraMax M5).
Determination of cell size
MEF2A and MEF2D double-knockdown and control HeLa cells were seeded into 60 mm culture dishes for 24 h under. Cells were washed once with PBS and harvested by trypsinization, then cells were resuspended with PBS and were subjected to cell diameter determination at the confluence of 2 ~ 4 × 106 cells/ml by the easy cell counter (JIMBIO FIL PLUS).
Cell proliferation and colony formation assays
For cell proliferation experiment, cell counting kit-8 (CCK-8) (MedChemExpress, HY-K0301) was used according to the manufacturer’s instructions. For colony formation assay, AsPC-1 and PANC-1 cells were seeded at appropriate concentrations in the 6-well plate with 2 ml complete medium. Cells were cultured for around 2 weeks for colony formation. Colonies were fixed with 4% paraformaldehyde and stained with crystal violet staining solution (Sangon Biotech, E607309).
Tumor xenograft studies
MEF2A and MEF2D double-knockdown and control AsPC-1 cells were harvested by trypsinization then washed twice with serum free culture medium and re-suspended to 1 × 106 cells per 50 μl in serum free DMEM. The cells were then mixed with an equal volume of Matrigel (Corning, 356230). One hundred microliter (μl) of the cell suspension were injected into the 8-week-old BALB/c nude mice (BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. and kept in a specific-pathogen-free facility). Xenografts were measured with a caliper every 3 days (tumor volume = width2 × length × 1/2) after one week of injection. Mice were euthanized when tumors reached about 15 mm and xenograft tumors were dissected for analysis. The protocol of the experiments using mice was carried out according to the Guidelines for Animal Care and Use of the Fourth Military Medical University (FMMU), Xi’an, China.
Clinical samples
Human pancreatic cancer samples were all from the Second Affiliated Hospital of FMMU. All the procedures were approved by the Institutional Review Board of the Fourth Military Medical University and conducted by the Declaration of Helsinki. Fresh pancreatic cancer samples (12 tumor samples and 12 paired adjacent normal tissues) were obtained from patients, quickly processed and stored at −80°C for further use. The clinical stages of pancreatic cancer were classified according to the 8th Edition of the AJCC Cancer Staging Manual.
Statistical analysis
The statistical analysis was performed using GraphPad version 8 software for Windows. Unpaired two-sided Student’s t test was used to compare two groups, and one-way or two-way ANOVA with Dunnett’s or Turkey’s post-hoc test was carried out for multiple comparisons. Experimental data are shown as the average ± SEM. Log-rank test was used to determine the statistical differences of the survival data. P < 0.05 was considered significant and details of reproducibility and statistics are indicated in the corresponding figure legends.
Supplementary Material
Acknowledgements
The authors gratefully thank all patients who participated in this work. We also sincerely thank Dr. Z. Mao (Departments of Pharmacology and Neurology, School of Medicine, Emory University) for providing Flag-tagged MEF2A, Flag-tagged MEF2D, Flag-tagged dnMEF2, MEF2-VP16, GFP-LC3 and MEF2 reporter constructs.
Funding Statement
Project of International Cooperation and Exchange [No. 81720108016] (QY); National Natural Science Foundation of China [No. 31930048] (QY); National Natural Science Foundation of China [No. 82221001] (QY); National Natural Science Foundation of China [No. 82201588] (TJN).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2259735
References
- [1].Blenis J. TOR, the gateway to cellular metabolism, cell growth, and disease. Cell. 2017 Sep 21;171(1):10–13. doi: 10.1016/j.cell.2017.08.019 [DOI] [PubMed] [Google Scholar]
- [2].Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007. Jul;12(1):9–22. doi: 10.1016/j.ccr.2007.05.008 [DOI] [PubMed] [Google Scholar]
- [3].Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020. Apr;21(4):183–203. doi: 10.1038/s41580-019-0199-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017 Mar 9;168(6):960–976. doi: 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011. Jan;12(1):21–35. doi: 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013. Jun;15(6):555–564. doi: 10.1038/ncb2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018. Dec;18(12):744–757. doi: 10.1038/s41568-018-0074-8 [DOI] [PubMed] [Google Scholar]
- [8].Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006. Sep;6(9):729–734. doi: 10.1038/nrc1974 [DOI] [PubMed] [Google Scholar]
- [9].Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014. Jul;24(7):400–406. doi: 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Durán RV, Hall MN. Regulation of TOR by small GTPases. EMBO Rep. 2012 Feb 1;13(2):121–128. doi: 10.1038/embor.2011.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Demetriades C, Plescher M, Teleman AA. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat Commun. 2016 Feb 12;7(1):10662. doi: 10.1038/ncomms10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003 Aug 1;17(15):1829–1834. doi: 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang H, Jiang X, Li B, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature. 2017 Dec 21;552(7685):368–373. doi: 10.1038/nature25023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008 Jun 13;320(5882):1496–1501. doi: 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by rag GTPases in nutrient response. Nat Cell Biol. 2008. Aug;10(8):935–945. doi: 10.1038/ncb1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002 Jul 26;110(2):177–189. doi: 10.1016/S0092-8674(02)00833-4 [DOI] [PubMed] [Google Scholar]
- [17].Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002 Jul 26;110(2):163–175. doi: 10.1016/S0092-8674(02)00808-5 [DOI] [PubMed] [Google Scholar]
- [18].Rogala KB, Gu X, Kedir JF, et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science. 2019 Oct 25;366(6464):468–475. doi: 10.1126/science.aay0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bos JL, Rehmann H, Wittinghofer A. Gefs and GAPs: critical elements in the control of small G proteins. Cell. 2007 Jun 1;129(5):865–877. doi: 10.1016/j.cell.2007.05.018 [DOI] [PubMed] [Google Scholar]
- [20].Hesketh GG, Papazotos F, Pawling J, et al. The GATOR–rag GTPase pathway inhibits mTORC1 activation by lysosome-derived amino acids. Science. 2020 Oct 16;370(6514):351–356. doi: 10.1126/science.aaz0863 [DOI] [PubMed] [Google Scholar]
- [21].Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid–dependent activation of rag GTPases. J Cell Bio. 2013 Sep 30;202(7):1107–1122. doi: 10.1083/jcb.201307084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shen K, Rogala KB, Chou HT, et al. Cryo-EM structure of the human FLCN-FNIP2-Rag-Ragulator complex. Cell. 2019 Nov 27;179(6):1319–1329.e8. doi: 10.1016/j.cell.2019.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Takagi Y, Kobayashi T, Shiono M, et al. Interaction of folliculin (birt-hogg-dubé gene product) with a novel Fnip1-like (FnipL/fnip2) protein. Oncogene. 2008 Sep 11;27(40):5339–5347. doi: 10.1038/onc.2008.261 [DOI] [PubMed] [Google Scholar]
- [24].Yang G, Humphrey SJ, Murashige DS, et al. RagC phosphorylation autoregulates mTOR complex 1. EMBO J. 2019 Feb 1;38(3). doi: 10.15252/embj.201899548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends in cell biology. Trends Cell Biol. 2015. Sep;25(9):545–555. doi: 10.1016/j.tcb.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cipponi A, Goode DL, Bedo J, et al. MTOR signaling orchestrates stress-induced mutagenesis, facilitating adaptive evolution in cancer. Science. 2020 Jun 5;368(6495):1127–1131. doi: 10.1126/science.aau8768 [DOI] [PubMed] [Google Scholar]
- [27].Chen J, Ou Y, Yang Y, et al. KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature. 2018. May;557(7706):585–589. doi: 10.1038/s41586-018-0128-9 [DOI] [PubMed] [Google Scholar]
- [28].Han J, Jiang Y, Li Z, et al. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997 Mar 20;386(6622):296–299. doi: 10.1038/386296a0 [DOI] [PubMed] [Google Scholar]
- [29].Pallavi SK, Ho DM, Hicks C, et al. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in drosophila. EMBO J. 2012 Jun 29;31(13):2895–2907. doi: 10.1038/emboj.2012.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development (Cambridge, England). Development. 2007. Dec;134(23):4131–4140. doi: 10.1242/dev.008367 [DOI] [PubMed] [Google Scholar]
- [31].Okamoto S, Li Z, Ju C, et al. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci, USA. 2002 Mar 19;99(6):3974–3979. doi: 10.1073/pnas.022036399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ornatsky OI, Andreucci JJ, McDermott JC. A dominant-negative form of transcription factor MEF2 inhibits myogenesis. J Biol Chem. 1997 Dec 26;272(52):33271–33278. doi: 10.1074/jbc.272.52.33271 [DOI] [PubMed] [Google Scholar]
- [33].Andrés V, Cervera M, Mahdavi V. Determination of the consensus binding site for MEF2 expressed in muscle and brain reveals tissue-specific sequence constraints. J Biol Chem. 1995 Oct 6;270(40):23246–23249. doi: 10.1074/jbc.270.40.23246 [DOI] [PubMed] [Google Scholar]
- [34].Cole CJ, Mercaldo V, Restivo L, et al. MEF2 negatively regulates learning-induced structural plasticity and memory formation. Nat Neurosci. 2012. Sep;15(9):1255–1264. doi: 10.1038/nn.3189 [DOI] [PubMed] [Google Scholar]
- [35].Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008 Feb 28;451(7182):1125–1129. doi: 10.1038/nature06607 [DOI] [PubMed] [Google Scholar]
- [36].Mao Z, Bonni A, Xia F, et al. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999 Oct 22;286(5440):785–790. doi: 10.1126/science.286.5440.785 [DOI] [PubMed] [Google Scholar]
- [37].Yu W, Huang C, Wang Q, et al. MEF2 transcription factors promotes EMT and invasiveness of hepatocellular carcinoma through TGF-β1 autoregulation circuitry. Tumor Biol. 2014. Nov;35(11):10943–10951. doi: 10.1007/s13277-014-2403-1 [DOI] [PubMed] [Google Scholar]
- [38].McDonald C, Karstegl CE, Kellam P, et al. Regulation of the Epstein-Barr virus zp promoter in B lymphocytes during reactivation from latency. J Gen Virol. 2010. Mar;91(3):622–629. doi: 10.1099/vir.0.017277-0 [DOI] [PubMed] [Google Scholar]
- [39].Nagel S, Meyer C, Quentmeier H, et al. MEF2C is activated by multiple mechanisms in a subset of T-acute lymphoblastic leukemia cell lines. Leukemia. 2008. Mar;22(3):600–607. doi: 10.1038/sj.leu.2405067 [DOI] [PubMed] [Google Scholar]
- [40].Clark RI, Tan SW, Péan CB, et al. MEF2 is an in vivo immune-metabolic switch. Cell. 2013 Oct 10;155(2):435–447. doi: 10.1016/j.cell.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol. 1995. Jun;15(6):2907–2915. doi: 10.1128/MCB.15.6.2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hara K, Yonezawa K, Weng QP, et al. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998 Jun 5;273(23):14484–14494. doi: 10.1074/jbc.273.23.14484 [DOI] [PubMed] [Google Scholar]
- [43].Liu B, Wang L, Jiang W, et al. Myocyte enhancer factor 2A delays vascular endothelial cell senescence by activating the PI3K/p-Akt/SIRT1 pathway. Aging. 2019 Jun 10;11(11):3768–3784. doi: 10.18632/aging.102015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Battaglioni S, Benjamin D, Wälchli M, et al. mTOR substrate phosphorylation in growth control. Cell. 2022 May 26;185(11):1814–1836. doi: 10.1016/j.cell.2022.04.013 [DOI] [PubMed] [Google Scholar]
- [45].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. Feb;13(2):132–141. doi: 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011 Jun 10;332(6035):1322–1326. doi: 10.1126/science.1199484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schmidt EK, Clavarino G, Ceppi M, et al. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009. Apr;6(4):275–277. doi: 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- [48].Fingar DC, Salama S, Tsou C, et al. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2022 Jun 15;16(12):1472–1487. doi: 10.1101/gad.995802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaya-Okur HS, Wu SJ, Codomo CA, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019 Apr 29;10(1):1930. doi: 10.1038/s41467-019-09982-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bar-Peled L, Schweitzer LD, Zoncu R, et al. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012 Sep 14;150(6):1196–1208. doi: 10.1016/j.cell.2012.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Molecular Cell. 2013 Nov 21;52(4):495–505. doi: 10.1016/j.molcel.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annual review of cell and developmental biology. Annu Rev Cell Dev Biol. 1997;13(1):513–609. doi: 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- [53].Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009. Oct;6(10):587–595. doi: 10.1038/nrclinonc.2009.129 [DOI] [PubMed] [Google Scholar]
- [54].Song L, Liu Z, Hu HH, et al. Proto-oncogene src links lipogenesis via lipin-1 to breast cancer malignancy. Nat Commun. 2020 Nov 17;11(1):5842. doi: 10.1038/s41467-020-19694-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gong X, Tang X, Wiedmann M, et al. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003 Apr 10;38(1):33–46. doi: 10.1016/S0896-6273(03)00191-0 [DOI] [PubMed] [Google Scholar]
- [56].Ornatsky OI, Cox DM, Tangirala P, et al. Post-translational control of the MEF2A transcriptional regulatory protein. Nucleic Acids Res. 1999 Jul 1;27(13):2646–2654. doi: 10.1093/nar/27.13.2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shalizi A, Gaudilliére B, Yuan Z, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006 Feb 17;311(5763):1012–1017. doi: 10.1126/science.1122513 [DOI] [PubMed] [Google Scholar]
- [58].Yang Q, She H, Gearing M, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009 Jan 2;323(5910):124–127. doi: 10.1126/science.1166088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011 Aug 5;146(3):408–420. doi: 10.1016/j.cell.2011.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017 Jun 16;356(6343):1188–1192. doi: 10.1126/science.aag2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhao M, New L, Kravchenko VV, et al. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999. Jan;19(1):21–30. doi: 10.1128/MCB.19.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ornatsky OI, McDermott JC. MEF2 protein expression, DNA binding specificity and complex composition, and transcriptional activity in muscle and non-muscle cells. J Biol Chem. 1996 Oct 4;271(40):24927–24933. doi: 10.1074/jbc.271.40.24927 [DOI] [PubMed] [Google Scholar]
- [63].Suzuki E, Satonaka H, Nishimatsu H, et al. Myocyte enhancer factor 2 mediates vascular inflammation via the p38-dependent pathway. Circ Res. 2004 Jul 9;95(1):42–49. doi: 10.1161/01.RES.0000134631.75684.4A [DOI] [PubMed] [Google Scholar]
- [64].Yin Y, She H, Li W, et al. Modulation of neuronal survival factor MEF2 by kinases in Parkinson’s disease. Front Physiol. 2012;3:171. doi: 10.3389/fphys.2012.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ralston R, Bishop JM. The product of the protooncogene c-src is modified during the cellular response to platelet-derived growth factor. Proc Natl Acad Sci, USA. 1985. Dec;82(23):7845–7849. doi: 10.1073/pnas.82.23.7845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011 Dec 1;480(7375):118–122. doi: 10.1038/nature10598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pal R, Palmieri M, Chaudhury A, et al. Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-rag GTPase interaction. Nat Commun. 2018 Oct 19;9(1):4351. doi: 10.1038/s41467-018-06844-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pon JR, Marra MA. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget. 2016 Jan 19;7(3):2297–2312. doi: 10.18632/oncotarget.6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang X, Tang X, Li M, et al. Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase a signaling pathway in neuronal survival. J Biol Chem. 2005 Apr 29;280(17):16705–16713. doi: 10.1074/jbc.M501819200 [DOI] [PubMed] [Google Scholar]
- [70].Abu-Remaileh M, Wyant GA, Kim C, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017 Nov 10;358(6364):807–813. doi: 10.1126/science.aan6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of bcr-abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant abl kinase domain mutants. Cancer Res. 2005 Jun 1;65(11):4500–4505. doi: 10.1158/0008-5472.CAN-05-0259 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.