

ABSTRACT
Although microglial activation is induced by an increase in chemokines, the role of mitophagy in this process remains unclear. This study aimed to elucidate the role of microglial mitophagy in CKLF/CKLF1 (chemokine-like factor 1)-induced microglial activation and neuroinflammation, as well as the underlying molecular mechanisms following CKLF treatment. This study determined that CKLF, an inducible chemokine in the brain, leads to an increase in mitophagy markers, such as DNM1L, PINK1 (PTEN induced putative kinase 1), PRKN, and OPTN, along with a simultaneous increase in autophagosome formation, as evidenced by elevated levels of BECN1 and MAP1LC3B (microtubule-associated protein 1 light chain 3 beta)-II. However, SQSTM1, a substrate of autophagy, was also accumulated by CKLF treatment, suggesting that mitophagy flux was reduced and mitophagosomes accumulated. These findings were confirmed by transmission electron microscopy and confocal microscopy. The defective mitophagy observed in our study was caused by impaired lysosomal function, including mitophagosome-lysosome fusion, lysosome generation, and acidification, resulting in the accumulation of damaged mitochondria in microglial cells. Further analysis revealed that pharmacological blocking or gene-silencing of mitophagy inhibited CKLF-mediated microglial activation, as evidenced by the expression of the microglial marker AIF1 (allograft inflammatory factor 1) and the mRNA of proinflammatory cytokines (Tnf and Il6). Ultimately, defective mitophagy induced by CKLF results in microglial activation, as observed in the brains of adult mice. In summary, CKLF induces defective mitophagy, microglial activation, and inflammation, providing a potential approach for treating neuroinflammatory diseases.
Abbreviation: 3-MA: 3-methyladenine; AIF1: allograft inflammatory factor 1; ANOVA: analysis of variance; BAF: bafilomycin A1; BSA: bovine serum albumin; CCCP: carbonyl cyanide m-chlorophenyl hydrazone; cGAMP: cyclic GMP-AMP; CGAS: cyclic GMP-AMP synthase; CKLF/CKLF1: chemokine-like factor 1; CNS: central nervous system; DMEM: Dulbecco’s Modified Eagle Medium; DNM1L: dynamin 1 like; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescence protein; IRF3: interferon regulatory factor 3; IgG: immunoglobulin G; LAMP1: lysosomal-associated membrane protein 1; LAPTM4A: lysosomal-associated protein transmembrane 4A; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; Mdivi-1: mitochondrial division inhibitor 1; mRFP: monomeric red fluorescent protein; mtDNA: mitochondrial DNA; MTORC1: mechanistic target of rapamycin kinase complex 1; OPTN: optineurin; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; PINK1: PTEN induced putative kinase 1; PLL: poly-L-lysine; PRKN: parkin RBR E3 ubiquitin protein ligase; qPCR: quantitative polymerase chain reaction; ROS: reactive oxygen species; SQSTM1: sequestosome 1; TBK1: TANK-binding kinase 1; TFEB: transcription factor EB; VDAC: voltage-dependent anion channel
KEYWORDS: Chemokine-like factor 1, inflammation, lysosomal function, microglia, mitophagosome formation, neuroinflammatory diseases
Introduction
Microglia, the predominant resident innate immune cells in the central nervous system (CNS), play a critical role in CNS homeostasis and development from embryonic stages to adulthood [1,2]. In the homeostatic state, microglia exhibit highly ramified morphologies with small cell bodies that contribute to maintaining CNS homeostasis and development [2]. Microglia also contribute to the pathogenesis of CNS disorders [3]. Upon activation by immunogenic stimuli, microglia transform into an activated response phenotype and perform inflammatory functions, including the enhanced production of cytokines and reactive oxygen species (ROS), which are considered responders and inducers of neuroinflammation that play a role in the pathogenesis of various CNS diseases [2,4].
Mitochondrial ROS are crucial regulators of inflammatory responses in microglia [5]. Mitochondria are the primary source of ROS and maintain mitochondrial integrity and cellular homeostasis [6,7]. Under pathological conditions, ROS production increases, leading to oxidative stress and various pathological processes, such as microglial overactivation, resulting in cell death and disease [8,9]. Correspondingly, cells have evolved mechanisms to mitigate mitochondrial damage [10]. Mitophagy, a selective form of autophagy, plays a crucial role in mitochondrial quality control by removing damaged and redundant mitochondria [11]. Several studies have demonstrated that mitophagy can limit the inflammatory response in microglial cells, whereas defective mitophagy facilitates microglial overactivation and promotes neuroinflammation, underscoring the importance of mitophagy in microglial fate [12–14].
Mitophagy is a process whereby phagophores engulf and encapsulate damaged mitochondria, subsequently maturing into double-membraned vesicles known as mitophagosomes, which are subsequently transported to lysosomes for proteolytic degradation [15]. Two primary categories of mitophagy pathways have been identified: ubiquitin-dependent and ubiquitin-independent [16]. Of these, the ubiquitin-dependent pathway has undergone the most comprehensive investigation and demonstrated a crucial role in numerous CNS diseases, such as stroke, Alzheimer disease, and Parkinson disease [17–19]. In general, damaged mitochondria are segregated from healthy parts by DNM1L/Drp1 (dynamin 1 like), followed by a decrease in mitochondrial membrane potential [20,21]. Subsequently, the altered membrane potential prevents the translocation of PINK1 (PTEN induced putative kinase 1) from the outer mitochondrial membrane to the mitochondrial matrix, thereby phosphorylating and recruiting cytosolic PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase) [22]. The recruited and activated PRKN executes mitophagy by ubiquitinating mitochondrial membrane proteins, followed by the recruitment of autophagic receptor proteins that bridge the ubiquitinated cargo into mitophagosomes, such as OPTN (optineurin; a crucial receptor for selective autophagy, in particular, mitophagy) and SQSTM1/p62 (sequestosome 1), to bind MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) and the ER membrane, ultimately leading to mitophagosome formation [9,16,23–25]. Mitophagosomes containing damaged mitochondria are subsequently transported to the lysosomes for efficient clearance [15]. However, defective mitophagy leads to the accumulation of dysfunctional mitochondria, thereby exacerbating oxidative stress and inflammation [15]. Most extant studies have focused on investigating the impact of pathological stimuli, such as lipopolysaccharide, morphine, and cocaine, on microglial mitophagy [26–28]. Although several studies have examined the influence of cytokines on microglial mitophagy [29–31], the effects of chemokines on this process have rarely been explored.
CKLF/CKLF1 (chemokine-like factor 1) is a CC-type chemokine that plays a critical role in various diseases, including ischemic stroke, hypersensitivity, tumors, cardio-cerebrovascular diseases, and respiratory diseases, with evidence demonstrating its involvement in inflammation [32–37]. C27 is a peptide obtained from secreted CKLF and has been validated as an active peptide of CKLF [38–40]. Our previous studies primarily focused on the role of CKLF in CNS diseases, and we have shown that CKLF C27 peptide exacerbates neuronal damage after cerebral ischemia/reperfusion by inducing microglial overactivation, resulting in aberrant mRNA expression of proinflammatory cytokines, such as TNF/TNF-α and IL1B/IL-1β [35]. Conversely, CKLF knockdown or pharmacological inhibition with anti-CKLF immunoglobulin G (IgG) lowers proinflammatory cytokine levels, such as TNF, IL1B, CXCL2/MIP-2 and IL8 and significantly improves neurobehavioral deficits [41,42]. The conditional knockout of neuronal CKLF inhibits microglial overactivation induced by ischemic stroke [32]. These findings indicated the crucial role of CKLF in microglia-mediated inflammation under various pathological conditions. Recently, we found that CKLF C27 peptide exposure led to microglial metabolism reprogramming, as evidenced by an enhanced extracellular acid rate, reduced oxygen consumption, and increased mitochondrial fission [43], suggesting that mitochondrial dysfunction induced by CKLF exposure may be linked to mitophagy in microglial cells.
To verify this hypothesis, the present study aimed to elucidate the role of microglial mitophagy in CKLF-induced microglial activation and neuroinflammation, as well as the underlying molecular mechanisms following CKLF treatment. Our study identified, for the first time, a correlation between mitochondrial dysfunction, defective mitophagy, and microglial activation in response to CKLF exposure.
Results
C27-induced defective mitophagy and mitochondrial dysfunction in microglial cells
To investigate how CKLF affects mitophagy, BV2 microglial cells were exposed to various concentrations of the CKLF active peptide C27 (250, 500, and 1000 nM) for 24 h. The levels of the mitophagy markers DNM1L, PINK1, PRKN and OPTN were detected by western blotting, with BAF used as the positive control [44]. C27 upregulated the mitophagy markers DNM1L (Figure 1B), PINK1 (Figure 1C), PRKN (Figure 1D), and OPTN (Figure 1E) in a concentration-dependent manner, with the most significant increase observed at 1000 nM. Therefore, this concentration of C27 was selected for subsequent experiments. The levels of mitophagy markers were examined at different time points (0, 3, 6, 12, and 24 h) after exposure to C27 (1000 nM). No significant differences in mitophagy markers were observed in the BV2 microglial cells without C27 exposure (Figure 1F–J). However, after 3 h of C27 exposure, a significant increase in the mitophagy marker DNM1L was observed (Figure 1G), whereas PINK1 levels increased significantly after 6 h (Figure 1H). All these four markers increased within 24 h of detection (Figure 1F–J). In addition, the cytosolic and mitochondrial fractions of BV2 microglial cells were isolated, and the levels of mitophagy markers were detected in each fraction. The expression levels of the mitophagy markers DNM1L, PINK1, PRKN, and OPTN in both the cytosol (Figure 1L,N,P,R) and mitochondria (Figure 1M,O,Q,S) also significantly increased in a time-dependent manner.
Figure 1.
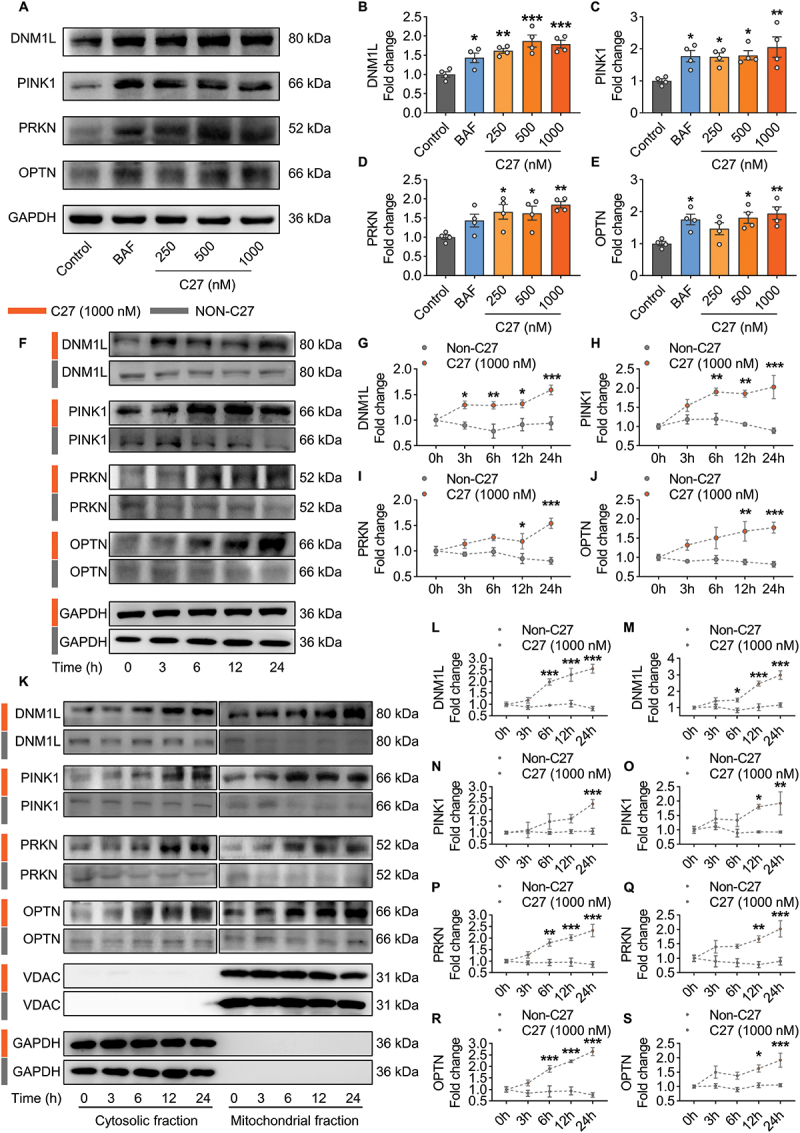
C27-induced mitophagy in microglial cells. (A–E) concentration-dependent upregulation of mitophagy markers in BV2 microglial cells exposed to C27. Representative immunoblot images (A) and the quantified results of DNM1L (B), PINK1 (C), PRKN (D), and OPTN (E) (n = 4). (F–J) time-dependent upregulation of mitophagy markers in BV2 microglial cells exposed to C27 (1000 nM). Representative immunoblot images (F) and the quantified results of DNM1L (G), PINK1 (H), PRKN (I), and OPTN (J) (n = 4). (K–S) C27 upregulated the levels of mitophagy markers in a time-dependent manner in the cytosolic fractions and mitochondrial fractions in BV2 microglial cells. Representative immunoblot images (K) and the quantified results of DNM1L (L, M), PINK1 (N, O), PRKN (P, Q) and OPTN (R, S) (n = 4). GAPDH (for total and cytosol) or VDAC (for mitochondria) was used as the protein loading control. Data are presented as the mean ± SEM. One-way ANOVA followed by Dunnett’s post hoc test (B–E) or two-way ANOVA followed by Sidak’s post hoc test (G–J, L–S) were used to determine statistical significance. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Mitochondrial membrane potential (Δψm) plays a crucial role in stabilizing PINK1, and the PINK1-PRKN mitophagy pathway is dependent on mitochondrial Δψm [21,45]. Therefore, whether C27 affects the mitochondrial membrane potential in microglial cells was examined. After exposing primary microglial cells to C27 for 3 h, mitochondrial Δψm was assessed using the JC-1 kit. In the control group, most mitochondria appeared healthy, with JC-1 forming J-aggregates in the mitochondrial matrix and emitting red fluorescence. However, microglial cells with low mitochondrial Δψm exhibited JC-1 as a monomer and green fluorescence [46]. As shown in Figure 2 (A, B), primary microglial cells in the control group displayed normal mitochondrial Δψm with red fluorescence, whereas the C27- and CCCP-positive control groups showed significantly increased green fluorescence. BV2 microglial cells also exhibited significantly increased green fluorescence after C27 treatment (Figure S1A, S1B), indicating that C27 significantly decreased mitochondrial Δψm in microglial cells.
Figure 2.
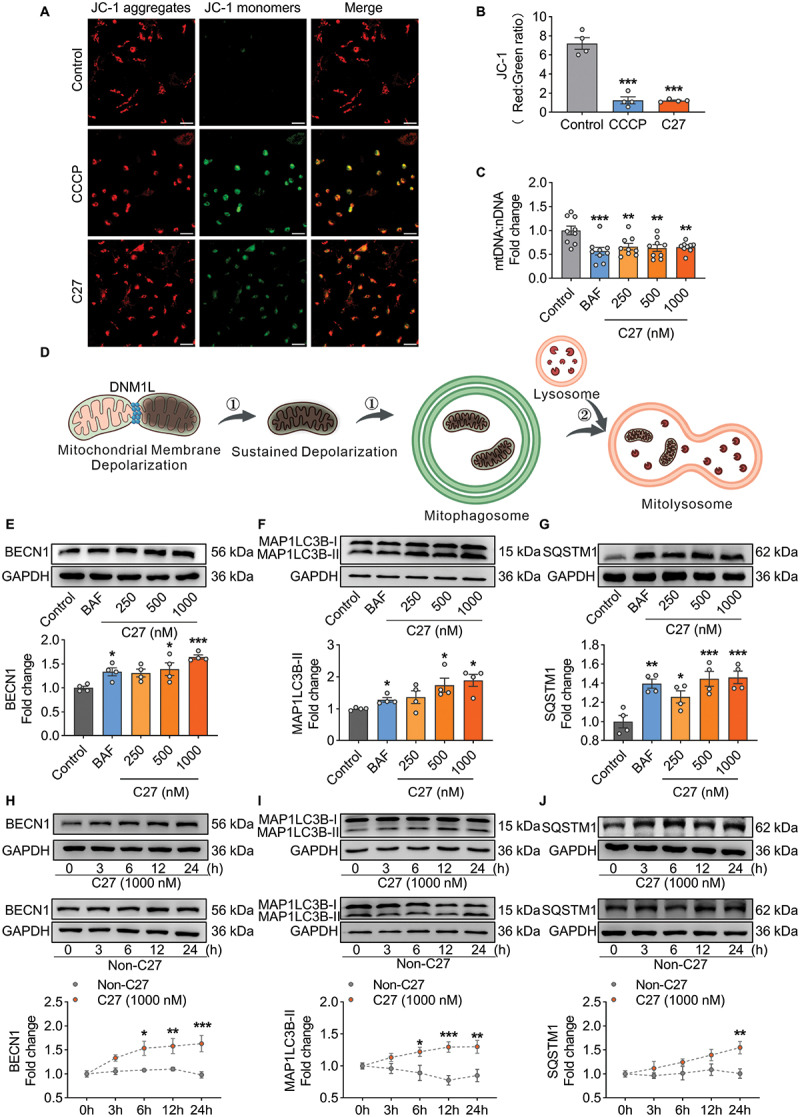
C27 induced mitochondrial dysfunction and increased autophagy marker proteins in microglial cells. (A–B) C27 decreased the mitochondrial membrane potential in mouse primary microglial cells. (A) exposure to C27 (1000 nM) and CCCP (20 μM) for 3 h resulted in a reduction in JC-1 dye aggregation in the mitochondria and decreased the ratio of the aggregate (red) to monomer JC-1 (green) in primary microglial cells. Scale bar: 100 µm. (B) quantification of JC-1 staining in primary microglial cells exposed to C27 (1000 nM) and CCCP (20 μM) for 3 h (n = 4). (C) the relative level of mitochondrial DNA (mtDNA) significantly decreased in BV2 microglial cells exposed to C27, as measured by mtDna/nDNA analysis (n = 9). (D) a schematic of mitophagy. (E–G) C27 upregulated the levels of autophagy markers, such as BECN1 (E), MAP1LC3B-II (F), and SQSTM1 (G), in BV2 microglial cells in a concentration-dependent manner (n = 4). One-way ANOVA followed by Dunnett’s post hoc test were employed in B–C, E, G. Non-parametric mann – Whitney U test were used to measure significance between two groups (F). (H–J) C27 upregulated autophagy markers, such as BECN1 (H), MAP1LC3B-II (I), and SQSTM1 (J), in a time-dependent manner in BV2 microglial cells. (n = 4). Data are presented as the mean ± SEM. Two-way ANOVA followed by Sidak’s post hoc test (H–J) were used to determine statistical significance. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Reduced mitochondrial DNA (mtDNA) content is a manifestation of mitochondrial dysfunction [47]. Considering that C27 decreased mitochondrial Δψm, the mtDNA copy number after C27 treatment was also measured. There was a significant decrease in mtDNA: nDNA ratios in BV2 microglial cells after 24 h of exposure to different concentrations of C27 (Figure 2C). Moreover, it is noteworthy that the CGAS-STING1 pathway can be triggered by cytosolic mtDNA [48]. Considering the observed reduction in mtDNA: nDNA ratios caused by C27, we propose that this decrease may be associated with the presence of the small fragments of mtDNA, resulting in its release into the cytosol. To explore this hypothesis, we quantified the extent of mtDNA release into the cytosol. As expected, treatment with C27 led to a notable increase in the levels of cytosolic mtDNA (Figure S2 [A – C]). Furthermore, the upregulation of CGAS and STING1 expression, along with the increased phosphorylation of TBK1 and IRF3, was observed in BV2 microglial cells (Figure S2 [D-H]), indicating the activation of the CGAS-STING1 signaling pathway in response to treatment with C27.
Given the observed impact of C27 exposure on mitochondrial Δψm and mtDNA in microglial cells, we aimed to investigate its effect on the mitochondrial oxygen consumption rate (OCR). OCR serves as an indicator of mitochondrial function related to aerobic glycolysis and oxidative phosphorylation. Notably, exposure to C27 resulted in significant alterations in OCR levels in BV2 microglial cells (Figure S1C, S1D), indicating the occurrence of mitochondrial dysfunction. Furthermore, our findings demonstrated a significant decrease in mitochondrial basal respiration, maximal respiration, proton leak, ATP-linked respiration, and spare capacity in BV2 microglial cells exposed to C27 when compared to the control group. This further supports the presence of mitochondrial dysfunction induced by C27 exposure in microglial cells.
Given that mitophagy involves both the formation of mitophagosomes and the subsequent degradation of mitophagosomes by lysosomes via the autophagic pathway (Figure 2D), we assessed the levels of the macroautophagy/autophagy markers BECN1/Beclin1 (an autophagy initiation marker), MAP1LC3B-II (an autophagy formation marker), and SQSTM1 (an autophagy degradation marker) in BV2 microglial cells exposed to varying concentrations of C27 for 24 h [9]. The expression levels of BECN1 (Figure 2E), MAP1LC3B-II (Figure 2F), and SQSTM1 (Figure 2G) significantly increased. Within 24 h of exposure to C27, BECN1 (Figure 2H), MAP1LC3B-II (Figure 2I), and SQSTM1 (Figure 2J) exhibited time-dependent increases, suggesting that lysosomal degradation was blocked by C27 exposure.
C27 increased mitophagosome formation and reduced mitophagy flux in microglial cells
Following the detection of upregulated mitophagy-related proteins, mitochondria were co-stained with MAP1LC3B fluorescence to confirm the relationship between C27-induced mitophagy protein upregulation and mitophagosome formation in microglial cells. After exposure to C27 or rotenone (a mitophagy inducer) for 6 h, the number of mitophagosomes (yellow puncta) and percentage of mitophagosomes (yellow puncta/green puncta %) significantly increased in both BV2 microglial cells (Figure S3 [A – C]) and primary microglial cells (Figure 3A–C), indicating a significant increase in mitophagosome formation induced by C27.
Figure 3.
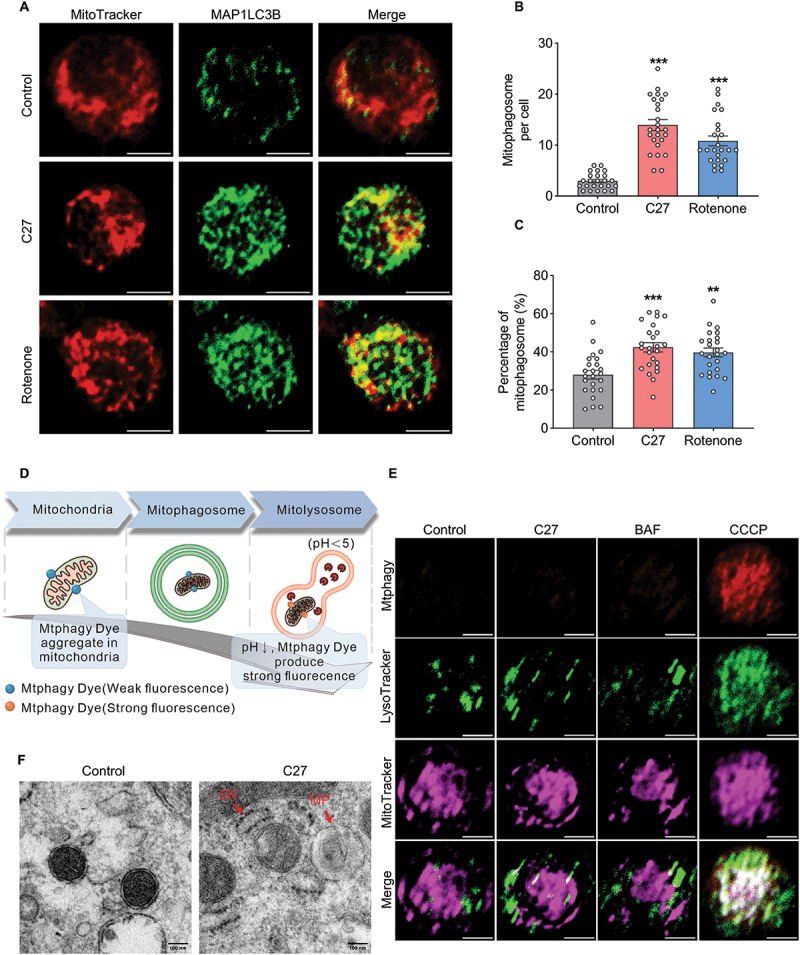
C27 increased mitophagosome formation and decreased mitophagic flux in microglial cells. (A) Representative images of MitoTracker-MAP1LC3B colocalization showed mitophagosome formation in primary microglia cells exposed to C27 (1000 nM) or rotenone (1000 nM) for 6 h. Scale bar: 5 μm. (B) quantification of the number of MitoTracker-MAP1LC3B colocalized puncta (n = 20–25 cells per group). Non-parametric mann – Whitney U test were used to measure significance between two groups. (C) quantification of the percentage of MitoTracker-MAP1LC3B co-localized puncta (n = 20–25 cells per group). One-way ANOVA followed by Dunn’s post-hoc test was used to determine statistical significance between multiple groups. (D, E) Representative images of mitophagy in BV2 microglial cells treated with C27. The colocalization of mitophagy dye, lysosome dye, and MitoTracker showing mitophagy flux in BV2 microglial cells exposed to C27 (1000 nM), BAF (400 nM), or CCCP (10 μM) for 6 h. Scale bar: 5 μm. (F) transmission electron microscopy images revealed the mitochondrial ultrastructure and mitophagosomes in adult microglial cells exposed to C27 (1000 nM) for 6 h. ER, endoplasmic reticulum; MP, mitophagosome. Data are presented as the mean ± SEM. **P < 0.01, ***P < 0.001 vs. control.
To verify the effect of C27 on mitophagic flux, we stained BV2 microglial cells using the Mitophagy Detection Kit (Figure 3D). Strong fluorescence was emitted only when mitophagosomes fused with lysosomes. While the colocalization of mitophagosomes and lysosomes was significantly increased in the CCCP treatment group, BV2 microglial cells in the control, C27 and BAF groups showed weak fluorescence and rare colocalization of mitophagosomes and lysosomes, indicating that fusion processing was blocked in the C27 treated group (Figure 3E). To further confirm these findings, we evaluated the formation and accumulation of mitophagosomes using transmission electron microscopy. As predicted, the mitochondria in the control adult microglial cells had normal, intact, and healthy morphologies. In contrast, exposure to C27 damaged mitochondria and mitophagosome formation (Figure 3F). Adult microglial cells exposed to C27 showed an unhealthy accumulation of mitophagosomes.
C27-mediated increased autophagosome formation and defective autophagic flux in microglial cells
To determine whether the accumulation of mitophagosomes was due to an increase in mitophagosome formation or a decrease in autophagic flux, the expression levels of MAP1LC3B-II and SQSTM1 were measured. MAP1LC3B-I is converted into MAP1LC3B-II during autophagy activation, which is required for autophagosome membrane formation [49]. Therefore, the expression level of MAP1LC3B-II directly reflects autophagosome formation. BV2 microglial cells were exposed to C27 (1000 nM) for 24 h with or without 400 nM of BAF (an autophagosome-lysosome fusion inhibitor) for the last 4 h. No significant difference in MAP1LC3B-II expression level was observed between BAF-treated and untreated BV2 microglial cells exposed to C27 (Figure 4A), suggesting that C27 May lead to decreased autophagy flux and block the fusion between mitophagosomes and lysosomes.
Figure 4.
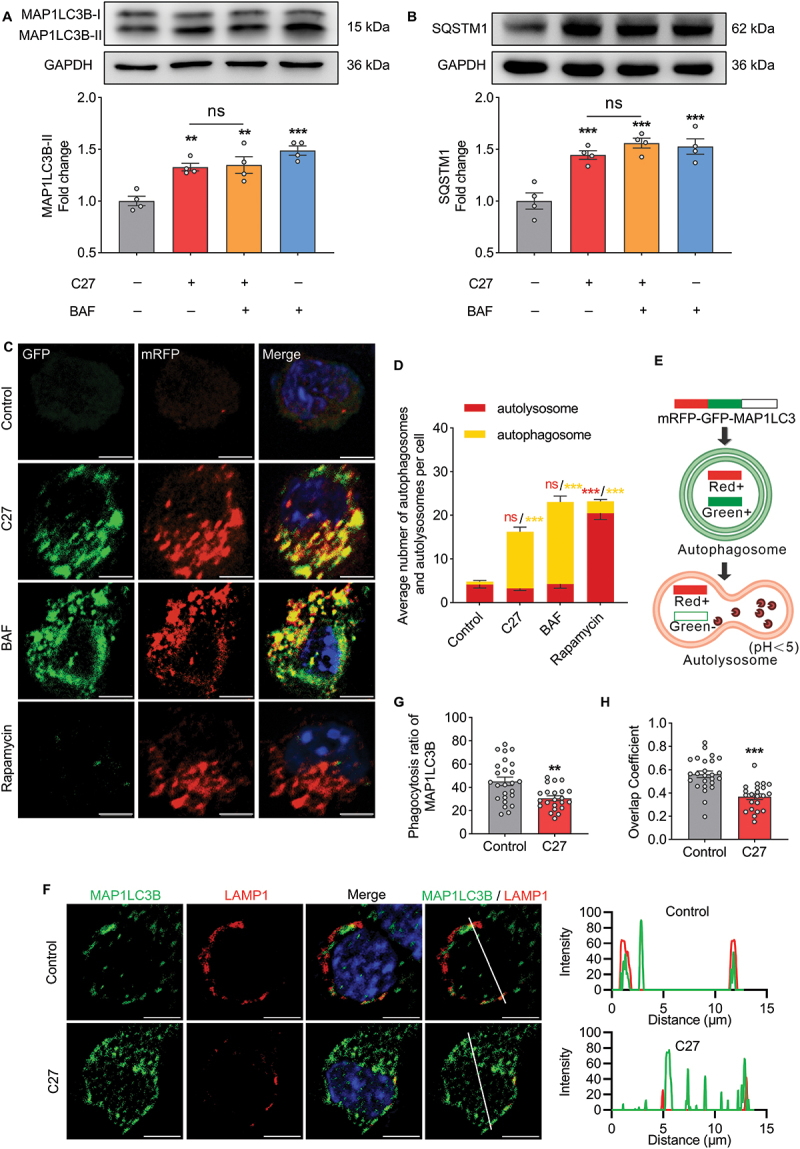
C27 increased autophagosome formation and decreased autophagic flux in microglial cells. (A, B) western blots showing the expression of MAP1LC3B-II (A) and SQSTM1 (B) in BV2 microglial cells exposed to C27 (1000 nM) for 24 h, followed by treatment with 400 nM of BAF for the last 4 h of the treatment period (n = 4). One-way ANOVA followed by Dunn’s post-hoc test was used to determine statistical significance between multiple groups. (C–E) BV2 microglial cells were infected with the mRFP-GFP-MAP1LC3B adenovirus and treated with C27 (1000 nM), BAF (100 nM), and rapamycin (10 nM) for 24 h. The number of autophagosomes (yellow puncta) and autolysosomes (red puncta) per cell was counted (n = 10–20 cells per group). Non-parametric mann – Whitney U test were used to measure significance between two groups. (F) Representative images of MAP1LC3B co-labeling with LAMP1 in BV2 microglial cells exposed to C27 (1000 nM) for 24 h. Graphs on the right show the corresponding MAP1LC3B and LAMP1 intensities along the white lines, with colors consistent with the merged images. Scale bar: 5 μm. (G, H) quantitative analysis of the lysosomal phagocytosis ratio of MAP1LC3B (G) and the overlap coefficient of MAP1LC3B and LAMP1 (H) (n = 20–25 cells per group). Data are presented as the mean ± SEM. Student’s t-test was used to measure the significance between two groups. **P < 0.01, ***P < 0.001 vs. control.
SQSTM1 is a receptor protein that links MAP1LC3B to ubiquitin and is selectively trapped in autophagosomes and degraded by lysosomes. Therefore, SQSTM1 expression negatively correlates with the rate of autophagic degradation [50,51]. Consistent with the above findings on MAP1LC3B-II levels, there was no significant change in SQSTM1 expression levels between BAF-treated and untreated BV2 microglial cells exposed to C27 (Figure 4B), suggesting that C27 inhibited autophagic degradation of microglial cells and reduced autophagy flux.
To further confirm that C27 induced the dysregulation of autophagic flux, BV2 microglial cells were transduced with an mRFP-GFP-MAP1LC3-expressing adenovirus and exposed to C27. This mechanism is shown in Figure 4E. BV2 microglial cells exposed to C27 (1000 nM) for 24 h exhibited accumulation of colocalized mRFP-GFP yellow puncta, indicating defective autophagosome degradation and a possible defect in lysosomal function. Similarly, increased numbers of yellow puncta were observed in BV2 microglial cells exposed to BAF (400 nM) in the last 4 h. In contrast, accumulated red puncta were observed in BV2 microglial cells exposed to rapamycin (100 nM, an autophagy inducer) for 24 h, suggesting increased autophagic flux (Figure 4C–E). Co-staining for MAP1LC3B and LAMP1 (a lysosomal marker) was performed. As predicted, C27 significantly increased the number of autophagosomes, decreased LAMP1 intensity, and reduced colocalization (Figures 4F and S4A). Furthermore, the lysosomal phagocytosis ratio of MAP1LC3B and overlap coefficient decreased after C27 treatment (Figures 4G,H and S4B). Overall, these results demonstrated that C27 induced increased autophagosome formation and defective autophagic flux, leading to mitophagosome accumulation.
C27-induced lysosomal dysfunction and inhibited fusion between mitophagosomes and lysosomes
To further validate the inhibition of mitochondrial-lysosomal crosstalk induced by C27, MitoTracker Green and LysoTracker Red were used to reveal the interaction between mitochondria and lysosomes. The morphology of the mitochondria was analyzed using the mitochondrial form factor, average perimeter, and average size. Higher levels of form factors indicate elongated mitochondria. In both primary microglial cells (Figure 5A–D) and BV2 microglial cells (Figure S5 [A – D]), the mitochondrial form factor, average perimeter, and average size decreased significantly after the C27 treatment compared to the control group. These results demonstrated that C27 facilitated mitochondrial fission, consistent with the increased expression of DNM1L. The colocalization of mitochondria and lysosomes declined after exposure to C27, indicating that C27 might inhibit the fusion of mitophagosomes and lysosomes. Based on these findings, the lysosomal diameters were investigated. Lysosomes could be categorized into three groups based on diameter: normal lysosomes (<1.0 μm), intermediate lysosomes (1.0–2.5 μm), and abnormal lysosomal vacuoles (>3.0 μm) [52]. In primary microglial cells (Figure 5A,E) and BV2 microglial cells (Figure S5A, S5E), most lysosomes in the control group had a diameter <1 μm, whereas larger lysosomes were detected after exposure to C27, indicating that C27 might lead to lysosomal dysfunction.
Figure 5.
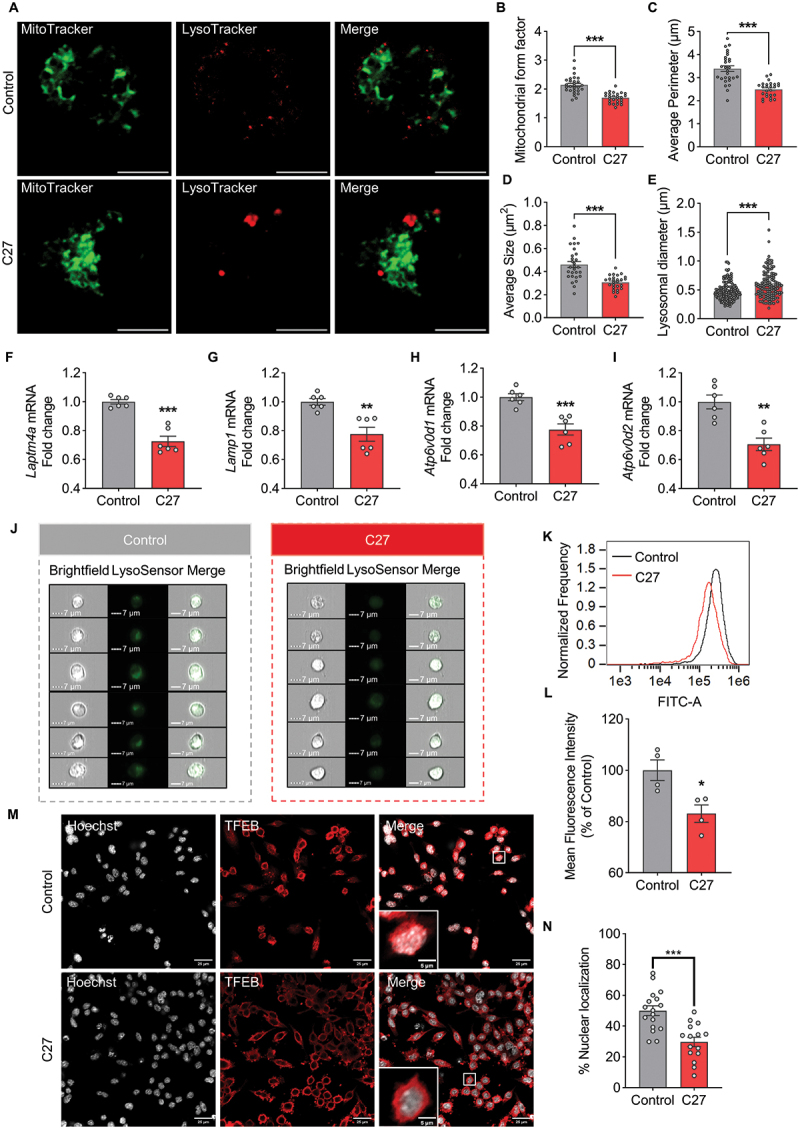
C27 impaired lysosomal function and induced defective mitophagy in microglia cells. (A) Representative fluorescence images of MitoTracker green and LysoTracker red in primary microglia cells exposed to C27 (1000 nM) for 24 h. Scale bar: 5 μm. Quantitative analysis of mitochondrial form factor (n = 26–30 cells per group) (B), average perimeter (n = 26–30 cells per group) (C), average size (n = 26–30 cells per group) (D), and lysosomal diameter (n = 10 cells per group) (E) were measured using ImageJ software. (F–I) the mRNA expressions of lysosome-related genes, such as Laptm4a (F), Lamp1 (G), Atp6v0d1 (H), and Atp6v0d2 (I), were measured by qPCR analysis in BV2 microglia cells treated with C27 (1000 nM) for 24 h (n = 6). (J–L) the representative images (J), histogram overlay (K), and quantitative analysis of the mean fluorescent intensity of LysoSensor green DND189 (L) were measured by imaging flow cytometry in BV2 microglia cells treated with C27 (1000 nM) for 24 h (n = 4). (M, N) BV2 microglial cells exposed to C27 (1000 nM) for 24 h were subjected to TFEB-Hoechst colocalization analysis. Representative images are shown in (M) (scale bar: 25 μm), and the quantified results of TFEB nuclear localization (n = 15–20 random fields) are presented in (N). Data are presented as the mean ± SEM. Student’s t-test or non-parametric mann – Whitney U test (E, G) was used to measure significance between two groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Moreover, we monitored lysosome-related genes to validate the effects of C27 on lysosomal function. The mRNA levels of Laptm4a (Figure 5F) and Lamp1 (Figure 5G) (promoting lysosome biogenesis), Atp6v0d1 (Figure 5H) (promotes lysosomal acidification), and Atp6v0d2 (Figure 5I), (promotes the fusion of mitophagosomes and lysosomes) were downregulated, demonstrating that C27 May induce lysosomal dysfunction [27]. Lysosomal function depends on lysosomal acidification [53]. Consistent with the results of the mRNA assay, the reduction in lysosomal acidification demonstrated that C27 led to lysosomal dysfunction (Figure 5J–L).
To further investigate the mechanism of C27-induced lysosomal dysfunction, we examined the nuclear localization of TFEB. TFEB is a key transcriptional master regulator of lysosomal biogenesis [54]. TFEB could normally be observed to be localized in both cytosol and nuclear to maintain metabolic homeostasis. However, C27 significantly inhibited the nuclear translocation of TFEB, indicating that TFEB was inactivated (Figure 5M,N). These results suggest that C27 induced lysosomal dysfunction and inhibited the fusion between mitophagosomes and lysosomes.
Restoring lysosomal function resulted in complete mitophagy in C27-treated microglial cells
In general, the inhibition of TFEB nuclear localization can be caused by the activation of the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) and subsequent phosphorylation. Moreover, MTOR Ser-2481 phosphorylation is closely associated with MTORC1 signaling [55,56]. Therefore, we hypothesized that C27 induced lysosomal dysfunction by mediating MTOR signaling. Notably, C27 induced MTORC1 activity while torin1, a potent MTORC1 inhibitor, significantly suppressed MTOR phosphorylation induced by C27 treatment (Figure 6A,B). To gain further insights into their involvement in the initiation and formation of autophagosomes, we conducted western blot analysis of autophagy markers, namely BECN1, MAP1LC3B-II, and SQSTM1, in both the C27 and C27+torin1 groups (Figure 6C). Both C27 and C27+torin1 exhibited increased expression of autophagy initiation marker BECN1 (Figure 6D) and autophagy formation marker MAP1LC3B-II (Figure 6E), with no significant difference in expression between the two groups. However, following torin1 treatment, the expression of the autophagy degradation marker SQSTM1 was significantly reduced, indicating an improvement in the blocked autophagy flux (Figure 6F). This outcome might be attributed to enhanced lysosome functionality. Consequently, we proceeded to investigate the alterations in lysosome function.
Figure 6.
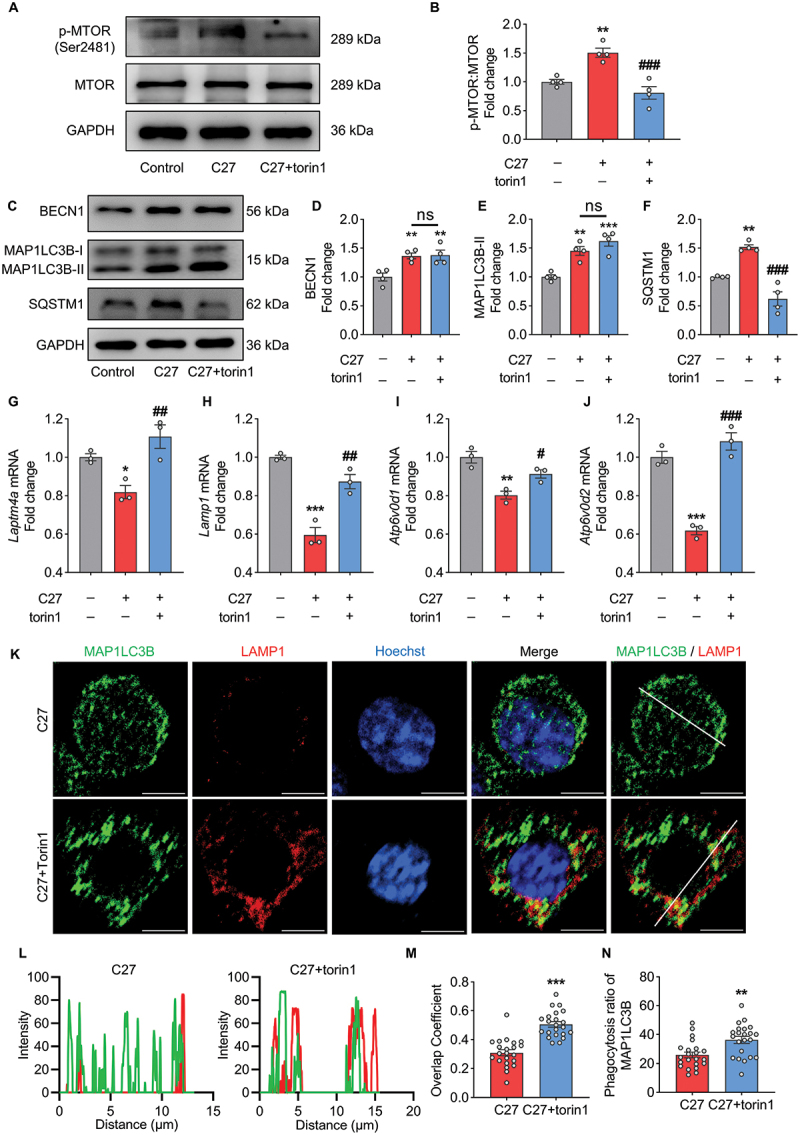
Torin1 treatment recovered the lysosomal function impaired by C27 in BV2 microglia cells. (A–B) the activation of MTOR was measured by western blotting (n = 4). (C–F) Representative immunoblots (C) and bar graphs (D–F) showing western blot analyses of BECN1 (D), MAP1LC3B-II (E) and SQSTM1 (F) in BV2 microglial cells pretreated with 2 μM of torin1 for 2 h, followed by exposure to C27 (1000 nM) for 24 h (n = 4). (G–J) the mRNA expressions of lysosome-related genes, such as Laptm4a (G), Lamp1 (H), Atp6v0d1 (I), and Atp6v0d2 (J), were measured by qPCR analysis in BV2 microglial cells pretreated with 2 μM of torin1 for 2 h, followed by exposure to C27 (1000 nM) for 24 h (n = 3). (K) Representative images of MAP1LC3B co-labeling with LAMP1 in BV2 microglial cells pretreated with 2 μM of torin1 for 2 h followed by exposure to C27 (1000 nM) for 24 h. (L) the corresponding MAP1LC3B and LAMP1 intensity along the white line, colors as in merged images. Scale bar: 5 μm. (M, N) quantitative analysis of overlap coefficient of MAP1LC3B and LAMP1 (M) and lysosomal phagocytosis ratio of MAP1LC3B (N) (n = 20–25 cells per group). Data are presented as the mean ± SEM. One-way ANOVA followed by Dunn’s post-hoc test was used to determine the statistical significance between multiple groups. Student’s t-test was used to measure significance between two groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. C27.
Torin1 restored the mRNA levels of lysosomal function-related genes downregulated by C27 (Figure 6G–J). Thereafter, to further investigate the role of MTORC1 in C27-induced lysosomal dysfunction, we performed co-staining for MAP1LC3B and LAMP1 in BV2 microglial cells treated with C27 with or without torin1. Compared to BV2 microglial cells treated with C27 alone, BV2 microglial cells treated with both C27 and torin1 exhibited significantly increased LAMP1 puncta and enhanced colocalization of MAP1LC3B and LAMP1 (Figure 6K,L). The overlap coefficients and lysosomal phagocytosis ratio of MAP1LC3B were enhanced; however, the formation of MAP1LC3B was not affected (Figure 6M,N, S4C, and S4D), which was consistent with the previous result (Figure 6E). Overall, our findings suggest that C27-induced defective mitophagy may be attributed to lysosomal dysfunction, which can be reversed by the inhibition of MTOR signaling.
Inhibition of mitophagosome initiation blocked C27-induced mitophagy
To elucidate the relationship between C27-mediated mitophagy and autophagy, BV2 microglial cells were treated with pharmacological autophagy inhibitors 3-methyladenine (3-MA) and wortmannin. The cells were pretreated with 500 μM of 3-MA or 1 μM of wortmannin for 1 h, followed by exposure to 1000 nM of C27 for 24 h. At the end of the experiment, the expression levels of mitophagy and autophagy markers were detected using western blotting.
Inhibition of autophagy with 3-MA or wortmannin significantly suppressed the C27-mediated increase in mitophagy markers, including DNM1L (Figure 7A), PINK1 (Figure 7B), and PRKN (Figure 7C), and increased the levels of autophagy markers, including BECN1 (Figure 7D), MAP1LC3B-II (Figure 7E), and SQSTM1 (Figure 7F). These results were further verified using a gene-silencing approach. Consistent with the findings mentioned above, C27 did not increase the expression levels of mitophagy (Figure 7G–I) and autophagy markers (Figure 7J–L) in BV2 microglial cells transfected with BECN1 siRNA. These findings suggest that the upregulation of mitophagy and autophagy induced by C27 could be inhibited by inhibiting autophagy initiation.
Figure 7.
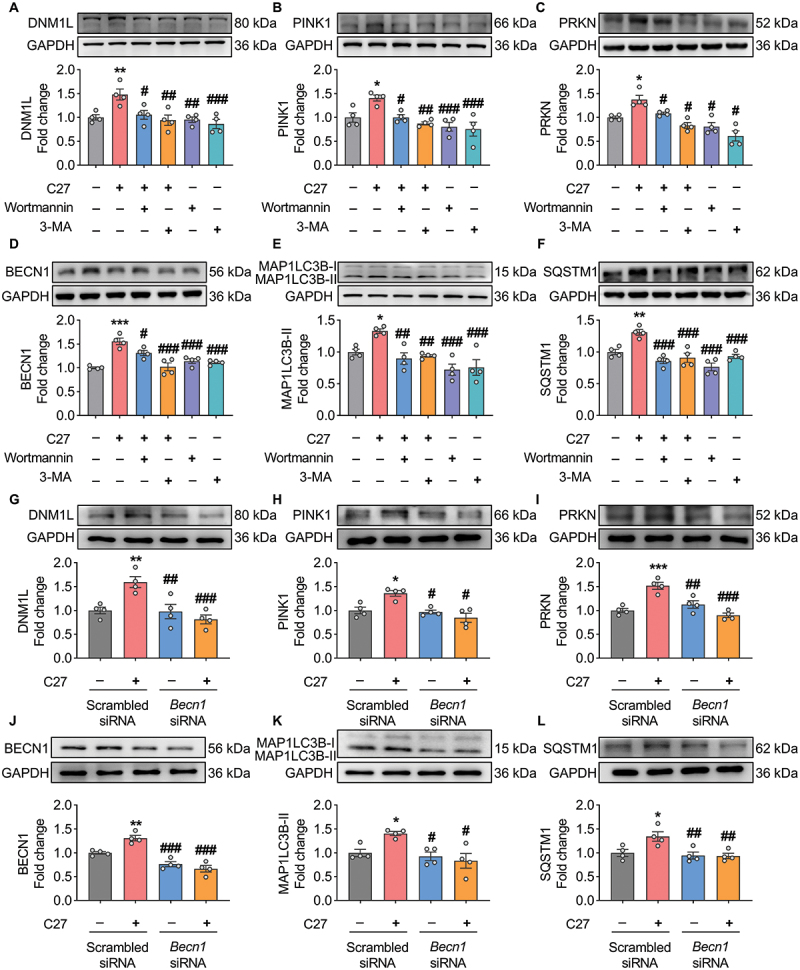
Pharmacological inhibition and gene silencing of autophagy blocked C27-induced mitophagy. (A–C) Representative western blots showed the expression of mitophagy markers, such as DNM1L (A), PINK1 (B), and PRKN (C) in BV2 microglial cells pretreated with 0.5 mM of 3-methyladenine (3-MA) or 1 μM of wortmannin for 1 h, followed by exposure to C27 (1000 nM) for 24 h (n = 4). (D–F) Representative western blots showed the expression of autophagy markers, such as BECN1 (D), MAP1LC3B-II (E), and SQSTM1 (F) in BV2 microglial cells pretreated with 0.5 mM of 3-methyladenine (3-MA) or 1 μM wortmannin for 1 h, followed by exposure to C27 (1000 nM) for 24 h (n = 4). (G–L) Representative western blots showed the expression of DNM1L (G), PINK1 (H), PRKN (I), BECN1 (J), MAP1LC3B-II (K), and SQSTM1 (L) in BV2 microglial cells transfected with either BECN1 siRNA or scrambled siRNA, following exposure to C27 (1000 nM) for 24 h (n = 4). Data are presented as the mean ± SEM. Non-parametric mann – Whitney U test was used to measure significance between two groups (C, H, K). One-way ANOVA followed by Dunn’s post-hoc test was used to determine the statistical significance between multiple groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. C27.
We used the mitophagy pharmacological inhibitor Mdivi-1 and a gene-silencing approach to further validate the relationship between autophagy and mitophagy. BV2 microglial cells were pretreated with Mdivi-1 (5 μM) for 1 h, followed by exposure to C27 for 24 h. The expression levels of mitophagy and autophagy markers were determined using western blotting.
As predicted, in BV2 microglial cells pretreated with Mdivi-1, C27 did not induce an increase in the mitophagy markers DNM1L (Figure 8A), PINK1 (Figure 8B), and PRKN (Figure 8C). Furthermore, the expression levels of autophagy markers, including MAP1LC3B-II (Figure 8D) and SQSTM1 (Figure 8E), were significantly inhibited, whereas the C27-induced autophagy initiation marker BECN1 was not inhibited by Mdivi-1 (Figure 8F). Similarly, BV2 microglial cells transfected with PINK1 siRNA inhibited the upregulation of mitophagy markers, including DNM1L (Figure 8G), PINK1 (Figure 8H), and PRKN (Figure 8I), and autophagy markers, including MAP1LC3B-II (Figure 8J) and SQSTM1 (Figure 8K). However, the upregulation of BECN1 induced by C27 was not inhibited by PINK1 siRNA (Figure 8L), suggesting that the initiation of autophagy occurred upstream of mitochondrial damage and that C27 induced the initiation of mitophagy and autophagy simultaneously.
Figure 8.
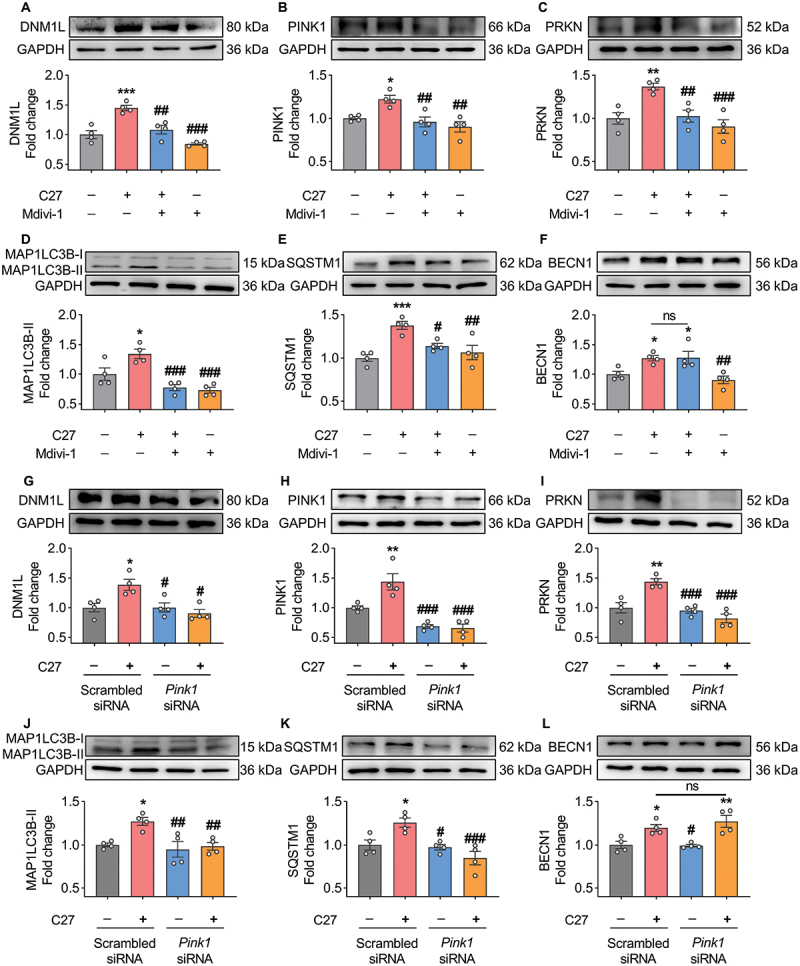
Pharmacological inhibition and gene silencing of mitophagy blocked C27-induced mitophagy. (A–C) Representative western blots showed the expression of mitophagy markers, such as DNM1L (A), PINK1 (B), and PRKN (C) in BV2 microglial cells pretreated with 5 μM of Mdivi-1 (a mitophagy inhibitor) for 1 h, followed by exposure to C27 (1000 nM) for 24 h (n = 4). (D–F) Representative western blots showing expression of autophagy markers, such as MAP1LC3B-II (D), SQSTM1 (E), and BECN1 (F) in BV2 microglial cells pretreated with 5 μM of Mdivi-1 for 1 h, followed by exposure to C27 (1000 nM) for 24 h (n = 4). (G–L) Representative western blots showed the expression of DNM1L (G), PINK1 (H), PRKN (I), MAP1LC3B-II (J), SQSTM1 (K), and BECN1 (L) in BV2 microglial cells transfected with either PINK1 siRNA or scrambled siRNA, following exposure to C27 (1000 nM) for 24 h (n = 4). Data are presented as the mean ± SEM. Non-parametric mann – Whitney U test was used to measure significance between two groups (G). One-way ANOVA followed by Dunn’s post-hoc test was used to determine the statistical significance between multiple groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. C27.
C27-mediated defective mitophagy promoted activation of microglial cells
The previous work of our group demonstrated that C27 induces microglial overactivation and excessive inflammation, leading to neuronal damage [35]. Knockout of CKLF inhibits microglial overactivation and reduces neuronal damage [32,57]. Initially, we assessed the activation status of microglia by examining the expression levels of key genes associated with microglia homeostasis, namely those encoding TMEM119, P2RY12, and TGFB/TGF-β, as well as pro-inflammatory genes such as IL1B, TNF, and NOS2/iNOS [58,59]. Through qPCR analysis, we observed a significant reduction in the expression of microglia homeostasis-related genes and a concurrent increase in the expression levels of pro-inflammatory genes following C27 treatment (Figure S6A). These findings provide compelling evidence of C27-induced microglial activation. Thereafter, to explore the cause of microglial overactivation, we investigated the involvement of defective mitophagy and mitophagosome accumulation. Activated microglia exhibit a characteristic genetic profile with an enhanced expression of AIF1 (allograft inflammatory factor 1) [60]. Therefore, western blotting was used to detect the expression levels of AIF1. As shown in Figure 9(A,E,I,M), C27 significantly increased the expression level of AIF1. However, when autophagy or mitophagy was pharmacologically inhibited or gene silenced, AIF1 expression did not increase. Consistent with these findings, the mRNA expression levels of proinflammatory cytokines, such as Il6 and Tnf, were also significantly inhibited in C27-exposed BV2 microglial cells after pharmacological blocking or gene silencing of autophagy or mitophagy (Figure 9C,D,G,H,K,L,O,P). Inhibition of mitophagy resulted in significant downregulation of the mRNA levels of proinflammatory cytokine Il1b (Figure 9F,N). Moreover, Il1b mRNA levels decreased in C27-exposed BV2 microglial cells with the inhibition of autophagy (Figure 9B,J). These findings indicated that C27-mediated defective mitophagy led to microglial overactivation and subsequent inflammation.
Figure 9.
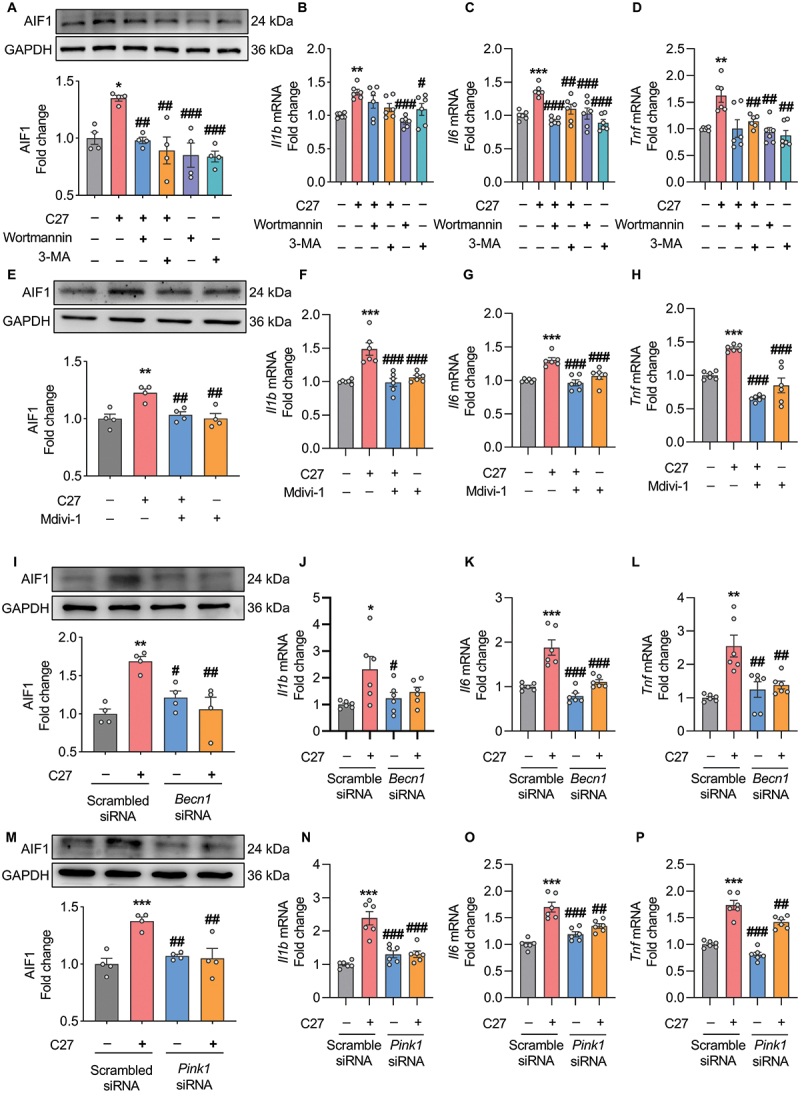
Defective mitophagy induced by C27 led to increased microglial activation and elevated proinflammatory cytokines. (A) Representative western blots showed the expression of AIF1 in BV2 microglial cells pretreated with 0.5 mM of 3-methyladenine (3-MA) or 1 μM of wortmannin for 1 h, followed by exposure to C27 for 24 h (n = 4). (B–D) qPCR was used to measure the mRNA expression of proinflammatory cytokines, such as Il1b (B), Il6 (C), and Tnf (D) in BV2 microglial cells pretreated with 0.5 mM of 3-MA or 1 μM of wortmannin for 1 h, followed by exposure to C27 for 24 h (n = 6). (E) Representative western blots showed the expression of AIF1 in BV2 microglial cells pretreated with 0.5 μM of Mdivi-1 for 1 h, followed by exposure to C27 for 24 h (n = 4). (F–H) qPCR was used to measure the mRNA expression of proinflammatory cytokines, such as Il1b (F), Il6 (G), and Tnf (H) in BV2 microglial cells pretreated with 0.5 μM of Mdivi-1, following exposure to C27 for 24 h (n = 6). (I) western blot analysis was used to measure the expression of AIF1 in BV2 microglial cells transfected with either BECN1 siRNA or scrambled siRNA, following exposure to C27 for 24 h (n = 4). (J–L) qPCR was used to measure the mRNA expression of proinflammatory cytokines, such as Il1b (J), Il6 (K), and Tnf (L) in BV2 microglial cells transfected with either BECN1 siRNA or scrambled siRNA, following exposure to C27 for 24 h (n = 6). (M) western blot analysis was used to measure the expression of AIF1 in BV2 microglial cells transfected with either PINK1 siRNA or scrambled siRNA, following exposure to C27 for 24 h (n = 4). (N–P) qPCR was used to measure the mRNA expression of proinflammatory cytokines, such as Il1b (N), Il6 (O), and Tnf (P) in BV2 microglial cells transfected with either PINK1 siRNA or scrambled siRNA, following exposure to C27 for 24 h (n = 6). Data are presented as the mean ± SEM. Non-parametric mann – Whitney U test was used to measure significance between two groups (D, L). One-way ANOVA followed by Dunn’s post-hoc test was used to determine the statistical significance between multiple groups. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. C27.
C27 increased the expression level of mitophagy markers and promoted microglial activation in vivo
To further validate that C27 induced defective mitophagy and microglial activation in vivo, mice were injected with C27 (10 μg) for 3 and 6 h via stereotaxic injection, and brain slices near the injection site were collected. We performed immunofluorescence co-staining of mitophagy markers (DNM1L and PINK1) and autophagy markers (MAP1LC3B and SQSTM1) with AIF1 (a microglial marker) to confirm the effect of C27 on mitophagy and the relationship between C27-mediated mitophagy and microglial activation. As shown in Figure 10A–D, compared with the sham group, the levels of microglial mitophagy markers DNM1L and PINK1 and autophagy markers MAP1LC3B and SQSTM1 were significantly increased 3 h after C27 treatment and remained at a high expression level 6 h after administration, suggesting that C27 induced defective mitophagy in vivo. In addition, we assessed the morphology of AIF1+ microglia to confirm microglial activation. Notably, in the sham operation group, microglial cells exhibited a highly ramified structure with multiple branched processes. In contrast, after the C27 treatment, the morphology of the microglial cells showed significant changes, including a reduction in endpoints (Figure 10E), branches (Figure 10F), junctions (Figure 10G), and cell process length (Figure 10H). In conclusion, these findings verified that C27 induced defective mitophagy and increased microglial overactivation in vivo.
Figure 10.
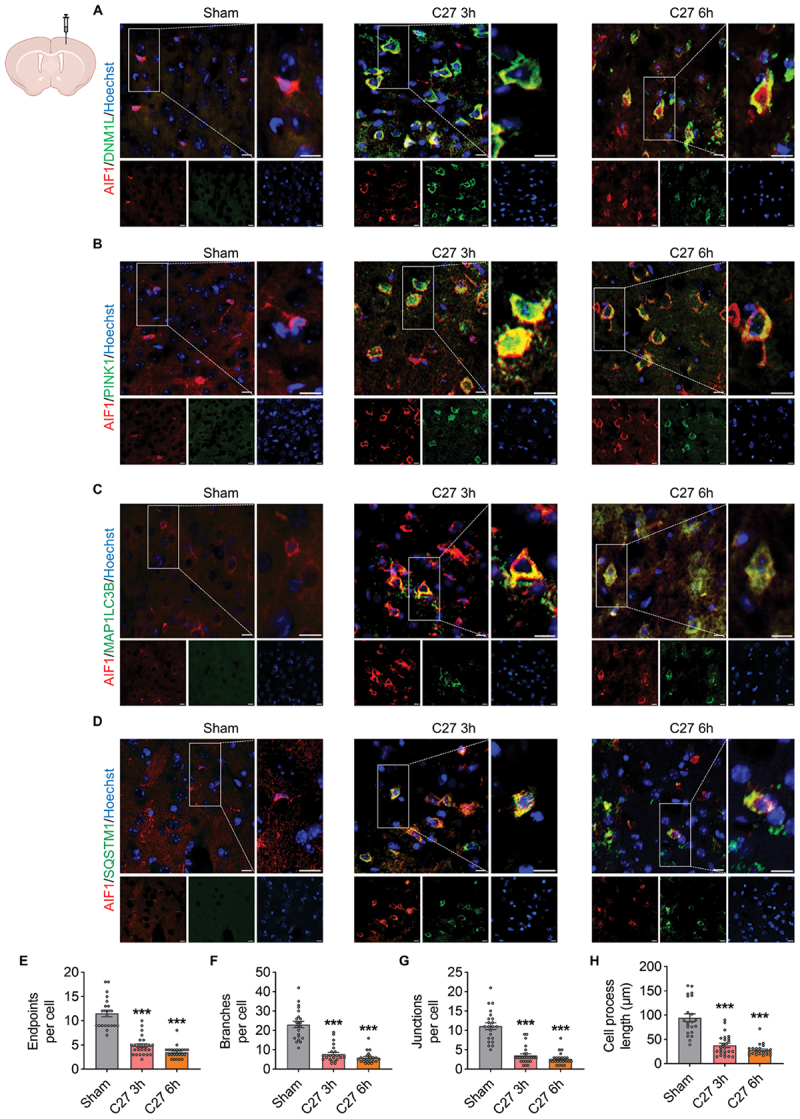
C27 induced upregulation of mitophagy markers and microglial activation in vivo in a time-dependent manner. (A) immunofluorescence staining for AIF1 (red), DNM1L (green), and Hoechst (blue) in the cortices near the injection site. (B) immunofluorescence staining for AIF1 (red), PINK1 (green), and Hoechst (blue) in the cortices near the injection site. (C) immunofluorescence staining for AIF1 (red), MAP1LC3B (green), and Hoechst (blue) in the cortices near the injection site. (D) immunofluorescence staining for AIF1 (red), SQSTM1 (green), and Hoechst (blue) in the cortices near the injection site. Scale bar: 10 μm. (E–H) C27 upregulated microglial activation in vivo. Morphological features, including endpoints (E), branches (F), junctions (G), and cell process length (H), were quantified using ImageJ software (n = 20–25 cells from three mice per group). The data are presented as the mean ± SEM. Non-parametric mann – Whitney U test was used to measure significance between two groups. ***P < 0.001 vs. control.
Discussion
As resident cells of the CNS, microglia have been implicated in brain homeostasis and disease [61,62]. Under pathological conditions, microglial activation is closely associated with neuroinflammation [4]. Mitophagy, a selective form of autophagy, is the fundamental mechanism of mitochondrial degradation. Evidence has shown that mitophagy and neuroinflammation interact and modulate each other in response to pathological events, and impaired mitophagy can induce exuberant inflammation [63–65]. Although several studies have investigated the influence of cytokines on mitophagy and inflammation, the contribution of chemokines in this regard remains largely understudied [30]. Our research group has extensively investigated the role of CKLF, a CC-type chemokine, in inducing aberrant inflammation in nervous system diseases [32,35,41,42]. However, the underlying mechanism remains unclear. In this study, we demonstrate that CKLF induces defective mitophagy in microglia, leading to microglial overactivation and inflammation.
We investigated the role of C27 (an active peptide of CKLF) in microglial mitophagy. These findings demonstrate that CKLF induces mitophagy in microglia, as evidenced by the significantly increased expression of the mitophagy markers DNM1L, PINK1, PRKN, and OPTN and the autophagy markers BECN1 and MAP1LC3B in a dose- and time-dependent manner. Transmission electron microscopy analysis further revealed that C27 induces the formation of mitophagy in adult microglial cells. Moreover, the detection of mitophagosomes by the co-location of MAP1LC3B and mitochondria demonstrate that the C27-mediated upregulation of these proteins was accompanied by the accumulation of mitophagosomes in primary microglial cells. The PINK1-PRKN mitophagy pathway is typically associated with mitochondrial dysfunction, with Δψm serving as a critical indicator of mitochondrial function [66,67]. Under pathological conditions, mitochondrial membrane depolarization leads to the stabilization of PINK1 in the outer mitochondrial membrane, which further activates the PINK1-PRKN mitophagy pathway [68]. As predicted, CKLF significantly decreases Δψm in microglial cells. This result was accompanied by a decrease in various aspects of mitochondrial function, including basal respiration, maximal respiration, proton leak, ATP-linked respiration, and spare capacity.
In addition, mitochondria possess their own genomes, known as mitochondrial DNA (mtDNA), and abnormalities in mtDNA, including deletions and copy number variations, contribute to mitochondrial dysfunction [69]. Both C27 and BAF treatment resulted in a relative decrease in the mtDNA:nDNA ratio. This decrease may be attributed to the presence of small fragments of mtDNA, which could potentially lead to a significantly lower enrichment of mtDNA compared to nuclear DNA [69,70]. Furthermore, an increase in cytosolic mtDNA levels was observed in BV2 microglial cells following exposure to C27, providing additional support for the aforementioned hypothesis. Previous studies have demonstrated that the release of small mtDNA fragments into the cytosol triggers the activation of the CGAS-STING1 pathway [71]. The cytosolic DNA sensor CGAS (cyclic GMP-AMP synthase) plays a crucial role in initiating innate immune responses by generating the second messenger cGAMP. This molecule, in turn, activates the protein STING1. Once activated, STING1 recruits and activates TBK1 (TANK-binding kinase 1), which phosphorylates STING1 and IRF3 (interferon regulatory factor 3) [72]. Our results demonstrate a significant upregulation of CGAS and STING1 expression, as well as a substantial increase in TBK1 and IRF3 phosphorylation, in BV2 microglial cells upon treatment with C27. These findings strongly suggest that C27 is capable of inducing the activation of the CGAS-STING1 pathway.
In general, the mitophagy pathway involves the formation of mitophagosomes and their degradation by lysosomes [73]. Therefore, we assessed the expression of SQSTM1, an autophagic cargo receptor protein recruited by phospho-ubiquitinated PRKN, and considered it an autophagy degradation marker [74]. Notably, our findings reveal that SQSTM1 is upregulated in a dose- and time-dependent manner, suggesting impaired degradation of accumulated mitophagosomes. This was further confirmed by the unaltered expression level of MAP1LC3B-II and SQSTM1 in BV2 microglial cells treated with C27 alone and in combination with the autophagosome-lysosome fusion inhibitor BAF. Mitophagic and autophagic fluxes were also reduced, thereby validating these results.
Mitophagy is an essential catabolic process whereby damaged mitochondria are sequestered by mitophagosomes and subsequently fused with lysosomes for degradation, thereby maintaining cellular homeostasis [16]. Lysosomes are considered the primary sites for degradation in most eukaryotic cells. Proper lysosomal physiology requires lysosomal acidification, lysosomal biogenesis, fusion between lysosomes and autophagosomes [75]. Consequently, impairing lysosomal functions significantly disrupts the flux of mitophagy. Considering that the impaired degradation of mitophagosomes may be due to lysosomal dysfunction, we assessed the effects of C27 on lysosomal function. Our results indicate that exposure to C27 significantly reduces mRNA levels related to lysosomal biogenesis, lysosomal acidification, and mitophagosome-lysosome fusion, suggesting that C27 disrupts lysosomal function [76]. In addition, C27 reduced the acidity of lysosomes, as evidenced by the lysosomal pH indicator. TFEB exerts comprehensive control on autophagy and lysosomal function by regulating genes involved in various stages of autophagosome formation, fusion between autophagosomes and lysosomes, and degradation within lysosomes [75]. Thus, TFEB serves as a pivotal regulator of autophagy by controlling the delivery of cargo and degradation of substrates [77]. Moreover, TFEB overexpression stimulates the generation of new lysosomes, highlighting the importance of TFEB in lysosomal regulation [78]. Our findings demonstrate that treatment with C27 inhibits TFEB activity and reduces its nuclear translocation, leading to a severe impairment of lysosomal function [79]. As MTORC1-mediated phosphorylation inhibits the nuclear translocation of TFEB, we hypothesized that C27 impairs lysosomal function via MTORC1-TFEB signaling [75,79]. As predicted, torin1 treatment inhibited C27-induced MTOR phosphorylation, which was accompanied by recovery of lysosomal function. Our findings suggest that C27-mediated defective mitophagy may result from lysosomal dysfunction via MTORC1-TFEB signaling. However, further investigations are needed to elucidate the interactions between lysosomes and MTORC1 following C27 treatment.
Previous studies have shown that PINK1 and BECN1 interact with and influence each other, thereby influencing mitophagy and autophagy [80,81]. Our findings illustrate that the inhibition of autophagy significantly reduces the expression of mitophagy markers. In contrast, blocking mitophagy did not reduce the expression of the autophagy initiation marker BECN1, suggesting that autophagy initiation occurs upstream of mitophagy.
Our previous study has provided evidence that CKLF is responsible for inducing acute microglial activation and inflammation and initiating metabolic reprogramming, leading to a shift from oxidative phosphorylation to glycolysis [43]. These findings underscore the critical involvement of CKLF in the pathogenesis of ischemic stroke. Dysregulated mitophagy may trigger inflammation associated with damaged mtDNA [65,82]. Consequently, the present study aimed to investigate the impact of CKLF-induced defective mitophagy on microglial overactivation and inflammation. In this study, we used both pharmacological inhibition and gene silencing approaches. The overactivation of microglia was reduced by inhibiting either pharmacological autophagy inhibitors (3-MA and wortmannin) and the mitophagy inhibitor Mdivi-1 or by silencing BECN1 and PINK1, as evidenced by the expression level of AIF1. Furthermore, the neuroinflammation caused by C27 treatment was reversed by both autophagy and mitophagy inhibition, as evidenced by the significantly decreased mRNA expression levels of proinflammatory cytokines Il6, Tnf, and Il1b. These results highlight the crucial role of C27-mediated defective mitophagy in microglial activation and inflammation.
Finally, we validated the in vivo findings. Immunofluorescence assays showed that the expression of mitophagy markers DNM1L and PINK1 and autophagy markers MAP1LC3B and SQSTM1 were significantly increased in the microglial cells of mice administered C27. These findings suggest enhanced induction of mitophagy in microglial cells, albeit with a concurrent blockade of the degradation process. The morphology of microglial cells transitions from a homeostatic to an activated response state, which enables their participation in immune responses [83]. In the sham group, microglia exhibited a highly ramified structure, whereas microglial cells in C27-treated mice showed reduced endpoints, branches, junctions, and cell process lengths, suggesting that defective mitophagy was accompanied by microglial activation in CKLF-treated mice, similar to the in vitro results.
In summary, we demonstrated for the first time that CKLF can facilitate defective mitophagy in microglial cells, leading to microglial overactivation and inflammation (Figure 11). Our study substantiates the new concept that CKLF-mediated defective mitophagy in microglial cells is associated with mitochondrial dysfunction, microglial activation, and inflammation in the brain. Given the crucial role of CKLF in inflammation, intervening in diseases exacerbated by CKLF may be possible by blocking mitochondrial dynamics and/or promoting the clearance of damaged mitochondria. This study provides a basis for further enriching our understanding of the mechanism of CKLF-mediated diseases and treatments based on this mechanism. However, the precise impact of CKLF on pathological conditions remains an area that requires further investigation.
Figure 11.
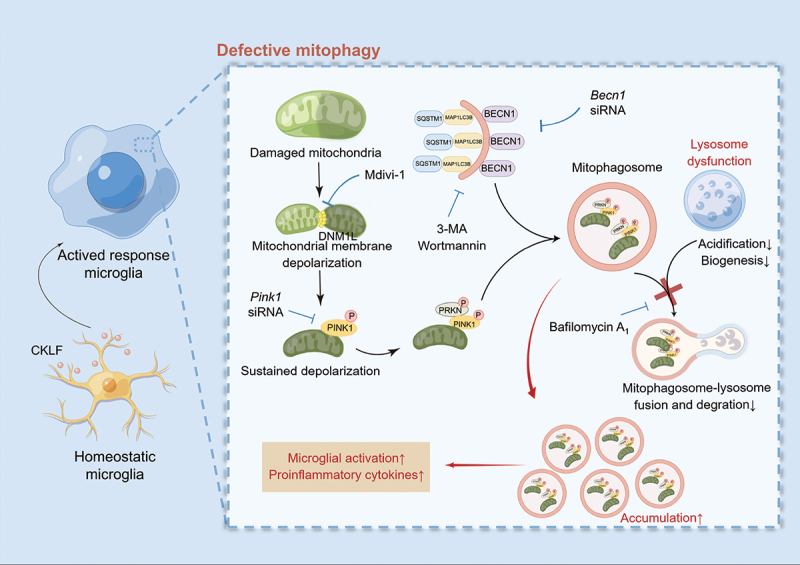
Schematic diagram of CKLF-mediated defective mitophagy and microglial activation (by Figdraw). CKLF decreased mitochondrial membrane potential and induced mitochondrial dysfunction, followed by initiation of PINK1-PRKN mitophagy and subsequent mitophagosome formation. However, an impaired lysosomal function disrupted mitophagosome-lysosome fusion and degradation, leading to mitophagosome accumulation. The defective mitophagy caused by CKLF resulted in microglial overactivation, which further triggered neuroinflammation.
Materials and methods
Reagents
CKLF peptide C27 (ALIYRKLLFNPSGPYQKKPVHEKKEVL) with > 99% purity was obtained from Guoping Pharmaceutical Co., LTD (Hefei, China). Wortmannin (T6283), 3-methyladenine (3-MA) (T1879), and mitochondrial division inhibitor 1/Mdivi-1 (T1907) were obtained from TargetMol. Rotenone (R8875) was obtained from Sigma-Aldrich. Bafilomycin A1 (BAF; S1413) was obtained from Selleck. Torin1 (SC0245) was obtained from Beyotime.
Animals
Male C57BL/6 (eight-weeks old) mice weighing 20–22 g were obtained from Vital River (Beijing, China). The animals were housed in sanitary cages with constant temperature (24 ± 2°C), humidity (45 ± 5%), and a 12–12 h day-night cycle. During the experiment, animals were provided free access to food and water. All animal experiments were conducted following the protocols approved by the Animal Care and Use Committee of the Peking Union Medical College, Chinese Academy of Medical Sciences, and the NIH Guide for the Care and Use of Laboratory Animals.
Cell cultures
Primary newborn microglial cells were isolated from the cerebral cortices of C57BL/6 neonatal mice within 24 h [35]. The cortex was separated at 4°C on Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium (Gibco 12,400,024) after removing the meninges and blood vessels. The cortex tissue was then cut into 1 mm3 and transferred to a digestion solution (10 ml of DMEM/F12 medium containing 1 ml of 2.5% trypsin-EDTA, 20 μl of 5 mg/ml of DNase I) at 37°C for 10 min. The collected cells were plated in poly-L-lysine (PLL)-precoated cell culture flasks (Sigma-Aldrich, P1399) in DMEM/F12 containing 10% FBS and 1% penicillin-streptomycin. Half of the medium was changed on the third day, and the medium was then changed every three days. Approximately 13 days later, when the mixture cells attained confluence, primary microglial cells were collected by shaking at 180 × g for 5 h at 37°C. The purity of the primary microglial cells was confirmed to be > 99% (Figure S7A, S7B).
Adult microglial cells were isolated from the cerebral cortices of male C57BL/6 (eight weeks old) mice using a modified procedure based on previous studies [84]. The brain tissue was dissociated using the Adult Brain Dissociation Kit following the instructions of the manufacturer (RWD, DHABE-5003). Adult microglial cells were then isolated using ITGAM/CD11b (microglia) MicroBeads (Miltenyi Biotec, 130-093-634). Purified adult microglial cells were plated in poly-D-lysine-precoated cell culture dishes in DMEM/F12 containing 10% FBS, 1% penicillin-streptomycin, 10 ng/ml of recombinant murine CSF1/M-CSF (Beyotime, P6015), and 50 ng/ml of TGFB1/TGF-β1 (Bioss, bs-10899P). Half of the medium was changed every three days, and the adult microglial cells were cultured for 9 d before use. The purity of the adult microglial cells was confirmed by immunofluorescence staining with AIF1 (Figure S7C, S7D).
BV2 microglial cells were cultured in DMEM supplemented with 10% G-FBS and 1% penicillin-streptomycin. All cells were incubated in a humified 37°C incubator with 5% CO2.
Mitochondria isolation
BV2 microglial cells were cultured in 100-mm culture dishes at a density of 7 × 106 cells per dish and incubated in the incubator for 24 h. Following exposure to 1000 nM of C27 for 0, 3, 6, 12, and 24 h, the mitochondrial fractions were extracted using a Mitochondria Isolation kit (Beyotime, C3601) according to the instructions of the manufacturer. The BV2 cells were collected and centrifuged at 600 × g for 6 min. The cells were then resuspended in a mitochondrial separation reagent and incubated on ice for 15 min. The suspensions were homogenized until the Trypan Blue positive cells exceeded 50%, and the homogenate was centrifuged at 1000 × g for 10 min at 4°C. The supernatant was transferred to another centrifuge tube and centrifuged at 3500 × g for 10 min at 4°C, resulting in the isolation of the mitochondrial fraction. The supernatant was further centrifuged at 12,000 × g for 12 min at 4°C to improve the purity of cytoplasmic proteins, resulting in the isolation of purified cytoplasmic proteins. The mitochondrial fraction was added to 50 μl of mitochondrial lysis solution and incubated on ice for 10 min. The suspensions were then centrifuged at 12,000 × g for 10 min at 4°C, and the resulting supernatant was used for subsequent western blot experiments. The isolated mitochondrial and cytoplasmic protein concentrations were measured using a BCA kit (Applygen, P1511).
Detection of mitophagy flux
To assess mitophagy flux, BV2 microglial cells were stained with a Mitophagy Detection Kit (Dojindo, MD01) following the instructions of the manufacturer with minor modifications. Briefly, BV2 microglial cells were seeded in confocal dishes at a density of 2 × 105 cells/dish and cultured in an incubator for 24 h. After staining with Mtphagy Dye (containing 25 nM of MitoTracker Deep Red [Yeasen, 40743ES50]) for 30 min at 37°C, the cells were washed twice and treated with C27 for 6 h. Thereafter, the cells were treated with Lyso Dye for 30 min at 37°C and washed with phosphate-buffered saline (PBS; Servicebio, G0002) twice. Fluorescent images were captured using a Cytation C10 Confocal Imaging Reader (Agilent BioTek, CA, USA).
Analysis of mitochondrial membrane potential
The effect of C27 on the mitochondrial membrane potential of microglia was detected using a JC-1 mitochondrial membrane potential detection kit (Beyotime, C2006) following the instructions of the manufacturer. Briefly, the primary microglial cells were seeded in 12-well plates at a density of 2 × 105 cells/well. The cells were exposed to 1000 nM of C27 24 h later; carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (10 μM) was used as a positive control. Subsequently, the cells were washed with PBS, and JC-1 staining solution (JC-1 working solution: DMEM serum-free medium = 1:3) was added and incubated for 20 min in a 37°C incubator. The cells were then washed twice with JC-1 staining buffer, and the fluorescence was observed using an inverted optical microscope.
Measurement of oxygen consumption rate (OCR)
OCR was assessed using a Seahorse XF24 analyzer (Agilent, USA). A total of 4 × 104 cells were seeded in XF24 cell culture microplates and incubated overnight at 37°C and 5% CO2. The following day, the cells were treated with C27 (1000 nM) for 12 h. Simultaneously, the “Flux Pak” cartridge was hydrated with XF Calibrant solution by overnight incubation in a non-CO2 incubator at 37°C. Prior to the assay, the culture medium was replaced with the assay medium (XF DMEM medium containing 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine). Using the Agilent Seahorse XF Cell Mito Stress Test Kit (Seahorse Bioscience 103,015–100), we prepared mitochondrial complex inhibitor solutions according to the manufacturer’s instructions. These solutions were loaded into the hydrated Flux Pak as follows: Port A: 56 μl oligomycin A ([stock] = 15 μM), Port B: 62 μl FCCP ([stock] = 20 μM), and Port C: 69 μl rotenone-antimycin A ([stock] = 5 μM). Each well on the cell culture plate was quantified, and the data were normalized to the cell count. Basal respiration, ATP production, maximal respiration, spare capacity, and proton leak were calculated.
Analysis of mitophagosome formation
Primary microglial cells were seeded at a density of 1 × 105 cells/well in a PLL-precoated 24-well plate with sterile glass coverslips and cultured in an incubator at 37°C for 24 h. The cells were then exposed to C27 (1000 nM) and the positive control rotenone (1000 nM) for 6 h. The medium was replaced with 200 nM of MitoTracker Red CMXRos (Beyotime, C1049B), and the cells were incubated at 37°C for 30 min. After being washed three times with PBS, the cells were fixed with 4% paraformaldehyde for 20 min at 25°C. The cells were then washed with PBS and permeabilized with 0.2% Triton X-100 (Rhawn Chemical Technology Co. Ltd., R009177) for 7 min. The cells were then blocked with 5% bovine serum albumin (BSA; Roche 10,738,328,103) for 60 min and incubated with primary antibody MAP1LC3B (Cell Signaling Technology, 3868S) overnight at 4°C. Alexa Fluor 488-labeled donkey Anti-Rabbit IgG (Invitrogen, A21206; 1:500) was used as the secondary antibody. Fluorescent images were captured using a confocal laser microscope. The number of puncta that colocalized with MAP1LC3B and MitoTracker, and the percentage of MAP1LC3B+ puncta that colocalized with MitoTracker, were quantified using the ImageJ software. A total of 20–25 cells were analyzed in three independent experiments per group.
siRNA transfection
BV2 microglial cells were transfected with either BECN1 siRNA (Santa Cruz Biotechnology, sc -29,798) or PINK1 siRNA (Santa Cruz Biotechnology, sc -44,599). Briefly, BV2 microglial cells were plated in 6-well plates at a density of 6 × 105 cells/well and cultured in an incubator at 37°C for 24 h. Thereafter, the INTERFERin Transfection Reagent (Polyplus Transfection, PT-409-10) was used to transfect the targeted siRNA or scrambled siRNA (100 nM), according to the instructions of the manufacturer. After 24 h, the transfected BV2 microglial cells were exposed to C27 for the next 24 h. Transfection efficiency was analyzed by western blotting.
Adenovirus monomeric red fluorescence protein-green fluorescence protein-MAP1LC3 infection
BV2 microglial cells were seeded in 8-well chamber slides at a density of 1 × 105 cells/well. The cells were infected by 100 multiplicity of infection/MOI adenovirus expressing monomeric red fluorescent protein (mRFP)‐green fluorescent protein (GFP)‐MAP1LC3 fusion protein (Hanbio, HBAD-1007) on the next day according to the instructions of the manufacturer. Fluorescent images were captured using a Cytation C10 Confocal Imaging Reader. A total of 10–20 cells per group from three independent experiments were analyzed using ImageJ software.
Transmission electron microscopy
Adult microglial cells were isolated as previously described [84]. The cells were seeded in 60 mm culture dishes at a density of 2 × 106 cells/dish and cultured as previously described. After treatment, adult microglial cells were fixed with 2.5% glutaraldehyde (Solarbio, P1126) for 30 min at 4°C. Subsequently, adult microglial cells were gently collected in 2.5% glutaraldehyde, and images were obtained using a transmission electron microscope (JEOL, Japan).
MitoTracker and LysoTracker co-staining and image quantification
Primary microglial cells were seeded at a density of 1 × 105 cells/well in a PLL-precoated 24-well plate with sterile glass coverslips and cultured for 24 h. Subsequently, the cells were exposed to C27 (1000 nM) for 24 h. After washing with PBS, the cells were stained with 500 nM of MitoTracker Green (Invitrogen, M7514) and 50 nM of LysoTracker Red (Beyotime, C1046) at 37°C for 30 min. The medium was then replaced with DMEM, and fluorescence images were captured using a laser confocal microscope. The morphology of mitochondria was analyzed using ImageJ software from 26–30 cells in three independent experiments per group [52,85]. In addition, the diameters of the lysosomes were calculated using ImageJ software, with ten cells in three independent experiments per group.
Analysis of lysosome acidity
BV2 microglial cells were cultured in 60-mm culture dishes at a density of 4 × 106 cells/dish and incubated for 24 h. After exposure to 1000 nM of C27 for 24 h, the cells were incubated with 1 μM of LysoSensor Green DND-189 (Yeasen, 40767ES50) for 30 min at 37°C. The cells were then harvested and suspended in 50 μl Flow Cytometry Staining Buffer with a concentration of 2 × 107 cells/ml for imaging flow cytometry. Subsequently, the mean fluorescence intensity of the cells was determined using the ImageStreamX MK II (Amnis Corporation, USA). The data were analyzed using IDEAS 6.2 software.
Stereotaxic injection
Male C57BL/6 (eight weeks old) mice were anesthetized using 1.5% isoflurane and placed in a stereotaxic frame (RWD 68,002). The body temperatures of the mice were maintained at 37°C with an electric heating pad. After identifying bregma, 1 μl of C27 (10 μg/μl) was injected into the brain tissue at 0.2 μl/min. The stereotaxic injection sites (from the bregma) were anterior to posterior (0 mm), mid to lateral (1.5 mm), and dorsal to ventral (−1.3 mm). After injection, the needle was left in position for 10 min to ensure the complete distribution of the reagent. The sham-operated mice were injected with PBS using the same procedure.
Western blotting
Cell suspensions were harvested and lysed on ice with radioimmunoprecipitation assay lysis buffer (Applygen, C1053) containing a protease inhibitor (Sigma-Aldrich, P8340) and a phosphatase inhibitor (APE × BIO, K1015) for 30 min. After centrifugation at 13,201 × g for 30 min at 4°C, the supernatant was collected for western blot analysis. Protein concentration was determined using a BCA protein quantitative kit, following the instructions of the manufacturer. The protein expression levels were detected by western blotting. Equal amounts of proteins were loaded onto SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes. After blocking with 5% BSA, the membrane was incubated with primary antibody overnight at 4°C. The primary antibodies used in this study included: PINK1 (Abcam, ab23707; 1:600), PRKN (Proteintech 14,060–1-AP; 1:3000), DNM1L (Santa Cruz Biotechnology, sc -271,583; 1:50), OPTN (Proteintech 10,837–1-AP; 1:6000), BECN1 (Abcam, ab207612; 1:2000), MAP1LC3B (Cell Signaling Technology, 3868S; 1:1000), SQSTM1 (Proteintech 18,420–1-AP; 1:1000), AIF1 (Wako Pure Chemical Industries, 019–19741; 1:1000), MTOR (Abcam, ab32028; 1:1000), p-MTOR (Abcam, ab137133; 1:1000), CGAS (Proteintech 26,416–1-AP; 1:300), STING1 (Proteintech 19,851–1-AP; 1:2000), TBK1 (Proteintech 28,397–1-AP; 1:1000), p-TBK1 (Cell Signaling Technology, 5483S; 1:1000), IRF3 (Proteintech 66,670–1-Ig; 1:5000), p-IRF3 (Cell Signaling Technology, 29047S; 1:1000), GAPDH (Proteintech 60,004–1-Ig; 1:10000), and VDAC (Beyotime, AF1027; 1:1000). The membrane was washed thrice with TBST (Tris Buffered Saline [Servicebio, G0001-2 L], 2 mL Tween-20 [Tianjin Fuchen Chemical Reagent Factory, 9005-64-5], pH 7.6 for 2 L) and incubated with species-specific horseradish peroxidase-conjugated sheep Anti-Rabbit IgG (SeraCare, 5220–0336; 1:5000) or horseradish peroxidase-polyclonal sheep Anti-Mouse IgG (SeraCare, 5220–0341; 1:5000) for 2 h at 25°C. The density of the bands was measured using an Image Quant LAS 4000 mini (GE Healthcare, WI, USA) and quantified using ImageJ software.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from BV2 microglial cells using TRIzol reagent. The extracted RNA was reverse transcribed into cDNA using the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen Biotech, AT311–04) following the instructions of the manufacturer. cDNA was amplified on a 7900 hT real-time quantitative polymerase chain reaction (PCR) System (Applied Biosystems) using the PerfectStart Green quantitative PCR (qPCR) Supermix kit (TransGen Biotech, AQ601–04). The following mouse primer sequences were utilized: Tnf forward 5”-AGAAGTTCCCAAATGGCCTC-3‘, Tnf reverse 5’-CCACTTGGTGGTTTGCTACG-3‘, Il1b forward 5’-GCCCATCCTCTGTGACTCAT-3‘, Il1b reverse 5’-AGCTCATATGGGTCCGACAG-3‘, Il6 forward 5’- AGTTGCCTTCTTGGGACTGA-3‘, Il6 reverse 5’-TCCACGATTTCCCAGAGAAC-3‘, Laptm4a forward 5’-TGCGTTCTTTTTGCCGTCTC-3‘, Laptm4a reverse 5’-GAATCAGCCAGCCCACTTGA-3‘, Lamp1 forward 5’-CCAGAGCGTTCAACATCAGC-3‘, Lamp1 reverse 5’-ACAGGCTAGAGCTGGCATTC-3‘, Atp6v0d1 forward 5’-CGCCACATGAGAAACCATGC-3‘, Atp6v0d1 reverse 5’-CTCAAAGCTGCCTAGCGGAT-3‘, Atp6v0d2 forward 5’-CTGGTTCGAGGATGCAAAGC-3‘, Atp6v0d2 reverse 5’-TCCAAGGTCTCACACTGCAC-3‘, Tmem119 forward 5’- CCTTCACCCAGAGCTGGTTC-3‘, Tmem119 reverse 5’- GGCTACATCCTCCAGGAAGG-3‘, P2ry12 forward 5’- ATGGATATGCCTGGTGTCAACA-3‘, P2ry12 reverse 5’- AGCAATGGGAAGAGAACCTGG-3‘, Tgfb forward 5’- TGCGCTTGCAGAGATTAAAA-3‘, Tgfb reverse 5’- CGTCAAAAGACAGCCACTCA-3‘, Nos2 forward 5’- GTTCTCAGCCCAACAATACAAGA-3‘, Nos2 reverse 5’- GTGGACGGGTCGATGTCAC-3‘, Actb forward 5’-GTGACGTTGACATCCGTAAAGA-3‘, and Actb reverse 5 ’-GCCGGACTCATCGTACTCC-3”. The results were analyzed by the 2−△△CT method, with β-actin serving as the internal control.
Immunofluorescence and immunocytochemistry staining and analysis
C57 BL/6 mice were infused with PBS and 4% paraformaldehyde, and their brain tissues were collected and fixed in 4% paraformaldehyde. The brains were then cut into thick slices (20 μm) using a Leica CM3050S cryostat after being subjected to gradient dehydration in sucrose. After antigen retrieval, the slides were washed with PBS, permeabilized with PBS containing 0.5% Triton X-100 at 25°C for 15 min, washed with PBS, and blocked with 5% BSA for 30 min. The slides were then incubated overnight with primary antibodies at 4°C. The following primary antibodies were used: AIF1 (Servicebio, GB12105; 1:100), PINK1 (Abclonal, A11435; 1:50), DNM1L (Santa Cruz Biotechnology, sc -271,583; 1:30), MAP1LC3B (Cell Signaling Technology, 3868S; 1:200), and SQSTM1 (Proteintech 18,420–1-AP; 1:200). After washing three times with PBST, the slices were incubated with the appropriate secondary antibodies at 25°C for 2 h. The secondary antibodies used were Alexa Fluor 488-labeled donkey Anti-Rabbit IgG (Invitrogen, A21206; 1:500) and Alexa Fluor 546-labeled donkey Anti-Mouse IgG (Invitrogen, A10036; 1:500), and the nuclei were labeled with Hoechst 33,342 (Dojindo, H342; 1:1000). Images were captured using a laser confocal scanning microscope, and the morphology of the microglia was analyzed using ImageJ software [86].
For immunocytochemistry, BV2 microglial cells were seeded at a density of 1 × 105 cells/well in a PLL-pre-coated 24-well plate containing sterile glass coverslips and cultured for 24 h. After treatment, cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min at 25°C. The cells were then washed with PBS and permeabilized with 0.2% Triton X-100 for 7 min. The cells were then blocked with 5% BSA for 60 min and incubated with primary antibodies overnight at 4°C. The primary antibodies used were TFEB (Beyotime, AF8130; 1:200), TOMM20 (Proteintech 11,802–1-AP; 1:200), DNA (PROGEN, AC-30-10; 1:200), MAP1LC3B (Cell Signaling Technology, 3868S; 1:200), and LAMP1 (GeneTex, GT25212; 1:200). The secondary antibodies used were Alexa Fluor 488-labeled donkey Anti-Rabbit IgG (Invitrogen, A21206; 1:500), Alexa Fluor 546-labeled donkey Anti-Mouse IgG (Invitrogen, A10036; 1:500), and Alexa Fluor 546-labeled donkey Anti-Rabbit IgG (Invitrogen, A10040; 1:500). The nucleus was labeled with Hoechst 33,342 (Dojindo, H342; 1:1000). Fluorescent images were captured using a Cytation C10 Confocal Imaging Reader. For the TFEB nuclear translocation analysis, 15–20 randomly selected fields from three independent experiments per group were analyzed. To analyze MAP1LC3B and LAMP1 colocalization, 20–25 randomly selected cells from three independent experiments per group were analyzed. All analyses were performed using ImageJ software.
Analysis of the mitochondrial DNA copy number
BV2 microglial cells were seeded in 6-well plates at a density of 4 × 105 cells/well and cultured in an incubator at 37°C for 24 h. The cells were then exposed to various concentrations of C27 for 24 h. Genomic DNA was extracted from the BV2 microglial cells using the EasyPure® Genomic DNA Kit (TransGen Biotech, EE101–01) according to the instructions of the manufacturer. DNA was amplified by Applied Biosystems 7900 hT real-time quantitative PCR using the PerfectStart Green qPCR Supermix Kit (TransGen Biotech, AQ601–04). The mitochondrial DNA copy number was measured as the relative ratio of mitochondrial DNA to nuclear DNA. The mitochondrial DNA level was reflected by the mt-Co1/cox1 gene encoded on the heavy strand with the forward primer 5 ”-CCACTTCGCCATCATATTCGTAGG-3‘ and the reverse primer 5 ’-TCTGAGTAGCGTCGTGGTATTCC-3‘ while Actb was used as the nuclear DNA control with the forward primer 5 ’-GCAGGAGTACGATGAGTCCG-3‘ and the reverse primer 5 ’-ACGCAGCTCAGTAACAGTCC-3”. The results were obtained by the 2−△△CT method.
Statistical analyses
All data are expressed as the mean ± SEM. A normal distribution test was conducted to assess the distribution of the data. Student’s t-test was used for the comparison of two groups for variables with normal distributions. Mann-Whitney U test was used for variables with non-normal distributions. One-way analysis of variance (ANOVA) followed by Dunnett’s test or Two-way ANOVA followed by Sidak’s multiple comparison test was used to compare data among multiple groups. All statistical analyses were performed using GraphPad Prism software (version 8.4.3). Values with P < 0.05 were considered statistically significant in all analyses.
Supplementary Material
Acknowledgements
We thank the Taylor & Francis Editing Services provided professional editing services.
Funding Statement
The work was supported by the National Key Research and Development Program of China [2022YFC3500300]; National Natural Science Foundation of China [U2202214, 82074044, U21A20410, 82130109, 81730096, 81973499]; Key R&D Program of Shanxi Province [201803D421006, 201903D421018]; CAMS Innovation Fund for Medical Sciences (CIFMS) [2021-I2M-1–020]; High-End Foreign Experts introduction program [G20200001485].
Disclosure statement
The authors declare no relevant or known conflicts of interest.
Data availability statement
We authors declare all data and materials are available on request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2276639
References
- [1].Prinz M, Jung S, Priller J.. Microglia biology: one century of evolving concepts. Cell. 2019 Oct 3;179(2):292–311. PubMed PMID: 31585077. doi: 10.1016/j.cell.2019.08.053 [DOI] [PubMed] [Google Scholar]
- [2].Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32(1):367–402. PubMed PMID: 24471431. doi: 10.1146/annurev-immunol-032713-120240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cserep C, Posfai B, Denes A. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron. 2021 Jan 20;109(2):222–240. PubMed PMID: 33271068. doi: 10.1016/j.neuron.2020.11.007 [DOI] [PubMed] [Google Scholar]
- [4].Woodburn SC, Bollinger JL, Wohleb ES. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J Neuroinflammation. 2021 Nov 6;18(1):258. PubMed PMID: 34742308. doi: 10.1186/s12974-021-02309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Simpson DSA, Oliver PL. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants. 2020 Aug 13;9(8):743. PubMed PMID: 32823544. doi: 10.3390/antiox9080743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Benard G, Karbowski M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin Cell Dev Biol. 2009. May;20(3):365–374. PubMed PMID: 19530306. doi: 10.1016/j.semcdb.2008.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Franklin JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal. 2011 Apr 15;14(8):1437–1448. PubMed PMID: 20812874. doi: 10.1089/ars.2010.3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Falabella M, Vernon HJ, Hanna MG, et al. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol Metab. 2021. Apr;32(4):224–237. PubMed PMID: 33640250. doi: 10.1016/j.tem.2021.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018. Jun;16:263–275. PubMed PMID: 29549824. doi: 10.1016/j.redox.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].MYW N, Wai T, Simonsen A. Quality control of the mitochondrion. Dev Cell. 2021 Apr 5;56(7):881–905. PubMed PMID: 33662258. doi: 10.1016/j.devcel.2021.02.009 [DOI] [PubMed] [Google Scholar]
- [11].Liu Y, Wang M, Hou XO, et al. Roles of microglial mitophagy in neurological disorders. Front Aging Neurosci. 2022;14:979869. PubMed PMID: 36034136. doi: 10.3389/fnagi.2022.979869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Han B, Jiang W, Cui P, et al. Microglial PGC-1alpha protects against ischemic brain injury by suppressing neuroinflammation. Genome Med. 2021 Mar 26;13(1):47. PubMed PMID: 33771213. doi: 10.1186/s13073-021-00863-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ahmed S, Kwatra M, Ranjan Panda S, et al. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of parkinson disease. Brain Behav Immun. 2021. Jan;91:142–158. PubMed PMID: 32971182. doi: 10.1016/j.bbi.2020.09.017 [DOI] [PubMed] [Google Scholar]
- [14].Thangaraj A, Periyasamy P, Liao K, et al. HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy. 2018;14(9):1596–1619. PubMed PMID: 29966509. doi: 10.1080/15548627.2018.1476810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lou G, Palikaras K, Lautrup S, et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020. Jan;26(1):8–20. PubMed PMID: 31375365. doi: 10.1016/j.molmed.2019.07.002 [DOI] [PubMed] [Google Scholar]
- [16].Lu Y, Li Z, Zhang S, et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13(2):736–766. PubMed PMID: 36632220. doi: 10.7150/thno.79876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Malpartida AB, Williamson M, Narendra DP, et al. Mitochondrial dysfunction and Mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem Sci. 2021. Apr;46(4):329–343. PubMed PMID: 33323315. doi: 10.1016/j.tibs.2020.11.007 [DOI] [PubMed] [Google Scholar]
- [18].Mao Z, Tian L, Liu J, et al. Ligustilide ameliorates hippocampal neuronal injury after cerebral ischemia reperfusion through activating PINK1/Parkin-dependent mitophagy. Phytomedicine. 2022. Jul;101:154111. PubMed PMID: 35512628. doi: 10.1016/j.phymed.2022.154111 [DOI] [PubMed] [Google Scholar]
- [19].Morton H, Kshirsagar S, Orlov E, et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: focus on aging, mitochondria and synapse. Free Radic Biol Med. 2021 Aug 20;172:652–667. PubMed PMID: 34246776. doi: 10.1016/j.freeradbiomed.2021.07.013 [DOI] [PubMed] [Google Scholar]
- [20].Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011 May 15;14(10):1939–1951. PubMed PMID: 21128700. doi: 10.1089/ars.2010.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol. 2018;9:1283. PubMed PMID: 29951054. doi: 10.3389/fimmu.2018.01283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Quinn PMJ, Moreira PI, Ambrosio AF, et al. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. 2020 Nov 9;8(1):189. PubMed PMID: 33168089. doi: 10.1186/s40478-020-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017. Apr;45:83–91. PubMed PMID: 28437683. doi: 10.1016/j.ceb.2017.03.013 [DOI] [PubMed] [Google Scholar]
- [24].Vargas JNS, Hamasaki M, Kawabata T, et al. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023. Mar;24(3):167–185. PubMed PMID: 36302887. doi: 10.1038/s41580-022-00542-2 [DOI] [PubMed] [Google Scholar]
- [25].Liu ZZ, Hong CG, Hu WB, et al. Autophagy receptor OPTN (optineurin) regulates mesenchymal stem cell fate and bone-fat balance during aging by clearing FABP3. Autophagy. 2021. Oct;17(10):2766–2782. PubMed PMID: 33143524. doi: 10.1080/15548627.2020.1839286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020. Jan;16(1):3–17. PubMed PMID: 30951392. doi: 10.1080/15548627.2019.1603547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Peng J, Pan J, Wang H, et al. Morphine-induced microglial immunosuppression via activation of insufficient mitophagy regulated by NLRX1. J Neuroinflammation. 2022 Apr 12;19(1):87. PubMed PMID: 35414088. doi: 10.1186/s12974-022-02453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thangaraj A, Periyasamy P, Guo ML, et al. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy. 2020. Feb;16(2):289–312. PubMed PMID: 30990365. doi: 10.1080/15548627.2019.1607686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tyrrell DJ, Blin MG, Song J, et al. Aging Impairs Mitochondrial Function and Mitophagy and Elevates Interleukin 6 Within the Cerebral Vasculature. J Am Heart Assoc. 2020. Dec;9(23):e017820. PubMed PMID: 33225820. doi: 10.1161/JAHA.120.017820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].WKE I, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017 May 5;356(6337):513–519. PubMed PMID: 28473584. doi: 10.1126/science.aal3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin YC, Lin YC, Tsai ML, et al. IL-33 regulates M1/M2 chemokine expression via mitochondrial redox-related mitophagy in human monocytes. Chem Biol Interact. 2022 May 25;359:109915. PubMed PMID: 35339432. doi: 10.1016/j.cbi.2022.109915 [DOI] [PubMed] [Google Scholar]
- [32].Zhou X, Zhang YN, Li FF, et al. Neuronal chemokine-like-factor 1 (CKLF1) up-regulation promotes M1 polarization of microglia in rat brain after stroke. Acta Pharmacol Sin. 2022. May;43(5):1217–1230. PubMed PMID: 34385606. doi: 10.1038/s41401-021-00746-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu Y, Liu L, Zhou Y, et al. CKLF1 Enhances inflammation-mediated Carcinogenesis and prevents Doxorubicin-induced apoptosis via IL6/STAT3 signaling in HCC. Clin Cancer Res. 2019 Jul 1;25(13):4141–4154. PubMed PMID: 30918019. doi: 10.1158/1078-0432.CCR-18-3510 [DOI] [PubMed] [Google Scholar]
- [34].Tian L, Li W, Wang J, et al. The CKLF1-C19 peptide attenuates allergic lung inflammation by inhibiting CCR3- and CCR4-mediated chemotaxis in a mouse model of asthma. Allergy. 2011. Feb;66(2):287–297. PubMed PMID: 21208220. doi: 10.1111/j.1398-9995.2010.02478.x [DOI] [PubMed] [Google Scholar]
- [35].Chen C, Chu SF, Ai QD, et al. CKLF1 Aggravates focal cerebral ischemia injury at early stage partly by modulating microglia/Macrophage toward M1 polarization through CCR4. Cell Mol Neurobiol. 2019. Jul;39(5):651–669. PubMed PMID: 30982091. doi: 10.1007/s10571-019-00669-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen HD, Liu YB, Ning N, et al. CKLF1 as a potential therapeutic target for ischemic stroke. Prog Biochem Biophys. 2022. Jan;49(1):126–138. PubMed PMID: WOS:000749853000004. [Google Scholar]
- [37].Liu DD, Song XY, Yang PF, et al. Progress in pharmacological research of chemokine like factor 1 (CKLF1). Cytokine. 2018. Feb;102:41–50. PubMed PMID: 29275012. doi: 10.1016/j.cyto.2017.12.002 [DOI] [PubMed] [Google Scholar]
- [38].Wang Y, Zhang Y, Han W, et al. Two C-terminal peptides of human CKLF1 interact with the chemokine receptor CCR4. Int J Biochem Cell Biol. 2008;40(5):909–919. PubMed PMID: 18069042. doi: 10.1016/j.biocel.2007.10.028 [DOI] [PubMed] [Google Scholar]
- [39].Cai X, Deng J, Ming Q, et al. Chemokine-like factor 1: a promising therapeutic target in human diseases. Exp Biol Med (Maywood). 2020. Oct;245(16):1518–1528. PubMed PMID: 32715782. doi: 10.1177/1535370220945225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Y, Yu H, Feng J. Role of chemokine-like factor 1 as an inflammatory marker in diseases. Front Immunol. 2023;14:1085154. PubMed PMID: 36865551. doi: 10.3389/fimmu.2023.1085154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kong LL, Wang ZY, Han N, et al. Neutralization of chemokine-like factor 1, a novel C-C chemokine, protects against focal cerebral ischemia by inhibiting neutrophil infiltration via MAPK pathways in rats. J Neuroinflammation. 2014 Jun 20;11(1):112. PubMed PMID: 24946684. doi: 10.1186/1742-2094-11-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen C, Chu SF, Ai QD, et al. CKLF1/CCR5 axis is involved in neutrophils migration of rats with transient cerebral ischemia. Int Immunopharmacol. 2020. Aug;85:106577. PubMed PMID: 32446198. doi: 10.1016/j.intimp.2020.106577 [DOI] [PubMed] [Google Scholar]
- [43].Ma WY, Wu QL, Wang SS, et al. A breakdown of metabolic reprogramming in microglia induced by CKLF1 exacerbates immune tolerance in ischemic stroke. J Neuroinflammation. 2023 Apr 25;20(1):97. PubMed PMID: 37098609. doi: 10.1186/s12974-023-02779-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bhansali S, Bhansali A, Dhawan V. Metformin promotes mitophagy in mononuclear cells: a potential in vitro model for unraveling metformin’s mechanism of action. Ann N Y Acad Sci. 2020. Mar;1463(1):23–36. PubMed PMID: 31225649. doi: 10.1111/nyas.14141 [DOI] [PubMed] [Google Scholar]
- [45].Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Bio. 2010 Apr 19;189(2):211–221. PubMed PMID: 20404107. doi: 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Perelman A, Wachtel C, Cohen M, et al. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012 Nov 22;3(11):e430. PubMed PMID: 23171850. doi: 10.1038/cddis.2012.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jeng JY, Yeh TS, Lee JW, et al. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem. 2008 Feb 1;103(2):347–357. PubMed PMID: 18072287. doi: 10.1002/jcb.21625 [DOI] [PubMed] [Google Scholar]
- [48].West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015 Apr 23;520(7548):553–557. PubMed PMID: 25642965. doi: 10.1038/nature14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang P, Shao BZ, Deng Z, et al. Autophagy in ischemic stroke. Prog Neurobiol. 2018. Apr-May;163-164:98–117. PubMed PMID: 29331396. doi: 10.1016/j.pneurobio.2018.01.001 [DOI] [PubMed] [Google Scholar]
- [50].Galluzzi L, Bravo-San Pedro JM, Blomgren K, et al. Autophagy in acute brain injury. Nat Rev Neurosci. 2016. Aug;17(8):467–484. PubMed PMID: 27256553. doi: 10.1038/nrn.2016.51 [DOI] [PubMed] [Google Scholar]
- [51].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008 Aug 15;283(33):22847–22857. PubMed PMID: 18524774. doi: 10.1074/jbc.M802182200 [DOI] [PubMed] [Google Scholar]
- [52].Singh K, Roy M, Prajapati P, et al. NLRX1 regulates TNF-alpha-induced mitochondria-lysosomal crosstalk to maintain the invasive and metastatic potential of breast cancer cells. Biochim Biophys Acta Mol Basis Dis. 2019 Jun 1;1865(6):1460–1476. PubMed PMID: 30802640. doi: 10.1016/j.bbadis.2019.02.018 [DOI] [PubMed] [Google Scholar]
- [53].Root J, Merino P, Nuckols A, et al. Lysosome dysfunction as a cause of neurodegenerative diseases: lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. 2021. Jul;154:105360. PubMed PMID: 33812000. doi: 10.1016/j.nbd.2021.105360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ahsan A, Liu M, Zheng Y, et al. Natural compounds modulate the autophagy with potential implication of stroke. Acta Pharm Sin B. 2021. Jul;11(7):1708–1720. PubMed PMID: 34386317. doi: 10.1016/j.apsb.2020.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tan A, Prasad R, Lee C, et al. Past, present, and future perspectives of transcription factor EB (TFEB): mechanisms of regulation and association with disease. Cell Death Differ. 2022. Aug;29(8):1433–1449. PubMed PMID: 35739255. doi: 10.1038/s41418-022-01028-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Soliman GA, Acosta-Jaquez HA, Dunlop EA, et al. mTOR ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem. 2010 Mar 12;285(11):7866–7879. PubMed PMID: 20022946. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen C, Ai Q, Chu S, et al. IMM-H004 protects against oxygen-glucose deprivation/reperfusion injury to BV2 microglia partly by modulating CKLF1 involved in microglia polarization. Int Immunopharmacol. 2019. May;70:69–79. PubMed PMID: 30785093. doi: 10.1016/j.intimp.2019.02.012 [DOI] [PubMed] [Google Scholar]
- [58].Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018. Oct;19(10):622–635. PubMed PMID: 30206328. doi: 10.1038/s41583-018-0057-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pan RY, He L, Zhang J, et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022 Apr 5;34(4):634–648 e6. PubMed PMID: 35303422. doi: 10.1016/j.cmet.2022.02.013. [DOI] [PubMed] [Google Scholar]
- [60].Pinto MV, Fernandes A. Microglial phagocytosis—rational but challenging therapeutic target in multiple sclerosis. Int J Mol Sci. 2020 Aug 19;21(17):5960. PubMed PMID: 32825077. doi: 10.3390/ijms21175960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Borst K, Dumas AA, Prinz MM. Microglia: Immune and non-immune functions. Immunity. 2021 Oct 12;54(10):2194–2208. PubMed PMID: 34644556. 10.1016/j.immuni.2021.09.014 [DOI] [PubMed] [Google Scholar]
- [62].Jie Z, Ko CJ, Wang H, et al. Microglia promote autoimmune inflammation via the noncanonical NF-kappaB pathway. Sci Adv. 2021 Sep 3;7(36):eabh0609. PubMed PMID: 34516909. doi: 10.1126/sciadv.abh0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Eshraghi M, Adlimoghaddam A, Mahmoodzadeh A, et al. Alzheimer’s disease pathogenesis: role of autophagy and mitophagy focusing in microglia. Int J Mol Sci. 2021 Mar 24;22(7):3330. PubMed PMID: 33805142. doi: 10.3390/ijms22073330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017. Dec;6(1):24. PubMed PMID: 28748360. doi: 10.1186/s40169-017-0154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yin X, Xin H, Mao S, et al. The role of autophagy in sepsis: protection and injury to organs. Front Physiol. 2019;10:1071. PubMed PMID: 31507440. doi: 10.3389/fphys.2019.01071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Shen L, Gan Q, Yang Y, et al. Mitophagy in cerebral ischemia and ischemia/reperfusion injury. Front Aging Neurosci. 2021;13:687246. PubMed PMID: 34168551. doi: 10.3389/fnagi.2021.687246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tomita K, Kuwahara Y, Igarashi K, et al. Mitochondrial dysfunction in diseases, longevity, and treatment resistance: tuning mitochondria function as a therapeutic strategy. Genes (Basel). 2021 Aug 29;12(9):1348. PubMed PMID: 34573330. doi: 10.3390/genes12091348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xian H, Liou YC. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021. Mar;28(3):827–842. PubMed PMID: 33208889. doi: 10.1038/s41418-020-00657-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fukunaga H. Mitochondrial DNA copy number and Developmental Origins of Health and disease (DOHaD). Int J Mol Sci. 2021 Jun 21;22(12):6634. PubMed PMID: 34205712. doi: 10.3390/ijms22126634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang X, Berkowicz A, King K, et al. Pharmacologic enrichment of exosome yields and mitochondrial cargo. Mitochondrion. 2022. May;64:136–144. PubMed PMID: 35398304. doi: 10.1016/j.mito.2022.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim J, Kim HS, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023. Mar;55(3):510–519. PubMed PMID: 36964253. doi: 10.1038/s12276-023-00965-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chen L, Dong J, Liao S, et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology. 2022. Nov;76(5):1389–1408. PubMed PMID: 35313046. doi: 10.1002/hep.32471 [DOI] [PubMed] [Google Scholar]
- [73].Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021 Feb 1;40(3):e104705. PubMed PMID: 33438778. doi: 10.15252/embj.2020104705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yao RQ, Ren C, Xia ZF, et al. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021. Feb;17(2):385–401. PubMed PMID: 32048886. doi: 10.1080/15548627.2020.1725377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020. Feb;21(2):101–118. PubMed PMID: 31768005. doi: 10.1038/s41580-019-0185-4 [DOI] [PubMed] [Google Scholar]
- [76].Wang J, Su Q, Wu Q, et al. Sanguinarine impairs lysosomal function and induces ROS-dependent mitophagy and apoptosis in human hepatocellular carcinoma cells. Arch Pharm Res. 2021. Nov;44(11):1025–1036. PubMed PMID: 34751932. doi: 10.1007/s12272-021-01356-0 [DOI] [PubMed] [Google Scholar]
- [77].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011 Jun 17;332(6036):1429–1433. PubMed PMID: 21617040. doi: 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011. Nov;7(11):1379–1381. PubMed PMID: 21785263. doi: 10.4161/auto.7.11.17166 [DOI] [PubMed] [Google Scholar]
- [79].Cora D, Bussolino F, Doronzo G. TFEB Signalling-related MicroRNAs and Autophagy. Biomolecules. 2021 Jul 4;11(7):985. PubMed PMID: 34356609. doi: 10.3390/biom11070985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ning J, Junyi T, Chang M, et al. TOM7 silencing exacerbates focal cerebral ischemia injury in rat by targeting PINK1/Beclin1-mediated autophagy. Behav Brain Res. 2019 Mar 15;360:113–119. PubMed PMID: 30468788. doi: 10.1016/j.bbr.2018.11.031 [DOI] [PubMed] [Google Scholar]
- [81].Michiorri S, Gelmetti V, Giarda E, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010. Jun;17(6):962–974. PubMed PMID: 20057503. doi: 10.1038/cdd.2009.200 [DOI] [PubMed] [Google Scholar]
- [82].Picca A, Calvani R, Coelho-Junior HJ, et al. Cell Death and inflammation: the role of mitochondria in health and disease. Cells. 2021 Mar 3;10(3):537. PubMed PMID: 33802550. doi: 10.3390/cells10030537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gober R, Ardalan M, Shiadeh SMJ, et al. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. 2022. Jan;32(1):e13003. PubMed PMID: 34297453. doi: 10.1111/bpa.13003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Carroll JA, Race B, Williams K, et al. Innate immune responses after stimulation with Toll-like receptor agonists in ex vivo microglial cultures and an in vivo model using mice with reduced microglia. J Neuroinflammation. 2021 Sep 6;18(1):194. PubMed PMID: 34488805. doi: 10.1186/s12974-021-02240-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chaudhry A, Shi R, Luciani DS. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2020 Feb 1;318(2):E87–E101. PubMed PMID: 31846372. doi: 10.1152/ajpendo.00457.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Young K, Morrison H. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J Vis Exp. 2018. Jun;5(136):57648. PubMed PMID: 29939190. doi: 10.3791/57648-v [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We authors declare all data and materials are available on request.