

ABSTRACT
Seneca Valley virus (SVV) causes vesicular disease in pigs, posing a threat to global pork production. OPTN (optineurin) is a macroautophagy/autophagy receptor that restricts microbial propagation by targeting specific viral or bacterial proteins for degradation. OPTN is degraded and cleaved at glutamine 513 following SVV infection via the activity of viral 3C protease (3C[pro]), resulting in N-terminal and a C-terminal OPTN fragments. Moreover, OPTN interacts with VP1 and targets VP1 for degradation to inhibit viral replication. The N-terminal cleaved OPTN sustained its interaction with VP1, whereas the degradation capacity targeting VP1 decreased. The inhibitory effect of N-terminal OPTN against SVV infection was significantly reduced, C-terminal OPTN failed to inhibit viral replication, and degradation of VP1 was blocked. The knockdown of OPTN resulted in reduced TBK1 activation and phosphorylation of IRF3, whereas overexpression of OPTN led to increased TBK1-IRF3 signaling. Additionally, the N-terminal OPTN diminished the activation of the type I IFN (interferon) pathway. These results show that SVV 3C[pro] targets OPTN because its cleavage impairs its function in selective autophagy and type I IFN production, revealing a novel model in which the virus develops diverse strategies for evading host autophagic machinery and type I IFN response for survival.
Abbreviations: Co-IP: co-immunoprecipitation; GFP-green fluorescent protein; hpi: hours post-infection; HRP: horseradish peroxidase; IFN: interferon; IFNB/IFN-β: interferon beta; IRF3: interferon regulatory factor 3; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MOI: multiplicity of infection; OPTN: optineurin; PBS: phosphate-buffered saline; SVV: Seneca Valley virus; SQSTM1: sequestosome 1; TAX1BP1: Tax1 binding protein 1; TBK1: TANK binding kinase 1; TCID50: 50% tissue culture infectious doses; UBAN: ubiquitin binding in TNIP/ABIN (TNFAIP3/A20 and inhibitor of NFKB/NF-kB) and IKBKG/NEMO; UBD: ubiquitin-binding domain; ZnF: zinc finger.
KEYWORDS: 3C protease (3C[pro]), cleavage, OPTN (optineurin), selective autophagy, Seneca Valley virus (SVV), type I interferon (IFN)
Introduction
Seneca Valley virus (SVV) was first discovered in the United States in 2002 and was isolated from a cell culture contaminant [1,2]. As an emerging pathogen of swine, it was not until 2014 that the association of etiological agents with vesicular disease in pigs was identified, and SVV has spread globally [3–8]. As an oncolytic virus, SVV is an attractive therapeutic modality for cancer treatment [9,10]. SVV is a single-stranded RNA virus belonging to the genus Senecavirus within the Picornaviridae family [1,11]. The genome of SVV comprises a 5′ untranslated region/UTR, a large open reading frame/ORF, a 3′ untranslated region, and a polyA tail of approximately 7.2 kb in length. The open reading frame encodes a polyprotein that is subsequently processed into four structural proteins (VP4, VP2, VP3, and VP1) and seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) [1]. Similar to other picornaviruses, SVV viral 3C protease (3C[pro]) possesses a conserved catalytic box with cysteine (160) and histidine (48) residues. Moreover, 3C[pro]-mediated cleavage and degradation of host proteins play an indispensable role in the viral life cycle [1,12–15]. SVV 3C[pro] degrades and cleaves numerous key innate immune molecules, including MAVS (mitochondrial antiviral signaling protein) [12], RIGI (RNA sensor RIG-I) [14], IRF3 (interferon regulatory factor 3) [16], IRF7 [16], NLRP3 (NLR family pyrin domain containing 3) [17], GSDMD (gasdermin D) [17], and NFKB/NF-κB subunit RELA/p65 [18].
Autophagy is a cellular strategy used to degrade damaged proteins and organelles and is involved in the antiviral immunity response by targeting viruses and specific viral components for degradation to eliminate invading pathogens [19–21]. To date, increased evidence has indicated that viruses have multiple strategies to evade or even exploit autophagic machinery for survival. Autophagy was initially regarded as a strictly nonselective catabolic process. Selective autophagy is modulated by a growing number of autophagy receptors that harbor a ubiquitin-binding domain (UBD) that recognizes ubiquitinated cytoplasmic cargo and a MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-interacting region (LIR), which links the cargo to autophagosomes for degradation. Xenophagy is a process in which autophagy receptors target the invading bacteria and viruses for degradation. Members of the family of SQSTM1/p62 (sequestosome 1)-like receptors (SLRs), including SQSTM1, OPTN (optineurin), NBR1 (NBR1 autophagy cargo receptor), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), and TAX1BP1 (Tax1 binding protein 1), harbor UBD and LIR domains to recognize ubiquitinated substrates in autophagosomes [22,23].
SQSTM1 and autophagy cargo receptor NBR1 were cleaved following coxsackievirus B3 (CVB3) infection, leading to impaired function of SQSTM1 and NBR1 in selective autophagy [24,25]. SQSTM1/p62 and CALCOCO2/NDP52 directly interact with the capsid protein VP1 of CVB3 to differentially regulate viral propagation [26]. Coxsackievirus 2A proteinase cleaves SQSTM1 and impairs its interaction with the viral capsid, while 3C proteinase targets CALCOCO2/NDP52 for cleavage to generate a C-terminal fragment that holds the full-length function [26]. SQSTM1/p62 was not degraded in a conventional manner during poliovirus infection but was cleaved in a manner similar to CVB3 [27]. SQSTM1/p62 is capable of binding to, and degrading avibirnavirus VP2 via autophagy, to limit viral replication [28]. NBR1 restricts viral infection by targeting the viral capsid protein and cauliflower mosaic virus (CaMV) [29]. SVV 3C[pro] targets selective autophagy SQSTM1 for cleavage [30,31], and the SQSTM1 cleavage fragments lose their capacity to mediate selective autophagy and suppress viral replication [30]. However, other autophagy receptors that regulate SVV replication remain unclear.
The autophagy receptor, OPTN, is a multifunctional protein participated in multiple cellular processes such as vesicular trafficking, autophagy, viral infection, antibacterial and antiviral responses, and innate immune responses [32,33]. OPTN facilitates the degradation of mitochondria, bacteria, and viral proteins [34–39]. The protein comprises an NFKB/NF-κB-essential molecule (NEMO)-like domain, two coiled-coil (CC) motifs, a leucine zipper (LZ), ubiquitin binding in TNIP/ABIN (TNFAIP3/A20 and inhibitor of NFKB/NF-kB) and IKBKG/NEMO (UBAN), LC3-interacting region (LIR), and zinc finger (ZnF) domain [32,40,41]. The UBAN domain links ubiquitinated cargo to autophagosomal membranes arrayed by microtubule-associated protein LC3 via its LIR domain [40]. Both endogenous and ectopically expressed OPTN can interact with itself to form homo-oligomers [42]. OPTN targets herpes simplex virus 1 (HSV-1) VP16 and glycoprotein B (gB) for degradation [38], whereas HSV-1 ICP0 protein downregulates SQSTM1 and OPTN expression at early stages of viral replication to counteract the host [43]. Phosphorylated OPTN enhanced selective autophagy of ubiquitin-coated Salmonella, TBK1 (TANK binding kinase 1) phosphorylated OPTN, increasing LC3 binding capacity and autophagic clearance of Salmonella [34]. Moreover, OPTN is required for host resistance to mycobacterial infection [39]. OPTN is involved in TBK1 activation and plays a pivotal role in antiviral type I IFN (interferon) production [33]. OPTN recruits ubiquitinated TBK1 and activates TBK1 to induce the phosphorylation of IRF3 and type I IFN production [33,44–46]. Bluetongue virus NS3 protein targets OPTN to decrease TBK1 activation and dampen IRF3 signaling [46].
In this study, we investigated how the selective autophagy receptor, OPTN, affects viral accumulation in response to SVV infection. Our results provide a novel mechanism for OPTN-mediated selective autophagy in host antiviral defense and reveal viral antagonistic actions to evade autophagic clearance.
Results
SVV infection cleaves and downregulates OPTN expression
We examined OPTN expression during SVV infection and found that OPTN was potentially cleaved after SVV infection in BHK-21 and PK-15 cells, producing two cleavage fragments using a specific OPTN antibody (Figure 1a,d), while OPTN was not cleaved in mock-infected cells (Figure 1a,d). Furthermore, OPTN was degraded during SVV infection (Figure 1a,b,d and e), suggesting that OPTN was cleaved and degraded during SVV infection. To determine whether OPTN was transcriptionally downregulated after SVV infection, qRT-PCR was used to detect the gene expression of OPTN. As shown in Figure 1c and 1f, the mRNA levels of OPTN were unaltered in response to SVV infection, suggesting that the decreased protein expression of OPTN was not related to gene expression. Overall, these results suggest that OPTN was cleaved and degraded in the SVV-infected cells.
Figure 1.
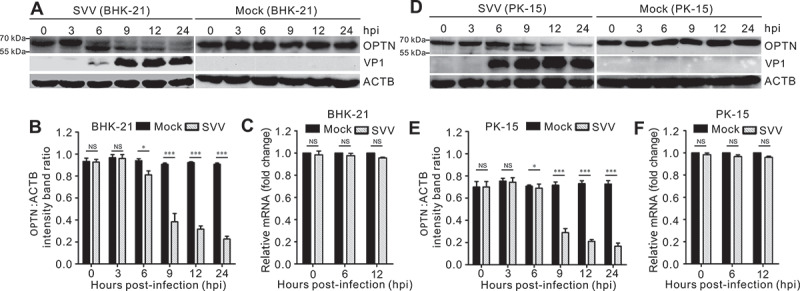
Cleavage and downregulation of OPTN after Seneca Valley virus (SVV) infection. Protein expression levels of OPTN after SVV infection. (a, d) BHK-21 cells and PK-15 cells were mock-infected with phosphate-buffered saline (PBS) or infected with SVV for 0, 3, 6, 9, and 12 h at an MOI of 5. Western blotting was performed to examine the expression of OPTN using an anti-OPTN antibody, SVV capsid protein VP1. ACTB served as a loading control. (b, e) quantification analysis of OPTN expression from figure (a, d) with ImageJ. Data are accumulated from three independent experiments. (*, P < 0.05; ***, P < 0.001; NS, not significant). (s, f) the mRNA expression of OPTN following SVV infection. BHK-21 cells and PK-15 cells were mock-infected with PBS or infected with SVV for 0, 6, and 12 h (MOI = 5). Real-time quantitative RT-PCR was used to analyze the transcriptional level of OPTN and normalized to ACTB mRNA. Statistical data from three independent infection experiments is shown (NS, not significant).
Cleavage and degradation of OPTN was mediated by SVV 3C[pro]
To investigate whether SVV proteases are responsible for OPTN cleavage, co-transfection of human influenza hemagglutinin (HA)-OPTN with previously constructed SVV protein-expressing plasmids were utilized [31]. Only 3C[pro] resulted in the production of cleavage fragments, similar to those observed during SVV infection (Figure 2a). We also found that 3C[pro] from other members of the Picornaviridae family downregulated OPTN expression, including EMCV, FMDV, CVB3, HRV, and EV71 (Figure 2b). As shown in Figure 2b, only SVV 3C[pro] cleaved OPTN, indicating the specificity of OPTN cleavage by 3C[pro] in different picornaviruses. SVV 3C[pro] directly cleaves host proteins based on its protease activity [12,13,17]. We cotransfected HA-OPTN with green fluorescent protein (GFP)-3C plasmids with mutated protease activity residues, including GFP-3CH48A, GFP-3CC160A, and GFP-3C[DM] H48A,C160A from our previous study [47]. The 3C[pro] mutant lacking conserved protease activity failed to mediate the cleavage of OPTN (Figure 2c). These results indicate that the cleavage of OPTN was mediated by the protease sites (3CH48 and 3CC160) of 3C[pro]. In the presence of the pan-caspase inhibitor Z-VAD-FMK, OPTN degradation was restored, but caspase inhibition did not prevent OPTN cleavage (Figure 2d, e), indicating that OPTN cleavage was caspase-independent.
Figure 2.
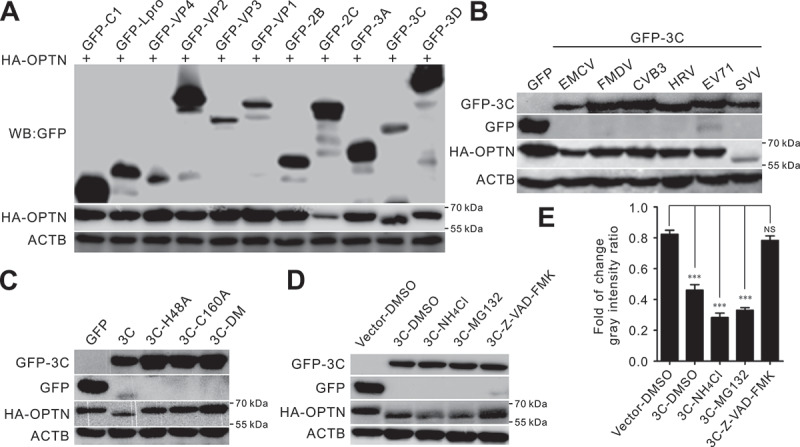
Seneca Valley virus (SVV) 3C protease (3C[pro]) targets OPTN for cleavage and degradation via its protease activity. (a) BHK-21 cells were cotransfected with plasmids encoding OPTN and viral proteins for 24 h, then western blotting was used to analyze OPTN expression with antibodies against green fluorescent protein (GFP) and HA. ACTB served as a loading control. (b) BHK-21 cells were cotransfected GFP-tagged 3C[pro] of EMCV, FMDV, CVB3, HRV, EV71, and SVV with human influenza hemagglutinin (HA)-OPTN. Cell lysates were collected at 24 h post-transfection (hpt), and analyzed by immunoblotting using anti-GFP and anti-HA antibodies. ACTB served as a loading control. (c) BHK-21 cells were cotransfected with plasmids encoding GFP-tagged 3C[pro], 3CH48A, 3CC160A, and 3C-DM H48A, C160A with HA-OPTN. At 24 hpt, cell samples were subjected to immunoblot analysis with antibodies against GFP and HA. ACTB served as a loading control. (d) BHK-21 cells were cotransfected with a GFP empty vector or GFP-3C with HA-OPTN. Dimethyl sulfoxide (DMSO), lysosome inhibitor (NH4Cl) (10 mM), proteasome inhibitor (MG132) (10 µM), and caspase inhibitor (Z-VAD-FMK) (50 µM) were added to cells at 12 hpt. Samples were collected at 24 h post-infection (hpi) and subjected to immunoblot analysis with antibodies against GFP and HA. ACTB served as a loading control. (e) the quantification relative to ACTB of three independent experiments of (D) (***, P < 0.001; NS, not significant). The gray intensity ratio was examined using ImageJ.
SVV 3C[pro] cleaves OPTN at residue Q513
To characterize the cleavage site of OPTN, we constructed truncation and point mutation plasmids based on the OPTN structure and molecular weight of the cleaved OPTN, including HA-OPTN (1–438), HA-OPTN (224–574), HA-OPTN (141–574), and HA-OPTN (1–483) (Figure 3a). Picornavirus 3C[pro] preferentially target glutamine-glycine (Q-G) or glutamic acid-glutamine (E-Q) pairs in cellular proteins for cleavage [48]. As shown in Figure 3b, the potential cleavage site of OPTN was located at residues 224–574. HA-OPTN (224–574) and HA-OPTN (141–574) were cleaved by GFP-3C, produced two bands. Two (E-Q) pairs within residues 224–574 were replaced with alanine (A). However, we found that HA-OPTNE368A, Q369A and HA-OPTNE483A,Q484A mutants were still cleavable after co-transfection with GFP-3C (Figure 3c). Next, we replaced all glutamic acid (E) and glutamine (Q) residues within residues 483–574 with alanine (A) of OPTN. Two triple mutant HA-OPTNQ488A,E495A, E500A and HA-OPTNQ507A,E511A,Q513A, two quadruple mutant were HA-OPTNQ525A,Q526A,Q531A,E535A and HA-OPTNQ541A,Q542A,Q543A,Q545A, one double mutant HA-OPTNE557A,Q566A were constructed. The results showed that the specific cleavage manner for SVV 3C[pro] disappeared in OPTNQ507A,E511A,Q513A, while all the other OPTN mutants were still cleaved after co-transfection with SVV 3C[pro] (Figure 3d), indicating that Q507, E511, and Q513 were potential targets of SVV 3C[pro] for OPTN cleavage. Therefore, we constructed three single-point mutants within Q507, E511, and Q513 of the OPTN. We found that the cleavage product disappeared in HA-OPTNQ513A cotransfected cells with 3C[pro] (Figure 3e), indicating that the Q513A mutation in OPTN was resistant to SVV 3C[pro] cleavage. GFP-3C exhibited diffuse distribution in the cytoplasm and nucleus, HA-OPTN and HA-OPTN (1–513) displayed puncta structures in the cytoplasm, HA-OPTN (514–574) was expressed in the cytoplasm and nucleus (Figure 3f). In co-transfected BHK-21 cells, GFP-3C formed punctua aggregates with HA-OPTN in the cytoplasm (Figure 3f), whereas GFP-3C lost the punctua structures with OPTN truncations. These results suggest that Q513 was a cleavage site on OPTN after SVV 3C[pro] expression.
Figure 3.
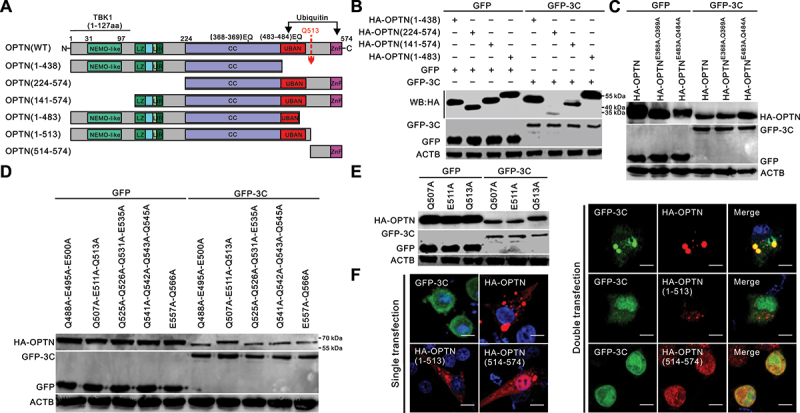
OPTN is cleaved at residue Q513 by Seneca Valley virus (SVV) 3C protease (3C[pro]). (a) schematic diagram of the structural domains and truncated OPTN mutants. IKBKG/NEMO, inhibitor of nuclear factor-kappa B kinase regulator subunit gamma; TBK1, TANK-binding kinase 1; LIR, LC3-interacting region; CC, coiled-coil domain; UBAN: ubiquitin binding in TNIP/ABIN (TNFAIP3/A20 and inhibitor of NFKB/NF-KB) and IKBKG/NEMO; ZnF, zinc finger. (b) BHK-21 cells were cotransfected with human influenza hemagglutinin (HA)-OPTN and its truncation constructs with green fluorescent protein (GFP)-3C for 24 h. Samples were analyzed by western blotting with antibodies against GFP, HA, and ACTB. (c) BHK-21 cells were cotransfected with GFP-3C and HA-OPTN (E368A-Q369A) and HA-OPTN (E483A-Q484A) for 24 h. Samples were analyzed by western blotting with antibodies against GFP, HA, and ACTB. (d) BHK-21 cells were cotransfected with GFP-3C with HA-OPTNQ488A,E495A,E500A, HA-OPTNQ507A,E511A,Q513A, HA-OPTNQ525A,Q526A,Q531A,E535A, HA-OPTNQ541A,Q542A,Q543A,E545A, and HA-OPTNE557A,Q566A for 24 h. Samples were analyzed by western blotting with antibodies against GFP, HA, and ACTB. (e) BHK-21 cells were cotransfected GFP-3C with HA-OPTNQ507A, HA-OPTNE511A, and HA-OPTNQ513A for 24 h. Samples were analyzed by western blotting with antibodies against GFP, HA, and ACTB. (f) BHK-21 cells were transfected or cotransfected GFP-3C, and HA-OPTN cleavage fragments with HA-OPTN. The cells were stained with antibodies to HA and examined under confocal microscopy. Samples were stained with HA antibody (red) and 4’,6-diamidino-2-phenylindole (DAPI) (blue), then examined by confocal microscopy. Scale bar: 10 μm.
OPTN targets VP1 for degradation
Next, we evaluated the colocalization of OPTN with viral proteins using confocal microscopy. The results indicated that GFP-tagged leader proteinase (Lpro), VP3, VP1, 2 B, 3C, and 3D colocalized with HA-tagged OPTN (Figure 4a) and exhibited puncta distribution in the cytoplasm after cotransfection. Co-immunoprecipitation (co-IP) assays were performed to assess the interaction between OPTN and the viral proteins (Figure 4b). As shown in Figure 4b, HA-OPTN strongly interacted with Lpro, VP3, VP1, and 3D and weakly interacted with VP4, 2C, and 3C. We further analyzed the interaction between OPTN and viral capsid proteins using GFP beads (Figure 4c). Consistent with the results shown in Figure 4a, OPTN interacted only with VP3 and VP1 (Figure 4C). Western blotting indicated that HA-OPTN degraded VP1, whereas the cleaved fragments lost their degradation ability (Figure 4d). The cleaved OPTN (1–513) fragment still interacted and colocalized with GFP-VP1 (Figure 4e,f). BHK-21 cells were infected with SVV for 12 h with cycloheximide (CHX) to inhibit protein synthesis, bafilomycin A1 (Baf. A1), chloroquine (CQ), 3-methyladenine (3-MA) to inhibit autophagy, proteasome inhibitor (MG132), lysosome inhibitor (NH4Cl), caspase inhibitor (Z-VAD-FMK), or dimethyl sulfoxide (DMSO) as a control. Compared to the MG132, NH4Cl, and Z-VAD-FMK treatment groups, the VP1 levels were significantly higher in the autophagy inhibitor Baf. A1-, CQ-, and 3-MA-treated groups (Figure 4g,f). These results showed that the degradation of SVV VP1 is autophagy-dependent. We further explored degradation in vitro. In GFP-VP1, HA-OPTN, or HA vector co-transfected cells, compared with DMSO treatment, the protein abundance of VP1 was slightly restored after treatment with an autophagy inhibitor (Baf. A1, CQ, 3-MA), whereas treatment with MG132, NH4Cl, and Z-VAD-FMK could not recover VP1 expression levels (Figure 4i,j). In autophagy inducer rapamycin (Rapa)-treated cells, the levels of VP1 were significantly reduced (Figure 4i,j). Collectively, these results demonstrated that VP1 was degraded in an autophagy-dependent manner, and OPTN targeted the VP1 protein for degradation.
Figure 4.
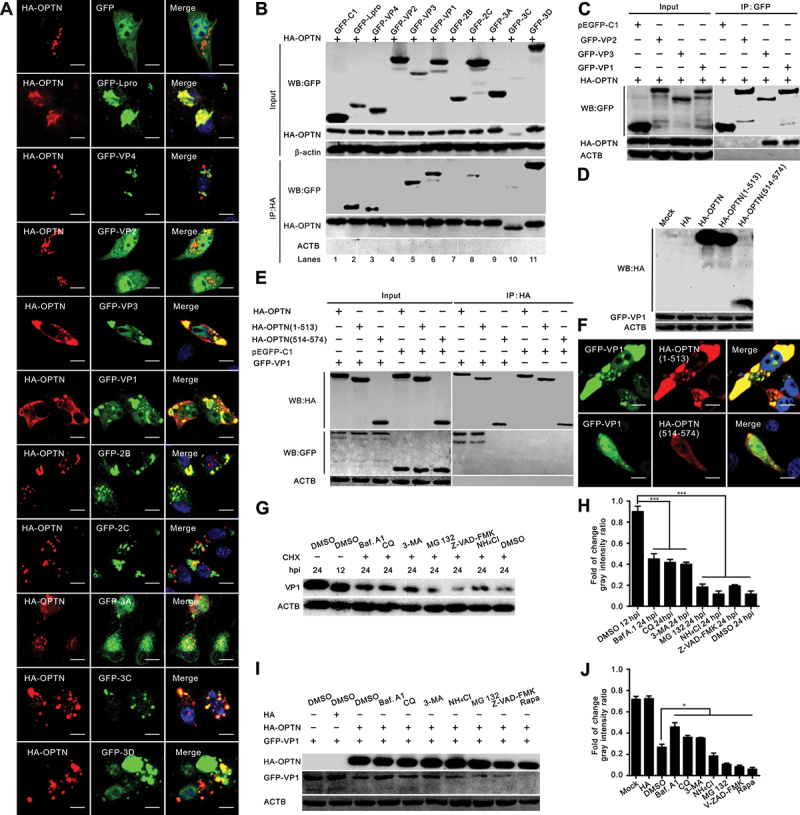
OPTN targets VP1 for degradation. (a) a series of green fluorescent protein (GFP)-tagged Seneca Valley virus (SVV) protein-expressing plasmids were cotransfected with human influenza hemagglutinin (HA)-OPTN in BHK-21 cells for 24 h. The cells were stained with antibodies to HA and examined under confocal microscopy. Samples were stained with HA antibody (red) and 4”,6-diamidino-2-phenylindole (DAPI) (blue), then examined by confocal microscopy. Scale bar: 10 μm. (b) a series of GFP-tagged SVV protein-expressing plasmids were cotransfected with HA-OPTN in BHK-21 cells for 24 h. Cell samples were prepared for co-IP analysis. (c) GFP-fused SVV capsid protein-expressing plasmids were cotransfected with HA-OPTN in BHK-21 cells for 24 h. Cell samples were subjected to co-IP analysis. (d) BHK-21 cells were cotransfected with GFP-VP1 with HA-OPTN constructs for 24 h. Cell samples were prepared for western blot analysis. (e) BHK-21 cells were cotransfected with GFP-VP1 and HA-OPTN constructs for 24 h. Cell samples were subjected to co-IP analysis. (f) BHK-21 cells were cotransfected with GFP-VP1 and HA-OPTN constructs for 24 h. The cells were stained with antibodies to HA and examined under confocal microscopy. Samples were stained with HA antibody (red) and 4”,6-diamidino-2-phenylindole (DAPI) (blue), then examined by confocal microscopy. Scale bar: 10 μm. (g) BHK-21 cells were infected with SVV (MOI = 0.5). At 12 h post-infection (hpi), cells were treated with cycloheximide (CHX) (100 µg/ml) and dimethyl sulfoxide (DMSO), bafilomycin A1 (Baf A1; 200 nM), chloroquine (CQ; 40 μM), 3-methyladenine (3-MA; 25 mM), proteasome inhibitor (MG132; 10 μM), lysosome inhibitor (NH4Cl; 10 mM), and caspase inhibitor (Z-VAD-FMK) (50 μM) for 12 h. Cell samples were prepared for western blot analysis at 24 hpi. (h) the quantification relative to ACTB of three independent experiments of (G) (***, P < 0.001; NS, not significant). The gray intensity ratio was examined using ImageJ. (i) BHK-21 cells were cotransfected GFP-VP1 with HA-OPTN. At 16 hpi, cells were treated with DMSO, Baf. A1 (200 nM), CQ (40 μM), 3-MA (25 mM), MG132 (10 μM), and NH4Cl (10 mM), Z-VAD-FMK (50 μM), and rapamycin (50 nM) for 12 h. Cell samples were prepared for western blot analysis at 28 hpt. (j) the quantification relative to ACTB of three independent experiments of (I) (***, P < 0.001; NS, not significant). The gray intensity ratio was examined using ImageJ.
Cleaved OPTN products attenuated its antiviral activity
We examined the effects of the autophagy receptor OPTN on SVV propagation using siRNA-mediated knockdown and OPTN overexpression. OPTN was considerably downregulated after transfection with specific siRNAs (Figure 5a), and silencing OPTN expression promoted SVV VP1 production and enhanced virus titers (Figure 5c,d). Knockdown and overexpression of OPTN had no obvious effect on cell viability (Figure 5b). In contrast to the RNAi-mediated knockdown, overexpression of OPTN inhibited SVV replication (Figure 5e,f). We also assessed the potential effects of the OPTN cleavage fragments and OPTN cleavage site mutants on viral replication. BHK-21 cells were transfected with HA-OPTN, HA-OPTN (1–513), HA-OPTN (514–574), HA-OPTNQ513A, or empty HA vectors, followed by SVV infection for 12 h (Figure 5g). Compared with the full-length OPTN, the inhibitory effect of OPTN (1–513) against SVV infection was markedly reduced, and the cleaved OPTN (514–574) fragments and cleavage site mutant HA-OPTNQ513 lost their ability to suppress SVV propagation (Figure 5g-i). Collectively, these results indicate that 3C[pro]-mediated cleavage of OPTN products dampened their antiviral activity.
Figure 5.
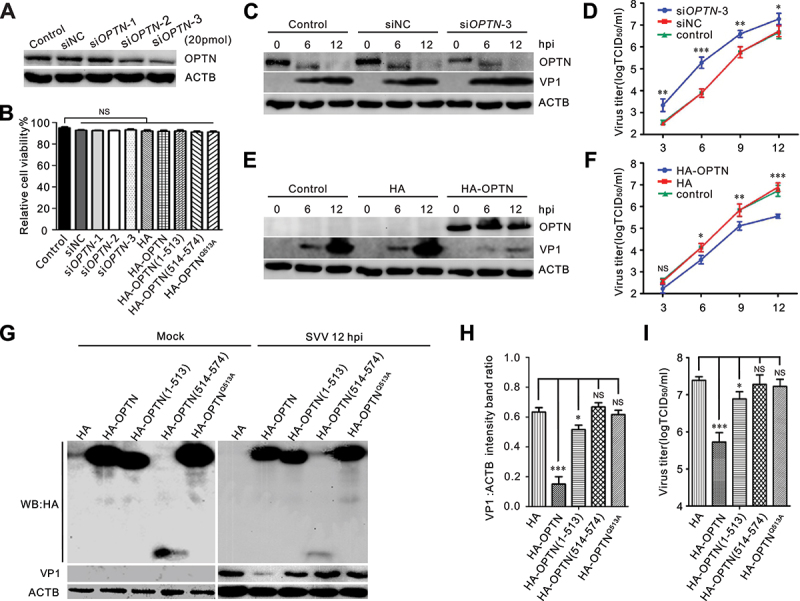
OPTN inhibits Seneca Valley virus (SVV) replication. (A) effects of knockdown of OPTN. BHK-21 cells were transiently transfected with siOPTN for 36 h. Western blotting was performed to examine the expression of OPTN using an anti-OPTN antibody. (B) Cell viability of BHK-21 was analyzed after transfection with siRnas and human influenza hemagglutinin (HA)-OPTN constructs via CCK-8 assay. (C, E) BHK-21 cells were transfected with siRNA or HA-OPTN for 36 h, then infected with SVV (MOI = 0.5) for 6 h and 12 h. Cell lysates were subjected to western blot analysis with antibodies to OPTN, HA, VP1, and ACTB. (D, F) growth analysis of siRnas or HA-OPTN transfected BHK-21 cells after SVV infection. BHK-21 cells in 12-well plates were infected with SVV (MOI = 0.5). At each indicated time point, the total viruses were titrated with the TCID50 assay. (G, I) influence of cleaved OPTN for SVV replication. BHK-21 cells were transfected with HA empty vector or HA-OPTN constructs for 36 h, then infected with SVV (MOI = 0.5) for 12 h. Samples were subjected to western blot analysis and TCID50 assay, respectively. (H) quantification displayed with graphs representing VP1 normalized against ACTB using ImageJ. Error bars indicate mean ± SD of three independent tests (***, P < 0.001; NS, not significant).
OPTN cleavage product impairs its function in selective autophagy
We examined the interaction of 3C[pro]-mediated OPTN cleavage products with ubiquitin and LC3 and found that both OPTN and N-terminal OPTN formed aggregates, and the C-terminal OPTN lost the ability to form aggregates (Figure 6a,b). Punctate colocalization of full-length OPTN with ubiquitin or LC3 was rarely observed for C-terminal OPTN (Figure 6c-f), and an impaired colocalization ratio was observed for N-terminal OPTN (Figure 6c-f). Co-IP results indicated that intact OPTN was able to bind to both ubiquitinated proteins and LC3, the OPTN (1–513) binding ability to ubiquitinated proteins, and LC3 was maintained, but attenuated (Figure 6g,h). OPTN (514–574), which lacked both LIR and UBAN domains, lost its ability to bind to ubiquitinated proteins and LC3 (Figure 6g,h). Additionally, we found that HA-ubiquitin, RFP-LC3, and MYC-OPTN colocalized with GFP-VP1 in the cytoplasm (Figure 6I,K). Compared with full-length OPTN, the colocalization ratios for OPTN (1–513) were markedly reduced (Figure 6j,l). The OPTN UBAN domain is essential for self-oligomerization in a ubiquitin-independent manner [42]. OPTN (1–513) maintained its ability to bind to full-length OPTN despite weakening, likely through its UBAN domain (Figure 6m). OPTN (514–574) failed to interact with WT OPTN (Figure 6M). In summary, the 3C[pro]-mediated cleavage of OPTN attenuated its ability to induce selective autophagy.
Figure 6.
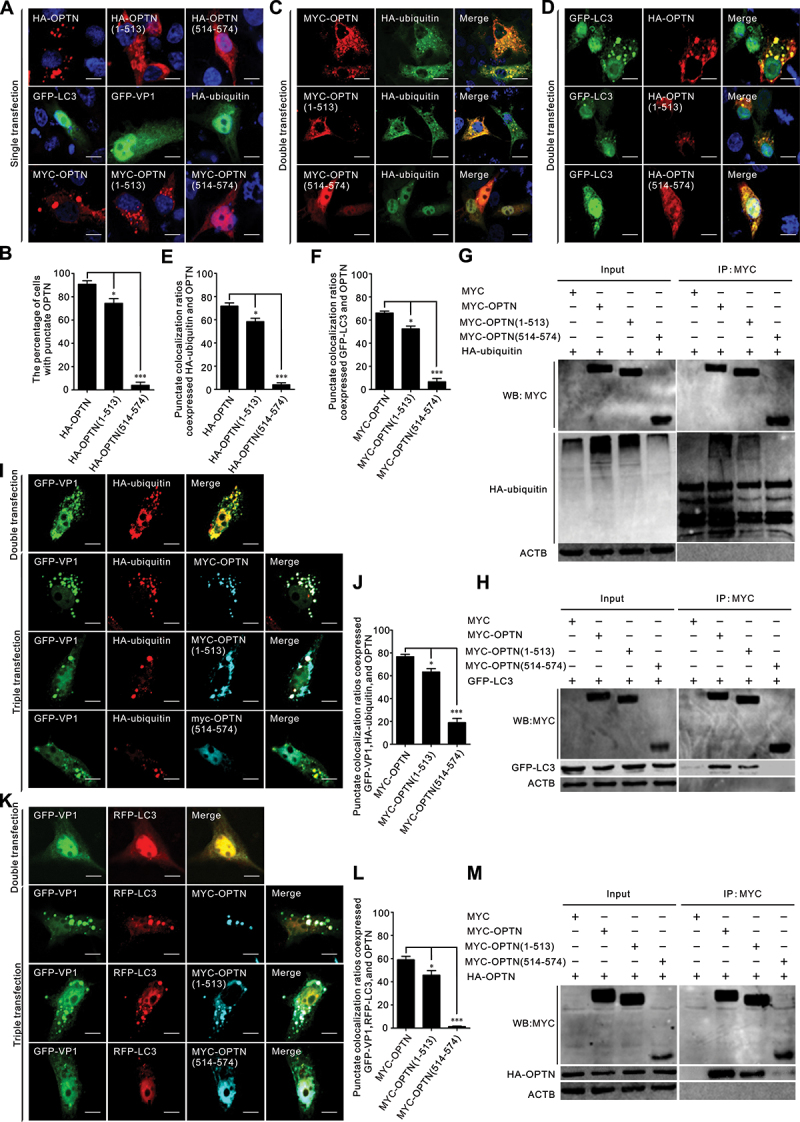
Binding activities impaired for OPTN cleavage fragments. (a, c, d) BHK-21 cells were transfected with indicated plasmids for 24 h. Samples were stained with human influenza hemagglutinin (HA) antibodies, MYC antibodies, and 4’,6-diamidino-2-phenylindole (DAPI), then examined by confocal microscopy. Scale bar: 10 μm. (b, e, f) the percentage of cells with a punctate structure was counted. Statistical data from three independent infection tests is shown (NS, not significant). (g-h) interaction of OPTN cleavage fragments with ubiquitin and LC3. BHK-21 cells were cotransfected with OPTN cleavage constructs and HA-ubiquitin (g), or green fluorescent protein (GFP)-LC3 (h) for 24 h. Cell samples were prepared for co-immunoprecipitation (co-IP) analysis. (i-l) colocalization of OPTN cleavage fragments with GFP-VP1, HA-ubiquitin, and RFP-LC3, respectively. BHK-21 cells were cotransfected with the indicated plasmids for 24 h. Samples were stained with HA antibody, MYC antibody, and DAPI (blue), then examined by confocal microscopy. Scale bar: 10 μm. The percentage of cells with punctate colocalization over the total number of cotransfected cells is counted. Statistical data from three independent infection tests is shown (NS, not significant). (m) interaction of OPTN cleavage fragments with wide-type OPTN. BHK-21 cells were cotransfected with OPTN cleavage constructs and full-length OPTN for 24 h. Cell samples were prepared for western blot analysis.
Cleaved OPTN dampened TBK1-IRF3 activation and IFNB/IFN-β production
We then investigated the role of OPTN cleavage products in IFN response. We found that SVV infection dramatically inhibited TBK1 and IRF3 phosphorylation (Figure 7a). Knockdown of OPTN impaired TBK1 activation and IRF3 phosphorylation (Figure 7b), and overexpression of OPTN promoted TBK1-IRF3 phosphorylation (Figure 7c,d). OPTN (1–513) attenuated the capacity to induce TBK1 activation and IRF3 phosphorylation (Figure 7d). To explore whether OPTN inhibited SVV replication through the innate immune pathway, we examined IFN expression using qRT-PCR and enzyme-linked immunosorbent assay and found that OPTN overexpression enhanced the mRNA and protein levels of IFN (Figure 7e,f). Consistently, OPTN (1–513) diminished its ability to induce IFN production (Figure 7e,f), OPTN (514–574) completely lost its function in IFN signaling (Figure 7d-f). To identify whether cleaved OPTN interacted with TBK1, co-IP and immunofluorescence staining indicated that OPTN (1–513) sustained its interaction and colocalization with TBK1 (Figure 7g,h). The level of OPTN was decreased when treated with the TBK1 inhibitor BX795, and the AKT signaling pathways were inactivated (Figure 7l). Previous studies have indicated that B×795treatment reduced OPTN expression, and B×795inhibited HSV infection by inhibiting AKT phosphorylation [38,49,50]. Taken together, these results reveal that OPTN promotes IFN production via the TBK1-IRF3 pathway, resulting in facilitation of the host antiviral innate immune response, thereby inhibiting SVV replication. To resist these effects, SVV 3C[pro]-cleaved OPTN antagonizes OPTN-mediated TBK1-IRF3 signaling and IFNB production.
Figure 7.
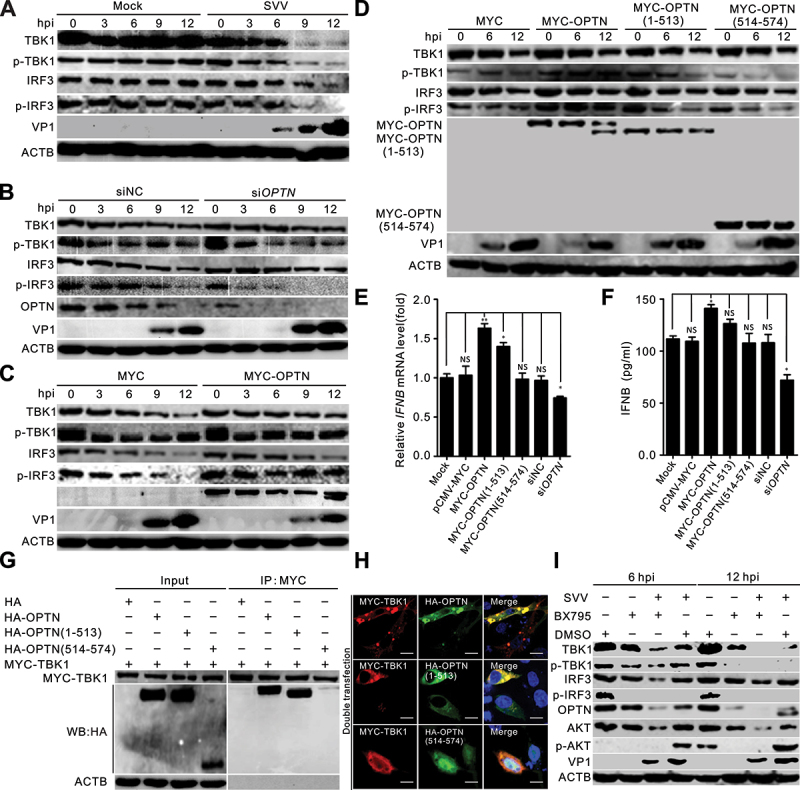
Cleaved OPTN mediating the TBK1-IRF3 signaling pathway was dampened. (a) BHK-21 cells were mock-infected with phosphate-buffered saline (PBS) or infected with Seneca Valley virus (SVV) for 0, 3, 6, 9, and 12 h (MOI = 5). Western blot was performed to examine the expression of TBK1, p-TBK1, IRF3, p-IRF3, SVV-VP1, and ACTB as an internal control. (b) BHK-21 cells were transfected with OPTN siRNA or siNC for 36 h, followed by SVV infection (MOI = 5). Western blot was performed to examine the expression of TBK1, p-TBK1, IRF3, p-IRF3, OPTN, SVV-VP1, and ACTB as an internal control. (c-d) BHK-21 cells were transfected with MYC-OPTN plasmids for 36 h, followed by SVV infection (MOI = 5). Western blot was performed to examine the expression of TBK1, p-TBK1, IRF3, p-IRF3, SVV-VP1, and ACTB as an internal control. (e) BHK-21 cells were transfected with MYC-OPTN plasmids, specific siRnas targeting OPTN, the IFNB mRNA levels were detected by quantitative RT-PCR and normalized to ACTB mRNA. Statistical data from three independent infection experiments is shown (NS, not significant). (f) the HEK293T cells were transfected with MYC-OPTN plasmids and specific siRnas targeting OPTN after 10 μg/ml poly (I:C) treatment, respectively. Concentrations of IFNB in the cell culture media were measured by enzyme-linked immunosorbent assay (ELISA) at 24 h post-transfection (hpt). (g) interaction of OPTN with TBK1. BHK-21 cells were cotransfected with OPTN cleavage constructs and full-length OPTN with MYC-TBK1 for 24 h. Cell samples were prepared for co-immunoprecipitation (co-IP) analysis. (h) BHK-21 cells were cotransfected with OPTN cleavage constructs and full-length OPTN with MYC-TBK1 for 24 h. The cells were stained with antibodies to HA and MYC, and examined under confocal microscopy. Samples were stained with HA antibody (green), MYC antibody (red) and 4’,6-diamidino-2-phenylindole (DAPI) (blue), then examined by confocal microscopy. Scale bar: 10 μm. (i) BHK-21 cells were pretreated with B×795(10 mM) or dimethyl sulfoxide (DMSO) for 1 h. After SVV (MOI = 5) adsorption for 1 h, BHK-21 cells were overlaid with new medium containing B×795(10 mM) or DMSO. Western blot was performed to examine the expression of TBK1, p-TBK1, IRF3, p-IRF3, AKT, p-AKT, OPTN, SVV-VP1, and ACTB as an internal control.
Discussion
Autophagy is a fundamental catabolic process that maintains cellular homeostasis by degrading damaged cytoplasmic and detrimental components in lysosomes [51]. Autophagy was originally considered a nonselective process. However, accumulating evidence has indicated its selective ability. Selective autophagy via autophagy receptors binds to cytoplasmic contents and LC3, thereby linking the cargo to autophagosomal membranes [52]. Members of the autophagy receptors function in autophagic host defense mechanisms and target many invading microbes, including Sindbis virus, cauliflower mosaic virus (CaMV), and CVB3 [29,53]. Accumulating evidence has demonstrated the effects of autophagy receptors and autophagic signaling pathways on the replication of Picornaviridae family members [24,31,54–56]. We previously revealed that SQSTM1/p62 is cleaved following SVV infection and that SVV infection activates the AKT-AMPK-MAPK-MTOR pathway for autophagy [31]. The present study investigated the interaction between the autophagy receptor OPTN and SVV. In this study, we showed that OPTN is also cleaved after SVV infection, and that cleavage is catalyzed by viral 3C[pro]. 3C[pro]-mediated OPTN cleavage products impaired its ability to induce selective autophagy and VP1 degradation and reduced its ability to inhibit SVV replication. Our results suggest a function for autophagy in anti-SVV infection: OPTN-mediated autophagic viral capsid protein clearance enhances host survival, and the virus adapts autophagic processes for survival.
CVB3 2A[pro] and 3C[pro] cleave SQSTM1/p62, impairing its capacity to associate with the viral capsid, whereas 3C[pro] cleaves CALCOCO2/NDP52 to generate cleavage fragments that retain the function of full-length CALCOCO2/NDP52 [24,26]. NBR1 is also cleaved following CVB3 infection, and cleavage is mediated by viral 2A[pro] and 3C[pro] at two different sites [25]. SQSTM1/p62 and NBR1 C-terminal cleavage products exhibit dominant-negative effects, and overexpression of the C-terminal cleavage fragments of NBR1 induces an accumulation of native SQSTM1/p62 [25]. SQSTM1/p62 and NBR1 are mutually regulated following CVB3 infection, and loss of function of one autophagy receptor after CVB3 infection cannot be compensated by the other [25]. SQSTM1/p62 and NBR1 interact and form oligomers that can function independently [57]. SQSTM1/p62 and CALCOCO2/NDP52 mediate Shigella and Listeria through different pathways of selective autophagy [58]. NBR1 is also recruited to Shigella and its reduction strikingly diminishes the recruitment of SQSTM1/p62 and CALCOCO2/NDP52 [58]. Furthermore, CVB3 2A[pro] cleaves autophagic receptor protein TAX1BP1, which separates the C-terminal UBD domain, causing disrupted ubiquitin association and impaired selective autophagy [59]. SQSTM1/p62 is also cleaved following poliovirus (PV) infection [27]. Similarly, SVV infection cleaved SQSTM1/p62 [31,54], and 3C[pro] targeted glutamic acid 355, glutamine 392, and glutamine 395 for cleavage [54]. OPTN targets Salmonella, Mycobacterium tuberculosis, and the HSV-1 VP16 and gB proteins to promote autophagy [34,39]. SQSTM1/p62, OPTN, and CALCOCO2/NDP52 are mutually dependent in promoting Salmonella xenophagy [34,60,61]. There may be a fine-tuned regulatory mechanism among the different autophagy receptors. Overall, these studies indicate that more than one autophagy adaptor is involved in counteracting these invading microbes, and some of them work together against intruding pathogens. Future work will aim to elucidate the roles of other autophagy receptors during SVV infection.
OPTN selectively targets the HSV-1 VP16 protein and fusion glycoprotein (gB) for autophagy degradation [38]. To counteract this, the ICP0 protein of HSV-1 degrades OPTN expression during the early stages of infection [43]. In the present study, we revealed that OPTN was cleaved and degraded during SVV infection of BHK-21 and PK-15 cells (Figure 1a, d). Further study indicated that the proteolytic cleavage and degradation of OPTN were mediated by viral 3C[pro] through its catalytic activity (Figure 2a, c). Additionally, we found that OPTN was downregulated by some 3C[pro] of picornaviruses, whereas only SVV 3C[pro] preferentially cleaved OPTN (Figure 2b). SVV 3C[pro] cleaves OPTN at glutamine 513, generating an N-terminal fragment that contains the LIR motif and UBAN domain and a short C-terminal ZnF domain (Figure 3a, e). The cleavage pattern of OPTN differs from that of other adaptors. For example, CVB3 2A cleaves SQSTM1/p62, generating a C-terminal LIR domain with a UBA domain; 3C cleaves CALCOCO2/NDP52, generating a C-terminal LIR motif with a UBA domain; and 2A and 3C cleave NBR1 at two distinct sites that generate a C-terminal LIR motif with a UBA domain, a middle LIR motif, and an N-terminal ZnF domain [24–26]. Therefore, the SVV 3C[pro]-medicated OPTN N-terminal cleavage fragment likely retains the function of the full-length OPTN. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) NSP5 targets SQSTM1/p62 for separation of the UBA domain from the LIR motif and disrupts the synergistic effect of the functional domains, then abolishes the ability to induce selective autophagic degradation of viral membrane proteins [62]. Although the cleavage sites in SQSTM1/p62 are different, cleavage of SQSTM1/p62 disrupts its function and thus promotes evasion of cellular autophagic degradation for viral survival.
We observed that Lpro, VP3, VP1, 2B, 3C, and 3D colocalized with HA-OPTN (Figure 4a), confirming the interaction between Lpro, VP1, VP3, 3D, and HA-OPTN in the co-IP assay (Figure 4b,c). OPTN degraded SVV VP1 in an autophagic manner (Figure 4d-j). Similarly, NBR1 targets CaMV capsid proteins to induce autophagy-dependent degradation [29]. The SARS-CoV-2 protease NSP5 cleaved SQSTM1/p62 to evade the autophagic degradation of viral M protein [62]. CVB3 2A[pro] mediated C-terminal SQSTM1/p62 cleavage fragment that contains both LIR and UBA domains maintained its binding capacity to LC3 but lost its interaction with ubiquitinated protein, and punctate colocalization between C-terminal SQSTM1/p62 and LC3 and ubiquitin was greatly decreased [24]. We found that the OPTN N-terminal cleavage product contains both LIR and UBAN domains and retained its ability to bind to ubiquitin and LC3 (Figure 6g-h), but the punctate colocalization ratios were markedly reduced (Figure 6b-f). Phosphorylated OPTN localizes to ubiquitin- and LC3B-positive Salmonella [34]. OPTN strongly colocalized with HSV-1 VP16 and LAMP1 (lysosomal associated membrane protein 1) [38]. Our results showed that OPTN strongly colocalized with SVV VP1, ubiquitin, and LC3 (Figure 6i,k), although the N-terminal OPTN cleavage product had a lower colocalization ratio (Figure 6J,L). OPTN restricts HSV-1 and HSV-2 infection, and OPTN-knockout mice are susceptible to herpesvirus infection [35,38]. In OPTN-deficient cells, intracellular replicating Salmonella, Mycobacterium marinum (Mm), and Listeria monocytogenes are increased [39,61,63]. Overexpression of OPTN by mRNA injection enhances GFP-LC3 puncta in association with Mm and inhibits Mm infection [39]. In this study, we observed that OPTN-depletion markedly enhanced SVV infection (Figure 5).
TBK1 functions in autophagic clearance of invading bacteria [64]. TBK1 directly phosphorylates OPTN after activation by invaders, thereby strengthening the interaction of OPTN with ATG8, which allows the autophagic machinery to recruit invading bacteria for elimination by lysosomes [64]. CALCOCO2/NDP52 recognizes Salmonella enterica coated with ubiquitin and recruits TBK1; downregulation of CALCOCO2/NDP52 and TBK1 promotes bacterial proliferation [64,65]. SVV infection barely induced IRF3 phosphorylation and IRF3 translocation to inhibit antiviral type I IFNs production [12]. This indicated an unknown mechanism for blocking type I IFNs signaling upstream of IRF3 activation. Downregulation of OPTN expression restricted phosphorylation of IRF3 and production of IFNB (Figure 7b,e,f), while overexpression of OPTN activated TBK1 and IRF3 phosphorylation, promoting IFNB production (Figure 7c-f). Previous studies have indicated that OPTN deficiency in vivo negatively regulates IFNB [33,46,66]. In contrast, Sendai virus (SeV) infection induces OPTN expression, which negatively regulates the induction of IFNB during SeV infection [67]. The apparent differences among these reports are possibly due to virus-specific and cell type discrepancies, but with the use of OPTN-deficient- or mutant mice, it has been demonstrated that OPTN is a positive inducer of IFNB responses via TBK1 activation [46,61,66,68]. The polyubiquitin binding-defective mutant OPTND477N/D477N mice markedly reduced phosphorylation of IRF3 and the production of IFNB mRNA and secretion [45], and ubiquitin-binding domain deletion in OPTN470T mice dampened TLR4-induced IFNB production, indicating that OPTN is a positive regulator of the IRF3 pathway [68]. The limitation in our study is without using OPTN-deficient mice or piglets to assess the roles of OPTN in SVV infection and IFNB production. CVB3 2A[pro] cleaves SQSTM1/p62, which disrupts selective autophagy and loses the ability to activate NFKB/NF-κB signaling [24]. The NS3 protein of the bluetongue virus (BTV) targets OPTN to inhibit IRF3 signaling [46]. Many other upstream factors of type I IFN signaling, including RIGI-IFIH1/MDA5, and MAVS, are targeted by SVV 3C[pro] [12,16]. Similar to previous studies, we found that SVV 3C[pro] cleaves OPTN, resulting in diminished selective autophagy, impaired IRF3 activation, and IFNB production (Figure 7d, f). The interaction between ubiquitin chains and OPTN is necessary for OPTN-TBK1 complex activation, leading to IRF3 phosphorylation and IFNB production [45,69]. The zinc finger domain (ZnF) is the ubiquitin binding site for CALCOCO2/NDP52 and TAX1BP1 [64,70]. CALCOCO2/NDP52 depends on its ubiquitin-binding ZnF domain to detect ubiquitin-coated Salmonella enterica and marks invading bacteria with an additional layer of host protein [64,71]. OPTN contains two ubiquitin-binding domains, the UBAN domain and ZnF domain [72,73]. Binding of the ZnF to ubiquitin is consistent with our study, SVV 3C[pro]-mediated cleavage of OPTN separated its ZnF domain, which lost one ubiquitin-interacting region, significantly reducing OPTN-mediated selective autophagy and IFNB production. This likely indicates that OPTN-mediated selective autophagy during SVV infection has a mechanism similar to that of CALCOCO2/NDP52-mediate autophagy in Salmonella enterica. TBK1 phosphorylates autophagy-relevant sites, including the ubiquitin- and LC3-binding domains of OPTN [74]. Constitutive interaction of TBK1 with OPTN and the ability of OPTN to bind to ubiquitin chains are essential for TBK1 recruitment and kinase activation on mitochondria [74]. Our studies demonstrated that OPTN lacking the ubiquitin-binding domain reduced TBK1 activation (Figure 7d), it indicated that OPTN has a dual role in IFN signaling and selective autophagy. Recent studies have indicated that 3C[pro]-mediated SQSTM1/p62 cleavage products completely lose their capacity to mediate selective autophagy and restrict SVV replication [30].
In summary, the present study proposes a model in which OPTN targets SVV VP1 for degradation to inhibit viral propagation. SVV 3C[pro] cleaves OPTN to impair its functions in selective autophagy, TBK1-IRF3 signaling, and type I IFN production (Figure 8). These findings provide further insights into the crosstalk between selective autophagy receptors and type I IFN signaling modulated by OPTN during SVV infection.
Figure 8.
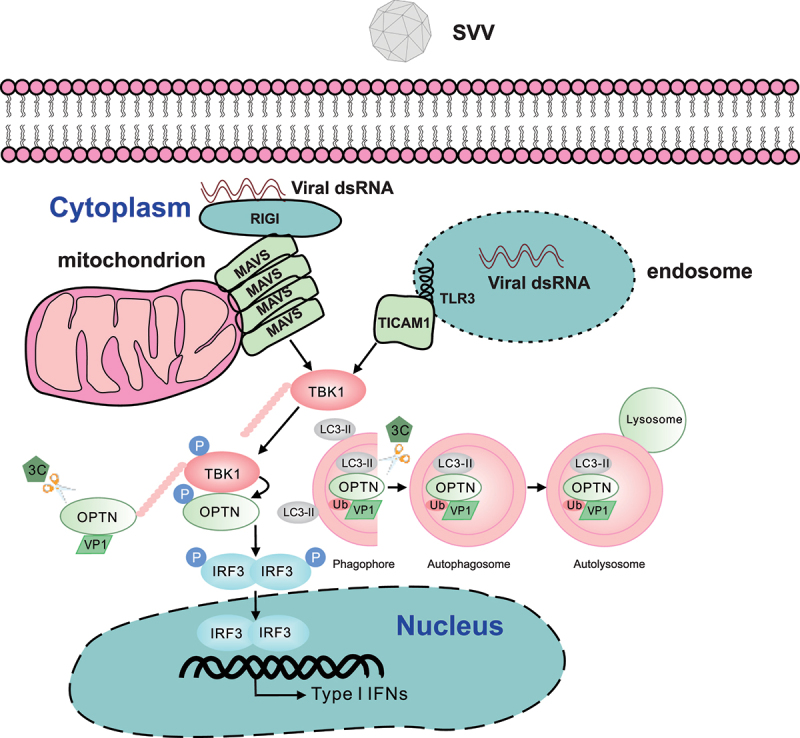
Proposed model of OPTN-mediated TBK1-IRF3 signaling pathway during Seneca Valley virus (SVV) infection. OPTN induces the activation of TBK1-IRF3 signaling pathway and targeted SVV-VP1 for degradation, thereby inhibiting viral replication. To facilitate survival, SVV (3C protease) 3C[pro] targets OPTN, since its cleavage impairs the antiviral activity of OPTN and the OPTN-mediated TBK1-IRF3 signaling pathway. Furthermore, the binding capacity to ubiquitinated proteins for N-terminal OPTN is attenuated, resulting in impaired selective autophagy.
Materials and methods
Cells, viruses, and antibodies, and chemical reagents
BHK-21 cells (ATCC, CCL-10), PK-15 cells (ATCC,CCL-33), and HEK-293T cells (ATCC, CRL-11268) were grown at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen 1,917,773) containing 10% fetal bovine serum (FBS; Invitrogen, 10099141C). The SVV CHhb17 strain was used in the study [56]. The mouse monoclonal antibody specific to SVV VP1 was prepared in our laboratory and has been reported previously [56]. Rabbit anti-OPTN (10837–1-AP) and rabbit anti-GFP (50430–2-AP) were obtained from Proteintech. Rabbit anti-HA (3724), TBK1 (3504), p-TBK1 (5483; Ser172), IRF3 (11904), p-IRF3 (29047; Ser396), AKT (4685), p-AKT (4085; Ser473) were obtained from Cell Signaling Technology. Mouse anti-GFP (ab127417) and mouse anti-ACTB/β-actin antibody (ab8226) were obtained from Abcam. Mouse anti-HA (H3663) monoclonal antibody, mouse anti-MYC (M4439) monoclonal antibody, and rabbit anti-MYC antibody (C3956) were obtained from Sigma-Aldrich. Alexa Fluor 568-conjugated goat anti-rabbit secondary antibody (11011), Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (11017), and Alexa Fluor 647-conjugated goat anti-mouse secondary antibody (A32728) were obtained from Invitrogen. Chemical reagents MG132 (S2619), Z-VAD-FMK (S7023), bafilomycin A1 (S1413), chloroquine (CQ; S6999), 3-methyladenine (3-MA; S2767), and B×795(S1274) were purchased from Selleck Chemicals.
Growth kinetics of SVV in BHK-21 cells
BHK-21 cells grown in 6-well plates were infected with SVV at the multiplicity of infection (MOI) indicated. After 1 h of incubation at 37°C, cells were washed with DMEM medium, then supplemented with DMEM containing 2% FBS. At the indicated times postinfection, the cells and supernatants were harvested, frozen, and thawed. Samples were titrated on BHK-21 cells by using TCID50 assay.
Plasmid construction
he OPTN gene from BHK-21 cells was amplified and recombined into the eukaryotic vectors pCMV-HA (Clontech 631,604) and pCMV-MYC (Clontech 631,604) by using ClonExpress One Step Cloning Kit (Vazyme, C112). LC3 was cloned from BHK-21 cells and inserted into pEGFP-C1 (Clontech, 6084–1) and pmCherry-C1 (Clontech 632,524). The genes coding for SVV structural and nonstructural proteins and engineered into the eukaryotic vectors pEGFP-C1 that have been used in our previous studies [31]. The mutants of GFP-3C plasmid, GFP-3CH48A (single point mutant), GFP-3CC160A (single point mutant) and GFP-3C[DM] (H48A and C160A double mutants), and 3C sequence of encephalomyocarditis virus (EMCV), foot-and-mouth disease virus (FMDV), human rhinovirus (HRV), Coxsackievirus type 3 (CVB3), and EV71 (enterovirus 71) were recombined into pEGFP-C1 that have been used in our previous studies [47]. The mutants of HA-OPTNE368A,Q369, HA-OPTNE483A,Q484A, HA-OPTNQ488A,E495,E500A, HA-OPTNQ507A,E511A,Q513A, HA-OPTNQ525A,Q526A,Q531A,E535A, HA-OPTNQ541A,Q542A,Q543A,Q545A, HA-OPTNE557A,Q566A were performed with mutagenesis PCR on HA-OPTN. Lipofectamine 3000 (Invitrogen, L3000015) was used for plasmid transfection. Primers used in this study are displayed in Table 1.
Table 1.
Primers used in this study.
| Primersa | Sequence (5’-3’)b | Restriction site |
|---|---|---|
| HA-myc-OPTN-F HA-myc-OPTN-R HA-OPTN(141–574)-F HA-OPTN(224–574)-F HA-OPTN(1–438)-R HA-myc-OPTN(1–513)-R HA-myc-OPTN(514–574)-F GFP-RFP-LC3-F GFP-RFP-LC3-R Q-OPTN-F Q-OPTN-R Q-β-actin-F Q-β-actin-R Q-IFN-β-F Q-IFN-β-R |
TGGCCATGGAGGCCCGAATTCGGATGTCCCATCAACCTCTGAGCT GATCCCCGCGGCCGCGGTACCTCAAATGATGCAATCCATGACA TGGCCATGGAGGCCCGAATTCGGGAGCAGCTGAAGATCCAGGTGA TGGCCATGGAGGCCCGAATTCGGGAATTAGCTGTGAGCCAACTCC GATCCCCGCGGCCGCGGTACCTCACATCTGAAGCTGTTTGGATGCC GATCCCCGCGGCCGCGGTACCTTACTGCATTTCCATCAAGGATTGC TGGCCATGGAGGCCCGAATTCGGAGCCGGCACGGGGCAAGAACCA TCGAGCTCAAGCTTCGAATTCTATGCCGTCCGAGAAGACTTTC TTATCTAGATCCGGTGGATCCTTACACAGCCAGTGCTGTTCCGA AGGCAATCCTTGATGGAAATGCAG AACTTCTCCACATTTGGGGCAAGA TTCAACACCCCAGCCATGTACG CAGCCAGGTCCAGACGCAGGAT AGAAGCAATTCAGAGTGTGAGGAC TGCTGGAGAAAGTGTTGTTGAAGA |
EcoRI KpnI EcoRI KpnI EcoRI KpnI EcoRI EcoRI BamHI |
aF denotes forward PCR primer; R denotes reverse PCR primer. bRestriction sites are underlined.
RNA interference (RNAi)
The small interfering RNAs (siRNAs) targeting OPTN were synthesized by GenePharma (Suzhou, China): siOPTN-1 (sense, 5’- GGAAUUAGCUGUGAGCCAATT-3’; antisense, 5’-UUGGCUCACAGCUAAUUCCTT-3’), siOPTN-2 (sense, 5’- GGAAGUGAAGUGGAAGCAUTT −3’; antisense, 5’-AUGCUUCCACUUCACUUCCTT-3’), siOPTN-3 (sense, 5’-GGACAAGAUGAUGCUGCAATT −3’; antisense, 5’-UUGCAGCAUCAUCUUGUCCTT-3’). The siNC (sense, 5’-UUCUCCGAACGUGUCACGUTT-3’; antisense, 5’-ACGUGACACGUUCGGAGAATT-3’) served as a negative control. Cells seeded in 6-well plates were transfected with siRNAs using Lipofectamine RNAiMAX transfection reagent (Invitrogen 13,778,150). Cells were lysed at 36 h post-transfection, the effects of gene-silenced for OPTN were analyzed by western blotting, and cell viability of siRNA-transfected cells was tested by CCK-8 assay. At 36 h post-transfection, cells were infected with SVV. The cells were collected at indicated times for analysis of VP1 production and virus titer.
Western blotting
The cell samples were prepared using lysis buffer containing 1% NP-40 (Beyotime, P0013F), 50 mM Tris, pH 7.4, 0.5 mM EDTA, 150 mM NaCl and protease inhibitor (Sigma, P8340), and clarified by centrifugation at 14,000 × g for 20 min at 4°C with rotation. Proteins were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis/SDS-PAGE and blotted onto nitrocellulose (NC) membranes (PALL 66,485), then blocked with 5% nonfat milk (BD 232,100) in phosphate-buffered saline (PBS; Solarbio, P1020). The membranes were incubated with specific primary antibodies, followed by an appropriate horseradish peroxidase-conjugated secondary antibody (Abcam, ab205718 and ab205719). Membrane-bound protein bands were detected using enhanced chemiluminescence detection reagents (Vazyme, E412).
Co-immunoprecipitation (co-IP)
BHK-21 cells were seeded on 6-well plates and cultured for 24 h before co-transfection with plasmids. Twenty-four hours after co-transfection, the whole cell lysates were prepared using NP-40 buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40, 0.5 mM EDTA) for 30 min at 4°C with rotation. The cell lysates were clarified by centrifugation at 14,000 × g for 20 min. The cell supernatants were incubated with anti-HA magnetic beads (Thermo Scientific 88,836), GFP-trap agarose (Chromotek, gta-20), or anti-MYC antibody in conjunction with protein A/G beads (Santa Cruz Biotechnology, SC-2003) overnight at 4°C with rotation. Immunoprecipitation pellets were washed three times with lysis buffer and boiled with 5 × SDS loading buffer for 5 min, and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis for western blot analysis.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen 15,596,026). RT SuperMix for qPCR (Vazyme, R122) was used to prepare the first strand cDNA. The generated cDNA was used as template, qRT-PCR was performed using SYBR Green Master Mix (Vazyme, Q111). The mRNA level of ACTB was used as the reference control, data are expressed as relative fold change that calculated using the 2−ΔΔCT method. Primers used in this study are displayed in Table 1.
Immunofluorescence assay (IFA)
BHK-21 cells were seeded on cover slips in 24-well plates. After 24 h of transfection, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 (Sigma, T8787) in PBS for 10 min. After that, the cells were blocked with 2% bovine serum albumin (BSA; Beyotime, ST023) in PBS for 30 min. Then, the cells were incubated with primary antibody for 1 h and secondary antibody for another 1 h. After washing with PBS, cells were incubated with 4’,6-diamidino-2-phenylindole (DAPI; Beyotime, C1006) for 5 min. The images were taken with a laser scanning confocal microscope (Nikon A1, Japan).
Statistical analysis
Statistical analysis was determined with GraphPad Prism version 6.00 (La Jolla, CA, USA). All experiments were performed in three independent experiments. Experimental data in the graphs represent means ± standard deviations (SD). Asterisks indicate statistically significant: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Funding Statement
This work was supported by the Outstanding Young Foundation of Beijing Academy of Agriculture and Forestry Sciences (YXQN202302), the Innovation Capacity of Beijing Academy of Agriculture and Forestry Sciences (KJCX20220411), the Reform and Development of Institute of Animal Husbandry and Veterinary Medicine, Beijing Academy of Agriculture and Forestry Sciences (XMS202312), Key Laboratory of Livestock Infectious Diseases, Ministry of Education, Shenyang Agricultural University (FKLID-2021-03), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure statement
The authors have no financial conflicts of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Hales LM, Knowles NJ, Reddy PS, et al. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol. 2008;89(Pt 5):1265–1275. doi: 10.1099/vir.0.83570-0 [DOI] [PubMed] [Google Scholar]
- [2].Leme RA, Alfieri AF, Alfieri AA.. Update on Senecavirus infection in pigs. Viruses. 2017;9(7):170. doi: 10.3390/v9070170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu Q, Zhao X, Bai Y, et al. The first identification and complete genome of Senecavirus a affecting pig with idiopathic vesicular disease in China. Transbound Emerg Dis. 2017;64(5):1633–1640. doi: 10.1111/tbed.12557 [DOI] [PubMed] [Google Scholar]
- [4].Pasma T, Davidson S, Shaw SL. Idiopathic vesicular disease in swine in Manitoba. Can Vet J. 2008;49(1):84–85. [PMC free article] [PubMed] [Google Scholar]
- [5].Leme RA, Zotti E, Alcantara BK, et al. Senecavirus A: An emerging vesicular infection in Brazilian pig herds. Transbound Emerg Dis. 2015;62(6):603–611. doi: 10.1111/tbed.12430 [DOI] [PubMed] [Google Scholar]
- [6].Wang L, Prarat M, Hayes J, et al. Detection and genomic characterization of Senecavirus a Ohio, US,2015. a. Emerg Infect Dis. 2016;22(7):1321–1323. doi: 10.3201/eid2207.151897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saeng-Chuto K, Rodtian P, Temeeyasen G, et al. The first detection of Senecavirus a in pigs in Thailand, 2016. Transbound Emerg Dis. 2018;65(1):285–288. doi: 10.1111/tbed.12654 [DOI] [PubMed] [Google Scholar]
- [8].Sun D, Vannucci F, Knutson TP, et al. Emergence and whole-genome sequence of Senecavirus a in Colombia. Transbound Emerg Dis. Transbound Emerg Dis. 2017;64(5):1346–1349. doi: 10.1111/tbed.12669 [DOI] [PubMed] [Google Scholar]
- [9].Kennedy EM, Denslow A, Hewett J, et al. Development of intravenously administered synthetic RNA virus immunotherapy for the treatment of cancer. Nat Commun. 2022;13(1):5907. doi: 10.1038/s41467-022-33599-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miles LA, Burga LN, Gardner EE, et al. Anthrax toxin receptor 1 is the cellular receptor for Seneca Valley virus. J Clin Invest. 2017;127(8):2957–2967. doi: 10.1172/JCI93472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Venkataraman S, Reddy SP, Loo J, et al. Structure of Seneca Valley virus-001: an oncolytic picornavirus representing a new genus. Structure. 2008;16(10):1555–1561. doi: 10.1016/j.str.2008.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qian S, Fan W, Liu T, et al. Seneca Valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J Virol. 2017;91(16):e00823. doi: 10.1128/JVI.00823-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xue Q, Liu H, Zhu Z, et al. Seneca Valley virus 3C(pro) cleaves PABPC1 to promote viral replication. Pathogens. 2020;9(6):443. doi: 10.3390/pathogens9060443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wen W, Yin M, Zhang H, et al. Seneca Valley virus 2C and 3C inhibit type I interferon production by inducing the degradation of RIG-I. Virology. 2019;535:122–129. doi: 10.1016/j.virol.2019.06.017 [DOI] [PubMed] [Google Scholar]
- [15].Xue Q, Liu H, Zhu Z, et al. Seneca Valley virus 3C protease negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. Antiviral Res. 2018;160:183–189. doi: 10.1016/j.antiviral.2018.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xue Q, Liu H, Zhu Z, et al. Seneca Valley virus 3C(pro) abrogates the IRF3- and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology. 2018;518:1–7. doi: 10.1016/j.virol.2018.01.028 [DOI] [PubMed] [Google Scholar]
- [17].Wen W, Li X, Wang H, et al. Seneca Valley virus 3C protease induces pyroptosis by directly cleaving porcine gasdermin D. J Immunol. 2021;207(1):189–199. doi: 10.4049/jimmunol.2001030 [DOI] [PubMed] [Google Scholar]
- [18].Fernandes MHV, Maggioli MF, Otta J, et al. Senecavirus A 3C protease mediates host cell apoptosis late in infection. Front Immunol. 2019;10:363. doi: 10.3389/fimmu.2019.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. doi: 10.1038/nrm2239 [DOI] [PubMed] [Google Scholar]
- [20].Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- [21].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi: 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi: 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [24].Shi J, Wong J, Piesik P, et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013;9(10):1591–1603. doi: 10.4161/auto.26059 [DOI] [PubMed] [Google Scholar]
- [25].Shi J, Fung G, Piesik P, et al. Dominant-negative function of the C-terminal fragments of NBR1 and SQSTM1 generated during enteroviral infection. Cell Death Differ. 2014;21(9):1432–1441. doi: 10.1038/cdd.2014.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mohamud Y, Qu J, Xue YC, et al. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019;26(6):1062–1076. doi: 10.1038/s41418-018-0185-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Corona Velazquez A, Corona AK, Klein KA, et al. Poliovirus induces autophagic signaling independent of the ULK1 complex. Autophagy. 2018;14(7):1201–1213. doi: 10.1080/15548627.2018.1458805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Y, Hu B, Ji G, et al. Cytoplasmic cargo receptor p62 inhibits avibirnavirus replication by mediating autophagic degradation of viral protein VP2. J Virol. 2020;94(24):e01255–20. doi: 10.1128/JVI.01255-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hafren A, Macia JL, Love AJ, et al. Selective autophagy limits cauliflower mosaic virus infection by NBR1-mediated targeting of viral capsid protein and particles. Proc Natl Acad Sci U S A. 2017;114(10):E2026–E2035. doi: 10.1073/pnas.1610687114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wen W, Li X, Yin M, et al. Selective autophagy receptor SQSTM1/p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy. 2021;17(11):3763–3775. doi: 10.1080/15548627.2021.1897223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Song J, Hou L, Quan R, et al. Synergetic contributions of viral VP1, VP3, and 3C to activation of the AKT-AMPK-MAPK-MTOR signaling pathway for Seneca Valley virus-induced autophagy. J Virol. 2022;96(2):e0155021. doi: 10.1128/JVI.01550-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Slowicka K, van Loo G, van Loo G. Optineurin functions for optimal immunity. Front Immunol. 2018;9:769. doi: 10.3389/fimmu.2018.00769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Outlioua A, Pourcelot M, Arnoult D. The role of optineurin in antiviral type I interferon production. Front Immunol. 2018;9:853. doi: 10.3389/fimmu.2018.00853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Patil CD, Suryawanshi R, Ames J, et al. Intrinsic antiviral activity of optineurin prevents hyperproliferation of a primary herpes simplex virus type 2 infection. J Immunol. 2022;208(1):63–73. doi: 10.4049/jimmunol.2100472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Patil CD, Shukla D. OPTN (optineurin)-mediated selective autophagy prevents neurodegeneration due to herpesvirus infection. Autophagy. 2022;18(4):944–945. doi: 10.1080/15548627.2022.2037223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ames J, Yadavalli T, Suryawanshi R, et al. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat Commun. 2021;12(1):5401. doi: 10.1038/s41467-021-25642-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang R, Varela M, Vallentgoed W, et al. The selective autophagy receptors Optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PLOS Pathog. 2019;15(2):e1007329. doi: 10.1371/journal.ppat.1007329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol. 2012;294:223–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–4448. doi: 10.1073/pnas.1405752111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gao J, Ohtsubo M, Hotta Y, et al. Oligomerization of optineurin and its oxidative stress- or E50K mutation-driven covalent cross-linking: possible relationship with glaucoma pathology. PLoS One. 2014;9(7):e101206. doi: 10.1371/journal.pone.0101206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Waisner H, Kalamvoki M, Sandri-Goldin RM. The ICP0 protein of herpes simplex virus 1 (HSV-1) downregulates major autophagy adaptor proteins sequestosome 1 and optineurin during the early stages of HSV-1 infection. J Virol. 2019;93(21):e01258–19. doi: 10.1128/JVI.01258-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Morton S, Hesson L, Peggie M, et al. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582(6):997–1002. doi: 10.1016/j.febslet.2008.02.047 [DOI] [PubMed] [Google Scholar]
- [45].Gleason CE, Ordureau A, Gourlay R, et al. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem. 2011;286(41):35663–35674. doi: 10.1074/jbc.M111.267567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pourcelot M, Zemirli N, Silva Da Costa L, et al. The golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016;14(1):69. doi: 10.1186/s12915-016-0292-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Song J, Quan R, Wang D, et al. Seneca Valley virus 3C(pro) mediates cleavage and redistribution of nucleolin to facilitate viral replication. Microbiol Spectr. 2022;10(2):e0030422. doi: 10.1128/spectrum.00304-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Blom N, Hansen J, Blaas D, et al. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 1996;5(11):2203–2216. doi: 10.1002/pro.5560051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jaishankar D, Yakoub AM, Yadavalli T, et al. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci Transl Med. 2018;10(428):eaan5861. doi: 10.1126/scitranslmed.aan5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Madavaraju K, Yadavalli T, Singh SK, et al. Prophylactic treatment with BX795 blocks activation of AKT and its downstream targets to protect vaginal keratinocytes and vaginal epithelium from HSV-2 infection. Antiviral Res. 2021;194:105145. doi: 10.1016/j.antiviral.2021.105145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23. doi: 10.1038/cr.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi: 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Orvedahl A, MacPherson S, Sumpter R Jr., et al. Autophagy protects against sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7(2):115–127. doi: 10.1016/j.chom.2010.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wen W, Li X, Yin M, et al. Selective autophagy receptor SQSTM1/p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy. 2021;2021(11):1–13. doi: 10.1080/15548627.2021.1897223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sun D, Kong N, Dong S, et al. 2AB protein of Senecavirus a antagonizes selective autophagy and type I interferon production by degrading LC3 and MARCHF8. Autophagy. 2022;18(8):1–13. doi: 10.1080/15548627.2021.2015740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hou L, Dong J, Zhu S, et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology. 2019;537:254–263. doi: 10.1016/j.virol.2019.08.029 [DOI] [PubMed] [Google Scholar]
- [57].Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–516. doi: 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- [58].Mostowy S, Sancho-Shimizu V, Hamon MA, et al. Cossart P: p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286(30):26987–26995. doi: 10.1074/jbc.M111.223610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mohamud Y, Xue YC, Liu H, et al. Autophagy receptor protein Tax1-binding protein 1/TRAF6-binding protein is a cellular substrate of enteroviral proteinase. Front Microbiol. 2021;12:647410. doi: 10.3389/fmicb.2021.647410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zheng YT, Shahnazari S, Brech A, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183(9):5909–5916. doi: 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- [61].Slowicka K, Vereecke L, Mc Guire C, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur J Immunol. 2016;46(4):971–980. doi: 10.1002/eji.201545863 [DOI] [PubMed] [Google Scholar]
- [62].Zhang Y, Liu S, Xu Q, et al. Cleavage of the selective autophagy receptor SQSTM1/p62 by the SARS-CoV-2 main protease NSP5 prevents the autophagic degradation of viral membrane proteins. Mol Biomed. 2022;3(1):17. doi: 10.1186/s43556-022-00083-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Puri M, La Pietra L, Mraheil MA, et al. Listeriolysin O regulates the expression of optineurin, an autophagy adaptor that inhibits the growth of Listeria monocytogenes. Toxins (Basel). 2017;9(9):273. doi: 10.3390/toxins9090273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thurston TL, Ryzhakov G, Bloor S, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–1221. doi: 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- [65].Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal. 2011;4(187):39. doi: 10.1126/scisignal.2002355 [DOI] [PubMed] [Google Scholar]
- [66].Meena NP, Zhu G, Mittelstadt PR, et al. Munitic I: the TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett. 2016;590(10):1498–1508. doi: 10.1002/1873-3468.12176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mankouri J, Fragkoudis R, Richards KH, et al. Optineurin negatively regulates the induction of IFNbeta in response to RNA virus infection. PLOS Pathog. 2010;6(2):e1000778. doi: 10.1371/journal.ppat.1000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Munitic I, Giardino Torchia ML, Meena NP, et al. Optineurin insufficiency impairs IRF3 but not NF-kappaB activation in immune cells. J Immunol. 2013;191(12):6231–6240. doi: 10.4049/jimmunol.1301696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bakshi S, Taylor J, Strickson S, et al. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon beta. Biochem J. 2017;474(7):1163–1174. doi: 10.1042/BCJ20160992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Iha H, Peloponese JM, Verstrepen L, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27(4):629–641. doi: 10.1038/emboj.2008.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].von Muhlinen N, Thurston T, Ryzhakov G, et al. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6(2):288–289. doi: 10.4161/auto.6.2.11118 [DOI] [PubMed] [Google Scholar]
- [72].Wagner S, Carpentier I, Rogov V, et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene. 2008;27(26):3739–3745. doi: 10.1038/sj.onc.1211042 [DOI] [PubMed] [Google Scholar]
- [73].Ryan TA, Tumbarello DA. Optineurin: A coordinator of membrane-associated cargo trafficking and autophagy. Front Immunol. 2018;9:1024. doi: 10.3389/fimmu.2018.01024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113(15):4039–4044. doi: 10.1073/pnas.1523926113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.