

ABSTRACT
Anaplastic thyroid carcinoma (ATC) is one of the most aggressive and lethal neoplasms in humans, and just limited progresses have been made to extend patient survival and decrease ATC-associated mortality. Thus, the identification of novel therapeutic strategies for treating ATC is needed. Recently, our group has identified two proteins with oncogenic activity, namely HMGA1 and EZH2, with pivotal roles in ATC cancer progression. Therefore, we tested the ability of trabectedin, a HMGA1-targeting drug, and GSK126, an inhibitor of EZH2 enzymatic activity, to impair cell viability of four ATC-derived cell lines. In the present study, we first confirmed the overexpression of HMGA1 and EZH2 in all ATC-derived cell lines and tissues compared to the normal primary thyroid cells and tissues. Then, treatment of the ATC cell lines with trabectedin and GSK126 resulted in a drastic induction of apoptotic cell death, which increased when the ATC cell lines were treated with a combination of both drugs. Conversely, normal primary human thyroid cells did not show any significant reduction in their viability when exposed to the same drugs. Noteworthy, both drugs induced the deregulation of EZH2- and HMGA1-controlled genes. Altogether, these findings propose the combination of trabectedin and GSK126 as possible novel strategy for ATC therapy.
KEYWORDS: HMGA, EZH2, Trabectedin, GSK126, anaplastic thyroid carcinoma
1. Background
Malignant tumors derived from thyroid gland present the highest incidence among those originating from the endocrine system [1]. Thyroid carcinomas (TCs) are classified, following their differentiation grade, in well-differentiated (WDTC), poorly differentiated (PDTC) and undifferentiated (UTC) ones [2,3].
The WDTCs are represented mainly by the papillary thyroid carcinomas (PTCs, about 80% of all TCs) and follicular thyroid carcinomas (FTCs, about 10% of TCs), while anaplastic thyroid carcinomas (ATCs, 1–2% of TCs) are the most characterized UTCs. Indeed, ATCs lose the main biological structures and functions of normal thyroid cells, thus acquiring a highly malignant phenotype with invariable lethality [4], since the loss of the ability to trap iodide, a feature of thyroid cells, by ATC cells does not allow the therapy based on radioiodide treatment that has a high efficacy in the treatment of differentiated TCs [5].
ATC incidence worldwide is very low, representing just 1–2% of all TCs, and its peak of incidence is between the sixth and seventh decades of life [6]. In spite of its low incidence, ATC accounts for 14–39% of all deaths related to TCs with a median survival rate of about 5–6 months [7]. Unfortunately, nowadays, no effective therapies are available for ATC treatment, since these carcinomas are resistant to chemo- and radiotherapy, and even innovative therapeutic approaches, such as immunotherapy or the treatment with tyrosine kinase inhibitors, have given just very slight improvements to the survival rate of ATC patients [8,9]. Therefore, the search for new approaches that could allow the identification of drugs potentially able to improve ATC patient outcome is a critical aim of scientific community.
During the last few years, our group has underlined the pivotal role of High Mobility Group A (HMGA) proteins in ATC carcinogenesis [10,11]. HMGA is represented by three different proteins, HMGA1 (namely HMGA1a and HMGA1b, encoded by alternative splicing) and HMGA2. These proteins act as architectural chromosomal factors, able to bind the minor groove of DNA, and, by interacting with the transcriptional apparatus, can regulate the expression of a number of genes in a positive or negative manner [10]. HMGA expression is extremely high during embryonic development, whereas it is low or absent in normal adult tissues. However, they are abundantly expressed during carcinogenesis process, representing a feature of human malignancies [12–14]. HMGA proteins are also highly expressed in ATC tissues [15] and cells and their silencing leads to death through apoptosis [16,17]. Moreover, our group has also reported that the block of HMGA expression in rat thyroid cells prevents neoplastic transformation induced by murine sarcoma retroviruses [10].
More recently, we have identified the role of the Enhancer of Zeste Homolog 2 (EZH2) protein, whose overexpression contributes to ATC carcinogenesis [18]. Indeed, it is the enzymatic subunit of the Polycomb Repressive Complex 2 (PRC2), a repressor of gene expression mainly acting via the trimethylation of H3 histone on lysine 27 (H3K27me3) [19,20]. EZH2 is usually not active in adult tissues, however it can be activated in aggressive carcinomas, resulting in abnormal gene expression pattern that promotes cancer progression and metastasis [21,22]. Interestingly, EZH2 is overexpressed in TC, and in particular ATC, contributing to transcriptional silencing of PAX8 gene, a transcription factor critical for thyroid differentiation [23–25]. Noteworthy, at least two microRNAs, namely mir-25 and mir-30d, are drastically downregulated in ATC and have EZH2 as target [24,26]. Moreover, the tumor suppressor of long non-coding RNA PAR5 is able to negatively regulate EZH2 activity in ATC cells [27], supporting the idea that the overexpression and the increased activity of EZH2 in ATC are also enhanced at epigenetic level. Furthermore, we have recently reported that HMGA1 is able to transcriptionally enhance EZH2 gene [28] and that HMGA1 pseudogenes, namely HMGA1P6 and HMGA1P7, which are overexpressed in ATC, are able to increase EZH2 protein levels by a competitive endogenous RNA (ceRNA) mechanism [29].
Recently, we have demonstrated that a novel antineoplastic agent, Trabectedin (Ecteinascidin-743 or ET-743), is able to interact with the minor groove of the DNA, displacing HMGA proteins, thus impairing transcriptional machinery and inducing cell death [30,31].
In the present study, we first confirmed the overexpression of HMGA1 and EZH2 in a set of ATC tissues and cells. Then, we treated four ATC cell lines (8505c, SW1736, FRO and ACT1) with ET-743 and GSK126, an inhibitor of EZH2 activity [32], alone or in combination. Both drugs drastically impaired the viability of the tested ATC cell lines, with a synergistic effect when these drugs are used in combination. In contrast, the viability of a normal primary thyroid culture was not significantly affected by these drugs even when they were used at high doses.
Therefore, taken together, our findings suggest that the use of ET-743 and GSK126 may represent a novel therapeutic strategy for ATC.
2. Materials and methods
2.1. Cells and cell culture
In this study, all of the ATC cell lines (8505c, FRO, SW1736 and ACT-1) [33,34] were grown in DMEM medium (Sigma-Aldrich, Saint Louis, MO), 10% fetal calf serum (Sigma-Aldrich, Saint Louis, MO). Cells were kept in a humidified environment with 5% CO2 at 37°C. At the Head and Neck Surgery Unit, Department of Anesthesiology, Surgical and Emergency Science, University of Campania “Luigi Vanvitelli” in Naples, Italy, a sample of a thyroid was taken for the primary culture. Prior consent was given by the donor before the collection, acquisition or use of human tissue. Thyroid tissue was rinsed with PBS (Sigma-Aldrich, Saint Louis, MO, USA) containing 500 U/ml penicillin and 0.5 mg/ml streptomycin (Sigma-Aldrich, Saint Louis, MO, USA), and then cut into small pieces. The fragments were then broken down with 1 mg/mL collagenase (Roche Diagnostics, Indianapolis, USA), 0.02 mg/mL DNAase (Sigma-Aldrich, Saint Louis, MO, USA), and 0.01 mg/mL hyaluronidase (Sigma-Aldrich, Saint Louis, MO, USA) dissolved in PBS, for 16–20 h at room temperature. The lysates were then grown in 6 H medium (modified F-12 M medium, Coon’s modification of Ham’s F-12 medium) (Thermo Fisher Scientific, Massachusetts, USA), 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich, Saint Louis, MO, USA), 2 mM glutamine (Thermo Fisher Scientific, Massachusetts, USA), 2.6 g/L NaHCO3 (Thermo Fisher Scientific, Massachusetts, USA), 5 μg/mL gentamicin (Thermo Fisher Scientific, Massachusetts, USA), 1% NEAA (Sigma-Aldrich, Saint Louis, MO, USA), and 10 mIU/mL of thyrotropin (TSH) (Sigma-Aldrich, Saint Louis, MO, USA), 10 mIU/mL of insulin (Sigma-Aldrich, Saint Louis, MO, USA), 1 nM of hydrocortisone (Roche Diagnostics, Indianapolis, USA), 2 ng/mL of glycyl-histidyl-L-lysine acetate (Sigma-Aldrich, Saint Louis, MO, USA), 5 μg/mL of transferrin (Roche Diagnostics, Indianapolis, USA), and 10 ng/mL of somatostatin (Sigma-Aldrich, Saint Louis, MO, USA).
2.2. Tumor specimens
Normal and anaplastic thyroid tumors (frozen tissue) were collected from Lyon Sud Hospital using common standardized operating procedures and stored in Biological Resources Center and Tumor Bank Platform of Hospices Civils de Lyon (BB-0033–00046).
2.3. Reagents
GSK126 and ET-743 were supplied by Selleckchem (Planegg, Germany). Dimethyl sulfoxide (DMSO) was used to prepare the GSK126 and ET-743 stocks. After the solubilization, they were stored at −80°C with a concentration of 1 mM for ET-743 and 10 mM for GSK126. The dilution tested were:
GSK126 from 1 μM to 256 μM
ET-743 from 0.006 nM to 1.6 nM
2.4. Cell viability
The antiproliferative and cytotoxic effects of GSK126 and ET-743 were first tested using the MTT method [3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (Sigma-Aldrich, Saint Louis, MO) as a sign of metabolically active cells. Two thousand ATC cells, resuspended in 100 μL of culture medium, were put into 96-well plates and left to grow for 24 h before the test substance was added. The cells were treated with these drugs for 72 h at 37°C. MTT was added at a maximum dose of 0.5 mg/mL. After 3 h, the crystals of formazan were dissolved in DMSO (Sigma-Aldrich, Saint Louis, MO, USA). A microplate-photometer (Bio-Tek Instruments, Colmar, France) with a test wavelength of 570 nm was used to measure how well the colored solution absorbed light. GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA) was used to figure out the amount of material needed to stop growth by 50% (IC50). For the trypan blue exclusion methods, the supernatant was discarded, and cells were trypsinized (Sigma-Aldrich, Saint Louis, MO, USA) and then resuspended with medium. The cell suspension was mixed with trypan blue dye (Sigma-Aldrich, Saint Louis, MO, USA), transferred to a hemocytometer, and counted under a phase-contrast microscope (Carl Zeiss, Oberkochen, Germany).
2.5. Trypan blue exclusion test
5 × 104 cells were cultivated into 6-well plates. After 24 h, GSK126 and ET-743 drugs were added to cells media. After 72-h treatment, cells were collected and dissolved in 50 μL of PBS. Next, 10 μL of cell suspension was mixed with 10 μL of trypan blue solution (Sigma-Aldrich, Saint Louis, MO, USA) and incubated for 5 min at room temperature. A total of 10 μL of cell – trypan blue solution mixture was applied to glass hemocytometer, and viable and nonviable cells were counted. Viable cells have a clear cytoplasm, whereas nonviable cells have a blue cytoplasm.
2.6. Cell cycle analysis
Fluorescence-activated cell sorting was used to examine cell cycle distribution and Cell Quest Pro software (BD Biosciences, San Jose, CA, USA) was used to interpret the results. 2 × 105 cells, resuspended in 2 mL of culture medium, were plated in 6-well plates, treated for 48 h with ET-743 or GSK126, and then trypsinized and centrifuged at 1500 g for 5 min. The cells were subsequently fixed with 70% ethanol (Carlo Erba, Milan, Italy) overnight at −20°C. The cells were removed from the ethanol solution and then given two PBS (Sigma-Aldrich, Saint Louis, MO, USA) washes. The cells were then treated with 300 μl of a solution containing ribonuclease and propidium iodide (PI) dye (Thermo Fisher Scientific, Massachusetts, USA). Cell Quest Pro software was used to evaluate the data, and the results were represented as a percentage of cells in each stage of the cell cycle.
2.7. Apoptosis
For apoptosis assessment, 1.5 × 105 cells were resuspended in 2 mL of culture medium and then seeded in 6-well plates. After 24 h, cells were treated with ET-743 and/or GSK126 for 72 h and exposed to FITC-Annexin V and PI according to the manufacturer’s protocol (BD Biosciences, San Jose, CA, USA). This allows us to distinguish Annexin V- positive cells in early apoptosis from Annexin V- and PI-positive cells in late apoptosis. The results were the sum of early and late apoptotic cells. Cells were analyzed by flow cytometry using FL1 for Annexin V and FL2 for PI. Flow cytometry (FACScan; BD Biosciences) data were analyzed with Cell Quest Pro software.
2.8. RNA extraction and quantitative real-time PCR (qRT-PCR)
Using the Trizol reagent (Thermo Fisher Scientific, Massachusetts, USA), total RNA was isolated from cell lines (1×106 cells) and normal or malignant thyroid tissues (20–30 mg). The QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) was used to produce double stranded cDNA from 1 μg of total RNA from each sample. Quantitative Real-Time PCR (qRT-PCR) experiments were carried out using CFX96 thermocycler (Bio-Rad, Hercules, CA, USA). Each PCR reaction required 10 μL of 2X SYBR Green (Bio-Rad, CA, USA), 200 nM of each primer, and 20 ng of the previously produced cDNA. Exon–exon junction-covering oligonucleotides for qRT-PCR were created using Primer-BLAST software. Comparative C(T) method was used to determine the relative gene expression, as described elsewhere [35], using the primers purchased from Integrated DNA Technologies (San Diego, CA, USA) (Table 1).
Table 1.
Primer list.
| HMGA1 | Fw5’-aaggggcagacccaaaaa-3’ Rev5’-tccagtcccagaaggaagc-3’ |
| EZH2 | Fw5’-gctgaccattgggacagtaa-3’ Rev5’-cagatggtgccagcaataga-3’ |
| CDKN1A | Fw5’-ccgaagtcagttccttgtgg-3’ Rev5’-catgggttctgacggacat-3’ |
| E-cadherin(CDH1) | Fw5’-gcccagaaaatgaaaaagg-3’ Rev5’-gtgtatgtggcaatgcgttc-3’ |
| CyclinB1(CCNB1) | Fw5’-acatggtgcactttcctcct-3’ Rev5’-gtaatgttgtagagttggtgtcc-3’ |
| BCL2 | Fw5’-tacctgaaccggcacctg-3’ Rev5’-gccgtacagttccacaaagg-3’ |
| PAX8 | Fw5’-cttcagaaggaggagagacacc −3’ Rev5’-tttacacctgcatcgtctgg −3’ |
| G6PD | Fw5’-acagagtgagcccttcttcaa-3’ Rev5’-ataggagttgcgggcaaag-3’ |
G6PD gene was used as housekeeping for the normalization of data.
2.9. Drug combination assay
In a 96-well plate, 100 μL of culture medium containing 4 × 104 8505c cells were inoculated and incubated overnight at 37°C. Different dilutions of ET-743 and GSK126 drugs were then combined in all possible drug pairs to generate a 5 × 6 drug combination matrix. Then, cells were exposed to either single-agent drugs or drug pairs from the drug combination matrix, with DMSO serving as the negative control (each treatment was conducted in triplicate). After 72 h of incubation at 37°C, cell viability was determined using MTT (Sigma-Aldrich, Saint Louis, MO, USA) at a final concentration of 0.5 mg/mL. After 3 h of incubation, formazan crystals were dissolved in DMSO (Sigma-Aldrich, Saint Louis, MO, USA). The absorbance of the colored solution was measured with a microplate-photometer (Bio-Tek Instruments, Colmar, France) at 570 nm. The ET-743/GSK126 drug interactions and expected drug responses were calculated with the Combenefit tool [36], using the Loewe additivity model.
2.10. Western blot
Western blot analyses were performed as previously described [37–39]. The primary antibodies used were as follows: anti-EZH2 (AC22) from Cell Signaling, Massachusetts, USA. Antibodies against HMGA1 are polyclonal Ab raised against a synthetic peptide located in the NH2-terminal region. Membranes were blocked with 5% TBS-BSA proteins and incubated with the primary antibodies. Immunoblotting experiments were carried out according to standard procedures and visualized using the ECL chemiluminescence system (Thermo Fisher Scientific, Massachusetts, USA). As a control for equal loading of protein lysates, blotted proteins were probed with antibodies against GAPDH sc-32,233 protein (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
2.11. Ethics
The study was conducted according to the criteria set by the declaration of Helsinki and each subject signed an informed consent before participating in the study. Informed consent was obtained from all patients. The activity of biological samples conservation was declared under the number DC-2011–1437 to the Ministry of Research, to the committee of people’s protection of south-east IV and to the Health Regional Agency. The activity of biological material cession was agreed upon by the Ministry of Health under the number AC-2013–1867.
2.12. Statistical analysis
Data are presented as mean ± standard deviation (SD). For comparisons between two groups, Student’s t test was used to determine differences between mean values for normal distribution. All data were analyzed for significance using GraphPad Prism 6 software (San Diego, CA, USA). p values less than 0.05 were considered significant.
3. Results
3.1. HMGA1 and EZH2 are overexpressed in ATC tissues and cell lines
We first analyzed the expression of HMGA1 and EZH2 in a panel of human ATC cell lines, including 8505c, SW1736, FRO and ACT1. The results, shown in Figures 1(a, b), confirmed HMGA1 and EZH2 overexpression, at mRNA (Figure 1(a)) and protein level (Figure 1(b)), respectively, in all the analyzed ATC cell lines with respect to normal human thyroid-derived primary cells. Then, we extended this analysis to a panel of surgically removed ATC tissues using qRT-PCR. The results shown in Figure 1(c) confirmed the overexpression of both the HMGA1 and EZH2 genes also in all the ATC tissues with respect to normal thyroids.
Figure 1.
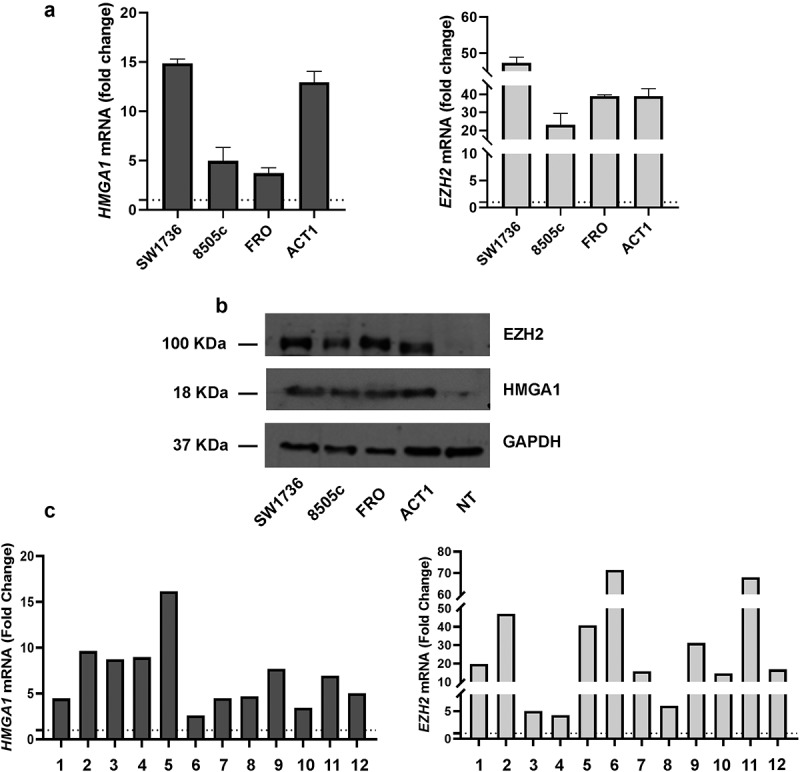
EZH2 and HMGA1 expression levels are upregulated in ATC-derived cell lines and tissues. (a) HMGA1 and EZH2 mRNA expression levels in a panel of ATC-derived cell lines (ACT1, FRO, 8505c and SW1736). The expression levels of normal human thyroid-derived primary cells were set equal to 1. Data are shown as mean ± SD (n = 3) (b) HMGA1 and EZH2 protein expression in the same ATC-derived cell lines and in normal human thyroid-derived primary cells. 50 μg of total cellular lysate was loaded per lane in reducing conditions. GAPDH was used as protein loading control. NT = normal thyroid tissue. (c) Twelve ATC tissues were analyzed for HMGA1 and EZH2 expression with respect to three normal thyroid tissues. The mean of normal samples was set equal to 1. Data are shown as mean ± SD (n = 3).
3.2. ET-743 and GSK126 induce strong cell death in ATC cell lines, but not in normal thyroid primary culture
Since the results shown above confirmed the overexpression in ATC of HMGA1 and EZH2, two proteins already reported to have a critical role in ATC cancer progression, we analyzed the effects of ET-743, a HMGA1-targeting drug, and GSK126, an inhibitor of EZH2 enzymatic activity, on the viability of the ATC cell lines after the exposure to increasing concentrations of ET-743 or GSK126.
Both drugs drastically reduced cell viability of the treated ATC cell lines, whereas primary thyroid cells were slightly affected only by very high concentrations of both the tested drugs (Figure 2(a)). Indeed, exposure to GSK126 induced low percentage of death in primary thyroid cells at concentrations 10-fold higher (128 μM) than the doses used for ATC-derived models. Equally, ET-743 did not affect normal primary thyroid cell viability even at the highest concentration used (1.6 nM), which is about eight-fold higher than the doses used for ATC-derived models. Subsequently, we calculated IC50 values for ET-743 and GSK126 in ATC cell lines. ET-743 induced high cytotoxicity in all of the cell lines, with IC50 values ranging from 0.046 nM to 0.15 nM. GSK126 showed lower cytotoxicity with respect to ET-743, with IC50 values ranging from 9.82 μM to 16.8 μM (Figure 2(b)).
Figure 2.
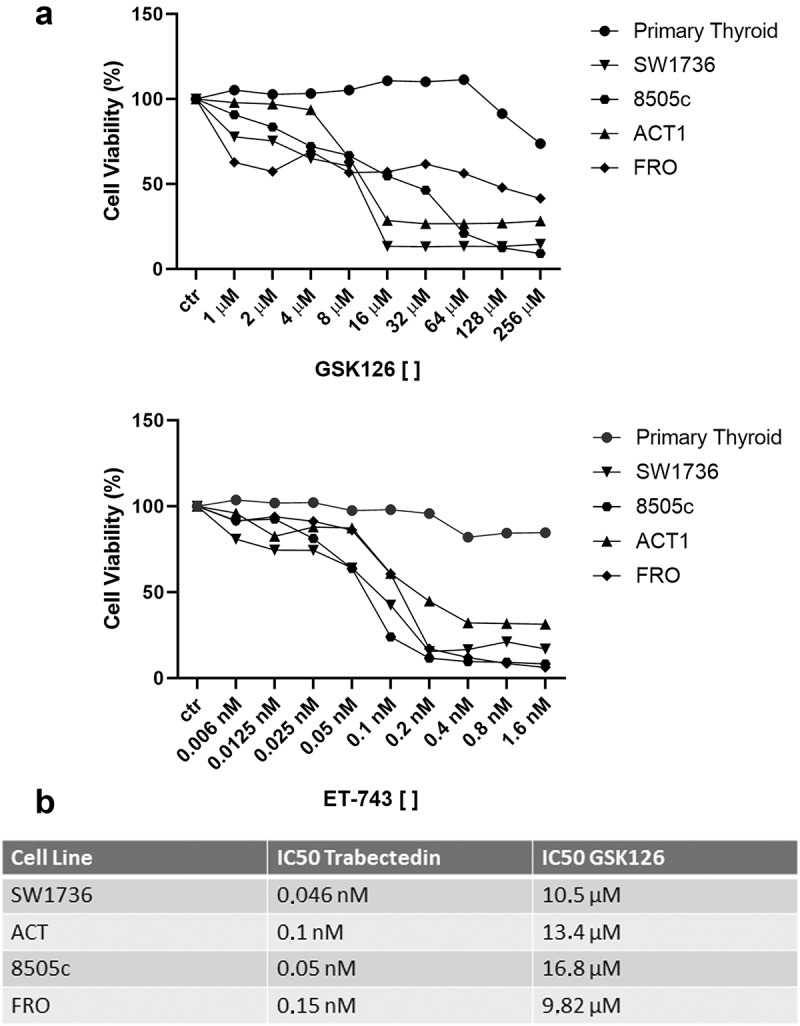
ET-743 and GSK126 affect cell viability of ATC-derived cell lines but not that of primary thyroid cells. (a) Dose–response curves of ACT1, FRO, 8505c, SW1736 ATC cell lines and normal primary thyroid cells treated with increasing concentrations of ET-743 and GSK126 for 72 h (n = 3). The cells were exposed to increasing concentrations of the drugs and then tested for viability by MTT assay. (b) ET-743 and GSK126 IC50 values for ATC cell lines.
Since exposure to GSK126 and ET-743 robustly affected ATC cell line viability, we treated them with a combination of both these drugs. Interestingly, in all the tested ATC cell lines, the mortality was further increased by this combined treatment (ACT1 and SW1736 ~80%, FRO ~90%, 8505c ~60%), and the induction of death was higher than that observed when the cells were exposed to just one drug (Figures 3(a), 1S). Noteworthy, primary thyroid cells were not affected at all by the treatment with drug combination at the same dosage (Figure 3(a)).
Figure 3.
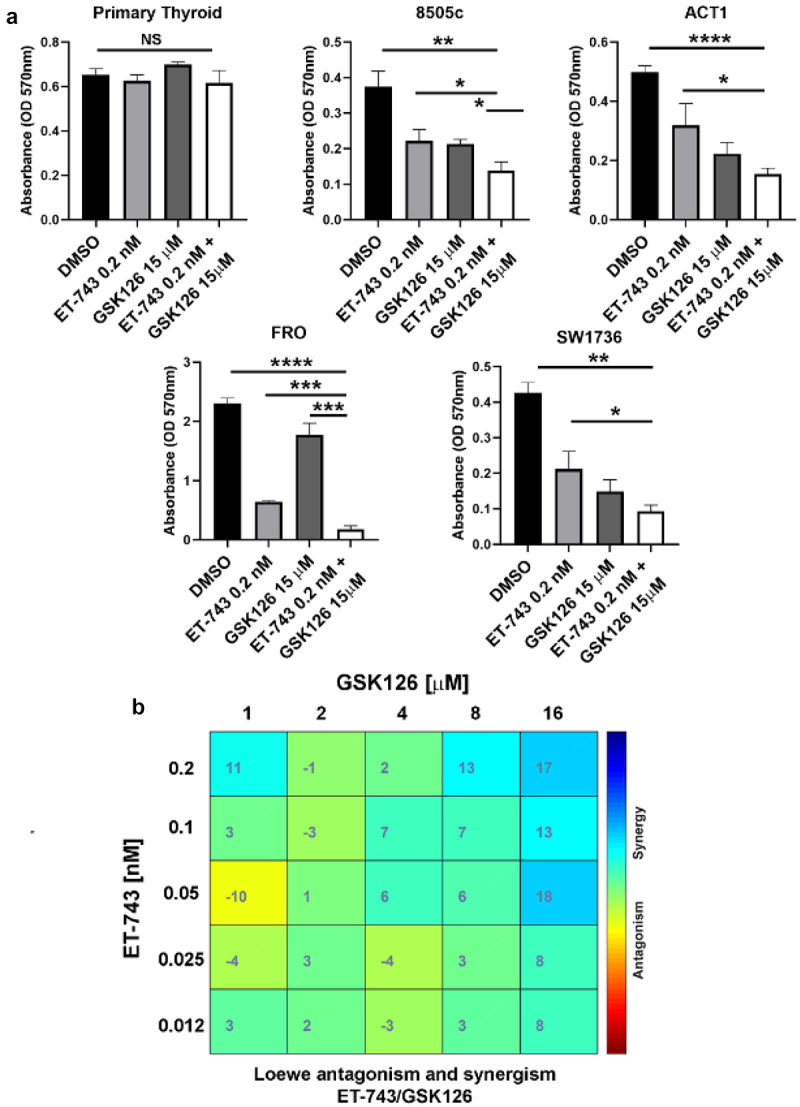
ET-743 and GSK126 synergistically induce ATC cell death. (a) effects of combination treatment with ET-743 and GSK126 on ATC cell viability by the MTT assay 72 h after exposure. (c) Combenefit mapped surface output for the drug combinations involving ET-743 and GSK126 using Loewe synergy model. These drugs synergistically inhibit 8505c cell growth. Cells were treated with ET-743 and GSK126 in a 5 × 5 concentration grid for 48 h, cell viability was determined by MTT assay. Data are shown as mean ± SD (n = 3) *p < 0.0.5; **p < 0.01; ***p < 0.001.
To evaluate the degree of synergy of the combined drugs, we used the Loewe reference model [40] on 8505c cell line after 72-h exposure to various combinations of the two agents. Interestingly, the combination indices revealed that ET-743 and GSK126 act synergistically at concentration of 0.2 nM and 16 μM, respectively (Figure 3(b)).
Then, we assessed the expression of some genes regulated by HMGA1 and EZH2 in the drug-treated cells compared to the DMSO treated cells. ATC cells treated with ET-743 showed decreased expression levels of two HMGA1-regulated genes, namely Cyclin B, a master regulator of cell cycle progression, and Bcl-2, an anti-apoptotic protein [10,30] (Figure 4(a)). Equally, in the GSK126-treated ATC cells, the expression levels of E-cadherin, an adhesion molecule whose expression loss represents a hallmark of epithelial–mesenchymal transition (EMT) and CDKN1A, a regulator of cell cycle, two EZH2-regulated genes [41,42] were drastically upregulated (Figure 4(b)). Interestingly, also PAX8, a pivotal transcription factor for thyroid differentiation, which is repressed by EZH2 [23], has been found upregulated in GSK126 treated cells (Figure 4(b)).
Figure 4.
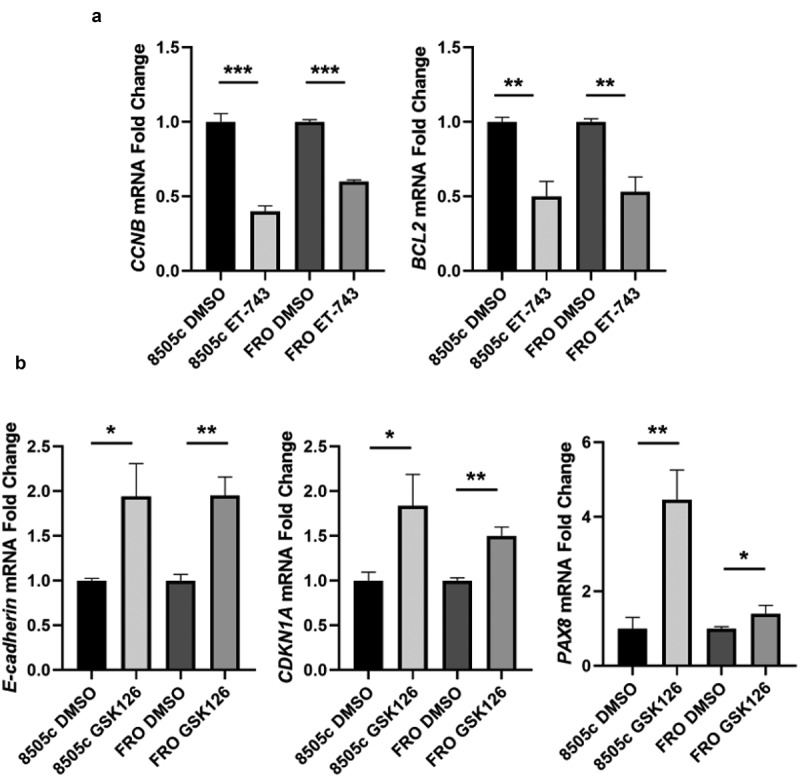
ET-743 and GSK126 treatment induces deregulation of HMGA1- and EZH2-target genes. (a) CCNB and BCL2 mRNA expression levels in 8505c cell treated or not with ET-743. (b) E-cadherin, CDKN1A and PAX8 mRNA expression levels in 8505c cell treated or not with GSK126. Data are shown as mean ± SD (n = 3) *p < 0.0.5; **p < 0.01; ***p < 0.001.
3.3. ET-743 and GSK126 induce cell cycle arrest and apoptosis in ATC cell lines
Subsequently, we investigated the effects of ET-743 and GSK126 on cell cycle and apoptosis. Figure 5(a) shows a strong G2/M accumulation peak after treatment with the ET-743, whereas a G0/G1 accumulation peak was observed in 8505c, ACT1, FRO and SW1736 cell lines treated with GSK126.
Figure 5.
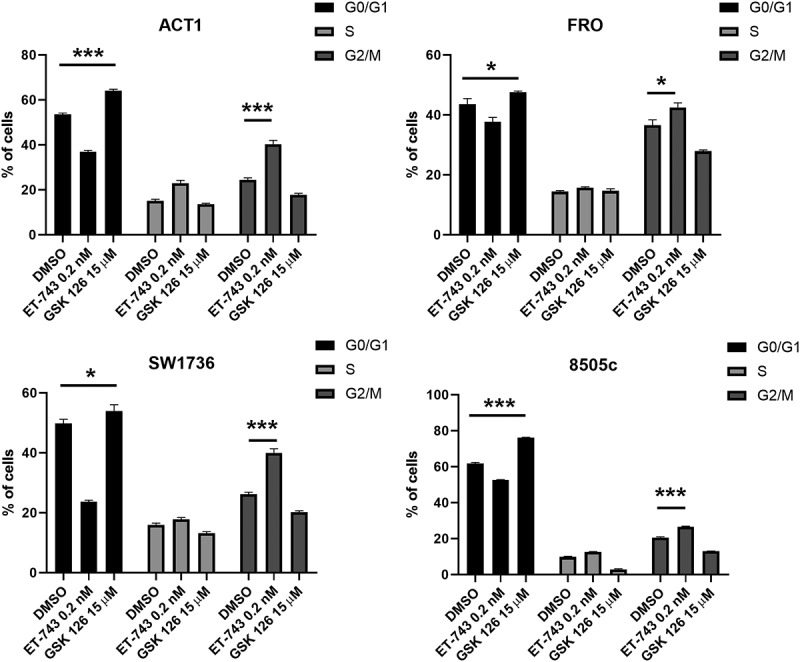
Effects of ET-743 and GSK126 on cell cycle progression on ATC cell lines. cell-cycle profile after 48 h of treatment with ET-743 and GSK126 analyzed by PI incorporation and flow cytometry in the ACT1, FRO, 8505c and SW1736 cell lines. Data are shown as mean ± SD (n = 3) *p < 0.0.5; **p < 0.01; ***p < 0.001.
Furthermore, we assessed the apoptotic rate induced by drug exposure by Annexin V test. Interestingly, the apoptotic rate has increased after exposure to ET-743 and GSK126 in all the analyzed ATC-derived cells (Figures 6(a,b)). Consistently, after 48 h of exposure, ET-743 and GSK126 combination (0.2 nM of ET-743 and 15 μM of GSK126) increased the apoptotic rate in comparison with the single-drug treatment in ATC cell lines (Figure 6(b)).
Figure 6.
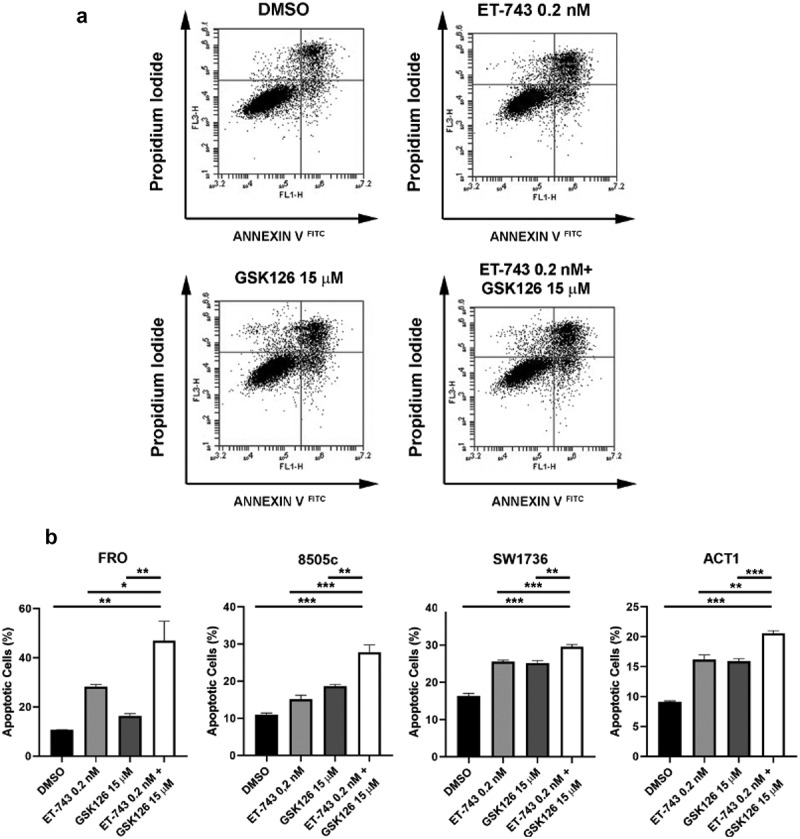
Effects of ET-743 and GSK126 combination on apoptosis. (a) Annexin V FITC-A vs. PI plots from the gated cells shows the populations corresponding to viable and non-apoptotic (annexin V – PI–), early (annexin V + PI–), and late (annexin V + PI+) apoptotic cells in SW1736 cell line. (B) Quantification of ACT1, 8505c, FRO and SW1736 apoptotic cells after 72 h of treatment with ET-743 and GSK126 alone or combination of the two drugs. Data are shown as mean ± SD (n = 3) *p < 0.05; **p < 0.01; ***p < 0.001.
4. Discussion
In order to propose novel therapeutic approaches for ATC, one of the most lethal cancers in mankind that lacks an effective therapy, we concentrate on the possibility to inhibit the activity of two proteins, EZH2 and HMGA, whose overexpression plays a critical role in ATC carcinogenesis [10,23].
Then, as an inhibitor of the HMGA function we used ET-743, the lead compound of ecteinascidins originally isolated from the extracts of the tunicate E. turbinate [43,44]. It has shown antineoplastic activity in human liposarcoma and leiomyosarcoma after primary anthracycline exposure and has a favorable toxicity profile [45,46]. Moreover, ET-743 was associated with substantially prolonged progression-free survival (PFS) in patients with platinum-sensitive recurrent ovarian cancer [47,48]. ET-743 forms adducts in the minor groove of DNA, generating single-strand breaks (SSBs) and double-strand breaks (DSBs) and setting off a cascade of events that results in cell cycle arrest and apoptosis [49]. Recent studies suggest that the anticancer activity of ET-743 may depend, at least in part, on the displacement of HMGA proteins from the minor groove of DNA, where they are bound [30]. Indeed, these chromatinic proteins are able to regulate the expression of several cancer-related genes by switching on or off their transcription [10]. In particular, it has been reported that ET-743 has a higher cytotoxic effect on thyroid and colon carcinoma cells expressing abundant levels of HMGAs in comparison with cells not expressing them, enhancing the sensitivity of these cells to Ionising Radiations [30].
During the last few years, several EZH2 inhibitors have been developed [50]. The first developed EZH2 inhibitor was 3-deazaneplanocin A (DZNep) [51]. DZNep is able to inhibit S-adenosyl-L-homocysteine (SAH) hydrolase activity, thus indirectly impairing the S-adenosyl-L-methionine-dependent histone methyltransferase activity. In this manner, DZNep is not a specific EZH2 inhibitor, but it globally blocks histone methylation [51]. Recently, Nakayama et al. reported that EZH2 inhibition by DZNep on four ATC-cell lines reduces the cell growth rate, having an interesting potential for a therapeutic approach [52].
Recently, several potent and highly selective S-adenosyl-methionine (SAM)-competitive inhibitors of EZH2 methyltransferase activity have been developed. GSK126 is a powerful, highly selective, SAM-competitive, small-molecule inhibitor of EZH2 methyltransferase activity. This inhibitor lowers H3K27me3 levels and reactivates the repressed PRC2 target genes [53]. It has been used to efficiently inhibit the proliferation of DLBCL cell lines bearing EZH2 mutation. In addition, animals that had Karpas-422 xenograft tumors experienced significant tumor regression following treatment with GSK126 [54]. Interestingly, Claro de Mello et al. reported that the EZH2 silencing in ATC cell models by CRISPR/Cas9 and EPZ6438, another SAM-competitive small-molecule, resulted in the reduction of cell growth, migration and invasion in vitro and in vivo mouse models [25]. These findings underline that the inhibition of HMGA1 and EZH2 activity is able to induce a drastic reduction of ATC vitality associated with cell cycle arrest and increased apoptotic rate.
Subsequently, we hypothesized that the combined impairment of these two ATC-master regulator genes would synergistically increase the sensitivity to the combination of ET-743 and GSK. Indeed, treatment with both ET-743 and GSK resulted in a further increase in apoptotic cell death of the ATC cell lines, whereas no significant effects were achieved after treatment with the same drugs of a primary thyroid culture where slight cytotoxic effects were observed at high doses only with GSK126. Conversely, even treatment with high doses of ET-743 did not result in cytotoxic effects on normal thyroid cells.
Furthermore, ATC cells treated with ET-743 showed decreased expression levels of two HMGA-regulated genes: Cyclin B, a master regulator of cell cycle progression, and Bcl-2, an anti-apoptotic protein deeply involved in the carcinogenesis process. Equally, GSK126 exposure, through EZH2 inhibition, increased the expression levels of EZH2-downregulated genes: E-cadherin, a surface protein whose loss is pivotal for the EMT, and CDKN1A, a potent cell cycle inhibitor, and PAX8, a thyroid-specific transcription factor that controls thyroid differentiation, whose expression is downregulated in ATC by EZH2 action [23].
Therefore, these findings provide valuable insights into the potential benefits of combining ET-743 and GSK126 in pre-clinical models of ATC, and their combination treatment may represent a promising strategy for ATC therapy as also supported by the lack of toxic effects on normal thyroid cells. Noteworthy since HMGA and EZH2 overexpression have been observed in almost all human malignancies, and in particular in the most aggressive forms [53,55], a therapy based on the combination of HMGA and EZH2 inhibitors can be taken into consideration also for other human malignant neoplasms.
The current successful use of both drugs in clinical routines for other human malignancies further supports the proposal of this novel therapy.
Supplementary Material
Acknowledgements
We thank Mrs. Giulia Speranza, Ms. Giuseppina Ippolito and Mrs. Mariarosaria Montagna from IEOS-CNR (Naples, Italy) for their technical support. This work was supported by Associazione Partenopea Ricerche Oncologiche (APRO).
Funding Statement
This research received no external funding
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
Conceptualization, M.D.M., A.F. and F.E.; methodology, M.D.M., S.P., M.D.P., D.T., N.M.D.C, P.P., P.C., A.F. and F.E.; software, M.D.M., S.P., M.D.P., N.M.D.C, F.E.; validation, M.D.M., S.P., N.M.D.C. and F.E.; formal analysis, M.D.M., S.P., N.M.D.C., P.P. and F.E.; investigation, M.D.M., S.P. and F.E.; resources, M.D.P., D.T. and A.F.; data curation, M.D.M., S.P., M.D.P., D.T., N.M.D.C., P.P., P.C., A.F. and F.E.; writing-original draft preparation, M.D.M., S.P., M.D.P., D.T., N.M.D.C., P.P., P.C., A.F. and F.E.; writing – review and editing M.D.M., S.P., M.D.P., D.T., N.M.D.C., P.P., P.C., A.F. and F.E.; supervision, P.C, A.F. and F.E.; project administration, P.C., A.F. and F.E. All authors have read and agreed to the published version of the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2023.2298027
References
- [1].Wells SA Jr. Progress in endocrine neoplasia. Clin Cancer Res. 2016 Oct 15;22(20):4981–4988. PubMed PMID: 27742784; PubMed Central PMCID: PMCPMC5111798. doi: 10.1158/1078-0432.CCR-16-0384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bai Y, Kakudo K, Jung CK.. Updates in the pathologic classification of thyroid neoplasms: a review of the world health organization classification. Endocrinol Metab (Seoul). 2020. Dec;35(4):696–715. PubMed PMID: 33261309; PubMed Central PMCID: PMCPMC7803616. doi: 10.3803/EnM.2020.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Baloch ZW, Asa SL, Barletta JA, et al. Overview of the 2022 WHO classification of thyroid neoplasms. Endocr Pathol. 2022. Mar;33(1):27–63. PubMed PMID: 35288841. doi: 10.1007/s12022-022-09707-3 [DOI] [PubMed] [Google Scholar]
- [4].Hu J, Yuan IJ, Mirshahidi S, et al. Thyroid carcinoma: phenotypic features, underlying biology and potential relevance for targeting therapy. Int J Mol Sci. 2021 Feb 16;22(4): PubMed PMID: 33669363; PubMed Central PMCID: PMCPMC7920269. doi: 10.3390/ijms22041950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tesselaar MH, Smit JW, Nagarajah J, et al. Pathological processes and therapeutic advances in radioiodide refractory thyroid cancer. J Mol Endocrinol. 2017. Nov;59(4):R141–R154. PubMed PMID: 28931558. doi: 10.1530/JME-17-0134 [DOI] [PubMed] [Google Scholar]
- [6].Nagaiah G, Hossain A, Mooney CJ, et al. Anaplastic thyroid cancer: a review of epidemiology, pathogenesis, and treatment. J Oncol. 2011;2011:542358. PubMed PMID: 21772843; PubMed Central PMCID: PMCPMC3136148. doi: 10.1155/2011/542358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jannin A, Escande A, Al Ghuzlan A, et al. Anaplastic thyroid carcinoma: an update. Cancers (Basel). 2022 Feb 19;14(4): 1061. PubMed PMID: 35205809; PubMed Central PMCID: PMCPMC8869821. doi: 10.3390/cancers14041061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Molinaro E, Romei C, Biagini A, et al. Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat Rev Endocrinol. 2017. Nov;13(11):644–660. PubMed PMID: 28707679. doi: 10.1038/nrendo.2017.76 [DOI] [PubMed] [Google Scholar]
- [9].Ratajczak M, Gawel D, Godlewska M. Novel inhibitor-based therapies for thyroid cancer—an update. Int J Mol Sci. 2021 Oct 31;22(21):11829. PubMed PMID: 34769260; PubMed Central PMCID: PMCPMC8584403. doi: 10.3390/ijms222111829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007. Dec;7(12):899–910. PubMed PMID: 18004397. doi: 10.1038/nrc2271 [DOI] [PubMed] [Google Scholar]
- [11].Palmieri D, Valentino T, D’Angelo D, et al. HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene. 2011 Jul 7;30(27):3024–35. PubMed PMID: 21339738. doi: 10.1038/onc.2011.21 [DOI] [PubMed] [Google Scholar]
- [12].Palumbo A Jr., Da Costa NM, Esposito F, et al. HMGA2 overexpression plays a critical role in the progression of esophageal squamous carcinoma. Oncotarget. 2016 May 3;7(18):25872–84. PubMed PMID: 27027341; PubMed Central PMCID: PMCPMC5041951. doi: 10.18632/oncotarget.8288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].De Martino M, Fusco A, Esposito F. HMGA and cancer: a review on patent literatures. Recent Pat Anticancer Drug Discov. 2019;14(3):258–267. PubMed PMID: 31538905. doi: 10.2174/1574892814666190919152001 [DOI] [PubMed] [Google Scholar]
- [14].De Martino M, Esposito F, Pellecchia S, et al. HMGA1-regulating microRnas let-7a and miR-26a are downregulated in human seminomas. Int J Mol Sci. 2020 Apr 24;21(8). PubMed PMID: 32344629; PubMed Central PMCID: PMCPMC7215726. doi: 10.3390/ijms21083014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Scala S, Portella G, Fedele M, et al. Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc Natl Acad Sci USA. 2000 Apr 11;97(8):4256–61. PubMed PMID: 10759549; PubMed Central PMCID: PMCPMC18219. doi: 10.1073/pnas.070029997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mansoori B, Mohammadi A, Shirjang S, et al. HMGI-C suppressing induces P53/caspase9 axis to regulate apoptosis in breast adenocarcinoma cells. Cell Cycle. 2016. Oct;15(19):2585–2592. PubMed PMID: 27245202; PubMed Central PMCID: PMCPMC5053549. doi: 10.1080/15384101.2016.1190892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fujikane R, Komori K, Sekiguchi M, et al. Function of high-mobility group a proteins in the DNA damage signaling for the induction of apoptosis. Sci Rep. 2016 Aug 19;6(1):31714. PubMed PMID: 27538817; PubMed Central PMCID: PMCPMC4990841. doi: 10.1038/srep31714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Masudo K, Suganuma N, Nakayama H, et al. EZH2 overexpression as a useful prognostic marker for aggressive behaviour in thyroid cancer. Vivo. 2018. Jan-Feb;32(1):25–31. PubMed PMID: 29275295; PubMed Central PMCID: PMCPMC5892628. doi: 10.21873/invivo.11200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ferrari KJ, Scelfo A, Jammula S, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014 Jan 9;53(1):49–62. PubMed PMID: 24289921. doi: 10.1016/j.molcel.2013.10.030 [DOI] [PubMed] [Google Scholar]
- [20].Hojfeldt JW, Laugesen A, Willumsen BM, et al. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol. 2018. Mar;25(3):225–232. PubMed PMID: 29483650; PubMed Central PMCID: PMCPMC5842896. doi: 10.1038/s41594-018-0036-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011 Jan 20;469(7330):343–9. PubMed PMID: 21248841; PubMed Central PMCID: PMCPMC3760771. doi: 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhao X, Wu X. Polycomb-group proteins in the initiation and progression of cancer. J Genet Genomics. 2021 Jun 20;48(6):433–443. PubMed PMID: 34266781. doi: 10.1016/j.jgg.2021.03.013 [DOI] [PubMed] [Google Scholar]
- [23].Borbone E, Troncone G, Ferraro A, et al. Enhancer of zeste homolog 2 overexpression has a role in the development of anaplastic thyroid carcinomas. J Clin Endocrinol Metab. 2011. Apr;96(4):1029–38. PubMed PMID: 21289264. doi: 10.1210/jc.2010-1784 [DOI] [PubMed] [Google Scholar]
- [24].Esposito F, Tornincasa M, Pallante P, et al. Down-regulation of the miR-25 and miR-30d contributes to the development of anaplastic thyroid carcinoma targeting the polycomb protein EZH2. J Clin Endocrinol Metab. 2012. May;97(5):E710–8. PubMed PMID: 22399519. doi: 10.1210/jc.2011-3068 [DOI] [PubMed] [Google Scholar]
- [25].de Mello DC, Saito KC, Cristovao MM, et al. Modulation of EZH2 activity induces an antitumoral effect and cell redifferentiation in anaplastic thyroid cancer. Int J Mol Sci. 2023 Apr 26;24(9):7872. PubMed PMID: 37175580; PubMed Central PMCID: PMCPMC10178714. doi: 10.3390/ijms24097872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pallante P, Battista S, Pierantoni GM, et al. Deregulation of microRNA expression in thyroid neoplasias. Nat Rev Endocrinol. 2014. Feb;10(2):88–101. PubMed PMID: 24247220. doi: 10.1038/nrendo.2013.223 [DOI] [PubMed] [Google Scholar]
- [27].Pellecchia S, Sepe R, Decaussin-Petrucci M, et al. The long non-coding RNA Prader Willi/Angelman Region RNA5 (PAR5) is downregulated in anaplastic thyroid carcinomas where it acts as a tumor suppressor by reducing EZH2 activity. Cancers (Basel). 2020 Jan 17;12(1):235. PubMed PMID: 31963578; PubMed Central PMCID: PMCPMC7017000. doi: 10.3390/cancers12010235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].De Martino M, Nicolau-Neto P, Ribeiro Pinto LF, et al. HMGA1 induces EZH2 overexpression in human B-cell lymphomas. Am J Cancer Res. 2021;11(5):2174–2187. PubMed PMID: 34094676; PubMed Central PMCID: PMCPMC8167683 [PMC free article] [PubMed] [Google Scholar]
- [29].Esposito F, De Martino M, Petti MG, et al. HMGA1 pseudogenes as candidate proto-oncogenic competitive endogenous RNAs. Oncotarget. 2014 Sep 30;5(18):8341–54. PubMed PMID: 25268743; PubMed Central PMCID: PMCPMC4226687. doi: 10.18632/oncotarget.2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].D’Angelo D, Borbone E, Palmieri D, et al. The impairment of the High Mobility Group a (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur J Cancer. 2013. Mar;49(5):1142–51. PubMed PMID: 23149213. doi: 10.1016/j.ejca.2012.10.014 [DOI] [PubMed] [Google Scholar]
- [31].Loria R, Laquintana V, Bon G, et al. HMGA1/E2F1 axis and NFkB pathways regulate LPS progression and trabectedin resistance. Oncogene. 2018. Nov;37(45):5926–5938. PubMed PMID: 29980789; PubMed Central PMCID: PMCPMC6224401. doi: 10.1038/s41388-018-0394-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen YT, Zhu F, Lin WR, et al. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol. 2016. Apr;77(4):757–65. PubMed PMID: 26898301. doi: 10.1007/s00280-016-2990-1 [DOI] [PubMed] [Google Scholar]
- [33].Namba H, Nakashima M, Hayashi T, et al. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab. 2003. Sep;88(9):4393–7. PubMed PMID: 12970315. doi: 10.1210/jc.2003-030305 [DOI] [PubMed] [Google Scholar]
- [34].Landa I, Pozdeyev N, Korch C, et al. Comprehensive genetic characterization of human thyroid cancer cell lines: a validated panel for preclinical studies. Clin Cancer Res. 2019 May 15;25(10):3141–3151. PubMed PMID: 30737244; PubMed Central PMCID: PMCPMC6522280. doi: 10.1158/1078-0432.CCR-18-2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vicini E, Loiarro M, Di Agostino S, et al. 17-beta-estradiol elicits genomic and non-genomic responses in mouse male germ cells. J Cell Physiol. 2006. Jan;206(1):238–245. PubMed PMID: 15991248. doi: 10.1002/jcp.20454 [DOI] [PubMed] [Google Scholar]
- [36].Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016 Sep 15;32(18):2866–8. PubMed PMID: 27153664; PubMed Central PMCID: PMCPMC5018366. doi: 10.1093/bioinformatics/btw230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].De Martino M, Palma G, Azzariti A, et al. The HMGA1 pseudogene 7 induces miR-483 and miR-675 upregulation by activating Egr1 through a ceRNA mechanism. Genes (Basel). 2017 Nov 17;8(11):330. PubMed PMID: 29149041; PubMed Central PMCID: PMCPMC5704243. doi: 10.3390/genes8110330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sinisi AA, Chieffi P, Pasquali D, et al. EPN: a novel epithelial cell line derived from human prostate tissue. Vitro Cell Dev Biol Anim. 2002. Mar;38(3):165–172. PubMed PMID: 12026165. doi: [DOI] [PubMed] [Google Scholar]
- [39].Pero R, Lembo F, Chieffi P, et al. Translational regulation of a novel testis-specific RNF4 transcript. Mol Reprod Dev. 2003. Sep;66(1):1–7. PubMed PMID: 12874792. doi: 10.1002/mrd.10322 [DOI] [PubMed] [Google Scholar]
- [40].Tang J, Wennerberg K, Aittokallio T. What is synergy? The saariselka agreement revisited. Front Pharmacol. 2015;6:181. PubMed PMID: 26388771; PubMed Central PMCID: PMCPMC4555011: doi: 10.3389/fphar.2015.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cao Q, Yu J, Dhanasekaran SM, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008 Dec 11;27(58):7274–84. PubMed PMID: 18806826; PubMed Central PMCID: PMCPMC2690514. doi: 10.1038/onc.2008.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fan T, Jiang S, Chung N, et al. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res. 2011. Apr;9(4):418–29. PubMed PMID: 21383005; PubMed Central PMCID: PMCPMC3078218. doi: 10.1158/1541-7786.MCR-10-0511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Manzanares I, Cuevas C, Garcia-Nieto R, et al. Advances in the chemistry and pharmacology of ecteinascidins, a promising new class of anti-cancer agents. Curr Med Chem Anticancer Agents. 2001. Nov;1(3):257–276. PubMed PMID: 12678757. doi: 10.2174/1568011013354561 [DOI] [PubMed] [Google Scholar]
- [44].Jimenez PC, Wilke DV, Branco PC, et al. Enriching cancer pharmacology with drugs of marine origin. Br J Pharmacol. 2020. Jan;177(1):3–27. PubMed PMID: 31621891; PubMed Central PMCID: PMCPMC6976878. doi: 10.1111/bph.14876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol. 2007. Jul;8(7):595–602. PubMed PMID: 17586092. doi: 10.1016/S1470-2045(07)70175-4 [DOI] [PubMed] [Google Scholar]
- [46].Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of Trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016 Mar 10;34(8):786–93. PubMed PMID: 26371143; PubMed Central PMCID: PMCPMC5070559 online at www.jco.org. Author contributions are found at the end of this article. doi: 10.1200/JCO.2015.62.4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Poveda A, Vergote I, Tjulandin S, et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer: outcomes in the partially platinum-sensitive (platinum-free interval 6-12 months) subpopulation of OVA-301 phase III randomized trial. Ann Oncol. 2011. Jan;22(1):39–48. PubMed PMID: 20643862; PubMed Central PMCID: PMCPMC3003616. doi: 10.1093/annonc/mdq352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pignata S, Scambia G, Villanucci A, et al. A European, observational, prospective trial of trabectedin plus pegylated liposomal doxorubicin in patients with platinum-sensitive ovarian cancer. Oncology. 2021. Apr;26(4):e658–e668. PubMed PMID: 33289956; PubMed Central PMCID: PMCPMC8018301. doi: 10.1002/onco.13630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010. Aug;9(8):2157–63. PubMed PMID: 20647340. doi: 10.1158/1535-7163.MCT-10-0263 [DOI] [PubMed] [Google Scholar]
- [50].Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020 Jul 28;13(1):104. PubMed PMID: 32723346; PubMed Central PMCID: PMCPMC7385862. doi: 10.1186/s13045-020-00937-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Miranda TB, Cortez CC, Yoo CB, et al. Dznep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009. Jun;8(6):1579–88. PubMed PMID: 19509260; PubMed Central PMCID: PMCPMC3186068. doi: 10.1158/1535-7163.MCT-09-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nakayama H, Saito N, Kasajima R, et al. Validation of EZH2 inhibitor efficiency in anaplastic thyroid carcinoma cell lines. Anticancer Res. 2023. Mar;43(3):1073–1077. PubMed PMID: 36854530. doi: 10.21873/anticanres.16252 [DOI] [PubMed] [Google Scholar]
- [53].Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016. Feb;22(2):128–134. PubMed PMID: 26845405; PubMed Central PMCID: PMCPMC4918227. doi: 10.1038/nm.4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012 Dec 6;492(7427):108–12. PubMed PMID: 23051747. doi: 10.1038/nature11606 [DOI] [PubMed] [Google Scholar]
- [55].Zhang D, Yang XJ, Luo QD, et al. EZH2 enhances the invasive capability of renal cell carcinoma cells via activation of STAT3. Mol Med Rep. 2018. Mar;17(3):3621–3626. PubMed PMID: 29286132; PubMed Central PMCID: PMCPMC5802166. doi: 10.3892/mmr.2017.8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.