

ABSTRACT
Triglycerides, phospholipids, and cholesterol are prominent lipid types that serve diverse biological functions. These lipids act as vital structural components of cell membranes, provide energy for the body, and serve as signaling molecules that regulate physiological and pathological processes, including regulated cell death (RCD). While the initiation signals, biochemical pathways, and effector molecules may differ, increased membrane permeabilization from plasma membranes or organelles is a shared characteristic of RCD. This process involves alterations in lipid species and functions, along with autophagic degradation. Thus, the lipid hypothesis presents a novel perspective that complements other theories elucidating the features of cell survival and cell death. In this review, we present a comprehensive summary of the latest findings on the role and potential mechanisms of diverse lipids in influencing RCD processes, encompassing apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy.
Abbreviations
ACSL: acyl-CoA synthetase long chain family; DISC: death-inducing signaling complex; DAMPs: danger/damage-associated molecular patterns; Dtgn: dispersed trans-Golgi network; FAR1: fatty acyl-CoA reductase 1; GPX4: glutathione peroxidase 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LPS: lipopolysaccharide; MUFAs: monounsaturated fatty acids; MOMP: mitochondrial outer membrane permeabilization; MLKL, mixed lineage kinase domain like pseudokinase; oxPAPC: oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine; OxPCs: oxidized phosphatidylcholines; PUFAs: polyunsaturated fatty acids; POR: cytochrome p450 oxidoreductase; PUFAs: polyunsaturated fatty acids; RCD: regulated cell death; RIPK1: receptor interacting serine/threonine kinase 1; SPHK1: sphingosine kinase 1; SOAT1: sterol O-acyltransferase 1; SCP2: sterol carrier protein 2; SFAs: saturated fatty acids; SLC47A1: solute carrier family 47 member 1; SCD: stearoyl-CoA desaturase; VLCFA: very long chain fatty acids
KEYWORDS: Apoptosis, autophagy, cell death, lipid metabolism, pathway
Introduction
Lipids play a critical role in various biological processes. One of their primary functions is as an energy source for cellular processes, with cells breaking down triglycerides [1] or sterol esters [2] from lipid droplets to generate metabolic energy. Lipid storage abnormalities are associated with diseases, such as obesity, diabetes, and atherosclerosis [3,4]. Additionally, lipids serve as the primary structural components of biological membranes, providing hydrophobic barriers that separate intracellular compartments and the extracellular environment. Sterols, fatty acids, and phospholipids are essential for membrane synthesis, and their varied compositions affect membrane charge density and surface tension, fundamental for membrane homeostasis [5]. Lipids also facilitate transmembrane protein function, serving as signaling molecules. For example, cardiolipin binds to specific sites within the membrane domain of the respiratory complex, providing stabilization for the highly tilted transmembrane helices of the subunit [6–8]. Moreover, lipids are substrates for post-translational modification. Proteins can be modified with fatty acids, which can regulate intracellular trafficking, subcellular localization, and protein-protein as well as protein-lipid interactions [9,10]. Palmitate is a common fatty acid modifying group that may contribute to metabolic diseases [9].
Regulated cell death (RCD) is a controllable biological process that extensively participates in the development or tissue homeostasis of multicellular organisms [11]. Linked to the pathogenesis of human diseases, including autoimmunity, viral infections, neurodegenerative diseases, and cancer, RCD is divided into apoptotic and nonapoptotic forms, each exhibiting different molecular mechanisms and signaling regulation [12]. While membrane permeabilization appears as a common theme in RCD, each form of cell death involves unique membrane-related changes that trigger and regulate their distinct biochemical machinery. Lipids are essential for signaling in cell death and macroautophagy/autophagy, a tightly regulated process involved in lysosomal degradation and recycling [13].
In this review, we discuss the role of different lipids in RCD and autophagy, and highlight the challenges and opportunities associated with activating cell death pathways through precision lipid intervention [14]. The post-genomic revolution and systems biology have made lipid biology a cutting-edge research field.
Lipid classification and nomenclature
Lipids are a group of organic compounds that are hydrophobic or amphiphilic, and are characterized by their complexity and heterogeneity, making a precise definition difficult. In 2005, the International Lipid Classification and Nomenclature Committee (ILCNC) developed a comprehensive classification system for lipids based on the concept of two fundamental “building blocks”: ketoacyl and isoprene groups [15]. This system has been widely adopted by the lipidomics community and divides lipids into eight categories, each with its own subclassification hierarchy that can be further divided into classes, subclasses, and, in some cases, 4th-level classes for prenol lipids. The classification system assigns each lipid a unique LIPID MAPS Structure Database ID (LM ID), which is based on the classification scheme and provides a systematic means of identifying each lipid molecule. For example, palmitic acid belongs to the category of fatty acyls (FA), main class of fatty acids and conjugates (FA01), subclass of straight chain fatty acids (FA0101), and has a unique LM ID of “LMFA01010001”. The LIPID MAPS Structure Database currently contains 47,817 unique lipid structures, making it the largest public lipid-only database in the world. LIPID MAPS offers an online website (https://lipidmaps.org/) to provide useful information on lipid molecules and structures [16,17].
Lipid nomenclature falls into two main categories: systematic names and common names. Common names include abbreviations that are a convenient way to define acyl/alkyl chains in acylglycerols, sphingolipids, and glycerophospholipids [18]. For example, “palmitic acid” is a common name, while “hexadecanoic acid” is the systematic name, with synonyms such as palmitate, C16:0, and cetylic acid. A standardized nomenclature for lipid species is important to promote and advance the field of lipidomics. The International Union of Pure and Applied Chemists and the International Union of Biochemistry and Molecular Biology (IUPAC-IUBMB) Commission on Biochemical Nomenclature has defined guidelines for systematic names to provide a comprehensible shorthand notation for commonly analyzed lipids. Table 1 lists some common names of example lipids involved in RCD, according to the LIPID MAPS web resources.
Table 1.
Lipids involved in regulated cell death.
| Category | Example (common name) | Synonyms | Subclass | RCD involved | |
|---|---|---|---|---|---|
| Fatty acyls (FA) | Palmitic acid | C16:0 | Straight chain fatty acids | Ferroptosis, pyroptosis | |
| Stearic acid | C18:0 | Straight chain fatty acids | Ferroptosis, pyroptosis | ||
| Arachidonic acid | AA | Unsaturated fatty acids | Ferroptosis | ||
| Adrenic Acid | C22:4n-6,9,12,15 | Unsaturated fatty acids | Ferroptosis | ||
| Oleic acid | C18:1n-9 | Unsaturated fatty acids | Ferroptosis | ||
| Cis-9-palmitoleic acid | Palmitoleic acid; C16:1n-7 | Unsaturated fatty acids | Ferroptosis | ||
| Linoleic acid | C18:2n-6,9 | Unsaturated fatty acids | Ferroptosis | ||
| Docosahexaenoic acid | C22:6n-3,6,9,12,15,18 | Unsaturated fatty acids | Ferroptosis; pyroptosis |
||
| Docosapolyenoic acid | C22:5n-3,6,9,12,15 | Unsaturated fatty acids | Ferroptosis | ||
| Eicosapentaenoic acid | C20:5n-3,6,9,12,15 | Unsaturated fatty acids | Ferroptosis; pyroptosis |
||
| Acetic acid | Acetate; C2:0 | Straight chain fatty acids | Pyroptosis | ||
| Propionic acid | Propionate; C3:0 | Straight chain fatty acids | Pyroptosis | ||
| Butyric acid | Butanoate; C4:0 | Straight chain fatty acids | Pyroptosis | ||
| Cetyl alcohol | 1-hexadecanol | / | Ferroptosis | ||
| Stearyl alcohol | 1-octadecanol | / | Ferroptosis | ||
| Glycerolipids (GL) | Triglycerides (16:0/16:0/16:0) | Tripalmitin | Triacylglycerols | / | |
| Category | Example (common name) | Synonyms | Subclass | RCD involved | |
| Glycerophospholipids (GP) | Cardiolipin (CL) | 1,3-bis (sn-3-phosphatidyl)-sn-glycerol | Diacylglycerophosphoglycero phosphodiradylglycerols |
Apoptosis, pyroptosis | |
| PS (P-16:0/12:0) | PS (P-28:0) | 1-(1Z-alkenyl),2-acylglycerophosphoserines | Apoptosis, necroptosis | ||
| PtdIns | PI | Diacylglycerophosphoinositols | / | ||
| PtdIns (12:0/13:0) | PI 25:0 | Diacylglycerophosphoinositols | / | ||
| PtdIns3P | / | Diacylglycerophosphoinositol monophosphates | Autophagy | ||
| PtdIns4P | / | Diacylglycerophosphoinositol monophosphates | Pyroptosis, autophagy | ||
| PC (16:0/20:4(5Z,8Z,10E,14Z) (12OH[S])) | PC (16:0_20:4(OH)) | Oxidized glycerophosphocholines | Pyroptosis | ||
| Sphingolipids (SP) | Sphingosine | 4-Sphingenine | Sphing-4-enines (Sphingosines) | Pyroptosis | |
| Sphingosine-1-phosphate | / | Sphingoid base 1-phosphates | Apoptosis | ||
| Ceramide | N-acylsphingosine | N-acylsphingosines (ceramides) | Apoptosis, pyroptosis, autophagy | ||
| Cer (d18:1/14:0) | C14 Cer | N-acylsphingosines (ceramides) | / | ||
| Category | Example (common name) | Synonyms | Subclass | RCD involved | |
| GD3 | Gangliosides | Autophagy | |||
| Sphingomyelin | Ceramide phosphocholines (sphingomyelins) | Apoptosis | |||
| Sterol Lipids (ST) | Cholesterol | Cholesterol and derivatives | Apoptosis, autophagy; Pyroptosis | ||
| 25-hydroxy-cholesterol | 25-OHC | Cholesterol and derivatives | Pyroptosis | ||
| 22:6 Cholesterol ester | CE (22:6) | Steryl esters | Ferroptosis | ||
| 22:5 Cholesterol ester | CE (22:5) | Steryl esters | Ferroptosis | ||
| Prenol Lipids | Coenzyme Q10 | Ubiquinone-10 | Ubiquinones | Ferroptosis | |
| alpha-tocopherol | Vitamin E | Vitamin E | Ferroptosis | ||
Lipid mechanisms in RCD
RCD heavily relies on the recruitment and activation of specific signaling pathways, and emerging evidence highlights the significant involvement of lipids in different forms of RCD. In this section, our focus centers on exploring the specific lipids that contribute to RCD pathways and we shed light on how these lipids drive the development of major forms of RCD, including apoptosis, necroptosis, pyroptosis, and ferroptosis.
Apoptosis
In 1972, apoptosis was initially characterized by distinct morphological features, including cell shrinkage, nuclear condensation, and the formation of apoptotic bodies [19]. The caspase activation cascade downstream of mitochondrial CYCS (cytochrome C, somatic) release involves two main pathways: the intrinsic and extrinsic pathways (Figure 1). In apoptosis, lipids play a crucial role in regulating either the intrinsic or extrinsic pathway. Investigations focus on understanding how lipid composition, fatty acid metabolism, and lipid transport proteins influence mitochondrial integrity, the release of apoptogenic factors, and the activation of downstream apoptotic signaling pathways.
Figure 1.
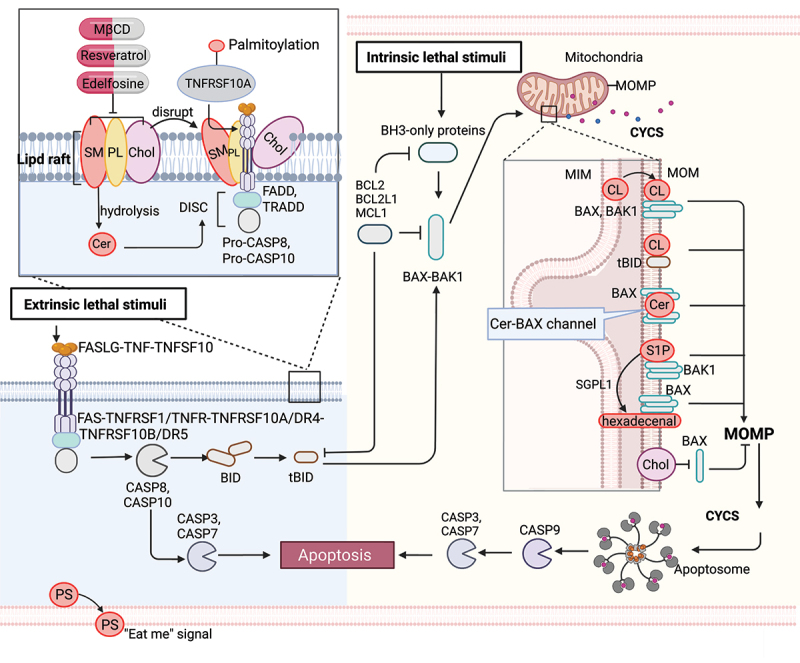
Lipid mechanism in apoptosis. Under intrinsic lethal stimuli, activated BAX or BAK1 oligomerizes and forms pores in the mitochondrial membrane, resulting in mitochondrial outer membrane permeabilization (MOMP) and the subsequent release of CYCS/cytochrome c into the cytosol. During this process, cardiolipin (CL) translocates to the mitochondrial outer membrane (MOM) and acts as a binding site for truncated BID (tBID), BAX or BAK1, triggering pore formation. Furthermore, mitochondrial ceramide (Cer) is necessary for BAX oligomerization. Cer and activated BAX together contribute to the formation of stable pores known as the ceramide channel (Cer-BAX channel). Additionally, sphingosine-1-phosphate (S1P) is degraded by SGPL1/S1P lyase to form hexadecenal. S1P and hexadecenal specifically cooperate with BAK1 and BAX, respectively. Conversely, cholesterol (Chol) accumulation in the mitochondrial membrane inhibits MOMP. Under extrinsic lethal stimuli, activated CASP8 can induce apoptosis directly by activating CASP3 or CASP7 or indirectly through the activation of the BH3-only protein BID, leading to the intrinsic apoptotic pathway. Lipid raft-disrupting agents such as methyl-beta cyclodextrin (MβCD), resveratrol, and edelfosine can trigger apoptosis. Cer, a hydrolyzed product of sphingomyelin (SM), induces coalescence of elementary rafts and enhances DISC formation, thereby amplifying FAS signaling. Furthermore, palmitoylation of TNFRSF10A/DR4 promotes TNFSF10/TRAIL-mediated apoptotic signaling. Phosphatidylserine (PS) exposure serves as an “eat me” signal for apoptotic cell clearance. [change TRAIL to “TNFSF10”.
Intrinsic pathway
The intrinsic pathway, also called the mitochondrial pathway, is triggered by various stress signals, such as toxic agents and DNA damage. Upon intrinsic apoptotic stimuli, pro-apoptotic BH3-only proteins (e.g., BCL2L11/BIM [BCL2 like 11], PMAIP1/NOXA [phorbol-12-myristate-13-acetate-induced protein 1], BBC3/PUMA [BCL2 binding component 3], and BAD [BCL2 associated agonist of cell death]) can be upregulated to activate BAX (BCL2 associated X, apoptosis regulator) and BAK1 (BCL2 antagonist/killer 1) at the mitochondrial outer membrane [20,21]. Activated BAX or BAK1 oligomerize and form pores in the mitochondrial membrane, causing mitochondrial outer membrane permeabilization (MOMP), which leads to the release of CYCS into the cytosol [22]. Extrinsic stimuli activate CASP8 (caspase 8), which cleaves the pro-apoptotic BH3-only protein BID (BH3 interacting domain death agonist) to generate truncated BID (tBID), which then translocates to mitochondria, also triggering MOMP. MOMP and the release of CYCS trigger the formation of apoptosomes, which recruit CASP9 (caspase 9) and thereby promote downstream CASP3 (caspase 3) and CASP7 (caspase 7) activation, leading to apoptosis and “the point of no return” [23,24].
Mitochondrial lipids, such as cardiolipin, ceramide, and sphingosine-1-phosphate, are involved in the regulation and propagation of intrinsic apoptosis. In healthy mitochondria, cardiolipin is predominantly found in the inner mitochondria membrane (IMM) and is involved in lipid-protein interactions, ensuring proper mitochondrial function [25,26]. During the apoptosis process, cardiolipin translocates to the mitochondrial outer membrane [27–29] and acts as a target site for binding tBID [21]. The tBID-cardiolipin interaction plays a critical role in the cristae remodeling essential for the release of CYCS into the cytosol and for the formation of fission sites in the mitochondria [30]. In the presence of cardiolipin, BAX and BAK1 then undergoes dimerization to form active oligomers and trigger pore-formation, whereas a lack of cardiolipin results in the formation of inactive BAX and BAK1 oligomers [31]. Moreover, mitochondrial ceramide is produced inside cells in response to pro-apoptotic stimuli [32,33], and the ceramide-enriched membrane is the primary requirement for BAX oligomerization [34–37]. The addition of exogenous ceramide to cultured cells induces the release of CYCS from mitochondria [38,39]. Ceramide and activated BAX together are responsible for forming stable pores known as the ceramide channel in the phospholipid bilayer, leading to the release of CYCS from the mitochondria and activating apoptosis [40,41]. In addition, sphingosine-1-phosphate is degraded by SGPL1 (sphingosine-1-phosphate lyase 1) to form hexadecenal under different stresses. Sphingosine-1-phosphate and hexadecenal cooperate specifically with BAK1 and BAX, respectively, promoting their oligomerization followed by the release of CYCS [42]. BAX activation is inhibited by cholesterol and by decreases in membrane fluidity [43]. Cholesterol accumulation in the mitochondrial membrane contributes to diminishing cellular capacity for inducing MOMP and resistance to chemotherapy in cancer cells [43,44]. Mechanistically, cholesterol can form ordered domains that inhibit the insertion and activation of BAX into the mitochondrial membrane, which ultimately prevents apoptosis [43,45].
While there are several reports suggesting that fatty acids are capable of inducing apoptosis in different cancer cells, the apoptotic regulation mechanism for different sources and bioactivities of fatty acids remains controversial [46]. For example, polyunsaturated fatty acids (PUFAs), such as docosahexaenoic acid, eicosapentaenoic acid, arachidonic acid, and linoleic acid, promote apoptosis of cancer cells through the upregulation of numerous pro-apoptotic proteins, such as BCL2 family proteins, CYCS, and different caspases [47–49]. Oleic acid serves as a potent anticancer agent by inducing autophagy and apoptotic responses via inhibiting the AKT (AKT serine/threonine kinase)-MTOR (mechanistic target of rapamycin kinase) pathway [50]. Oleic acid induces different apoptotic markers, such as TP53 (tumor protein p53) and cleaved CASP3, while decreasing the expression of CCND1 (cyclin D1) and BCL2. Additionally, stearic acid inhibits invasion and proliferation and induces apoptosis in various human cancer cell types, although the underlying mechanism remains elusive [51]. Given the rich variety of fatty acids, more specific mechanisms of their roles in apoptosis need to be carefully evaluated. Understanding how lipid signaling pathways intersect with intrinsic pathways can provide new insights into apoptotic regulation and potential therapeutic targets.
Extrinsic pathway
The extrinsic apoptotic pathway is initiated by the activation of cell surface death receptors, such as TNFRSF1A/TNFR1 (TNF receptor superfamily member 1A) and TNFRSF1B/TNFR2) [52,53], FAS [54,55], TNFRSF10A/DR4 (TNF receptor superfamily member 10a), and TNFRSF10B/DR5 (TNF receptor superfamily member 10b) [56,57]. Upon binding of their ligands (e.g., FASLG [Fas ligand], TNFSF10/TRAIL [TNF superfamily member 10], and TNF [tumor necrosis factor]), the death receptors oligomerize and recruit adapter proteins (TRADD [TNFRSF1A associated via death domain] and FADD [Fas associated via death domain]) [58] and pro-CASP8, resulting in the formation of the death-inducing signaling complex (DISC) [58], which then activates CASP8. Activated CASP8 induces apoptosis either directly by proteolytically activating an effector caspase (CASP3 or CASP7) or indirectly through proteolytic activation of the BH3-only protein BID into tBID, which then links to the intrinsic apoptotic pathway [59] (Figure 1).
Apoptotic death receptors can be translocated into lipid rafts and trigger downstream apoptotic signals, making lipid rafts behave as scaffolds for coupling adaptor proteins (e.g., FADD and TRADD) and effector proteins (e.g., pro-CASP8 and pro-CASP10) involved in the extrinsic pathway of apoptosis [60–64]. Mechanistically, lipid rafts enhance the apoptotic signaling capacity by: i) promoting death receptor trimerization, which is required for signal transduction, ii) acting as concentrating platforms for DISC assembly and the recruitment of death domains, and iii) protecting death receptors from internalization or enzymatic degradation [65].
Lipid rafts are dynamic membrane microdomains that consist of cholesterol, phospholipids, and sphingomyelin and are key regulators of apoptosis [64]. Altering their composition affects raft behavior. Because cholesterol is usually enriched in rafts, methyl-beta cyclodextrin (MβCD) , a lipid raft-disrupting agent that selectively and efficiently removes cholesterol from membranes, has been widely used [66]. At high concentrations, MβCD can induce cytotoxicity by depleting cholesterol from lipid rafts, triggering apoptosis in cancer cells through the inhibition of the PI3K-Akt-Bad pathway [67,68]. Furthermore, the modification of MβCD with mannose enhances its anticancer activity [69]. In addition, mitochondria-associated lipid microdomains play a role in regulating apoptosis. The disruption of these lipid microdomains in isolated mitochondria by MβCD prevents mitochondrial depolarization and sensitizes lymphoblastoid CEM cells to apoptosis induced by disialoganglioside GD3 and t-BID [70]. However, resveratrol promotes the formation of cholesterol- and sphingolipid-enriched lipid rafts that significantly alter TNFRSF10A/DR4-raft colocalization and sensitize TNFSF10-induced apoptosis in colorectal cancer cells [61]. Edelfosine, a synthetic alkyl lipid, was reported as the first anticancer drug that acts through its interaction with lipid raft domains in cell membranes, promoting their reorganization and eventually leading to the triggering of apoptosis [71]. Edelfosine treatment of multiple myeloma cells leads to FAS-DISC formation and apoptosis [72,73]. Ceramide, a hydrolyzed product of sphingomyelin, induces coalescence of elementary rafts, leading to the formation of large platforms that trap FAS complexes and enhance DISC formation, thereby potentiating FAS signaling [74,75].
Besides lipid composition, protein-lipid interactions also play key roles in raft regulation. For instance, palmitoylation increases a protein’s affinity for rafts [76,77]. Both FAS and TNFRSF10A/DR4 receptor are palmitoylated, and palmitoylation is mandatory for TNFRSF10A/DR4 oligomerization, lipid raft localization, and TNFSF10-induced apoptotic signaling [61]. Inhibiting palmitoylation with 2-bromopalmitate, a non-metabolizable palmitate analog that blocks palmitate incorporation onto proteins, abrogates TNFRSF10A/DR4-mediated TNFSF10 signaling in colorectal cancer cells. Thus, targeting lipid rafts in cancer treatment has the potential to disrupt signaling platforms, inhibit oncogenic receptors, modulate cholesterol homeostasis, disrupt cell adhesion and migration, and overcome drug resistance.
Apoptotic cell clearance
Apoptosis and the clearance of apoptotic cells are essential processes in body development and homeostasis. Phosphatidylserine is one of the major phospholipids present in the plasma membrane, which is normally confined to the cytoplasmic leaflet of the membrane by a “flippase”. However, during apoptosis, a “scramblase” is activated, which quickly exposes phosphatidylserine (PS) on the cell surface [78]. PS exposure was discovered in 1985 as an “eat me” signal that prompts phagocytes to engulf red blood cells [79]. Living cells exhibit phosphatidylserine (PS) exposure when the flippases, specifically TMEM30A (transmembrane protein 30A), are fully inactivated [80]. PS exposure also occurs when cells are transformed with a constitutively active form of ANO6 (anoctamin 6) [81]. However, it’s important to note that PS recognition alone is not enough to trigger engulfment [82]. Cells lacking flippase activity due to a null mutation of TMEM30A or expressing an active form of ANO6 scramblase continuously expose phosphatidylserine without undergoing apoptosis [80,82]. In co-culture experiments with thioglycollate-elicited macrophages, cells lacking TMEM30A-mediated flippase activity are engulfed, while cells expressing active ANO6 are not engulfed [80]. When apoptotic cells cannot expose the phagocytic signal or cannot be swiftly recognized by macrophages, they undergo secondary necrosis and release danger/damage-associated molecular patterns (DAMPs) that activate the immune system. Mice deficient in the phosphatidylserine-binding protein MFGE8 (milk fat globule EGF and factor V/VIII domain containing) develop a systemic lupus erythematosus-type autoimmune disease [83].
In summary, phosphatidylserine plays a critical role in the signal transduction system for apoptotic cell engulfment. Its exposure on apoptotic cells facilitates their recognition and clearance by phagocytes, leading to the maintenance of immune homeostasis, suppression of inflammation, induction of immune tolerance, and resolution of inflammation. Understanding the importance of phosphatidylserine in these processes is essential for elucidating the mechanisms of immune regulation and developing strategies to modulate immune responses and treat immune-related disorders.
Necroptosis
Necroptosis is a caspase-independent form of regulated necrosis, orchestrated by necrosomes and governed by kinases [84,85]. This pathway exhibits morphological features similar to necrosis, such as organelle swelling and plasma membrane rupture, and can be triggered by stimuli shared with apoptosis, such as the activation of death receptors (e.g., TNFRSF1A and TNFRSF1B [86], FAS [87]) or pathogen recognition receptors (e.g., TLR3 [toll like receptor 3] and TLR4 [toll like receptor 4] [88]), in the presence of caspase inhibition [89]. Under these conditions, the activity of CASP8 determines whether a cell will undergo apoptosis or necroptosis. If CASP8 is active, it cleaves RIPK1 (receptor interacting serine/threonine kinase 1) [90] and RIPK3 (receptor interacting serine/threonine kinase 3) [91,92], preventing their activation, and apoptosis proceeds. If CASP8 is inactive, however, a complex called the necrosome containing RIPK1, RIPK3, and MLKL (mixed lineage kinase domain like pseudokinase) forms [93,94]. Within this complex, RIPK3-mediated phosphorylation induces a conformational change that activates MLKL and drives its oligomerization and translocation to the plasma membrane, a step that is essential for the execution of necroptosis [95,96]. MLKL forms pores or cation channels at the plasma membrane that disrupt osmotic balance, leading to the eventual rupture of the plasma membrane [97]. Current models propose that MLKL binds to negatively charged phosphatidylinositol phosphate (PIP) phospholipids via a patch of positively charged amino acids on the surface of a four-helical bundle domain located in its N-terminal region [98] (Figure 2).
Figure 2.
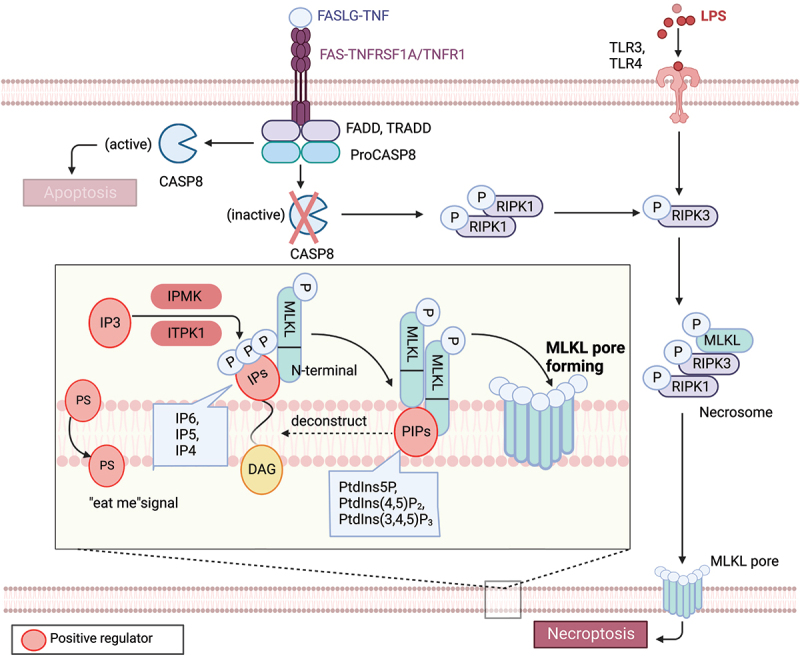
Lipid mechanism in necoroptosis. In the presence of caspase inhibition, necroptosis can be triggered by death receptor stimuli (e.g., TNFRSF1A/TNFR1 and FAS) or pathogen recognition receptor stimuli (e.g., TLR3 and TLR4). When CASP8 is inactive, a complex called the necrosome, comprised of RIPK1, RIPK3, and MLKL, forms. Within this complex, RIPK3-mediated phosphorylation induces a conformational change that activates MLKL, leading to its oligomerization and translocation to the plasma membrane. Subsequently, MLKL forms pores or cation channels at the plasma membrane, resulting in necroptosis. MLKL binds to PtdIns5P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, but not to unphosphorylated phosphatidylinositol (PtdIns) or other phospholipids. PIPs can be enzymatically deconstructed into diacylglycerol and inositol phosphates. MLKL interacts with highly phosphorylated inositol products (e.g., IP6, IP5, IP4) generated by IP kinases (IPMK and ITPK1). Necroptotic cells also expose phosphatidylserine (PS) on the outer leaflet of the plasma membrane, acting as an “eat-me” signal that can be recognized and phagocytosed.
Phosphoinositide lipids consist of seven naturally occurring phosphorylated derivatives of phosphatidylinositol (PtdIns), namely phosphatidylinositol-3-phosphate (PtdIns3P), phosphatidylinositol-4-phosphate (PtdIns4P), phosphatidylinositol-5-phosphate (PtdIns5P), phosphatidylinositol-3,4-bisphosphate (PtdIns [3,4]P2), phosphatidylinositol-3,5-bisphosphate (PtdIns [3,5]P2), phosphatidylinositol-4,5-bisphosphate (PtdIns [4,5]P2), and phosphatidylinositol-3,4,5-trisphosphate (PtdIns [3–5]P3) [99]. Lipid array analyses have shown that recombinant MLKL binds to PtdIns5P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, but not to unphosphorylated phosphatidylinositol PtdIns or other phospholipids [98]. However, experiments with MLKL in micelles suggest a binding preference for PtdIns(4,5)P2 in the presence of PtdIns and PtdIns4P [100]. Interfering with the formation of PtdIns5P or PtdIns(4,5)P2 inhibits TNF-induced necroptosis, rather than apoptosis. Therefore, PIPs, including PtdIns5P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, act as lipid receptors of MLKL in the inner leaflet of the plasma membrane.
PIPs are enzymatically deconstructed into diacylglycerol and inositol phosphates (IPs) [100]. A current model of MLKL activation assumes that its interaction with soluble inositol phosphates and membrane lipids is also important for MLKL to exert its membrane-destabilizing function [101]. MLKL binds to certain PIPs, in part via recognition of their IP polar head group. For example, MLKL directly binds inositol hexakisphosphate (IP6) with high affinity and does not require phospholipids to mediate structural rearrangements in MLKL. Mutagenesis of IP-binding sites in the N-terminal domain impairs necroptosis. Beginning with an unbiased genetic screen in haploid human cell knockouts, necroptosis specifically requires the IP kinase activity of IPMK (inositol polyphosphate multikinase) and ITPK1 (inositol-tetrakisphosphate 1-kinase) for producing highly phosphorylated IPs [101]. However, IP6, but not IP3, promotes an active conformation of MLKL that allows it to undergo necroptosis by binding MLKL and displacing the auto-inhibitory region of its N-terminal bundle. MLKL binds to highly phosphorylated inositol products (e.g., IP6, IP5, and IP4) of the IP kinases. The IP kinases IPMK and ITPK1 are required for TNF-induced necroptosis. Hence, all results indicate an important role for highly phosphorylated IPs as activators of MLKL function. Engagement of IPs and PIPs may occur at distinct stages of MLKL activation, promoting MLKL oligomerization, membrane recruitment, and downstream effector function during necroptosis. Future studies are needed to dissect the precise dynamics of these regulatory mechanisms.
In addition to PIPs, the functional involvement of very long chain fatty acids (VLCFAs) in necroptosis has been demonstrated [102]. In a necroptosis model of colorectal adenocarcinoma cell line HT-29, the accumulation of VLCFAs promotes membrane permeabilization and necroptosis, while perturbing VLCFA biosynthesis via FASN (fatty acid synthase) inactivation delays necroptosis. However, it is not clear whether VLCFAs are directly related to the machinery of necroptosis induction, or whether they are required for the membrane compositions that facilitate cell death by changing the organization of lipids and/or proteins in the membrane. Inactivation of FASN will affect the fatty acid pools and other downstream complex lipids. Therefore, it is important to carefully evaluate whether there are other downstream lipids involved in necroptosis besides VLCFAs.
Similar to apoptosis, necroptotic cells also expose phosphatidylserine on the outer leaflet of the plasma membrane, which acts as an “eat-me” signal and can be recognized and phagocytosed [103,104]. Phosphatidylserine-exposure is inhibited by necrosulfonamide, which binds phosphorylated MLKL and inhibits phosphorylated-MLKL membrane translocation prior to induction of necroptosis. However, it is currently unclear whether the “eat-me” signal involved in necroptosis differs from the one observed in apoptosis. Further research will be necessary to fully comprehend the underlying mechanisms and physiological significance of this necroptotic “eat-me” signal.
Ferroptosis
Ferroptosis is a form of iron-dependent regulated cell death that is characterized by uncontrolled lipid peroxidation and abnormal autophagic activity, ultimately leading to membrane rupture and cell lysis [105–109]. In 2003, during a high-throughput synthetic lethal screening, a novel compound called erastin was discovered to selectively kill engineered tumorigenic cells with an oncogenic RAS mutation [110]. Subsequently, in 2012, the term “ferroptosis” was coined to describe this iron-dependent, non-apoptotic form of RCD [105]. Ferroptosis can be inhibited by various antioxidant enzymes, especially GPX4 (glutathione peroxidase 4) or AIFM2/FSP1 (apoptosis inducing factor mitochondria associated 2) [111–113], which reduces coenzyme Q10 (CoQ10), one of the most significant lipid antioxidants [114]. In contrast, inhibition of GPX4 or AIFM2 sensitizes ferroptosis by driving lipid peroxidation [115]. Lipid peroxidation products play a crucial role as the primary mediators of ferroptosis [116], and the regulation of lipid metabolism plays a central role in determining the ferroptotic response [117]. Alpha-tocopherol, a prenol lipid and potent biological antioxidant, is widely recognized as the most active form of vitamin E in humans, capable of blocking GPX4 inhibitor RSL3-induced ferroptosis [118]. Numerous lipid-associated molecules have been discovered to control cell sensitivity to ferroptosis [117]. In this section, we summarize the impact of lipid metabolism on ferroptosis from two perspectives: lipid substrates and lipid metabolic enzymes (Figure 3).
Figure 3.
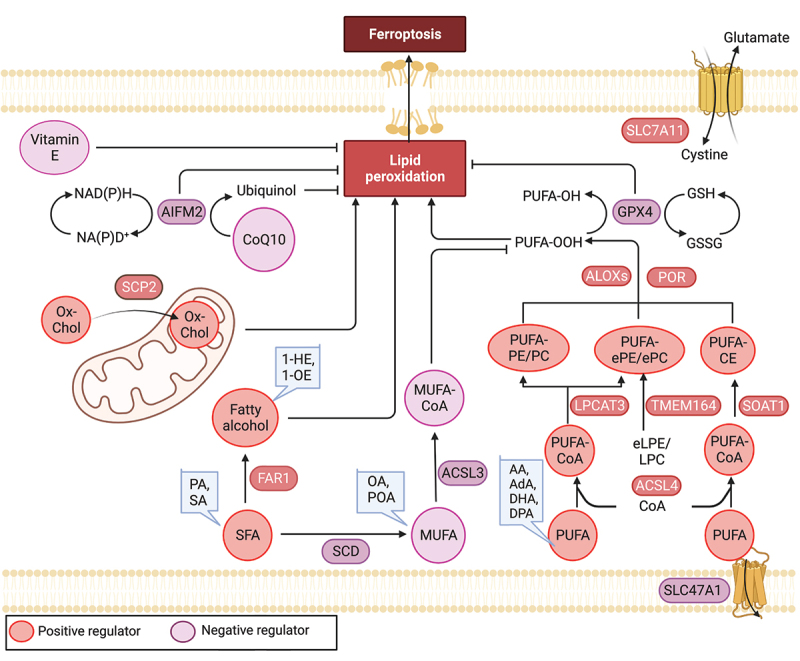
Lipid mechanism in ferroptosis. Ferroptosis is regulated by antioxidant enzymes, including GPX4 and AIFM2, which play a role in reducing ubiquinone (coenzyme Q10) and inhibiting ferroptosis. Among lipids, polyunsaturated fatty acids (PUFAs) are highly susceptible to peroxidation during ferroptosis. Long-chain saturated fatty acids (SFAs), such as palmitic acid (PA, C16:0) and stearic acid (SA, C18:0), as well as their downstream fatty alcohols (1-hexadecanol and 1-octadecanol), sensitize RSL3-induced ferroptosis. Conversely, exogenous monounsaturated fatty acids (MUFAs), including oleic acid (OA, C18:1) and palmitoleic acid (POA, C16:1), suppress ferroptosis. The regulation of key lipid metabolic enzymes involved in lipid synthesis, degradation, storage, transformation, and utilization is crucial in the context of ferroptosis. AA: arachidonic acid, AdA: adrenic acid, DHA: docosahexaenoic acid, DPA: docosapolyenoic acid, 1-HE: 1-hexadecanol, 1-OE: 1-octadecanol, eLPE/LPC: ether lysophosphatidylethanolamine/lysophosphocholine, ePe/epc: ether phosphatidylethanolamine/phosphocholine.
Lipid substrates
The oxidation of polyunsaturated fatty acids (PUFAs) plays a critical role in executing ferroptosis [119]. Lipidomics studies have revealed that, in human fibrosarcoma cells (HT1080) treated with erastin, PUFAs are the most susceptible lipids to peroxidation during ferroptosis [120]. Exogenous PUFAs enhance erastin-induced ferroptosis, while deleting genes that exchange PUFAs with deuterated PUFAs inhibit ferroptosis [121,122]. For example, adding PUFAs to culture media restores sensitivity to cell death in a ferroptosis-tolerant melanoma cell line (UACC-257) [123]. Mechanistically, PUFAs with weak C-H bonds are particularly prone to undergo autoxidation and can be converted into reactive free radicals after being oxidized by free radicals, thus propagating the lipid peroxidation chain reaction. In addition, the double bonds adjacent to methylene groups in PUFAs weaken the hydrogen bond energy of the bis-allylic methylene groups, resulting in an increased susceptibility to hydrogen abstraction and consequent oxygenation [124]. Interestingly, in HT1080 cells long-chain saturated fatty acids (SFAs), including palmitic acid (C16:0) and stearic acid (C18:0), as well as their downstream fatty alcohols (1-hexadecanol and 1-octadecanol), sensitize to RSL3-induced ferroptosis [125]. Interestingly, fatty alcohol demonstrates a more pronounced effect on the induction of ferroptosis [125]. However, the underlying reasons and mechanisms for the heightened impact of fatty alcohol on ferroptosis are yet to be further examined.
PUFAs are especially vulnerable to oxidation but must first be esterified into membrane lipids and undergo oxidation to serve as ferroptotic signals [122]. Lipidomic studies in ferroptotic Pfa1 cells indicate that phosphatidylethanolamines (PEs) containing arachidonic acid (C20:4) or its elongation product, adrenic acid (C22:4), are key phospholipids that undergo oxidation and drive cells toward ferroptosis [122]. Moreover, a genome-wide CRISPR-Cas9 screen in human renal and ovarian carcinoma cells, combined with lipidomic profiling analysis, reveals that peroxisomes synthesize PUFA-containing ether phospholipids/PUFA-ePLs that act as substrates for lipid peroxidation during ferroptosis [126]. In contrast, in the C. elegans model, ether lipids can protect against ferroptosis instead of promoting it [127]. This could be due to the relatively lower presence of PUFAs in ether lipids of C. elegans compared to mammalian cells.
Another study demonstrated that oxidized phosphatidylcholines can induce cardiomyocyte cell death through a ferroptotic pathway during reperfusion injury [128]. In addition to phospholipids, the plasma membrane of mammalian cells contains cholesterol, which affects membrane fluidity. Our cohort has shown that accumulating cholesteryl esters containing PUFAs, including docosahexaenoic acid (C22:6) and docosapolyenoic acid (C22:5), also increases RSL3- or erastin-induced ferroptosis in human pancreatic cancer cell lines [129].
In contrast to PUFAs and SFAs, which promote ferroptosis, exogenous monounsaturated fatty acids (MUFAs), such as oleic acid (C18:1) and palmitoleic acid (C16:1), suppress ferroptosis [130]. Treatments that impede MUFA uptake, activation, or de novo synthesis have the potential to overcome the ferroptosis-resistant state and enhance the effectiveness of existing proferroptotic agents. Mechanistically, MUFAs oppose PUFA activation and inhibit the accumulation of lipid-based reactive oxygen species/ROS within the plasma membrane [130]. Nevertheless, the mechanism by which MUFAs decrease cellular levels of PUFA-phospholipids remains unclear. Monitoring the interplay between PUFAs, SFAs, and MUFAs can provide insights into predicting the susceptibility of cells to ferroptosis.
Additional resources of lipids for ferroptosis can be obtained from lipophagy-mediated lipid degradation [131] or the activation of peroxisomes [126]. Peroxisomes are involved in the biosynthesis of ether-linked phospholipids, which are particularly susceptible to lipid peroxidation-induced ferroptosis.
Lipid metabolic enzymes
Emerging evidence suggests that specific lipid substrates play a crucial role in ferroptosis, and the regulation of key lipid metabolic enzymes is essential in governing processes such as lipid synthesis, degradation, storage, transformation, and utilization [117]. ACSL (acyl-CoA synthetase long chain family) enzymes play a critical role in activating free fatty acids to fatty acyl-CoAs, which are necessary for their incorporation into phospholipids. Among the five human ACSL enzymes (ACSL1, and ACSL3 to ACSL6; ACSL2 does not exist), ACSL4 has a marked preference for activating PUFAs. As a result, the deletion of ACSL4 prevents the incorporation of PUFAs into membrane phospholipids, effectively blocking the execution of ferroptosis [121,122,132]. In contrast, ACSL3 mainly activates MUFAs and exerts an inhibitory function in ferroptosis [130]. Once activated by ACSL4, PUFAs are esterified to lysophospholipids through LPCAT3 (lysophosphatidylcholine acyltransferase 3), an enzyme that transfers the fatty acyl chain from fatty acyl-CoA to 1-acyl lysophospholipid to form various classes of phospholipids. LPCAT3 functions as a ferroptosis prompter downstream of ACSL4, most likely by modulating membrane phospholipid remodeling [121]. Additionally, the lack of LPCAT3 leads to extreme reductions in membrane arachidonate levels during ferroptosis [133]. SCD (stearoyl-CoA desaturase), the lipid desaturase in the synthesis of MUFAs, catalyzes the desaturation of saturated fatty acids and thereby interferes with lipid homeostasis and protects against ferroptosis [134,135]. Conversely, inhibition of SCD decreases CoQ10 during ferroptosis [134]. Furthermore, the peroxisomal enzyme FAR1 (fatty acyl-CoA reductase 1) is critical for SFA-induced ferroptosis by the conversion of fatty acyl-CoA molecules to fatty alcohols, utilizing NADPH as a cofactor. Elimination of FAR1 expression in HT1080 cells results in robust resistance to erastin- or RSL3-induced ferroptosis [125].
Lipid peroxidation can occur via enzymatic or nonenzymatic processes. Nonenzymatic peroxidation of lipids is mediated by carbon- and oxygen-centered radicals, is also known as autoxidation, and requires iron for the initiation step [136]. In contrast, lipid peroxidation during ferroptosis can be catalyzed by cellular enzymes, such as ALOXs (arachidonate lipoxygenases) [120] and POR (cytochrome p450 oxidoreductase) [123,137], in a controlled manner.
ALOXs are dioxygenases that contain nonheme iron and catalyze the stereospecific oxygenation of PUFAs [138]. In humans, there are six members of the ALOX family (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3), named based on the specific carbon position where they introduce oxygen into their PUFA substrates [139]. The involvement of ALOX in ferroptosis is context dependent, as different family members exhibit distinct tissue and cell expression profiles. For instance, the overexpression of ALOX5 [140], ALOX12 [141], or ALOX15 [142] sensitizes human embryonic kidney cells to ferroptosis.
POR belongs to a class of oxidoreductases residing in the endoplasmic reticulum, and it plays a crucial role in xenobiotic detoxification, cellular metabolism, and homeostasis. In genome-wide suppressor screens utilizing CRISPR-Cas9 technology, POR was identified as a promoter of ferroptotic cell death in cancer cells. It achieves this by upregulating the peroxidation of membrane polyunsaturated phospholipids independently of ALOX enzymes [123]. Similarly, an siRNA screening study demonstrates that oxidoreductases, including POR and CYB5R1 (cytochrome b5 reductase 1), promote ferroptosis in HeLa cells by facilitating the transfer of electrons from NAD(P)H to oxygen, resulting in the production of hydrogen peroxide [137]. While the exact threshold of reactive oxygen species for triggering ferroptosis remains undetermined, there exist numerous mechanisms through which lipid peroxidation occurs during this process.
The proper transport of phospholipids from their sites of synthesis to specific organelle membranes is crucial for many biological processes, and this function is carried out by lipid transport proteins. One such protein is SCP2 (sterol carrier protein 2), which is involved in propagating oxidative stress between compartments by trafficking cholesterol hydroperoxides to mitochondria [143]. SCP2 promotes Gpx4 depletion-induced ferroptosis, leading to acute renal failure in mice [144]. Another lipid transporter, SLC47A1 (solute carrier family 47 member 1), has recently been identified as a novel repressor of ferroptosis. In SLC47A1-deficient cells, SOAT1 (sterol O-acyltransferase 1), a rate-limiting enzyme that catalyzes the conversion of cholesterol and fatty acids into cholesteryl esters, acts downstream of ACSL4 to mediate PUFA-CE production, promoting ferroptosis [129]. Further research is needed to determine whether other lipid transporters play a similar or different role in ferroptosis.
TMEM164 is a transmembrane protein that has been identified as a positive regulator of ferroptosis in cancer cells [145]. This protein can induce autophagosome formation or act as an acyltransferase that forms ferroptotic C20:4 ether phospholipids [146]. As a result, targeting TMEM164 May have therapeutic potential for diseases associated with ferroptosis, including cancer and neurodegenerative disorders.
Pyroptosis
Pyroptosis was first identified in 2001, but the effector of pyroptosis remained unknown until 2015 when GSDMD (gasdermin D) was identified as an executor of pyroptosis [147]. Pyroptosis is a proinflammatory form of RCD characterized by membrane pore formation, which causes osmotic imbalance and cell lysis, and allows the release of intracellular inflammatory cytokines (e.g., IL1B [interleukin 1 beta] and IL18 [interleukin 18]) and DAMPs (e.g., HMGB1 [high mobility group box 1] and SQSTM1/p62 [sequestosome 1]) [148–153]. The pore-forming process is executed by the inflammasome-mediated cleavage and activation of GSDMD [154,155]. Additionally, lipids, including oxysterols, fatty acids, and phospholipids, play intricate roles in pyroptosis (Figure 4).
Figure 4.
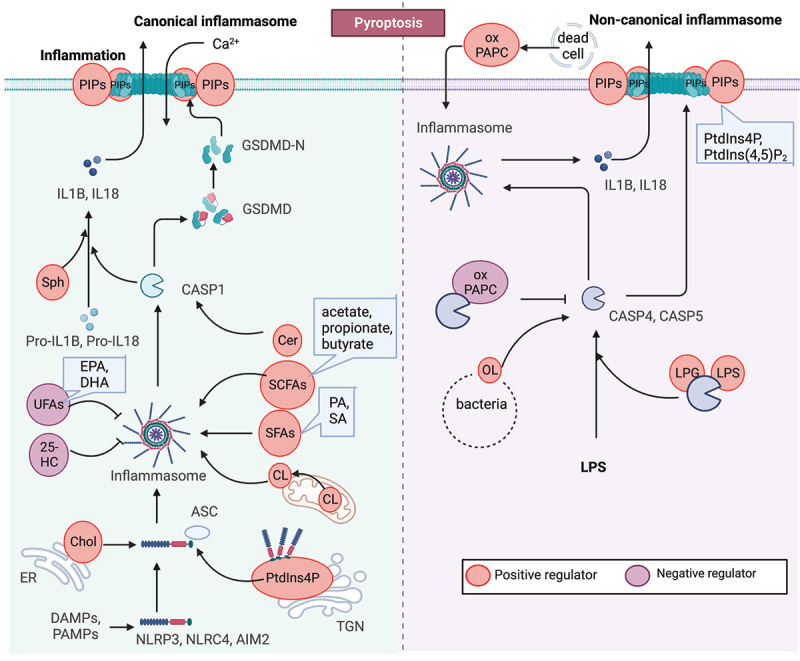
Lipid mechanism in pyroptosis. Pyroptosis is a proinflammatory form of programmed cell death characterized by the formation of membrane pores. The process of pore formation is executed through the inflammasome-mediated cleavage and activation of GSDMD. Lipids, including oxysterols, fatty acids, and phospholipids, play complex and critical roles in pyroptosis. For instance, the activation of the NLRP3 inflammasome requires binding to cholesterol in the endoplasmic reticulum, while the stable oligomerization of NLRP3 is facilitated by phosphoinositides (PIPs), and cardiolipin promotes NLRP3 inflammasome formation. Moreover, the functionality of GSDMD pores is dependent on phosphoinositides, and the enrichment of phosphatidylinositol-4,5-bisphosphate (PtdIns [4,5]P2) directly enhances their dynamics.
Inflammasome activation
Inflammasomes are multi-protein complexes that regulate the cleavage of cysteine protease CASP1 or CASP4/CASP11, secretion of inflammatory cytokines, and induction of pyroptosis [156,157]. There are two inflammasome pathways. In the canonical inflammasome pathway, bacterial and viral pathogen-associated stimuli, including pathogen-associated molecular pattern/PAMP molecules and DAMPs that are released from damaged or dying cells, activate cytosolic sensor proteins such as NLRP3 (NLR family pyrin domain containing 3), NLRC4 (NLR family CARD domain containing 4), or AIM2 (absent in melanoma 2). Once activated, these sensors oligomerize and recruit the adapter protein PYCARD/ASC (PYD and CARD domain containing) to form inflammasomes [158]. This leads to the activation of CASP1 through the assembly of the inflammasome complex [147,159,160].
Numerous lipids have been linked to the activation of inflammatory processes, including:
PtdIns4P. One of the cytosolic sensor proteins, NLRP3, localizes to cellular compartments with exposed negatively charged lipids such as PtdIns4P in dispersed trans-Golgi network (dTGN) vesicles, where it adopts a stable oligomeric structure [161]. Disruption of the interaction between NLRP3 and PtdIns4P on the dTGN impairs NLRP3 aggregation and downstream signaling. In vitro assays with lipidic strips have shown that a purified NLRP3 fragment containing the KKKK motif, as well as inactive NLRP3 cages, bind to several phosphorylated phosphatidylinositols and phosphatidic acid, but not unphosphorylated phosphatidylinositols, suggesting that NLRP3 is recruited to dTGN via binding to PtdIns4P. This process is essential for NLRP3 inflammasome activation [161].
Cardiolipin. Lipopolysaccharide (LPS)-induced exposure of cardiolipin in the mitochondrial membrane is necessary for functional NLRP3 inflammasome assembly in macrophages, creating a platform for the formation of the NLRP3 inflammasome. Cardiolipin binds to NLRP3 directly, and interference with cardiolipin synthesis specifically inhibits NLRP3 inflammasome activation [162–164].
Ceramide and sphingosine. Lipotoxicity increases intracellular ceramide, which can be sensed by the NLRP3 inflammasome, inducing CASP1 cleavage in macrophages and adipose tissue. Therefore, NLRP3 inflammasome senses ceramide and contributes to obesity-induced insulin resistance and inflammation in a mouse model [165]. Sphingosine, a downstream metabolite of ceramide, induces NLRP3-dependent IL1B secretion [166].
Fatty acids. Stimulation of macrophages with ω-3 fatty acids, including eicosapentaenoic acid and docosahexaenoic acid, prevents NLRP3 inflammasome activation, inhibiting subsequent CASP1 activation and IL1B secretion [167,168]. In contrast, SFAs (such as palmitic and stearic acids) activate the NLRP3 inflammasome and promote IL1B release in human and murine monocytes and macrophages [169,170]. Short-chain fatty acids (SCFAs), such as acetate, propionate, and butanoate, promote inflammasome activation in macrophages by binding to the PYRIN domain of PYCARD. Administration of SCFAs or dietary fibers, which are fermented to SCFAs by gut bacteria, significantly prolongs the survival of Salmonella enterica serovar Typhimurium-infected mice through PYCARD-mediated inflammasome activation. Activated inflammasomes facilitate neutrophil recruitment to promote bacterial elimination and suppress survival of S. Typhimurium in macrophages by pyroptosis [158].
Cholesterol. Cholesterol plays a critical role in modulating the activation of the NLRP3 inflammasome in macrophages. Depletion of cholesterol specifically in the endoplasmic reticulum of murine macrophages strongly affects the association of NLRP3 with the adapter protein PYCARD, leading to the abrogation of CASP1 activation and IL1B secretion. This effect is not observed when cholesterol is depleted from the plasma membrane [171,172]. Mechanistically, cholesterol crystals induce phagolysosomal damage, resulting in the release of lysosomal contents, including cathepsins, into the cytoplasm. This activates the NLRP3 sensor protein, triggering the inflammasome cascade [173]. However, an oxysterol called 25-hydroxycholesterol suppresses the activation of both NLRP3 and AIM2 inflammasomes [174,175]. In response to bacterial infection or LPS stimulation, macrophages upregulate the expression of CH25H (cholesterol 25-hydroxylase) to maintain repression of SREBF2/SREBP2 (sterol regulatory element binding transcription factor 2) activation and cholesterol synthesis. The accumulation of excess cholesterol, which is mediated by deficiency in CH25H, can trigger IL1B release in an AIM2-dependent manner.
The non-canonical inflammasome pathway is activated by cytoplasmic LPS directly binding to mouse CASP4/CASP11 or human CASP4 or CASP5, leading to the cleavage of GSDMD [176]. CASP4 and CASP5 recognize cytosolic LPS by binding to the lipid A moiety [177]. Accumulating evidence indicates that CASP4 and CASP5 are activated by binding to bacterial LPS or certain endogenous oxidized phospholipids.
Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (oxPAPC), a complex mixture of oxidized lipids released from dying cells, can translocate to the cytoplasm and directly bind to CASP4, inducing NLRP3 inflammasome activation and IL1B release in dendritic cells, bypassing pyroptosis [178,179]. However, targeting the non-canonical inflammasome in macrophages, oxPAPC protects against septic shock [180]. High concentrations of extracellular oxPAPC may activate the alternative NLRP3 pathway, whereas intracellularly delivered oxPAPC acts as an antagonist of CASP4 [181].
In addition, pathogenic lipids, such as E. coli LPS and Leishmania glycolipid lipophosphoglycan/LPG, can bind and activate CASP4 and CASP5, inducing non-canonical NLRP3 inflammasome activation [182]. Delivery of lipophosphoglycan into macrophages triggers CASP4 activation, whereas infections performed with Lpg1-/- parasites reduce CASP4-NLRP3 activation [182]. Ornithine lipids, found in bacteria such as Vibrio cholerae and Pseudomonas aeruginosa, can activate CASP4 and induce non-canonical NLRP3 inflammasome activation in murine macrophages [182,183].
Lipids play a crucial role in the entire process of inflammasome activation. For instance, NLRP3 activation requires binding to cholesterol in the endoplasmic reticulum, whereas the stable oligomerization of NLRP3 requires PIPs, and cardiolipin favors NLRP3 inflammasome formation. However, some lipid regulation models can induce NLRP3 inflammasome activation and IL1B release while bypassing pyroptosis, such as oxPAPC. Interestingly, the function of oxPAPC in modulating NLRP3 activation appears to be cell-type and location-dependent, and the underlying mechanism requires further investigation.
Membrane pore formation
The activation of the inflammasome cleaves GSDMD, leading to the production of the N-terminal fragment (GSDMD-N), which mediates pyroptosis by forming pores on the plasma membrane [148]. During pore formation, GSDMD-N oligomerizes and binds phosphoinositides on the inner leaflet of the plasma membrane [184], allowing the release of mature IL1B and IL18. GSDMD-N domains bind preferentially to liposomes containing 10–20% PtdIns(4,5)P2, but not unphosphorylated phosphatidylinositol [185].
Mycobacterium tuberculosis, a pathogen that causes tuberculosis, secretes an effector protein called PtpB (protein-tyrosine-phosphatase) that dephosphorylates host plasma membrane PtdIns4P and PtdIns(4,5)P2, inhibiting the membrane localization of GSDMD-N and preventing pyroptosis and cytokine release. This strategy helps the bacteria evade host immunity and survive intracellularly [186].
GSDMD-N can also bind cardiolipin, a lipid present in bacterial membranes, resulting in bacterial destruction outside of the cell. The N domains of GSDMD, GSDMA or GSDMA3 are efficiently and specifically precipitated by cardiolipin liposomes. In contrast, the binding efficiency of GSDMD-N decreases when the concentration of cardiolipin is reduced from 20% to 10% [185].
In addition to phosphoinositides and cardiolipin, peroxidation products of phospholipids such as PtdIns4P, PtdIns(4,5)P2, diacylglycerol, and phosphatidic acid enhance the activity of GSDMD-N pores and contribute to cell death. Oxidation of these phospholipids promotes GSDMD-N-mediated pyroptosis in lethal polymicrobial sepsis, whereas GPX4 blocks GSDMD cleavage during inflammasome activation [187]. Overall, membrane pore formation is a key event in pyroptosis, and lipids play a pivotal role in both the activation and execution of this form of cell death.
Lipid mechanisms in autophagy
Autophagy is a highly regulated and evolutionarily conserved cellular process that facilitates the degradation and recycling of unnecessary or dysfunctional cellular components. It serves as a critical quality control mechanism implicated in various human diseases and aging [188,189]. During autophagy, double-membrane structures called autophagosomes are formed, enclosing portions of the cytoplasm, organelles, or protein aggregates [190]. These autophagosomes then fuse with lysosomes, forming autolysosomes, where the engulfed contents are degraded by lysosomal enzymes. The breakdown products are subsequently recycled and utilized to generate new molecules and provide energy for the cell.
Autophagy encompasses both nonselective and selective processes, exerting complex effects on cellular stress and homeostasis [145,191,192]. In many cases, an increase in nonselective autophagy can promote cell survival in the context of environmental stresses, such as nutrient deprivation, infection, and cellular damage [191,193]. However, selective autophagy exhibits context-dependent roles in RCD. For example, selective degradation of ferritin, known as ferritinophagy, leads to iron release and promotes ferroptosis [194]. Autophagic degradation of PTPN13 (protein tyrosine phosphatase non-receptor type 13), a negative regulator of extrinsic apoptosis, promotes apoptosis [195], while selective removal of mitochondria via mitophagy by autophagy inhibits apoptosis [196]. Moreover, the targeted autophagic degradation of BIRC2/cIAP1 (baculoviral IAP repeat containing 2), a negative regulator of necroptosis, facilitates necroptosis [197].
Various lipid species, including phospholipids, sphingolipids, and sterols, play crucial roles at different stages of autophagy (Figure 5). Particularly, phosphoinositides, a subgroup of phospholipids, have emerged as key modulators of autophagy, influencing various signaling processes involved in autophagy initiation, autophagosome biogenesis, and maturation. PtdIns(3,4,5)P3 is involved in the regulation of autophagy by activating the MTOR signaling pathway, which negatively regulates autophagy initiation [198]. PTEN (phosphatase and tensin homolog), a 3-phosphatase responsible for dephosphorylating PtdIns(3,4,5)P3, triggers autophagy by inactivating the PI3K-AKT-MTOR pathway [199].
Figure 5.
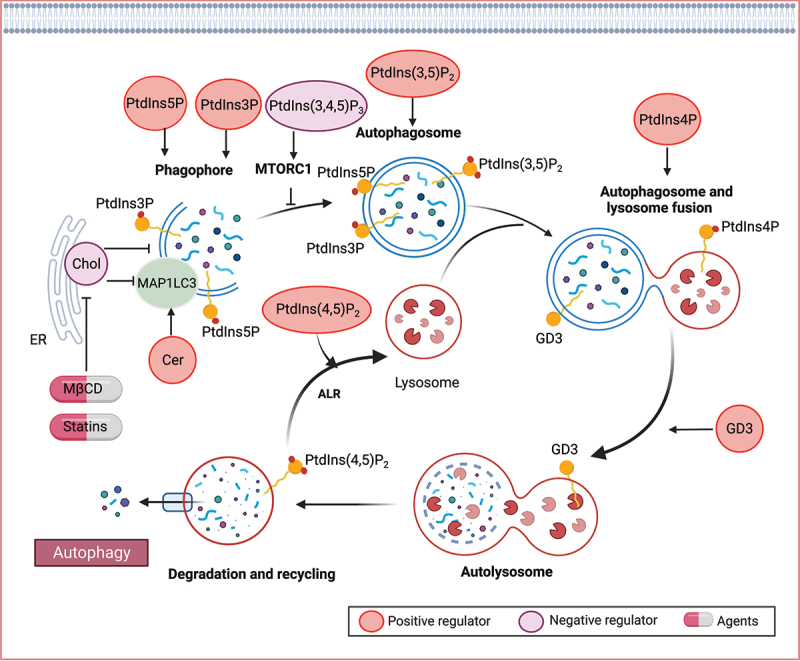
Lipid mechanisms in autophagy. Various lipid species, such as phospholipids, sphingolipids, and sterols, play crucial roles in different stages of autophagy. Specifically, phosphoinositides have been identified as key modulators of autophagy, involved in signaling processes that govern autophagy initiation, autophagosome biogenesis, and maturation. Ceramide (Cer) has also been implicated in the induction of autophagy. Furthermore, gangliosides, including GD3 gangliosides, can be recruited to autophagosomes and contribute to morphogenic remodeling.
During the autophagic process, the phagophore emerges from specialized PtdIns3P-enriched sites, which act as signaling molecules for the recruitment of various PtdIns3P-binding proteins. This process allows for the interaction between PtdIns3P and MAP1LC3 (microtubule associated protein 1 light chain 3) lipidation, which is essential for phagophore function. PtdIns3P generated by the class III PtdIns 3-kinase is critical for autophagosome formation [198]. PtdIns5P synthesis is also required for autophagosome formation that acts via a similar pathway as that utilizing PtdIns3P for the formation of the autophagosome [200]. In addition, PtdIns (3,5)P2, generated by PIKFYVE/FAB1 from PtdIns3P, is required for the maturation of autophagosomes in mammals [201]. PtdIns4P, generated by mammalian PI4KA (phosphatidylinositol 4-kinase alpha), promotes autophagosome-lysosome fusion [202,203]. Depression of PI4K2A (phosphatidylinositol 4-kinase type 2 alpha), which generates PtdIns4P from PtdIns, reduces the autophagic flux by blocking autophagosome and lysosome fusion. Besides, the conversion of PtdIns4P to PtdIns (4,5)P2 is important in a late step of mammalian autophagy, termed autophagic lysosome reformation, which is critical for maintaining lysosome homeostasis [204].
Sphingolipids play a bioactive role in the induction of autophagy [205]. Among them, ceramide, a major species that regulates diverse cellular processes including survival, differentiation, and senescence, has been implicated in autophagy induction across various models [206–209]. Ceramide levels are upregulated during stress-induced autophagy, and ceramide itself activates the autophagic process. In response to the increased ceramide levels, JUN/c-JUN upregulates the transcription of MAP1LC3, thereby promoting autophagy [210]. Ceramide-1-phosphate (C1P) is a bioactive sphingolipid that plays a pivotal role in the regulation of diverse physiological processes. In both human epithelial cells and monocytes, the depletion of the C1P transfer protein CPTP (ceramide-1-phosphate transfer protein), or the introduction of specific point mutants designed to target the C1P binding site, triggers a significant increase in the assembly of ATG9A-dependent phagophores and the formation of MAP1LC3-dependent autophagosomes [211]. This cascade of events further culminates in heightened inflammasome activation [211]. Similarly, the elevation of intracellular C1P levels through exogenous C1P treatment (rather than CPTP inhibition) also induces both autophagy and inflammasome activation [211]. Gangliosides, sialic-acid-containing sphingolipids, also contribute to autophagy. For instance, GD3 gangliosides are recruited to the autophagosome and play a role in morphogenic remodeling, including changes in membrane curvature and fluidity, ultimately facilitating the maturation of autophagosomes into autolysosomes [212].
The autophagosome membrane is characterized by a high content of unsaturated fatty acids [213,214] and low levels of cholesterol [215,216]. The removal of cholesterol plays a crucial role in autophagosome initiation. For example, depleting cholesterol using MβCD and statins enhances MAP1LC3 lipidation and promotes autophagosome formation [217–219]. Furthermore, the endoplasmic reticulum cholesterol transfer protein GRAMD1C (GRAM domain-containing 1C) is a negative regulator of starvation-induced autophagy. Autophagosomes form at endoplasmic reticulum-associated sites, and the loss of GRAMD1C leads to increased mitochondrial cholesterol levels and reduced endoplasmic reticulum cholesterol. These elevated mitochondrial cholesterol levels in GRAMD1C-depleted cells affect mitochondrial bioenergetics [215,220]. On the other hand, the cholesterol-transfer protein GRAMD1A (GRAM domain-containing protein 1A) accumulates at autophagosome initiation sites in response to starvation [221]. GRAMD1A appears to play a role in maintaining the elevated cholesterol content of autophagosomal membranes, thus facilitating autophagosome maturation [221].
Notably, human ACSL4, which localizes to autophagic membranes, plays a role in autophagosome formation, further establishing a connection between autophagy and lipid metabolism in the context of ferroptosis [222]. Given the intricate interplay between autophagy and lipid metabolism, elucidating the feedback mechanisms in response to different stresses can be a challenging endeavor.
Moreover, the internalization of cellular free fatty acids stands as a fundamental mechanism, particularly activated in response to starvation, for instigating the autophagic process. Conversely, within cultured hepatocyte models, autophagy assumes a pivotal role in finely modulating lipid content. This reciprocity implies an intricate interplay. However, it is essential to underscore that an abnormal accumulation of intracellular lipids can significantly impede the efficient clearance of materials targeted for autophagic degradation [223]. In harmony with an alternative hypothalamic model focused on starvation, heightened levels of fatty acids induced by fasting orchestrate a swift uptake of these molecules within the hypothalamus. This, in turn, promptly triggers the initiation of autophagy [224]. Notably, the introduction of exogenous oleic or palmitic acid amplifies the flux of autophagic activity in hypothalamic GT1–7 cells [224]. Nevertheless, in contrast to the acute lipid-driven stimulus that actively fosters autophagy, a chronic condition of lipid overload, as manifested in prolonged overnutrition or sustained high-fat diets, ultimately leads to a regression in autophagic functionality. This situation is characterized by a compromised capacity for autolysosome formation and overall autophagic activity [223].
In summary, lipids and lipid-related processes exert pivotal roles throughout all stages of phagophore formation, autophagosome development, and autolysosome maturation, finely tuning the intricate activity of the autophagic pathway.
Conclusion and outlook
In recent years, mounting evidence has highlighted the intricate connections among lipids, autophagy, and RCD. Lipids serve as structural components in cell membranes, providing scaffolds for proteins such as BAX, MLKL, and GSDMD to regulate cellular death processes, including pore formation. Moreover, lipids can act as substrates for peroxidation, triggering ferroptosis. However, lipids also play a crucial role in specific protein-lipid interactions and signaling events necessary for the activation of the RCD machinery. Lipids are essential for autophagosome biogenesis, and lipid metabolism as well as lipid-modifying enzymes such as phospholipases, acyltransferases, and lipid kinases, are crucial for the regulation of autophagy and cellular homeostasis.
The emerging concepts of the lipid hypothesis in governing RCD not only provide insights into the intricate lipid molecular networks involved in RCD but also open up new avenues for the development of innovative therapeutic strategies targeting defective lipid metabolism-associated diseases. For instance, nanoliposomes containing C6-ceramide have been identified as a chemotherapeutic agent capable of suppressing metastasis in a mouse xenograft model of ovarian cancer by inducing MLKL-dependent necrotic cell death [225]. Similarly, in the case of M. tuberculosis infection, bacterial-secreted PtpB utilizes host ubiquitin to inhibit pyroptosis by altering host membrane phosphoinositide composition, thereby presenting potential directions for the development of anti-M. tuberculosis therapies.
Moreover, targeting cholesterol metabolism for cancer treatment has gained significant recognition. For instance, cholesterol deficiency contributes to T cell exhaustion by inhibiting T cell proliferation and triggering autophagy-mediated apoptosis. This connection between cholesterol deficiency and antitumor immunity highlights the importance of cholesterol in regulating immune responses [226]. On the other hand, cholesterol depletion using MβCD has shown potential anticancer effects in various types of cancer cells. Studies have reported the potential anticancer activity of MβCD in non-small cell lung cancer (NSCLC) cells [227], melanoma cells [228], colorectal cancer cells [69] and others. These findings suggest that manipulating cholesterol levels holds promise as a therapeutic approach in cancer treatment.
However, as discussed here, certain lipids can exhibit similar or even opposing functions in different modes of RCD, and the precise lipid molecular effectors, crosstalk, and potential switches between distinct lipid sub-pathways in RCD remain largely unknown. Addressing these unresolved questions is crucial for the development of lipid-targeted therapies and interventions in lipid metabolic processes, as dysregulation of the cellular lipidome undeniably has an impact upon various human pathologies associated with RCD.
Delineating the precise landscape of lipid-regulated RCD and autophagy encounters several challenges. First, the inherent flexibility and heterogeneity of lipid structures often lead to limited resolution, leaving uncertainties regarding their specific identities. Second, our quantitative understanding of lipid biology, particularly concerning transient and precarious lipid species, remains limited. Third, the absence of accurate tools to determine absolute lipid concentrations in living cells, in vivo models, or with subcellular resolution presents a significant obstacle. Fourth, lipid metabolism and autophagy are dynamic processes influenced by diverse cellular and environmental factors, including cellular stress, nutrient availability, and signaling pathways. Capturing and comprehending the dynamic nature of lipid metabolism and autophagy pose challenges as experimental conditions must faithfully replicate physiological or pathological contexts. Furthermore, the presence of redundancy and compensatory mechanisms within lipid metabolism and autophagy pathways further complicates research efforts. Collectively, these challenges impede our understanding of how lipids precisely undergo alterations under specific stresses and, conversely, how lipids affect the regulation of RCD and autophagy at the cellular, tissue, or organismal levels. Acquiring such knowledge is crucial for elucidating the specific mechanisms through which lipids contribute to these processes.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81974000, 82303255 and 82270185], the Natural Science Foundation of Hunan Province [2021JJ10077, 2023JJ40894 and 2023JJ40894] and the National Institutes of Health [GM131919].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Wilfling F, Wang H, Haas JT, et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell. 2013;24(4):384–399. doi: 10.1016/j.devcel.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thiam AR, Farese RV Jr., Walther TC.. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol. 2013;14(12):775–786. doi: 10.1038/nrm3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lionetti L, Mollica MP, Lombardi A, et al. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr Metab Cardiovasc Dis. 2009;19(2):146–152. doi: 10.1016/j.numecd.2008.10.010 [DOI] [PubMed] [Google Scholar]
- [4].Krahmer N, Farese RV Jr., Walther TC. Balancing the fat: lipid droplets and human disease. EMBO Mol Med. 2013;5(7):973–983. doi: 10.1002/emmm.201100671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 2018;19(5):281–296. doi: 10.1038/nrm.2017.138 [DOI] [PubMed] [Google Scholar]
- [6].Jussupow A, Di Luca A, Kaila VRI. How cardiolipin modulates the dynamics of respiratory complex I. Sci Adv. 2019;5(3):eaav1850. doi: 10.1126/sciadv.aav1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arias-Cartin R, Grimaldi S, Pommier J, et al. Cardiolipin-based respiratory complex activation in bacteria. Proc Natl Acad Sci, USA. 2011;108(19):7781–7786. doi: 10.1073/pnas.1010427108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277(46):43553–43556. doi: 10.1074/jbc.C200551200 [DOI] [PubMed] [Google Scholar]
- [9].Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8(1):74–84. doi: 10.1038/nrm2084 [DOI] [PubMed] [Google Scholar]
- [10].Resh MD. Fatty acylation of proteins: the long and the short of it. Progress Lipid Res. 2016;63:120–131. doi: 10.1016/j.plipres.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi: 10.1038/s41423-020-00630-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi: 10.1038/s41422-019-0164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xie Y, Li J, Kang R, et al. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431. doi: 10.3389/fcell.2020.00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fahy E, Subramaniam S, Brown HA, et al. A comprehensive classification system for lipids. J Lipid Res. 2005;46(5):839–861. doi: 10.1194/jlr.E400004-JLR200 [DOI] [PubMed] [Google Scholar]
- [16].Sud M, Fahy E, Cotter D, et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35(Database):D527–32. doi: 10.1093/nar/gkl838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fahy E, Sud M, Cotter D, et al. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007;35(Web Server):W606–12. doi: 10.1093/nar/gkm324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liebisch G, Fahy E, Aoki J, et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J Lipid Res. 2020;61(12):1539–1555. doi: 10.1194/jlr.S120001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bleicken S, Jeschke G, Stegmueller C, et al. Structural model of active Bax at the membrane. Molecular Cell. 2014;56(4):496–505. doi: 10.1016/j.molcel.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–342. doi: 10.1016/S0092-8674(02)01036-X [DOI] [PubMed] [Google Scholar]
- [22].Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi: 10.1016/S0092-8674(00)80085-9 [DOI] [PubMed] [Google Scholar]
- [23].Cain K, Brown DG, Langlais C, et al. Caspase activation involves the formation of the aposome, approximately 700 kDa) caspase-activating complex. J Biol Chem. 1999;274(32):22686–22692. doi: 10.1074/jbc.274.32.22686 [DOI] [PubMed] [Google Scholar]
- [24].Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA. 1999;96(20):10964–10967. doi: 10.1073/pnas.96.20.10964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Horvath SE, Daum G. Lipids of mitochondria. Progress Lipid Res. 2013;52(4):590–614. doi: 10.1016/j.plipres.2013.07.002 [DOI] [PubMed] [Google Scholar]
- [26].Harwood JL. [44] phosphoglycerides of mitochondrial membranes. In: Methods in Enzymology. Academic Press; 1987. pp. 475–485. doi: 10.1016/0076-6879(87)48046-4 [DOI] [Google Scholar]
- [27].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. doi: 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Connerth M, Tatsuta T, Haag M, et al. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science. 2012;338(6108):815–818. doi: 10.1126/science.1225625 [DOI] [PubMed] [Google Scholar]
- [29].Garcia Fernandez M, Troiano L, Moretti L, et al. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ: Mol Biol J Am Ass Cancer Res. 2002;13:449–455. [PubMed] [Google Scholar]
- [30].Lutter M, Fang M, Luo X, et al. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2(10):754–761. doi: 10.1038/35036395 [DOI] [PubMed] [Google Scholar]
- [31].Lai YC, Li CC, Sung TC, et al. The role of cardiolipin in promoting the membrane pore-forming activity of BAX oligomers. Biochim Biophys Acta - Biomembr. 2019;1861(1):268–280. doi: 10.1016/j.bbamem.2018.06.014 [DOI] [PubMed] [Google Scholar]
- [32].Deng X, Yin X, Allan R, et al. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science. 2008;322(5898):110–115. doi: 10.1126/science.1158111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Siskind LJ. Mitochondrial ceramide and the induction of apoptosis. J Bioenerg Biomembr. 2005;37(3):143–153. doi: 10.1007/s10863-005-6567-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Birbes H, Luberto C, Hsu YT, et al. A mitochondrial pool of sphingomyelin is involved in TNFα-induced Bax translocation to mitochondria. Biochem J. 2005;386(3):445–451. doi: 10.1042/BJ20041627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee H, Rotolo JA, Mesicek J, et al. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One. 2011;6(6):e19783. doi: 10.1371/journal.pone.0019783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim HJ, Oh JE, Kim SW, et al. Ceramide induces p38 MAPK-dependent apoptosis and Bax translocation via inhibition of Akt in HL-60 cells. Cancer Lett. 2008;260(1–2):88–95. doi: 10.1016/j.canlet.2007.10.030 [DOI] [PubMed] [Google Scholar]
- [37].Sawada M, Nakashima S, Banno Y, et al. Influence of Bax or bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene. 2000;19(31):3508–3520. doi: 10.1038/sj.onc.1203699 [DOI] [PubMed] [Google Scholar]
- [38].von Haefen C, Wieder T, Gillissen B, et al. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–4019. doi: 10.1038/sj.onc.1205497 [DOI] [PubMed] [Google Scholar]
- [39].Sawada M, Nakashima S, Banno Y, et al. Ordering of ceramide formation, caspase activation, and Bax/Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Diff. 2000;7(9):761–772. doi: 10.1038/sj.cdd.4400711 [DOI] [PubMed] [Google Scholar]
- [40].Ganesan V, Perera MN, Colombini D, et al. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis. 2010;15(5):553–562. doi: 10.1007/s10495-009-0449-0 [DOI] [PubMed] [Google Scholar]
- [41].Siskind LJ, Feinstein L, Yu T, et al. Anti-apoptotic bcl-2 family proteins disassemble ceramide channels. J Biol Chem. 2008;283(11):6622–6630. doi: 10.1074/jbc.M706115200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chipuk JE, McStay GP, Bharti A, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148(5):988–1000. doi: 10.1016/j.cell.2012.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Diff. 2008;15(3):484–493. doi: 10.1038/sj.cdd.4402280 [DOI] [PubMed] [Google Scholar]
- [44].Montero J, Morales A, Llacuna L, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008;68(13):5246–5256. doi: 10.1158/0008-5472.CAN-07-6161 [DOI] [PubMed] [Google Scholar]
- [45].Westphal D, Dewson G, Menard M, et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci, USA. 2014;111(39):E4076–85. doi: 10.1073/pnas.1415142111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Biswas P, Datta C, Rathi P, et al. Fatty acids and their lipid mediators in the induction of cellular apoptosis in cancer cells. Prostaglandins Other Lipid Mediat. 2022;160:106637. doi: 10.1016/j.prostaglandins.2022.106637 [DOI] [PubMed] [Google Scholar]
- [47].Zhang C, Yu H, Shen Y, et al. Polyunsaturated fatty acids trigger apoptosis of colon cancer cells through a mitochondrial pathway. Arch Med Sci. 2015;11(5):1081–1094. doi: 10.5114/aoms.2015.54865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kwon JI, Kim GY, Park KY, et al. Induction of apoptosis by linoleic acid is associated with the modulation of bcl-2 family and Fas/FasL system and activation of caspases in AGS human gastric adenocarcinoma cells. J Med Food. 2008;11(1):1–8. doi: 10.1089/jmf.2007.073 [DOI] [PubMed] [Google Scholar]
- [49].Tsai CH, Shen YC, Chen HW, et al. Docosahexaenoic acid increases the expression of oxidative stress-induced growth inhibitor 1 through the PI3K/Akt/Nrf2 signaling pathway in breast cancer cells. Food Chem Toxicol. 2017;108:276–288. doi: 10.1016/j.fct.2017.08.010 [DOI] [PubMed] [Google Scholar]
- [50].Jiang L, Wang W, He Q, et al. Oleic acid induces apoptosis and autophagy in the treatment of tongue squamous cell carcinomas. Sci Rep. 2017;7(1):11277. doi: 10.1038/s41598-017-11842-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Evans LM, Cowey SL, Siegal GP, et al. Stearate preferentially induces apoptosis in human breast cancer cells. Nutr Cancer. 2009;61(5):746–753. doi: 10.1080/01635580902825597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gon S, Gatanaga T, Sendo F. Involvement of two types of TNF receptor in TNF-α induced neutrophil apoptosis. Microbiol Immunol. 1996;40(6):463–465. doi: 10.1111/j.1348-0421.1996.tb01095.x [DOI] [PubMed] [Google Scholar]
- [53].Pennica D, Nedwin GE, Hayflick JS, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. 1984;312(5996):724–729. doi: 10.1038/312724a0 [DOI] [PubMed] [Google Scholar]
- [54].Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66(2):233–243. doi: 10.1016/0092-8674(91)90614-5 [DOI] [PubMed] [Google Scholar]
- [55].Schneider P, Bodmer JL, Holler N, et al. Characterization of Fas (apo-1, CD95)-Fas ligand interaction. J Biol Chem. 1997;272(30):18827–18833. doi: 10.1074/jbc.272.30.18827 [DOI] [PubMed] [Google Scholar]
- [56].Pan G, O’Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. doi: 10.1126/science.276.5309.111 [DOI] [PubMed] [Google Scholar]
- [57].Schneider P, Thome M, Burns K, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-Dependent apoptosis and activate NF-κB. Immunity. 1997;7(6):831–836. doi: 10.1016/S1074-7613(00)80401-X [DOI] [PubMed] [Google Scholar]
- [58].Vanden Berghe T, van Loo G, Saelens X, et al. Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein FADD. J Biol Chem. 2004;279(9):7925–7933. doi: 10.1074/jbc.M307807200 [DOI] [PubMed] [Google Scholar]
- [59].Li H, Zhu H, Xu CJ, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi: 10.1016/S0092-8674(00)81590-1 [DOI] [PubMed] [Google Scholar]
- [60].Marconi M, Ascione B, Ciarlo L, et al. Constitutive localization of DR4 in lipid rafts is mandatory for TRAIL-induced apoptosis in B-cell hematologic malignancies. Cell Death Dis. 2013;4(10):e863. doi: 10.1038/cddis.2013.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Greenlee JD, Lopez-Cavestany M, Ortiz-Otero N, et al. Oxaliplatin resistance in colorectal cancer enhances TRAIL sensitivity via death receptor 4 upregulation and lipid raft localization. Elife. 2021;10:10. doi: 10.7554/eLife.67750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lotocki G, Alonso OF, Dietrich WD, et al. Tumor necrosis factor receptor 1 and its signaling intermediates are recruited to lipid rafts in the traumatized brain. J Neurosci. 2004;24(49):11010–11016. doi: 10.1523/JNEUROSCI.3823-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Legler DF, Micheau O, Doucey MA, et al. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity. 2003;18(5):655–664. doi: 10.1016/S1074-7613(03)00092-X [DOI] [PubMed] [Google Scholar]
- [64].Sezgin E, Levental I, Mayor S, et al. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18(6):361–374. doi: 10.1038/nrm.2017.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Greenlee JD, Subramanian T, Liu K, et al. Rafting down the metastatic cascade: the role of lipid rafts in cancer metastasis, cell death, and clinical outcomes. Cancer Res. 2021;81(1):5–17. doi: 10.1158/0008-5472.CAN-20-2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mahammad S, Parmryd I. Cholesterol depletion using methyl-β-cyclodextrin. Methods mol biol (Clifton, NJ). 2015;1232:91–102. [DOI] [PubMed] [Google Scholar]
- [67].Motoyama K, Kameyama K, Onodera R, et al. Involvement of PI3K-Akt-bad pathway in apoptosis induced by 2,6-di-O-methyl-β-cyclodextrin, not 2,6-di-O-methyl-α-cyclodextrin, through cholesterol depletion from lipid rafts on plasma membranes in cells. Eur J Pharm Sci. 2009;38(3):249–261. doi: 10.1016/j.ejps.2009.07.010 [DOI] [PubMed] [Google Scholar]
- [68].Onodera R, Motoyama K, Okamatsu A, et al. Involvement of cholesterol depletion from lipid rafts in apoptosis induced by methyl-β-cyclodextrin. Int J Pharmaceut. 2013;452(1–2):116–123. doi: 10.1016/j.ijpharm.2013.04.071 [DOI] [PubMed] [Google Scholar]
- [69].Ohno Y, Toshino M, Mohammed AFA, et al. Mannose-methyl-β-cyclodextrin suppresses tumor growth by targeting both colon cancer cells and tumor-associated macrophages. Carbohydr Polym. 2023;305:120551. doi: 10.1016/j.carbpol.2023.120551 [DOI] [PubMed] [Google Scholar]
- [70].Garofalo T, Giammarioli AM, Misasi R, et al. Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Diff. 2005;12(11):1378–1389. doi: 10.1038/sj.cdd.4401672 [DOI] [PubMed] [Google Scholar]
- [71].Jaffrès PA, Gajate C, Bouchet AM, et al. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol Ther. 2016;165:114–131. doi: 10.1016/j.pharmthera.2016.06.003 [DOI] [PubMed] [Google Scholar]
- [72].Gajate C, Mollinedo F. The antitumor ether lipid ET-18-OCH(3) induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood. 2001;98(13):3860–3863. doi: 10.1182/blood.V98.13.3860 [DOI] [PubMed] [Google Scholar]
- [73].Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood. 2007;109(2):711–719. doi: 10.1182/blood-2006-04-016824 [DOI] [PubMed] [Google Scholar]
- [74].Grassmé H, Cremesti A, Kolesnick R, et al. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene. 2003;22(35):5457–5470. doi: 10.1038/sj.onc.1206540 [DOI] [PubMed] [Google Scholar]
- [75].Gupta S, Natarajan R, Payne SG, et al. Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J Biol Chem. 2004;279(7):5821–5828. doi: 10.1074/jbc.M310979200 [DOI] [PubMed] [Google Scholar]
- [76].Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052 [DOI] [PubMed] [Google Scholar]
- [77].Bhattacharyya R, Barren C, Kovacs DM. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J Neurosci. 2013;33(27):11169–11183. doi: 10.1523/JNEUROSCI.4704-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Segawa K, Nagata S. An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25(11):639–650. doi: 10.1016/j.tcb.2015.08.003 [DOI] [PubMed] [Google Scholar]
- [79].Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J Biol Chem. 1985;260(8):5131–5138. doi: 10.1016/S0021-9258(18)89189-X [DOI] [PubMed] [Google Scholar]
- [80].Segawa K, Kurata S, Yanagihashi Y, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164–1168. doi: 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- [81].Suzuki J, Umeda M, Sims PJ, et al. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–838. doi: 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- [82].Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci, USA. 2011;108(48):19246–19251. doi: 10.1073/pnas.1114799108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304(5674):1147–1150. doi: 10.1126/science.1094359 [DOI] [PubMed] [Google Scholar]
- [84].Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- [85].Xie Y, Zhu S, Zhong M, et al. Inhibition of Aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153(5):1429–43 e5. doi: 10.1053/j.gastro.2017.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol (Baltimore, Md : 1950). 1988;141(8):2629–2634. doi: 10.4049/jimmunol.141.8.2629 [DOI] [PubMed] [Google Scholar]
- [87].Vercammen D, Brouckaert G, Denecker G, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919–930. doi: 10.1084/jem.188.5.919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].He S, Liang Y, Shao F, et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc Natl Acad Sci, USA. 2011;108(50):20054–20059. doi: 10.1073/pnas.1116302108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Vercammen D, Beyaert R, Denecker G, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–1485. doi: 10.1084/jem.187.9.1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308 [DOI] [PubMed] [Google Scholar]
- [92].He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021 [DOI] [PubMed] [Google Scholar]
- [93].Hsu H, Huang J, Shu HB, et al. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/S1074-7613(00)80252-6 [DOI] [PubMed] [Google Scholar]
- [94].Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi: 10.1016/j.cell.2009.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi: 10.1016/j.cell.2011.11.031 [DOI] [PubMed] [Google Scholar]
- [96].Chen X, Li W, Ren J, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–121. doi: 10.1038/cr.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Molecular Cell. 2014;54(1):133–146. doi: 10.1016/j.molcel.2014.03.003 [DOI] [PubMed] [Google Scholar]
- [98].Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–981. doi: 10.1016/j.celrep.2014.04.026 [DOI] [PubMed] [Google Scholar]
- [99].Phan TK, Bindra GK, Williams SA, et al. Combating human pathogens and cancer by targeting phosphoinositides and their metabolism. Trends Pharmacol Sci. 2019;40(11):866–882. doi: 10.1016/j.tips.2019.09.006 [DOI] [PubMed] [Google Scholar]
- [100].Quarato G, Guy CS, Grace CR, et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Molecular Cell. 2016;61(4):589–601. doi: 10.1016/j.molcel.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Dovey CM, Diep J, Clarke BP, et al. MLKL requires the inositol phosphate code to execute necroptosis. Molecular Cell. 2018;70(5):936–48.e7. doi: 10.1016/j.molcel.2018.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Parisi LR, Li N, Atilla-Gokcumen GE. Very long chain fatty acids are functionally involved in necroptosis. Cell Chem Biol. 2017;24(12):1445–54.e8. doi: 10.1016/j.chembiol.2017.08.026 [DOI] [PubMed] [Google Scholar]
- [103].Gong YN, Guy C, Olauson H, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169(2):286–300.e16. doi: 10.1016/j.cell.2017.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zargarian S, Shlomovitz I, Erlich Z, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15(6):e2002711. doi: 10.1371/journal.pbio.2002711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Chen X, Kang R, Kroemer G, et al. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28(10):2843–2856. doi: 10.1038/s41418-021-00859-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;17(11):3361–3374. doi: 10.1080/15548627.2021.1872241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035. doi: 10.1080/15548627.2019.1659623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dai E, Meng L, Kang R, et al. ESCRT-III–dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522(2):415–421. doi: 10.1016/j.bbrc.2019.11.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Dolma S, Lessnick SL, Hahn WC, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296. doi: 10.1016/S1535-6108(03)00050-3 [DOI] [PubMed] [Google Scholar]
- [111].Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi: 10.1038/s41586-019-1705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi: 10.1038/s41586-019-1707-0 [DOI] [PubMed] [Google Scholar]
- [113].Xie Y, Kang R, Klionsky DJ, et al. GPX4 in cell death, autophagy, and disease. Autophagy. 2023;1–18. doi: 10.1080/15548627.2023.2233846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi: 10.1080/15548627.2020.1810918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022;289(22):7038–7050. doi: 10.1111/febs.16059 [DOI] [PubMed] [Google Scholar]
- [116].Chen X, Huang J, Yu C, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13(1):6318. doi: 10.1038/s41467-022-34096-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Lin Z, Liu J, Kang R, et al. Lipid metabolism in ferroptosis. Adv Biol. 2021;5(8):e2100396. doi: 10.1002/adbi.202100396 [DOI] [PubMed] [Google Scholar]
- [118].Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Diff. 2016;23(3):369–379. doi: 10.1038/cdd.2015.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci, USA. 2016;113(34):E4966–75. doi: 10.1073/pnas.1603244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi: 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309. doi: 10.1038/s41589-020-0472-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: new mechanistic insights and the development of peroxyl radical clocks. Acc Chem Res. 2011;44(6):458–467. doi: 10.1021/ar200024c [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Cui W, Liu D, Gu W, et al. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021;28(8):2536–2551. doi: 10.1038/s41418-021-00769-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zou Y, Henry WS, Ricq EL, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–608. doi: 10.1038/s41586-020-2732-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Perez MA, Magtanong L, Dixon SJ, et al. Dietary lipids induce ferroptosis in caenorhabditiselegans and human cancer cells. Dev Cell. 2020;54(4):447–54 e4. doi: 10.1016/j.devcel.2020.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Stamenkovic A, O’Hara KA, Nelson DC, et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2021;320(3):H1170–h84. doi: 10.1152/ajpheart.00237.2020 [DOI] [PubMed] [Google Scholar]
- [129].Lin Z, Liu J, Long F, et al. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat Commun. 2022;13(1):7965. doi: 10.1038/s41467-022-35707-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Magtanong L, Ko PJ, To M, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420–32.e9. doi: 10.1016/j.chembiol.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. doi: 10.1016/j.bbrc.2018.12.039 [DOI] [PubMed] [Google Scholar]
- [132].Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. doi: 10.1016/j.bbrc.2016.08.124 [DOI] [PubMed] [Google Scholar]
- [133].Hashidate-Yoshida T, Harayama T, Hishikawa D, et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife. 2015;4:4. doi: 10.7554/eLife.06328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Tesfay L, Paul BT, Konstorum A, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79(20):5355–5366. doi: 10.1158/0008-5472.CAN-19-0369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Long F, Lin Z, Long Q, et al. CircZBTB46 protects acute myeloid leukemia cells from ferroptotic cell death by upregulating SCD. Cancers. 2023;15(2):15. doi: 10.3390/cancers15020459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944–5972. doi: 10.1021/cr200084z [DOI] [PubMed] [Google Scholar]
- [137].Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Molecular Cell. 2021;81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024 [DOI] [PubMed] [Google Scholar]
- [138].Kuhn H, Saam J, Eibach S, et al. Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem Biophys Res Commun. 2005;338(1):93–101. doi: 10.1016/j.bbrc.2005.08.238 [DOI] [PubMed] [Google Scholar]
- [139].Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297–310. doi: 10.1016/j.redox.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kuang F, Liu J, Xie Y, et al. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol. 2021;28(6):765–75.e5. doi: 10.1016/j.chembiol.2021.01.006 [DOI] [PubMed] [Google Scholar]
- [141].Chu B, Kon N, Chen D, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–591. doi: 10.1038/s41556-019-0305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628–41.e26. doi: 10.1016/j.cell.2017.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kriska T, Pilat A, Schmitt JC, et al. Sterol carrier protein-2 (SCP-2) involvement in cholesterol hydroperoxide cytotoxicity as revealed by SCP-2 inhibitor effects. J Lipid Res. 2010;51(11):3174–3184. doi: 10.1194/jlr.M008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi: 10.1038/ncb3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Liu J, Liu Y, Wang Y, et al. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy. 2022;19(3):1–12. doi: 10.1080/15548627.2022.2111635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Reed A, Ware T, Li H, et al. TMEM164 is an acyltransferase that forms ferroptotic C20: 4 ether phospholipids. Nat Chem Biol. 2023;19(3):378–388. doi: 10.1038/s41589-022-01253-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi: 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- [148].Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi: 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Place DE, Kanneganti TD. Cell death–mediated cytokine release and its therapeutic implications. J Exp Med. 2019;216(7):1474–1486. doi: 10.1084/jem.20181892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Zhou B, Liu J, Zeng L, et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol. 2020;5(12):1576–1587. doi: 10.1038/s41564-020-00795-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Zou B, Liu J, Klionsky DJ, et al. Extracellular SQSTM1 as an inflammatory mediator. Autophagy. 2020;16(12):2313–2315. doi: 10.1080/15548627.2020.1843253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Chen F, Wu R, Liu J, et al. The STING1-MYD88 complex drives ACOD1/IRG1 expression and function in lethal innate immunity. iScience. 2022;25:104561. doi: 10.1016/j.isci.2022.104561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Yang L, Xie M, Yang M, et al. PKM2 regulates the warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5(1):4436. doi: 10.1038/ncomms5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Zhang H, Zeng L, Xie M, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;27(4):556–70 e6. doi: 10.1016/j.chom.2020.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Zou J, Zheng Y, Huang Y, et al. The versatile gasdermin family: their function and roles in diseases. Front Immunol. 2021;12:751533. doi: 10.3389/fimmu.2021.751533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Wu R, Wang N, Comish PB, et al. Inflammasome-dependent coagulation activation in sepsis. Front Immunol. 2021;12:641750. doi: 10.3389/fimmu.2021.641750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Chen R, Zeng L, Zhu S, et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci Adv. 2019;5(5):eaav5562. doi: 10.1126/sciadv.aav5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Tsugawa H, Kabe Y, Kanai A, et al. Short-chain fatty acids bind to apoptosis-associated speck-like protein to activate inflammasome complex to prevent salmonella infection. PLoS Biol. 2020;18(9):e3000813. doi: 10.1371/journal.pbio.3000813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–673. doi: 10.1038/s41586-019-1769-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Tang D, Wang H, Billiar TR, et al. Emerging mechanisms of immunocoagulation in sepsis and septic shock. Trends Immunol. 2021;42(6):508–522. doi: 10.1016/j.it.2021.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–76. doi: 10.1038/s41586-018-0761-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Elliott EI, Miller AN, Banoth B, et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is Initiated at priming. J Immun (Baltimore, Md : 1950). 2018;200(9):3047–3052. doi: 10.4049/jimmunol.1701723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol. 2020;30(11):892–903. doi: 10.1016/j.tcb.2020.09.004 [DOI] [PubMed] [Google Scholar]
- [165].Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Med. 2011;17(2):179–188. doi: 10.1038/nm.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Luheshi NM, Giles JA, Lopez-Castejon G, et al. Sphingosine regulates the NLRP3-inflammasome and IL-1β release from macrophages. Eur J Immunol. 2012;42(3):716–725. doi: 10.1002/eji.201142079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Yan Y, Jiang W, Spinetti T, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38(6):1154–1163. doi: 10.1016/j.immuni.2013.05.015 [DOI] [PubMed] [Google Scholar]
- [168].L’Homme L, Esser N, Riva L, et al. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res. 2013;54(11):2998–3008. doi: 10.1194/jlr.M037861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Gianfrancesco MA, Dehairs J, L’Homme L, et al. Saturated fatty acids induce NLRP3 activation in human macrophages through K(+) efflux resulting from phospholipid saturation and na, K-ATPase disruption. Biochimica et biophysica acta Mol Cell Biol Lipids. 2019;1864(7):1017–1030. doi: 10.1016/j.bbalip.2019.04.001 [DOI] [PubMed] [Google Scholar]
- [170].Karasawa T, Kawashima A, Usui-Kawanishi F, et al. Saturated fatty acids undergo intracellular crystallization and activate the NLRP3 inflammasome in macrophages. Arteriosclerosis Thrombosis Vasc Biol. 2018;38(4):744–756. doi: 10.1161/ATVBAHA.117.310581 [DOI] [PubMed] [Google Scholar]
- [171].Rajamäki K, Lappalainen J, Oörni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765. doi: 10.1371/journal.pone.0011765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].de la Roche M, Hamilton C, Mortensen R, et al. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J Cell Bio. 2018;217(10):3560–3576. doi: 10.1083/jcb.201709057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Reboldi A, Dang EV, McDonald JG, et al. 25-hydroxycholesterol suppresses interleukin-1–driven inflammation downstream of type I interferon. Science. 2014;345(6197):679–684. doi: 10.1126/science.1254790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Dang EV, McDonald JG, Russell DW, et al. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171(5):1057–71.e11. doi: 10.1016/j.cell.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. doi: 10.1038/nature15541 [DOI] [PubMed] [Google Scholar]
- [177].Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi: 10.1038/nature13683 [DOI] [PubMed] [Google Scholar]
- [178].Zanoni I, Tan Y, Di Gioia M, et al. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity. 2017;47(4):697–709.e3. doi: 10.1016/j.immuni.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zanoni I, Tan Y, Di Gioia M, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352(6290):1232–1236. doi: 10.1126/science.aaf3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Chu LH, Indramohan M, Ratsimandresy RA, et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun. 2018;9(1):996. doi: 10.1038/s41467-018-03409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Oskolkova OV, Afonyushkin T, Preinerstorfer B, et al. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J Immunol (Baltimore, Md : 1950). 2010;185(12):7706–7712. doi: 10.4049/jimmunol.0903594 [DOI] [PubMed] [Google Scholar]
- [182].de Carvalho RVH, Andrade WA, Lima-Junior DS, et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 2019;26(2):429–37.e5. doi: 10.1016/j.celrep.2018.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Pizzuto M, Hurtado-Navarro L, Molina-Lopez C, et al. Ornithine lipid activates both TLR4 and the non-canonical NLRP3 inflammasome. bioRxiv 2022:2022.01.28.477396.
- [184].Cruz Garcia AB S, Schnur KP, Malik AB, et al. Gasdermin D pores are dynamically regulated by local phosphoinositide circuitry. Nat Commun. 2022;13(1):52. doi: 10.1038/s41467-021-27692-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi: 10.1038/nature18590 [DOI] [PubMed] [Google Scholar]
- [186].Chai Q, Yu S, Zhong Y, et al. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin. Science. 2022;378(6616):eabq0132. doi: 10.1126/science.abq0132 [DOI] [PubMed] [Google Scholar]
- [187].Kang R, Zeng L, Zhu S, et al. Lipid peroxidation drives gasdermin D-Mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24(1):97–108.e4. doi: 10.1016/j.chom.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Li J, Chen X, Kang R, et al. Regulation and function of autophagy in pancreatic cancer. Autophagy. 2021;17(11):3275–3296. doi: 10.1080/15548627.2020.1847462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [191].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. doi: 10.1126/sciadv.aaw2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Kriel J, Loos B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Diff. 2019;26(4):640–652. doi: 10.1038/s41418-018-0267-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi: 10.1038/nature13148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Gump JM, Staskiewicz L, Morgan MJ, et al. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014;16(1):47–54. doi: 10.1038/ncb2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Chen X, Zeh HJ, Kang R, et al. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18(11):804–823. doi: 10.1038/s41575-021-00486-6 [DOI] [PubMed] [Google Scholar]
- [197].He W, Wang Q, Srinivasan B, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33(23):3004–3013. doi: 10.1038/onc.2013.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Dall’armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23(1):R33–45. doi: 10.1016/j.cub.2012.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Arico S, Petiot A, Bauvy C, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):35243–35246. doi: 10.1074/jbc.C100319200 [DOI] [PubMed] [Google Scholar]
- [200].Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Molecular Cell. 2015;57:219–234. doi: 10.1016/j.molcel.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009;18(24):4868–4878. doi: 10.1093/hmg/ddp460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Wang H, Sun HQ, Zhu X, et al. Gabaraps regulate PI4P-dependent autophagosome: lysosome fusion. Proc Natl Acad Sci, USA. 2015;112(22):7015–7020. doi: 10.1073/pnas.1507263112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Yamashita S, Oku M, Wasada Y, et al. PI4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J Cell Bio. 2006;173(5):709–717. doi: 10.1083/jcb.200512142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14(9):924–934. doi: 10.1038/ncb2557 [DOI] [PubMed] [Google Scholar]
- [205].Yamagata M, Obara K, Kihara A. Sphingolipid synthesis is involved in autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2011;410(4):786–791. doi: 10.1016/j.bbrc.2011.06.061 [DOI] [PubMed] [Google Scholar]
- [206].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831–838. doi: 10.1038/nchembio.1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Scarlatti F, Bauvy C, Ventruti A, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279(18):18384–18391. doi: 10.1074/jbc.M313561200 [DOI] [PubMed] [Google Scholar]
- [208].Peralta ER, Edinger AL. Ceramide-induced starvation triggers homeostatic autophagy. Autophagy. 2009;5(3):407–409. doi: 10.4161/auto.5.3.7809 [DOI] [PubMed] [Google Scholar]
- [209].Daido S, Kanzawa T, Yamamoto A, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64(12):4286–4293. doi: 10.1158/0008-5472.CAN-03-3084 [DOI] [PubMed] [Google Scholar]
- [210].Sun T, Li D, Wang L, et al. C-jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J Transl Med. 2011;9(1):161. doi: 10.1186/1479-5876-9-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Mishra SK, Gao YG, Deng Y, et al. CPTP: a sphingolipid transfer protein that regulates autophagy and inflammasome activation. Autophagy. 2018;14:862–879. doi: 10.1080/15548627.2017.1393129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Matarrese P, Garofalo T, Manganelli V, et al. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy. 2014;10(5):750–765. doi: 10.4161/auto.27959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Laczkó-Dobos H, Maddali AK, Jipa A, et al. Lipid profiles of autophagic structures isolated from wild type and Atg2 mutant drosophila. Biochim Biophys Acta Mol Cell Biol. 2021;1866(3):158868. doi: 10.1016/j.bbalip.2020.158868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Schutter M, Giavalisco P, Brodesser S, et al. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell. 2020;180(1):135–49 e14. doi: 10.1016/j.cell.2019.12.005 [DOI] [PubMed] [Google Scholar]
- [215].MYW N, Charsou C, Lapao A, et al. The cholesterol transport protein GRAMD1C regulates autophagy initiation and mitochondrial bioenergetics. Nat Commun. 2022;13(1):6283. doi: 10.1038/s41467-022-33933-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Punnonen EL, Pihakaski K, Mattila K, et al. Intramembrane particles and filipin labelling on the membranes of autophagic vacuoles and lysosomes in mouse liver. Cell Tissue Res. 1989;258(2):269–276. doi: 10.1007/BF00239447 [DOI] [PubMed] [Google Scholar]
- [217].Liao Y, Zhang P, Yuan B, et al. Pravastatin protects against avascular necrosis of femoral head via autophagy. Front Physiol. 2018;9:307. doi: 10.3389/fphys.2018.00307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Cheng J, Ohsaki Y, Tauchi-Sato K, et al. Cholesterol depletion induces autophagy. Biochem Biophys Res Commun. 2006;351(1):246–252. doi: 10.1016/j.bbrc.2006.10.042 [DOI] [PubMed] [Google Scholar]
- [219].Gao S, Zhang ZM, Shen ZL, et al. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen Res. 2016;11(6):977–982. doi: 10.4103/1673-5374.184498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Charsou C, Ng MYW, Simonsen A. Regulation of autophagosome biogenesis and mitochondrial bioenergetics by the cholesterol transport protein GRAMD1C. Autophagy. 2022;19(7):1–3. doi: 10.1080/15548627.2022.2155020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Laraia L, Friese A, Corkery DP, et al. The cholesterol transfer protein GRAMD1A regulates autophagosome biogenesis. Nat Chem Biol. 2019;15(7):710–720. doi: 10.1038/s41589-019-0307-5 [DOI] [PubMed] [Google Scholar]
- [222].Yang L, Ye F, Liu J, et al. Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. Autophagy. 2022;19(6):1–12. doi: 10.1080/15548627.2022.2152209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Kaushik S, Rodriguez-Navarro JA, Arias E, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14(2):173–183. doi: 10.1016/j.cmet.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Zhang X, Kitatani K, Toyoshima M, et al. Ceramide Nanoliposomes as a MLKL-Dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol Cancer Ther. 2018;17(1):50–59. doi: 10.1158/1535-7163.MCT-17-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Yan C, Zheng L, Jiang S, et al. Exhaustion-associated cholesterol deficiency dampens the cytotoxic arm of antitumor immunity. Cancer Cell. 2023;41(7):1276–1293.e11. doi: 10.1016/j.ccell.2023.04.016 [DOI] [PubMed] [Google Scholar]
- [227].Mohammadalipour A, Showalter CA, Muturi HT, et al. Cholesterol depletion decreases adhesion of non-small cell lung cancer (NSCLC) cells to E-selectin. Am J Physiol Cell Physiol. 2023;325(2):C471–C482. doi: 10.1152/ajpcell.00197.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Fedida-Metula S, Elhyany S, Tsory S, et al. Targeting lipid rafts inhibits protein kinase B by disrupting calcium homeostasis and attenuates malignant properties of melanoma cells. Carcinogenesis. 2008;29(8):1546–1554. doi: 10.1093/carcin/bgn146 [DOI] [PubMed] [Google Scholar]