

Summary
Background:
Lynch syndrome is an autosomal dominant familial condition caused by a pathogenic variant (PV) in a DNA mismatch repair (MMR) gene, which then predisposes carriers to various cancers.
Aim:
To review the pathogenesis, clinical presentation, differential diagnosis, and clinical strategies for detection and management of Lynch syndrome (LS).
Methods:
A narrative review synthesizing knowledge from published literature, as well as current National Comprehensive Cancer Network guidelines for management of LS was conducted.
Results:
LS tumors are characterized by unique pathogenesis, ultimately resulting in hypermutation, microsatellite instability, and high immunogenicity that has significant implications for cancer risk, clinical presentation, treatment, and surveillance. LS is one of most common hereditary causes of cancer, and about 1 in 279 individuals carries a PV in a LS gene that predisposes to associated cancers. Individuals with LS have increased risks for colorectal, endometrial, and other cancers, with significant variation in lifetime risk by LS-associated gene.
Conclusions:
As genetic testing becomes more widespread, the number of individuals identified with Lynch syndrome is expected to increase in the population. Understanding the pathogenesis of LS informs current strategies for detection and clinical management, and also guides future areas for clinical innovation. Unraveling the mechanisms by which these tumors evolve may help to more precisely tailor management by the gene involved.
Introduction
Lynch syndrome (LS) is the hereditary predisposition to colorectal cancer (CRC), endometrial cancer (EC) and certain other cancers, with a population frequency of about 1 in 2791. It is inherited through pathogenic variants (PVs) in one of four DNA mismatch repair (MMR) genes: MSH2, MLH1, MSH6, and PMS22. LS can also be due to a pathogenic variant in EPCAM, a gene immediately upstream of MSH2, which leads to MSH2 promoter methylation and silencing3. The presence of a PV in a Lynch syndrome-associated gene results in abnormal loss of expression of one or more DNA mismatch repair proteins. When loss of DNA mismatch repair proteins occurs, errors in DNA replication that commonly develop are less likely to be repaired. When DNA replication errors go unrepaired in genes important to carcinogenesis, such as tumor suppressor genes, the risk for cancer development increases, and results in the observed lifetime risks for multiple cancers associated with LS, particularly CRC and EC. Cancers develop at earlier ages in the setting of LS, and multiple synchronous or metachronous tumors may occur4. This review provides an update on the mechanisms for developing LS-associated tumors, cancer risks associated with LS, and strategies for identifying and managing individuals with LS. A glossary of key genetics terms and definitions used throughout this review is provided in Table 1.
Table 1.
Key genetics terms and definitions.
| Lynch syndrome | The hereditary predisposition to multiple cancers caused by having a pathogenic variant in one of 4 DNA mismatch repair genes (MLH1, MSH2, MSH6, PMS2) or EPCAM. Formerly known as Hereditary Nonpolyposis Colorectal Cancer (HNPCC) |
| Germline mutation | A mutation in a gene on one of the inherited chromosomes. Some germline mutations predispose to cancer. |
| Somatic mutation | A mutation in a gene that was not inherited and developed after conception. |
| Germline pathogenic variant (PV) | A mutation in a germline gene that causes predisposition to disease such as cancer. |
| DNA Mismatch Repair (MMR) | The process by which cells in the body recognize and repair errors in DNA replication. A key aspect of DNA mismatch repair that is managed by LS-associated genes is to recognize when correct base pairing (adenine with thymine, and guanine with cytosine) does not occur during DNA replication, and repair these errors. Failure of DNA mismatch repair leads to inactivation of tumor suppressor genes and promotes carcinogenesis. |
| Hypermutated Tumor | A tumor that carriers a high burden of mutations. LS often causes hypermutated tumors, but not all hypermutated tumors are attributable to LS. Patients with a hypermutated tumor should be evaluated for LS. |
| Microsatellite Instability (MSI) | A tumor that carriers a high burden of errors in repetitive sections of DNA sequence known as microsatellites. LS often causes tumors with microsatellite instability, but not all microsatellite unstable tumors are attributable to LS. Patients with a microsatellite unstable tumor should be evaluated for LS. |
| Haploinsufficiency | The state of having one inactivated or deleted one copy of a gene where the remaining copy is insufficient for carrying out the gene's function for a normal phenotype. Classically, it had been thought that expression from just one allele of a gene was sufficient for normal function, but there are instances (such as for the PV gene in patients with LS) where the presence of just one normal allele together with the deletion or mutation of the other allele can alter normal cell functioning, and this is called 'haploinsufficiency.' |
| Loss of Heterozygosity | The irreversible loss of one parental allele (allelic loss). When starting with one lost allele (either due to germline or acquired somatic mutation), losing the remaining allele is a case of “loss of heterozygosity”. In LS, LOH of the MMR PV gene within a tumor or polyp is associated with predisposition to more rapid growth and carcinogenesis. |
The DNA MMR system
DNA MMR is the process by which cells recognize and repair errors in DNA replication that occur during S phase5. A key aspect of DNA MMR is to recognize when correct base pairing does not occur during replication, and repair these errors. In the face of irreparable DNA damage, the MMR system signals mechanisms that result in cell death6. In the normal state, the protein products of LS-related DNA MMR genes work together (as heterodimers) in a coordinated fashion to recognize and repair base pair mismatches (Figure 1). Specifically, the MSH2 protein pairs with the MSH6 protein to form a heterodimer called MutSα. MutSα binds to single base-pair mismatches in the newly synthesized DNA strand, such as when guanine is incorrectly paired with thymine, or adenine is incorrectly paired with cytosine. MutSα then recruits a heterodimer of the proteins MLH1 and PMS2 called MutLa that enlists the additional proteins needed to repair the mismatch. The MSH2 protein can also heterodimerize with the MSH3 protein, forming MutSβ (Figure 1A), that recognizes different DNA mismatches, and works with MutLa to achieve DNA MMR.
Figure 1:
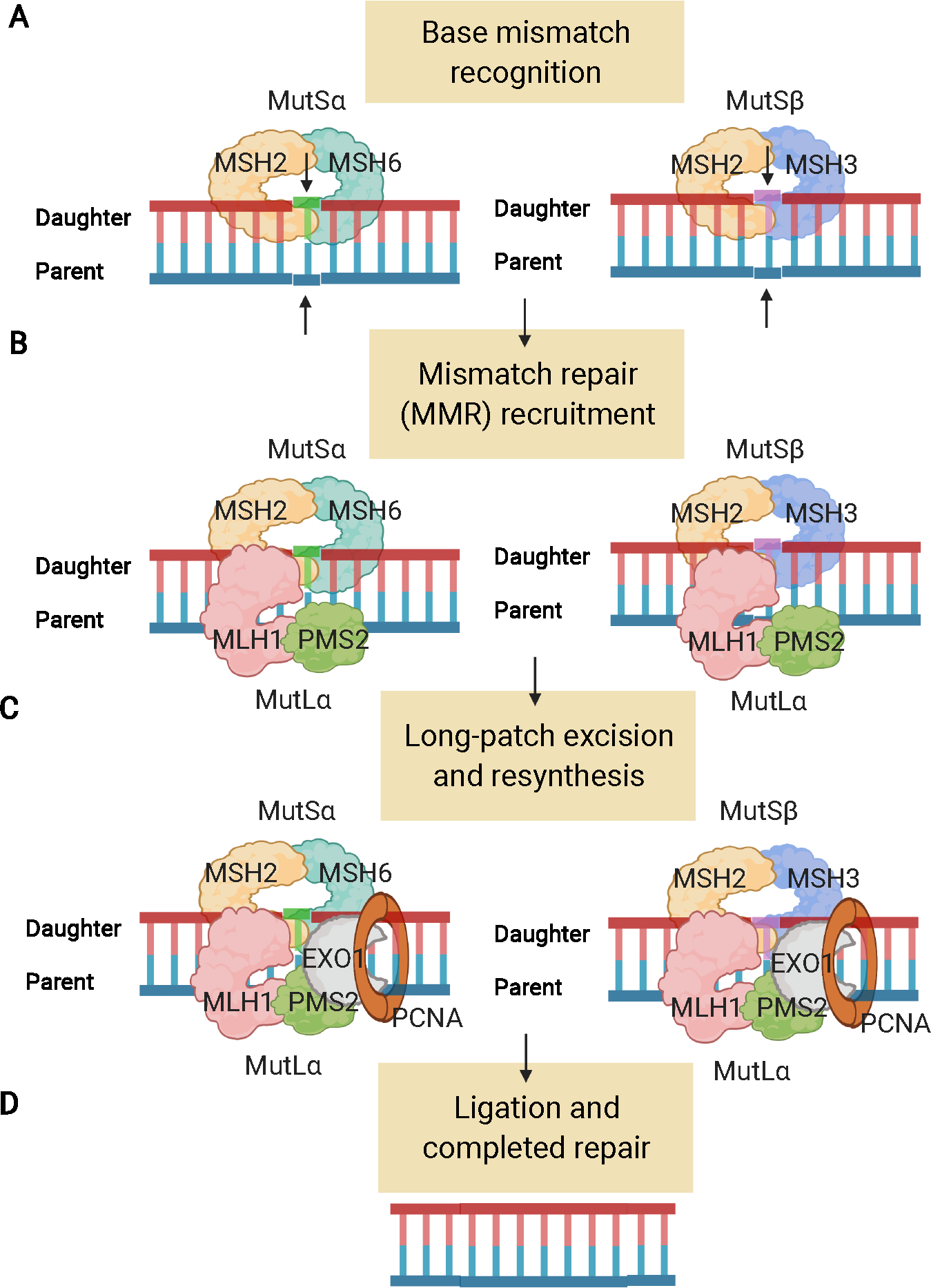
The DNA mismatch repair (MMR) system. A) The MutS MMR proteins recognize DNA errors (mispairs and loop-outs) made during S phase. They are only stable as heterodimers (MSH2 + MSH6 [MutSα] or MSH2 + MSH3 [MutSβ]). B) MutL (MutLa here) heterodimers bind to the MutS proteins and act as recruitment proteins for the DNA repair process. C) Enzymes such as exonuclease 1 (EXO1), DNA polymerase, and proliferating cell nuclear antigen (PCNA) serving as a sliding clamp, perform long patch excision to remove the error, and then enable resynthesis and ligation. D) The daughter strand is resynthesized, which results in correction of the DNA error. Figure created using BioRender.com.
When does defective MMR activity occur in a Lynch syndrome neoplasm?
The classic Knudson hypothesis speculated that neoplastic behavior caused by a germline PV in a familial cancer gene requires the loss of the “wild type” copy of the gene inherited from the unaffected parent to initiate neoplastic growth, the so-called “second hit”7. However, evidence from a mouse model suggests that excessive mutations begin to accumulate even before the second hit occurs8. The presence of one inactivated gene (with at least some PVs) results in a lower effective dose of the MMR protein, and a small degree of hypermutability in otherwise normal tissues. This situation is called “haploinsufficiency”. Then, when the true second hit takes place (with an inactivating mutation or loss of heterozygosity of the “wild type” gene), full blown MMR-deficiency takes place, which results in microsatellite instability (MSI). Then, for example, the TGFB1R2 gene, a tumor suppressor gene, by virtue of a coding microsatellite sequence (ten adenines in a row), is particularly susceptible to mutations during replication that are more likely to go uncorrected in the setting of MMR-deficiency, and this gene is frequently mutated in tumors with MMR-deficiency and associated MSI.
MMR associated with LS can be identified in several ways within cancers such as CRC. A PV in a LS gene results in “abnormal absence” of the corresponding gene protein product within the tumor9. For example, a germline PV in MLH1 results in absent expression of the MLH1 protein when assessed with immunohistochemistry techniques. Typically, both proteins of a heterodimer pair (MLH1 and PMS2; MSH2 and MSH6) show abnormally absent protein expression, because the absence of one protein results in instability of the other, unpaired heterodimer protein, even if it is fully functional (Table 2). Mutations may occur anywhere, but are particularly common at microsatellite repeat loci. Some genes important for preventing cancer development are particularly susceptible to somatic mutations that result from suboptimal MMR activity10.
Table 2:
Abnormal immunohistochemistry (IHC) for DNA MMR proteins in human tissue. + denotes the protein is normally expressed in the nucleus.
| Germline PV | MSH2 protein | MSH6 protein | MLH1 protein | PMS2 protein |
|---|---|---|---|---|
| MSH2/EPCAM | negative | negative | + | + |
| MLH1 | + | + | negative | negative |
| MSH6 | + | negative | + | + |
| PMS2 | + | + | + | negative |
MSI was originally identified – almost by accident - as a feature present in about 15% of colorectal cancers in 199211–13; soon thereafter, MSI was identified as a feature of LS associated with MSH214,15 and MLH116,17. Detection of MSI is easily achieved using PCR of tumor DNA and recognizing the alterations in the homopolymeric DNA sequence compared to that individual’s non-neoplastic tissues (although the current assays do not require normal tissue to perform the diagnostic assays)10. Multiple panels of target microsatellite sequences, such as the Bethesda marker panel, have been identified and validated over the years5. More recently, evidence of MMR deficiency has been detected by identifying the “hypermutator” mutational signatures specific to MSI within tumors based on next generation sequencing, which is likely to be the preferred approach in the future18,19.
How do tumors develop in Lynch syndrome?
Based upon studies on sporadic CRCs, it was proposed in the late 1980s that CRCs evolve through the sequential accumulation of somatic mutations, beginning in the pre-cancerous and then tumor tissue, that activate oncogenes and inactivate tumor suppressor genes20,21. It has been subsequently documented that most colorectal neoplasms begin with mutations in the WNT signaling system, most often loss-of-function variants in the APC gene, leading to unregulated proliferation. In fact, APC is the only gene among the 15 most common somatic mutations in CRCs that occur in both hypermutated CRCs (i.e., with MSI) and non-hypermutated tumors (microsatellite stable – MSS), being found in 51% of the former and 81% of the latter CRCs22. Also, The Cancer Genome Atlas Network collaborative study counted the numbers of mutations per megabase (MB) of DNA in a collection of 276 CRCs, and found an inflection point between the hypermutated tumors (>10 mutations/MB, up to as many as 400 mutations/MB) and the non-hypermutated CRCs (all <8 mutations/MB, some with <1/MB). The hypermutated CRC group included all of the tumors with MSI, and the “ultramutated” tumors were associated with alterations in the DNA polymerase proofreading genes22. These significant genetic differences potentially explain the differences in clinical behavior between MSI and non-MSI tumors.
More critical to this disease is to understand the implications of the observation that half of MSI CRCs have APC mutations and the other half do not22. For example, APC mutations are the most common initiator of advanced adenomas; thus if half of MSI tumors do not carry a clonal APC mutation, then alternative pathways for premalignant progression must be considered in order to develop effective preventive measures. Indeed, to disrupt WNT signaling for neoplastic proliferation, MSI-H tumors that lack APC mutations have been found to have other WNT activating mutations through either RNF43 (among most common CRC mutations), beta-catenin mutations, or PTPRK-RSPO3 fusions23–26.
In a study of adenomatous polyps from individuals with Lynch syndrome, it has been reported that most begin as microsatellite stable (MSS) lesions, but develop MSI when the polyp grows to >8–10 in diameter27. Moreover, a study from Japan reported that 79% of all adenomas and 100% of CRCs in LS showed MSI24. A mutational analysis revealed frequent frameshift alterations and that MMR-deficiency precedes adenoma formation24. Additionally, a group from Germany has found that most colons from patients with Lynch syndrome that manifest CRCs also have foci of defective MMR (dMMR) activity in normal-appearing colonic mucosa, including loss of MMR protein expression, the presence of MSI, and mutations at certain target driver genes28. The mucosa in these dMMR crypts does not appear to be dysplastic microscopically, which one finds in “aberrant crypt foci”29. Observations of both late development of MMR in adenomas that have had initial loss of APC, as well as early MMR deficiency in normal-appearing mucosa, has led to the hypothesis that neoplastic lesions in Lynch syndrome may emerge through more than one genetic pathway. A 2018 systematic literature review of previous molecular studies found that less than a quarter (23.3%) of LS adenomas were MMR-proficient. 30 This indicates that the most common of the potential genetic pathways of Lynch carcinogenesis is likely one that begins with MMR-deficiency in normal cells before APC loss and development of adenomas.
Models for CRC carcinogenesis in Lynch syndrome
Conceptual models for CRC carcinogenesis have been developed taking into account current basic science that has characterized potential pathogenesis pathways, as well as clinical observations regarding variation in the occurrence of adenomas and CRC across Lynch syndrome subtypes (Figure 2). Interestingly, clinical observations suggest that patients with Lynch syndrome who have PVs in MSH2, MLH1 or MSH6 all develop adenomas at about the same rate over 20 years of surveillance31. However, patients with Lynch syndrome associated with MSH2 are more likely to develop advanced adenomas than individuals with MLH1 or MSH6, and CRCs are more likely to develop in MSH2 or MLH1 PV carriers than those with a PV in MSH631 (Table 3). Furthermore, patients with Lynch syndrome associated with PMS2 have relatively few advanced adenomas or CRCs when under colonoscopic surveillance32. Therefore, risk-based surveillance strategies should be gene-specific and models for CRC carcinogenesis must be broad enough to capture the clinical variations that have been observed based upon the genetic PV involved.
Figure 2:
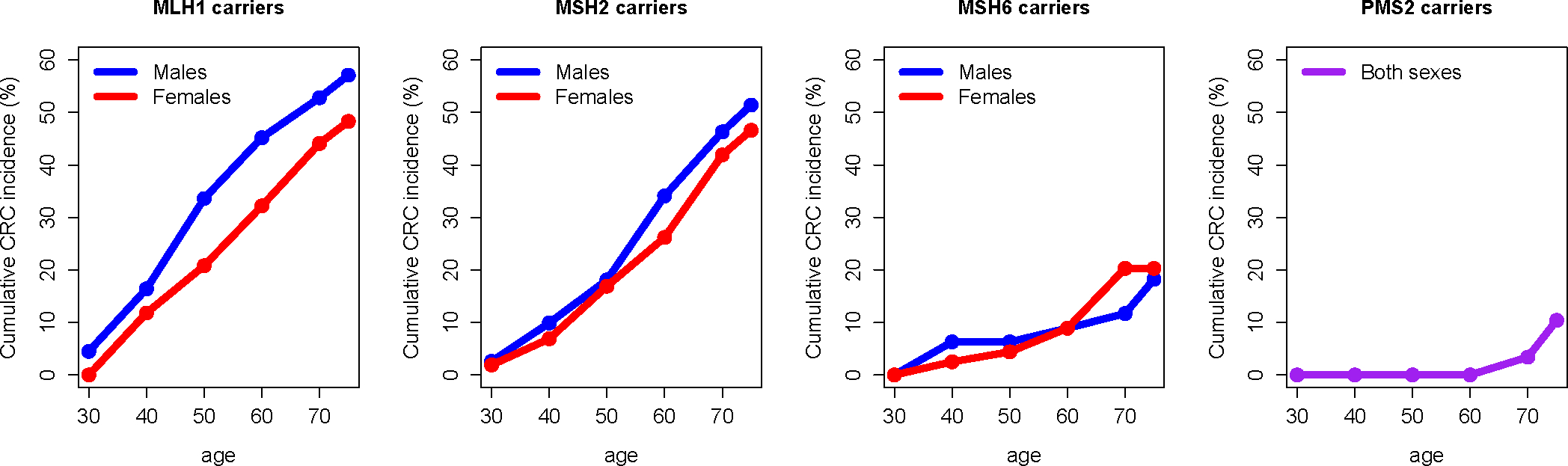
Cumulative colorectal cancer (CRC) incidence by age, shown for males (blue) and females (red) in separate panels for each specified MMR gene mutation, as determined using the Prospective Lynch Syndrome Database (PLSD). Data for carriers of PMS2 PVs were combined for males and females (purple) due to limited total follow-up years within the data42
Table 3: 10-year cumulative incidence of adenoma, advanced adenoma, and colorectal cancer during surveillance since index colonoscopy.
Data for cohort from Engel et al31 consisting of 2747 patients with Lynch syndrome (LS) who had at least 2 surveillance colonoscopies as part of LS registries in Germany, the Netherlands, and Finland. See ref 31 for full survival analysis and Kaplan-Meier curves from baseline (index colonoscopy) through 20 years of patient follow-up. CI: confidence interval.
| LS gene mutation: | 10-year adenoma risk | 10-year advanced adenoma risk | 10-year colorectal cancer risk |
|---|---|---|---|
| MLH1 | 32.2% (95% CI: 29.2%–35.2%) | 7.7% (95% CI: 6.0%–9.4%) | 11.3% (95% CI: 9.4%–13.2%) |
| MSH2 | 44.2% (95% CI: 40.0%–48.4%) | 17.8% (95% CI: 14.6%–21.0%) | 11.4% (95% CI: 8.9%–14.0%) |
| MSH6 | 38.4% (95% CI: 30.8%–45.9%) | 9.4% (95% CI: 5.4%–13.4%) | 4.7% (95% CI: 1.8%–7.7%) |
Accordingly, the Heidelberg group led by Kloor33 and the Cardiff group led by Frayling34 have proposed the following model with 3 potential pathways (Figure 3). As described above, the least frequently observed scenario is a ‘classical’ initiation of adenomas via an APC inactivating mutation causing WNT pathway activation in Lynch syndrome, which then lose DNA MMR activity as they grow, and evolve through a pathway through polypoid lesions. These polyps may be detected and removed colonoscopically, delaying the development of CRC in these patients.
Figure 3:
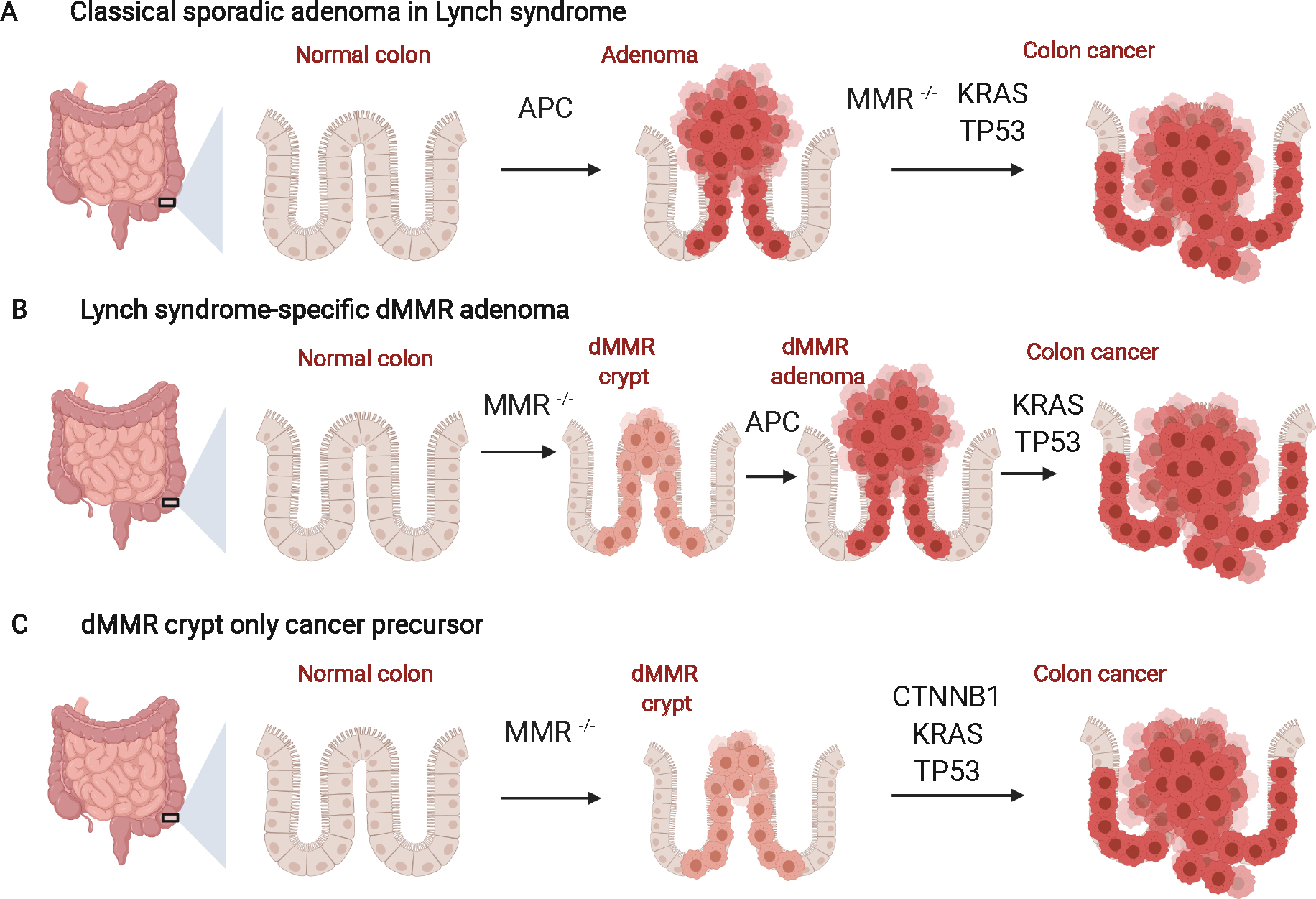
Three potential pathways for CRC evolution in LS 33,34. Based on molecular and clinical evidence (see text for details), multiple pathways for CRC carcinogenesis in LS have been suggested that vary in placement of the timing of mismatch repair (MMR) deficiency and the growth of polyp precursors.
A) Adenomas can first develop via traditional mutations such as APC inactivation followed by MMR loss and accumulations of additional driver mutations leading to CRC. The theoretical basis of this is that APC inactivation leads to the formation of an elevated lesion (benign polyp) that can be detected and removed by colonoscopy. This pathway is proposed to be common in LS associated with germline PVs in MSH6 and PMS2, but is the rarest scenario found in molecular studies of Lynch associated-adenomas.
B) Inactivating mutations in the MMR gene can occur as the first step producing MMR-deficient (dMMR) crypts, followed by inactivating mutations in APC, leading to an elevated lesion in the colon. Subsequent accumulation of mutations lead to CRCs, and this pathway is proposed to be most common in LS-MSH2.
C) MMR-deficient crypts may accumulate driver mutations such as activation of CTNNB1 and serve as the precursor to a flat CRC, without necessarily initiating precursor elevated adenomas, confounding the beneficial effects of surveillance colonoscopy, as may occur in LS-MLH1. This figure was adapted from “Colon Cancer Progression” by BioRender.com (2022)
It has been proposed that in the setting of MSH2 Lynch syndrome, the adenomas may evolve through the classic pathway or from flat dMMR crypts (perhaps through an activating mutation in β-catenin rather than an inactivating mutation in APC) and the subsequent accumulation of somatic mutations leads to the emergence of a polypoid lesion that can be removed through surveillance, as occurs also in the setting of MSH6 and PMS2 Lynch syndrome (Figure 3a, 3b)35. However, in the case of MLH1 Lynch syndrome, it is proposed that these lesions are more likely to evolve as a flat lesion from dMMR crypts, which then accumulate multi-hit mutations (e.g., CTNNB1, TP53, KRAS) in normal-appearing colonic mucosa, making it more difficult to detect and remove the premalignant lesion and prevent CRC (Figure 3c). This remains somewhat speculative and more molecular studies of normal colonic epithelium in LS are needed, but this reasonable hypothesis would help explain some of the clinical heterogeneity based upon which gene is involved, and provides a conceptual framework for precision prevention in the future.
Conceptual models of Lynch-associated CRC carcinogenesis have been formally extended into mathematical models that seek to explicitly consider these molecular changes in order to infer the potential dynamics underlying biological data and clinical observations. For example, Komarova and colleagues previously quantified genomic instability in LS by solving ordinary differential equations (ODEs) for a Moran process that includes inactivating APC mutations as the mechanism governing dysplastic crypt initiation36. This type of model can take parameters such as the number of crypts in a colon and mutation rates as input and predict useful quantities such as expected number of APC-mutated crypts by age 40, which is not possible from clinical data alone. Expanding on these assumptions, Haupt and colleagues recently developed a dynamical system model that allows for the multiple pathways of cancer formation in LS as outlined above, and exploits a useful Kronecker structure for solutions to ODEs37. Given certain parameter inputs such as the number of point mutations per cell division, an example output for this model is the expected proportion of crypts in a typical MLH1 carrier for which MMR deficiency was the initial event during carcinogenesis before additional mutations such as KRAS activation accrue. Other recent efforts include crypt-based simulations for modeling monoclonal conversion rates of MMR deficiency and driver mutations37, as well as modeling the expected protective effects of aspirin by modification of cell fitness or crypt fission rates that could be used to inform clinical trial design38. Mathematical models allow estimation of evolutionary dynamics that cannot currently be directly measured in vivo, and further advancements will be enabled upon availability of more comprehensive, prospective data that will inform unknown parameters such as probability of spontaneous crypt death given mutation type. Importantly, this requires temporal data from patients without detected adenomas or cancers along with accompanying colonoscopy quality data (e.g., adenoma detection rates) for each time point. Overall, novel models may help us understand progression rates of particular pathways to be used in risk prediction and future cost-benefit analyses.
Lynch syndrome prevalence
In the US, Australia, and Canada, the prevalence of Lynch syndrome caused by germline PVs in one of the four main MMR genes was estimated to be 1 in 279 people1. Heterogeneity was also found in the population prevalence for carrying a specific MMR gene mutation (1 in 1,946 for MLH1, 1 in 2,841 for MSH2, 1 in 758 for MSH6, 1 in 714 for PMS21). In Iceland, the prevalence was slightly greater (1/226) partly due to the higher prevalence of lower-penetrance founder mutations in MSH6 and PMS2 “imputed” in this population39. Notably, genetic models used for prevalence estimates of LS must rely on comparison of observed cancer incidence among confirmed PV carriers to the expected incidence based on the general population; LS prevalence has not been directly measured as yet with population sampling. Population-level screening for LS in healthy individuals is not performed in practice, and consequently it is estimated that only ~1% of the true population with Lynch syndrome in the US are diagnosed during their lifetimes, signifying drastic under-diagnosis40.
Variable penetrance for cancer in Lynch syndrome by gene involved
Cancers of the colorectum, uterus, ovary, urinary tract, stomach, pancreas, bile duct, small intestine, brain (usually glioblastoma), and skin (sebaceous carcinomas) more commonly occur in individuals with LS compared to the general population, though each of the MMR mutation types confers a different future risk of cancer for the patient41. As part of the Prospective Lynch Syndrome Database (PLSD), 6350 pathogenic MMR gene carriers, including 3480 females and 2870 males with mean age = 46.8 years (range 25–74) at study inclusion, were followed up to age 75 and incident cancers were recorded to calculate cumulative age- and sex-specific penetrance42 (Figure 2). For women, cumulative incidence of any cancer by age 75 was 81.0% for MLH1, 84.3% for MSH2, and 61.8% for MSH6. For men, cumulative incidence of any cancer by age 75 was 71.4% for MLH1, 75.2% for MSH2, and 41.7% for MSH6. For both sexes combined, carriers of a PV in PMS2 had a cumulative cancer incidence of 34.1% by age 75. In women with LS, EC risk was substantial; by age 75, the cumulative incidence was estimated to be 37.0% for MLH1, 48.9% for MSH2, 41.1% for MSH6, and 12.8% for PMS2. Over the total 15,646 patient-years of follow-up, 580 of the 1808 (32.1%) incident cancers diagnosed in this patient cohort were CRCs. For women, cumulative incidence of CRC by age 75 was 48.3% for MLH1, 46.6% for MSH2, and 20.3% for MSH6. For men, cumulative incidence of CRC by age 75 was 57.1% for MLH1, 51.4% for MSH2, and 18.2% for MSH6. For both sexes combined, carriers of a pathogenic variant of PMS2 had a cumulative CRC incidence of 10.4% by age 75 42. Importantly, these were patients undergoing surveillance and thus most CRC were caught at an early stage, resulting in high 5-year survival rates (95%).
Variable penetrance for precursor colorectal lesions in Lynch syndrome
Beyond CRC risk during surveillance, differential risk for CRC precursors – adenomas and advanced adenomas - has also been calculated using colonoscopy follow-up data since study entry by MMR gene mutation type31. In a prospective cohort of 2747 patients with LS, the 10-year cumulative incidence (under surveillance) of any adenoma was 32.2% in MLH1 carriers, 44.2% in MSH2 carriers, and 38.4% in MSH6 carriers. For advanced adenomas, the 10-year cumulative incidence was 7.7% in MLH1 and 9.4% in MSH6 carriers, which was significantly lower than the 17.8% detected in MSH2 carriers31. Although the prospective CRC risk was nearly identical in MLH1 and MSH2 in this study (~11% by year 10), more somatic APC mutations in the CRCs and higher risk of advanced adenoma was found in MSH2 versus MLH1 PV carriers. Notably, MLH1 PV carriers were more likely to have more β-catenin (CTNNB1) mutations in their CRCs, indicating a subtle difference in the pathways of tumor development between MLH1 and MSH2 carriers. Interestingly, MLH1 carriers had a lower cumulative risk of advanced adenoma than risk of CRC on surveillance, as opposed to MSH2 carriers who had a higher cumulative risk for advanced adenomas than CRC (Table 3).
As mentioned previously, one hypothesis proposed is that in patients with PVs in MSH2 (as well as MSH6 and PMS2), the accumulation of somatic mutations more frequently follows a classical model that begins with mutations in APC leading to the formation of a polypoid adenoma, while MLH1 carriers may develop CRCs through an alternative pathway from flat dMMR crypts without a detectable adenoma precursor34.
VARIATIONS IN PHENOTYPE
Muir-Torre Syndrome
Beyond developing conventional colorectal adenomas, it has long been noted that some patients with Lynch syndrome are also prone to develop specific cutaneous lesions including sebaceous adenomas, sebaceous carcinomas and keratoacanthomas, constituting the Muir-Torre syndrome (MTS) variant of LS. These lesions occur anywhere on the body, and the sebaceous lesions are particularly common on the face. About 28% of families and 9% of individuals in Lynch syndrome families have MTS43. The skin lesions have MSI, but this is also true of sporadic sebaceous neoplasms, so looking for dMMR activity is not sufficient for making the diagnosis of LS44. MTS is more commonly associated with germline variants in MSH2 and MLH1 than with the other DNA MMR genes, and more than half of patients with MTS present with a cutaneous lesion prior to the diagnosis of an internal malignancy43. In clinical practice, some dermatologists routinely assess MMR activity within sebaceous neoplasms, and refer patients with evidence of MMR to gastroenterologists for cancer surveillance and other genetics professionals for follow up. In these cases, it is critical to establish whether or not the patient has LS by confirmatory genetic evaluation and testing that includes assessment for PVs in LS-associated genes.
Biallelic MMR gene mutations: constitutional mismatch repair deficiency (CMMRD)
Rarely, PVs in the same DNA MMR gene are inherited from both parents, and the child is born with no MMR activity, which is called constitutional mismatch repair deficiency (CMMRD) syndrome45. These patients develop a variety of cancers in childhood, including adenomas and cancers of the small bowel and colon, brain tumors, lymphomas, leukemia, urinary tract cancers, endometrial cancers and other malignancies. CMMRD is most commonly associated with biallelic PVs in PMS2 and MSH6, perhaps reflecting the relative population prevalence of these genes and the possibility that biallelic inactivating mutations in MSH2 and MLH1 are more likely to be embryonically lethal. Because of the genes most commonly involved have lower penetrance and later onset of tumors compared to other LS patients, it is not unusual for the child to present with one or more neoplastic lesions without a family history of cancer. CMMRD is more common in populations with higher incidences of consanguineous marriages.
CONFOUNDERS OF LYNCH SYNDROME
Acquired methylation of MLH1
About 15% of unselected CRCs have MSI5, but only 3% of all CRCs occur in individuals with Lynch syndrome. In about 12% of all CRCs5, the MSI is due to the acquired methylation-induced silencing of the MLH1 gene, which is a reflection of the genome-wide methylation of promoters (in the tumor cells), called the “CpG island methylator phenotype”, or CIMP46. The loss of MLH1 protein expression also leads to the loss of PMS2 protein expression, for the reasons described above. The acquired form of genome-wide methylation occurs more frequently in older patients and in women, and the appearance of MSI in the setting of CIMP changes the genetic drivers of the CRC, so these tumors behave more like other CRCs with MSI rather than those with CIMP and MSS47. CIMP and the presence of biallelic methylation of MLH1 is typically (but not universally) accompanied by the V600E mutation in the BRAF oncogene to drive tumor progression, which appears to be mechanistically facilitated by the aging-like acquisition of promoter DNA hypermethylated genes48,49,50. There is evidence that the CRCs with CIMP and somatic BRAF mutations begin as serrated polyps; consequently, this is referred to as the “serrated pathway” of colorectal carcinogenesis51. The proposed pathway is that serrated polyps develop in the setting of CIMP, and if biallelic methylation of MLH1 occurs, MSI develops and the tumor growth is accelerated.
“Constitutional methylation” of MLH1
There are “constitutional” forms of methylation of MLH1 in which the gene is silenced in all cells of the body. The mechanism underlying this is unknown. This disorder clinically behaves like LS, but there are no germline PVs in any DNA MMR gene. There is methylation of MLH1 in DNA taken from all three embryonic germ layers (typically hair follicles, blood and cheek swabs)52, indicating that the constitutional hypermethylation occurred very early in embryogenesis. There is also a rare variant in the MLH1 promoter that can create a familial form of constitutional methylation of MLH153. All of these will mimic LS in the proband, but only those with the rare promoter variant will present with a family history of cancer.
Lynch-Like syndrome (LLS)
When screening CRCs for MSI or abnormal IHC to detect LS, some proportion of the tumors show abnormalities using either modality, but there is no PV found after germline genetic evaluation. Twenty years ago, when this was encountered, it was often assumed that there was actually a germline PV present, but that it was undetectable for technical reasons. Large deletions, balanced inversions, promoter alterations, and other variants outside of the genetic coding region were not always assayed; year by year, more of these variants have been detected and recognized as causes of LS. However, deep sequencing of tumor tissue has revealed the surprising fact that some patients with dMMR tumors had no germline PVs, but actually had acquired double somatic mutations in one MMR gene in the tumor cells54,55. Lynch-like syndrome (LLS) has been found for all four of the Lynch syndrome genes55. Interestingly, the age of CRC in LLS is younger than that in the general population but older than that in LS, and many appear to have family history of LS-associated cancers: 11% fulfill the Amsterdam criteria for LS56, and 65% meet the Bethesda guidelines55,57. So, there would appear to be more to learn about this entity; presumably some proportion may still have “hidden” germline PVs. Also, it is not clear whether there can be an underlying abnormality that permits the tumor to accumulate somatic mutations (in the MMR genes) at an accelerated rate, or whether the presence of double somatic mutations is due to chance. Notably, if bi-allelic somatic mutations within a tumor can be confirmed in a patient with CRC, they may be treated and followed using guidelines for follow up of patients with sporadic CRC, provided that the family history does not fit a Lynch syndrome like pattern.
Familial CRC – Type X
The discovery of the pathogenetic basis of LS in the 1990s was associated with enthusiasm that dMMR activity was the cause of most familial non-polyposis CRC. However, many familial clusters of CRC are unrelated to LS, do not show MSI or abnormal IHC for the MMR genes, and represent something different. Lindor et al recognized this in 2005 and referred to these familial clusters of CRC as “Familial CRC-Type X”58. By definition, the families met the Amsterdam I criteria for a familial cluster of CRC59, but the tumors lacked the features of defective DNA MMR. There was initial hope that one or a small number of genes might be responsible for this clinical phenotype. However, there have been multiple reports of a large number of unrelated genetic defects that are responsible for “Syndrome X”. The genetic basis of these unrelated syndromes ranges from regulation of gene expression, DNA repair processes, developmental genes and regulation of protein and RNA metabolism60. The number of genes is large enough (and the cases sporadic enough) to complicate the development of a genetic testing platform for families with a non-LS cluster of cancer cases (see Table 4).
Table 4:
Genes proposed to cause Familial CRC- Type X, their corresponding proteins, function, and evidence provided for each2.
| Gene | Protein | Function | Evidence |
|---|---|---|---|
| GALNT12 | GalNAC transferase type 12 | Adds GalNAc (a simple sugar) to proteins (O-linked) | 7/272 CRC patients; Low penetrance |
| RSP20 | Ribosomal protein S20 | Ribosomal biosynthesis | 4/118 probands, 2 Type X families |
| BRF1 | RNA pol II subunit | Initiates 5S RNA synthesis | 11/503 Early-onset; 3 Type X families |
| FAF1 | Fas-associated factor 1 | Tumor suppressor gene | 2 Spanish Type X |
| SEMA4A | Semaphorine A | Multiple functions | Multiple families |
| POLD1/POLE | DNA polymerase proofreading | Proof-reads DNA polymerase | Ultramutated tumors (not MSI) |
| HNRNPA0+WIF1 | Interactions of 2 proteins | Cell signaling | Multiple cancers, 1 family |
CLINICAL ASPECTS OF LYNCH SYNDROME
Behavior of CRC in patients with Lynch syndrome
CRCs with MSI differ biologically from non-MSI CRCs (e.g., fewer chromosomal gains and losses) and fortunately for patients with LS who develop MSI-high (MSI-H) CRCs, this translates to better clinical outcomes. For example in a study of 607 CRCs (17% MSI-H) diagnosed under the age of 50, a statistically significant decrease in overall mortality was found for those patients with MSI tumors versus those patients with microsatellite stable tumors (hazard ratio = 0.42, 95% CI 0.27–0.6)61. The reason for improved survival is likely due in part to high immunogenicity as a product of the mutator phenotype characterizing MSI tumors. Due to germline mutation in a MMR gene, patients with LS accumulate large numbers of frameshift mutations leading to creation of an abundance of neoantigens that can elicit an immune response and eventually lead to elimination of precancerous mutated cells during carcinogenesis. However, creation of frameshift mutations in other genes can also permit immune evasion (specifically, HLA Class I defects and β2-microglobulin mutations) but this is seen in the primary tumor mass rather than in circulation; thus tumors are less likely metastasize early and most often only invade locally. In particular for immune evasion, loss of β2-microglobulin was found in 28% of LS CRCs (but not seen LS adenomas), and upregulation of PD-L1 on tumor cells was found in 76% of LS CRCs62.
As a consequence of an increased burden of tumor-infiltrating lymphocytes, patients with LS CRCs generally respond well to immune-checkpoint therapies. In an early trial of 86 patients treated with anti-PD-1 therapy, 21% achieved complete response and 52% exhibited significant tumor regression63. In contrast, patients with LS-associated CRCs do not respond well to some chemotherapeutic regimens since cytotoxic chemotherapy depends upon functioning MMR mechanisms to signal apoptosis in tumor cells64,65.
Detection of familial cancer syndromes and Lynch syndrome
A genetic evaluation for LS may be indicated based on the presence of a known PV in a LS gene in a family member, family history of LS-associated cancers, a personal history of a LS-associated cancer, or by screening CRCs for MMR-deficiency66. As a guiding principle, genetic testing should only be pursued if it would change clinical management for an individual or their family members, if a PV were to be identified.
Evaluation based on known Lynch syndrome associated PV in family
Because risks for LS-associated cancers are inherited in an autosomal dominant manner, all first-degree relatives of an individual with a known PV have a 50% chance of carrying the same variant, and should have genetic testing performed. Generally, testing for first-degree relatives is performed after age 18 years, given that specialized surveillance is not recommended prior to this age. In some cases, a second degree relative (e.g. grandparent, aunt, or uncle) might be known to have a PV in a family where the intervening first degree relative is unable or unwilling to have genetic evaluation. In such cases, given the potential 25% chance of carrying the familial variant, genetic testing for the familial PV is recommended66.
Evaluation based on family and personal history of Lynch associated cancers
Strategies for evaluation of LS have been in evolution for over 20 years (Table 5), with movement over time towards criteria that increase the sensitivity for identification of individuals with LS. Initially, genetic testing was reserved for patients who met criteria based mainly on the number and age of family members with LS-associated cancers, as with the 1999 Amsterdam II criteria. These evolved to be less stringently based on the number and age of relatives, and to also include tumor characteristics such as the presence of MSI, as with the 2004 Bethesda criteria. With studies showing increased sensitivity and yield with universal tumor testing to screen for MMR deficiency in CRCs and ECs, compared to family history only based criteria for detecting LS, many have recommended this approach for selection of individuals for genetic evaluation67,68.
Table 5: Selection strategies for genetic evaluation for Lynch syndrome (LS).
NCCN, National Comprehensive Cancer Network66.
| 1999 Amsterdam II Criteria 56 | Family with ≥3 members affected by
CRC or another LS-associated cancer* and meeting the following criteria: • 1 relative should be a 1st degree relative of the other two • ≥2 successive generations affected • ≥1 LS-associated cancer diagnosed* under age 50 • Familial adenomatous polyposis excluded • Tumors verified by pathological evaluation |
| 2004 Revised Bethesda Criteria 57 | • CRC diagnosed in a person younger
than age 50 • Synchronous, metachronous, colorectal, or LS associated tumor** • CRC with microsatellite high type histology (i.e., presence of tumor-infiltrating lymphocytes, Crohn's-like lymphocytic reaction, mucinous/signet-ring differentiation, medullary growth pattern) in a patient younger than 60 years • CRC in a patient with a family history of a LS-associated cancer diagnosed earlier than age 50 years. If more than one relative was diagnosed with a Lynch syndrome-associated cancer, then the age criterion is not needed. |
| 2021 National Comprehensive Cancer Network® (NCCN®) 66 | • Known LS pathogenic variant in the
family • Personal history of a tumor with mismatch repair deficiency determined by polymerase chain reaction, next generation sequencing, or immunohistochemistry diagnosed at any age. NCCN recommends tumor screening for mismatch repair deficiency for all CRCs and endometrial cancers regardless of age, and consideration of tumor screening for mismatch repair deficiency for sebaceous neoplasms as well as the following adenocarcinomas: small bowel, gastric, pancreas, biliary tract, brain, bladder, urothelial, and adrenocortical. • An individual with colorectal or endometrial cancer and any of the following: • Diagnosed < age 50 years • A synchronous or metachronous LS-related cancer*** regardless of age • 1 1st or 2nd-degree relative with an LS-related cancer diagnosed < age 50 y • ≥2 1st or 2nd-degree relatives with an LS-related cancer regardless of age • Family history of any of the following: • ≥1 1st-degree relative with a colorectal or endometrial cancer diagnosed <50 y • ≥1 1st-degree relative with a colorectal or endometrial cancer and a synchronous or metachronous LS- related cancer regardless of age • ≥2 1st-degree or 2nd-degree relatives with LS-related cancers, including ≥1 diagnosed <50 y • ≥3 1st-degree or 2nd-degree relatives with LS-related cancers regardless of age • Increased model-predicted risk for Lynch syndrome • An individual with a ≥5% risk of having an MMR gene pathogenic variant based on predictive models (i.e., PREMM5, MMRpro, MMRpredict) • Individuals with a personal history of colorectal and/or endometrial cancer with a PREMM5 score of ≥2.5% should be considered for multi-gene panel testing. • For individuals without a personal history of colorectal and/or endometrial cancer, some data have suggested using a PREMM5 score threshold of ≥2.5% rather than ≥5% to select individuals for MMR genetic testing. Based on these data, it is reasonable for testing to be done based on the ≥2.5% score result and clinical judgment. Of note, with the lower threshold, there is an increase in sensitivity, but a decrease in specificity. |
| Possible Future State | • Known LS pathogenic variant in
family • Personal history of CRC or other LS associated cancer, without pre-selection based on family history or tumor testing • Increased family history model-predicted risk for Lynch syndrome and other cancer risk genes |
LS associated cancers for Amsterdam II were defined as cancers of the endometrium, small bowel, ureter, or renal pelvis;
LS associated cancers under revised Bethesda criteria were defined as colorectal, endometrial, stomach, ovarian, pancreas, ureter, renal pelvis, biliary tract, brain (usually glioblastoma), sebaceous gland adenomas and keratoacanthoma, small bowel;
NCCN defined LS associated cancers as colorectal, endometrial, gastric, ovarian, pancreas, urothelial, brain (usually glioblastoma), biliary tract, and small intestinal cancers, as well as sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas
Risk prediction models utilizing information on personal and family history of LS-associated cancers have also been developed, validated, and recommended as another approach to identifying individuals for genetic evaluation69,70. For example, the PREMM5 model, readily available online, has been shown to have a sensitivity of 89% for identification of individuals with a PV in a LS-associated gene at a cutoff selected to achieve a 2.5% or higher probability of finding a PV. The 2021 NCCN (National Comprehensive Cancer Network) Clinical Practice Guidelines in Oncology guidelines (NCCN Guidelines®) recommend genetic evaluation for individuals meeting one or more diverse criteria based on: 1) a known PV in the family; 2) personal history of a tumor with dMMR activity; 3) testing for and finding dMMR activity in a LS-associated cancer; 4) the number, relationship, and age of diagnosis of relatives with LS-associated cancers; and 5) increased prediction model-based risk for having LS (Table 5). These criteria are complex and represent the full spectrum of strategies that have been developed and applied for identifying individuals with LS over the last 20 years.
A future simplified paradigm for genetic evaluation for Lynch syndrome and other cancer risk genes
Given complexity of current criteria, decreasing costs of clinically available germline multigene panel testing (MGPT), and emergence of prediction models for LS, there is increasing interest in simplifying our approach to selecting individuals for evaluation for cancer risk genes associated with LS and other syndromes. We envision a future state that could continue to recommend gene-specific testing for individuals with a known familial PV, and also have a simplified approach to testing based on personal or family history of cancer consisting of: 1) MGPT for individuals with any personal history of CRC or other LS-associated cancer, without pre-selection based on family history or tumor testing, and 2) MGPT for individuals with increased prediction model-based risk for LS and other cancer risk genes. Here, we define MGPT as testing that includes evaluation for PVs in genes associated with LS (MLH1, MSH2, EPCAM, MSH6, and PMS2), as well as other cancer risk genes. Typical panels test 20 or more genes, include genes that have been associated with increased risk for CRC, and may include genes associated with risk for other cancer types or syndromes (e.g. BRCA1).
A simplified strategy of offering germline MGPT to evaluate for LS-associated and other cancer risk genes, without preselection based on family history or tumor characteristics has been best studied for individuals with a personal history of CRC. Available evidence suggests that MGPT testing has comparable, or even higher yield for identifying individuals with a personal history of CRC and LS compared to tumor and family history-based selection for screening70–72. Further, MGPT identifies PVs in cancer susceptibility genes beyond those associated with LS. For example, in a population-based study in Ohio of 3,310 individuals with CRC, MGPT with a panel including at least 25 cancer susceptibility genes was offered to those with MMR deficiency in their tumors, age <50 at diagnosis, multiple primary tumors, or a first-degree relative with CRC or EC71. Among the 1,462 individuals who had testing based on these criteria, 16% had a PV in a cancer risk gene; 142 had a PV in a LS-associated gene, and 101 in a non-LS associated gene. Other studies have been consistent in demonstrating among individuals with CRC that MGPT has comparable or even higher yield for detection of LS compared to tumor-based testing, and that an additional 3–13% of individuals with a personal history of CRC tested with this approach will have a PV in a non-LS associated cancer risk gene71–74. Of note, all studies to date are subject to potential selection bias that could have resulted in higher yield for cancer risk gene detection, as these studies either enrolled patients with CRC at tertiary referral centers or included some preselection criteria based on age, number of LS-associated tumors, or family history. Additional limitations and challenges to the use of MGPT for detection of LS and other cancer risk genes among individuals with CRC include: 1) ensuring correct management of the 29–64% who may have variants of uncertain significance (VUS) detected by MGPT; 2) including cancer risk genes for which the clinical management is uncertain on many multigene panels; 3) the need for more data on yield for PVs and VUSs across populations with diverse race and ethnicity; 4) clarifying whether, for patients with CRC, germline MGPT has any implications beyond tumor-based testing such as for treatment planning; and 5) identifying the best approaches to ensure high rates of familial cascade testing when a PV is identified.
Selection of patients for evaluation for LS and other cancer risk syndromes based on family history also has potential to be simplified and made more feasible through risk prediction calculators. For example, nearly half of patients included in one study evaluating the simple online PREMM5 risk calculator had no personal history of LS, yet accounted for 20% of the individuals with LS-associated PVs identified in the study75 (risk calculator available at: https://premm.dfci.harvard.edu/). More research is needed to determine the potential clinical yield and utility of online risk calculators such as PREMM5 among individuals with a family but not a personal history of LS-associated cancer at the time of evaluation.
Overall, we anticipate that current and anticipated future discoveries on the yield and clinical utility of genetic evaluation of patients for LS and other cancer risk genes are likely to herald a new era in which the criteria for genetic evaluation become more simplified and more easily implemented in usual practice (Table 5).
Current management recommendations for Lynch syndrome
Recommendations for early detection and prevention of cancers in LS have been issued by a range of groups, and are in constant evolution. For example, in the United States, NCCN Guidelines® for management of patients with LS are updated annually based on evolving evidence and expert opinion66. Recommendations are generally based on observational studies that describe yield and outcomes of various surveillance strategies, assessment of cumulative risk for specific cancers, and observations regarding the average or median age of presentation with cancer (Table 6). Based on increasing evidence supporting differences in cancer site-specific risks based on the gene that is mutated, NCCN Guidelines have moved towards providing specific risk and management guidelines for each gene, and encouraging referral of patients with LS based on their specific PV (e.g. MLH1-LS vs PMS2-LS). The goal of this effort is to raise awareness among patients and clinicians that cancer risks and management should be informed by the specific gene that is mutated.
Table 6. Average age of cancer diagnosis and cumulative cancer risk to age 80 by Lynch syndrome gene and cancer site.
Surveillance epidemiology, and end results cumulative incidence data are provided for comparison. Adapted with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Genetic/Familial High-Risk Assessment: Colorectal, Version 1.2022. © 2021 National Comprehensive Cancer Network, Inc. All rights reserved. The NCCN Guidelines® and illustrations herein may not be reproduced in any form for any purpose without the express written permission of NCCN. To view the most recent and complete version of the NCCN Guidelines, go online to NCCN.org. The NCCN Guidelines are a work in progress that may be refined as often as new significant data becomes available. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.66
| MLH1 | MLH2/EPCAM | MSH6 | PMS2 | Cumulative lifetime risk, general population # | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cancer Risk Site | Estimated Average Age of Presentation | Cumulative risk for diagnosis through age 80 y | Estimated Average Age of Presentation | Cumulative risk for diagnosis through age 80 y | Estimated Average Age of Presentation | Cumulative risk for diagnosis through age 80 y | Estimated Average Age of Presentation | Cumulative risk for diagnosis through age 80 y | |
| Colorectal | 44 y | 46%–61% | 44 y | 33%–52% | 42–69 y | 10%-44% | 61–66 y | 8.7%-20% | 4.2% |
| Endometrial | 49 y | 34%–54% | 47–48 y | 21%–57% | 53–55 y | 16%-49% | 49–50 y | 13%-26% | 3.1% |
| Ovarian | 46 y | 4%–20% | 43 y | 8%–38% | 46 y | <1%-13% | 51–59 y | 1.3%-3% | 1.3% |
| Renal pelvis/and or ureter | 59–60 y | 0.2%–5% | 54–61 y | 2.2%–28% | 65–69 y | 0.7%-5.5% | Uncertain* | ≤1%-3.7% | **** |
| Bladder | 59 y | 2%–7% | 59 y | 4.4%–12.8% | 71 y | 1.0%-8.2% | 71 y | ≤1%-2.4% | 2.4% |
| Gastric | 52 y | 5%–7% | 52 y | 0.2%–9.0% | Uncertain** | ≤1%-7.9% | Uncertain* | Uncertain* | 0.9% |
| Small bowel | 47 y | 0.4%–11% | 48 y | 1.1%-10% | 54 y | ≤1%-4% | *** | 0.1%-0.3% | 0.3% |
| Pancreas | Uncertain* | 6.2% | Uncertain* | 0.5%–1.6% | Uncertain* | 1.4%-1.6% | Uncertain* | ≤1%--1.6% | 1.6% |
| Biliary Tract | 50 y | 1.9%–3.7% | 57 y | 0.02%-1.7% | Uncertain* | 0.2%-≤1% | Uncertain* | 0.2%-≤1% | 0.2% |
| Prostate | 63 y | 4.4%–13.8% | 59–63 y | 3.9%-23.8% | 63 y | 2.5%-11.6% | No data | 4.6%-11.6% | 11.6% |
| Brain | Uncertain* | 0.7%–-1.7% | Uncertain* | 2.5%-7.7% | 43–54 y | 0.8—1.8% | 40 y | 0.6%--≤1% | 0.6% |
y, years;
Cumulative risk for the general population represents cumulative incidence reported by the Surveillance, Epidemiology, and End Results 21 program data, y, years; 2014–2016, accessed October 8, 2019 at SEER*Explorer (https://seer.cancer.gov/)
NCCN reported as “uncertain”;
NCCN reported as 2 cases reported at age 45 and 81;
NCCN reported as: single case - 59 y;
NCCN noted cumulative incidence for general population for this category not available through SEER*Explorer
2021 NCCN Guidelines for surveillance by cancer risk site are specified in Table 7. Several similarities and differences are of note across the LS genes. Based on current and emerging evidence supporting higher CRC risk among individuals with MLH1-LS and MSH2-LS compared to MSH6-LS and PMS2-LS, the recommended ages to initiate colonoscopy and frequency of surveillance differ. Colonoscopy surveillance is recommended every 1–2 years, with initiation at age 20–25 years (or 2–5 years earlier to the earliest CRC in the family if diagnosed under age 25) for MLH1 and MSH2 LS, and initiation at age 30–35 years (or 2–5 years prior to earliest CRC in family if diagnosed under age 30) for MSH6 and PMS2 LS.
Table 7:
Summary of 2021 National Comprehensive Cancer Network Recommendations for cancer surveillance for individuals with Lynch syndrome by pathogenic variant 66.
| MLH1 | MSH2 and EPCAM | MSH6 | PMS2 | |
|---|---|---|---|---|
| Cancer Risk Site | ||||
| Colorectal | • Colonoscopy every 1–2 y, initiated at age 20–25 y, or 2–5 y prior to earliest CRC in family if diagnosed before age 25 | • Colonoscopy every 1–2 y, initiated at age 30–35 y, or 2–5 y prior to earliest CRC in family if diagnosed before age 30 | ||
| Endometrial | • Patient education to
monitor for abnormal uterine bleeding or postmenopausal bleeding, which
can allow for early detection of endometrial cancer. NCCN recommends
evaluation of these symptoms should include endometrial
biopsy. • Total hysterectomy can be considered as risk reducing option to reduce incidence, with timing individualized based on childbearing plans, comorbidities, and family history. • NCCN notes that screening via endometrial biopsy does not have proven benefit, but that this strategy is highly sensitive and specific as a diagnostic procedure, and that screening with endometrial biopsy every 1–2 y, starting at age 30–35 y can be considered. • Transvaginal ultrasound may be considered for postmenopausal women at clinician discretion, but sensitivity may be limited. Ultrasound is not recommended as a screening tool in premenopausal women. |
• NCCN notes that PMS2 Lynch
syndrome appears to be associated with only modestly increased risk of
endometrial cancer in contrast to MLH1, MSH2, and MSH6 Lynch
syndrome. • Considerations for surveillance are otherwise the same as for MLH1, MSH2, and MSH6 Lynch syndrome |
||
| Ovarian | • Individualized
consideration for bilateral salpingo-oopherectomy (BSO) based on
childbearing plans, menopause status, comorbidities, and family history
to reduce ovarian cancer incidence • Patient education to monitor for persistent pelvic/abdominal pain, bloating, increased abdominal girth, early satiety, or urinary frequency/urgency • NCCN currently states data do not support routine screening, and that transvaginal ultrasound has not been shown to be sufficiently sensitive or specific to support a routine recommendation, but may be considered at clinician's discretion. A similar conclusion was reached for serum CA-125-based screening. |
• NCCN has noted insufficient evidence to make a specific recommendation for risk-reducing BSO in MSH6 Lynch syndrome, and guidance for those considering prophylactic surgery or screening is otherwise the same as for MLH1 and MSH2 Lynch syndrome | • NCCN has noted insufficient
evidence to make a specific recommendation for risk-reducing BSO for
PMS2 Lynch syndrome, and that PMS2 pathogenic variant carriers appear to
be at no greater than average risk for ovarian cancer and therefore may
consider deferring surveillance and may reasonably elect against
oophorectomy • For PMS2 pathogenic variant carriers who consider surveillance or surgery, recommendations are the same as for MLH1 and MSH2 Lynch syndrome |
|
| Urothelial Cancer (Renal pelvis, ureter, and/or bladder) | • No clear evidence to support
surveillance • Surveillance options may include annual urinalysis starting at age 30–35 y, but NCCN stated there is insufficient evidence to recommend a particular surveillance strategy |
• NCCN has recommended the same strategy as for MLH1 Lynch syndrome, but has noted that those with MSH2 Lynch syndrome (especially males) appear to be at higher risk for urothelial cancer | • No clear evidence to
support surveillance • Surveillance options may include annual urinalysis starting at age 30–35 y, but NCCN stated there is insufficient evidence to recommend a particular surveillance strategy |
|
| Gastric and small bowel cancer | • NCCN states no
clear data exist to support surveillance for gastric, duodenal, and
more distal small bowel cancer for LS. Individuals with a family
history of these tumors may have increased risk but the benefit of
surveillance is unknown. • Consider baseline EGD with random biopsy of the proximal and distal stomach to evaluate for H. Pylori, autoimmune gastritis, and intestinal metaplasia beginning at age 40 y and surveillance EGD every 3–5 y in those with risk factors for gastric cancer such as: male sex; MLH1 or MSH2 pathogenic variants; first degree relative; Asian ethnicity; residing in or immigrant from country with high background gastric cancer incidence; autoimmune gastritis; gastric intestinal metaplasia; gastric adenoma • Consider testing for H. pylori, and treat if detected. |
|||
| Pancreas | • For individuals with exocrine pancreatic cancer in ≥1 first- or second-degree relatives from the same side of the family as the identified pathogenic/likely pathogenic germline variant, consider pancreatic cancer screening beginning at age 50 years (or 10 years younger than the earliest exocrine pancreatic cancer diagnosis in the family, whichever is earlier) with annual contrast-enhanced MRI/MRCP and/or EUS at a center of expertisea | • NCCN has specifically noted there
are limited data on pancreatic cancer risk for MSH6 Lynch
syndrome • Recommendations are otherwise the same as for MLH1 and MSH2 Lynch syndrome |
• Current NCCN guidelines conclude that PMS2 carriers have not been shown to be at increased risk for pancreatic cancer, and that patients with family history of pancreatic cancer should be managed based on careful assessment and clinical judgement | |
| Prostate | • The version 1.2021 NCCN Guidelines for Prostate Cancer Early Detection recommend it is reasonable for men with Lynch syndrome to consider beginning shared decision-making about screening at age 40 and to consider screening at annual intervals. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Prostate Cancer Early Detection V1.2021 © National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed March 22, 2021. To view the most recent and complete version of the guideline, go online to NCCN.org | |||
| Breast (female) | • NCCN concluded there is insufficient evidence to support increased screening above average-risk breast cancer screening recommendations or those based on personal/family history of breast cancer | |||
| Brain | • Patient education regarding signs and symptoms of neurologic cancer and the importance of prompt reporting of abnormal symptoms to their physicians | |||
| Skin | • Consider skin exam every 1–2 years to evaluate for sebaceous adenocarcinomas, sebaceous adenomas, and keratoacanthomas; age to initiate surveillance is uncertain and can be individualized | • NCCN noted that elevated risk of sebaceous tumors and keratoacanthoma has not been documented for PMS2 Lynch syndrome, and that surveillance can be considered using the same strategies as for MLH1, MSH2, and MSH6 Lynch syndrome | ||
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) are updated annually; see nccn.org for most current NCCN recommendations.
The NCCN recommends that such screening only take place after an in-depth discussion about the potential limitations to screening, including cost, the high incidence of benign or indeterminate pancreatic abnormalities, and uncertainties about the potential benefits of pancreatic cancer screening
Notably, since the 2021 NCCN Guidelines were issued, Kastrinos et al. evaluated the effectiveness and cost-effectiveness of a range of gene-specific surveillance strategies among individuals with LS. Specifically, the authors calibrated a Markov state-transition model to cancer incidence data (natural history42) and adenoma progression risk31 to then simulate trial data outcomes for hypothetical patients from age 25 to age of death given 20 different surveillance strategies. Their modeling analyses suggested the optimal strategies for initiating and repeating colonoscopy are age 25 years repeated every year for MLH1 Lynch syndrome, age 25 repeated every two years for MSH2 Lynch syndrome, age 35 years repeated every three years for MSH6 Lynch syndrome, and age 40 years repeated every 3 years for PMS2 Lynch syndrome76. As such, evidence to support PV-specific colonoscopy surveillance strategies is still emerging and recommendations are expected to continue to evolve over time.
Based on the aforementioned guideline and modeling study, the initiation age, frequency, and approach to surveillance needs to be individualized. Considerations favoring earlier initiation or more frequent colonic surveillance include patient preferences77, as well as the youngest age of cancer diagnosis in the family as a guide, though evidence supporting this strategy requires further examination. Individuals who might benefit from a shorter versus longer interval between colonoscopies include those with one or more of the following risk factors: history of CRC, male sex, having a MLH1/MSH2 pathogenic variant, age >40 years, and personal history of adenoma. Notably, there have not been clinical trials directly comparing 1 vs 2 year intervals for surveillance, stratified by LS PV carrier status. Routine use of chromendoscopy has not been recommended by NCCN Guidelines, and data from a meta-analysis of three studies did not show superior adenoma detection with use of dye-assisted chromoendoscopy versus white light examination alone among individuals with LS78. We recommend surveillance colonoscopy in patients with LS follow best practices for ensuring a high quality exam, including having an excellent bowel preparation, an evaluation complete to the cecum, detection and complete removal of all polyps, and performance by a colonoscopist with a high adenoma detection rate and skill in the detection of flat and subtle lesions.
Across all groups, NCCN Guidelines for endometrial surveillance are similar, with emphasis placed on educating patients to monitor for abnormal uterine bleeding, including postmenopausal uterine bleeding, with a note that PMS2 Lynch syndrome is associated with only modestly increased risk for endometrial cancer, compared to MLH1-, MSH2-, EPCAM-, and MSH6-LS. For ovarian cancer surveillance, NCCN Guidelines note insufficient evidence to make a firm recommendation for risk-reducing bilateral salpingo-oopherectomy (BSO) for MSH6- and PMS2-LS. Further, NCCN Guidelines specifically point out that PMS2 PV carriers appear to be at no greater than average risk for ovarian cancer, potentially supporting deferral of surveillance and election against BSO. For individuals with MLH1- or MSH2-LS, NCCN Guidelines recommend individualized consideration for BSO based on childbearing plans, menopause status, and family history.
A lack of evidence for the best strategies for surveillance for urothelial, gastric, and small bowel cancers has been noted. For urothelial cancers, NCCN Guidelines note that there is no clear evidence to support surveillance, and points out that options that could be considered include annual surveillance with urinalysis to identify microscopic blood starting at age 30 to 35. Note that MSH2 PV carriers, particularly males, appear to be at higher risk for urothelial cancers than other individuals with LS. NCCN Guidelines have also recommended consideration for surveillance upper endoscopy every 3–5 years beginning at age 40 in those with risk factors for gastric cancer, including: male sex, older age, MLH1 or MSH2 PVs, a first-degree relative with gastric cancer, being born in a country with significant background incidence rates of gastric cancer (as is case for many countries in Asia), chronic autoimmune gastritis, gastric intestinal metaplasia, or gastric adenoma. Note that data are particularly limited on whether the risk for gastric cancer is indeed elevated in individuals with MSH6- and PMS2-LS. At the time of upper endoscopy, enteroscopy to the ligament of Treitz may be considered, as duodenal cancers have been identified at time of upper endoscopic surveillance79.
At last update, NCCN Guidelines concluded insufficient evidence to support increased breast cancer surveillance beyond screening criteria available for average risk or based on personal/family history of breast cancer.
For pancreatic cancer, surveillance was recommended if the observed spectrum of LS-associated cancers within a given family included pancreatic cancer for MLH1-, MSH2-, and MSH6-LS, with specific note that data supporting increased pancreatic cancer risk among MSH6 carriers are limited. Additionally, NCCN observed that PMS2 carriers have not been shown to have increased risk for pancreatic cancer. If pancreatic cancer surveillance is implemented, NCCN Guidelines recommend initiation at age 50 years or 10 years younger than the earliest exocrine pancreatic cancer diagnosis in the family for individuals with exocrine pancreatic cancer in one or more first-or second-degree relatives from the same side of the family that carries the identified LS PV. The recommended approach includes annual endoscopic ultrasound (EUS) and/or contrast-enhanced MRI/MRCP. Biopsy of concerning lesions or even more intense surveillance should be guided by imaging findings; most small cystic lesions, will not require biopsy or resection.
Regarding surveillance for sebaceous adenocarcinomas, sebaceous adenomas, and keratoacanthomas, consideration for a skin exam every 1 to 2 years was recommended for MLH1-, MSH2-, and MSH6-LS, with specific note that elevated risks for these lesions have not been documented for PMS2-LS.
NCCN Guidelines for early detection of prostate cancer have suggested that men with LS consider shared decision-making about screening starting at age 40.
Beyond cancer-site specific surveillance strategies, current 2021 NCCN Guidelines recommend that all individuals with LS consider using daily aspirin to reduce future CRC risk, with evidence stemming from a randomized trial and a large observational study. The CAPP2 study randomized 861 individuals with MLH1, MSH2, and MSH6 LS 1:1 to placebo vs 600 mg aspirin daily for at least 2 years80,81. At a mean 10 years of follow up, a relative 35% reduction in risk for incident CRC among individuals assigned to aspirin vs placebo was noted on intention to treat analysis (hazard ratio 0.65, 95% confidence interval: 0.43–0.97, p=0.035). Adverse events in both groups were similar, and no effect on risk for extracolonic cancers was observed. These data are complemented by an observational study of 1858 individuals with LS that reported a 57% relative reduced risk for incident CRC (hazard ratio 0.43, 95% confidence interval: 0.25 to 0.75, p=0.003) among subjects who reported taking aspirin at least twice a week for a month or longer, with even greater relative reduction in risk among those who took aspirin regularly for five years or more82. The optimal dose for chemoprevention in usual clinical practice is uncertain, as many physicians and patients may be uncomfortable with committing a patient to 600 mg of aspirin daily, and NCCN Guidelines have suggested that dose selection can be guided by patient-specific factors (age, concurrent medications) that may impact risk. Studies evaluating the efficacy of lower doses of aspirin are on-going at this time. Further, it should be noted that aspirin is pregnancy category D (positive evidence of human fetal risk), such that young women with LS should be cautioned to avoid use if sexually active and not using contraception or if pregnant. In our clinical practice (CRB, SG) we recommend all patients with LS consider aspirin, starting at a dose of 325 mg per day. Dose is adjusted lower if the patient reports side effects, and we routinely screen for and treat H. pylori prior to initiating aspirin therapy to reduce risk for peptic ulcer disease on aspirin therapy.
Conclusions
As genetic testing for LS becomes more widespread, the recognized prevalence of this condition is expected to increase in the general population in future years, and effective management is an increasingly important clinical need. Patients with LS are at higher risk than the general population of developing MMR deficiency in cells throughout the body which then predisposes carriers to various cancers at early ages. Several future research needs exist. Longitudinal prospective studies that characterize molecular changes in normal tissue of patients with LS before they progress to cancer are critical to increase our mechanistic understanding of cancer initiation pathways. Prevalence of CRC precursors in patients without CRC, and their corresponding rates of malignant transformation need to be elucidated to allow for better risk prognostication. Improved cancer risk prediction models that integrate molecular variables beyond MMR gene type are needed, including models that might inform management. For example, clinical trials for optimal dosing of aspirin as a chemopreventative agent are still ongoing (CAPP3) but operationalizing information on immune activity and driver mutations (e.g., CTNNB1), along with clinical features such as adenoma detection and removal, in individual patients may enable tailoring surveillance intensity over time in an increasingly patient-specific manner. Future studies of prospective surveillance outcomes in LS need to carefully report quality of exams (e.g., adenoma detection rates) in order to best assess both the effectiveness of surveillance and the validity of stages in proposed models of carcinogenesis (e.g., evidence for or against the necessity of adenoma precursors in patients with MLH1 mutation for cancer progression). Importantly, such complete data on surveillance serves as model inputs in studies of cost-effectiveness of differing potential strategies and thus must be reliable. For example, the model of Kastrinos et al. accounts for state-transitions through granular categories (e.g., ‘advanced adenoma’ which encompasses all sizes and types of adenomas) fit to the surveillance data. Overall, there are further modeling improvements to be made such as including more detailed evolutionary pathways by gene type, including timescales of growth of adenomas. From a clinical perspective, more data are needed on the optimal strategies to implement for cancer surveillance.
As knowledge increases of intricate differences in cancer risk between the four specific MMR genes, clinical management strategies for diagnosis and subsequent surveillance of patients with LS will continue to evolve accordingly to improve cancer prevention and early detection.
Reference List
- 1.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. Mar 2017;26(3):404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peltomaki P. Lynch syndrome genes. Familial cancer. 2005;4(3):227–232. [DOI] [PubMed] [Google Scholar]
- 3.Cuatrecasas M, Gorostiaga I, Riera C, et al. Complete Loss of EPCAM Immunoexpression Identifies EPCAM Deletion Carriers in MSH2-Negative Colorectal Neoplasia. Cancers (Basel). Sep 29 2020;12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boland PM, Yurgelun MB, Boland CR. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin. May 2018;68(3):217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. Jun 2010;138(6):2073–2087 e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawn MT, Umar A, Carethers JM, et al. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. Sep 1 1995;55(17):3721–3725. [PubMed] [Google Scholar]
- 7.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. Apr 1971;68(4):820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrestha KS, Aska EM, Tuominen MM, Kauppi L. Tissue-specific reduction in MLH1 expression induces microsatellite instability in intestine of Mlh1(+/-) mice. DNA Repair (Amst). Oct 2021;106:103178. [DOI] [PubMed] [Google Scholar]
- 9.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. Jul 2008;10(4):293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. Nov 15 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 11.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. Jun 10 1993;363(6429):558–561. [DOI] [PubMed] [Google Scholar]
- 12.Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. May 7 1993;260(5109):812–816. [DOI] [PubMed] [Google Scholar]
- 13.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. May 7 1993;260(5109):816–819. [DOI] [PubMed] [Google Scholar]
- 14.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. Dec 3 1993;75(5):1027–1038. [DOI] [PubMed] [Google Scholar]
- 15.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. Dec 17 1993;75(6):1215–1225. [DOI] [PubMed] [Google Scholar]
- 16.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. Mar 17 1994;368(6468):258–261. [DOI] [PubMed] [Google Scholar]
- 17.Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. Mar 18 1994;263(5153):1625–1629. [DOI] [PubMed] [Google Scholar]
- 18.Chung J, Maruvka YE, Sudhaman S, et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. May 2021;11(5):1176–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia P, Yang X, Guo L, et al. MSIsensor-pro: Fast, Accurate, and Matched-normal-sample-free Detection of Microsatellite Instability. Genomics Proteomics Bioinformatics. Feb 2020;18(1):65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. Sep 1 1988;319(9):525–532. [DOI] [PubMed] [Google Scholar]
- 21.Vogelstein B, Fearon ER, Kern SE, et al. Allelotype of colorectal carcinomas. Science. Apr 14 1989;244(4901):207–211. [DOI] [PubMed] [Google Scholar]
- 22.Comprehensive molecular characterization of human colon and rectal cancer. Nature. Jul 19 2012;487(7407):330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilkens J, Timmer NC, Boer M, et al. RSPO3 expands intestinal stem cell and niche compartments and drives tumorigenesis. Gut. Jun 2017;66(6):1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sekine S, Mori T, Ogawa R, et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch Syndrome-Associated colorectal tumorigenesis. Mod Pathol. Aug 2017;30(8):1144–1151. [DOI] [PubMed] [Google Scholar]
- 25.Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. Aug 30 2012;488(7413):660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer. Jan 2021;21(1):5–21. [DOI] [PubMed] [Google Scholar]
- 27.Yurgelun MB, Goel A, Hornick JL, et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev Res (Phila). Apr 2012;5(4):574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kloor M, Huth C, Voigt AY, et al. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol. Jun 2012;13(6):598–606. [DOI] [PubMed] [Google Scholar]
- 29.Takayama T, Ohi M, Hayashi T, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. Sep 2001;121(3):599–611. [DOI] [PubMed] [Google Scholar]
- 30.Ahadova A, Gallon R, Gebert J, et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int J Cancer. Jul 1 2018;143(1):139–150. [DOI] [PubMed] [Google Scholar]
- 31.Engel C, Ahadova A, Seppala TT, et al. Associations of Pathogenic Variants in MLH1, MSH2, and MSH6 With Risk of Colorectal Adenomas and Tumors and With Somatic Mutations in Patients With Lynch Syndrome. Gastroenterology. Apr 2020;158(5):1326–1333. [DOI] [PubMed] [Google Scholar]
- 32.Moller P, Seppala T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. Mar 2017;66(3):464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahadova A, Seppala TT, Engel C, et al. The “unnatural” history of colorectal cancer in Lynch syndrome: Lessons from colonoscopy surveillance. International journal of cancer. Feb 15 2021;148(4):800–811. [DOI] [PubMed] [Google Scholar]
- 34.Cerretelli G, Ager A, Arends MJ, Frayling IM. Molecular pathology of Lynch syndrome. The Journal of pathology. Apr 2020;250(5):518–531. [DOI] [PubMed] [Google Scholar]
- 35.Ahadova A, von Knebel Doeberitz M, Blaker H, Kloor M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: a model of interval cancers in Lynch syndrome. Familial cancer. Oct 2016;15(4):579–586. [DOI] [PubMed] [Google Scholar]
- 36.Komarova NL, Lengauer C, Vogelstein B, Nowak MA. Dynamics of genetic instability in sporadic and familial colorectal cancer. Cancer Biol Ther. Nov-Dec 2002;1(6):685–692. [DOI] [PubMed] [Google Scholar]
- 37.Haupt S, Zeilmann A, Ahadova A, et al. Mathematical modeling of multiple pathways in colorectal carcinogenesis using dynamical systems with Kronecker structure. PLoS Comput Biol. May 2021;17(5):e1008970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Boland CR, Goel A, Wodarz D Komarova NL The protective effect of aspirin in colorectal carcinogenesis: a multiscale computational study from mutant evolution to age induced curves. bioRxiv. Vol 10.1101/2121.05.11.4436712021:1-20. [DOI] [Google Scholar]
- 39.Haraldsdottir S, Rafnar T, Frankel WL, et al. Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun. May 3 2017;8:14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hampel H, de la Chapelle A. The search for unaffected individuals with Lynch syndrome: do the ends justify the means? Cancer Prev Res (Phila). Jan 2011;4(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moller P, Seppala T, Bernstein I, et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective Lynch syndrome database. Gut. Sep 2017;66(9):1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominguez-Valentin M, Sampson JR, Seppala TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genetics in medicine : official journal of the American College of Medical Genetics. Jan 2020;22(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. Journal of the National Cancer Institute. Feb 20 2008;100(4):277–281. [DOI] [PubMed] [Google Scholar]
- 44.Lamba AR, Moore AY, Moore T, Rhees J, Arnold MA, Boland CR. Defective DNA mismatch repair activity is common in sebaceous neoplasms, and may be an ineffective approach to screen for Lynch syndrome. Fam Cancer. Jun 2015;14(2):259–264. [DOI] [PubMed] [Google Scholar]
- 45.Durno C, Boland CR, Cohen S, et al. Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. May 2017;152(6):1605–1614. [DOI] [PubMed] [Google Scholar]
- 46.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. Jul 20 1999;96(15):8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goel A, Boland CR. Epigenetics of colorectal cancer. Gastroenterology. Dec 2012;143(6):1442–1460 e1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang M, Ou J, Hutchinson L, Green MR. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol Cell. Sep 18 2014;55(6):904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao Y, Kang B, Petkovich DA, et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf(V600E)-Induced Tumorigenesis. Cancer Cell. Feb 11 2019;35(2):315–328 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fennell L, Kane A, Liu C, et al. Braf mutation induces rapid neoplastic transformation in the aged and aberrantly methylated intestinal epithelium. Gut. Jul 6 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young J, Jenkins M, Parry S, et al. Serrated pathway colorectal cancer in the population: genetic consideration. Gut. Oct 2007;56(10):1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goel A, Nguyen TP, Leung HC, et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. Feb 15 2011;128(4):869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. Feb 15 2007;356(7):697–705. [DOI] [PubMed] [Google Scholar]
- 54.Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. Dec 2014;147(6):1308–1316 e1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pico MD, Castillejo A, Murcia O, et al. Clinical and Pathological Characterization of Lynch-Like Syndrome. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. Feb 2020;18(2):368–374 e361. [DOI] [PubMed] [Google Scholar]
- 56.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. Jun 1999;116(6):1453–1456. [DOI] [PubMed] [Google Scholar]
- 57.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. Feb 18 2004;96(4):261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. Apr 27 2005;293(16):1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boland CR. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam Cancer. 2005;4(3):211–218. [DOI] [PubMed] [Google Scholar]
- 60.Peltomaki P, Olkinuora A, Nieminen TT. Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Rev Gastroenterol Hepatol. Aug 2020;14(8):707–720. [DOI] [PubMed] [Google Scholar]
- 61.Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. Jan 13 2000;342(2):69–77. [DOI] [PubMed] [Google Scholar]
- 62.Walkowska J, Kallemose T, Jonsson G, et al. Immunoprofiles of colorectal cancer from Lynch syndrome. Oncoimmunology. 2019;8(1):e1515612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. Jul 28 2017;357(6349):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carethers JM, Hawn MT, Chauhan DP, et al. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N'-nitro-N-nitrosoguanidine. J Clin Invest. Jul 1 1996;98(1):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. Jul 1999;117(1):123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Genetic/ Familial High-Risk Assessment: Colorectal V.1.2021. © National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed May 11, 2021. To view the most recent and complete ver- sion of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
- 67.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). The New England journal of medicine. May 5 2005;352(18):1851–1860. [DOI] [PubMed] [Google Scholar]
- 68.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. Oct 17 2012;308(15):1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khan O, Blanco A, Conrad P, et al. Performance of Lynch syndrome predictive models in a multi-center US referral population. The American journal of gastroenterology. Oct 2011;106(10):1822–1827; quiz 1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kastrinos F, Ojha RP, Leenen C, et al. Comparison of Prediction Models for Lynch Syndrome Among Individuals With Colorectal Cancer. Journal of the National Cancer Institute. Feb 2016;108(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pearlman R, Frankel WL, Swanson BJ, et al. Prospective Statewide Study of Universal Screening for Hereditary Colorectal Cancer: The Ohio Colorectal Cancer Prevention Initiative. JCO Precis Oncol. 2021;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang W, Li L, Ke CF, et al. Universal germline testing among patients with colorectal cancer: clinical actionability and optimised panel. Journal of medical genetics. Feb 9 2021. [DOI] [PubMed] [Google Scholar]
- 73.Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. Apr 1 2017;35(10):1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uson PLS Jr., Riegert-Johnson D, Boardman L, et al. Germline Cancer Susceptibility Gene Testing in Unselected Patients With Colorectal Adenocarcinoma: A Multicenter Prospective Study. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. Apr 20 2021. [DOI] [PubMed] [Google Scholar]
- 75.Kastrinos F, Uno H, Ukaegbu C, et al. Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. Jul 1 2017;35(19):2165–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kastrinos F, Ingram MA, Silver ER, et al. Gene-Specific Variation in Colorectal Cancer Surveillance Strategies for Lynch Syndrome. Gastroenterology. Aug 2021;161(2):453–462 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biller LH, Horiguchi M, Uno H, Ukaegbu C, Syngal S, Yurgelun MB. Familial Burden and Other Clinical Factors Associated With Various Types of Cancer in Individuals With Lynch Syndrome. Gastroenterology. Jul 2021;161(1):143–150 e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Houwen B, Mostafavi N, Vleugels JLA, et al. Dye-Based Chromoendoscopy in Patients With Lynch Syndrome: An Individual Patient Data Meta-Analysis of Randomized Trials. Am J Gastroenterol. Apr 2021;116(4):825–828. [DOI] [PubMed] [Google Scholar]
- 79.Kumar S, Dudzik CM, Reed M, Long JM, Wangensteen KJ, Katona BW. Upper Endoscopic Surveillance in Lynch Syndrome Detects Gastric and Duodenal Adenocarcinomas. Cancer Prev Res (Phila). Dec 2020;13(12):1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. Dec 17 2011;378(9809):2081–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. Jun 13 2020;395(10240):1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ait Ouakrim D, Dashti SG, Chau R, et al. Aspirin, Ibuprofen, and the Risk of Colorectal Cancer in Lynch Syndrome. Journal of the National Cancer Institute. Sep 2015;107(9). [DOI] [PMC free article] [PubMed] [Google Scholar]