

Abstract
Reliably scoring and ranking candidate models of protein complexes and assigning their oligomeric state from the structure of the crystal lattice represent outstanding challenges. A community-wide effort was launched to tackle these challenges. The latest resources on protein complexes and interfaces were exploited to derive a benchmark dataset consisting of 1677 homodimer protein crystal structures, including a balanced mix of physiological and non-physiological complexes. The non-physiological complexes in the benchmark were selected to bury a similar or larger interface area than their physiological counterparts, making it more difficult for scoring functions to differentiate between them. Next, 252 functions for scoring protein-protein interfaces previously developed by 13 groups were collected and evaluated for their ability to discriminate between physiological and non-physiological complexes. A simple consensus score generated using the best performing score of each of the 13 groups, and a cross-validated Random Forest (RF) classifier were created. Both approaches showed excellent performance, with an area under the Receiver Operating Characteristic (ROC) curve of 0.93 and 0.94 respectively, outperforming individual scores developed by different groups. Additionally, AlphaFold2 engines were shown to recall the physiological dimers with significantly higher accuracy than the non-physiological set, lending support for the pertinence of our benchmark dataset. Optimizing the combined power of interface scoring functions and evaluating it on challenging benchmark datasets appears to be a promising strategy.
1. Introduction
Protein-protein interactions and multi-protein assemblies, which often include other macromolecular components such as DNA or RNA, play crucial roles in cellular processes [1] their disruption or deregulation often leads to disease [2,3]. Consequently, charting these interactions and elucidating their functions at the molecular and cellular levels have been important goals in molecular biology and medicine.
Of crucial importance to these endeavors are atomic-resolution 3D structures of these assemblies. These data are produced by experimental techniques such as X-ray crystallography, cryo-electron microscopy (cryo-EM), with the resulting structural models deposited into the worldwide Protein Data Bank (wwPDB) [4]. More recently, predicted structures of protein complexes have also been produced by deep learning-based methods [5–7]. Moreover, recent technical advances are facilitating the study of different states of macromolecular assemblies [8,9], varying in compositions and conformational properties, thereby helping to derive deeper insights into the underlying functional mechanisms. This, in turn, requires better characterisation of the structural data for these assemblies in terms of their structural states, their binding interfaces, and the molecular function of their components.
The formation of dimers and higher order protein assemblies is dependent on the concentrations of the component proteins and conditions such as pH and ionic strength. Both protein concentrations and experimental conditions, used to determine the structures of these assemblies by methods such as X-ray crystallography, often differ from the physiological conditions under which these assemblies operate in cells. In some cases, the 3D structures of the solved assemblies deposited in the wwPDB may not represent the physiologically meaningful assembly.
For protein assemblies solved by X-ray diffraction, these problems are compounded by the fact that proteins form many contacts whose sole function is to stabilize the crystal lattice. Deciding which of these contacts are physiologically meaningful can be difficult. When the crystal contains only a single protein chain, the problem of assigning the physiologically meaningful oligomeric state of the protein (i.e., does it form a homodimer, homotrimer, etc, and through which interfaces?) can be even more challenging. In general, these assignments are provided by the authors during the deposition process with the PDB, but nevertheless remain error prone, as they require independent biophysical/biochemical characterisation, which may yield ambiguous results or be missing altogether. A significant fraction of protein assemblies solved by X-ray crystallography in the PDB (18%) are not even associated with a publication [10].
To remedy this problem, computational methods have been developed to infer the oligomeric state of a protein directly from the intermolecular contacts made by the protein in the solved structure of the crystal lattice. These methods exploit results from a large body of previous studies that systematically evaluated the interface properties of native protein complexes and compared them to those of crystal contacts, considered as representing weak nonspecific interactions [11,12]. The most widely used methods are PISA [13] and EPPIC [12]. PISA evaluates chemical and structural properties of interfaces, whereas EPPIC uses geometric measures and sequence conservation, which is often associated with regions involved in biological function [14].
The corollary of the problem of defining physiological association modes, is to define association modes that are non-physiological, i.e., that have not been selected by evolution, and would not form in the cell. As mentioned above, crystal contacts are commonly considered as representing non-physiological interactions.
The last 20 years have witnessed the development of efficient algorithms to predict the 3D structure of protein-protein complexes. These include ab-initio docking methods [15,16] and template-based methods that model the structure of a target complex, using as template the known structure of a complex formed by related proteins [17,18]. An important component of both procedures are the scoring functions, These evaluate binding interfaces to single out native complexes from non-native ‘decoy’ candidates [19–22]. The parameters of these scoring functions have typically been optimized on various benchmark datasets of protein complexes from the PDB.
Prominent examples are the community-wide Docking Benchmark [23–25] which contains the structures of native protein complexes and those of the corresponding unbound protein components. An updated version of this benchmark also integrates binding affinity data for a subset of the complexes [26]. More recently, various datasets of X-ray and modeled structures, suitable for testing most aspects of docking algorithms are offered by the Dockground resource [27], whereas customized benchmarks that include large sets of crystal contacts representing non-physiological association modes in addition to native complexes have been developed to train and test scoring functions such as EPPIC [12] and PRODIGY [28]. Non-native binding modes of a given protein pair that are extensively sampled by docking algorithms are a typical category of non-physiological association modes that scoring functions need to select against. Two recent benchmarks include a large set of such ‘decoy’ poses in addition to native complexes. The Scoreset [29] includes non-native docking poses computed by participants of blind prediction challenges in CAPRI (Critical Assessment, of Predicted Interaction [30,31]) in addition to the experimentally determined structure of the target complex. The second benchmark comprises 230 structures of native complexes from the community-wide benchmark-V5 [26] and 30,000 non-native docking poses generated for each of the native complexes [32]. Datasets of this type, which contain as many as possible examples of both native and non-native protein complexes, are a prerequisite for optimizing scoring functions, including AI-based inference models, capable of distinguishing between the two categories of complexes.
Here we present a novel approach for defining a benchmark dataset composed of high confidence examples of 3D structures of both physiological and non-physiological protein complexes, limited to homodimers at this point. Physiological homodimers are defined using two complementary methods. These methods rely on the conservation of the 3D structure of the interface between two proteins in PDB entries, respectively, across crystal forms of PDB entries as defined by the ProtCID resource [33] and across protein homologs as defined by QSalign [34]. Non-physiological homodimers are defined using two complementary criteria. In one, such dimers are defined as protein structures whose assigned biological unit in the PDB is a homodimer, but the corresponding interface is deemed as incorrect because another interface, which is conserved across homologs, is formed by the same protein (QSalign approach). In the second, non-physiological dimers are defined as those forming a unique interface that bears no similarity to any interface across all crystal forms in ProtCID, including crystals of homologous proteins, when sufficient numbers of such crystal forms exist.
Physiological protein complexes, including homodimers, on average engage in higher affinity interactions than their non-physiological counterparts, as reflected in their buried interface area, known to positively correlate to binding affinity [35]. Therefore, a sizable fraction of the complexes in each category can be correctly assigned solely on the basis of the interface area, a parameter that the majority of the scoring functions include. At the same time, the significant overlap of the interface area size distributions of complexes from both categories [11,12] is a clear indication that other factors are at play in distinguishing between the two types of complexes. To better focus on these factors, we generated a set of non-physiological dimers that display on average similar or larger interface areas than the physiological counterparts in the benchmark.
The resulting benchmark includes 1677 homodimers (836 physiological, and 841 non-physiological) and is unique in several important ways. The definition of our negative examples (non-physiological dimers) is less error prone because they feature interfaces that are unlike any other protein interface across structures of protein families or across crystals of the entire known structural database. The set of positive examples of our homodimer dataset is more reliable than the biological unit definitions used by the PDB, because quaternary structure conservation across homologs proves highly accurate at identifying physiological homomers, reducing the error rate from 10% (based on PDB definitions) to less than 5% [34].
In the second part of the study, we compute specialized scores for the dimers of our benchmark dataset. These scores are commonly used to rank interfaces, notably in blind prediction challenges [30,36]. We then evaluate the performance of these scores in discriminating between the physiological homodimers and their non-physiological counterparts. Two types of scores are considered. Simple scores or ‘raw scores’ that quantify specific properties (features) of the interaction interface, and complex scores that combine multiple simple scores (‘integrated scores’). Integrated scores represent complex functions previously optimized or trained on datasets of known protein complexes [20–22,37].
In addition to analyzing the discriminatory power of raw and integrated scores, we derive a community-wide consensus score and train a Random Forest (RF)-based classifier [38] on the raw scores as features. The consensus score and the RF classifier are shown to outperform raw scores as well as the independently derived integrated scores evaluated here.
Lastly, AlphaFold2, the deep learning-based inference engine developed by DeepMind [5], has shown a remarkable ability to accurately predict the 3D structure of protein monomers [39]. Follow up studies have indicated that the same engine can accurately predict the 3D structure of protein complexes by concatenating the sequences of the two protein chains, separated by a gap [40,41]. Following up on these reports we applied AlphaFold v2.0 and AlphaFold v2.1.1 - a subsequent version of AlphaFold which includes a ‘multimer’ mode directly trained on protein complexes [7] - to predict the 3D structure of the dimers of our benchmark dataset. Hypothesizing that physiological dimers will be predicted more accurately than non-physiological dimers, the accuracy of predicted AlphaFold models was used to classify the dimers into the two categories and the results were compared to those obtained here with the community-wide classifiers.
2. Material & Methods
2.1. The benchmark dataset of homodimers
The physiological dimers encompass 836 structures selected using two complementary methods. These methods rely on the conservation of the 3D structure of the interface across crystal forms in the PDB, as defined by the ProtCID resource [33], and across homologs, based on the QSalign resource [34,42].
The non-physiological set was defined using two approaches (Figure 1A). A first group of homodimers was identified based on QSalign. Those structures contained an interface not observed in homologues, and exhibited another distinct and conserved interface (Figure 1A). Dimers corresponding to these characteristics were further pruned to yield 141 assemblies with a similar interface area distribution as for the physiological dimers (Figure 1B). This additional criterion was imposed to create a set of non-physiological dimers that cannot be readily segregated from their physiological counterparts based on lower interface area. Application of these criteria yielded significantly fewer non-physiological dimers (141 in total) than those in the physiological set.
Figure 1.
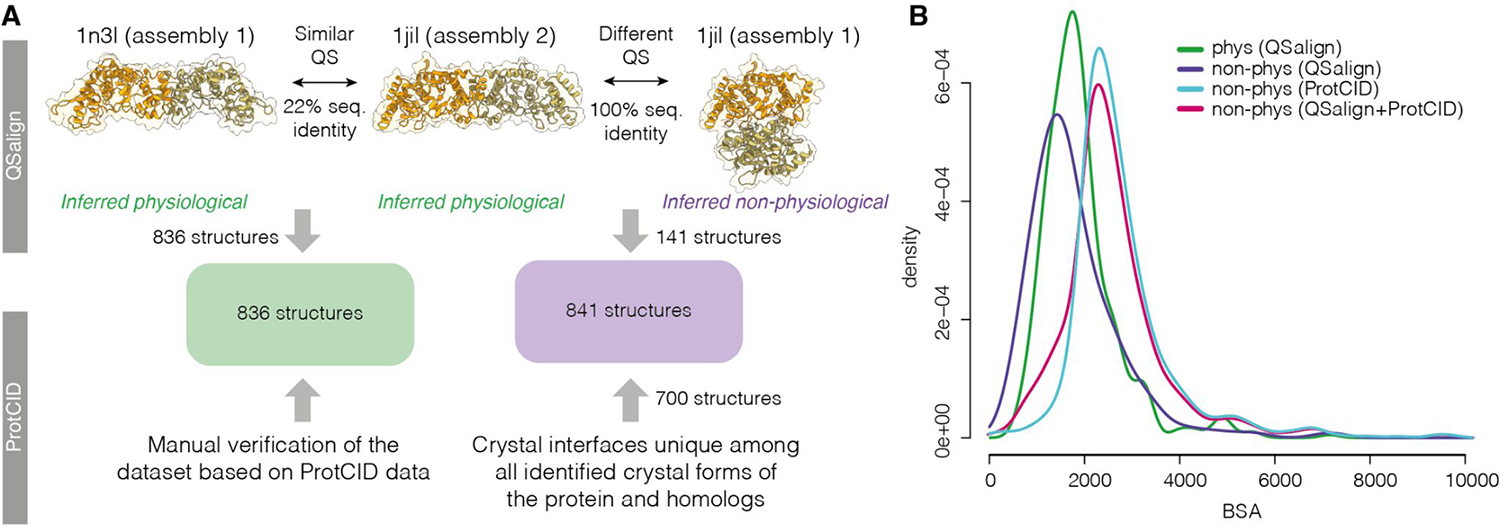
Approach used to define the benchmark dataset. A. Physiological dimers (836 in total) were identified based on the QSalign resource [34] and have their interface structure conserved across homologues (e.g., 1n3i vs 1jil). A manual curation of that set was also performed based on the ProtCID resource [33]. A subset of non-physiological dimers (141 structures) were identified based on QSalign as structures exhibiting a different and conserved interface. For example, two assemblies are available in the PDB for structure 1jil. Assembly 2 shows a conserved interface, which lets us infer that Assembly 1 is non-physiological. This set was expanded to also include dimers forming interfaces that are unique among all interfaces across crystal forms or across the crystal forms of homologs, as defined in ProtCID, yielding an additional 700 structures. B. Interfaces area distributions of the physiological and non-physiological homodimers and the sets described in panel A.
To balance the number of both types of assemblies, the set of non-physiological dimers was expanded using a second method. Here, non-physiological dimers were identified among structures where sufficient numbers of crystal forms exist. Specifically, we selected interfaces that are unique among all interfaces across crystal forms (CFs) or across the crystal forms of homologs, as defined in the ProtCID database (see Supplementary methods for details). The choice of these interfaces was further biased towards dimers featuring similar or larger interface areas than their physiological counterparts (Figure 1B). This procedure yielded an additional 700 non-physiological interfaces, bringing the total number of these interfaces to 841, in good balance with the physiological interfaces of the dataset (see Supplementary Table S1).
2.2. Scoring functions for evaluating protein-protein interfaces
Functions or scores capable of identifying native-like protein-protein association modes are a major component of methods for predicting the structures of protein-protein complexes. These methods usually sample very large numbers of putative binding modes between proteins, which need to be efficiently ranked. The development of such scoring schemes has therefore been a major focus over the past two decades. These scoring schemes focus on evaluating the properties of the binding interface formed between the two interacting partners. They include atom and residue pair potentials, sometimes in the context of classical physics-based potential energy terms, and increasingly implement different flavours of knowledge-based potentials, representing functions derived from experimentally determined structures in the PDB (wwPDB). More recently, these methods have been enriched with evolutionary information, augmented with deep learning models, or replaced with such models.
Here we evaluate the performance of methods developed by the community for scoring and ranking interfaces in discriminating between the physiological and non-physiological homodimers of our benchmark dataset. Two main types of scores are considered. Simple scores or ‘raw scores’ that quantify specific properties (features) of the binding interface, and ‘integrated scores’ that integrate multiple raw scores, usually representing complex functions optimized or trained on datasets of known structures.
The evaluated scoring functions are summarized in Table 1. These functions are described in further detail in the individual reports by the 13 groups, representing the developers or expert users of these functions (see Supplementary Material). Ten of these groups are longstanding participants in CAPRI, including in the CAPRI blind scoring challenges [30,43], which offered ample opportunities to test and optimize new scoring functions. In this study, each of the 13 groups independently applied their functions (raw scores and/or integrated scores) to the benchmark dataset and uploaded the results to the project GitHub repository, accompanied with relevant information enabling further analysis and comparison of the uploaded values (see description of the Github data in Supplementary Material). After filtering out redundant scores (computed independently by different teams) a total 221 raw scores, and 31 integrated scores were evaluated.
Table 1.
Summary of scoring functions used to score or classify the homodimer interfaces of the benchmark dataset, and to compute consensus scores.
Column 1 lists individual groups (PI name -group). Column 2 includes a high-level description of the methods with literature references and links to servers provided whenever appropriate. Column 3 lists more detailed descriptions of the scores. Additional information can be found in the reports of individual groups collated in the Supplementary Material.
| Group | Feature type / source | Brief scoresFeature description |
|---|---|---|
| Fernandez-Recio | CCharPPI features [58] ---- Integrated scores I_pyDock_Desolv_VDW [59] |
- Residue contact/step potentials - Residue distance-dependent potentials - Atomic contact/step potentials - Atomic distance-dependent potentials - Statistical potential constituent terms - Interface packing: F_NIPacking - Composite scoring functions - Solvation energy functions - Hydrogen bonding - Van der Waals and electrostatics --------- pyDock [59]: Desolvation, electrostatics and Van derWaals scores |
| MOBI | Three descriptors for each interface (F_shape, F_hydro and F_tails) and one integrated score (I_shape_hydro_tails) [60,61] (see MOBI group Report in Supplementary Material) | - I=F_shape+F_hydro(Ftails) - F_shape (Sum of # of hits/residues) - F_hydro (Frac. of surface hydrophobicity) - F_tails (Y/N of chain ends) |
| Venclovas | Multiple Voronoi tessellation-derived features computed using the Voronota software (https://kliment-olechnovic.github.io/voronota/) [21,62] | - Voronoi tessellation-derived interface contact areas, - Solvent contact areas, - All the contact areas. - Voronoi tessellation-derived volumes. - VoroMQA energy values (of interface contacts, solvent contacts, all the contacts) - VoroMQA-light and VoroMQA-dark scores . |
| Wolfson | All-atoms scores [63] --------- Deep-Learning scores [64] |
- F_SOAP: interaction score (all atoms) - F_FireDockScore (all atoms) - F_Network_binding_0 - F_Network_binding_1 (DL NN, P_residues in interface) - F_Network_full (DL, NN, P_residues in contact) --------- - I_Proba_Consensus (integrated score) |
| Zou | [65,66] | - ITScorePP (atomic-level, statistical potentials) --------- - DLScoreBC (DL/CNN model for interface prediction) |
| Bonvin | Two different classifiers [22,67] Scoring method used in HADDOCK [68] DeepRank-GNN [69]: The PPI interfaces were converted into residue graphs and each node was assigned PSSM information only (i.e. 20 features per node). |
- PRODIGY-CRYSTAL [67]: RF classifier (residue contacts; residue contacts per amino acid type, contact density/interface, trained on the MANY dataset [12]) --------- - DeepRank-GNN (DL/GNN, trained using PSSM features only on the MANY dataset [12]) --------- - HADDOCK score and its raw components [68] (not trained as a classifier) |
| Furman | Combined docking and local refinement with RosettaDock, InterfaceAnalyzer protocol (multiple features) [70,71], RosettaCommon* | - I_sc: interface score (‘Interaction energy_1’) - dG_cross/dSASA* (‘Interfaces energy_ 2’) - sc_value: shape complementarity - fa_intra_sol_xover4: intra-residue LK solvation - dG_separated/dSASA: binding energy of separated components/unit interface area - fa_dun: Internal energy of sidechain rotamers - dSASA_polar: polar components of interface area - fa_intra_rep: Lennard-Jones repulsive between atoms in the same residue |
| Oliva | COCOMAPS/CONSRANK features [72] and BSA calculated with NACCESS (http://www.bioinf.manchester.ac.uk/naccess/) ----------- Integrated scores: I_NN_all, I_NN_sel, I_RF_all, I_RF_sel [32] (see Oliva Group Report in Supplementary Material) |
- Residue-residue contact stats, including the total number of contacts at the interface, the number of contacts per physico-chemical class of amino acids involved in the contacts (polar, apolar, aliphatic, aromatic and charged) and the relative fraction over the total number of contacts per complex. - BSA upon complex formation plus the polar and apolar components calculated by NACCESS [73] and FreeSASA [74] --------- - Probability for a dimer to be physiological and predicted class (TRUE/FALSE) from Neural Network(NN)- and RF-based classifiers, using 148 (_all) and 42 selected (_sel) features |
| Kihara | A range of scores and potential including scores published by the Kihara group and other groups (See Kihara Group report in Supplementary Material). | Examples of scores: - DFIRE: all-atom statistical potential [75] - GOAP : all-atom statistical potential [75] - Dove: DL (3D CNN) model [76] - GNN-DOVE : Graph Neural Networks Model [77] - ITScore : knowledge-based scoring function [65] - PhysicsScore: physics-based score in Multi-LZerd [78] - RosettaInterfaceEnergy : Interface Energy of Rosetta Energy Function [79] - VoroMQA [21] |
| Casadio | ISPRED4 predictor of protein interaction sites (https://ispred4.biocomp.unibo.it) [80] | - Support vector machines (SVMs) and grammatical-restrained hidden conditional random fields (GRHCRFs) incorporating 46 features: - Sequence profiles (MSA) - Residue physical-chemical properties: - PSICOV: intra-chain coevolution scores (Jones et al. 2012) - Interface residue propensity - Difference between predicted and observed solvent exposure - Structural/geometric features: secondary structure, DPX, CX (computed using PSAIA (Mihel et al. 2008)) |
| SWISS-MODEL | QSQE score from SWISS-MODEL[46,81] | - QSQE: composite score (values 0–1); ML(SVM)-based (interface conservation, structural clustering, and other template features); depends on availability of templates in the SWISS-MODEL template library (not trained as a classifier but to rank templates for modelling). |
| Guerois | Scores from InterEvDock [82] | - SPPh.10seq and SPPh.10seq.normsize: novel version of SOAP-PP [63] using coevolutionary information at atomic level, using information from a set of 10 homologous complexes (.normsize: score normalized by interface size). - IESh.10 seq and IESh.10seq.normsize: same as above but using InterEvScore [83] as a base scoring function instead of SOAP-PP |
| Correia | DL MaSIF model [84] | Integrated score, combining chemical (electrostatics, H-bonds, hydrophobicity) and geometric (shape and curvature) features, in a surface descriptor for the interacting surface patch of each protein. --------- Computed the following quantities: - Descriptor Distance Score: complementarity of the 2-interacting surface patches. - Neural Network Alignment Score (0–1): calculates the alignment quality between the interacting surface patches. |
As a representative sample of the scoring functions developed by the community, the evaluated scores span a wide range of characteristics. Examples of raw scores listed in Table 1 include residue-residue contact potentials (Fernández-Recio), residue-residue contact statistics broken down by contributions from different residue types (polar apolar, aliphatic, aromatic, charged) (Oliva), or energy values for contacts at the interface, with solvent, and all contacts, computed using Voronoi tessellation (Venclovas). Residue-residue distance-dependent potentials, as well as atomic contacts and distance-dependent potentials are also used (Fernández-Recio). Many scores also account for the desolvation energy, often approximated by evaluating the solvent accessible area buried in the binding interface, with contributions broken down by residue types (Fernández-Recio, MOBI, Venclovas, HADDOCK, Oliva). Several groups use classical physics-based potentials, which include VdW and electrostatic terms (Fernández-Recio, Kihara). Different terms from the Rosetta InterfaceAnalyzer energy function are used by the Furman and Kihara groups (Table 1). The Furman group computed these terms after local redocking and model refinement with the RosettaDock software.
Examples of complex ‘integrated scores’ include ISPRED4 (Casadio), which combines information from co-evolution with various other terms optimized using Machine learning. Information on co-evolution is also incorporated in the scores of the Guerois group, whereas Kihara uses a set of published scores developed by other groups, as well as integrated scores developed in-house using Machine Learning (ML). ML-based (integrated) scores are also used by the groups of Zou, Bonvin, Correia, Oliva and Schwede, with the QSQE composite score from SWISS-MODEL being trained to rank template structures for modelling. Admittedly, this definition of integrated scores is biased towards scores derived using ML, or by combining scores developed by other groups. Scores such as the Rosetta energy functions, which approximate potential energy contributions and usually contain multiple terms, are considered as raw scores.
2.3. AUC-based analysis
Selection of the best performing score for each group
Five-fold cross validated receiver operating characteristic (ROC) curves were generated using the values of individual scores (simple or integrated score), computed for the dimers of our benchmark dataset by each group. The best performing score of each group was selected as the score yielding the largest area under the curve (AUC). The threshold used to classify a dimer as physiological (positive example) or non-physiological (negative example) was defined as the point on the ROC most distant from the diagonal. Figure S1 illustrates the 5-fold validation approach used to compute the ROC, associated AUC, and the threshold value for a score computed by a given group.
Computing a consensus score
After analyzing the performance of each group separately, we computed a consensus score that averages the classifications of the best scoring method of each group as follows:
For each score, the optimal threshold (thr) on the ROC curve (the point of the ROC farthest away from the diagonal in Figure S1B) was computed. Each score was then binarized with a score equal to 1 for values >thr, and 0 for values <=thr. A consensus score was then calculated for each structure as the average of the binary-scores for each structure. The values of the resulting consensus score range between 0 and 1, with 1 meaning that all the scores predicted the interface of as physiological, and 0 meaning that all the scores predicted it as non-physiological.
2.4. Random Forest based classification
To further investigate how effective the 221 unique raw features computed for the dimers of our benchmark dataset are in discriminating between the two categories of dimer, we used a RF classifier [38], as implemented in the Scikit-learn package [44]. The classifier, subjected to a leave-one-out cross-validation, assigned to each homodimer an average probability to carry out a given label (physiological, or non-physiological) that was used to compute the ROC curve. RFs are protected against overfitting, since the computed probabilities are averaged over many random decision-trees, thereby reducing variance without increasing bias.
Lastly, the contribution of individual features to the performance of the RF classifier was estimated using the mean Gain/Gini importance index [45].
2.5. Predicting and scoring homodimer structures using AlphaFold2 and AlphaFold-Multimer
For each dimer, we obtained the full sequence of the protein to be used as input for AlphaFold2. A few proteins included modified or unknown amino acids in their sequence, which we replaced with alanines. The sequence was then run through two versions of AlphaFold2 (“Gap” [40,41] and “Multimer” [7] versions) each resulting in 5 predicted models for each dimer.
For the “Gap” approach, the original AlphaFold2 pipeline was fed as input the concatenated sequences of the interacting protein chains separated by a 200-residue gap [40,41]. We used the original AlphaFold2 pipeline to produce the multiple sequence alignments (MSA) for the target proteins which were trivially duplicated to produce the input for both protein chains. For this case, no template structures were used, no model relaxation was performed, and all five “monomer_ptm” model parameter sets were used. We note that this version was not trained on any protein complex [5]. Due to various technical issues, 60 targets failed to be predicted using the “Gap” version.
The analysis was repeated using AlphaFold v2.1.1 (Multimer version), which has been trained on protein complexes [7]. This version includes a template search for single chains. This search was restricted to use only templates whose coordinates were deposited before those of the PDB entry in the benchmark dataset. We also performed model relaxation for some targets to evaluate its effect on model accuracy. Five models were also produced using the AlphaFold-Multimer parameter sets which were trained using complexes available in the PDB release of 2018-04-30. In this case, 4 targets could not be predicted, 160 targets were not relaxed due to the large runtime needed and 2 targets could not be scored using DockQ.
The 5 models produced for each dimer (“Gap” or “Multimer” approaches) were compared with the corresponding structure in the benchmark dataset, using the QS-score, a measure that quantifies the similarity between interfaces as a function of shared interfacial contacts [46,47] and DockQ [48], a composite score which integrates the evaluation criteria used by CAPRI [49]. In both cases the QS-scoring routines of OpenStructure [50] were used to find the optimal mapping between model and reference chains. ROC AUCs were computed using the highest (best) values for QS-score and DockQ over the 5 predicted structures for each dimer. Additionally, we report the mean and median of these best scores for the physiological dimers of the benchmark.
3. Results and Discussion
3.1. The benchmark dataset of homodimer protein assemblies
The benchmark dataset derived in this study comprises 1677 homodimeric protein assemblies predicted to be ‘physiological’ (836) or ‘non-physiological’ (841). Physiological dimers were identified using two complementary methods relying, respectively, on the conservation of the 3D structure of the interface between the subunits across crystal forms and across homologs in the PDB as defined by the ProtCID [33] and QSalign [42] resources. The non-physiological dimers identified by QSalign correspond to assemblies for which a different and conserved interface exist (and therefore the non-conserved interface was presumed incorrect). Those identified using ProtCID correspond to interfaces found in only one crystal form of several available (see Methods and Supplementary Methods for details). The selection of dimers in this category was biased towards dimers displaying similar or larger interface areas than the physiological dimers (Figure 1B).
The full list of the homodimers of the benchmark dataset, the characteristics of the corresponding proteins and their interfaces can be found in Table S1 of the Supplementary Material. The summary of these characteristics is given in Table 2, which indicates that the non-physiological homodimers display on average larger interface areas, involve more polar residues, as well as more polar residue-residue contacts than their physiological counterparts. The non-physiological dimers also include more extreme cases comprising dimers forming very few contacts (≤ 5 in entries 5ytb; 6g5g; 6ehy), or a dimer such as in entry 2znh, forming a very large interface (10985 Å2) due to significant intertwining between the subunits. Dimers of both categories feature a similar distribution in terms of the CATH fold classification [51,52] (Figure 2). Based on these characteristics, our benchmark dataset may be considered as particularly challenging for scoring methods.
Table 2:
Summary statistics of the interfaces of the benchmark dataset of homodimers. The full dataset and major properties and annotations associated with each entry can be found in the Supplementary Table S1. The calculation performed with freesasa-2.0.3 (Mitternacht 2016).
| Min. | 1st Qu. | Median | Mean | 3rd Qu. | Max. | set | property |
|---|---|---|---|---|---|---|---|
| 1 | 49 | 65 | 72.2 | 83 | 663 | all | number of contacts* |
| 100.62 | 1612.79 | 2097.57 | 2273.57 | 2646.31 | 10984.55 | all | BSA |
| 38.08 | 540.63 | 791.64 | 873.09 | 1100.79 | 4540.25 | all | BSA polar |
| 22.2 | 994.15 | 1260.71 | 1396.86 | 1632.15 | 7170.96 | all | BSA apolar |
| 0.09 | 0.31 | 0.38 | 0.38 | 0.45 | 0.86 | all | fraction BSA polar |
| 0.14 | 0.55 | 0.62 | 0.62 | 0.69 | 0.91 | all | fraction BSA apolar |
| 13 | 45 | 57 | 62.71 | 72 | 262 | physio | number of contacts* |
| 589.93 | 1421.01 | 1778.8 | 1918.41 | 2150.92 | 7202.51 | physio | BSA |
| 114.73 | 435.52 | 582.5 | 653.9 | 780.13 | 2721.74 | physio | BSA polar |
| 101.16 | 944.95 | 1167.95 | 1264.51 | 1458.69 | 4480.77 | physio | BSA apolar |
| 0.09 | 0.28 | 0.34 | 0.34 | 0.39 | 0.86 | physio | fraction BSA polar |
| 0.14 | 0.61 | 0.66 | 0.66 | 0.72 | 0.91 | physio | fraction BSA apolar |
| 1 | 57 | 72 | 81.63 | 92 | 663 | non-physio | number of contacts* |
| 100.62 | 2074.61 | 2426.91 | 2626.61 | 2959.75 | 10984.55 | non-physio | BSA |
| 38.08 | 796.62 | 1033.73 | 1090.98 | 1284.65 | 4540.25 | non-physio | BSA polar |
| 22.2 | 1091.63 | 1366.69 | 1528.42 | 1768.84 | 7170.96 | non-physio | BSA apolar |
| 0.09 | 0.36 | 0.43 | 0.42 | 0.49 | 0.78 | non-physio | fraction BSA polar |
| 0.22 | 0.51 | 0.57 | 0.57 | 0.63 | 0.91 | non-physio | fraction BSA apolar |
Residue-Residue contacts, using a distance threshold of 5 Å.
Figure 2.
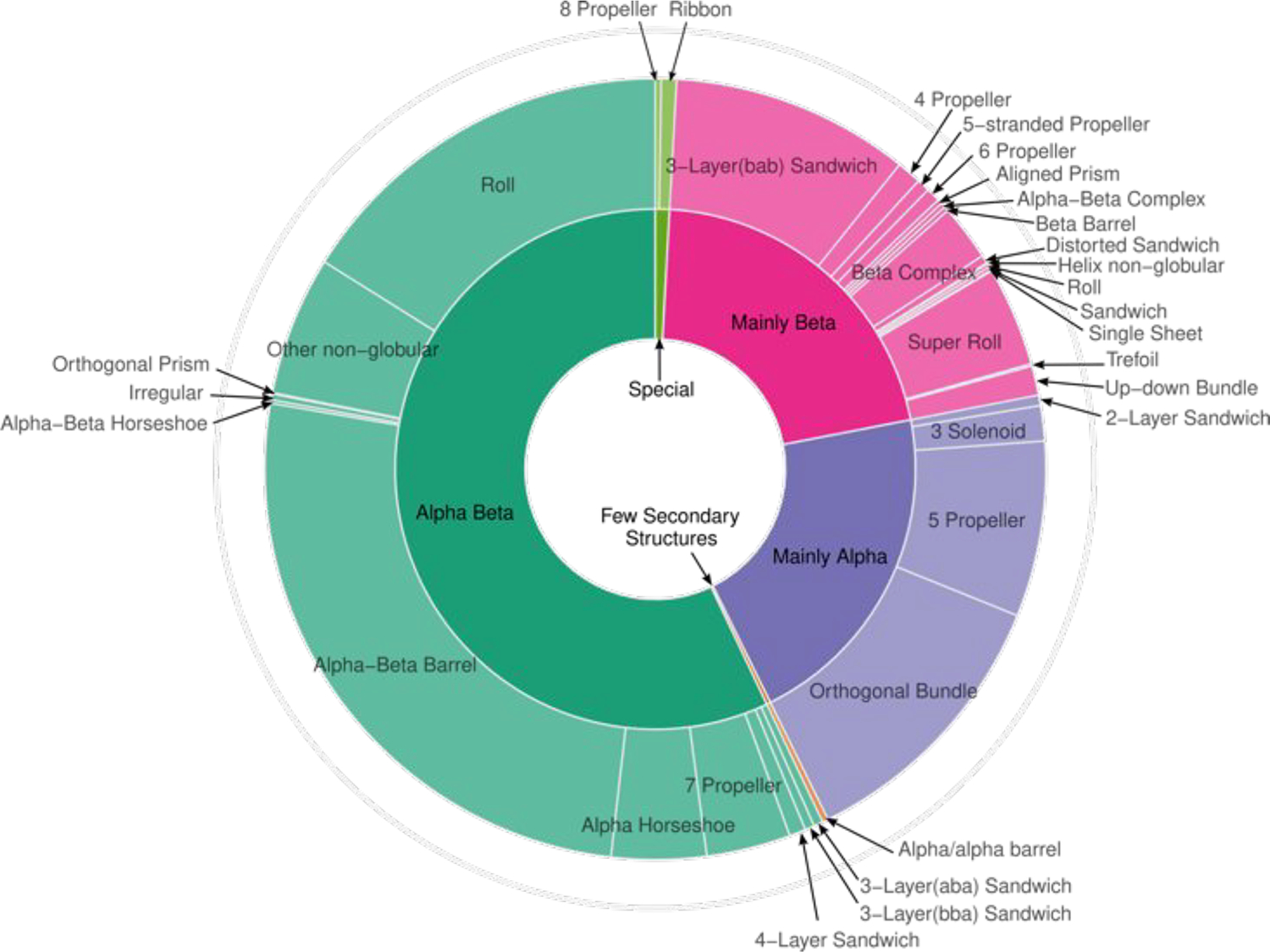
CATH classification of proteins of the physiological homodimer subset. The Sunburst plot was generated using the R package ggvenn [57].
3.2. Performance of scoring methods
Two main approaches were used to evaluate the performance of methods for scoring interfaces in segregating physiological from non-physiological dimers.
The first approach, which we denote as ROC-based, focuses on the groups participating in this effort and highlights the complementarity between their best score. Each group submitted values for raw scores (features) or integrated scores. Importantly, the latter were derived prior to this study and were not optimized on this benchmark dataset. ROCs were computed from these scores using 5-fold cross validation to select the best performing score for each group. For each of these ROCs, a threshold corresponding to the value farthest from the diagonal was used to assign interfaces as being physiological or non-physiological. Lastly, a simple consensus (average) score was derived from the 13 optimally thresholded ROCs (see Methods).
The second approach, which we denote RF-based, focuses on all the computed raw scores (features) (221 unique features in total) computed by the 13 participating groups (see Methods).
ROC-based approach: best performing scores from each group
Table 3 lists the number of scores from each category (raw or integrated) evaluated for each of the 13 participating groups, and the AUC values computed from the ROCs of the corresponding best performing scores. The AUC values, generally used to gauge performance, range from 0.71 to 0.85, with the best score obtained for Deeprank-GNN, a DL-based classifier, closely followed by two raw scores (features): F_DG.cross.DSASAx100 (AUC=0.84) from the Furman group, which evaluates the binding energy per unit interface (including only cross-interface energy terms), and F_Voronota_iface_expanded_area (AUC=0.82) from the Venclovas group, representing a sum of all the tessellation-derived interface contact areas and the SASAs (solvent accessible surface area) of interface atoms (see Table 2, and Collated Methods in Supplementary Material). The remaining 10 best scores of Table 3 exhibit significantly lower AUC values. Furthermore, of the 13 scores in Table 3, eight are integrated scores. Five of these are from groups that computed only integrated scores, and 3 are from groups that computed both types of scores, suggesting that applied individually, integrated scores confer in general little advantage over raw scores.
Table 3.
Areas under the curve (AUC) of the ROC curves of the best performing score of each group participating in the study.
| Group | Number of raw/integrated scores | Score | AUC |
|---|---|---|---|
| group Bonvin | 4/4 | Deeprank-GNN* | 0.85 |
| group Furman | 20/0 | F_dG.cross.dSASA100 | 0.84 |
| group Venclovas | 74/0 | F_Voronota_iface_expanded_area | 0.82 |
| group Zou | 0/2 | I_P_Biological* | 0.81 |
| group Guerois | 0/11 | I_IESh.10seq.normsize* | 0.81 |
| group Fernandez-Recio | 87/2 | F_NIPacking | 0.79 |
| group SWISS-MODEL | 0/1 | I_QSQE* | 0.79 |
| group Bologna | 0/2 | I_physiological_score_MCC* | 0.76 |
| group Kihara | 8/0 | F_GOAP | 0.75 |
| group Correia | 0/3 | I_descriptor_distance_score* | 0.75 |
| group Oliva | 37/4 | F_CP | 0.75 |
| group MOBI | 3/1 | I_shape_hydro_tails* | 0.72 |
| group Wolfson | 5/1 | I_Proba_Consensus* | 0.71 |
The corresponding descriptor is an integrated score or a classifier, rather than a raw score (see text for definitions). Descriptions of these scores can be found in Table 2, and in the detailed descriptions of individual groups (see Supplementary Material)
The classification of individual structures of our benchmark dataset into physiological and non-physiological dimers according to the best performing scores of each group is illustrated using 1D heat plots (Figure 3).
Figure 3.
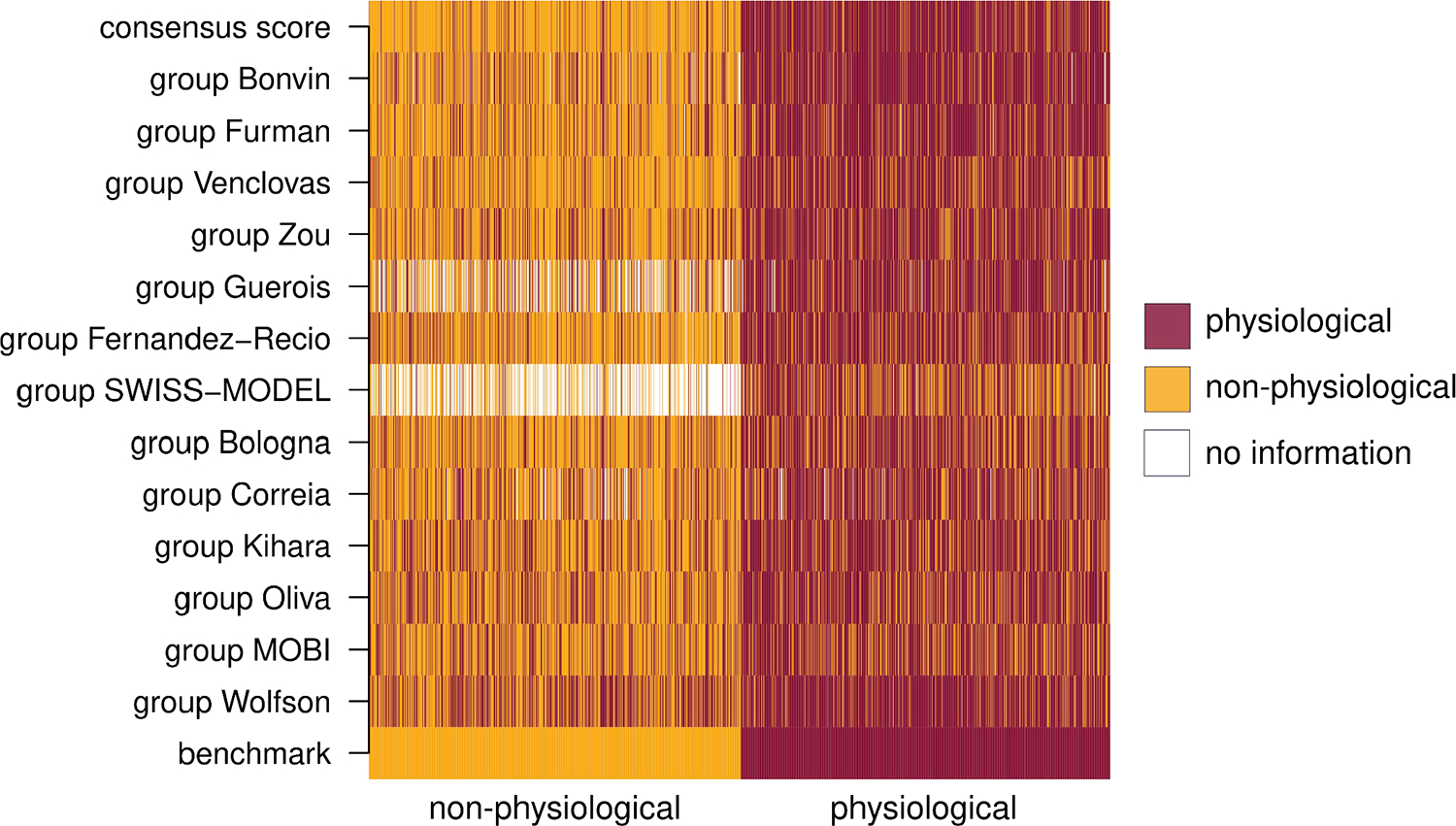
Heatmap of the consensus score and individual contributions. The bottom row groups non-physiological entries (left, orange); and physiological ones (left, dark red). Subsequent rows show the predictions of the best performing score from each group (Methods). Benchmark entries predicted as physiological are colored in dark red, and those predicted as non-physiological appear orange. Whenever no prediction was available, entries are shown in white. The top row shows the consensus score computed as described in Methods.
ROC-based community-wide consensus score.
A consensus score was computed using the 13 best-performing individual scores and applying an optimal threshold to each score for segregating the two types of dimers as described in Methods. The top row of the heat plot of Figure 3, illustrates the classification of individual structures of our benchmark dataset into physiological and non-physiological dimers produced by the consensus score.
Figure 4a displays the ROC obtained by applying the consensus score to all the structures of our benchmark dataset. The AUC of this ROC is 0.93, significantly higher than the highest AUC obtained for ROCs of individual scores (0.85), highlighting that the different scores include complementary features that help optimize performance. Interestingly, consensus scores computed from sets of fewer best performing scores displayed slightly higher performance as judged by their AUCs (0.92 for the top 5).
Figure 4.
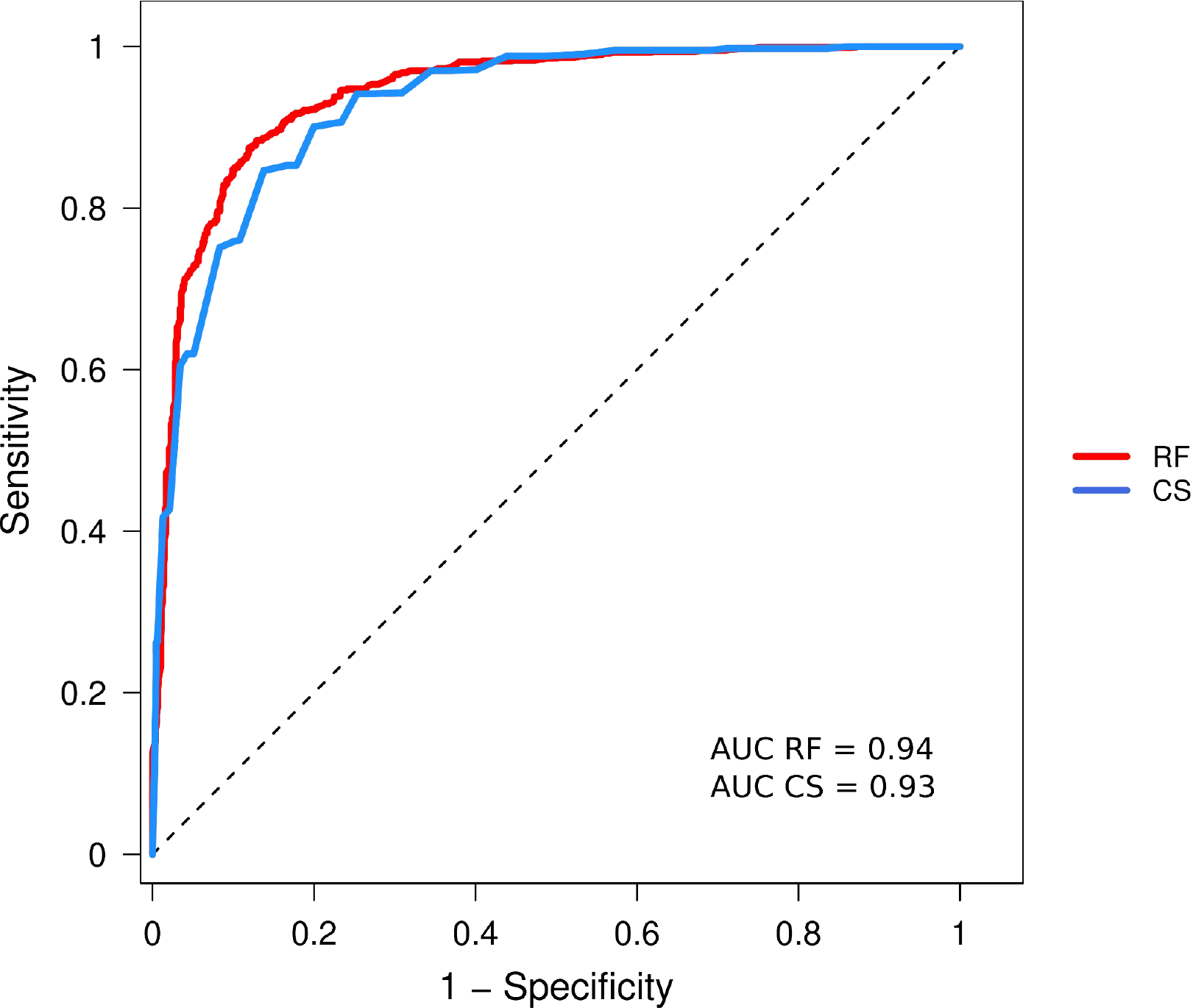
ROC curves computed using the consensus score and the RF procedure.
RF-based approach.
The RF classifier was derived using a total of 221 raw scores (features) computed by the participating groups on dimers of our benchmark dataset and applying leave-one-out cross-validation (Methods). Figure 4b displays the ROC obtained by applying this RF classifier to all the structures of our benchmark dataset (RF_221). The AUC of this ROC is 0.942, somewhat higher than the AUC of the ROC-based consensus score (0.93).
Using Gain/Gini analysis [45], we evaluated the average impact of individual scores on the prediction performance and found a smooth decay of the impact of these features. This is illustrated in Figure 5, which displays the Gain/Gini importance index for the 50 raw scores with the highest impact.
Figure 5.
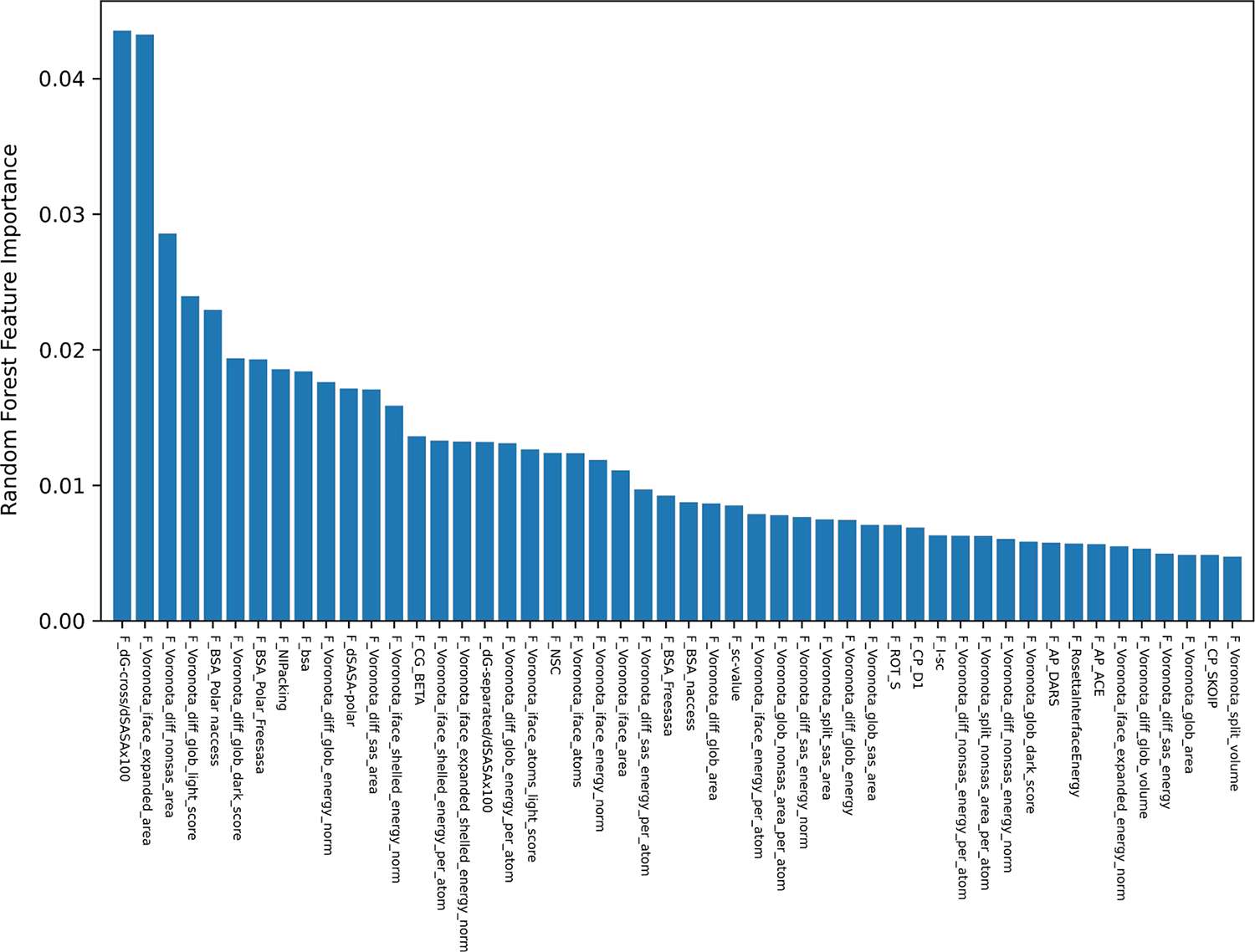
The 50 raw scores with the highest impact on the performance of the RF classifier. The plot displays the Gain/Gini importance index [45] for the 50 raw scores with highest impact on the RF classification.
Interestingly, ROCs computed using the RF classifier derived using only the 50 most impactful scores, or the 20 most impactful ones yielded an identical AUC to that obtained using the full set of raw scores, whereas the AUC dropped to 0.92 for the ROC computed using only 5 top scores (see Supplementary Figure S2). Thus, the RF model with all 221 raw scores, and those with 50 and 20 highest impact scores, outperform the best performing individual features of Table 3 (which include 8 integrated scores or classifiers), and display superior performance to that obtained with the consensus score derived using the ROC-based approach (0.93).
These results indicate that the RF classification and the Gain/Gini importance index prioritization of the 20 most impactful features outperform the greedy single feature per group, prioritization (of only 13 features) by the ROC-based approach. The fact that ~10% of all the raw scores, (20/221) is sufficient to closely approximate the optimal performance of the RF classifier is interesting. Thus, the combination of the 20 most impactful scores listed in Table 4, seems particularly efficient in capturing the key features that characterize the differences between physiological and non-physiological homodimer of our dataset.
Table 4.
list of the 20 raw scores with highest impact on the RF classification. The listed scores are the top 20 scores contributing to the RF classification of the benchmark dataset (see Figure 6).
| Feature (raw score) | Participant | References |
|---|---|---|
| ‘F_dG-cross/dSASAx100’ | Furman | RosettaCommon*, [71] |
| ‘F_Voronota_iface_expanded_area’ | Venclovas | [21,85] |
| ‘F_Voronota_diff_nonsas_area’ | ||
| ‘F_Voronota_diff_glob_light_score’ | ||
| ‘F_bsa’ | Oliva | NACCESS*, [86] |
| ‘F_Voronota_diff_sas_area’ | Venclovas | [21,85] |
| ‘F_BSA_Polar naccess’ | Oliva | NACCESS*, [86] |
| ‘F_NIPacking’ | Fernandez-Recio | [58,87] |
| ‘F_BSA_Polar_Freesasa’ | Oliva | NACCESS*, [86] |
| ‘F_Voronota_diff_glob_dark_score’ | Venclovas | [21,85] |
| ‘F_dG-separated/dSASAx100’ | Furman | |
| ‘F_Voronota_iface_shelled_energy_norm’ | Venclovas | [21,85] |
| ‘F_Voronota_iface_expanded_shelled_energy_norm’ | ||
| ‘F_Voronota_diff_glob_energy_norm’ | ||
| ‘F_dSASA-polar’ | Furman | RosettaCommon*, [71] |
| ‘F_Voronota_iface_area’ | Venclovas | [21,85] |
| ‘F_NSC’ | Fernandez-Recio | [58] |
| ‘F_Voronota_iface-Atoms_light_score’ | Venclovas | [21,85] |
| ‘F_CG_BETA’ | Fernandez-Recio | [58] |
| -F_Voronota_iface_enery_norm’ | Venclovas | [21,85] |
3.3. Correlations between raw scores for interfaces of protein-protein complexes
Raw scores developed by different groups tend to use different methods to quantify the same properties of a given interface. To gain insight into the level of redundancy or complementarity between different scores, we computed the correlations between the sets of values computed by pairs of scores for our benchmark dataset.
The heatmap of the pairwise (Pearson) correlations between the 50 most impactful scores in the RF classifier are displayed in Figures 6. The heatmap displaying the correlations between all 221 raw scores analyzed in this study is depicted in Figure S3. These scores are referred to by the acronyms provided by their authors. For further detail on the quantities that are being computed, the reader is referred to the summary descriptions in Table 1, as well as the reports of individual groups and the publications therein (see Collated Methods in the Supplementary Material).
Figure 6.
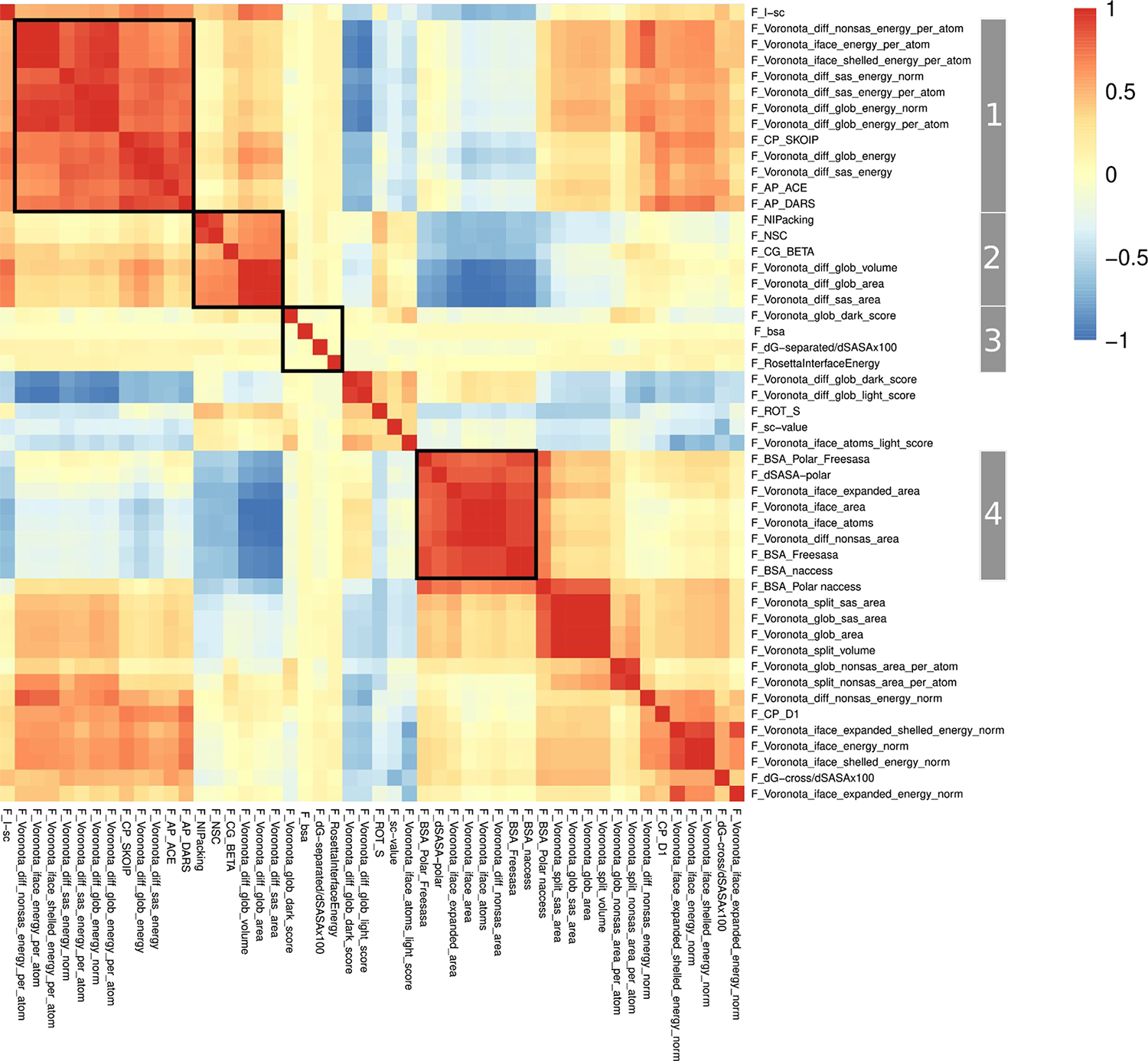
Heatmap of the pairwise (Pearson) correlations matrix between the 50 raw scores with the highest impact on the RF classification, clustered along columns and rows. The patterns of correlations of groups of scores (Groups 1–4) highlighted on the right-hand vertical dimension of the matrix are discussed in detail.
Both heatmaps display salient patterns of correlations between groups of scores. Examples of such patterns are highlighted for the groups labelled 1–4 in the heatmap of Figure 6. These groups comprise highly correlated scores that display weaker correlations (positively or negatively) with other score groups. Most of the scores in these four groups are Voronoi tessellation-based scores from the Venclovas team. Interestingly, scores from this team make up ~30% of all raw scores (69/221) but represent over 60% (31/50) of the 50 most impactful scores in the RF classifier. This indicates that while the individual features form the Venclovas team are poor discriminators between the physiological and non-physiological dimers (F-Voronota-iface_expanded_area with an AUC of 0.82, being the best performing individual tessellation-based score form this team), the combination of these scores is much more effective. These results are consistent with the outstanding performance of the Venclovas team in recent blind prediction challenges [43,49,53].
Furthermore the tessellation-based scores of this team display interesting patterns of correlations. For example, Group 1 (Figure 6) includes individual F_Voronota scores describing the normalized (per-atom) VoroMQA pseudo-energy of the inter-chain interface, computed using interface and global descriptors. Scores in this group are highly positively correlated to each other (red square, upper left end of the diagonal in Figure 6) and more weakly correlated to two other groups of scores. One of the latter groups contains positively correlated scores related to those in Group 1 such as the VoroMQA pseudo-energy of the inter-chain interface, normalized by the associated surface area terms, and the dG_cross interface energy term of the Rosetta suite from the Furman team (orange square at the upper right diagonal of Figure 6). The other group includes scores mostly weakly negatively correlated to those in Group1. These are F_Voronota scores representing global features of the dimers (pale blue regions, top middle of Figure 6). Other correlation patterns are observed for scores in Group 2 and 4, which contain a mix of Voronoi tessellation-bases scores and scores of other teams, whereas the four scores of Group 3, are the only scores that are uncorrelated to all other 46 scores (P_corr ~0).
Analogous patterns of correlations between groups of scores are observed in the heatmap of the full set of 221 raw scores. Interestingly, this larger set includes over 30 scores that are uncorrelated to any other score (see Supplementary Figure S3)
Taken together, these observations indicate that multiple factors govern the impact of a set of features on the classification performance, and that the combination of both highly correlated and weakly correlated but meaningful scores that capture somewhat different properties is important. Occasionally, particularly pertinent scores that are uncorrelated to any other score in the set can also be impactful. This seems to be the case of the dG_separated/dSASA and RosettaInterfaceEnergy scores of the Rosetta suite computed by the Furman group (see Table 1). Both scores are uncorrelated to all other scores of the analyzed set and are part of the 50 most impactful scores for the RF classification, with dG_separated/dSASA being the top-ranking individual score overall (Figure 5).
3.4. Classification based on similarity scores of structures predicted by AlphaFold
AlphaFold2 was used to predict the 3D structures of the benchmark dimers from their sequence, and the predicted structures were compared to those of the benchmark set. If Alphafold2 has learned the characteristics of physiological association modes, it should predict structures matching those of physiological dimers. At the same time, we expect the predictions of non-physiological dimer structures to be different, as measured by the QS-score and DockQ metrics. For example, AlphaFold might predict alternative models for non-physiological dimers (if the protein truly assembles into an alternative homodimer), or predict a different random interface if the protein is monomeric in solution. In both cases, we expect low QS-scores and DockQ metrics.
Figure 7 displays letter-value plots of the distributions of the QS-score and DockQ for a subset comprising 1480 dimers of the benchmark dataset, which were successfully predicted using both the gap and multimer versions, and which were relaxed using the multimer version. The dimers are split into the physiological entries identified using QSalign (836 dimers), and the non-physiological entries identified using QSalign (141 dimers) and ProtCID (700 dimers), respectively (see Methods for details). Both the QS- and DockQ-scores range from 0 to 1, with 1 indicating optimal fit to the target structure. We see indeed that the physiological dimers are predicted at significantly higher accuracy than the non-physiological dimers of our dataset and that the distributions are highly distinct.
Figure 7.
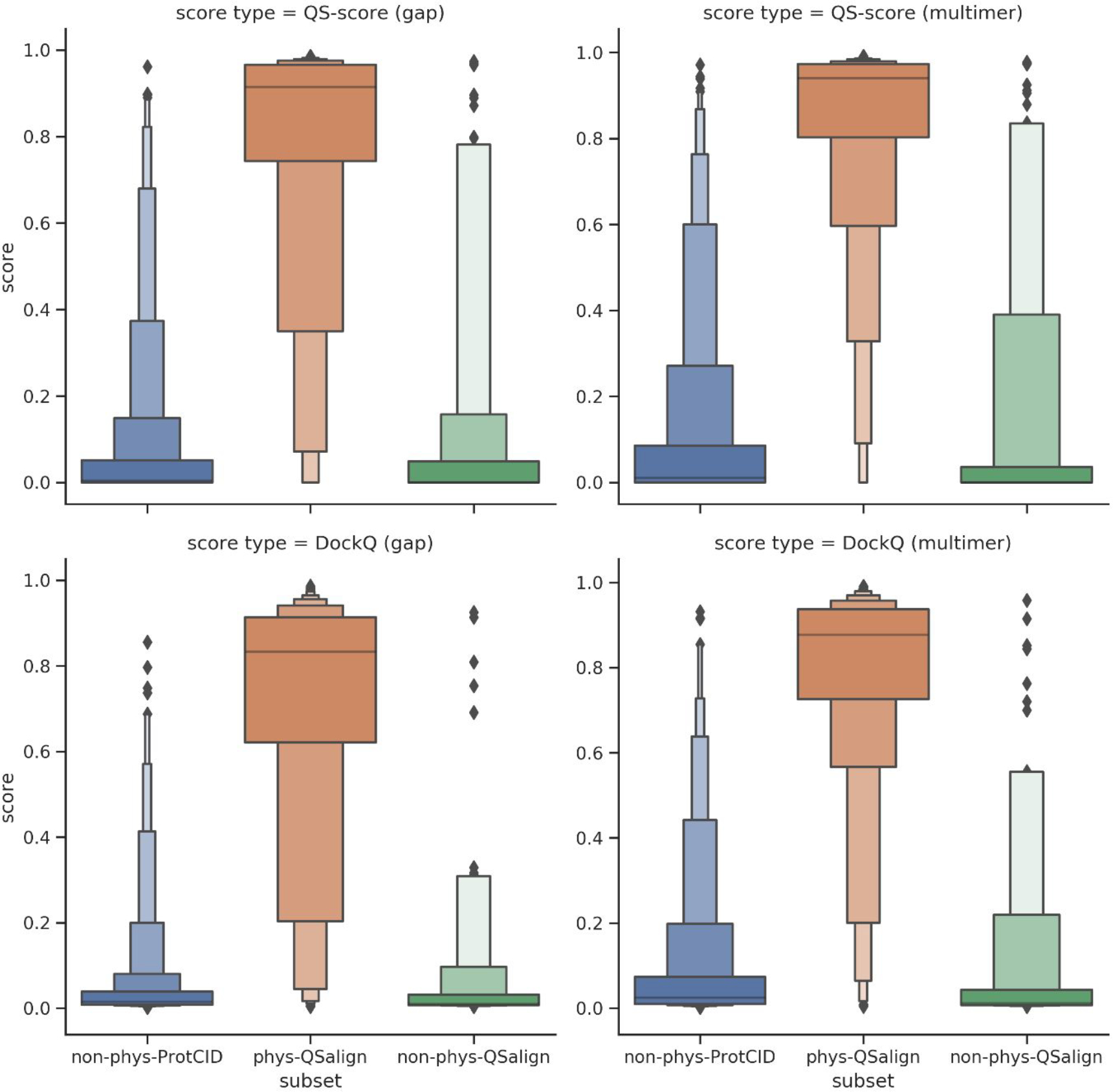
Letter-value plots of the distribution of QS- and DockQ-scores for AlphaFold2 models. The distributions are evaluated on the subset of 1480 dimers for which results were produced with all AlphaFold2 versions (gap, multimer and multimer with model relaxation). The dimers are split into physiological (identified using QSalign) and non-physiological (identified using QSalign and ProtCID).
The ability of the DockQ and QS-score of the predicted models to classify the benchmark complexes into the corresponding categories was evaluated by a ROC analysis and by computing the AUC. In addition to evaluating the ROC AUC, we also computed the mean and median accuracy of the best scoring model (among the five given by Alphafold2) predicted for each of the physiological dimers, as measured by the QS-score and DockQ (see Methods). The results listed in Table 5 indicate a high classification accuracy reaching an AUC of 0.954 with the gap version (using DockQ). This is higher than the ones from any individual score (Table 3) or the consensus and RF scores (Figure 4) even though no complexes were in AlphaFold’s training data for the gap version. AlphaFold-multimer, on the other hand, should benefit from having complexes included in its training set, but it may also have been “misled” by the non-physiological dimers in the QSalign set which used to be in the PDB. In the end, the classification accuracy increased with the multimer version (AUC of 0.964 for DockQ). The accuracy with which the structures of the physiological dimers are predicted is also generally high and increases significantly for models produced by AlphaFold2-multimer. The data in Table 5, also suggests that a somewhat higher prediction accuracy was displayed by the QS-score than by DockQ for the physiological dimers, likely indicating that DockQ is a somewhat more stringent similarity measure. The model relaxation option available for the multimer version had no effect on classification performance or prediction accuracy.
Table 5.
Classification performance of AlphaFold2 models predicted for the physiological and non-physiological homo dimers of the benchmark dataset. This is evaluated on the subset of 1480 targets for which results were produced with all AlphaFold2 versions.
Column 1 lists the version of AlphaFold used for the predictions, and the score used to quantify the similarity between the AlphaFold2 predicted model, and the homodimer structures of the corresponding benchmark entry (see Main text for detail). Column 2 lists the Area Under the Curve (AUC) of the ROCs computed using the listed scores. Columns 3 and 4 list the mean and median values for the computed scores considering only the physiological dimers. Those for the non-physiological dimers are not reported, as AlphaFold2 tends to predict alternative association modes for a significant fraction of these dimers, as expected.
| Score | ROC AUC | Model Accuracy Mean (Physiological dimer) | Model Accuracy Median (Physiological dimers) |
|---|---|---|---|
| QS-score (AF-gap) | 0.938 | 0.778 | 0.914 |
| QS-score (AF-multimer) | 0.957 | 0.833 | 0.940 |
| QS-score (AF_multimer relaxed) | 0.957 | 0.832 | 0.938 |
| DockQ (AF-gap) | 0.954 | 0.707 | 0.833 |
| DockQ (AF-multimer) | 0.964 | 0.786 | 0.877 |
| DockQ (AF-multimer relaxed) | 0.964 | 0.787 | 0.876 |
The multimer version of AlphaFold2 also outputs a predicted model confidence score ipTM, for each predicted structure. This score, computed in absence of any information on the target structure is derived in the form a predicted TM-score [5], modified to score interactions between residues from different chains [7]. To evaluate how ipTM relates to the QS-score and DockQ scores, we compared the 3 scores for the model with highest ipTM score for each of the 1671 targets successfully evaluated with the multimer version (Figure S4). We find that the ipTM distributions for structures predicted for the different categories of dimers of our dataset show distinct behaviours. The distribution for physiological dimers is unimodal peaking at high values (0.5–1.0), indicating that interfaces of these dimers are predicted at high confidence, whereas the ipTM distribution for predicted structures of non-physiological dimers is bimodal, with a main peak around low values (~0.2) (indicating low confidence models) and a secondary peak around values of ~0.8 (indicating high confidence models). Dimer models with high ipTM values likely correspond to non-physiological dimers for which an alternative dimerization mode is correctly identified by Alphafold2. As a result, the ipTM values for the non-physiological dimers display poorer Pearson correlation with the structure similarity measures (0.17 with QS-score; 0.15 with DockQ) than for their physiological counterparts (0.68 with QS-score; 0.73 with DockQ). QS-score and DockQ are generally highly correlated (Pearson correlation of 0.97).
3.5. Comparing the performance of different scoring methods.
Using the AUC to gauge performance clearly indicates that the DockQ and QS similarity scores of the models predicted by AlphaFold2 (AUC: 0.954–0.964) outperform the RF and the consensus score community-wide scoring/classification methods (AUC: 0.94, and 0.91, respectively) on our benchmark dataset of homodimers (Supplementary Figure S5).
In line with these differences in performance, the similarity scores of AlphaFold2-predicted models yield the lowest number (156) of misclassified entries (physiological dimers predicted to be non-physiological, and vice versa). The RF classifier misclassified 202 dimers, whereas the consensus score misclassified the largest number of dimers (244) (Venn diagram of Figure 8). Interestingly however, the overlap between the sets of misclassified entries by the 3 procedures is remarkably small. Only 23 entries are misclassified by all three procedures, and 150 entries are misclassified by 2 or more procedures. These 150 misclassified entries include 68 dimers from the non-physiological set against 82 from the physiological set (Supplementary Table S2). Part of these misclassifications may be due to incorrect assignments of the dimers to this category when building the benchmark dataset.
Figure 8.
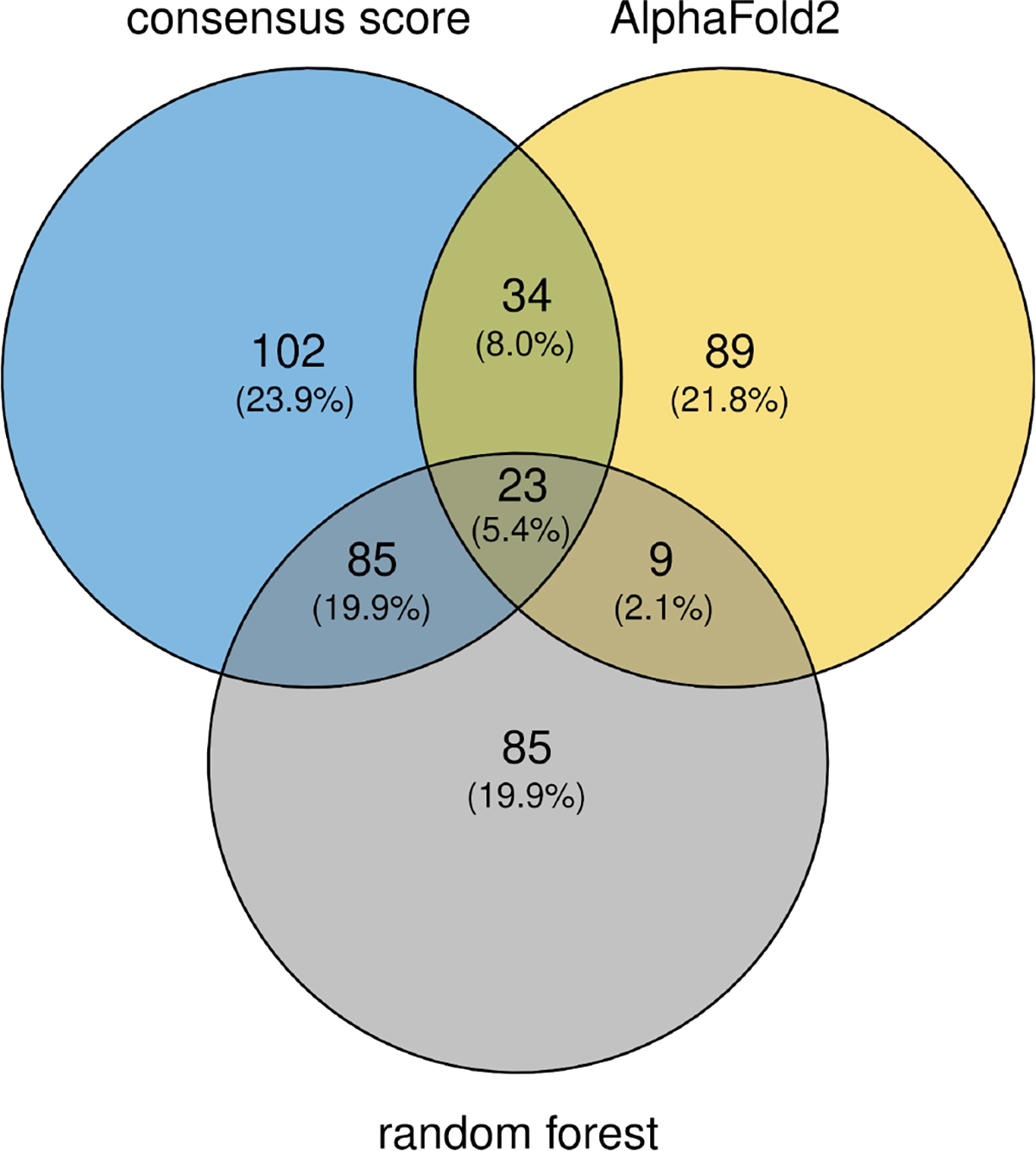
Venn Diagram illustrating the limited overlap of misclassified dimers by the three classifiers. CS stands for the ROC-based consensus score; RF stands for the Random Forest classifier, and AF stands for the classification performed using the DockQ score of structures predicted by AlphaFold2 (gap). Numbers indicate the number of misclassified dimers, the fraction (%) that these numbers represent of the total number of misclassified entries by all three methods (428) are given in parentheses. Classification results were determined by thresholding the 3 ROCs using the point of the curve most distant from the diagonal.
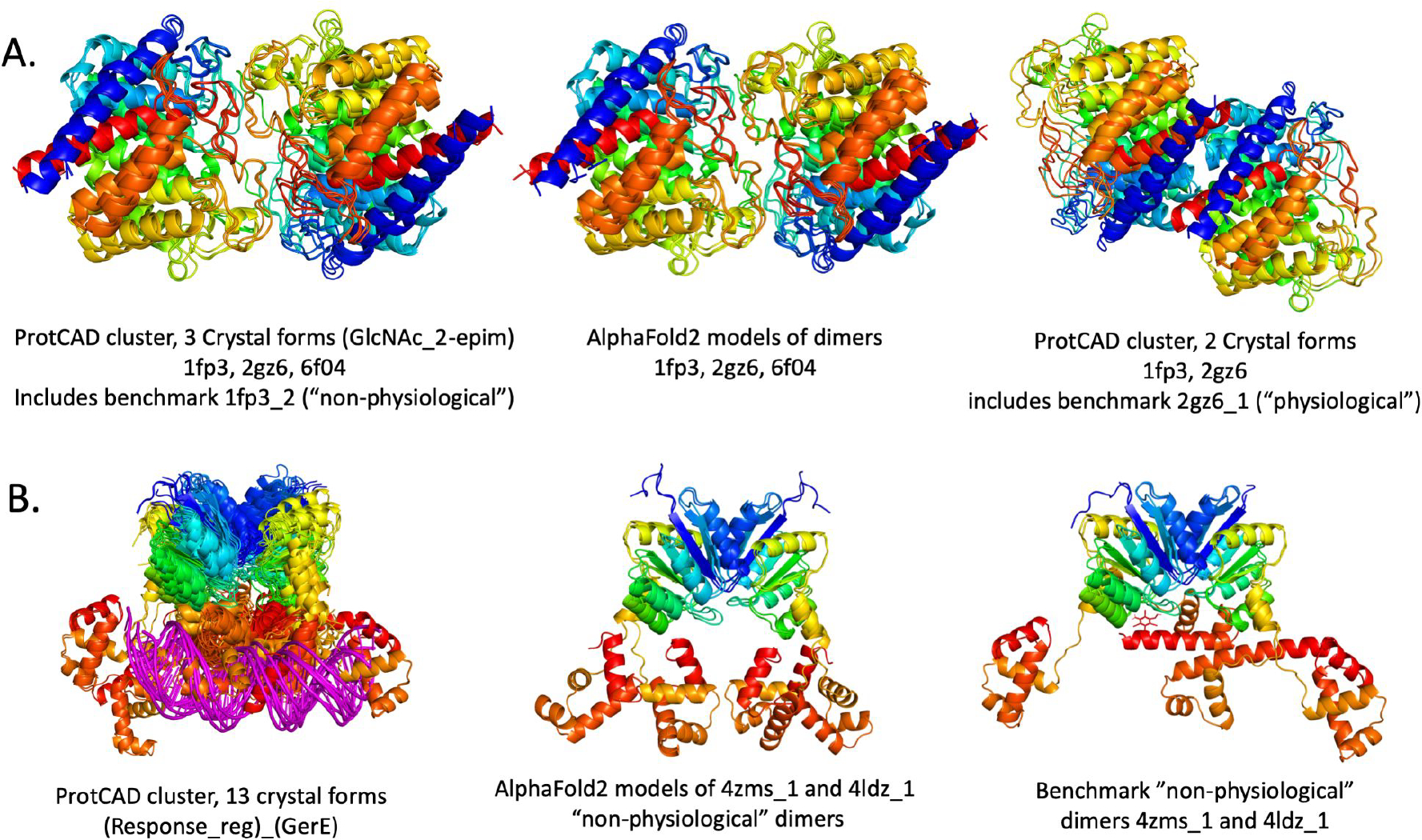
The assignments of the 23 dimer misclassified by all three methods were therefore manually re-evaluated using the updated ProtCID and ProtCAD resources. ProtCAD is similar to ProtCID but consists of viable symmetric biological assemblies (as determined by EPPIC [12]) instead of pairwise protein-protein interfaces [54]. This re-evaluation found evidence for 6 of the 12 misassigned non-physiological dimers to represent physiological dimers, and 4 of the 11 misassigned physiological dimers are likely to form other dimers than those in the benchmark, and hence to be in agreement with the prediction results for these dimers. Examples of misassigned non-physiological dimers that were correctly classified as physiological are illustrated in Figure 9. Interestingly, the non-physiological homodimers featuring large interface areas (such as 4pbc_1, 5cb2_3) are not systematically misclassified, but correctly classified as such by two out of the three classification procedures.
Figure 9.
Dimers misassigned in benchmark.
A. The benchmark contains a “non-physiological” dimer, 1fp3_2 and a “physiological” dimer 2gz6_1 that are very similar. ProtCAD/ProtCID contains two clusters of symmetric homodimers of Pfam GlcNAc_2-epim. The first one (left) contains 3 crystal forms and 3 different UniProts, including interfaces from 2gz6 and 6f04 (89% sequence identity), as well as the benchmark dimer 1fp3_2 (38% and 37% sequence identity respectively). AlphaFold2 reproduces all three of these dimers (center). At right is a ProtCAD/ProtCID cluster of 2 crystal forms, including an interface from 1fp3 and the benchmark dimer 2gz6_1. The structures are superposed on one monomer in each Figure. The data indicate that the larger cluster is likely to be physiological. B. Two “non-physiological” dimers in the benchmark belong to Pfam architecture (Response_reg)_(GerE), which has a cluster of 13 crystal forms (out of a total of 22 in the PDB), shown at left, some of which contain DNA. These two dimers, 4zms_1 (center) and 4ldz_1 (right) are therefore likely to be physiological.
4. Concluding remarks
This community-wide study exploited specialized resources on protein complexes and their interfaces to derive a carefully crafted benchmark dataset of 1677 homodimer proteins crystal structures comprising about an equal proportion of physiological and non-physiological dimers. We believe our definitions of the dimers in each category to be less error prone than in other available benchmark datasets because physiological dimers were required to have an interface that is well conserved across homologs, whereas non-physiological dimers were required to form an interface unlike any other interface across the known structural proteome. Interestingly, requiring non-physiological dimers to display on average similar or larger interface areas than their physiological counterparts in the benchmark dataset, had no detectable detrimental effect on the performance of the classification procedures. These procedures were furthermore robust enough to enable the detection of misassignments, i.e. dimers mistakenly assigned to the wrong category, based on earlier versions of the ProtCID and ProtCAD resources. Regularly updating these resources is hence important for improving the benchmark quality.
In the second part of the study, over 252 scoring functions developed by 13 different groups from the community were compiled and annotated. These functions were then used to evaluate the dimers of our benchmark dataset, and their performance in discriminating between the physiological and non-physiological complexes was quantified. Additionally, a greedy community-wide consensus score was derived using the best performing score of each of the 13 groups, and a RF classifier was trained on a subset of 221 scores, representing single features of the evaluated interfaces (the ‘raw scores’). Evaluating the performance by the AUC/ROC metrics revealed the community-wide consensus score to achieve a commendable performance (AUC 0.93). The latter was marginally surpassed by the more elaborate RF classifier using either all 221 raw scores or the subsets of 50 or 20 top performing raw scores (AUC 0.94). Both classifiers significantly outperformed individual scores previously developed by the community. These scores include raw scores, integrated scores (those combining multiple features of the interface), and integrated scores trained by deep learning methods on various earlier benchmark datasets. The best performing scores of each group yield significantly lower AUC values ranging from 0.71 to 0.85, and it is noteworthy that the two top ranking scores with AUC values of 0.85 and 0.84 were achieved with a deep learning-based Integrated score (Deeprank-GNN), and the physics-based F_dG.cross.dSASA100 score from the Rosetta suite, respectively (Table 3). None of the individual scores were trained or optimized using our newly derived benchmark dataset.
Taken together, these results indicate that most individual scores are rather ineffective in ranking physiological homodimers more highly than their non-physiological counterparts and therefore poorly discriminate between the two categories of dimers. The performance improves very significantly when individual scores are combined/integrated using increasingly sophisticated optimization methods or classifiers. A singularly effective combination was shown to comprise a set of scores derived using Voronoi-based descriptions to evaluate purely geometric features as well as contact surface areas and energy features. This is very encouraging considering the challenging nature of our benchmark dataset, which includes several examples of non-physiological dimers with very large interfaces that were nonetheless correctly classified as such by both the consensus score and RF classifiers.
Substantial progress was recently reported in CASP15 for the prediction of native protein complexes from sequence using deep learning methods such as AlphaFold2 [7,40]. Various modifications of the AlphaFold2 inference engine [41,55] and significant expansions of the MSA [56] were shown to enable AlphaFold2 more extensive sampling of protein models predicted with high confidence compatible with the evolutionary information extracted from the MSA. In several instances however, difficulties were reported in discriminating between models based on the confidence scores of the deep learning procedure. The analysis of the interface descriptors presented in this work could mitigate this problem, either through analyses/scoring of the structures generated, or perhaps by guiding the design of new deep learning architectures, capable of extracting relevant descriptors from the structure encoding.
In the last part of our study, AlphaFold2 and AlphaFold-multimer were used to predict the 3D structures of the dimers in our benchmark, and the structural similarity between the predicted and benchmark structures was used to score candidate models. As expected, models for the physiological dimers scored higher by the DockQ and QS similarity metrics, than for the non-physiological set, with corresponding AUC values of 0.954 – 0.964. This indicates that the AlphaFold2 engines were able to ‘recall’ the physiological dimers with high accuracy, whereas lower accuracy models often represent alternative association modes were predicted for a significant fraction of the non-physiological dimers, in line with the fact that these dimers are rarely observed in crystallized proteins and not conserved among homologs. Thus, while this analysis is orthogonal to the main goal of our study, the global convergence of the predictions by the different methods as well as Alphafold2, provides strong support for the definitions of physiological and non-physiological dimers of our benchmark dataset.
The benchmark dataset structures, the scores computed for the benchmark dimers, the results of the classifications and all other analyses reported in this study are available on GitHub (https://github.com/vibbits/Elixir-3DBioInfo-Benchmark-Protein-Interfaces)
Supplementary Material
Acknowledgements
This study (SJW coordinator) was performed as part of the Implementation Plan (2019–2021) of Activity-2 of the 3DBioInfo Elixir Community (https://elixir-europe.org/communities/3d-bioinfo). We thank Alexander Botzki (VIB Technology Training, Flanders, Belgium) for helping with installing and hosting the project GitHub. G.T., L.P., and T.S. acknowledge the contributions of Gabriel Studer in setting up the AlphaFold pipeline for our analysis, sciCORE at the University of Basel for providing computational resources and system administration support, and funding from the SIB Swiss Institute of Bioinformatics and the Biozentrum PhD Fellowships. D.K. acknowledges supports by the National Institutes of Health (R01GM133840) and the National Science Foundation (DMS2151678, DBI2003635, CMMI1825941, MCB2146026, and MCB1925643). X.W. is recipient of the MolSSI graduate fellowship. A.M.J.J.B. and M.R. acknowledge financial support from the Netherlands eScience Center (ASDI.2016.043), from the Netherlands Organization for Scientific Research (NWO) (TOP-PUNT grant 718.015.001) and from the European Union Horizon 2020 project BioExcel (823830). J.F.-R. acknowledges support by Spanish Ministry of Science (grant PID2019-110167RB-I00 / AEI / 10.13039/501100011033). B. J.-G. is employed by Zymvol Biomodeling on a project which received funding from the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement No. 801342 (Tecniospring INDUSTRY) and the Government of Catalonia’s Agency for Business Competitiveness (ACCIÓ). EDL acknowledges support from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 819318), by a research grant from A.-M. Boucher, by research grants from the Estelle Funk Foundation, the Estate of Fannie Sherr, the Estate of Albert Delighter, the Merle S. Cahn Foundation, Mrs. Mildred S. Gosden, the Estate of Elizabeth Wachsman, the Arnold Bortman Family Foundation.
References
- [1].Alberts B, The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [DOI] [PubMed] [Google Scholar]
- [2].Ideker T, Sharan R, Protein networks in disease. Genome Res. 2008, 18, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barabási A-L, Gulbahce N, Loscalzo J, Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Berman H, Henrick K, Nakamura H, Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [DOI] [PubMed] [Google Scholar]
- [5].Jumper J, Evans R, Pritzel A, Green T, et al. , Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baek M, DiMaio F, Anishchenko I, Dauparas J, et al. , Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Evans R, O’Neill M, Pritzel A, Antropova N, et al. , Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022, 2021.10.04.463034. [Google Scholar]
- [8].Malhotra S, Träger S, Dal Peraro M, Topf M, Modelling structures in cryo-EM maps. Curr. Opin. Struct. Biol. 2019, 58, 105–114. [DOI] [PubMed] [Google Scholar]
- [9].Tüting C, Kyrilis FL, Müller J, Sorokina M, et al. , Cryo-EM snapshots of a native lysate provide structural insights into a metabolon-embedded transacetylase reaction. Nat. Commun. 2021, 12, 6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA, 3D complex: a structural classification of protein complexes. PLoS Comput. Biol. 2006, 2, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Janin J, Rodier F, Protein-protein interaction at crystal contacts. Proteins 1995, 23, 580–587. [DOI] [PubMed] [Google Scholar]
- [12].Baskaran K, Duarte JM, Biyani N, Bliven S, Capitani G, A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 2014, 14, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Krissinel E, Henrick K, Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [DOI] [PubMed] [Google Scholar]
- [14].Glaser F, Pupko T, Paz I, Bell RE, et al. , ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [DOI] [PubMed] [Google Scholar]
- [15].Wodak SJ, Janin J, Structural basis of macromolecular recognition. Adv. Protein Chem. 2002, 61, 9–73. [DOI] [PubMed] [Google Scholar]
- [16].Wodak SJ, Méndez R, Prediction of protein-protein interactions: the CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol. 2004, 14, 242–249. [DOI] [PubMed] [Google Scholar]
- [17].Singh A, Dauzhenka T, Kundrotas PJ, Sternberg MJE, Vakser IA, Application of docking methodologies to modeled proteins. Proteins 2020, 88, 1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA, Structural templates for comparative protein docking. Proteins 2015, 83, 1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kozakov D, Brenke R, Comeau SR, Vajda S, PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [DOI] [PubMed] [Google Scholar]
- [20].Nadaradjane AA, Guerois R, Andreani J, Protein-Protein Docking Using Evolutionary Information. Methods Mol. Biol. 2018, 1764, 429–447. [DOI] [PubMed] [Google Scholar]
- [21].Olechnovič K, Venclovas Č, VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins 2017, 85, 1131–1145. [DOI] [PubMed] [Google Scholar]
- [22].Renaud N, Geng C, Georgievska S, Ambrosetti F, et al. , DeepRank: a deep learning framework for data mining 3D protein-protein interfaces. Nat. Commun. 2021, 12, 7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hwang H, Pierce B, Mintseris J, Janin J, Weng Z, Protein-protein docking benchmark version 3.0. Proteins 2008, 73, 705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hwang H, Vreven T, Janin J, Weng Z, Protein-protein docking benchmark version 4.0. Proteins 2010, 78, 3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guest JD, Vreven T, Zhou J, Moal I, et al. , An expanded benchmark for antibody-antigen docking and affinity prediction reveals insights into antibody recognition determinants. Structure 2021, 29, 606–621.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vreven T, Moal IH, Vangone A, Pierce BG, et al. , Updates to the Integrated Protein-Protein Interaction Benchmarks: Docking Benchmark Version 5 and Affinity Benchmark Version 2. J. Mol. Biol. 2015, 427, 3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kundrotas PJ, Anishchenko I, Dauzhenka T, Kotthoff I, et al. , Dockground: A comprehensive data resource for modeling of protein complexes. Protein Sci. 2018, 27, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vangone A, Bonvin AMJJ, PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio Protoc 2017, 7, e2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lensink MF, Wodak SJ, Score_set: a CAPRI benchmark for scoring protein complexes. Proteins 2014, 82, 3163–3169. [DOI] [PubMed] [Google Scholar]
- [30].Lensink MF, Wodak SJ, Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [DOI] [PubMed] [Google Scholar]
- [31].Janin J, Henrick K, Moult J, Eyck LT, et al. , CAPRI: a Critical Assessment of PRedicted Interactions. Proteins 2003, 52, 2–9. [DOI] [PubMed] [Google Scholar]
- [32].Barradas-Bautista D, Cao Z, Vangone A, Oliva R, Cavallo L, A random forest classifier for protein–protein docking models. Bioinformatics Advances 2022, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xu Q, Dunbrack RL Jr, ProtCID: a data resource for structural information on protein interactions. Nat. Commun. 2020, 11, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dey S, Ritchie DW, Levy ED, PDB-wide identification of biological assemblies from conserved quaternary structure geometry. Nat. Methods 2018, 15, 67–72. [DOI] [PubMed] [Google Scholar]
- [35].Janin J, Principles of protein-protein recognition from structure to thermodynamics. Biochimie 1995, 77, 497–505. [DOI] [PubMed] [Google Scholar]
- [36].Lensink MF, Velankar S, Wodak SJ, Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins 2017, 85, 359–377. [DOI] [PubMed] [Google Scholar]
- [37].Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, et al. , The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hastie T, Tibshirani R, Friedman J, The Elements of Statistical Learning, Springer; New York, n.d. [Google Scholar]
- [39].Jumper J, Evans R, Pritzel A, Green T, et al. , Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bryant P, Pozzati G, Elofsson A, Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mirdita M, Schütze K, Moriwaki Y, Heo L, et al. , ColabFold: making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dey S, Prilusky J, Levy ED, QSalignWeb: A Server to Predict and Analyze Protein Quaternary Structure. Front Mol Biosci 2021, 8, 787510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lensink MF, Nadzirin N, Velankar S, Wodak SJ, Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 2020, 88, 916–938. [DOI] [PubMed] [Google Scholar]
- [44].Pedregosa F, Varoquaux G, Gramfort A, Michel V, et al. , Scikit-learn: Machine Learning in Python. arXiv [cs.LG] 2012, 2825–2830. [Google Scholar]
- [45].Lundberg SM, Lee S-I, in:, Proceedings of the 31st International Conference on Neural Information Processing Systems, Curran Associates Inc., Red Hook, NY, USA; 2017, pp. 4768–4777. [Google Scholar]
- [46].Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T, Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xu Q, Canutescu AA, Wang G, Shapovalov M, et al. , Statistical analysis of interface similarity in crystals of homologous proteins. J. Mol. Biol. 2008, 381, 487–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Basu S, Wallner B, DockQ: A Quality Measure for Protein-Protein Docking Models. PLoS One 2016, 11, e0161879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lensink MF, Brysbaert G, Mauri T, Nadzirin N, et al. , Prediction of protein assemblies, the next frontier: The CASP14-CAPRI experiment. Proteins 2021, 89, 1800–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Biasini M, Schmidt T, Bienert S, Mariani V, et al. , OpenStructure: an integrated software framework for computational structural biology. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sillitoe I, Bordin N, Dawson N, Waman VP, et al. , CATH: increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lewis TE, Sillitoe I, Dawson N, Lam SD, et al. , Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res. 2018, 46, D435–D439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lensink MF, Brysbaert G, Nadzirin N, Velankar S, et al. , Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 2019, 87, 1200–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xu Q, Dunbrack RL, The protein common assembly database (ProtCAD)-a comprehensive structural resource of protein complexes. Nucleic Acids Res. 2023, 51, D466–D478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Johansson-Åkhe I, Wallner B, Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front Bioinform 2022, 2, 959160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang C, Zheng W, Mortuza SM, Li Y, Zhang Y, DeepMSA: constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 2020, 36, 2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yan L, ggvenn: Venn Diagram by ggplot2, with really easy-to-use API, Github, n.d. [Google Scholar]
- [58].Moal IH, Jiménez-García B, Fernández-Recio J, CCharPPI web server: computational characterization of protein-protein interactions from structure. Bioinformatics 2015, 31, 123–125. [DOI] [PubMed] [Google Scholar]
- [59].Cheng TM-K, Blundell TL, Fernandez-Recio J, pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [DOI] [PubMed] [Google Scholar]
- [60].Martin J, Lavery R, Arbitrary protein-protein docking targets biologically relevant interfaces. BMC Biophys. 2012, 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Martin OMF, Etheve L, Launay G, Martin J, Implication of Terminal Residues at Protein-Protein and Protein-DNA Interfaces. PLoS One 2016, 11, e0162143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Olechnovič K, Venclovas Č, VoroMQA web server for assessing three-dimensional structures of proteins and protein complexes. Nucleic Acids Res. 2019, 47, W437–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dong GQ, Fan H, Schneidman-Duhovny D, Webb B, Sali A, Optimized atomic statistical potentials: assessment of protein interfaces and loops. Bioinformatics 2013, 29, 3158–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tubiana J, Schneidman-Duhovny D, Wolfson HJ, ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 2022, 19, 730–739. [DOI] [PubMed] [Google Scholar]
- [65].Huang S-Y, Zou X, An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008, 72, 557–579. [DOI] [PubMed] [Google Scholar]
- [66].Huang S-Y, Zou X, Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins 2011, 79, 2648–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jiménez-García B, Elez K, Koukos PI, Bonvin AM, Vangone A, PRODIGY-crystal: a web-tool for classification of biological interfaces in protein complexes. Bioinformatics 2019, 35, 4821–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, et al. , The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- [69].Réau M, Renaud N, Xue LC, Bonvin AMJJ, DeepRank-GNN: a graph neural network framework to learn patterns in protein-protein interfaces. Bioinformatics 2023, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Stranges PB, Kuhlman B, A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013, 22, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gray JJ, Moughon S, Wang C, Schueler-Furman O, et al. , Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [DOI] [PubMed] [Google Scholar]
- [72].Oliva R, Vangone A, Cavallo L, Ranking multiple docking solutions based on the conservation of inter-residue contacts. Proteins 2013, 81, 1571–1584. [DOI] [PubMed] [Google Scholar]
- [73].Hubbard SJ and Thornton JM, NACCESS. computer program. Department of Biochemistry and Molecular Biology, University College, London. 1993. [Google Scholar]
- [74].Mitternacht S, FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Res. 2016, 5, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhou H, Skolnick J, GOAP: a generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys. J. 2011, 101, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang X, Terashi G, Christoffer CW, Zhu M, Kihara D, Protein docking model evaluation by 3D deep convolutional neural networks. Bioinformatics 2020, 36, 2113–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wang X, Flannery ST, Kihara D, Protein Docking Model Evaluation by Graph Neural Networks. Front Mol Biosci 2021, 8, 647915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Esquivel-Rodríguez J, Yang YD, Kihara D, Multi-LZerD: multiple protein docking for asymmetric complexes. Proteins 2012, 80, 1818–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Marze NA, Roy Burman SS, Sheffler W, Gray JJ, Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics 2018, 34, 3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Savojardo C, Fariselli P, Martelli PL, Casadio R, ISPRED4: interaction sites PREDiction in protein structures with a refining grammar model. Bioinformatics 2017, 33, 1656–1663. [DOI] [PubMed] [Google Scholar]
- [81].Waterhouse A, Bertoni M, Bienert S, Studer G, et al. , SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Quignot C, Rey J, Yu J, Tufféry P, et al. , InterEvDock2: an expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs. Nucleic Acids Res. 2018, 46, W408–W416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Andreani J, Faure G, Guerois R, InterEvScore: a novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution. Bioinformatics 2013, 29, 1742–1749. [DOI] [PubMed] [Google Scholar]
- [84].Gainza P, Sverrisson F, Monti F, Rodolà E, et al. , Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17, 184–192. [DOI] [PubMed] [Google Scholar]
- [85].Olechnovič K, Venclovas C, Voronota: A fast and reliable tool for computing the vertices of the Voronoi diagram of atomic balls. J. Comput. Chem. 2014, 35, 672–681. [DOI] [PubMed] [Google Scholar]
- [86].Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R, COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 2011, 27, 2915–2916. [DOI] [PubMed] [Google Scholar]
- [87].Mitra P, Pal D, New measures for estimating surface complementarity and packing at protein-protein interfaces. FEBS Lett. 2010, 584, 1163–1168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.