

Abstract
X‐linked muscular dystrophy in cats (FXMD) is an uncommon disease, with few reports describing its pathogenic genetic variants. A 9‐year‐old castrated male domestic shorthair cat was presented with persistent muscle swelling and breathing difficulty from 3 years of age. Serum activity of alanine aminotransferase, aspartate transaminase, and creatine kinase were abnormally high. Physical and neurological examinations showed muscle swelling in the neck and proximal limb, slow gait, and occasional breathing difficulties. Electromyography showed pseudomyotonic discharges and complex repetitive discharges with a “dive‐bomber” sound. Histopathology revealed muscle necrosis and regeneration. Whole‐genome sequencing identified a novel and unique hemizygous nonsense genetic variant, c.8333G > A in dystrophin (DMD), potentially causing a premature termination codon (p.Trp2778Ter). Based on a combination of clinical and histological findings and the presence of the DMD nonsense genetic variant, this case was considered FXMD, which showed mild clinical signs and long‐term survival, even though immunohistochemical characterization was lacking.
Keywords: Becker, Duchenne, hypertrophic feline muscular dystrophy, precision medicine, rare disease
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- BMD
Becker muscular dystrophy
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- DMD
dystrophin
- EMG
electromyography
- FPR
false‐positive rate
- FXMD
X‐linked muscular dystrophy in cats
- LoF
loss‐of‐function
- TIVA
total intravenous anesthesia
1. INTRODUCTION
Dystrophin‐deficient muscular dystrophy, also called dystrophinopathy, is a hereditary muscular disease caused by abnormality of the dystrophin (DMD) gene, which is located on the X chromosome and encodes dystrophin. 1 In human medicine, there are 2 main forms of muscular dystrophy related to the DMD gene: Duchenne muscular dystrophy (DMD), which is characterized by early‐onset, rapidly progressive muscle weakness and reported median life expectancy below 30 years 2 , 3 ; and Becker muscular dystrophy (BMD), which is clinically heterogeneous but usually has a later onset and slower progression. 4 Unlike DMD, BMD has variable severity, ranging from severe to extremely mild. 4 In cats, several cases of dystrophin‐deficient muscular dystrophy are reported. Such cases are characterized by muscle hypertrophy and designated as X‐linked muscular dystrophy in cats (FXMD). 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Disease progression varies depending on the case; for example, one had a juvenile‐onset and severe clinical course, 14 and the other showed an adult‐onset that produces relatively mild clinical course. 15
In humans, numerous genetic variants are associated with dystrophinopathy, and our understanding of the genotype‐to‐phenotype correlation is increasing. However, in FXMD, few reports have identified the associated DMD variants. 14 , 15 , 16 Those reports identified the nonsense variant c.1180C > T (p.Arg394Ter), associated with a juvenile‐onset, severe phenotype in the Maine coon cat, 14 the missense variant c.4186C > T (p.His1396Tyr) in an adult‐onset, mildly affected domestic cat, 15 and the nonsense variant, c.4849C > T (p.Gln1617Ter) in a juvenile‐onset, severely affected domestic cat. 16
In human patients with muscular dystrophy, complications associated with volatile anesthesia are reported, 17 as halothane, isoflurane, sevoflurane, and desflurane might cause anesthesia‐related rhabdomyolysis, although the exact mechanism remains unclear. 18 , 19 Similarly, in veterinary practice, the association between volatile anesthesia and rhabdomyolysis in FXMD is reported, even causing deaths. 7 , 8 Although in humans, total intravenous anesthesia (TIVA) is a safe alternative for muscular dystrophy patients, 20 to date, no studies have investigated its use in cats.
2. CASE DESCRIPTION
2.1. Clinical history and findings and histology
A 9‐year‐old, castrated, male, domestic shorthair cat presented at Otakibashi Animal Hospital (Tokyo, Japan) with chronic muscle hypertrophy. From 3 years of age, the owner had noticed cervical muscle swelling and occasional breathing difficulty. Blood chemistry showed higher activity of alanine aminotransferase (ALT) and aspartate transaminase (AST) at 1 year and 7 months of age (creatine kinase [CK] activity was not measured then). At 2 years and 9 months of age and thereafter, the activity of ALT and AST were measured repeatedly, in addition to CK activity. At 3 years and 9 months of age, a serum sample was obtained from the cat and submitted to a commercial laboratory (Monolith; Tokyo, Japan) to analyze CK isoenzyme activities. CK isoenzyme activity analyses revealed 93% CK‐MM (from skeletal muscle), 2% CK‐BB (from brain), 3% CK‐MB (from myocardium), and 2% CK‐subtype. At 4 years of age, the cat showed dyspnea and underwent computed tomography, which revealed skeletal muscle hypertrophy in the whole body; however, a definitive diagnosis was not made. Initially, the cat was prescribed 0.5‐1.0 mg/kg Q24h of prednisolone, which stabilized the respiratory status but exerted no relevant effect on muscle swelling. At 9 years of age, dyspnea recurred, and a computed tomography scan was performed, without anesthesia. Computed tomography findings showed skeletal muscle hypertrophy, but no respiratory abnormalities were identified. Eventually, the cat was referred to Otakibashi Animal Hospital, and reevaluation and further clinical work‐up were performed.
Physical examination revealed cervical muscle and proximal limb muscle hypertrophy. The cat also showed tongue protrusion, although the tongue was not hypertrophic (Figure 1; Video 1). Neurological examination showed normal postural and spinal reflexes, without muscle stiffness, cramping, or dimpling after palpation/percussion. However, the cat's gait was slightly slower than normal. A thoracic X‐ray showed normal heart size and lung fields (Figure S1). Echocardiography could not be performed as the cat showed breathing difficulty associated with the stress of the examination. Abnormal blood chemistry was found at all ages, with higher ALT, AST, and CK activity (Table 1). Feline coronavirus antibody titer and the levels of N‐terminal prohormone of brain natriuretic peptide (NT‐proBNP) concentration, thyroxine, and fasting and postprandial serum bile acid were within the reference range.
FIGURE 1.
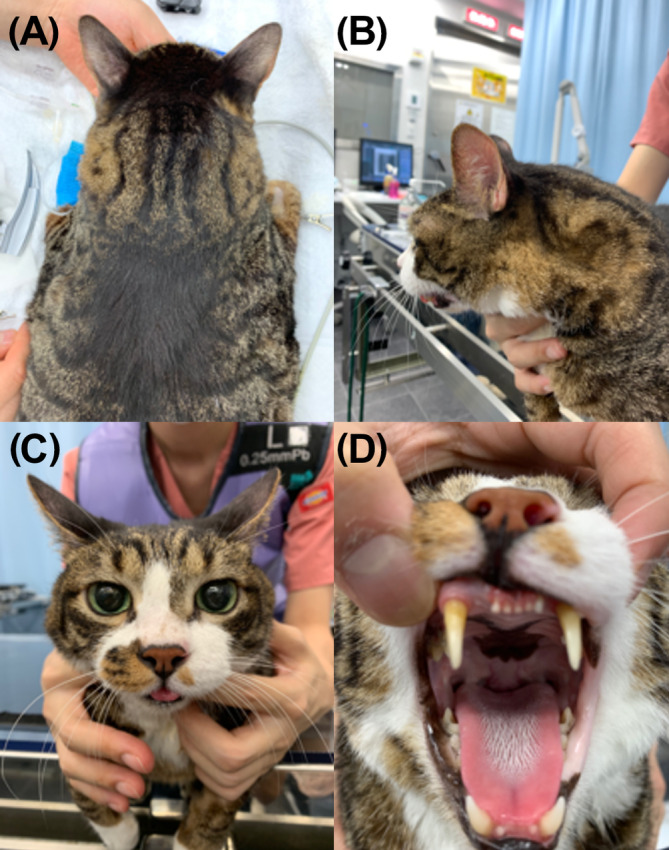
The cat presented with muscular hypertrophy of the neck muscles: (A,B) marked muscular hypertrophy of the neck muscle; (B,C) tongue protrusion; and (D) unobvious tongue hypertrophy.
VIDEO 1.
Appearance of the present case, revealing cervical muscle hypertrophy. Tongue enlargement or abnormal function were not observed. Labored breathing was often seen.
TABLE 1.
Serum alanine aminotransferase (ALT), aspartate transaminase (AST), and creatine kinase (CK) activity.
| Reference ranges | 19 months old a | 33 months old | 43 months old | 55 months old | 107 months old | 108 months old | |
|---|---|---|---|---|---|---|---|
| ALT (U/L) | 22‐84 | 410 | 463 | 325 | 97 | 88 | 122 |
| AST (U/L) | 18‐51 | 841 | 650 | 856 | 222 | 225 | 220 |
| CK (U/L) | 87‐309 | NA | 2000 | >20 000 | 17 707 | >20 000 | 14 292 |
Abbreviation: NA, not available.
Age at the case's initial examination.
Electromyography (EMG) and muscle biopsy were performed under TIVA, thus avoiding the risk of rhabdomyolysis associated with volatile anesthesia, malignant hyperthermia associated with volatile anesthesia, or both. The cat was premedicated with butorphanol (0.25 mg/kg) and maropitant (1 mg/kg) IV while being oxygenated. Continuous rate infusion was started with medetomidine hydrochloride (5 μg/kg/h) and propofol (10 mg/kg/h). Thereafter, propofol (1 mg/kg) was injected IV, and tracheal intubation was performed after sufficient sedation was obtained. During the EMG and muscle biopsy, medetomidine hydrochloride was increased (10 μg/kg/h), and the propofol was decreased (5 mg/kg/h). Electromyography and muscle biopsy performed under TIVA had no obvious adverse events during the perioperative period. Once the procedures were completed, the continuous rate infusion was terminated, and anesthesia was reversed with atipamezole hydrochloride 100 μg/kg IV. The cat recovered without hyperthermia or other complications.
Electromyography was performed, and the triceps brachii, vastus lateralis, anterior tibial, and semispinalis capitis muscles on the right side were evaluated. Electromyography findings included pseudomyotonic discharges from the triceps brachii, vastus lateralis, and anterior tibial muscles. Complex repetitive discharges were recorded from the semispinalis capitis (Figure 2). The recorded discharges generated a “dive‐bomber” sound.
FIGURE 2.
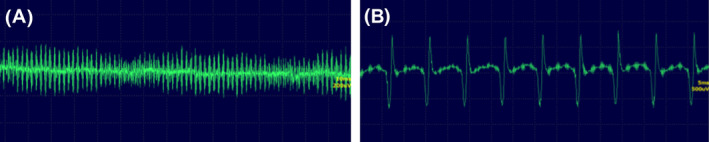
Electromyography of the right‐side muscles: (A) triceps brachii muscle presenting with pseudomyotonic discharges with a “dive‐bomber” sound; (B) longus colli muscle presenting with complex repetitive discharges.
Muscle biopsy specimen samples were taken from the triceps brachii, vastus lateralis, and anterior tibial muscles, contralateral to the EMG to avoid artifacts caused by needle insertion. Two samples were taken from the vastus lateralis and were shipped directly to the veterinary pathology laboratory at the University of Tokyo (Tokyo, Japan). Formalin‐fixed vastus lateralis tissue exhibited a marked variation in the size of muscle fibers, as small, angulated fibers were mixed with large, round, hypertrophic fibers. Some fibers presented internal nuclei suggestive of regeneration. Muscle fiber necrosis, macrophage infiltration, and mild fibrosis were also observed (Figure 3).
FIGURE 3.
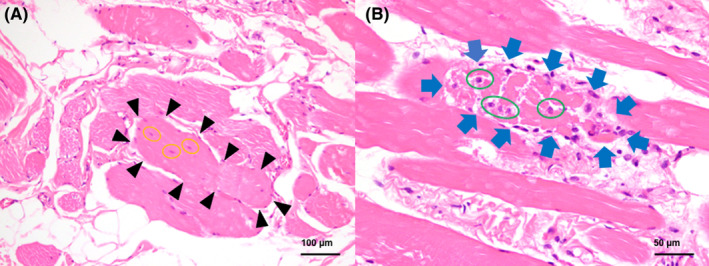
Histology findings in the present case. (A) Large fibers were seen, and one of them was indicated by black arrowheads. Muscle fibers exhibiting internal nuclei (yellow circles). Hematoxylin and eosin, ×200. (B) Muscle fiber necrosis (blue arrows) and infiltration of macrophages (green circles). Hematoxylin and eosin, ×400.
2.2. Whole‐genome sequencing
Informed consent was obtained from the owner for genomic investigation. DNA was extracted from blood by phenol‐chloroform method and submitted to the University of Missouri Genomics Technology Core (Columbia, MO). Library preparation, whole‐genome sequencing, and data analysis were performed, as previously described. 16 The cat in the present case corresponds to accession number SAMN35992016. The present case's genome data were compared with other 361 cats' whole‐genome sequencing and 52 cats' whole‐exome sequencing dataset in the 99 Lives Cat Genome Consortium (Appendix S1).
Both dominant and recessive inheritance were considered for variant filtering, without a specific candidate gene. The numbers of unique variants for this case are presented in Table S1. Sixty‐one homozygous/hemizygous and unique variants were identified, including a nonsense stop gain in DMD (Table S2). The DMD stop gain loss‐of‐function (LoF) variant (XM_023249210.1:c.8333G > A; ENSFCAT00000068370:c.8333G > A; p.Trp2778Ter) is at position NC_018741.3: chromosome X: 27361452 in Felis_catus_9.0 (Table S2) and at position NC_058386.1: chromosome X:27110574 in F.catus_Fca126_mat1.0. This variant had 15× sequencing reads for the case, suggesting good sequencing coverage for a region on the X chromosome of a male cat. Considering the present case as heterozygous, 824 variants were identified including 34 LoF variants and 252 missense variants (Table S3). All DMD variants detected in the 99 Lives dataset are presented in Table S4.
2.3. Candidate variant validation
The region of DMD including the variant, NC_058386.1: chromosome X: 27110467‐27 110 663 in F.catus_Fca126_mat1.0, was sequenced to confirm the stop gained variant uniquely identified in the case. The forward and reverse primer sequences used were 5′‐TGAAAATGGCCAGAAAGTCC‐3′ and 5′‐GTATCAATCGGCTCCGTACC‐3′, respectively. PCR was performed at an annealing temperature of 61°C. PCR amplicons were purified, and Sanger sequencing and analysis were performed as described previously. 21 An internal primer (5′‐CTGGCTTTTTTTTTTTTTTACC‐3′) was developed for the reverse sequencing after an intronic poly‐A region.
Identified variants were validated by producing a single PCR product of the expected size, 311 bp. The male control cat was hemizygous for the reference allele, whereas the female control cat was homozygous for the reference allele, and the affected male cat was hemizygous for the nonsense LoF variant (Figure 4).
FIGURE 4.
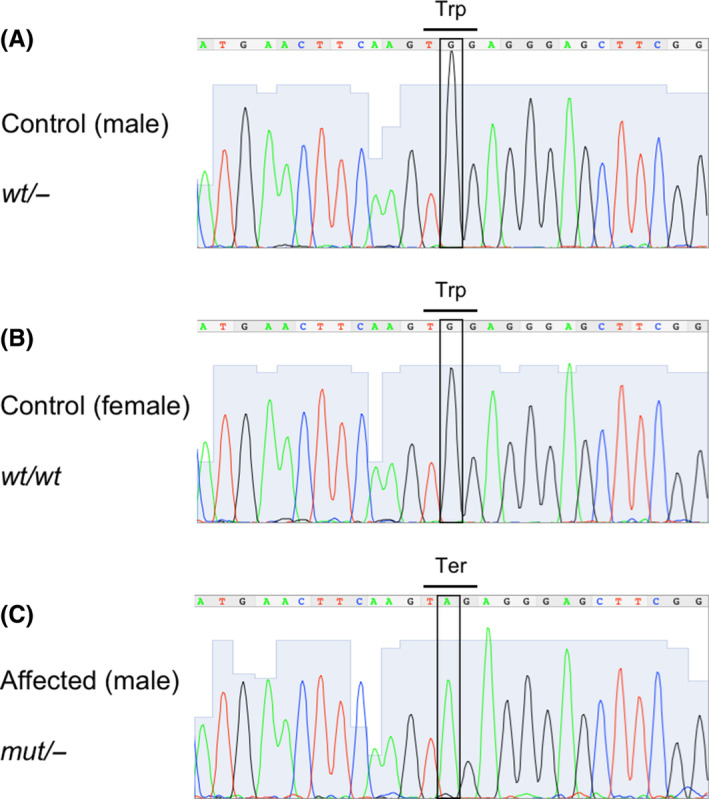
Sanger sequencing electropherograms for the DMD region presenting the identified nonsense genetic variant: (A) male control cat; (B) female control cat; (C) the cat of the present case. The boxes indicate either the reference alleles (A,B) or the variant allele (C). Because of the presence of the variant allele, Trp (TGG) is shown to be converted to Ter (TAG) in the affected cat (C), compared with control cats (A,B). wt/− or mut/− indicates hemizygote for the reference allele or the mutant allele (the nonsense genetic variant), respectively. wt/wt indicates homozygotes for the reference allele. mut, mutant; wt, wild type.
2.4. In silico analyses
Multispecies sequence alignment of the exon containing the variant was performed by CLC Sequence Viewer 8 (QIAGEN, Hilden, Germany) and included human, cat, dog, cow, pig, mouse, and zebrafish genomic sequences. That showed high sequence homology with the reference allele guanine (G) conserved across the 6 mammal species included in the analysis (Figure S2).
The cat DMD c.8333G > A; p.Trp2778Ter variant was analogous to the human p. Trp2786Ter (NM_004006.3[DMD]:c.8357G > A; ClinVar 22 , 23 Variation ID: 217214), which is located at NC_000023.11; X: 31507314 in the GRCh38.p14 human genome assembly and NC_000023.10; X: 31525431 in the GRCh37 human genome assembly, respectively. In humans, this truncating variant is considered pathogenic and has been observed in a DMD patient. 24 Similar to the cat variant, the human variant is also a guanine to adenine transition, changing the codon from TGG to TAG, which is an amber termination codon. This variant is not present in the Genome Aggregation Database (gnomAD). 25
The MutPred‐LOF web application 26 was used to evaluate the effect of the LoF variant in cats and humans. For MutPred‐LOF output, 3 score thresholds were suggested at different levels of the false‐positive rate (FPR): 0.40 (10% FPR), 0.50 (5% FPR), 0.70 (1% FPR). The predicted complete cat mRNA (XM_023249210.1) was compared with the predicted mutant mRNA with the stop termination Trp2778Ter. The predicted score from MutPred‐LOF was 0.4915 as compared with the amino acid change, p.Trp2786Ter, introduced into human DMD isoform X1 XP_006724531.1, which produced a score of 0.48435. Although scores >0.7 are considered the most deleterious with a 1% FPR by this prediction application, score thresholds of 0.40 with a 10% FPR and 0.5 with a 5% FPR were also suggested. Therefore, the MutPred‐LOF score of 0.4915 was considered pathogenic when a score threshold of 0.40 with a 10% FPR was used.
2.5. Outcome
The cat was prescribed oral prednisolone (1 mg/kg Q24h), which improved tongue swelling and breathing difficulty. Thereafter, prednisolone was continued at 0.5 mg/kg Q48h. At 10 years of age, the cat developed iatrogenic diabetes caused by the long‐term administration of prednisolone, which was well‐controlled with insulin treatment, thus maintaining sufficient quality of life. In the present case, clinical and histological findings, in addition to the presence of a unique hemizygous nonsense genetic variant in the DMD gene, suggested that it was consistent with FXMD. At the time of submission of this manuscript, the cat remains alive at 11.8 years of age.
3. DISCUSSION
X‐linked muscular dystrophy in cats, formerly known as hypertrophic feline muscular dystrophy, 6 is characterized by skeletal muscle hypertrophy in cats. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 X‐linked muscular dystrophy in cats is diagnosed based on clinical presentation, blood chemistry, EMG, histopathology, and immunohistochemistry. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 To date, genetic characterization has been performed in few cases. 14 , 15 , 16 The most prominent clinical features of cats with FXMD include symmetrical skeletal muscular hypertrophy, slow gait, and dyspnea, with some cats also showing megaesophagus concurrent with hypertrophy of the diaphragm and tongue, leading to feeding difficulties. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 15 Relevant increases in CK and AST activity are usually observed. Cases with juvenile‐onset often have a poor outcome, leading to disease‐related death 6 , 9 , 14 or humane euthanasia. 5 , 6 , 11 , 12 , 13 These cases often develop rhabdomyolysis when exposed to volatile anesthesia. 7 , 8 , 9
The current case presented with symmetrical muscle hypertrophy, slow gait, and occasional difficult breathing. Blood chemistry showed increased CK and AST activities. Additional CK isoenzyme analyses revealed an exclusive increase in serum CK‐MM activity (93%), reflecting leakage from mature or somewhat immature myofibers. 27 Furthermore, the onset of clinical signs occurred at 3 years of age (late‐onset), with a mild clinical course compared with previously reported cases. 5 , 6 , 8 , 9 , 11 , 12 , 13 , 14 The case did not present with megaesophagus associated with diaphragmatic hypertrophy. Despite presenting with tongue protrusion, tongue dysfunction leading to eating disorders was not observed. The EMG findings of this case were consistent with those of a previous report, 10 revealing frequent recordings of pseudomyotonic discharges and complex repetitive discharges with a “dive‐bomber” sound. Histopathological examination revealed muscle fiber necrosis and regeneration, which are characteristic of FXMD. 9 Unfortunately, comprehensive immunohistochemistry, including dystrophin, laminin alpha 2, and beta‐sarcoglycan, was not available to assess sarcolemmal integrity.
Tongue swelling and respiratory distress occurred occasionally and responded well to prednisolone treatment (1 mg/kg Q24h). Although the cat maintains a slow gait and low activity level, its overall quality of life is acceptable. In human medicine, prednisolone is recommended for DMD patients 28 ; however, its effectiveness in FXMD remains to be investigated. The present case showed an improvement of tongue swelling and respiratory distress after continuous oral administration of prednisolone, suggesting a beneficial effect of prednisolone in FXMD; however, further studies are needed to confirm this effect.
In cats, the BMD‐like phenotype, characterized by mild clinical signs, is associated with a missense variant. 15 Usually, the mRNA corresponding to the nonsense variant is completely deleted by nonsense‐mediated mRNA decay, resulting in the absence of dystrophin expression associated with severe phenotypes. 29 , 30 In humans, approximately 10% of DMD patients with a nonsense genetic variant exhibit mild BMD or intermediate between DMD and BMD phenotypes, caused by the expression of alternatively spliced transcripts, based on the disruption/creation of splicing regulatory elements. 31 Interestingly, a human DMD variant (NM_004006.3:c.8357G > A; p.Trp2786Ter; ClinVar Variation ID, 217214), which is analogous to the feline DMD genetic variant identified in this study, was reported in a DMD patient, and the variant was identified as a truncating variant by reverse transcription PCR, 24 which suggests that truncated dystrophin expression might also be present in this case. However, any analysis assessing how the DMD variants affected mRNA or protein was not performed in this case.
In humans, there is a known risk of rhabdomyolysis associated with volatile anesthesia in DMD patients, 17 which can be avoided by TIVA. 20 A similar effect has been reported in FXMD, 7 , 8 , 9 although the safety of TIVA has not yet been demonstrated in cats. Although no anesthesia‐related adverse events were observed in the present case, the cat had a mild clinical course. Therefore, the safety of TIVA for severe cases remains to be determined. We consider that volatile anesthesia should be avoided whenever FXMD is considered as a differential diagnosis based on clinical and laboratory findings.
The DMD nonsense variant (c.8333G > A; p.Trp2778Ter) was unique to this cat in the 99 Lives Cat Genome Sequencing Consortium database; thus, it can be considered rare. However, segregation analyses were not available, and it remains unknown if the variant was inherited or de novo. The short‐read sequencing had good coverage, and validity was confirmed by Sanger sequencing. High sequence homology was observed across multiple species. The MutPred‐LOF result supports that the genetic variant found in this study likely caused the cat's phenotype.
This study has several limitations. The pedigree of this case was unknown. Comprehensive immunohistochemistry for proteins, including dystrophin, laminin alpha 2, and beta‐sarcoglycan, was not performed in this case. No mRNA analysis and Western blotting was performed to assess how the DMD genetic variant identified in this case affected expression levels of mRNA and protein and expression of truncated mRNA and protein. Nevertheless, the nonsense genetic variant identified in the DMD gene was considered as the likely causative variant for this cat's phenotype. Population screening for the identified genetic variant was also not performed in this study, although the same variant was absent in other 413 cats of the 99 Lives Cat Genome Sequencing Consortium dataset.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
The work involved the use of non‐experimental animals only. Established internationally recognized high standards (“best practice”) of individual veterinary patient care were followed. Dr. Lyons has an exemption ACUC protocol 9178 at the University of Missouri for the receiving of samples for genomic applications.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Appendix S1. List of cats included in the 99 Lives Cat Genome Sequencing Consortium (domestic cat analysis comprising the whole‐genome sequencing data from 362 cats and whole‐exome sequencing data from 52 cats).
Figure S1. Inspiratory lateral (right‐side) thoracic X‐ray of the present case. No cardiac enlargement, diaphragmatic irregularity, or associated megaesophagus were observed. The cat presented with neck muscle hypertrophy (arrows).
Figure S2. (A) Multispecies sequence alignment of the exon containing the variant, including feline (Felis catus), human (Homo sapiens), canine (Canis lupus familiaris), bovine (Bos taurus), porcine (Sus scrofa), murine (Mus musculus), and zebrafish (Danio rerio), as well as the affected cat. The nonsense genetic variant found in this study (c.8333G > A in feline DMD) and the corresponding nucleotides of other species are surrounded by a black line and indicated by an arrow. The nucleotide G is conserved in all species' sequence alignment; (B) amino acids harboring the termination site (arrowhead). All mammals presented with relatively high amino acid conservation around the termination site.
Table S1. The numbers of variants identified in 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset (comprising the whole genome sequencing data from 362 cats and whole exome sequencing data from 52 cats), and unique variants identified in this case.
Table S2. Private homozygous/hemizygous genetic variants identified uniquely in the case within 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, including whole genome sequencing data from 362 cats and whole exome sequencing data from 52 cats.
Table S3. Private heterozygous genetic variants identified uniquely in the case within 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, including whole‐genome sequencing from 362 cats and whole‐exome sequencing data from 52 cats.
Table S4. List of all DMD genetic variants identified in the 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, comprising the whole‐genome sequencing data from 362 cats and whole‐exome sequencing data from 52 cats. The information on variants identified in the present case, such as Variant Allele Freq and Allelic Depths (AD), is also listed. Information pertaining to the nonsense genetic variant identified in this case (X:27361452) is shown in bold.
ACKNOWLEDGMENT
Funding was provided in part by the University of Missouri, College of Veterinary Medicine, Gilbreath McLorn Endowment, donations to the 99 Lives Cat Genome Sequencing project, and funding from the Winn Feline (EveryCat Health) Foundation and the George and Phyllis Miller (MT‐13‐010, MTW15‐017; MT18‐009; W16‐030; MTW18‐009; MT19‐001; LAL). The authors thank the 99 Lives Cat Genome Consortium (felinegenetics.missouri.edu) for sharing domestic cat variant frequency information. We appreciate the laboratory assistance of Thomas R. Juba, MS. A part of this case study was presented as an oral presentation at the annual congress of the Japanese Society of Veterinary Neurology in Fujisawa, Japan, June 2023.
Muto H, Yu Y, Chambers JK, et al. Association of a novel dystrophin (DMD) genetic nonsense variant in a cat with X‐linked muscular dystrophy with a mild clinical course. J Vet Intern Med. 2024;38(2):1160‐1166. doi: 10.1111/jvim.17024
REFERENCES
- 1. Monaco AP, Neve RL, Colletti‐Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323(6089):646‐650. [DOI] [PubMed] [Google Scholar]
- 2. Bushby K, Bourke J, Bullock R, Eagle M, Gibson M, Quinby J. The multidisciplinary management of Duchenne muscular dystrophy. Curr Paediatr. 2005;15(4):292‐300. [Google Scholar]
- 3. Broomfield J, Hill M, Guglieri M, Crowther M, Abrams K. Life expectancy in Duchenne muscular dystrophy: reproduced individual patient data meta‐analysis. Neurology. 2021;97(23):e2304‐e2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. England SB, Nicholson LVB, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343(6254):180‐182. [DOI] [PubMed] [Google Scholar]
- 5. Carpenter JL, Hoffman EP, Romanul FC, et al. Feline muscular dystrophy with dystrophin deficiency. Am J Pathol. 1989;135(5):909‐919. [PMC free article] [PubMed] [Google Scholar]
- 6. Gaschen FP, Hoffman EP, Gorospe JRM, et al. Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci. 1992;110(1–2):149‐159. [DOI] [PubMed] [Google Scholar]
- 7. Winand NJ, Edwards M, Pradhan D, Berian CA, Cooper BJ. Deletion of the dystrophin muscle promoter in feline muscular dystrophy. Neuromuscul Disord. 1994;4(5–6):433‐445. [DOI] [PubMed] [Google Scholar]
- 8. Gaschen F, Gaschen L, Seiler G, et al. Lethal peracute rhabdomyolysis associated with stress and general anesthesia in three dystrophin‐deficient cats. Vet Pathol. 1998;35(2):117‐123. [DOI] [PubMed] [Google Scholar]
- 9. Gaschen F, Burgunder JM. Changes of skeletal muscle in young dystrophin‐deficient cats: a morphological and morphometric study. Acta Neuropathol. 2001;101(6):591‐600. [DOI] [PubMed] [Google Scholar]
- 10. Howard J, Jaggy A, Busato A, Gaschen F. Electrodiagnostic evaluation in feline hypertrophic muscular dystrophy. Vet J. 2004;168(1):87‐92. [DOI] [PubMed] [Google Scholar]
- 11. van Soens I, Mols N, van Meervenne S, et al. A case of hypertrophic feline muscular dystrophy in a Belgian domestic shorthair cat. Vlaams Diergeneeskd Tijdschr. 2009;78(2):111‐116. [Google Scholar]
- 12. Gambino AN, Mouser PJ, Shelton GD, Winand NJ. Emergent presentation of a cat with dystrophin‐deficient muscular dystrophy. J Am Anim Hosp Assoc. 2014;50(2):130‐135. [DOI] [PubMed] [Google Scholar]
- 13. Eiras‐Diaz A, Prisco F, Paciello O, et al. Unusual clinical presentation of dystrophin‐deficient feline muscular dystrophy in the UK. Vet Rec Case Rep. 2020;8(1):e000983. [Google Scholar]
- 14. Beckers E, Cornelis I, Bhatti SFM, et al. A nonsense variant in the DMD gene causes X‐linked muscular dystrophy in the Maine coon cat. Animals. 2022;12(21):2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hilton S, Christen M, Bilzer T, Jagannathan V, Leeb T, Giger U. Dystrophin (DMD) missense variant in cats with Becker‐type muscular dystrophy. Int J Mol Sci. 2023;24(4):3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shelton GD, Tucciarone F, Guo LT, Coghill LM, Lyons LA. Precision medicine using whole genome sequencing identifies a novel dystrophin (DMD) variant for X‐linked muscular dystrophy in a cat. J Vet Intern Med. 2024;38(1):135‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller ED, Sanders DB, Rowlingson JC, Berry FA Jr, Sussman MD, Epstein RM. Anesthesia‐induced rhabdomyolysis in a patient with Duchenne's muscular dystrophy. Anesthesiology. 1978;48(2):146‐148. [DOI] [PubMed] [Google Scholar]
- 18. Smelt WLH. Cardiac arrest during desflurane anaesthesia in a patient with Duchenne's muscular dystrophy. Acta Anaesthesiol Scand. 2005;49(2):268‐269. [DOI] [PubMed] [Google Scholar]
- 19. Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109(4):1043‐1048. [DOI] [PubMed] [Google Scholar]
- 20. Muenster T, Mueller C, Forst J, Huber H, Schmitt HJ. Anaesthetic management in patients with Duchenne muscular dystrophy undergoing orthopaedic surgery: a review of 232 cases. Eur J Anaesthesiol. 2012;29(10):489‐494. [DOI] [PubMed] [Google Scholar]
- 21. Yu Y, Grahn RA, Lyons LA. Mocha tyrosinase variant: a new flavour of cat coat coloration. Anim Genet. 2019;50(2):182‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(D1):D980‐D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landrum MJ, Chitipiralla S, Brown GR, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835‐D844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whittock NV, Roberts RG, Mathew CG, Abbs SJ. Dystrophin point mutation screening using a multiplexed protein truncation test. Genet Test. 1997;1(2):115‐123. [DOI] [PubMed] [Google Scholar]
- 25. Chen S, Francioli LC, Goodrich JK, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625(7993):92‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pagel KA, Pejaver V, Lin GN, et al. When loss‐of‐function is loss of function: assessing mutational signatures and impact of loss‐of‐function genetic variants. Bioinformatics. 2017;33(14):i389‐i398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zweig MH, Adornato B, van Steirteghem AC, Engel WK. Serum creatine kinase BB and MM concentrations determined by radioimmunoassay in neuromuscular disorders. Ann Neurol. 1980;7(4):324‐328. [DOI] [PubMed] [Google Scholar]
- 28. Gloss D, Moxley RT, Ashwal S, Oskoui M. Practice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy: report of the guideline development Subcommittee of the American Academy of Neurology. Neurology. 2016;86(5):465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Brogna S, Wen J. Nonsense‐mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol. 2009;16(2):107‐113. [DOI] [PubMed] [Google Scholar]
- 30. Torella A, Zanobio M, Zeuli R, et al. The position of nonsense mutations can predict the phenotype severity: a survey on the DMD gene. PLOS One. 2020;15(8):e0237803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Juan‐Mateu J, González‐Quereda L, Rodríguez MJ, et al. Interplay between DMD point mutations and splicing signals in dystrophinopathy phenotypes. PloS One. 2013;8(3):e59916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. List of cats included in the 99 Lives Cat Genome Sequencing Consortium (domestic cat analysis comprising the whole‐genome sequencing data from 362 cats and whole‐exome sequencing data from 52 cats).
Figure S1. Inspiratory lateral (right‐side) thoracic X‐ray of the present case. No cardiac enlargement, diaphragmatic irregularity, or associated megaesophagus were observed. The cat presented with neck muscle hypertrophy (arrows).
Figure S2. (A) Multispecies sequence alignment of the exon containing the variant, including feline (Felis catus), human (Homo sapiens), canine (Canis lupus familiaris), bovine (Bos taurus), porcine (Sus scrofa), murine (Mus musculus), and zebrafish (Danio rerio), as well as the affected cat. The nonsense genetic variant found in this study (c.8333G > A in feline DMD) and the corresponding nucleotides of other species are surrounded by a black line and indicated by an arrow. The nucleotide G is conserved in all species' sequence alignment; (B) amino acids harboring the termination site (arrowhead). All mammals presented with relatively high amino acid conservation around the termination site.
Table S1. The numbers of variants identified in 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset (comprising the whole genome sequencing data from 362 cats and whole exome sequencing data from 52 cats), and unique variants identified in this case.
Table S2. Private homozygous/hemizygous genetic variants identified uniquely in the case within 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, including whole genome sequencing data from 362 cats and whole exome sequencing data from 52 cats.
Table S3. Private heterozygous genetic variants identified uniquely in the case within 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, including whole‐genome sequencing from 362 cats and whole‐exome sequencing data from 52 cats.
Table S4. List of all DMD genetic variants identified in the 414 cats of the 99 Lives Cat Genome Sequencing Consortium dataset, comprising the whole‐genome sequencing data from 362 cats and whole‐exome sequencing data from 52 cats. The information on variants identified in the present case, such as Variant Allele Freq and Allelic Depths (AD), is also listed. Information pertaining to the nonsense genetic variant identified in this case (X:27361452) is shown in bold.