

To the editor
During platelet activation, arachidonic acid (AA) is released from membrane phospholipids and metabolized to thromboxane A2 (TXA2) through the actions of cyclooxygenase-1 (COX-1) and TXA2 synthase. TXA2 binds to the platelet TXA2 receptor, causing shape change, secretion and platelet aggregation.1 COX-1 (599aa; 70kDa) has cyclooxygenase and peroxidase activities and it is functionally active as a homodimer, with each COX-1 monomer consisting of four highly conserved domains: an N-terminal signal peptide, a dimerization domain, a membrane-binding domain (MBD) and a large C-terminal catalytic domain2 (Figure 1A). Irreversible COX-1 inhibition by aspirin is a widely established anti-platelet therapy in cardiovascular disease.3
Figure 1. The novel PTGS1 variant p.Asn143Ser affecting the N-glycosylation residue 143 in COX-1, results in aspirin- like impaired platelet aggregation and expression of a hypo-glycosylated and dysfunctional COX-1 enzyme in human platelet and cell models.
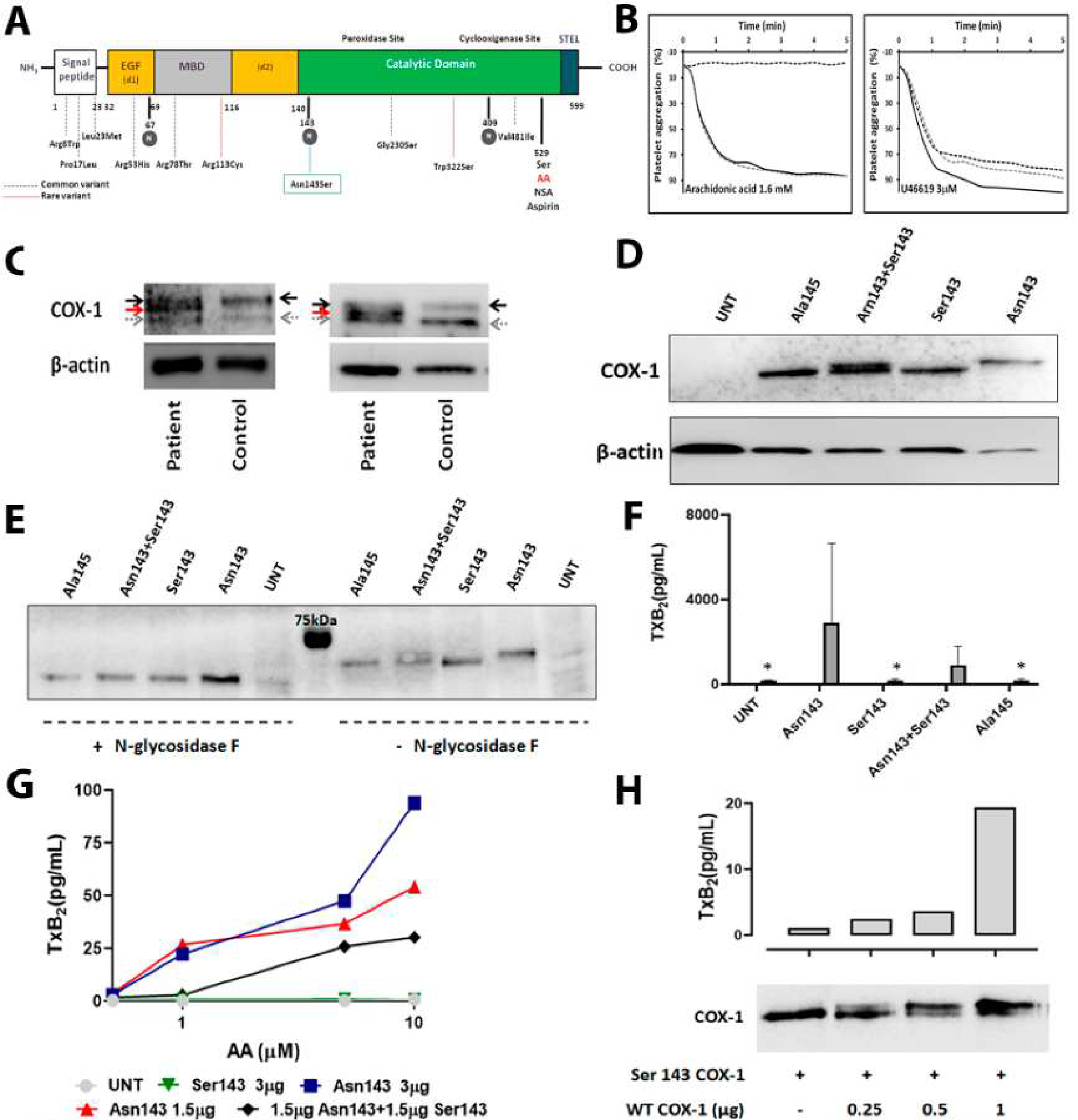
A) Schematic representation for COX-1 showing the different domains and the localization of the novel p.Asn143Ser genetic variant as well as all previously reported common (dotted blue lines) and rare (dotted red lines) variants. N-glycosylated residues are highlighted in grey circles. B) Platelet aggregation in response to arachidonic acid and U46619, a TxA2 analog, was evaluated in PRP from the proband (black dotted line) and from two healthy controls (grey dotted line and black line). C) Two independent western blots representing COX-1 in platelets from the proband and time-matched control. Platelet lysates were resolved in 8% SDS- polyacrylamide gel for 90 min. In addition to wild-type COX-1 (black arrow), a second COX-1 band with lower molecular weight (red arrow) was detected in the patient. Grey arrow identified a non-specific band. β-actin was used as an internal control. D) Western blot determination of COX-1 expression in 293 T HEK cells untransfected (UNT), transfected with wild-type Asn143 COX-1 vector (Asn143), mutant Ser143 COX-1 vector (Ser143), co-transfected with both vectors (Asn143+Ser143), or transfected with mutant Ala145 COX-1 vector (Ala145), β-actin was used as an internal control. E) Western blots of COX-1 expression in untransfected (UNT) or variant transfected 293 T HEK cells treated (+) or not (−) with N-glycosidase F. F) 293 T HEK cells were untransfected (UNT), transfected with wild-type Asn143 COX-1 vector (Asn143), mutant Ser143 COX-1 vector (Ser143), co-transfected with both vectors (Asn143+Ser143), or transfected with mutant Ala145 COX-1 vector (Ala145). The enzymatic activity of COX-1 was then assessed by using ELISA for TXB2 to assess the formation of TXA2 following incubation with 500 μM arachidonic acid. Plots show mean plus standard deviation from values obtained in three 3 independent experiments. ”*” denotes P value=0.05 vs. Asn143 (U de Mann-Whitney test). G) 293 T HEK cells untransfected (UNT), transfected with different amounts of wild-type Asn143 COX-1 vector, transfected with mutant Ser143 COX-1 vector, or co-transfected with the same amount of both vectors (Asn143+Ser143), were stimulated with different concentrations of AA (0.5 −10 μM) and the formation of TXA2 was assessed by using ELISA. The plot is representative of 2 independent experiments. H) HEK 293 cells were transfected with vector expressing Ser143 mutant COX-1 and the neomicine resistance gen and grown in the presence of geneticine. Resistant and stable transfected cell clones were isolated and then transfected with different amounts of vector (0–1 μg) expressing native Asn143 COX-1. Cells were washed and split for preparation of protein lysates and for arachidonic acid (1 μM) stimulation and TXA2 production measurement by ELISA. A double COX-1 protein is seen in co-transfected cells. Native COX-1 recovers TXA2 production, but following a non-linear dose-dependent pattern.
Despite the physiological and clinical relevance of platelet COX-1, few patients with congenital COX-1 defect (Bleeding Disorder Platelet Type 12; OMIM: 605735) have been characterized2. In the 1970’s some patients with mild bleeding diathesis, impaired aggregation and blunted TXA2 synthesis were described, but with no genetic confirmation. To date, only a few cases with uncommon genetic variants in PTGS1, the gene encoding COX-1, have been reported, without detailed study of their associated platelet phenotype. A patient with severe bleeding carried the PTGS1 single nucleotide polymorphisms (SNPs) p.Arg8Trp and p.Pro17Leu.2 Heterozygosity for the p.Pro17Leu SNP also aggravated hemorrhage in a pedigree with hemophilia A.2 Another patient showing bleeding and reduced TXA2 synthesis was heterozygous for the rare PTGS1 variant c.337C>T (p.Arg113Cys) and the common variant c.1003G>A (p.Val481Ile).2 Recently, the BRIDGE Consortium reported a pedigree with an autosomal recessive variant c.965G>C (p.Trp322Ser) that abrogated COX-1 expression resulting in aspirin-like platelet dysfunction.4
Here, we report a patient presenting with a lifelong mild bleeding tendency and platelet dysfunction, associated to heterozygosity for the novel PTGS1 variant c.428A>G (p.Asn143Ser). The distinguished feature of this mutation is that it disturbs one N-glycosylation sequon at the catalytic domain of COX-1 causing the expression of a dysfunctional hypo-glycosylated COX-1 protein with an apparent dominant-negative effect. The methodological details of this investigation are provided in Supplemental Material.
The proband was a 13-year-old teen from Asiatic origin, adopted by a Spanish family, presenting with a lifelong moderate bleeding diathesis (bruising and petechiae, being more frequent after minor traumas; recurrent epistaxis sometimes associated to nonsteroidal anti-inflammatory drug (NSAIDs) intake; menorrhagia, once requiring tranexamic acid and desmopressin treatment; excessive bleeding after surgical procedures such as tonsillectomy)(ISTH-BAT=6). On repeated testing, she showed normal blood cell counts including platelet number and volume (Supplemental Table 1) and no visible abnormalities in blood smears. Blood biochemical and coagulation parameters were within normal ranges. No clinical signs of relevant organ/tissue, immune or cognitive dysfunction were present. Detailed platelet phenotyping was performed that showed: i) PFA-100 closure times slightly and pathologically extended for collagen/ADP and collagen/epinephrine cartridges, respectively (Supplemental Table 1); ii) normal expression of major platelet adhesive receptors (Supplemental Figure 1A); iii) fibrinogen binding was reduced by about 50% in probandś platelets stimulated with several agonists but PAR4 (Supplemental Figure 1B); iv) impaired agonist-induced α- (10–30%) and δ- (25–50%) granule secretion, as evaluated by measurement of P-selectin and CD63 expression (Supplemental Figure 1C); v) light transmission aggregometry confirmed an aspirin-like platelet dysfunction in the proband, characterized by normal aggregation in response to PAR1p and ristocetin, and reduced aggregation with ADP and collagen (Supplemental Figure 1D). Moreover, the proband’s platelets showed absent AA-induced aggregation, but normal aggregation to U46619, a stable TXA2 analog, indicating unaffected signaling downstream the TXA2 receptor (Figure 1B); vi) markedly reduced platelet synthesis of TXA2 in the supernatant of the LTA experiments (<10% of the control levels) (Supplemental Figure 2A); and vii) reduced synthesis of all COX-1-dependent eicosanoids in whole blood stimulated with collagen or PAR1p (Supplemental Figure 2B)(Supplemental Table 2). Overall, this clinical and platelet phenotype resembles that of the few previously reported cases with COX-1 defect.2
Analysis of proband’s DNA with a HTS-gene panel,5 revealed an heterozygous variant c.428A>G (p.Asn143Ser) in the exon 5 of the PTGS1 gene (ENST00000362012), which was confirmed by Sanger sequencing. The proband’s biological parents were not available to assess the inheritance pattern of the variant. This variant is not reported in gnomAD or 1000 genomes project databases, but a c.429C>G [p.Asn143Lys] change is listed in the gnomAD collection with a very low allele frequency of 3.98×10−6. As highlighted in Figure 1A, the novel p.Asn143Ser substitution is located at the N-terminal of the COX-1 catalytic domain and affects a highly conserved residue (Genomic Evolutionary Rate Profiling conservation score=5.59; https://varsome.com/) (Supplemental Figure 3A). Remarkably, Asn143 is one of the three predicted N-glycosylation sites in COX-1 (Figure 1A). The recent and firstly resolved human COX-1 crystal by Scilimati A et al. (https://www.rcsb.org/structure/6Y3C) (Supplemental Figure 3B) confirmed that Asn143 is decorated by an N-glycan chain. Changing Asn to Ser would theoretically abolish this N-glycosylation resulting in an hypo-glycosylated protein with unknown consequences in folding, secretion, stability and enzymatic activity. Following the ACMG standards, the current verdict for the p.Asn143Ser variant is of Uncertain Significance, despite most computational analysis in silico predict a deleterious effect (https://varsome.com/) (Supplemental Table 3). Immunofluorescence analysis of platelets and leukocytes showed no apparent impairment in COX-1 expression in the proband (Supplemental Figure 4). However, 8% SDS–PAGE of platelet lysates and immunoblotting analysis revealed a second COX-1 protein band with lower molecular weight in the proband, which is consistent with the predicted hypo-glycosylated COX-1 variant (Figure 1C). In contrast to these findings, a recent study has showed that recessive inheritance of the 965G>C (p.Trp322Ser) missense variant in PTGS1, results in loss of COX-1 platelet expression instead of COX-1 loss-of function.4
Strong evidence of the deleterious effect of p.Ans143Ser variant in COX-1 glycosylation and its enzyme activity was obtained in HEK 293 T cells transfected with either wild-type (Asn143) or mutated (Ser143) enzyme vector (Supplemental Table 4). In this cell model, the variant Ser143 COX-1 displayed higher electrophoretic mobility than wild-type Asn143 COX-1, as was expected for a hypo-glycosylated enzyme with reduced molecular mass. Moreover, cells co-transfected with both Asn143 and Ser143 COX-1, simulating heterozygosis status, had a COX-1 double protein band similar to that observed in patientś platelet lysates (Figure 1D). Importantly, this COX-1 double band disappeared upon treatment of the lysates from co-transfected cells with N-glycosidase F (Figure 1E). As shown in Figure 1F, cells expressing wild-type COX-1 (Asn143) responded to 500 μM AA stimulation with a notable TXB2 production whilst cells transfected with mutant Ser143 COX-1 displayed almost no TXB2 synthesis (194±48.2 pg/mL), resembling untransfected cells (UNT) (169.0±24.8 pg/mL). Interestingly, cells co-transfected with equal amounts of wild-type (Asn143) and mutant Ser143 COX-1 (1.5 μg of each vector) showed 70% reduced TxB2 synthesis in comparison to cells transfected with 1.5 μg of wild-type COX-1 vector (Asn143+Ser143, 907.5±888.9 pg/mL vs. Asn143, 2906.9±3736.1) (Figure 1F). These results were confirmed in a separate series of experiments in which cells transfected with Asn143 COX-1 vector (1.5 or 3 μg) produced TXB2 in a dose-dependent manner when stimulated with increasing concentrations of AA (0.5–10 μμM), while in cells transfected with the mutant Ser143 this synthesis was abrogated. Remarkably, co-transfected cells (Asn143+Ser143) displayed higher than 50% reduction in TXB2 synthesis as compared to cells solely transfected with an equivalent dose of Asn143 COX-1 (Figure 1G). These data strongly support that the p.Ans143Ser variant has a dominant-negative effect in COX-1 activity. To further test this, we established a second model in which HEK 293 cells were first stably transfected with either mutant Ser143 or wild-type Asn143 COX-1, and then they were transiently transfected with the alternative construct. The TXB2 synthesis was abolished in cells stably expressing Ser143 COX1 and co-expression of wild-type Asn143 COX-1 partially recovered TXB2 production, but following a non-linear dose-dependent pattern (Figure 1H). Similar results were found in the opposite cell model (Supplemental Figure 5). Noteworthy, introduction in HEK 293 T cells of a different mutation, p.Ser145Ala, disrupting the same consensus N-glycosylation sequence (Asn-X-Ser/Thr) in COX-1, also resulted in null AA-induced TXA2 synthesis (Figure 1F) and in the expression of the hypo-glycosylated COX-1 protein also disappearing after treatment with N- glycosidase F (Figure 1D–E).
Overall, these results obtained in the patient platelets and in transfected cells reflected that the p.Asn143Ser variant has a dominant-negative effect and its deleterious action exceeded that of simply haploinsufficiency of functional COX-1 protein. Since COX-1 is active as a homodimer, we can speculate that a mechanism contributing for such a dominant negative effect is an impaired activity of both wild-type/mutant and mutant/mutant dimers. Indeed, dimerization of glycosylated and non-glycosylated monomers of COX-2, an isoform of COX-1, has been reported and this is more energetically favorable than the dimerization between two N-glycosylated monomers.6 Additionally, it may also be possible that the loss of N-glycans reduces the steric hindrance favoring the access of AA into the catalytic domain of the non-functional mutant COX-1, as suggested by the blunted TXA2 synthesis, at low AA concentration, in cells co-transfected with equivalent levels of mutant Ser143 COX-1 and wild-type COX-1. Of note, a similar dominant negative mechanism has been described for the London variant of antithrombin.7
To our knowledge, three N-glycosylation sequons are predicted in human COX-1 at positions 67, 143 and 409,8 and the recent resolution of a human COX-1 crystal structure by Scilimati et al. (PDB:6y3) provided the first experimental evidence that Asn143 is decorated by an N-glycan chain. Our results demonstrate, for the first time, that the N-glycosylation at 143 is functionally relevant as its disruption, by mutation of Asn143 or Ser145 residues, results in a smaller and dysfunctional COX-1 variant. In agreement with our results, Otto et al.9 has demonstrated that ovine COX-1 is N-glycosylated at positions Asn68, Asn144 and Asn410, the orthologous residues to the human ones, and removal of these N-glycans also abolishes the cyclooxygenase and peroxidase activity of hypo-glycosylated ovine COX-1 mutants.9
Mechanistically, glycans can influence protein folding, cellular trafficking and secretion, substrate binding affinity, proteolysis or clearance and the catalytic activity of proteins10. Indeed, the relevance of glycosylation in the function of proteins of the haemostatic system such as FXI,11,12 antithrombin,13 VWF10, protein S14 or the platelet P2Y12 ADP receptor,15 has been shown.
In summary, this work extends the genetic spectrum of COX-1 defects, by the characterization of a patient with a novel variant c.428A>G in PTGS1. Studies performed on the patient’s platelets and in cell models showed that this variant affects the post-transductional COX-1 N-glycosylation and leads to expression of a hypo-glycosylated protein with a dominant-negative effect in COX-1 enzymatic activity. COX-1 hypo-glycosylation is unveiled as a new dominant-negative mechanism causing congenital platelet function disorder. More broadly, our study strengthens the relevance of N-glycans in human health and disease.
Supplementary Material
Key Points.
A novel genetic variant, p.Asn143Ser, in COX-1 was identified in heterozygosity in a patient with lifelong moderate bleeding diathesis
This mutation disrupts N-Glycosylation of COX-1 and causes an aspirin-like platelet dysfunction with a dominant-negative effect
Funding and Acknowledgements
This work was partially supported by grants from Instituto de Salud Carlos III (ISCIII) & Feder (PI17/01311, PI20/00926, PI18/00598, PI17/01966 and CB15/00055), Fundación Séneca (19873/GERM/15), Gerencia Regional de Salud (GRS 2061A/19 and 1647/A/17), Fundación Mutua Madrileña (FMM, AP172142019) and Sociedad Española de Trombosis y Hemostasia (SETH-FETH; Premio López Borrasca 2019 and Ayuda a Grupos de Trabajo en Patología Hemorrágica). TDW research is supported by a grant from the British Heart Foundation (PG/17/40/33028). VPB has predoctoral contract from CIBERER. MEMB has a postdoctoral contract from University of Murcia. AMQ holds a predoctoral grant from the Junta de Castilla y León.
The corresponding author research on Inherited Platelet Disorders is conducted in accordance with the aims of the Functional and Molecular Characterization of Patients with Inherited Platelet Disorders Project, which is supported by the Hemorrhagic Diathesis Working Group of the Spanish Society of Thrombosis and Haemostasis (SETH).
Footnotes
Disclosure of Conflicts of Interest
The authors declare no conflict of interest. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
References
- 1.Rivera J, Lozano ML, Navarro-Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94(5):700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palma-Barqueros V, Bohdan N, Revilla N, Vicente V, Bastida JM, Rivera J. PTGS1 gene variations associated with bleeding and platelet dysfunction. Platelets. 2020:1–7. [DOI] [PubMed] [Google Scholar]
- 3.Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan MV, Hayman MA, Sivapalaratnam S, et al. Identification of a homozygous recessive variant in PTGS1 resulting in a congenital aspirin-like defect in platelet function. Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bastida JM, Lozano ML, Benito R, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103(1):148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vann-Victorino DDC, Cunanan J, Chen M, Chan R, Hall RW, Sevigny MB. Effect of glycosylation of cyclooxygenase-2 (COX-2) on homodimerization. The FASEB Journal. 2017;31(S1):lb79–lb79. [Google Scholar]
- 7.Raja SM, Chhablani N, Swanson R, et al. Deletion of P1 arginine in a novel antithrombin variant (antithrombin London) abolishes inhibitory activity but enhances heparin affinity and is associated with early onset thrombosis. J Biol Chem. 2003;278(16):13688–13695. [DOI] [PubMed] [Google Scholar]
- 8.Majed BH, Khalil RA. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev. 2012;64(3):540–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto JC, DeWitt DL, Smith WL. N-glycosylation of prostaglandin endoperoxide synthases-1 and −2 and their orientations in the endoplasmic reticulum. J Biol Chem. 1993;268(24):18234–18242. [PubMed] [Google Scholar]
- 10.Preston RJS, Rawley O, Gleeson EM, O’Donnell JS. Elucidating the role of carbohydrate determinants in regulating hemostasis: Insights and opportunities. Blood. 2013;121(19):3801–3810. [DOI] [PubMed] [Google Scholar]
- 11.McVey JH, Lal K, Imanaka Y, Kemball-Cook G, Bolton-Maggs PH, Tuddenham EG. Characterisation of blood coagulation factor XI T475I. Thromb Haemost. 2005;93(6):1082–1088. [DOI] [PubMed] [Google Scholar]
- 12.Linssen M, Mohamed M, Wevers RA, Lefeber DJ, Morava E. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab. 2013;109(1):107–111. [DOI] [PubMed] [Google Scholar]
- 13.de la Morena-Barrio ME, Martínez-Martínez I, de Cos C, et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absence of a SERPINC1 gene defect. Journal of Thrombosis and Haemostasis. 2016;14(8):1549–1560. [DOI] [PubMed] [Google Scholar]
- 14.Denis CV, Roberts SJ, Hackeng TM, Lenting PJ. In vivo clearance of human protein S in a mouse model: influence of C4b-binding protein and the Heerlen polymorphism. Arterioscler Thromb Vasc Biol. 2005;25(10):2209–2215. [DOI] [PubMed] [Google Scholar]
- 15.Zhong X, Kriz R, Seehra J, Kumar R. N-linked glycosylation of platelet P2Y12 ADP receptor is essential for signal transduction but not for ligand binding or cell surface expression. FEBS Letters. 2004;562(1–3):111–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.