

Abstract
Background
The incidence of syphilis continues to increase in the United States, yet little is known about Treponema pallidum genomic epidemiology within American metropolitan areas.
Methods
We performed whole-genome sequencing and tprK deep sequencing of 28 T. pallidum–containing specimens, collected mostly from remnant Aptima swab specimens from 24 individuals from Seattle Sexual Health Clinic during 2021–2022.
Results
All 12 individuals infected with Nichols-lineage strains were men who have sex with men, while a specific SS14 cluster (mean, 0.33 single-nucleotide variant) included 1 man who has sex with women and 5 women. All T. pallidum strains sequenced were azithromycin resistant via 23S ribosomal RNA A2058G mutation. Identical T. pallidum genomic sequences were found in pharyngeal and rectal swab specimens taken concurrently from the same individuals. The tprK sequences were less variable between patient-matched specimens and between epidemiologically linked clusters. We detected a 528–base pair deletion in the tprK donor site locus, eliminating 9 donor sites, in T. pallidum genomes of 3 individuals with secondary syphilis, associated with diminution of TprK diversity.
Conclusions
We developed an end-to-end workflow for public health genomic surveillance of T. pallidum from remnant Aptima swab specimens. tprK sequencing may assist in linking cases beyond routine T. pallidum genome sequencing. T. pallidum strains with deletions in tprK donor sites currently circulate and are associated with diminished TprK antigenic diversity.
Keywords: genomic epidemiology, immune evasion, syphilis, tprK, Treponema pallidum
Using a novel end-to-end workflow for genomic surveillance of Treponema pallidum from remnant Aptima swab specimens, we report universal genotypic azithromycin resistance and find circulating T. pallidum strains with deletions in tprK donor sites associated with diminished TprK antigenic diversity.
The incidence of syphilis in the United States has increased for the last 2 decades and remains a major public health concern [1]. Among all individuals, the incidence of primary and secondary syphilis in the United States was 16.2 cases per 100 000 persons in 2021, with higher incidence among vulnerable populations including men who have sex with men (MSM) and persons experiencing homelessness [1, 2]. Notably, despite routine screening during prenatal care, the incidence of congenital syphilis has risen more than 219% from 2017 to 2021, to 77.9 cases per 100 000 live births [1]. Improved diagnostics, public health surveillance including genomic epidemiology, and outreach to and education of vulnerable and general populations as well as medical providers, are all necessary tools to combat this epidemic.
Syphilis is caused by the spirochete bacterium Treponema pallidum subspecies pallidum. Few putative outer membrane proteins are encoded within its small (1.13-Mb) genome, and of those, most appear to be poorly and not consistently expressed on the bacterial surface throughout its life cycle [3–5], facilitating evasion of the adaptive immune system. An additional, and likely more important, immune evasion strategy used by T. pallidum involves continuous antigenic variation of 7 predicted surface-exposed variable (V) loop regions of the putative TprK outer membrane porin by nonreciprocal recombination of DNA sequences from donor sites flanking the tprD (tp0131) locus [6–10]. Recent studies in rabbits using a T. pallidum strain genetically modified to ablate 96% of donor sites demonstrated that the inability to vary the antigen composition of TprK leads to reduced V region diversity, attenuated disease manifestations, and decreased ability of T. pallidum to persist in the host, highlighting the importance of this virulence factor in pathogen persistence [11].
Despite an increase in global T. pallidum sequencing efforts over the past 5 years, significantly less genomic data are available for this bacterial pathogen compared with many others owing to the paucity of molecular testing and, to date, none of these sequencing efforts has described the genomic epidemiology focused on an American metropolitan area [12–16]. Here, using a novel workflow for public health genomics from mostly remnant Aptima swab specimens, we performed whole-genome sequencing (WGS) and tprK deep sequencing of 28 swab specimens from 24 individuals attending the Seattle Sexual Health Clinic during 2021–2022.
METHODS
Patient Specimens and T. pallidum Detection
Specimens came from 2 studies conducted in the Seattle King County Sexual Health Clinic. Both studies were approved by the University of Washington Institutional Review Board, and participants submitted written informed consent. The first study tested remnant rectal, pharyngeal, urine, vaginal, and serum specimens obtained from patients with a serological diagnosis of syphilis seeking clinic care in the clinic using an Aptima-based experimental transcription-mediated assay (TMA). The second study included swab specimens from anogenital lesions (if present) and oropharyngeal mucosa samples from patients with active syphilis. Samples were collected from approximately February 2021 to May 2022.
DNA was purified from 200-μL Aptima tube specimens using the MagNA Pure Total Nucleic Acid Isolation Kit I (Roche Applied Science) external lysis protocol on the automated MagNA Pure 24 Instrument and 50-µL elution. Swab specimens from non-TMA positive specimens were collected in 1 mL of 10 mmol/L Tris-hydrochloride, 0.1 mol/L ethylenediaminetetraacetic acid, and 0.5% sodium dodecyl sulfate, and extraction was performed with 200 µL of sample buffer using the QIAamp DNA Mini Kit (Qiagen) with a 100-µL elution.
Tp47 Quantification, T. pallidum Genome and tprK Library Generation, Donor Site Deletion Polymerase Chain Reaction, and Sequencing
Treponemal load quantification was performed using TaqMan quantitative (q) polymerase chain reaction (PCR) targeting the tp47 locus and multiplexed with human β-globin, as described elsewhere [17]. T. pallidum sequencing libraries were prepared using hybridization capture, as described elsewhere [15]. Both are described in greater detail along with tprK library generation and donor site PCR in the Supplementary Methods, Supplementary Table 2.
Genome Assembly
Fastq files were processed and genomes assembled using a custom pipeline (available at https://github.com/greninger-lab/T.Pallidum-Seattle). Briefly, paired-end reads were adapter and quality trimmed using Trimmomatic software (version 0.35) [18], kmer filtered to match T. pallidum with bbduk software (version 38.86) [19], and then mapped to the T. pallidum SS14 reference genome (NC_021508.1), using Bowtie2 software (version 2.4.1) [20], with default parameters followed by deduplication using Picard software (version 2.23.3). Separately, de novo assembly was performed with Unicycler software (version 0.4.4) [21], using reads filtered with bbduk software [19] to remove repetitive regions of the genome, including the repeat regions of the arp and TP0470 genes, as well as tprC/D/F/I and tprE/G/J loci. A hybrid fasta combining reference mapping and de novo sequences was generated, and deduplicated reads were remapped to this hybrid using default Bowtie2 settings, local misalignments corrected with Pilon software (version 1.23.0) [22], and a final Bowtie2 remapping to the Pilon consensus used as input to a custom R script to close gaps and generate a final consensus sequence. Raw data files and consensus genomes have been deposited to sequence read archive and GenBank (under BioProject PRJNA961304) (Supplementary Table 1).
Phylogenetic Analysis
Genomes masked at repetitive loci [15] were aligned, using MAFFT (version 7.490) and iqtree (version 2.0.3) software to generate a whole-genome maximum likelihood phylogeny, with 1000 ultrafast bootstraps and automated selection of the best substitution model.
tprK Bioinformatic Analysis
Paired-end tprK reads were adapter trimmed and merged in bbmerge [19] using maximum stringency and processed with our custom pipeline [16] to identify V regions (https://github.com/greninger-lab/T.Pallidum-Seattle). Sequences that resulted in frameshift or nonsense mutations were discarded. In-frame reads were randomly downsampled to an equivalent number of sequences (3471) per V region per replicate. Concordance between replicates was confirmed by determining that the Pearson correlation coefficient between replicates exceeded 0.85, and then reads per V region were averaged across both replicates. Sequences appearing only in a single replicate or below the sequencing error threshold (0.5%; 17 reads) were discarded. High-confidence short reads were then locally aligned, using the basic local alignment search tool (BLAST; version 2.13.0) [23], to the sample-matched donor site locus determined during WGS, using a word size 10 and 100% identity. The top BLAST hit for each V region was then annotated as belonging to 1 of 53 known donor sites [9].
Statistics and Visualization
All analyses were performed with R software (version 4.2.3), using the ggplot2 (version 3.4.2) [24] and ggtree (version 3.7.2) packages [25].
RESULTS
Descriptive Epidemiology of Sequenced T. pallidum Specimens
As part of a clinical study of T. pallidum testing in King County, we had available 85 oropharyngeal and rectal swab specimens in Aptima buffer positive for T. pallidum by Hologic real-time transcription mediated amplification [26, 27]. Of the 85 positive Aptima swab specimens, 13 specimens did not test positive by tp47 qPCR owing to low treponemal load. Among samples positive by both tp47 qPCR and TMA, comparison of the qPCR-derived treponemal loads with real-time TMA (rTMA) time revealed a robust negative correlation (r = −0.78), with successful genome recovery for specimens for which >600 genome copies were available for capture library preparation using a 17.5-µL input volume (Figure 1 and Supplementary Table 3).
Figure 1.
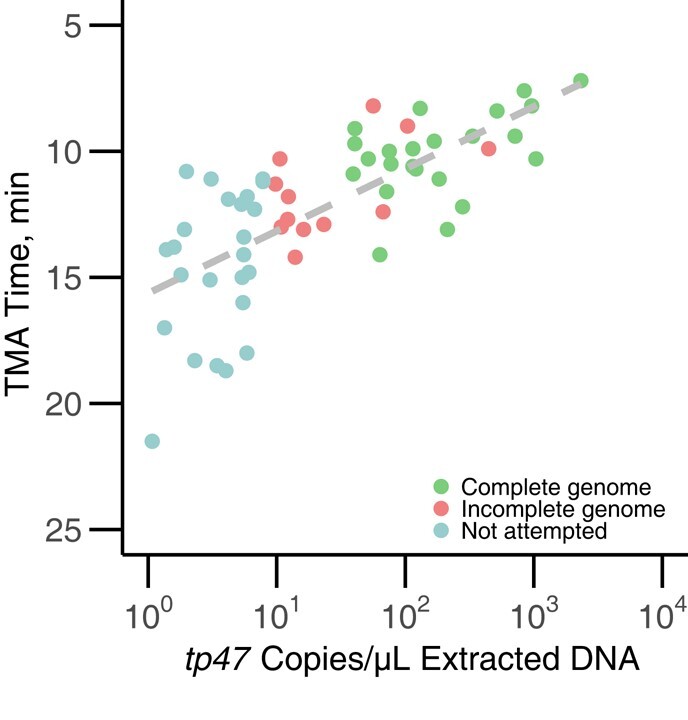
Relationship between Treponema pallidum genome copies in extracted DNA, rTMA time on Aptima assay, and ability to recover complete genome. T. pallidum genome copies, as determined by tp47 quantitative polymerase chain reaction, were highly correlated with rTMA time taken directly from the Hologic Aptima assay (r = −0.78). Complete genomes were defined as having <5.3% missing data in the genome (green), while incomplete genomes had >5.3% missing data (red). We did not attempt capture on specimens that had <10 copies/µL extracted DNA, based on prior experience (light blue). Abbreviation: TMA, transcription-mediated assay.
We initially attempted hybridization capture sequencing on 41 specimens with treponemal loads of >10 tp47 copies/µL extracted genomic DNA, consisting of DNA from 35 Aptima and 6 non-Aptima swab specimens. Separately, we attempted genome sequencing on 1 specimen at 3.9 copies/µL DNA (UW15993O). Twenty-eight near-complete T. pallidum genomes (<5.3% missing data) were recovered from 24 patients (Table 1). Of the patients, 75% were male, and 89% of male patients were MSM. Patients ranged in age from 20 to 64 years, with the most common age range 30–34 years (25% of patients). Eighty percent of patients had a clinical diagnosis of primary or secondary syphilis. None were human immunodeficiency virus positive, and only 2 patients had a prior history of syphilis. All patients with positive tp47 qPCR were also positive by treponemal test (Supplementary Table 4).
Table 1.
Demographic and Clinical Characteristics of 24 Patients From Whom a Treponema pallidum Genome was Recovered
| Characteristic | Patients, No. (%) |
|---|---|
| Biological sex | |
| Male | 18 (75) |
| Female | 6 (25) |
| MSM (among male patients only) | |
| Yes | 16 (89) |
| No | 2 (11) |
| Age category, y | |
| 20–24 | 3 (12) |
| 25–29 | 4 (17) |
| 30–34 | 6 (25) |
| 35–44 | 5 (21) |
| 45–54 | 4 (17) |
| 55–64 | 2 (8) |
| Race/ethnicity | |
| White | 12 (50) |
| Asian | 3 (12) |
| Black | 2 (8) |
| Hispanic | 5 (21) |
| NHPI | 1 (4) |
| Unknown | 1 (4) |
| Disease stage | |
| Primary | 10 (42) |
| Secondary | 9 (38) |
| Early latent | 2 (8) |
| Late or unknown duration | 3 (12) |
| Intravenous drug use in past year | |
| Yes | 6 (25) |
| No | 3 (12) |
| Unknown | 15 (62) |
| Methamphetamine use in past year | |
| Yes | 7 (29) |
| No | 11 (46) |
| Unknown | 6 (25) |
| Swab sample location (total N = 28 samples) | |
| Throat | 13 (46) |
| Rectal | 7 (25) |
| Other lesion | 4 (14) |
| Vaginal | 4 (14) |
Abbreviations: MSM, men who have sex with men; NHPI, Native Hawaiian and Pacific Islander.
Phylogenetic Clustering of T. pallidum Genomes Associated With Likely Transmission Chains
Phylogenetic analysis revealed that 15 genomes (representing 12 individuals) belonged to the Nichols lineage, while the remaining 13 genomes (12 individuals) were from the SS14 lineage (Figure 2 and Supplementary Table 3). All strains were azithromycin resistant via the A2058G mutation in the 23S ribosomal RNA locus. All 4 patient-matched sample pairs had identical genomes outside of tprK. Disease stage was significantly associated with lineage (P = .003; χ2 test). Nichols-lineage strains were more likely to be obtained from patients with secondary syphilis (n = 9; 89% Nichols vs 11% SS14), and SS14 lineage strains more likely to be obtained from patients with primary syphilis (n = 10; 20% Nichols vs 80% SS14).
Figure 2.

Maximum likelihood phylogeny of Treponema pallidum strains in Seattle from 2021 to 2022. Red nodes labeled with letters indicate high-confidence bootstrap values >0.9. Strains sequenced from paired pharyngeal (beginning with P or ending with O) and rectal (R) swab specimens obtained at the same time from 3 separate individuals are highlighted in pink, green, and yellow. Node C (P−22−20006 and UW15977O) consists of paired pharyngeal swab samples from the same individual on the same day and is highlighted in purple. Samples collected for study 1 begin with UW, and those collected for study 2 begin with P, R, or V. Reference sequences for cultured strains Nichols (NC_021490) and SS14 (NC_021508) are used for orientation. Abbreviations: AZ, azithromycin; MSM, men who have sex with men; NA, not applicable; SNVs, single-nucleotide polymorphisms.
Notably, we found that all 12 individuals with Nichols-lineage samples were MSM, consistent with data from Japan and our prior global sequencing efforts [15, 28], though contrasting with recent genetic surveillance in Amsterdam [29]. In contrast, only 4 of the 12 individuals with SS14 samples were MSM; the remainder were men who have sex with women (MSW) (n = 2) or women (n = 6) (P < .001; χ2 test).
Within the SS14 lineage, a cluster comprising 5 of the 6 female patients and 1 MSW (node F) separated by a median of 0 single-nucleotide variants (SNVs; mean, 0.33 SNV) is suggestive of local circulation of a single SS14-lineage strain among heterosexual individuals. Strains comprising node F were separated from the SS14 strains derived from MSM (R-21-10091, R-21-20052, P-21-20116, and UW16057L) by an average of 10.2 SNVs (median, 11 SNVs; range, 2-16) and from the other female-derived strain (P-21-20120) by 14 or 15 SNVs, which itself was separated from the 4 MSM strains by a median of 11.5 SNVs (range, 7–20). The 4 MSM strains differed each other by a median of 11 SNVs (range, 8–21). However, low diversity and/or undersampling limits the interpretability of phylogenetic and epidemiologic relationships between the node F cluster and the other SS14 strains derived from MSM.
Within the Nichols lineage, very limited diversity was seen, with a median of 2 SNVs (mean, 6.3 SNVs) (Supplementary Table 4 and 5) separating strains within node B (range, 0–26 SNVs). Interestingly, strains comprising node D (R-22-10139 and P-22-20198) taken from the same individual were more distantly related to other Nichols genomes sequenced here (mean, 140.5 SNVs), and were most closely related to Nichols subclades C and D [15], though they would likely constitute a separate Nichols subclade based on genetic distance.
Correlated tprK Sequences Associated With Likely Transmission Chains
We next performed deep sequencing of the tprK gene to examine V region composition and donor site usage by strain. We were particularly interested in how similar the pattern of use of V region sequences were within epidemiologically linked local clusters and in patients with samples obtained from different sites at the same time. We first examined potential confounders and found no relationship between the number of unique high-confidence V regions per sample and the number of T. pallidum genome copies input to the tprK library prep replicates (Pearson coefficient r = 0.204; P = .27) (Supplementary Figure 1A). There was also no difference in the number of unique V regions by lineage or clinical stage (P > .05; Welch's t test) (Supplementary Figure 1B), despite previous studies of full-length haplotypes showing increased diversity of tprK in secondary samples [9, 30]. Furthermore, we did not see associations between the number of unique tprK V regions and patient race, biological sex, sexual preference, age, swab sample location, intravenous drug use, or methamphetamine use.
We hypothesized that epidemiologically linked T. pallidum strains would have a more similar V region composition than distantly related strains. Because traditional phylogenetic methods reconstruct relationships based on single SNV evolution, they are not suitable to compare sequences from tprK, which changes its 7 variable regions via recombination of up to 10s of bases at a time. Instead, we calculated the correlation between the proportions of tprK V region sequences between sample pairs, epidemiologically linked node F, and unlinked samples in the same or opposite lineage. As expected, we found high (median, 0.93) correlation coefficients between paired samples from the same individual (Figure 3A and 3B), including a matched sample (UW15993O) from a tprK donor site deletion strain (UW15993L) for which a whole genome could not be recovered. Although lower than for paired samples, we found that the likely heterosexual cluster defined by node F (Figure 2) had significantly higher pairwise Pearson coefficients than between more distantly related samples either in the same or the opposite lineage (P < .001; 1-way analysis of variance with the Tukey honestly significant difference test). This is strongly suggestive of close relationships among samples in the cluster, though the background rate of tprK gene conversion in human syphilis infection is unknown.
Figure 3.

tprK V region sequence proportion correlations from strains in this study. A, tprK V region sequences and proportions were compared pairwise, and Pearson correlation coefficients were determined. More similar patterns of V region use result in higher coefficients. The outline of the phylogeny from Figure 2 (slightly modified to include only strains yielding tprK sequence data) is shown for orientation. Patient-matched samples are shown in the same color. B, Pearson coefficients are significantly different among patient-matched, node F cluster–associated, and within the same or opposite lineages. *P < .05; ***P < .001 (1-way analysis of variance with the Tukey honestly significant difference test).
Identification of a Large Deletion in tprK Donor Site Locus Associated With Reduced tprK Diversity
Intriguingly, 3 Nichols specimens (P-21-20167, P-22-20168 and UW15993L; Figure 2) contained a 528–base pair (bp) deletion, bounded by a 16-bp direct repeat, within the donor site locus for tprK, resulting in loss of 9 of the 53 known tprK donor sites (Figure 4). The deletion was confirmed by PCR band size (Supplementary Figure 2) and Sanger sequencing. There were no differences in the T. pallidum genome sequence between the 3 samples, strongly suggestive of transmission of a single deletion-bearing strain rather than independent deletion events. Although experimental elimination of 96% of donor sites results in reduced tprK diversity, severe attenuation of early lesions, and diminished ability to persist in the rabbit host [11], ablation of only 9 donor sites did not appear to prevent lesion appearance or prevent transmission in humans as these 3 patients had clinically evident secondary syphilis.
Figure 4.

Nichols-lineage strain with a 528–base pair (bp) deletion in tprK donor site locus. Sequencing read depth is depicted across the tprK donor site locus for 3 T. pallidum strains sequenced in this study, using SS14 genome reference NC_021508.1. The 528-bp deletion that removes 9 donor sites is denoted by red dotted lines in strain P-22-20168. A 51-bp deletion present in all sequenced Nichols strains is depicted in strain P-21-20135. SS14-lineage strains have neither deletion, as indicated by sequencing depth for strain UW15970L.
We next examined V region composition between strains with or without the tprK donor site deletion. Owing to the potential confounding effects of lineage and stage on the analysis, we compared the total V region diversity (number of unique V regions recovered) from deletion strains only to the diversity in other Nichols secondary syphilis samples. Strains harboring the tprK donor site deletion had significantly reduced diversity (P < .05; Welch's t test) (Figure 5A), consistent with the phenotype seen in rabbits infected with T. pallidum with tprK donor sites knocked out [11].
Figure 5.

tprK variable region composition and donor site usage in Treponema pallidum strains with donor site deletion. A, Among Nichols-lineage strains isolated from secondary syphilis lesions, diversity, as measured by the total number of unique V regions detected, is lower in strains bearing a deletion of 9 donor sites (P < .05; Welch's t test). B, As a proportion of total V regions, tprK V regions that incorporate the deleted donor sites are less abundant in strains with a deletion of 9 donor sites. C, Map of predicted donor site usage by strain. Donor sites are arranged relative to genomic coordinates; the x-axis is not to scale. Seven donor sites not found in any strain are excluded from the visualization. Green squares indicate the number of tprK V region sequences in which each donor site is found. Gray squares indicate the sum of use of each donor site across all samples, with darker gray indicating higher frequency. The deleted region is boxed in red in the affected strains.
We hypothesized that tprK V regions recovered from deletion strains would have fewer sequence fragments deriving from the deleted donor sites owing to their not being available to generate new variants following adaptive immune-mediated elimination of sequence remnants in place at the time of the deletion. To determine whether deletion of the tprK donor sites had an effect on the usage of those donor sites within each tprK, we performed a BLAST search using the tprK donor site locus as the subject and each tprK V region as the query. The top hit for each tprK sequence variant was retained, and the pattern of donor site use among samples was calculated. We found that in strains harboring the donor site deletion, the number of unique V regions incorporating the eliminated 9 donor sites was significantly lower than in other strains (P < .05; Welch's t test). (Figure 5B) Indeed, only 1 of the 9 deleted donor sites is present in tprK sequences from any of 4 strains carrying the deletion (Figure 5C).
DISCUSSION
T. pallidum genomic surveillance in the United States has been significantly limited by the availability of molecular testing for diagnosis and relatively low treponemal loads in many patient samples collected for molecular analysis [12]. Here, we offer a preview of a robust end-to-end platform for T. pallidum public health genomic surveillance using mostly remnant Aptima swab specimens [26, 27]. Our analysis demonstrates the feasibility of using specimens from TMA media for WGS, the correlation between TMA time and qPCR measures of treponemal load, differences in the predominant strains circulating in MSM and MSW/women with syphilis, the ubiquity of azithromycin resistance among circulating T. pallidum strains despite no azithromycin being prescribed by clinicians at the Seattle Sexual Health Clinic, and the existence of a large deletion in tprK donor site locus associated with reduced tprK diversity.
Our study sheds light on the nature of the syphilis epidemic in Seattle during 2021–2022. In particular, we noted some unique clustering of T. pallidum strains by MSM and MSW/women, with 1 tight MSW/woman-associated SS14 lineage cluster separate from the MSM-associated SS14 lineage and a separate tight MSM-associated Nichols-lineage cluster. At the same time, there were a number of unique T. pallidum strains in both SS14 and Nichols lineages that had >10 SNVs to phylogenetically closest strain, suggesting greater diversity in the community beyond these 2 clusters.
A large Australian study found an enrichment of MSW and women in a particular SS14 subclade [12]. Previous studies of broader geographic regions have seen some enrichment of MSM in Nichols lineage [15, 28], although another study conducted in the Amsterdam area [29] did not confirm this trend. This may be due in part to differences between localities in the populations in which the different lineages circulate, rather than any biological reason. Importantly, whole-genome analysis of cultured isolates collected in Seattle between 2001 and 2011 revealed only 2 of 60 (3.3%) belonging to Nichols lineage, with the remainder belonging to the SS14 lineage [13].
Although it is possible that differences in populations sampled during the study periods are driving the shift toward an increased proportion of Nichols-lineage strains, the possibility of a large outbreak of Nichols-lineage syphilis and population expansion among MSM in Seattle is consistent with the very low diversity (median, 2 SNVs) among node B Nichols strains included in this study and should be explored further. More work and finer sampling is required to understand the degree to which transmission occurs between MSM and MSW/women.
Our data support the use of oropharyngeal or rectal swab specimens for T. pallidum genome recovery, based on the identical genomes recovered. This work extends T. pallidum–multilocus sequence typing (MLST) data reported in 2022 that found identical MLST types among pharyngeal, urogenital, and/or anal swab specimens contemporaneously collected from 12 individuals [31]. Prior work also demonstrated a lack of SNVs within the recoverable portion (54%) of the genome in T. pallidum subspecies pertenue in banked skin lesion swab specimens obtained 2 years apart, consistent with the low intrahost diversity of T. pallidum genomes seen here [32].
Previous studies have shown no or few tprK haplotypes shared among cross-sectional patient samples, gradual accrual of tprK V region diversity in immunocompromised hosts and cultured isolates, as well as the requirement for tprK donor sites to create this antigenic diversity [7–9, 28]. Here, we showed that tprK sequence is correlated with T. pallidum genome relatedness while also serving as a more discriminating locus than the rest of the T. pallidum genome. Given the slow evolutionary rate of the T. pallidum genome and faster rate of tprK gene conversion, it may be worthwhile to include tprK deep sequencing data in public health genomic investigations to achieve greater temporal resolution of T. pallidum relatedness. Greater tprK diversity is associated with secondary syphilis cases [9, 30], and tprK diversity may be indicative of the time elapsed since infection and helpful for public health investigations. However, correlation with more granular clinical history and epidemiological information is required.
By combining T. pallidum genomics with tprK profiling, we showed contemporaneous circulation of a T. pallidum strain with a significant deletion in the tprK donor site locus that was associated diminished tprK antigen diversity. Such deletions are relatively rare, as searching of National Center for Biotechnology Information (NCBI) GenBank recovered 1 additional MexicoA-like T. pallidum strain (MD18Be; NCBI GenBank accession no. CP073487.2) with a unique 508-bp deletion in the tprK donor site locus removing 6 donor sites and creating a novel V7 chimeric donor site [15]. Intriguingly, the tprK gene in 3 of the 4 isolates with this donor site deletion contained a V1 region comprised entirely of the deleted V1-DS15 donor site. The V1 variable region is enriched in variant sequences comprising a complete donor site sequence, and our best interpretation is that this V1 sequence was likely present when the tprK donor site locus deletion occurred and had been recombined in only 1 of the 4 specimens [9]. Certainly, more work is required to understand the mechanism of variable region creation both in vitro and in vivo [10, 11, 33].
Our study was limited by the limited clinical history and lack of known transmission partners to train our T. pallidum and tprK genomic data. Limited clinical history also curtails our current ability to validate tprK diversity as a measure of duration of infection. The small sample set does not capture the full diversity of sexual behaviors and racial diversity of Seattle. Treponemal genome recovery is limited by the relatively low treponemal loads seen in human specimens, as illustrated by the recovery of complete genomes from fewer than one-third of the T. pallidum–positive swab specimens here, though partial genomes could be recovered from almost one-half of specimens.
We offer a specific molecular testing to T. pallidum genome analysis workflow amenable to public health genomic epidemiology for a pathogen with rapidly increasing incidence in the United States and elsewhere. Our data highlight the potential for T. pallidum genomics to inform public health epidemiology, as well as the ability of public health genomics to recover variants that bulwark our scientific understanding of this mercurial pathogen.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
Contributor Information
Nicole A P Lieberman, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Carlos C Avendaño, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Shah A K Mohamed Bakhash, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Ethan Nunley, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Hong Xie, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Lorenzo Giacani, Department of Medicine, University of Washington Medical Center, Seattle, Washington, USA; Department of Global Health, University of Washington, Seattle, Washington, USA.
Anna Berzkalns, Public Health—Seattle & King County HIV/STD Program, Seattle, Washington, USA.
Olusegun O Soge, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA; Department of Medicine, University of Washington Medical Center, Seattle, Washington, USA; Department of Global Health, University of Washington, Seattle, Washington, USA; Center for AIDS and STD, University of Washington, Seattle, Washington, USA.
Tara B Reid, Department of Medicine, University of Washington Medical Center, Seattle, Washington, USA.
Matthew R Golden, Department of Medicine, University of Washington Medical Center, Seattle, Washington, USA; Public Health—Seattle & King County HIV/STD Program, Seattle, Washington, USA; Department of Epidemiology, University of Washington, Seattle, Washington, USA; Center for AIDS and STD, University of Washington, Seattle, Washington, USA.
Alexander L Greninger, Department of Laboratory Medicine, University of Washington Medical Center, Seattle, Washington, USA; Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, Washington, USA.
Notes
Acknowledgments. The authors thank Sheila Lukehart for helpful comments.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grants R01 AI155442 to M. R. G. and U19 AI144133 to L. G. and A. L. G. Funding to pay the Open Access publication charges for this article was provided by departmental funds.
References
- 1. Centers for Disease Control and Prevention. National overview of STDs, 2021. https://www.cdc.gov/std/statistics/2021/overview.htm. Accessed 11 July 2023.
- 2. De Voux A. State-specific rates of primary and secondary syphilis among men who have sex with men—United States, 2015. MMWR Morb Mortal Wkly Rep 2017; 66:349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radolf JD, Kumar S. The Treponema pallidum outer membrane. Curr Top Microbiol Immunol 2018; 415:1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giacani L, Lukehart S, Centurion-Lara A. Length of guanosine homopolymeric repeats modulates promoter activity of subfamily II tpr genes of Treponema pallidum ssp. pallidum. FEMS Immunol Med Microbiol 2007; 51:289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Lay BD, Cameron TA, De Lay NR, Norris SJ, Edmondson DG. Comparison of transcriptional profiles of Treponema pallidum during experimental infection of rabbits and in vitro culture: highly similar, yet different. PloS Pathog 2021; 17:e1009949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. LaFond RE, Molini BJ, Van Voorhis WC, Lukehart SA. Antigenic variation of TprK V regions abrogates specific antibody binding in syphilis. Infect Immun 2006; 74:6244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Centurion-Lara A, Godornes C, Castro C, Van Voorhis WC, Lukehart SA. The tprK gene is heterogeneous among Treponema pallidum strains and has multiple alleles. Infect Immun 2000; 68:824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stamm LV, Bergen HL. The sequence-variable, single-copy tprK gene of Treponema pallidum Nichols strain UNC and street strain 14 encodes heterogeneous TprK proteins. Infect Immun 2000; 68:6482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Addetia A, Lin MJ, Phung Q, et al. Estimation of full-length TprK diversity in Treponema pallidum subsp. pallidum. mBio 2020; 11:e02726-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin MJ, Haynes AM, Addetia A, et al. Longitudinal TprK profiling of in vivo and in vitro-propagated Treponema pallidum subsp. pallidum reveals accumulation of antigenic variants in absence of immune pressure. PloS Negl Trop Dis 2021; 15:e0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Romeis E, Lieberman NAP, Molini B, et al. Treponema pallidum subsp. pallidum with an artificially impaired TprK antigenic variation system is attenuated in the rabbit model of syphilis. PloS Pathog 2023; 19:e1011259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taouk ML, Taiaroa G, Pasricha S, et al. Characterisation of Treponema pallidum lineages within the contemporary syphilis outbreak in Australia: a genomic epidemiological analysis. Lancet Microbe 2022; 3:e417–26. [DOI] [PubMed] [Google Scholar]
- 13. Beale MA, Marks M, Sahi SK, et al. Genomic epidemiology of syphilis reveals independent emergence of macrolide resistance across multiple circulating lineages. Nat Commun 2019; 10:3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beale MA, Marks M, Cole MJ, et al. Global phylogeny of Treponema pallidum lineages reveals recent expansion and spread of contemporary syphilis. Nat Microbiol 2021; 6:1549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lieberman NAP, Lin MJ, Xie H, et al. Treponema pallidum genome sequencing from six continents reveals variability in vaccine candidate genes and dominance of Nichols clade strains in Madagascar. PloS Negl Trop Dis 2021; 15:e0010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Addetia A, Tantalo LC, Lin MJ, et al. Comparative genomics and full-length Tprk profiling of Treponema pallidum subsp. pallidum reinfection. PloS Negl Trop Dis 2020; 14:e0007921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lieberman NAP, Armstrong TD, Chung B, et al. High-throughput nanopore sequencing of Treponema pallidum tandem repeat genes arp and tp0470 reveals clade-specific patterns and recapitulates global whole genome phylogeny. Front Microbiol 2022; 13:1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bushnell B. BBMap: a fast, accurate, splice-aware aligner. 2014. https://escholarship.org/uc/item/1h3515gn. Accessed 11 July 2023.
- 20. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput Biol 2017; 13:e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 2014; 9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wickham H. Ggplot2: elegant graphics for data analysis. New York, NY: Springer-Verlag, 2016. https://ggplot2.tidyverse.org. Accessed 11 July 2023. [Google Scholar]
- 25. Yu G. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinforma 2020; 69:e96. [DOI] [PubMed] [Google Scholar]
- 26. Golden M, O’Donnell M, Lukehart S, et al. Treponema pallidum nucleic acid amplification testing to augment syphilis screening among men who have sex with men. J Clin Microbiol 2019; 57:e00572-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Getman D, Lin M, Barakat N, et al. Analytical performance characteristics of a new transcription-mediated amplification assay for Treponema pallidum. J Clin Microbiol 2021; 59:e0051121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishiki S, Lee K, Kanai M, Nakayama S, Ohnishi M. Phylogenetic and genetic characterization of Treponema pallidum strains from syphilis patients in Japan by whole-genome sequence analysis from global perspectives. Sci Rep 2021; 11:3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zondag HCA, Zwezerijnen-Jiwa FH, de Vries HJC, De Baetselier I, Bruisten SM. Treponema pallidum strains among women and men who have sex with women in Amsterdam, the Netherlands and Antwerp, Belgium between 2014–2020. Sex Transm Dis 2023; 50:e5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu D, Liu LL, Zheng X-Q, et al. Genetic profiling of the full-length tprK gene in patients with primary and secondary syphilis. Microbiol Spectr 2023; 11:e0493122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zondag HCA, Nieuwenburg SA, Himschoot M, et al. Treponema pallidum subspecies pallidum intrapatient homogeneity at various body locations in men with infectious syphilis. Microbiol Spectr 2022; 10:e0248221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beale MA, Noguera-Julian M, Godornes C, et al. Yaws re-emergence and bacterial drug resistance selection after mass administration of azithromycin: a genomic epidemiology investigation. Lancet Microbe 2020; 1:e263–71. [DOI] [PubMed] [Google Scholar]
- 33. Romeis E, Tantalo L, Lieberman N, Phung Q, Greninger A, Giacani L. Genetic engineering of Treponema pallidum subsp. pallidum, the syphilis spirochete. PloS Pathog 2021; 17:e1009612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.