

Abstract
Optogenetics is a widely used technology with potential for translational research. A critical component of such applications is the ability to track the location of the transduced opsin in vivo. To address this problem, we engineered an excitatory opsin, ChRERα (hChR2(134R)-V5-ERα-LBD), that could be visualized using positron emission tomography (PET) imaging in a noninvasive, longitudinal, and quantitative manner. ChRERα consists of the prototypical excitatory opsin channelrhodopsin-2 (ChR2) and the ligand-binding domain (LBD) of the human estrogen receptor α (ERα). ChRERα showed conserved ChR2 functionality and high affinity for [18F]16α-fluoroestradiol (FES), an FDA-approved PET radiopharmaceutical. Experiments in rats demonstrated that adeno-associated virus (AAV)–mediated expression of ChRERα enables neural circuit manipulation in vivo and that ChRERα expression could be monitored using FES-PET imaging. In vivo experiments in nonhuman primates (NHPs) confirmed that ChRERα expression could be monitored at the site of AAV injection in the primary motor cortex and in long-range neuronal terminals for up to 80 weeks. The anatomical connectivity map of the primary motor cortex identified by FES-PET imaging of ChRERα expression overlapped with a functional connectivity map identified using resting state fMRI in a separate cohort of NHPs. Overall, our results demonstrate that ChRERα expression can be mapped longitudinally in the mammalian brain using FES-PET imaging and can be used for neural circuit modulation in vivo.
INTRODUCTION
Optogenetics is a widely used technology that enables cell type–specific manipulations of neuronal activity with millisecond precision (1–3). In addition to utility for preclinical neuroscience, optogenetics has a strong clinical potential (4–7) as evidenced recently by the partial recovery of visual function in a blind patient using optogenetic-based retinal therapy (8).
The vast majority of preclinical optogenetic experiments to date have used rodents and contributed valuable insights to the fields of behavioral neuroscience, neurology, and psychiatry (9). Nevertheless, findings from such studies would be further strengthened if performed in nonhuman primates (NHPs) that share strong neuroanatomical similarities to humans and can perform complex, human-like behaviors and tasks (10, 11). NHP research is essential for understanding relationships between nervous system abnormalities and maladaptive behaviors and for the development of brain-targeted human therapies (12, 13). Notwithstanding, NHP research has stronger ethical and economic constraints than rodent research. Such factors hinder feasibility of applications such as optogenetics in NHPs because of the need for postmortem validation of transgene expression that requires high numbers of animals. Therefore, transduction vectors cannot be easily optimized in NHPs, and long lags often occur between in vivo experimentation and postmortem validation of transgene expression.
Although progress in NHP optogenetics has been made in recent years (14), a variety of practical obstacles preclude widespread adoption of such applications (15–18). One critical factor limiting NHP optogenetic applications is the inability to monitor the anatomical distribution of opsin expression in a quantitative, noninvasive, and longitudinal manner. Except for NHP optogenetic studies performed in superficial brain regions (via a cranial window), current NHP optogenetics is mostly limited to targeting optical fibers to opsins expressed at the virus injection site. The main reason for this is that one cannot effectively predict opsin expression at distal terminal projection sites in deep brain regions, which is necessary for achieving pathway-specific optogenetic neuromodulation. In this way, pathway-specific optogenetic applications in NHPs are hindered by the need to rely on indirect anatomical knowledge (brain atlases) and do not account for intersubject anatomical variability or subject-specific transgene expression. Last, the inability to monitor opsin expression is a barrier to potential clinical applications where treatment plans and patient outcomes would benefit from noninvasive, longitudinal tracking of the transduced construct in the human body.
The molecular imaging capabilities of positron emission tomography (PET) can address the above challenges of in vivo opsin localization. The use of selective PET radiotracers has enabled noninvasive localization and quantification of several exogenous proteins introduced by viral transduction in rodents, NHPs, and humans (19–23). Furthermore, PET affords the highest detection sensitivity of any molecular imaging modality, and recent breakthroughs in PET detector technology have considerably improved spatial/temporal resolutions and radiation dose requirements (24–26). Hence, PET is ideal for visualizing the location and quantifying the expression of exogenous transgenic proteins across multiple translational applications (22, 27).
Because many opsins have already been created (28, 29), developing dedicated high-affinity and selective PET radioligands for any given opsin is impractical. A key reason for this is that other than the binding site for the covalently linked retinal, opsins lack targets for known small-molecule ligands. An alternate approach is to develop a PET-based reporter system that could visualize the distribution of any opsin (or other proteins of interest), making it adaptable to a variety of gene therapy strategies by fusing a small protein tag to the protein of interest. Such a “universal” PET reporter system would need to satisfy four major requirements: (i) The radioligand used to detect it should exhibit high binding affinity and high in vivo selectivity for the specific fusion tag, (ii) the radioligand-tag reporter system should be scalable to different classes of opsins/proteins, (iii) the tag should be small enough to fit into a viral vector backbone and preferably of human origin (to limit immunogenicity), and (iv) the tag should not interfere with the exogenous protein’s function or with endogenous physiology.
The human estrogen receptor α (ERα) is a multidomain nuclear receptor. ERα is mainly found in reproductive tissues as well as in the kidney, liver, breast tissue, and bone, but its expression in the brain relative to these other organs is low (30). The ligand-binding domain (LBD) of ERα is in the C-terminal part of the protein, and its binding properties are retained when detached from the DNA binding domain (31). A U.S. Food and Drug Administration (FDA)–approved PET radioligand, [18F]16α-fluoroestradiol ([18F]FES), has been used to visualize ERα in animals and humans and shows low brain accumulation (32, 33). We posited that the favorable anatomical distribution profile of ERα (low brain expression), the low brain uptake of [18F]FES, and the high affinity of the ERα-LBD for [18F]FES would enable longitudinal visualization of opsins in the mammalian brain.
RESULTS
Development and characterization of ChRERα binding and function in HEK-293 cells
To generate the ChRERα construct, the complementary DNA (cDNA) for ERα-LBD was appended to the C terminus of the open-reading frame of hChR2(H134R) using a V5 epitope as a linker. The fusion product, hChR2(134R)-V5-ERα-LBD or “ChRERα,” was inserted downstream of the cytomegalovirus–immediate early (CMV-IE) promoter in a pcDNA3.1 vector (Fig. 1A). This plasmid was transfected along other vectors containing V5-ERα-LBD and hChR2(H134R)–enhanced yellow fluorescent protein (EYFP) into human embryonic kidney (HEK) 293 cells. To examine cellular distribution, we lysed the cells and separated the lysates into cytosolic and membrane fractions. We used an anti-V5 antibody and Western blot to detect antigen expression. As expected, no immunoreactivity was detected for hChR2(H134R)-EYFP, whereas V5-ERα-LBD (a soluble protein) was strongly detected in the cytosolic and membrane fraction (Fig. 1B). ChRERα was only detected in the membrane fraction with a molecular weight (~70 kDa) compatible with the fusion construct (Fig. 1B).
Fig. 1. Development and characterization of ChRERα in HEK-293 cells.
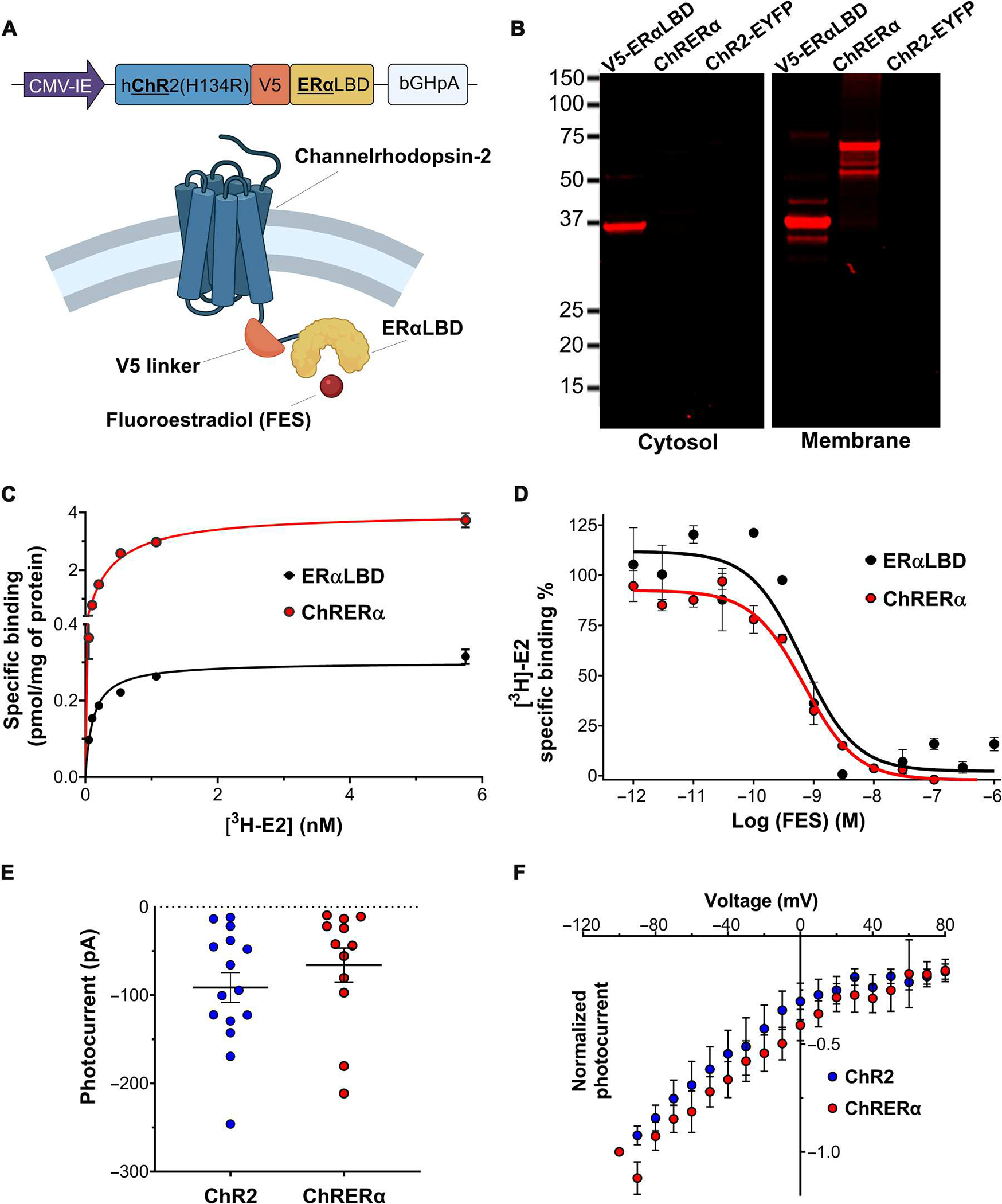
(A) Schematic illustrations of the encoding sequence for ChRERα (top) and the protein structure of ChRERα (bottom) are shown. (B) Representative Western blots verifying the subcellular localization of the V5 epitope in cytosolic or membrane fractions of HEK-293 cells transfected with V5-ERα-LBD, ChRERα, or ChR2-EYFP are shown. (C) [3H]E2 binding saturation curves in membrane homogenates from HEK-293 cells transfected with ERα-LBD (black) or ChRERα (red). (D) [3H]E2 competition binding curves with FES in membrane homogenates from HEK-293 cells transfected with ERα-LBD or ChRERα. Values for fitted parameters are described in the main text. (E) Photocurrent amplitudes in picoamperes (pA) and (F) light-induced voltage-current curves in millivolts (mV) of HEK-293 cells transfected with ChR2 (blue) or ChRERα (red). All data are shown as means ± SEM except that dots in (E) are individual cells.
Next, we tested whether ChRERα retained its binding affinity to 17β-estradiol (E2) and to FES using saturation and competition radioligand binding assays with estradiol labeled with tritium ([3H]E2) in HEK-293 cells transfected with ERα-LBD or ChRERα. The membrane fractions of cells transfected with ChRERα bound [3H]E2 with similar affinity [dissociation constant (Kd) = 0.39 ± 0.03 nM] as those transfected with ERα-LBD (Kd = 0.35 ± 0.03 nM) (Fig. 1C). In addition, [3H]E2 competition binding assays with FES showed that cells transfected with ChRERα bound FES [inhibition constant (Ki) = 0.4 ± 0.1 nM] with similar affinity as those transfected with the ERα-LBD (Ki = 0.3 ± 0.1 nM) (Fig. 1D), indicating that fusion to hChR2(134R) did not affect the binding properties of ERα-LBD.
To assess its ion channel function, we transfected ChRERα or hChR2(134R)-EYFP into HEK-293 cells and performed whole-cell patch-clamp recordings in response to blue light (473 nm) stimulation. Steady-state photocurrent amplitudes (ChRERα, −65.9 ± 19.32 pA; ChR2 −91.4 ± 17.02 pA; Fig. 1E) and voltage-current curves (Fig. 1F) of cells transfected with ChRERα did not significantly differ from those observed in ChR2-transfected cells, demonstrating that ChRERα retains opsin function (Student’s t test, t25 = 0.99, P = 0.33).
ChRERα trafficking and subcellular localization in the brain of rats
To assess its neuronal trafficking and subcellular localization profile, ChRERα was packaged into an adeno-associated viral (AAV) vector (serotype 2/5) with the human synapsin 1 (hSyn1) promoter to facilitate neuronal transduction [AAV-ChRERα: AAV2/5-hSyn1-ChR2(H134R)-V5-ERα-LBD; 1.32 × 1014 genome copies (gc)/ml; Boston Children’s Hospital Viral Vector Core]. AAV-ChRERα was injected into the right prelimbic/dorsal anterior cingulate cortex (PrL/ACd) area in rats (Fig. 2A). About 5 weeks later, rats were perfused with 4% paraformaldehyde (PFA), and ChRERα localization was assessed using immunohistochemistry (IHC) with an anti-ChR2 antibody. ChRERα was expressed in cell bodies and axonal processes at the site of AAV injection (Fig. 2, B to D). In a subset of rat brain slices, we used immuno–electron microscopy (immuno-EM) to visualize the subcellular localization of ChRERα. We observed plasma membrane ChRERα expression at the soma, dendrites, and synaptic axon terminals in the right PrL/Acd AAV injection area (Fig. 2, E to G, and fig. S1). Besides the injection location, we also observed ChRERα expression at downstream projection sites [right mediodorsal thalamus (MDT), zona incerta, and striatum] (Fig. 2, H to K, and fig. S1) where expression was also observed at the soma and synaptic axon terminals (Fig. 2, L to N, and fig. S1), confirming the subcellular membrane localization, anterograde trafficking, and retrograde AAV transduction of ChRERα.
Fig. 2. ChRERα trafficking and subcellular localization in the rat brain.
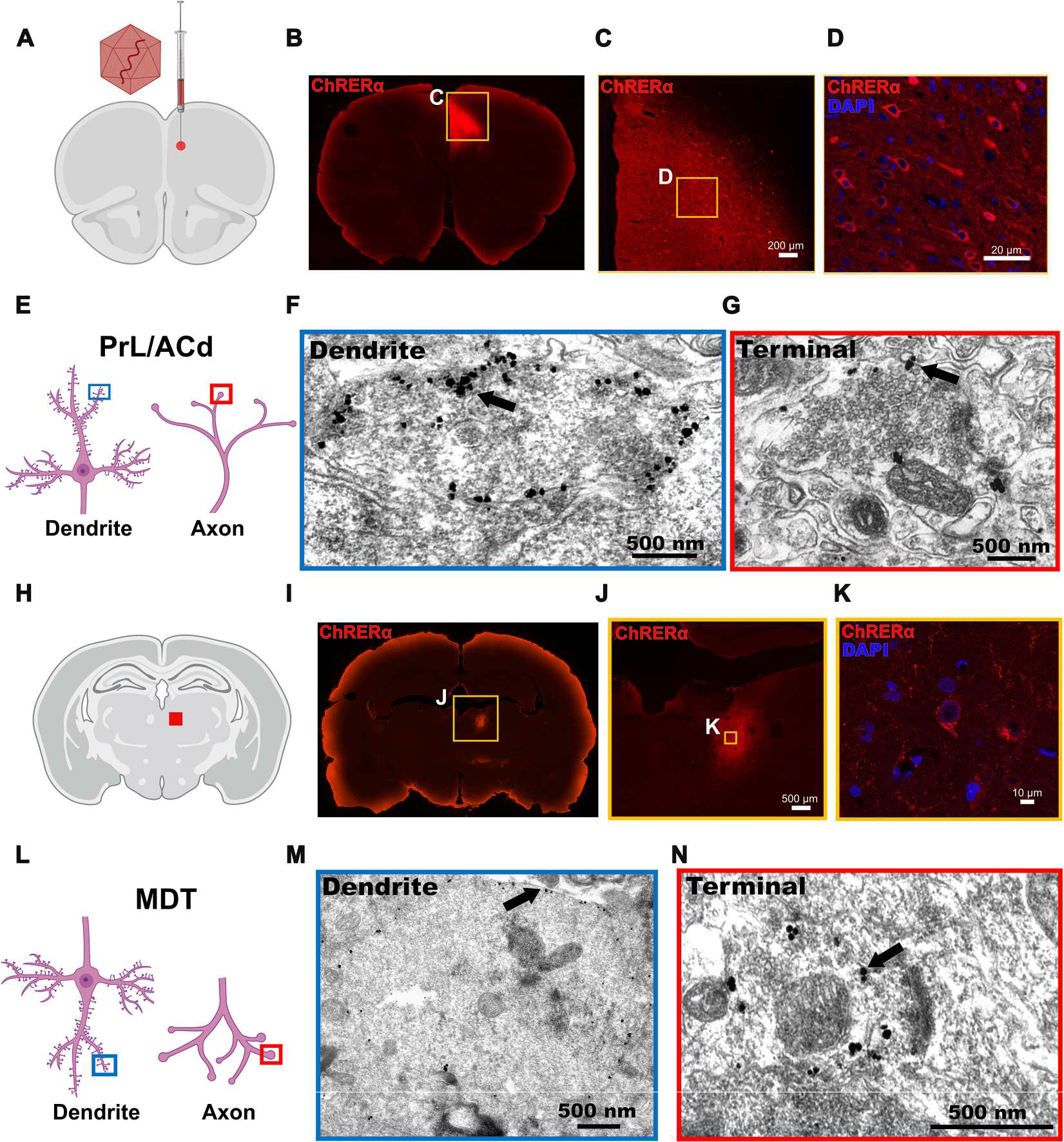
(A) Illustration of the AAV-ChRERα injection site in the right PrL/ACd (AP = 3.0, ML = 0.6, DV = −3.5) is shown. (B to D) Representative IHC images at three different levels of magnifications illustrate the expression of ChRERα in cells in the right PrL/ACd (red = anti-ChR2 and blue = DAPI). (E) Schematic of dendrite (left, blue box) and axon terminal (right, red box) illustrates the localization of immuno-EM images. (F and G) Immunogold labeling of ChRERα expression in (F) right PrL/ACd dendrite and (G) axon terminal. (H) Schematic of right MDT projection site (red square). (I to K) Representative IHC images at three different levels of magnification illustrate the expression of ChRERα in cells in the right MDT (red = anti-ChR2 and blue = DAPI). (L) Schematic of dendrite localization (left, blue box) after retrograde transduction and anterograde axon terminal (right, red box) trafficking in the MDT. (M and N) Immunogold labeling of ChRERα expression in (M) MDT dendrite and (N) axon terminal.
Noninvasive localization of ChRERα in the brain of individual rats
Next, we tested whether ChRERα expression could be visualized noninvasively with [18F]FES in the rat brain. We first obtained [18F]FES-PET scans in naïve male and female rats to establish a baseline of [18F]FES binding in the brain. Anesthetized rats were given a bolus injection of [18F]FES (intravenously, ~0.5 mCi) and scanned for 90 min using a Mediso nanoScan PET/computerized tomography (CT) scanner. Acquisition data were first converted to standard uptake values (SUVs; gram per milliliter) to correct for animal body weight and injected radiotracer dose. SUV ratios (SUVRs) and binding potentials (BPND) were calculated using the cerebellum as a nonspecific reference region (see Materials and Methods for details) because of its large size and low endogenous ERα expression (34). As expected, the greatest amount of [18F]FES binding was detected in the pituitary gland (Fig. 3A), which has high endogenous ERα expression (34). Low binding was observed in the rest of the brain, including the PrL/ACd (Fig. 3A). After characterizing baseline [18F]FES brain uptake in AAV-naïve rats (controls, n = 5), we injected a separate group of rats (ChRERα, n = 6) with AAV-ChRERα (1 μl) into the right PrL/ACd and scanned them with [18F]FES 3 to 5 weeks after AAV injection (Fig. 3B). Compared with controls at baseline, rats injected with AAV-ChRERα showed significantly higher binding of [18F]FES at the PrL/ACd AAV injection site and in downstream projection areas (ipsilateral MDT) (t = 1.89, P < 0.05) (Fig. 3, C to F, and fig. S2). [18F]FES-PET localized ChRERα expression in individual rats with similar efficacy in females (n = 3) and males (n = 3) (fig. S2). Furthermore, the pattern of [18F]FES binding in the brain coincided with ChRERα expression as confirmed via postmortem IHC (Fig. 3G). In addition, [18F]FES-PET was performed in a separate cohort of rats injected with a smaller volume of AAV-ChRERα (0.5 μl) in the left primary motor cortex (M1) (Fig. 3H). As expected, [18F]FES showed high binding in the left M1 (Fig. 3, I to K), and the ChRERα expression at this site was confirmed using postmortem IHC (Fig. 3L). To evaluate whether the expression of ChRERα was accompanied by an additional inflammatory response, we performed IHC against Iba1 (ionized calcium-binding adapter molecule 1; a microglial marker) and GFAP (glial fibrillary acidic protein; an astrocyte activation marker) in rats injected with AAV-ChRERα. We did not find any relation between Iba1 and GFAP immunolabeling when comparing the areas with or without expression of ChRERα near the injection site or in contralateral regions (fig. S3). ChRERα expression showed regional specificity, because the right PrL/ACd-injected group did not show above background binding of [18F]FES in M1 (Fig. 3E), and the left M1–injected group did not show higher [18F]FES in the PrL/ACd compared to controls (Fig. 3J). Both observations confirm the capacity of the technique to differentiate between injection sites that are anatomically close (<1 to 2 mm apart). To exclude the possibility that the [18F]FES signal reflected effects of the surgery or AAV transduction, we performed PET scans in rats (n = 3) previously injected with AAV vectors encoding constructs unrelated to ChRERα (not expressing ChR2 or the ERα-LBD). As expected, these rats did not show any focal increase in [18F]FES signal at the injection site nor in other brain areas (fig. S4), indicating that the [18F]FES signal in rats injected with AAV-ChRERα represents binding of [18F]FES to ChRERα.
Fig. 3. Noninvasive localization of ChRERα in the brain of individual rats with [18F]FES.
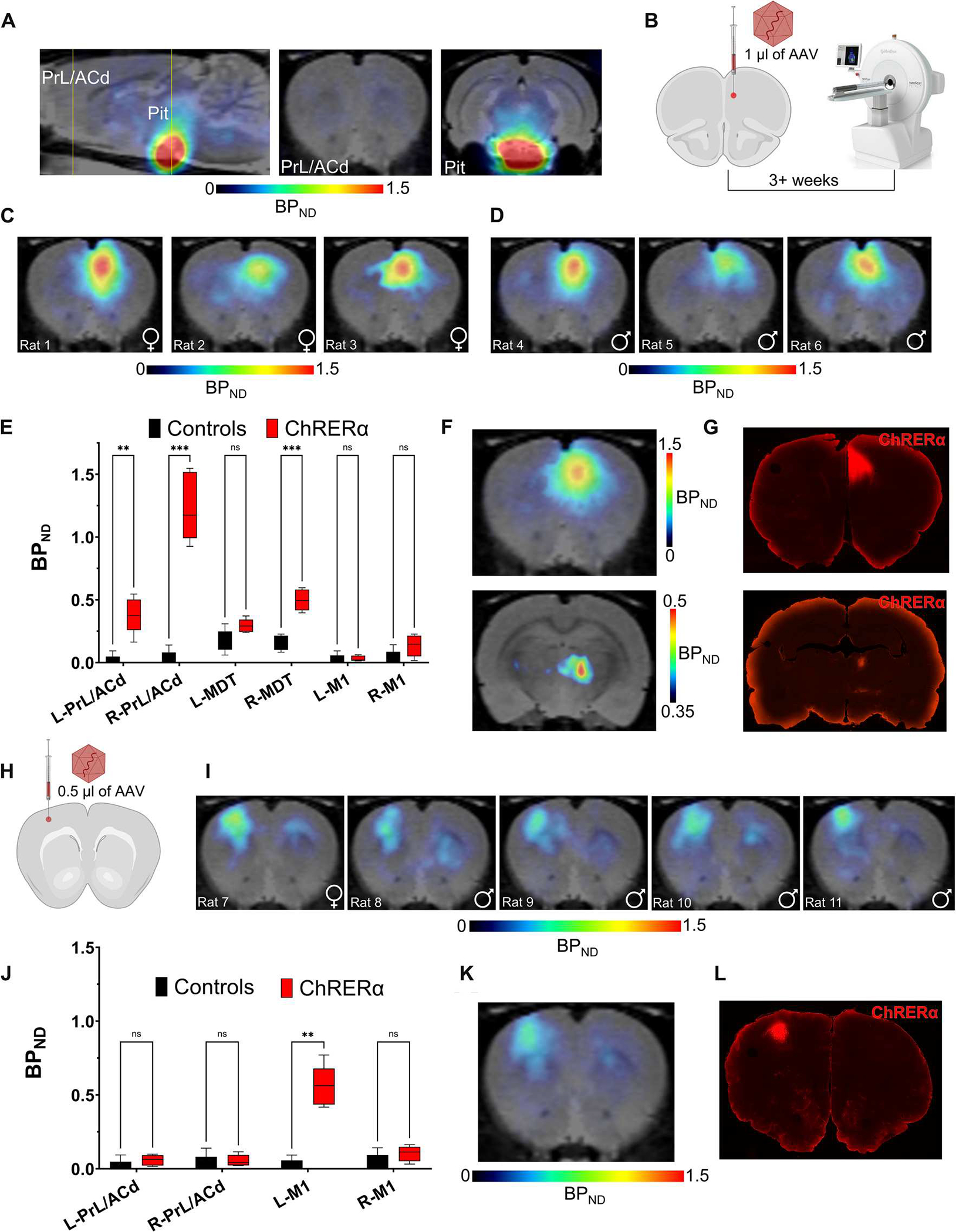
(A) Baseline [18F]FES-PET representative image in control rats (no AAV) shows high endogenous binding in the pituitary but low binding in the rest of the brain (yellow lines illustrate location of coronal cross sections). (B) Schematic of right PrL/ACd injection site for AAV-ChRERα in rats 3+ weeks before [18F]FES-PET scan. (C and D) [18F]FES binding localizes ChRERα near the AAV injection site in right PrL/ACd of (C) female (n = 3) and (D) male (n = 3) rats 3 to 5 weeks after receiving AAV-ChRERα (representative images from individual rats). (E) Group BPND plot reveals higher binding in the right PrL/ACd and MDT in AAV-ChRERα (n = 6) versus control (n = 5) rats. ***P < 0.001 and **P < 0.01. (F) Group average BPND of [18F]FES in the right PrL/ACd (top) and MDT (bottom) in ChRERα rats (n = 6). (G) IHC confirms ChRERα expression in the right PrL/ACd and MDT (red = anti-ChR2). (H) Schematic of the left M1 injection site for AAV-ChRERα in rats (n = 5). (I) Individual post-AAV [18F]FES-PET scans of rats injected with AAV-ChRERα in the left M1 (n = 5). (J) Group BPND plot of AAV-ChRERα (left M1 AAV, n = 5) versus control (n = 5) rats shows higher binding in the left M1 but not in PrL/ACd. **P < 0.01. (K) Group average BPND of [18F]FES in the left M1 of AAV-ChRERα rats (n = 5). (L) IHC confirms ChRERα expression (red = anti-ChR2) corresponding with the left M1 area localized with [18F]FES-PET. ns, not significant.
ChRERα activation alters brain activity and behavior in rodents
To further examine the functional properties of ChRERα, we performed ex vivo slice electrophysiology in a cohort of mice injected with the same AAV-ChRERα vector [AAV2/5-hSyn1-ChR2(H134R)-V5-ERα-LBD] in the PrL/ACd. Coronal brain slices were collected 2 to 4 weeks after AAV-ChRERα injection, and whole-cell patch-clamp recordings were acquired from pyramidal cells near the AAV injection site during blue light stimulation (Fig. 4, A to D). Recordings were performed in the absence of a fluorescent reporter, and ~40% of recorded cells showed consistently robust light-evoked depolarizations in response to 5-ms blue light pulses (Fig. 4B), and a 500-ms light pulse produced the steady-state current induction characteristic of opsin function (Fig. 4, C and D).
Fig. 4. ChRERα activation alters brain activity and behavior in rodents.
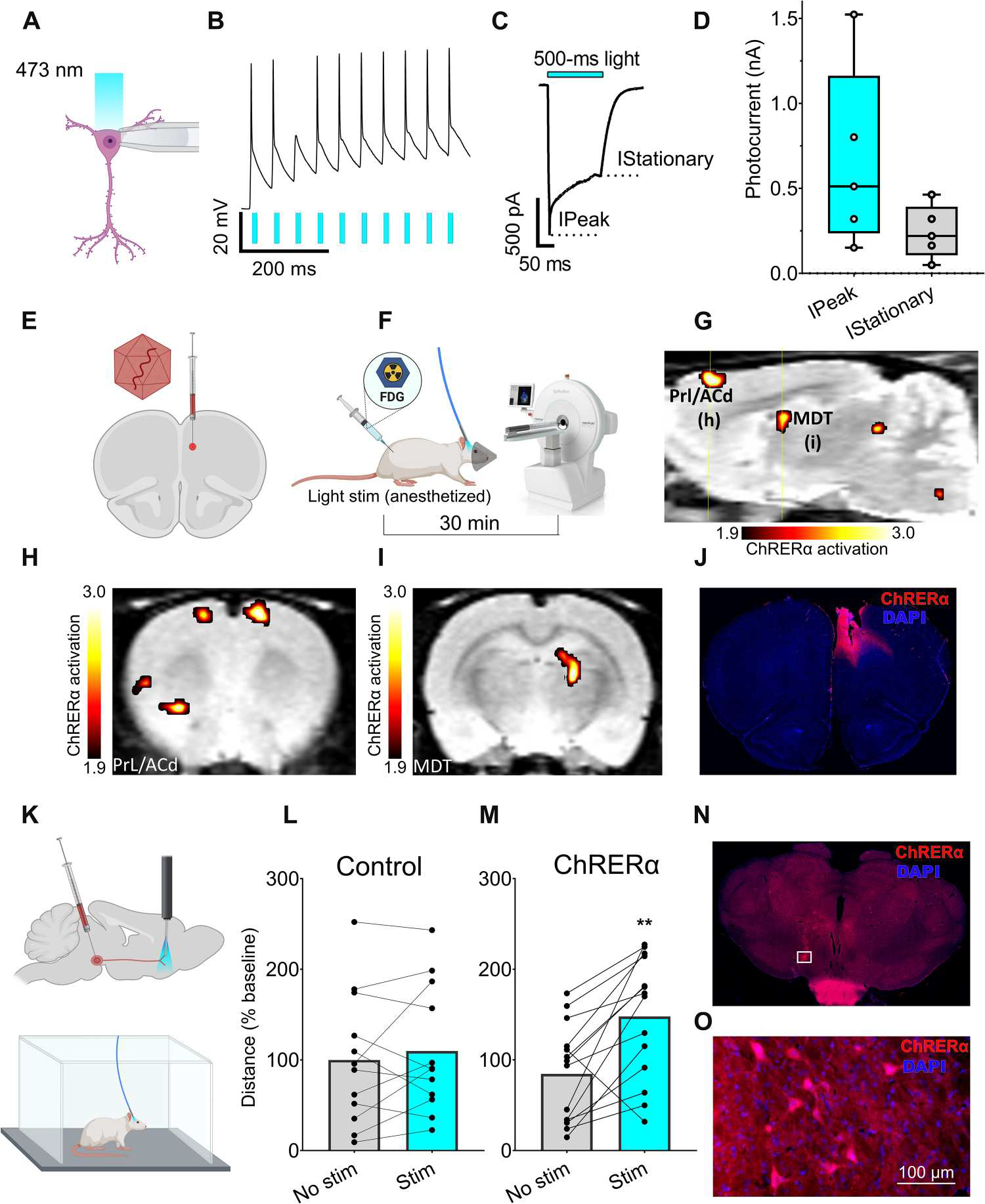
(A to D) Slice electrophysiology in mice. (A) Patch-clamp recordings of pyramidal neurons from layer 5 of the ventromedial prefrontal cortex during stimulation with 473-nm light at 10 megawatt (mW) power using 5- or 500-ms-long pulses. (B) Representative voltage trace of neuronal firing in responses (black trace) to a train of light pulses (blue bars). (C) Representative current trace in response to a 500-ms light pulse. (D) Average peak and stationary photocurrent values in nanoampere (nA) (five cells per three mice). (E to I) FDG-PET imaging in rats. (E) Schematic of AAV-ChRERα injection and optic fiber target in right PrL/ACd (ChRERα n = 5, control n = 4). (F) FDG-PET experimental design—bolus intraperitoneal. FDG injection at start of the 30-min light stimulation protocol (anesthetized), followed by PET/CT scan. (G) Sagittal view (right hemisphere) with regions of greater FDG uptake (maps thresholded at t1,7 = 1.89, P < 0.05) in ChRERα versus control rats [group analysis, yellow lines indicate level of coronal images shown in (H) and (I)]. (H and I) Coronal view with regions of significantly greater FDG uptake (ChRERα > control) that was observed in (H) bilateral PrL/ACd, left insula and striatum, and (I) right MDT. (J) IHC confirms expression of ChRERα and optic fiber placement in the right PrL/ACd (red = anti-ChR2 and blue = DAPI). (K to M) Locomotor behavior in rats. Each dot represents an individual trial from five different rats. (K) Schematic of unilateral AAV-ChRERα injection in VTA and optic fiber placement in NAc (top) and open-field optogenetic stimulation in awake rats (bottom). (L and M) Light stimulation significantly increased (**P = 0.01) distance traveled in (M) AAV-ChRERα rats but not in (L) control rats. (N to O) IHC confirms ChRERα expression in rat VTA (red = anti-ChR2 and blue = DAPI), and white box indicates area of high magnification (O) in VTA.
Next, we used optogenetics combined with PET imaging of [18F]fluorodeoxyglucose (FDG) (27, 35) to evaluate the metabolic activity in response to ChRERα stimulation in vivo in rats. We injected AAV-ChRERα in the right PrL/ACd of rats (Fig. 4E) and implanted an optic fiber of 0.5 mm above this site about 3 weeks later. A separate group of rats without AAV-ChRERα were also implanted with optic fibers in right PrL/Acd to serve as controls. On the day of the experiment, rats were anesthetized with ~2% isoflurane, and a bolus injection of FDG (~0.5 mCi, i.p.) was administered followed immediately by light stimulation (473 nm, 3-ms pulse trains at 50 Hz; 300 ms on, 700 ms off) for 30 min. After this tracer uptake period, rats were then scanned using PET (Fig. 4F). When compared with non-ChRERα controls, light stimulation in AAV-ChRERα rats induced significantly greater metabolic activity (t1,7 = 1.89, P < 0.05) in the right PrL/Acd and in the ipsilateral MDT (Fig. 4, G to I), which is consistent with ChRERα expression patterns seen with IHC after AAV injection in the right PrL/Acd (Fig. 2). Light stimulation in AAV-ChRERα rats also increased metabolic activity bilaterally in the inferior colliculi; contralaterally in PrL/Acd, nucleus accumbens (Nac), and insular cortex; and ipsilaterally in the lateral thalamus and brainstem (Fig. 4, G to I, and fig. S5). In the same group, light stimulation significantly decreased metabolic activity (t 1,7 = 1.89, P < 0.05) bilaterally in striatum and cerebellum; ipsilaterally in infralimbic cortex, motor cortex, and septum; and contralaterally in ventral pallidum, lateral thalamus, hippocampus, and entorhinal cortex (fig. S5). Significant changes in metabolic activity outside the PrL/Acd likely reflect downstream recruitment in response to ChRERα activation in the PrL/Acd. Postmortem IHC confirmed ChRERα expression and optic fiber placement in the right PrL/Acd (Fig. 4J).
Last, to test whether ChRERα activation could modulate behavior, we injected AAV-ChRERα into the ventral tegmental area (VTA) of rats. Four weeks later, an optical fiber was implanted into the Nac (Fig. 4K), where VTA dopaminergic fibers are known to project and activate Nac terminal fields to induce locomotion (36). A separate group of rats without AAV-ChRERα were also implanted with optic fibers to serve as controls. On the day of the experiment, rats were tethered to an optic fiber in an open-field chamber (Fig. 4K), and locomotor activity was recorded during periods with and without light stimulation (see Materials and Methods for protocol details). Light stimulation significantly increased locomotor activity in the AAV-ChRERα group as compared with the control group (Fig. 4, L and M). The two-way analysis of variance (ANOVA) of the distance traveled, which included two factors (AAV group and light stimulation) matched by subject, showed an interaction between AAV transduction and light stimulation (F 1,24 = 6.74, P = 0.016); a Šidák’s post hoc multiple comparisons test showed significant effects of light stimulation in the AAV-ChRERα group (t 24 = 4.52, P = 0.003) but not in the control group (t24 = 0.65, P = 0.77) (Fig. 4M). Postmortem IHC confirmed expression of ChRERα in the VTA (Fig. 4, N and O).
[18F]FES-PET localizes ChRERα and predicts structural and functional brain connectivity in NHPs
We next tested whether AAV-mediated ChRERα expression could be localized using [18F]FES-PET in the NHP brain. Two male squirrel monkeys underwent structural magnetic resonance imaging (MRI) and before AAV [18F]FES PET (Fig. 5A) to map the AAV stereotaxic injection location and determine the baseline distribution of [18F]FES binding. As observed in rats, the highest baseline [18F]FES binding in NHPs was detected in the pituitary (Fig. 5B). Additional [18F]FES signal, but of much lower extent, was observed in the brain along white matter tracts (Fig. 5B). For AAV injections, NHPs were placed in a stereotactic apparatus and injected with AAV-ChRERα in the forelimb region of the left M1 [(three 1.33-μl injections spaced 0.75 mm apart, at 1.0, 1.75, and 2.5 mm below the dura, infused at 0.33 μl/min) AAV titer of 1.32 × 1014 gc/ml]. The left M1 injection site was first approximated using individual structural MRIs, and then specific sites for injection within the forelimb representation were determined with intracortical microstimulation. In squirrel monkey #1, [18F]FES binding (BPND) in the left M1 increased from 0.12 at pre-AAV to 0.47 at 5 weeks after AAV injection (~4-fold increase; Fig. 5, C and D, and fig. S6). In addition, we observed a ~10-fold increase of [18F]FES (BPND from 0.1 to 1.1) in a region of the ipsilateral posterior parietal cortex (PPC) (Fig. 5, D to F), an area known to have anatomical connections with M1 (37). In squirrel monkey #2, a ~3.5-fold increase in [18F]FES was observed in the left M1 7 weeks after AAV injection (BPND from 0.15 before AAV injection to 0.53 after AAV injection) (Fig. 5, E and F). However, we did not see the same large increase in the ipsilateral PPC of squirrel monkey #2 (BPND from 0.1 before AAV injection versus 0.15 at 7 weeks after AAV injection). [18F]FES scans were also quantified as SUV (grams per milliliter) and SUVR [SUVRCB (cerebellum reference)], which showed distribution patterns consistent with BPND (fig. S6), suggesting that both methods are suitable for localizing ChRERα expression in individual animals.
Fig. 5. Noninvasive localization of ChRERα in squirrel monkeys with [18F]FES PET imaging.
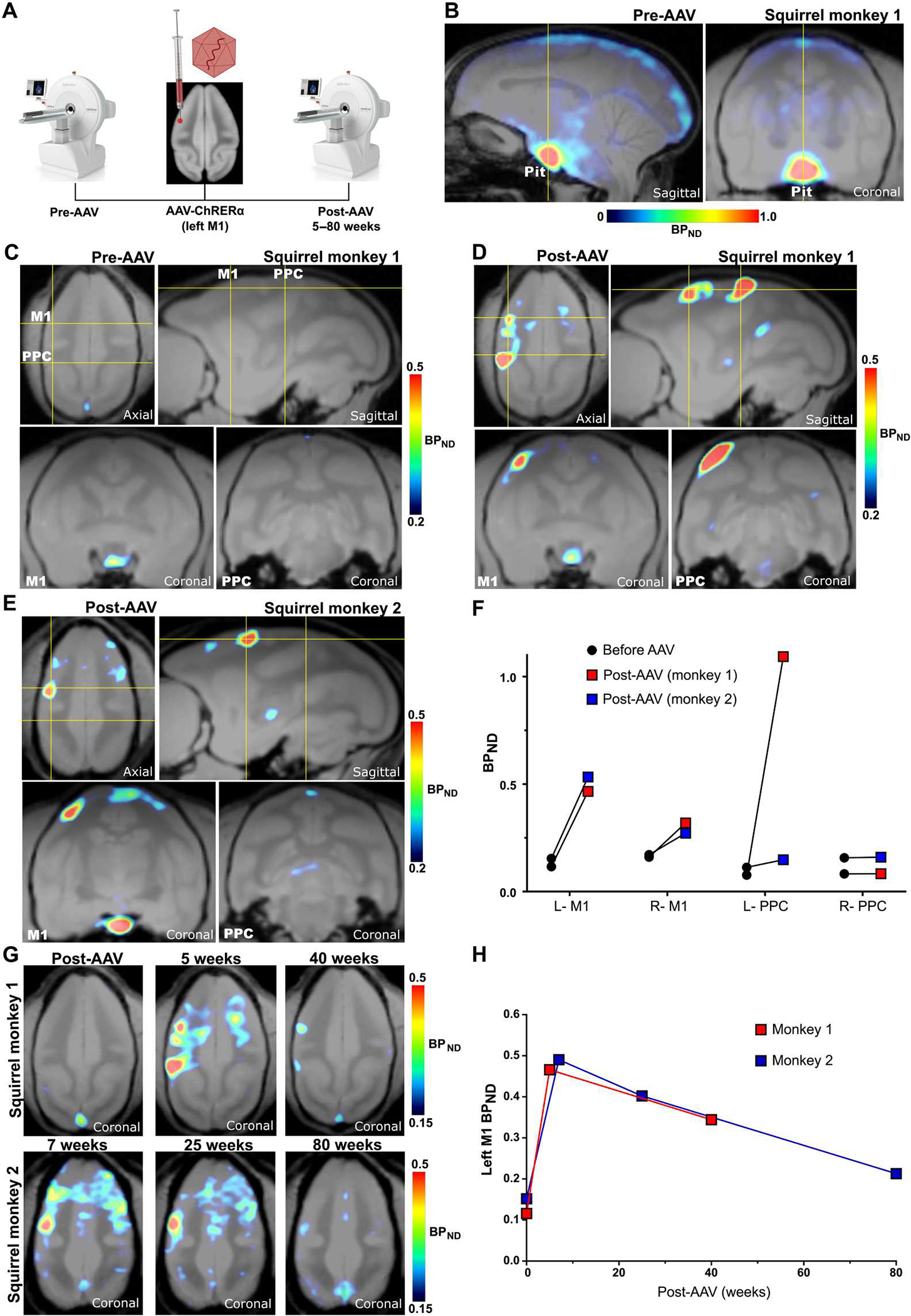
(A) Schematic of PET experimental design and the left M1 injection site for AAV-ChRERα. (B) Pre-AAV baseline [18F]FES-PET in a squirrel monkey (monkey 1) shows high endogenous binding in the pituitary but low binding in the rest of the brain. Yellow lines indicate the cross-sectional planes corresponding to the subsequent coronal images. (C) Pre-AAV baseline [18F]FES-PET in squirrel monkey 1 at the level of M1 and PPC. (D) Post-AAV [18F]FES in squirrel monkey 1 localizes ChRERα near the left M1 AAV injection site and in the ipsilateral PPC 5 weeks after receiving AAV. (E) [18F]FES localizes ChRERα near the left M1 AAV injection site in a second squirrel monkey (monkey 2) 7 weeks after AAV. (F) Pre-AAV and post-AAV BPND in left and right M1 and PPC for each animal. (G) Longitudinal comparisons of [18F]FES-PET in squirrel monkey 1 (top, before AAV, 5 and 40 weeks after AAV) and squirrel monkey 2 (bottom, 7, 25, and 80 weeks after AAV). (H) Longitudinal comparisons of average BPND in left M1 in squirrel monkey 1 (red) and squirrel monkey 2 (blue).
To longitudinally track ChRERα expression, we performed additional [18F]FES-PET scans for each NHP at different time points (25 to 80 weeks after AAV injection). The localized increases of [18F]FES in the left M1 observed in the initial post-AAV scans steadily declined over time in both animals, but they were still observable at 40 weeks after AAV injection in squirrel monkey #1 and at 25 and 80 weeks after AAV injection in squirrel monkey #2 (Fig. 5, G and H, and fig. S6).
To confirm that areas with increased [18F]FES binding reflect ChRERα expression and trafficking, IHC and immuno-EM with anti-ChR2 primary antibody were used for postmortem assessment of ChRERα expression in squirrel monkey #2 after the last scan at 80 weeks after AAV injection. In line with the increased [18F]FES signal in M1 and PPC (Fig. 6A), IHC revealed that ChRERα expression was observed in cell bodies both in the left M1 (Fig. 6, B, C, and I) and in the ipsilateral PPC (Fig. 6, D and O). We also observed increased [18F]FES signal deeper in the brain, in the ipsilateral thalamus (Fig. 6E), and IHC revealed ChRERα expression in cell bodies in the ventral lateral thalamus (VLT) (Fig. 6, F to H). Last, [18F]FES binding and IHC ChRERα expression were observed in the right M1 (fig. S7, A, B, and D) but not in the right VLT (fig. S7C). The [18F]FES signal and the number of cell bodies expressing ChRERα showed a positive, albeit nonsignificant correlation (R2 = 0.44, P = 0.15), although it is important to note that ChRERα expression on terminals would also contribute to the [18F]FES signal (fig. S7E). After euthanasia, immunogold EM was performed in the left M1, left PPC, and right M1 to visualize subcellular localization of ChRERα. Immunogold labeling was localized in the endoplasmic reticulum and at the plasma membrane of soma, dendrites, myelinated axons, and synaptic axon terminals in the left M1 (Fig. 6, J to N), left PPC (Fig. 6, P to R), and right M1 (fig. S7, F to H). These findings confirm ChRERα’s anterograde axon terminal trafficking, retrograde AAV-ChRERα transduction and expression, and plasma membrane localization in cells at cortical injection sites and at distal cortical and subcortical projection sites in squirrel monkeys. To evaluate whether the expression of ChRERα would have produced any additional inflammatory response, we performed IHC against Iba1 and GFAP in brain tissue from squirrel monkey #2. We did not find any differences in Iba1 or GFAP between tissue with ChRERα expression at the AAV-injected compared to noninjected sites (fig. S8, A and B).
Fig. 6. Postmortem validation of [18F]FES-PET ChRERa imaging in a nonhuman primate.
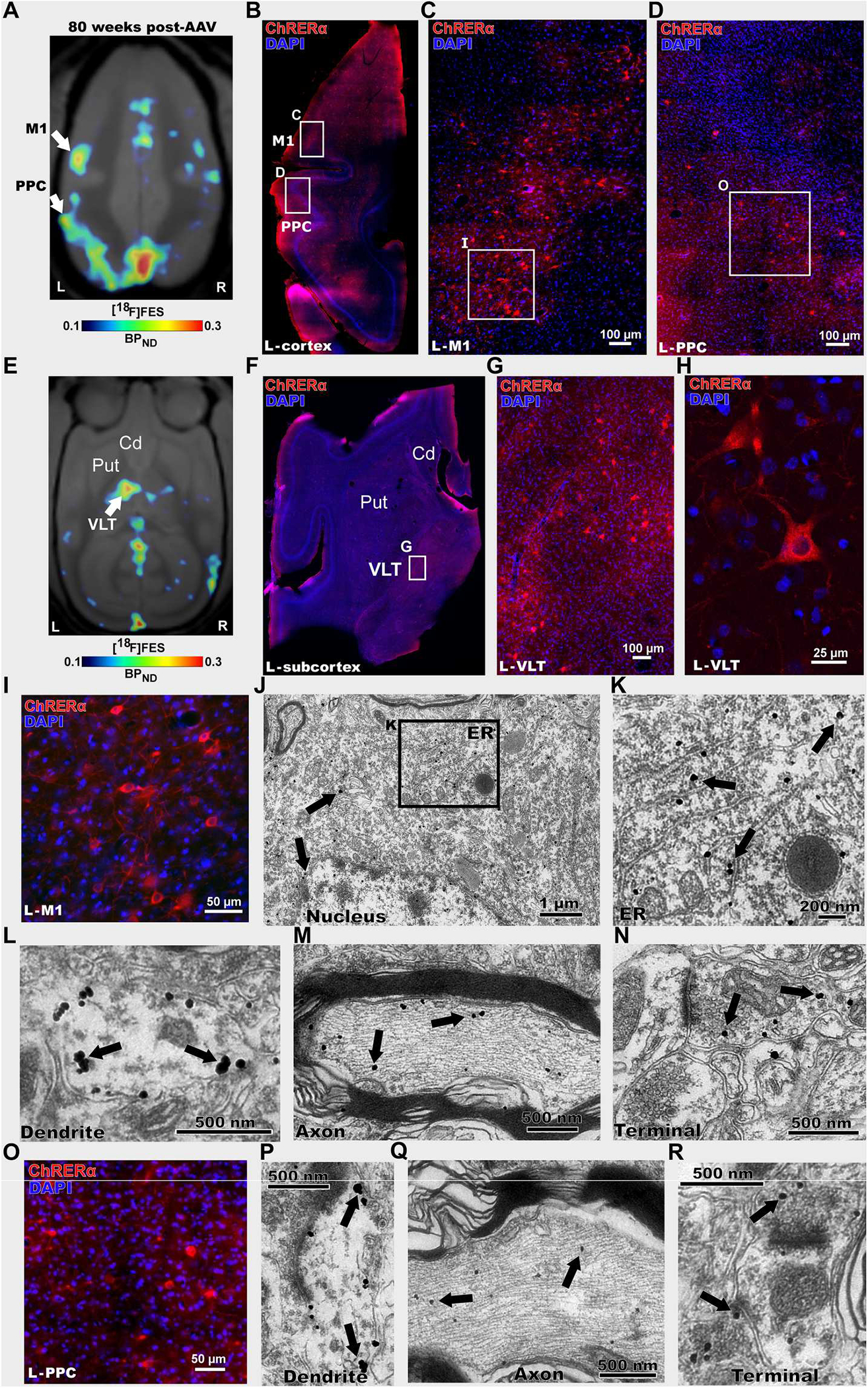
(A) [18F]FES-PET imaging 80 weeks after AAV-ChRERα injection in squirrel monkey 2 to localize ChRERα expression (BPND scaled to optimize [18F]FES signal). Arrows indicate the left M1 and PPC (B to R). (B) IHC in a left hemisphere horizontal brain slice confirms ChRERα expression in the left M1 and PPC (red = anti-ChR2 and blue = DAPI), white rectangles highlight regions in left M1 and PPC. (C and D) High magnification of images of insets shown in (B). (E) [18F]FES-PET 80 weeks after AAV-ChRERα injection in squirrel monkey 2 localized ChRERα expression in left ventral lateral thalamus (VLT) (BPND scaled to optimize [18F]FES signal). Arrow indicates left VLT (F) IHC in a left hemisphere horizontal brain slice confirms ChRERα expression in left VLT (red = anti-ChR2 and blue = DAPI), white rectangle highlights region in left VLT. (G and H) High-magnification IHC images of insets shown in (F). (I) High-magnification IHC image of left M1 (J to N) Immuno-EM in left M1 reveals subcellular localization of ChRERα expression in (J) cell body (black rectangle highlights location of (K), (K) ER, (L) dendrite, (M) myelinated axon, and (N) axon terminal. (O) High-magnification IHC image of left PPC. (P to R) Immuno-EM images in the left PPC showing ChRERα located in (P) dendrite, (Q) myelinated axon, and (R) axon terminal. Cd, caudate; ER, endoplasmic reticulum; Put, putamen.
Last, to explore the functional relevance of the individual anatomical connectivity identified by [18F]FES-PET (Fig. 7A), we acquired resting-state functional MRI (rsfMRI) data from an independent cohort of nine non–AAV-injected squirrel monkeys and performed whole-brain functional connectivity analysis using a seed (2-mm sphere) centered at each NHP’s individual peak [18F]FES signal in the left M1 (Fig. 7B). Functional connectivity analysis identified regions in the ipsilateral PPC and contralateral M1 that showed significant increased connectivity with the left M1 seed (Fig. 7, B to D). This functional circuit overlapped with regions identified using [18F]FES-PET in squirrel monkey #1 (Fig. 7, A and D; r = 0.23; P = 0 with Fisher’s z transformation; 27,871 voxels; and Dice overlap index = 0.38), which showed the greatest [18F]FES signal in the PPC, as well as with the smaller [18F]FES PPC signal observed in squirrel monkey #2 (fig. S8; r = 0.08; P = 0 with Fisher’s z transformation; 29,529 voxels; and Dice overlap index = 0.44) and is consistent with known anatomical and functional connectivity of M1 in New World monkeys (37–40). Collectively, these findings highlight the utility of [18F]FES-PET and ChRERα for in vivo mapping of anatomical projections for informing the structural basis of functional connectivity maps.
Fig. 7. [18F]FES-PET and ChRERα correlate with functional brain connectivity in NHPs.
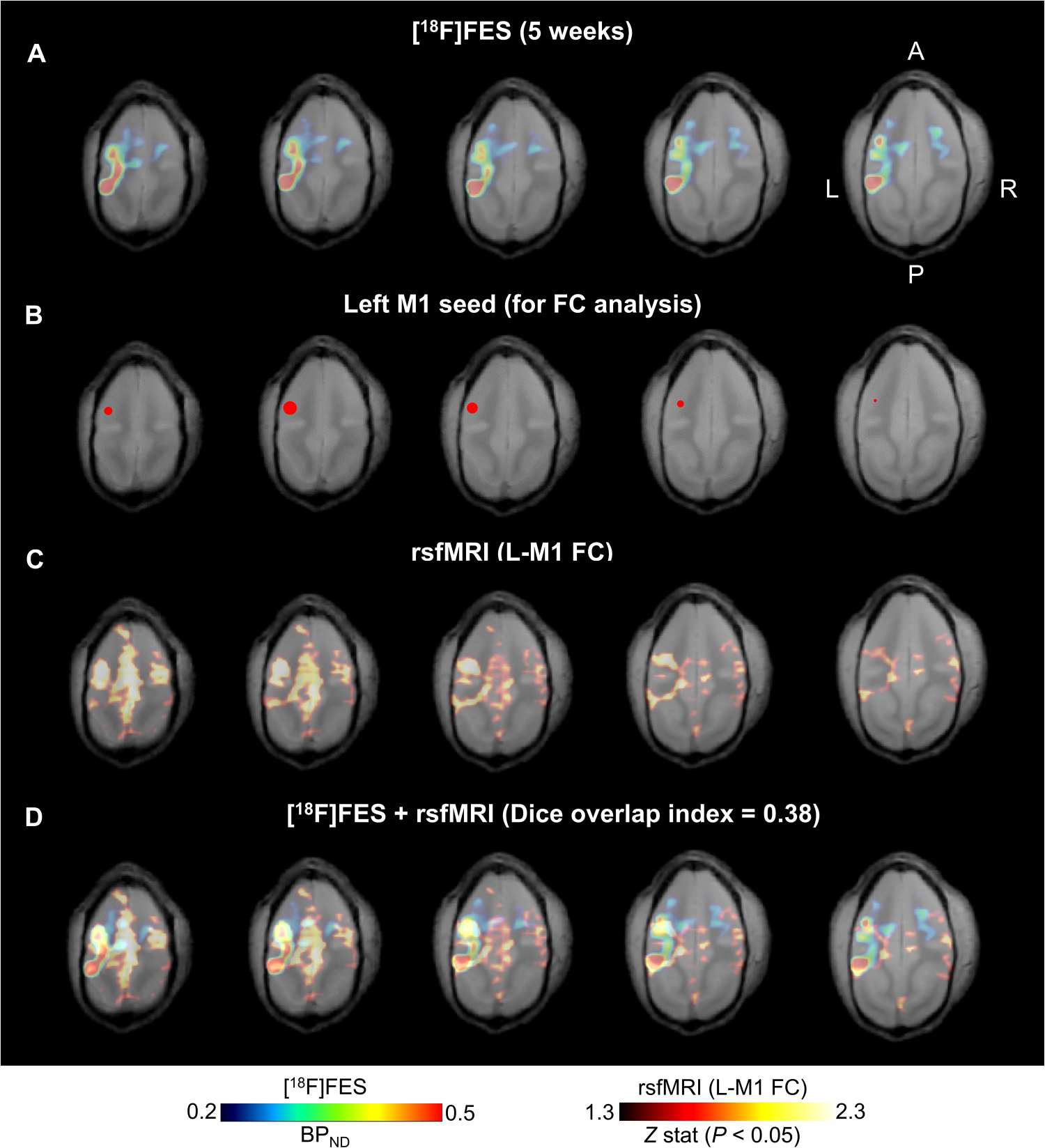
(A) Horizontal sections (left, most dorsal; right, most ventral) of [18F]FES-PET from squirrel monkey 1 at 5 weeks after AAV. (B) Left M1 seed (red 2-mm sphere centered at peak [18F]FES signal) for functional connectivity analysis of resting state functional MRI (rsfMRI). (C) rsfMRI functional connectivity patterns of the left M1 seed in an independent group of squirrel monkeys (n = 9, 35 total scans) coregistered to the squirrel monkey 1 structural MRI. (D) Overlapping patterns of [18F]FES binding (ChRERα expression) and rsfMRI suggest structural and functional connectivity between left M1 and ipsilateral PPC and in the contralateral hemisphere (r = 0.23; P = 0 with Fisher’s z transformation; 27,871 voxels; and Dice overlap index = 0.38).
DISCUSSION
Here, we describe the development of ChRERα, an opsin whose expression in the brain can be localized noninvasively and longitudinally in both the rat and NHP using the FDA-approved PET tracer [18F]FES. ChRERα retained the ligand-binding properties of the ERα-LBD and the functional properties of ChR2. When transduced into the brain, ChRERα displayed plasma membrane localization in cell bodies, dendrites, and axon terminals in rats and NHPs, indicating appropriate subcellular trafficking of the opsin to enable light-induced neuronal activation. Because ChRERα lacks the DNA binding domain of ERα and is tethered to the cell membrane by the transmembrane domains of the opsin (it cannot enter the cell nucleus), we expected this construct to be inert under nonstimulated conditions. We did not observe any effects when ChRERα was expressed in vitro or in vivo in the absence of light stimulation, whereas blue light effectively activated it to produce changes in physiology, metabolic brain activity, and behavior.
[18F]FES-PET effectively localized ChRERα in individual rats and NHPs at both the local AAV injection site and at distal cortical and subcortical brain regions with known connectivity to the cortical injection site. We did not observe meaningful differences in the efficacy of [18F]FES to detect ChRERα in male versus female rats, suggesting that even if sex differences existed in basal uptake of [18F]FES, these did not affect detection of virally driven expression of ChRERα.
Current NHP optogenetic applications have inherent limitations because they do not permit noninvasive longitudinal monitoring of opsin expression except in very specific circumstances: Cranial windows have been successfully used for monitoring fluorescent reporters through an artificial dura (41, 42), but the preparation would still be considered invasive given the need for craniotomy and durotomy. Moreover, the visual monitoring practiced in these conditions is limited to the cortical surface. Here, we used repeated within-subject scans in NHPs to follow whole-brain ChRERα expression over a 1.5-year period after AAV injection. This is particularly important because a histological snapshot of opsin expression at the end of the study would miss the expression pattern of ChRERα during the preceding time period when experiments would have been performed.
Although we targeted the same anatomical area in both NHPs, the ChRERα expression patterns detected using [18F]FES at the AAV injection site and at projection sites differed between the two animals. We also identified a temporal difference in [18F]FES signal within each NHP, indicating that ChRERα expression is a dynamic process that changes over time. Unfortunately, we could not perform postmortem validation experiments in one of the NHPs and had to exclusively rely on the postmortem assessment (a single time point) of squirrel monkey #2 for the validation of these expression patterns. Nevertheless, the postmortem data from squirrel monkey #2 matched the [18F]FES signal from the last PET scan performed on that animal shortly before euthanasia, confirming the utility of the ChRERα-[18F]FES reporter system.
AAV serotype 2/5 is mostly considered an anterograde tracer (it transduces cell bodies at the injection site rather than axon terminals) (43, 44). However, it is known that it transduces ChR2 in the squirrel monkey in a retrograde manner (45). In rats, we observed mostly anterograde transduction, although some degree of retrograde labeling was also identified. However, the NHP data showed substantial retrograde labeling because the [18F]FES signal in the contralateral M1 and ipsilateral PPC was stronger at certain time points than at the left M1 injection site. Because immunolabeling of inflammatory markers in postmortem tissue did not show any increase in microglia or activated astrocytes, it is not likely that the decrease in expression at later time points is due to an immune response against ChRERα. However, we cannot discard the possibility that an initial reaction against the AAV capsid was a factor in the unexpected lower expression at the injection site. Hence, these tropism effects are likely unrelated to the transduced construct (in this case, ChRERα) and are most likely related to the vector used, the target area, and/or the injection conditions. Hence, independent of its function as an opsin and its capacity to inform brain connectivity, ChRERα can effectively be used as a noninvasive reporter system and tool to personalize and optimize the transduction conditions of different gene delivery methods according to specific experimental needs. This property underscores the value of our technology, which permits tracking of the transduced opsin over time in individual subjects. These results also highlight the utility of the present reporter system for translational optogenetic studies where knowledge regarding the visualization of the spatial and temporal profiles of opsin expression in individual subjects would have major implications for successful translational and potential clinical implementation.
Translational and clinical optogenetic applications necessitate the development of combined optogenetic and noninvasive brain imaging, especially in NHPs. Specifically, the ability to localize and quantify opsin expression at discrete anatomical sites in a longitudinal manner is critical for ascribing neural circuit involvement to normal behavior and disease states across the life span. Coupled with optogenetic manipulation, the approach we present here can help to establish causality for brain region associations observed from human functional brain connectivity studies performed in normal subjects (46) or in the context of disease, disease diagnosis, or recovery (47, 48). When combined with multimodal imaging ([18F]FES-PET combined with [18F]FDG-PET or fMRI), our ChRERα data informed brain structure-function relationships in rats and NHPs. This noninvasive circuit mapping allowed us to correlate ChRERα activation with changes in brain regional metabolic activity in rats and ChRERα expression with functional connectivity in NHPs, illustrating how ChRERα can potentially be used to define relationships between brain anatomical and functional connectivity.
The use of the ERα-LBD reporter system is not expected to be limited to ChR2 and may provide a modular solution to image other opsins and potentially other effector proteins. In this way, [18F]FES/ERα-LBD may comprise a universal reporter system for noninvasive brain localization of transgenic constructs in rodents, NHPs, and potentially in clinical applications. This system could also facilitate standardization/optimization in gene therapy, which would facilitate comparisons between different gene therapy approaches in a personalized manner and across species.
Our study has limitations. A potential limitation of this technique is the intense accumulation of [18F]FES within the pituitary and other areas (visual cortex) that likely reflects nonspecific tracer accumulation due to the blood flow. Because of partial volume effects in PET, this may limit the use of this reporter system in animals with small brains, especially if the targeted region is close to these regions. However, as we show here, this is not a meaningful limitation in rats or squirrel monkeys and would have even less bearing in larger brains with relatively small pituitary glands (macaque and human). Another potential limitation is that [18F]FES accumulated, albeit to a low extent, in white matter. Therefore, it may show higher nonspecific accumulation in brains with high white matter content. This latter limitation can potentially be addressed, if necessary, by designing less lipophilic steroid analogs and by optimizing viral vector transduction to drive the optimal amount of ChRERα expression for its detection. In addition, the ERα-LBD may be mutated to further enhance [18F]FES affinity. Last, [18F]FES radio metabolite analyses and arterial blood–derived input functions, which we did not perform here because of the small size of the animals used but which are routinely performed in larger NHPs such as rhesus macaques or in humans, can likely aid to further minimize the [18F]FES signal in blood and improve ChRERα detection.
Other limitations of these experiments are that we did not assess the capacity for ChRERα to induce light-mediated optogenetic neuromodulation in NHPs and the small number of NHPs we used. As a first pass, our primary goal in the NHP experiments was to showcase the capacity for ChRERα/[18F]FES to serve as a PET reporter system. Therefore, to avoid compromising the integrity of the brain tissue with fiber implants and light stimulation, we prioritized the PET experiments and the postmortem validation of ChRERα expression over the functional aspects of optogenetic stimulation in this species. Although the in vitro characterization and in vivo experiments in rats suggest that ChRERα behaves equivalently to ChR2, assessment of ChRERα function in NHPs needs to be established in subsequent studies. Given the difficulty of conducting NHP optogenetic studies, subsequent studies using [18F]FES should aim to develop other opsin-ERα-LBD fusions that may have a more favorable capacity for optogenetic neuromodulation in NHPs (more potent, red-shifted opsins). In any case, ChRERa represents a proof of principle that is expected to facilitate functional optogenetics experiments in NHPs and, at the very least, the development of other opsins for use in NHPs. In summary, with the modular technology described here, we introduce a tool to monitor the expression of transgenic proteins that lack a known LBD and analyze their neuroanatomical distribution in a noninvasive, quantitative, and longitudinal manner.
MATERIALS AND METHODS
Study design
We designed this study to test our hypothesis that the ERα-LBD and [18F]FES could serve as a PET reporter system for visualizing opsins in the brains of rodents and NHPs in a noninvasive and longitudinal manner. To achieve this aim, we engineered ChRERα, an opsin that consists of ChR2 and the LBD of the human ERα. We performed biochemistry in HEK-293 cells and patch-clamp recordings in HEK-293 cells and mouse brain slices to validate the membrane expression and functionality of ChRERα. Patch-clamp experiments in mice and in vivo optogenetics in rats were performed to show that ChRERα can be used to manipulate neuronal circuits in vivo. FES-PET imaging studies in rats and NHPs (n = 2) were conducted to show that the expression of ChRERα could be traced in vivo. We selected group sizes on the basis of historical data where we used similar procedures. Animals were randomly assigned to experimental groups. Researchers were not blinded to animal group assignments or treatments. All animal experiments were conducted after approval by the National Institute on Drug Abuse Intramural Research Program Institutional Animal Care and Use Committee and complied with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and ethical regulations.
Plasmid cDNA and AAV production
We generated a plasmid encoding a chimeric protein consisting of channelrhodopsin ChR2(H134R) and ERα-LBD tethered by a linker containing a V5 epitope (ChR2–V5–ERα-LBD or “ChRERα”) and a plasmid encoding the V5-tagged ERα-LBD without the ChR2 domain (“V5–ERα-LBD”). Polymerase chain reaction was used to amplify ChR2 and ERα-LBD (Q5 Hotstart polymerase, New England Biolabs) with linkered primers. Amplicons were cloned into pcDNA3.1 using ligation-independent cloning (LIC; In-Fusion Cloning Mix, Takara Inc.). The open-reading frame for ERα-LBD was amplified with a V5-linker from pCK025 [a gift from J. Stuttmann (Addgene, plasmid # 105415; http://n2t.net/addgene:105415; RRID: Addgene_105415)]. The open-reading frame for hChR2(H134R) was amplified from pAAV-hSyn-hChR2(H134R)-EYFP [a gift from K. Deisseroth (Addgene, plasmid #26973; http://n2t.net/addgene:26973; RRID: Addgene_26973)]. These constructs were transfected in vitro in HEK-293 cells for radioligand binding assays and patch-clamp recordings during light stimulation. The coding region for ChRERα was also cloned into a AAV packaging vector driven by the promoter for human synapsin 1 (Syn1) to facilitate expression in neurons. This plasmid was packaged into AAV serotype 5 viral particles (AAV2/5-hSyn1-ChRERα; 1.32 × 1014 gc/ml; Boston Children’s Hospital Viral Vector Core).
Cell culture and transfection
HEK-293 cells were grown in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) with 5% bovine growth serum (HyClone, Logan, UT) and antibiotic/antimycotic supplements (Gibco) in an incubator at 37°C and 5% CO2. HEK-293 cells were seeded on 150-mm dishes and transfected with plasmids expressing ChRERα or the control constructs V5-ERα-LBD or ChR2(H134R)-EYFP using calcium phosphate precipitation (National Institute on Drug Abuse Intramural Research Program and Genetic Engineering and Viral Vector Core, NIDA-IRP GEVVC Project 19042). Cell harvesting for Western blot and radioligand binding assays was performed about 48 hours after transfection.
Western blot
Transfected HEK-293 cells were harvested and frozen at −80°C until undergoing cytosolic and membrane fraction preparation for Western blot analysis. Frozen cell pellets were thawed and homogenized using a polytron in 10 ml of ice-cold buffer [50 mM tris-HCl (pH 7.4) with protease inhibitor cocktail (Sigma-Aldrich)]. The mixture was then centrifuged for 50 min at 4°C (20,000 rcf), and the supernatant was collected as the cytosolic fraction. The remaining pellet was resuspended with a polytron in 10 ml of ice-cold tris-HCl buffer and centrifuged again for 50 min at 4°C (20,000 rcf). The supernatant was then discarded, and the remaining pellet (membrane fraction) was stored at −80°C until further use as the membrane fraction. Protein concentration was determined using the bicinchoninic acid assay kit (Pierce).
On the first day of the Western blot, the frozen membrane fraction was thawed and resuspended in ice-cold buffer [50 mM tris-HCl (pH 7.4) with protease inhibitor cocktail] to achieve a concentration of ~2 mg of protein/ml. Aliquots containing 20 μg of protein of the cytosolic and resuspended membrane fractions were mixed with 2× Laemmli sample buffer (1:1 volume), and β-mercaptoethanol was added as a reducing agent. Samples were then incubated at 95°C for 5 min. After incubation, samples were vortexed and placed on ice. An electrophoresis tank was filled with ice-cold running buffer (Bolt, Thermo Fisher Scientific) and a precast gel (NuPage Bis-Tris with a 4 to 12% polyacrylamide gradient, Thermo Fisher Scientific). A molecular weight protein ladder (LI-COR Biosciences Chameleon Duo prestained protein ladder) was loaded into a well of the gel along with the prepared cytosolic and membrane samples (~20 μg) after quick centrifuge at 4°C. Electrophoresis was run for 15 min at 50 V, and then the voltage was increased to ~125 V for about 1 hour. After the run, the gel was removed, rinsed in double-distilled water (ddH2O), then placed in a transfer stack, and run in an iBlot2 Western blotting system (Thermo Fisher Scientific). The transferred stack membrane was rinsed in ddH2O and then incubated in LI-COR Biosciences blocking buffer (LI-COR Biosciences) for 1 hour at room temperature (RT). The blocking buffer was then removed and replaced with primary antibody (V5 Rabbit Polyclonal, Invitrogen PA1-993) diluted in LICOR blocking buffer (1:1500) and incubated overnight at 4°C. On the second day, the membrane was rinsed with washing buffer [1× phosphate-buffered saline (PBS) + 0.1% Tween 30; 3× for 5 min) and then incubated in secondary antibody (IRDye 800CW, donkey anti-rabbit, LI-COR, 926-32213) for 2 hours at RT. After secondary antibody incubation, the membrane was washed with washing buffer (3× for 5 min, RT), rinsed in ddH2O, air-dried on filter paper, and then imaged using a LI-COR Odyssey CLx scanner (LI-COR Biosciences).
Radioligand binding assays
Transfected HEK-293 cells were harvested 48 hours after transfection. Cells were suspended in 50 mM tris-HCl (pH 7.4) supplemented with protease inhibitor cocktail (1:100; Sigma-Aldrich, St. Louis, MO, USA). HEK-293 cells were disrupted with a polytron homogenizer (Kinematica, Basel, Switzerland). Homogenates were centrifuged at 48,000g (50 min, 4°C) and washed twice under the same conditions to isolate the membrane fraction. Protein was quantified by the bicinchoninic acid method (also known as Pierce). For saturation experiments, membrane suspensions (50 μg of protein/ml) were incubated for 2 hours at RT in 50 mM tris-HCl (pH 7.4) containing 10 mM MgCl2 and increasing concentrations of [3H]E2 (0.1 to 5 nM, 88 Ci/mmol; PerkinElmer). Nonspecific binding was determined under the same conditions with the presence of cold E2 (10 μM). For competition experiments, membrane suspensions (50 μg of protein/ml) were incubated for 2 hours at RT in 50 mM tris-HCl (pH 7.4) containing 10 mM MgCl2, [3H]E2 (1 nM, 88 Ci/mmol; PerkinElmer), and increasing concentrations of cold E2 and FES (0.1 nM to 1 mM). Nonspecific binding was determined in the presence of 10 μM cold E2. In all cases, free and membrane-bound radioligands were separated by rapid filtration of 500-μl aliquots in a 96-well plate harvester (Brandel, Gaithersburg, MD, USA) and then washed with 2 ml of ice-cold tris-HCl buffer. MicroScint-20 scintillation liquid (65 μl per well; PerkinElmer) was added to the filter plates. Plates were incubated overnight at RT, and radioactivity counts were determined in a MicroBeta2 plate counter (PerkinElmer, Boston, MA, USA) with an efficiency of 41%. Saturation curves and one-site competition curves were fitted using Prism 9 (GraphPad Software, La Jolla, CA, USA). Ki values were calculated using the Cheng-Prusoff equation (49).
Electrophysiology in cell culture
HEK-293 cells transfected with either ChRERα or ChR2(H134R)-EYFP were exposed to blue light stimulation (473 nm, 1-s pulses) during single-cell patch-clamp recordings. Light stimulation was applied while holding cells at incremental voltages (−120 to 80 mV) during patch-clamp recordings in extracellular solution [140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, and 10 mM glucose (pH 7.4)]. Steady-state photocurrent amplitudes were measured for ChRERα or ChR2(H134R)-EYFP and plotted against holding voltage.
Experimental animal subjects
Wild-type C57BL/6J mice (Charles River Laboratories, 20- to 25-g weight, three males) and wild-type Sprague-Dawley rats (Charles River Laboratories, 250- to 500-g weight, 12 males and 8 females) were used. The three mice were group-housed. Rats were housed two per cage until surgery; from then, they were single-housed. Two male squirrel monkeys (Saimiri sciureus; ~17 to 20 years, 850 and 950 g) were also used. Monkeys were pair-housed, were provided water ad libitum in their home cage, and received a daily ration of high-protein monkey diet (LabDiet 5045, St. Louis, MO). In addition, fresh fruits and vegetables were provided as daily enrichment. All animals were housed in a temperature- and humidity-controlled vivarium that followed a 12-hour light/12-hour dark cycle (lights on at 7:00 a.m.). One monkey was euthanized, and its brain was processed for IHC and immunocytochemistry. The other monkey died of natural causes (potential heart failure).
AAV injections
Rat surgeries
Animals were anesthetized with isoflurane and/or a mix of ketamine/xylazine and positioned in a stereotaxic apparatus (Kopf). A custom-made AAV with 2/5 serotype carrying the ChRERα construct (ERα ligand ChR2 (hChR2-H134R) fused to ERα ligand-binding domain through a V5 linker under the hSyn1 promoter (AAV2/5-Syn1-ChRERα) was intracranially injected. Stereotaxic coordinates were determined from the Rat Atlas (Paxinos, George, and Watson, 6th ed.) to target one of the following regions: PrL/ACd [anteroposterior (AP) = 3.0, mediolateral (ML) = 0.6, dorsoventral (DV) = −3.5], VTA (AP: −5.5, ML: 0.8, DV: −8.2), or left M1 (AP = 2.0, ML = 3.0, DV = −2.0 and − 2.1). Rats received 0.5 μl of AAV (1.32 × 1014 gc/ml) at a flow rate of 100 nl/min using a Hamilton Neuros 33-gauge syringe driven with a Neurostar robotic injection system. After the surgery, the incision site was sutured, and analgesics were administered. Three additional control rats were injected in either the right PrL/ACd (n = 1 male) or left M1 (n = 2, 1 female and 1 male) with non-ChRERα AAVs (AAV2/5-Syn1-PSAM-GlyR-3XFLAG (2.61 × 1013 gc/ml; Addgene) and AAV2/5-Syn1-HA-hM3Dq (3.5 × 1013 gc/ml; Addgene) for use in assessing potential effects of intracranial surgery on FES-PET imaging. The same surgery/injection procedures were used (a total of 500 μl of virus at a flow rate of 100 nl/min using Hamilton 33-gauge syringe/Neurostar injection system). Control animals received identical postoperative care as AAV-ChRERα rats and underwent [18F]FES-PET scans 3 weeks after AAV surgery.
Nonhuman primate surgeries
Two adult male squirrel monkeys received intracranial injections of the AAV2/5-hSyn-ChRERα (1.32 × 1014 gc/ml) in the left M1 forelimb region. Animals were sedated with ketamine [10 mg/kg, intramuscularly)] and then received atropine (0.02 to 0.05 mg/kg, intramuscularly) or glycopyrrolate (0.005 to 0.01 mg/kg, intramuscularly). Vital signs were continuously monitored (body temperature, heart and respiration rate, and oxygen and CO2 concentration). Isoflurane was delivered (1.0 to 2.0% in O2) by mask until intubation and positioning in a stereotaxic apparatus. Injection sites were first estimated from individual structural MRIs and stereotaxic coordinates relative to center of fixed ear bars (AP = 14.2, ML = 10.6, DV = 26.0). Sites were then precisely defined with intracortical microstimulation. A tungsten microelectrode was lowered to the approximate depth of layer 5 (1.8 mm below surface). Trains of microstimulation pulses were delivered at each site (18 pulses, 0.2-ms pulse width, 300 Hz). Current amplitude was increased until a movement was evoked (up to 80 μA). The motor response was classified according to the location of muscle twitches (shoulder, elbow, etc). During motor mapping, anesthesia was maintained with ketamine infusion (~5 to 6 mg/kg per hour, intravenously) and isoflurane (0.25 to 0.5% in nitrous oxygen mixed 50:50). Injections proceeded once the target sites were finalized. Three injections separated by 0.75 mm (1.33 μl per injection) were made in each hemisphere at depths of 1.0, 1.75, and 2.5 mm below the cortical surface. A 10-μl Hamilton Neuros syringe was used (flow rate of 0.33 μl/min, 5 min between injections). After injections, the site was covered with artificial dura and sutured closed. Animals received postoperative care, and their health was closely monitored, allowing a minimum of 5 weeks before any further experimentation.
Immunohistochemistry
Rats
Rats were anesthetized with pentobarbital and transcardially perfused with PBS, followed by 4% PFA. The brains were stored in 4% PFA overnight at 4°C and then placed in PBS with 30% sucrose for 3 to 4 days at 4°C. Brains were frozen and sectioned on a cryostat (30 to 40 μm; Leica, Germany), and slices were collected in PBS. On the first day of IHC, brain slices were washed in PBS with 0.1% Triton X-100 (washing buffer) for 30 min (3 × 10 min washes at RT). Slices were then blocked with 3% bovine serum albumin (BSA) in washing buffer (blocking buffer, 1 hour at RT) and incubated with specific primary antibodies, anti-ChR2 primary antibody (1:500 in blocking buffer; mouse monoclonal American Research Products, 03-651180), a rabbit anti-GFAP (1:500; Agilent Technologies Singapore, Z0334), or a rabbit anti-Iba1 (1:700; Wako Pure Chemical Industries, 019-19741) at 4°C overnight. Slices were soaked in washing buffer (3× for 10 min at RT) and then incubated in a secondary antibody mix in PBS: anti-mouse Alexa Fluor 488 (1:500), Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG) (H+L) (1:300; Invitrogen, A-11034), or goat anti-mouse IgG H+L (Cy3, 1:300; Abcam, ab97035) and 4′,6-diamidino-2-phenylindole (DAPI; 1:1000) for 2 hours at RT (protected from light). After secondary antibody incubation, slices were washed once more and then mounted onto glass slides using aqueous mounting medium [90% glycerol + 30 mM tris-HCl (pH 8.0)] with a glass coverslip. Images were taken using a Leica DFC7000T microscope and processed with LasX software.
Nonhuman primates
One squirrel monkey was deeply anesthetized and transcardially perfused with PBS, followed by a fixative solution containing 4% PFA, 0.15% glutaraldehyde, and 0.15% picric acid. The brain was removed and separated into blocks. These blocks were stored at 4°C in 4% PFA for 3 days (fresh PFA solution was changed daily) and then rinsed in PBS before transferring to a solution with 10% sucrose. The blocks were subsequently transferred to PBS with 20% sucrose and then 30% sucrose over the following 2 days and stored at 4°C until sectioning. Blocks were sectioned using a cryostat (30 to 40 μm) and stored in a cryoprotectant solution at −80°C or in PBS with 0.1% sodium azide at 4°C. On the first day of IHC treatment, slices were transferred to fresh PBS and then rinsed with washing buffer (PBS with 0.1% Triton X-100) at RT (3× for 10 min). Slices were then incubated in blocking buffer (3% BSA in PBS) for 1 hour at RT, rinsed with PBS (3× for 10 min wash), and soaked in a background suppressor solution (Biotium-TrueBlack IF Background Suppressor) for 10 min at RT. Slices were then transferred to a primary antibodies (mouse anti-ChR2 solution, 1:300; rabbit anti-GFAP, 1:500; and rabbit anti-Iba1, 1:700) in TrueBlack IF Blocking Buffer and incubated overnight at 4°C. On the second day of IHC, treated slices were rinsed with PBS (3× for 10 min) and transferred to a secondary antibody solution (anti-mouse Alexa Fluor 594 or Cy3; 1:500 or anti-rabbit Alexa Fluor 488 and DAPI, 1:1500 in TrueBlack IF Background Suppressor) for 2 hours at RT (protected from light). After the secondary antibody incubation, slices were rinsed with PBS (3× for 10 min) and mounted onto a glass slide. The slice was then covered with Biotium-TrueBlack lipofuscin quencher (1:40 in PBS, 175 μl) for 10 min at RT. The slide was then rinsed briefly in PBS (3× for 10 s) and covered with aqueous mounting media [90% glycerol + 30 mM tris-HCl (pH 8.0)] and a glass coverslip (product information). Images were taken using a Leica DFC7000T microscope and processed with LasX software.
Confocal fluorescence microscopy.
Free-floating coronal sections (40 μm) from rats with viral injection of pAAV2/5-Syn1-ChRERα into the right PrL/ACd or left M1 and free-floating horizontal sections (80 μm) from squirrel monkey with viral injection of pAAV2/5-hSyn-ChRERα into the left M1 were incubated for 1 hour in phosphate buffer (PB) supplemented with 4% BSA and 0.3% Triton X-100. Sections were then incubated with the specific primary antibody, mouse anti-ChR2 (1:250; American Research Products Inc., 03-651180), overnight at 4°C. After rinsing 3× for 10 min in PB, sections were incubated in the fluorescence secondary antibody Alexa Fluor 594 AffiniPure donkey anti-mouse IgG (H+L) (1:100; Jackson ImmunoResearch Laboratories Inc., 715-585-151) for 2 hours at RT. After rinsing, sections were mounted with mounting medium with DAPI on slides. Fluorescent images were collected with the Zeiss LSM880 with Cy7.5 Confocal System (Zeiss). Images were taken sequentially with different lasers with a 20× objective. This experiment was successfully repeated three times.
Electron microscopy.
Vibratome tissue sections (40 μm) from rats with viral injection of pAAV2/5-hSyn-ChRERα into the right PrL/ACd or left M1 and vibratome tissue sections (80 μm) from squirrel monkey with viral injection of pAAV2/5-hSyn-ChRERα into the left M1 were rinsed and incubated with 1% sodium borohydride to inactivate free aldehyde groups, rinsed, and then incubated with blocking solution (1% normal goat serum and 4% BSA in PB) supplemented with 0.02% saponin. Sections were then incubated with primary antibody mouse anti-ChR2 (1:250; American Research Products Inc., 03-651180) diluted in blocking solution for 24 hours at 4°C. Sections were rinsed and incubated in the secondary antibody Nanogold-Fab′ goat anti-mouse IgG (H+L) (1:100; Nanoprobes, 2002) overnight at 4°C. Sections were rinsed in PB and then in ddH2O, followed by silver enhancement of the gold particles with the Nanoprobe Silver Kit (2012, Nanoprobes) for 7 min at RT. Sections were rinsed with PB and fixed with 0.5% osmium tetroxide in PB for 25 min, washed in PB, followed by ddH2O, and then contrasted in freshly prepared 1% uranyl acetate for 35 min. Sections were dehydrated through a series of graded alcohols and with propylene oxide. Afterward, they were flat embedded in Durcupan ACM epoxy resin (14040, Electron Microscopy Sciences). Resin-embedded sections were polymerized at 60°C for 2 days. Sections of 60 nm were cut from the outer surface of the tissue with an ultramicrotome UC7 (Leica Microsystems) using a diamond knife (Diatome). The sections were collected on formvar-coated single-slot grids and counterstained with Reynold’s lead citrate. Sections were examined and photographed using a Tecnai G2 12 transmission electron microscope (Thermo Fisher Scientific) equipped with the OneView digital micrograph camera (Gatan). Serial ultrathin sections were analyzed. Synaptic contacts were classified according to their morphology and immunolabel and photographed at a magnification of ×6800 to ×13,000. The morphological criteria used for identification and classification of cellular components or type of synapse observed in these thin sections were as previously described (50). In the serial sections, a terminal, dendrite, or cell body containing greater than five immunogold particles was considered as immunopositive terminal, dendrite, or cell body. Pictures were adjusted to match contrast and brightness by using Adobe Photoshop (Adobe Systems Incorporated, Seattle, WA). This experiment was successfully repeated three times. EM and confocal analysis quantification were blinded and performed as described previously (50).
Mouse brain slice electrophysiology
Slice electrophysiology experiments were carried out as previously described (51, 52). Briefly, mice were injected with AAV2/5-Syn1-ChRERα in PrL/ACd and 3 weeks later were anesthetized with euthasol (Butler-Schein) before decapitation. Brains were rapidly removed and placed in ice-cold N-methyl-d-glucamine (NMDG)–based cutting solution containing 92 mM NMDG, 20 mM Hepes, 25 mM glucose, 30 mM NaHCO3, 2.5 mM KCl, 1.2 mM NaPO4, 5 mM sodium ascorbate, 3 mM sodium pyruvate, and 2 mM thiourea saturated with 95% O2/5% CO2 with an osmolarity of 303 to 306 mosM. Coronal sections (300 μm) containing the ventromedial prefrontal cortex (vmPFC) were obtained by using a Leica VT1200 vibratome. Then, slices were transferred to modified holding artificial cerebrospinal fluid (aCSF) saturated with 95% O2/5% CO2 containing 92 mM NaCl, 20 mM Hepes, 25 mM glucose, 30 mM NaHCO3, 2.5 mM KCl, 1.2 mM NaPO4, 5 mM sodium ascorbate, 3 mM sodium pyruvate, and 2 mM thiourea (303 to 306 mosM) at RT. Slices were allowed to incubate at RT in this solution for at least 1 hour before being transferred to the recording chamber.
The recording chamber was kept at 32°C and perfused with a pump at a flow rate of 1.5 to 2.0 ml/min with aCSF containing 126 mM NaCl, 2.5 mM KCl, 1.4 mM NaH2PO4, 1.2 mM MgCl2, 2.4 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose (303 to 305 mosM) for recordings. Whole-cell recordings were made using glass microelectrodes (2 to 3 megohms) containing 135 mM K-gluconate, 10 mM Hepes, 4 mM KCl, 4 mM Mg–adenosine 5′-triphosphate, and 0.3 mM Na–guanosine 5′-triphosphate. Cells were identified using IR-DIC optics on an inverted Olympus BX5iWI microscope. Optogenetically evoked responses were elicited using a 473-nm laser directed at the brain slice (ThorLabs). Data were filtered at 2 kHz and digitized at 20 kHz using a 1440A Digidata Digitizer (Molecular Devices). For current-clamp recordings, train pulses (1 ms) were applied. Neurons were voltage-clamped at −70 mV using a MultiClamp 700B amplifier (Molecular Devices), and a 500-ms pulse of light was delivered. Photocurrents mediated by opsins in response to light exhibited different biophysical phases reflecting distinct events during the photocycle. During light stimulation, the peak current (Ipeak), largely voltage independent, was first observed. Then, such current decayed to a lower stationary amount (Istationary) in a voltage-dependent manner. The latter represents the rate-limiting step of the pump cycle. The amplitude of the current evoked at the peak of the response (IPeak) and the amplitude of the current evoked at the end of the pulse (IStationary) were quantified. Series resistance (10 to 20 megohms) was monitored using a −5-mV voltage step. Cells with >20% change in series resistance were discarded from further analysis.
FDG-PET with light stimulation
A group of rats (n = 5) were injected with AAV2/5-Syn1-ChRERα in PrL/ACd (AP = 3.0, ML = 0.6, DV = −3.3) and were later implanted with an optic fiber (ThorLabs) targeting just above the AAV injection site (AP = 3.0, ML = 0.6, DV = −3.1). For fiber implantation, a hole was made in the skull above the AAV site, and an optic fiber cannula (2.5-mm ferrule, 200-μm core, ThorLabs) was stereotaxically lowered until the optic fiber was just above the injection site (AP = 3.0, ML = 0.6, DV = −3.1). The optic fiber cannula was secured with dental cement and supporting screws in the skull. A group of control rats (AAV naïve, n = 4) were also implanted with an optic fiber at these coordinates. These rats were anesthetized with isoflurane (~2%) in prone position and hooked up to an optic fiber patch cable connected to a controllable light-emitting diode (LED) light source (473 nm; Plexon Inc.). Anesthetized rats received an intraperitoneal injection of [18F]-FDG (~0.5 mCi in 0.5 ml of saline) and then underwent a 30-min light stimulation protocol (473 nm, 3-ms pulse trains at 50 Hz; 300 ms on, 700 ms off for 30 min) controlled by Radiant software (Plexon Inc). After the [18F]-FDG uptake stimulation period, rats were scanned in a Mediso micro-PET/CT scanner for ~25 min (20 min PET plus ~5 min CT). Scan data were reconstructed using Nucline software with CT-based attenuation correction. The 20-min PET acquisitions were processed as static reconstructions for each animal and then analyzed using PMOD (PMOD Technologies). Each animal’s scan was registered to a template rat MRI brain (Schiffer) in PMOD. A pixelwise comparison was performed using MATLAB (Mathworks) SPM12 (University College London) to assess potential differences in uptake of [18F]-FDG between ChRERα versus controls.
Locomotor behavior in rats
A group of rats (n = 5) were injected with AAV2/5-Syn1-ChRERα unilaterally in the VTA (AP: −5.5, ML: 0.8, DV: −8.2) and were later implanted with an optic fiber (ThorLabs), as described previously, but this time targeting ipsilateral projections in the NAc (AP: 1.3, ML: −1.0, DV: −6.5). A group of rats with no AAV were also implanted with optic fibers to serve as controls. During locomotor behavior experiments, awake rats were hooked up to an optic fiber patch cable connected to a controllable LED light source (473 nm; Plexon Inc.) in an open-field chamber. Locomotor behavior was acquired with video recording using CinePlex software (Plexon Inc.), and the total distance traveled was calculated during blocks with and without light stimulation (473 nm, 3-ms pulse trains at 50 Hz; 300 ms on, 700 ms off for 5 min). The statistical analysis of distance traveled included two factors (AAV group and light stimulation) matched by subject to assess effects of light stimulation and the interaction effect between AAV-ChRERα transduction and light stimulation.
[18F]FES-PET acquisition
All PET/CT scans were acquired using a Mediso nanoScan PET/CT scanner, and data reconstruction was performed with Nucline nanoScan software (3.04.101.0000). In rat scans, male and female Sprague Dawley rats (n = 10 male and 6 female) were anesthetized and maintained on isoflurane (1.5 to 2.5%) during the whole procedure. Tail vein catheters were inserted for intravenous radiotracer delivery. [18F]FES was injected (0.5 to 1 mCi in ~0.5 ml, followed by 0.5 ml of saline) immediately at the start of scan, and scan duration lasted 90 to 120 min. Baseline scans were performed in a group of rats with no AAV (n = 5) and also in rats with non-ChRERα AAV (n = 3, details in AAV injection procedures, scanned 3 weeks after surgery) to serve as controls in FES-PET scan analysis. Post-AAV scans were acquired at least 3 weeks after injection in the group of rats receiving AAV-ChRERα in right PrL/ACd (n = 6) or left M1 (n = 5). In squirrel monkey scans, animals were anesthetized first with ketamine (intramuscularly) and then intubated and maintained on isoflurane (1.5 to 2.5%) oxygen mixture. An intravenous line with injection port was established in the hindlimb, and the animals were placed supine in the PET scanner. A CT scan was performed before radioligand injection and PET acquisition. [18F]FES (1.5 to 2 mCi) was diluted in sterile saline (~0.5 ml) and injected into the port, followed by saline (~1 ml). The animals were scanned for 90 min while physiological parameters (heart rate, body temperature, respiration rate, and oxygen saturation) were continuously monitored throughout the procedure. The animals were given fluids and carefully monitored during recovery after the scan.
[18F]FES-PET analysis
Rats
Each PET/CT scan was coregistered to a template rat MRI brain (Schiffer) in PMOD. PET acquisition values were first converted to SUVs (grams per milliliter) to correct for injected dose and body weight of the animal. We then normalized scans using the cerebellar gray matter atlas volume of interest (VOI) to calculate SUVRs. This VOI was chosen as a reference region because of the low endogenous binding observed at baseline and relatively large size. Time activity curves of SUV and SUVR were plotted, and a period of 20 to 60 min after injection was chosen (highest signal to noise) to calculate average VOI SUVRs. In addition, binding potential (BPND) was calculated using the pixel-based Logan reference tissue method, using PMOD, and the gray matter cerebellum (CB) was again used as the reference region (53). VOIs were determined using a 2-mm sphere centered at the site of interest with a 50% isocontour filter. In squirrel monkeys, each PET/CT scan was coregistered to their individual structural MRIs. SUV, SUVR, and BPND were calculated in the same way as performed in rats. Individual VOIs were determined using a 3-mm sphere centered at site of interest with a 50% isocontour filter. For defining the AAV injection site VOI, the 3-mm sphere was centered at the middle of the stereotaxic coordinate injection sites, and a 50% isocontour filter was applied.
fMRI functional connectivity acquisition and analysis
MRI data were acquired with a Bruker Biospin 9.4-T scanner on a Paravison 6.0.1 platform equipped with an active-shielded gradient coil and a quadrature transmitter/receiver RF coil (Bruker Medizintechnik, Karlsruhe, Germany). Functional images were acquired using a gradient-recalled echo-planar imaging (EPI) sequence (TR = 1.5 s, TE = 15 ms, with an isotropic resolution of 1 mm). Geometric distortions in EPI images were corrected using a reverse k-space trajectory EPI sequence developed in-house. Thirty-five functional scans (N = 9 squirrel monkeys) were acquired under intravenous dexmedetomidine (10 to 16 μg/kg per hour) and 0.5% inhaled isoflurane (54). Each functional connectivity acquisition lasted for 7.5 min. After manual skull stripping, the preprocessing was done by a set of homemade scripts based on three data analysis packages: AFNI (version: 20200809), FSL (5.0.9), and FreeSurfer (version: stable 5, 20130513). In detail, images were processed by discarding the first four temporal samples, slice-timing correction, head motion correction, physiological noise correction [CompCor; (55)], bandpass filtering (0.01 to 0.1 Hz), nonlinear alignment to the VALiDATe29 template, and smoothing to 2 mm using AFNI’s 3dBlurToFWHM. Anatomical normalization was performed using linear robust alignment by FreeSurfer’s mri_robust_register (56), followed by FSL’s nonlinear alignment (FNIRT). After Pearson correlation between the mean voxel signal within the brain mask with the mean time series of the 2-mm M1 region of interest sphere (centered at the maximum intensity peak of the [18F]FES signal in the left M1), we converted the Pearson correlation coefficient to z score by Fisher’s transformation. The 35 z-scored functional connectivity data points were fed into a permutation-based one-sample t test to identify significant group-level correlated brain regions. Group level functional connectivity was based on voxel-wise inference and a statistical threshold determined using permutation testing [PALM; (57)] with threshold free cluster enhancement [TFCE; (58, 59)] and multiple comparisons correction maintaining FWE <0.05. We also calculated the Pearson correlation and Dice overlap index (Sorensen-Dice index) between the PET binding and the fMRI functional connectivity spatial spread.
Statistics
Data were analyzed using GraphPad Prism 9, San Diego, CA (www.graphpad.com), unless otherwise noted, using two-way ANOVA taking repeated measures into account when appropriate or with independent t tests and paired-sample t tests, as defined in the text. Post hoc tests were performed using the Šidák multiple comparison correction. Voxel-wise data were corrected for multiple comparisons [probabilistic TFCE; (59)]. For correlation analyses, we used the Pearson correlation with Fisher’s z transformation.
Supplementary Material
Acknowledgments:
We thank R. Ye and K. Yu of the Confocal and Electron Microscopy Core, NIDA IRP, for confocal and immuno-EM images used in this study.
Funding:
This work was supported by the National Institute on Drug Abuse Intramural Research Program (ZIADA000069 to M. Michaelides and ZIADA000622 to C.B.), the National Institute of Biomedical Imaging and Bioengineering P41 (EB024495 to M.G.P.), and Spain’s Ministerio de Ciencia e Innovación (RYC-2019-027371-I to J.B.).
Footnotes
Competing interests: M. Michaelides has received research funding from AstraZeneca, Redpin Therapeutics, and Attune Neurosciences. All other coauthors report no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The plasmids for ChRERα can be obtained directly from Addgene (https://addgene.org/; plasmid IDs: 201818, 201819, and 201820).
REFERENCES AND NOTES
- 1.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K, Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Wang L-P, Boyden ES, Deisseroth K, Channelrhodopsin-2 and optical control of excitable cells. Nat. Methods 3, 785–792 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Zhang F, Wang L-P, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K, Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Chow BY, Boyden ES, Optogenetics and translational medicine. Sci. Transl. Med. 5, 177ps5 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Iyer SM, Vesuna S, Ramakrishnan C, Huynh K, Young S, Berndt A, Lee SY, Gorini CJ, Deisseroth K, Delp SL, Optogenetic and chemogenetic strategies for sustained inhibition of pain. Sci. Rep. 6, 30570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simunovic MP, Shen W, Lin JY, Protti DA, Lisowski L, Gillies MC, Optogenetic approaches to vision restoration. Exp. Eye Res. 178, 15–26 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Walker MC, Kullmann DM, Optogenetic and chemogenetic therapies for epilepsy. Neuropharmacology 168, 107751 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Sahel J-A, Boulanger-Scemama E, Pagot C, Arleo A, Galluppi F, Martel JN, Esposti SD, Delaux A, de Saint Aubert J-B, de Montleau C, Gutman E, Audo I, Duebel J, Picaud S, Dalkara D, Blouin L, Taiel M, Roska B, Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 27, 1223–1229 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Deubner J, Coulon P, Diester I, Optogenetic approaches to study the mammalian brain. Curr. Opin. Struct. Biol. 57, 157–163 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Kaas JH, The evolution of brains from early mammals to humans. Wiley Interdiscip. Rev. Cogn. Sci. 4, 33–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wise SP, Forward frontal fields: Phylogeny and fundamental function. Trends Neurosci. 31, 599–608 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalin NH, Shelton SE, Nonhuman primate models to study anxiety, emotion regulation, and psychopathology. Ann. N. Y. Acad. Sci. 1008, 189–200 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Garbarini N, Primates as a model for research. Dis. Model. Mech. 3, 15–19 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Tremblay S, Acker L, Afraz A, Albaugh DL, Amita H, Andrei AR, Angelucci A, Aschner A, Balan PF, Basso MA, Benvenuti G, Bohlen MO, Caiola MJ, Calcedo R, Cavanaugh J, Chen Y, Chen S, Chernov MM, Clark AM, Dai J, Debes SR, Deisseroth K, Desimone R, Dragoi V, Egger SW, Eldridge MAG, El-Nahal HG, Fabbrini F, Federer F, Fetsch CR, Fortuna MG, Friedman RM, Fujii N, Gail A, Galvan A, Ghosh S, Gieselmann MA, Gulli RA, Hikosaka O, Hosseini EA, Hu X, Hüer J, Inoue K-I, Janz R, Jazayeri M, Jiang R, Ju N, Kar K, Klein C, Kohn A, Komatsu M, Maeda K, Martinez-Trujillo JC, Matsumoto M, Maunsell JHR, Mendoza-Halliday D, Monosov IE, Muers RS, Nurminen L, Ortiz-Rios M, O’Shea DJ, Palfi S, Petkov CI, Pojoga S, Rajalingham R, Ramakrishnan C, Remington ED, Revsine C, Roe AW, Sabes PN, Saunders RC, Scherberger H, Schmid MC, Schultz W, Seidemann E, Senova Y-S, Shadlen MN, Sheinberg DL, Siu C, Smith Y, Solomon SS, Sommer MA, Spudich JL, Stauffer WR, Takada M, Tang S, Thiele A, Treue S, Vanduffel W, Vogels R, Whitmire MP, Wichmann T, Wurtz RH, Xu H, Yazdan-Shahmorad A, Shenoy KV, DiCarlo JJ, Platt ML, An open resource for nonhuman primate optogenetics. Neuron 108, 1075–1090.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bliss-Moreau E, Costa VD, Baxter MG, A pragmatic reevaluation of the efficacy of nonhuman primate optogenetics for psychiatry. Oxf. Open Neurosci 1, kvac006 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng C, Yuan H, Dai J, Behavioral manipulation by optogenetics in the nonhuman primate. Neuroscientist 24, 526–539 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Galvan A, Stauffer WR, Acker L, El-Shamayleh Y, Inoue KI, Ohayon S, Schmid MC, Nonhuman primate optogenetics: Recent advances and future directions. J. Neurosci. 37, 10894–10903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue K-I, Matsumoto M, Takada M, in Optogenetics: Light-Sensing Proteins and Their Applications in Neuroscience and Beyond, Yawo H, Kandori H, Koizumi A, Kageyama R, Eds. (Springer, 2021), pp. 345–358. [Google Scholar]
- 19.Bonaventura J, Eldridge MAG, Hu F, Gomez JL, Sanchez-Soto M, Abramyan AM, Lam S, Boehm MA, Ruiz C, Farrell MR, Moreno A, Galal Faress IM, Andersen N, Lin JY, Moaddel R, Morris PJ, Shi L, Sibley DR, Mahler SV, Nabavi S, Pomper MG, Bonci A, Horti AG, Richmond BJ, Michaelides M, High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat. Commun. 10, 4627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magnus CJ, Lee PH, Bonaventura J, Zemla R, Gomez JL, Ramirez MH, Hu X, Galvan A, Basu J, Michaelides M, Sternson SM, Ultrapotent chemogenetics for research and potential clinical applications. Science 364, eaav5282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagai Y, Miyakawa N, Takuwa H, Hori Y, Oyama K, Ji B, Takahashi M, Huang X-P, Slocum ST, DiBerto JF, Xiong Y, Urushihata T, Hirabayashi T, Fujimoto A, Mimura K, English JG, Liu J, Inoue K-I, Kumata K, Seki C, Ono M, Shimojo M, Zhang M-R, Tomita Y, Nakahara J, Suhara T, Takada M, Higuchi M, Jin J, Roth BL, Minamimoto T, Deschloroclozapine, a potent and selective chemogenetic actuator enables rapid neuronal and behavioral modulations in mice and monkeys. Nat. Neurosci. 23, 1157–1167 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Yaghoubi SS, Campbell DO, Radu CG, Czernin J, Positron emission tomography reporter genes and reporter probes: Gene and cell therapy applications. Theranostics 2, 374–391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J, Gambhir SS, Noninvasive detection of therapeutic cytolytic T cells with 18F–FHBG PET in a patient with glioma. Nat. Clin. Pract. Oncol. 6, 53–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mikhaylova E, Tabacchini V, Borghi G, Mollet P, D’Hoe E, Schaart DR, Vandenberghe S, Optimization of an ultralow-dose high-resolution pediatric PET scanner design based on monolithic scintillators with dual-sided digital SiPM readout: A simulation study. Phys. Med. Biol. 62, 8402–8418 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Hutton BF, Erlandsson K, Thielemans K, Advances in clinical molecular imaging instrumentation. Clin. Transl. Imaging 6, 31–45 (2018). [Google Scholar]
- 26.Du J, Bai X, Gola A, Acerbi F, Ferri A, Piemonte C, Yang Y, Cherry SR, Performance of a high-resolution depth-encoding PET detector module using linearly-graded SiPM arrays. Phys. Med. Biol. 63, 035035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boehm MA, Bonaventura J, Gomez JL, Solís O, Stein EA, Bradberry CW, Michaelides M, Translational PET applications for brain circuit mapping with transgenic neuromodulation tools. Pharmacol. Biochem. Behav. 204, 173147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duebel J, Marazova K, Sahel J-A, Optogenetics. Curr. Opin. Ophthalmol. 26, 226–232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen NT, Ma G, Zhou Y, Jing J, Optogenetic approaches to control Ca2+-modulated physiological processes. Curr. Opin. Physiol. 17, 187–196 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia M, Dahlman-Wright K, Gustafsson J-Å, Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 29, 557–568 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, Singh R, Bhasin S, Jasuja R, The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 812540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katzenellenbogen JA, The quest for improving the management of breast cancer by functional imaging: The discovery and development of 16α-[18F]fluoroestradiol (FES), a PET radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 92, 24–37 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paquette M, Ouellet R, Archambault M, Croteau É, Lecomte R, Bénard F, [18F]-fluoroestradiol quantitative PET imaging to differentiate ER+ and ERα-knockdown breast tumors in mice. Nucl. Med. Biol. 39, 57–64 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Khayum MA, de Vries EFJ, Glaudemans AWJM, Dierckx RAJO, Doorduin J, in vivo imaging of brain estrogen receptors in rats: A 16α-18F-fluoro-17β-estradiol PET study. J. Nucl. Med. 55, 481–487 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Thanos PK, Robison L, Nestler EJ, Kim R, Michaelides M, Lobo M-K, Volkow ND, Mapping brain metabolic connectivity in awake rats with μPET and optogenetic stimulation. J. Neurosci. 33, 6343–6349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boekhoudt L, Omrani A, Luijendijk MCM, Wolterink-Donselaar IG, Wijbrans EC, van der Plasse G, Adan RAH, Chemogenetic activation of dopamine neurons in the ventral tegmental area, but not substantia nigra, induces hyperactivity in rats. Eur. Neuropsychopharmacol. 26, 1784–1793 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Gharbawie OA, Stepniewska I, Kaas JH, Cortical connections of functional zones in posterior parietal cortex and frontal cortex motor regions in New World monkeys. Cerebral Cortex 21, 1981–2002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stepniewska I, Preuss TM, Kaas JH, Architectionis, somatotopic organization, and ipsilateral cortical connections of the primary motor area (M1) of owl monkeys. J. Comp. Neurol. 330, 238–271 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Stepniewska I, Gharbawie OA, Burish MJ, Kaas JH, Effects of muscimol inactivations of functional domains in motor, premotor, and posterior parietal cortex on complex movements evoked by electrical stimulation. J. Neurophysiol. 111, 1100–1119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Card NS, Gharbawie OA, Cortical connectivity is embedded in resting state at columnar resolution. Prog. Neurobiol. 213, 102263 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruiz O, Lustig BR, Nassi JJ, Cetin A, Reynolds JH, Albright TD, Callaway EM, Stoner GR, Roe AW, Optogenetics through windows on the brain in the nonhuman primate. J. Neurophysiol. 110, 1455–1467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nassi JJ, Avery MC, Cetin AH, Roe AW, Reynolds JH, Optogenetic activation of normalization in alert macaque visual cortex. Neuron 86, 1504–1517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salegio EA, Samaranch L, Kells AP, Mittermeyer G, San Sebastian W, Zhou S, Beyer J, Forsayeth J, Bankiewicz KS, Axonal transport of adeno-associated viral vectors is serotype-dependent. Gene Ther. 20, 348–352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Zhang L, Retrograde axonal transport property of adeno-associated virus and its possible application in future. Microbes Infect. 23, 104829 (2021). [DOI] [PubMed] [Google Scholar]
- 45.O’Shea DJ, Kalanithi P, Ferenczi EA, Hsueh B, Chandrasekaran C, Goo W, Diester I, Ramakrishnan C, Kaufman MT, Ryu SI, Yeom KW, Deisseroth K, Shenoy KV, Development of an optogenetic toolkit for neural circuit dissection in squirrel monkeys. Sci. Rep. 8, 6775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerits A, Farivar R, Rosen BR, Wald LL, Boyden ES, Vanduffel W, Optogenetically induced behavioral and functional network changes in primates. Curr. Biol. 22, 1722–1726 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Lao-Kaim NP, Roussakis A-A, Martín-Bastida A, Valle-Guzman N, Paul G, Soreq E, Daws RE, Foltynie T, Barker RA, Hampshire A, Piccini P, Longitudinal functional connectivity changes related to dopaminergic decline in Parkinson’s disease. NeuroImage Clin. 28, 102409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boon LI, Hillebrand A, Potters WV, de Bie RMA, Prent N, Bot M, Schuurman PR, Stam CJ, van Rootselaar A-F, Berendse HW, Motor effects of deep brain stimulation correlate with increased functional connectivity in Parkinson’s disease: An MEG study. NeuroImage Clin. 26, 102225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yung-Chi C, Prusoff WH, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973). [DOI] [PubMed] [Google Scholar]
- 50.Zhang S, Qi J, Li X, Wang H-L, Britt JP, Hoffman AF, Bonci A, Lupica CR, Morales M, Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat. Neurosci. 18, 386–392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pignatelli M, Tejeda HA, Barker DJ, Bontempi L, Wu J, Lopez A, Palma Ribeiro S, Lucantonio F, Parise EM, Torres-Berrio A, Alvarez-Bagnarol Y, Marino RAM, Cai Z-L, Xue M, Morales M, Tamminga CA, Nestler EJ, Bonci A, Cooperative synaptic and intrinsic plasticity in a disynaptic limbic circuit drive stress-induced anhedonia and passive coping in mice. Mol. Psychiatry 26, 1860–1879 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pignatelli M, Umanah GKE, Ribeiro SP, Chen R, Karuppagounder SS, Yau HJ, Eacker S, Dawson VL, Dawson TM, Bonci A, Synaptic plasticity onto dopamine neurons shapes fear learning. Neuron 93, 425–440 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL, Distribution volume ratios without blood sampling from graphical analysis of PET data. J. Cereb. Blood Flow Metab. 16, 834–840 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Brynildsen JK, Hsu L-M, Ross TJ, Stein EA, Yang Y, Lu H, Physiological characterization of a robust survival rodent fMRI method. Magn. Reson. Imaging 35, 54–60 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Behzadi Y, Restom K, Liau J, Liu TT, A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage 37, 90–101 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuter M, Rosas HD, Fischl B, Highly accurate inverse consistent registration: A robust approach. Neuroimage 53, 1181–1196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE, Permutation inference for the general linear model. Neuroimage 92, 381–397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith SM, Nichols TE, Threshold-free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44, 83–98 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Spisák T, Spisák Z, Zunhammer M, Bingel U, Smith S, Nichols T, Kincses T, Probabilistic TFCE: A generalized combination of cluster size and voxel intensity to increase statistical power. Neuroimage 185, 12–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The plasmids for ChRERα can be obtained directly from Addgene (https://addgene.org/; plasmid IDs: 201818, 201819, and 201820).