

Abstract
Background
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and is clinically characterized by the presence of motor (bradykinesia, rigidity, rest tremor and postural instability) and non-motor symptoms (cognitive impairment, autonomic dysfunction, sleep disorders, depression and hyposmia). The aetiology of PD is unknown except for a small but significant contribution of monogenic forms.
Sources of data
No new data were generated or analyzed in support of this review.
Areas of agreement
Up to 15% of PD patients carry pathogenic variants in PD-associated genes. Some of these genes are associated with mendelian inheritance, while others act as risk factors. Genetic background influences age of onset, disease course, prognosis and therapeutic response.
Areas of controversy
Genetic testing is not routinely offered in the clinical setting, but it may have relevant implications, especially in terms of prognosis, response to therapies and inclusion in clinical trials. Widely adopted clinical guidelines on genetic testing are still lacking and open to debate. Some new genetic associations are still awaiting confirmation, and selecting the appropriate genes to be included in diagnostic panels represents a difficult task. Finally, it is still under study whether (and to which degree) specific genetic forms may influence the outcome of PD therapies.
Growing points
Polygenic Risk Scores (PRS) may represent a useful tool to genetically stratify the population in terms of disease risk, prognosis and therapeutic outcomes.
Areas timely for developing research
The application of PRS and integrated multi-omics in PD promises to improve the personalized care of patients.
Keywords: Parkinson’s disease, genetics, tailored therapies, Polygenic Risk Scores
Background
PD general features, diagnosis and therapy
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease. PD is uncommon among individuals younger than 50 years, increases in prevalence with age and it is twice as common in men than in women in most populations.1
Characteristic features of PD include neuronal loss in specific areas of the substantia nigra and widespread intracellular protein (α-synuclein) accumulation, mainly in the form of Lewy bodies and Lewy neurites. Although neither the loss of pigmented dopaminergic neurons in the substantia nigra nor the deposition of α-synuclein in neurons is specific for PD, these two major neuropathologies are specific for a definitive diagnosis of PD when applied together.2
PD is clinically defined by the presence of motor and non-motor symptoms.1 Motor symptoms consist of bradykinesia, rigidity, rest tremor and postural instability. Bradykinesia is defined as slowness and progressively smaller movements (hypokinesia).1 Rigidity manifests as involuntary, velocity-independent resistance to the passive movement of a joint. Rest tremor is a 4–6 Hz tremor in a fully resting limb, which temporarily disappears when the limb is outstretched. Postural instability refers to balance impairment affecting the ability to change or maintain postures such as walking or standing and typically manifests as a late PD feature.
Non-motor symptoms such as cognitive impairment, autonomic dysfunction (especially constipation and orthostatic hypotension), sleep disorders, depression and impaired smell are part of the disease and add considerably to the overall burden.3
PD diagnosis is usually based on history and physical examination.4 Clinical diagnostic criteria require the presence of parkinsonism, defined as bradykinesia with rest tremor, rigidity or both. Dopamine transporter single-photon emission computed tomography (DaT-SPECT) identifies, with high accuracy, the presynaptic dopamine neuronal dysfunction presents in PD and parkinsonisms by demonstrating reduced uptake of a radioactive tracer that binds to dopamine transporters in the basal ganglia. However, DaT-SPECT is generally useful when the presence of parkinsonism is uncertain on examination but cannot differentiate between PD and other parkinsonisms (e.g. multiple system atrophy, progressive supranuclear palsy).5
Pharmacological treatments for PD motor symptoms are primarily dopamine-based.6 Levodopa preparations, dopamine agonists and monoamine oxidase-B (MAO-B) inhibitors are useful initial therapies. Therapy adjustments are often required during the disease course.
Advanced therapies for motor symptoms include deep brain stimulation (DBS), MRI-guided focused ultrasound and therapy with levodopa-carbidopa enteral suspension. These approaches are appropriate for PD patients with off periods or dyskinesias not responsive to medication adjustments.
Etiopathogenesis of PD
Early studies on PD etiopathogenesis demonstrated that mitochondrial dysfunction, protein accumulation and ageing are major disease contributors. Moreover, the identification of several families exhibiting a Mendelian inheritance pattern and twin studies provided evidence on the contribution of genetics in PD development, which culminated around mid-90’s in the discovery of the first PD-associated gene, namely α-synuclein (SNCA).7
In the following years, the molecular characterization of PD patients and families led to the identification of several disease-associated genes. Some genes are associated with a Mendelian inheritance pattern, while others increase the risk of PD development with ageing. Several studies in the last decade tried to disentangle the underlying molecular pathogenic mechanisms (Fig. 1).
Fig. 1.
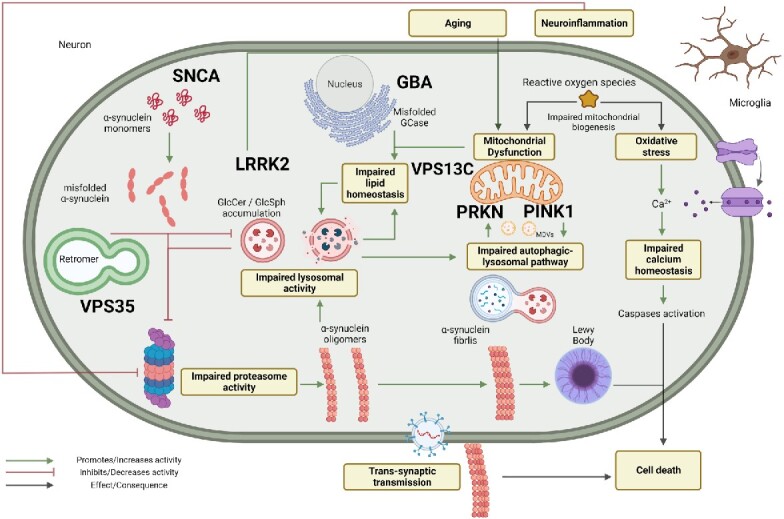
Schematic representation of the pathways involved in the pathogenesis of Parkinson’s disease. Created with BioRender.com.
Areas of agreement
Genes definitely associated with PD
The most important and/or frequent monogenic PD forms (SNCA, LRRK2, PRKN, PINK1, VPS35, VPS13C) and the major genetic risk factors (GBA1, LRRK2) are described in Table 1. Very rare forms of early-onset parkinsonism exist and, in the majority of cases, display a more complex phenotype than classical PD (e.g. ATP13A2, PLA2G6, FBOX7, DNAJC6, SYNJ1).
Table 1.
Major genetic determinants of PD
| Genes | Inheritance | Prevalence | Phenotype | Lewy bodies |
|---|---|---|---|---|
| GBA1 | Risk factor/AD | 5–15% of all PD (up to 20% in Ashkenazi Jews) | Early and late onset PD. Increased risk of dementia and dysautonomia | + |
| LRRK2 | Risk factor/AD | 1–5% of all PD (up to 15% in Ashkenazi Jews and 40% in North African Berbers) | Late onset typical | ± |
| SNCA | AD | Rare | Early and late onset | + |
| VPS35 | AD | Extremely rare | Late onset typical | ? |
| PRKN | AR | ~10% of early-onset PD | Early onset typical | − |
| PINK1 | AR | ~5 of early-onset PD | Early onset typical | ± |
| PARK7 (DJ-1) | AR | Rare | Early onset atypical | + |
| VPS13C | AR | Extremely rare | Early onset atypical | + |
‘Rare’ means <1% of all PD patients, ‘Extremely rare’ means <0.1% of all PD patients, early-onset PD = onset <50 years of age, late-onset >50 years of age.
Genes involved in monogenic PD and genotype–phenotype correlations
SNCA
SNCA gene encodes for α-synuclein, a neuronal protein involved in vesicular trafficking, docking and priming, fusion, neurotransmitters release and axonal transport.7
Pathogenic variants in SNCA are rare but contribute unequivocally to autosomal dominant (AD) inherited PD. Both missense and copy number variants (CNV) have been described in PD patients with earlier age of onset than idiopathic forms. The severity of the phenotype and the penetrance depends on the type of SNCA variant. Patients with four copies of the gene (triplicated locus) have younger age of onset and faster cognitive decline.8
Pathogenic variants in SNCA result in a misfolded protein, which promotes its structural conversion to crossed β-sheets monomers and oligomers. This, in a self-sustained impaired cycle that affects protein clearance and cellular quality control, leads to the aggregation of misfolded proteins that accumulate in Lewy bodies—the pathological hallmark of PD. Misfolded proteins impair vesicles assembly and transport, lysosomal activity and chaperone-mediated autophagy, which altogether contribute to impair cellular dynamics, neuronal propagation of fibrils and Lewy bodies leading to neuronal death8 (Fig. 1).
SNCA-PD patients usually have disease onset in the fourth or fifth decade. More than 30% of SNCA-PD patients have an even earlier age at onset, especially when they carry SNCA triplication. Cognitive decline is highly prevalent, described in 70% of patients. Atypical features, such as early anterocollis/retrocollis, pyramidal signs and alien limb syndrome, can also be present. These patients may display a good response to dopaminergic treatments and some cases undergo DBS with benefit on motor skills.9
LRRK2
LRRK2 gene (Leucine Rich Repeat Kinase 2) encodes for a protein kinase functioning as a key regulator of RAB GTPases that phosphorylates a broad range of proteins involved in multiple processes such as neuronal plasticity, autophagy and vesicular trafficking.10,11
The LRRK2 gene is highly polymorphic; however, only few variants (i.e. p.N1437H, p.R1441C/G/S/H, p.Y1699C, p.G2019S and p.I2020T) have been classified as definitely pathogenic so far.11,12 Some of them are very common within specific ethnic groups. The p.G2019S accounts for up to 29 and 37% of familial PD patients, in Ashkenazi Jewish and North African Berbers, respectively. The p.R1441G is found in 46% in Basque PD patients due to founder effect.11 LRRK2 pathogenic variants are AD inherited with incomplete penetrance (about 30% at 50 years and 70% at 80 years).13
In addition, p.G2385R and p.R1628P variants represent common genetic risk factors for late-onset PD in the Asian population. More recently, several other LRRK2 Single Nucleotide Polymorphisms (SNPs) have been proposed as genetic risk factors (e.g. p.M1646T and rs76904798).12
LRRK2 pathogenic variants increase the kinase activity.14 This gain of function influences different cellular pathways. In particular, the altered RAB GTPase regulation has been associated with the impairment of vesicle trafficking. A relationship has been established with impairment of cytoskeleton dynamics and a proinflammatory activity.11 Furthermore, recent studies described an interaction between α-synuclein and mutated LRRK2 in which the impairment of vesicle trafficking, protein clearance and neuroinflammation sustain α-synuclein misfolding. This interaction reinforces the pathological cascade and exacerbates cellular damage15 (Fig. 1).
LRRK2-PD patients show clinical features and disease progression which closely resemble idiopathic PD. They often manifest late-onset PD with typical clinical features of asymmetrical, tremor-dominant parkinsonism with bradykinesia and rigidity. Dystonia, especially painful off-period foot dystonia, is common after starting dopaminergic treatment. Cognitive impairment and hyposmia are less common in these patients,13,14 while sleep complaints are frequent.16 The majority has an excellent response to dopaminergic treatment, compared with non-carriers.13
PRKN
PRKN encodes for Parkin, a component of a multiprotein E3-Ubiquitin ligase complex. It mediates the targeting of substrate proteins for ubiquitination and proteasomal degradation. Along with other proteins (e.g. PINK1), Parkin regulates mitochondrial quality control, promoting the degradation of defective mitochondria and participating in signalling or trafficking pathways involving non-degradative ubiquitination.17
PRKN is the most common autosomal recessive (AR) PD gene, accounting for up to 40% of very early-onset PD (<40 years of age).18 The pathogenic variants, both SNVs and CNVs, act with a loss of function mechanism. The role of monoallelic PRKN variants as a risk factor is still debated;19 however, recent data seem to exclude their role in PD etiopathogenesis.20
Mutated PRKN results in a dysfunctional protein, which loses its ability to correctly identify and tag misfolded, disrupted or dysfunctional proteins, leading to a decrease in ubiquitination and to an impairment of proteasomal degradation. This activity is dysregulated especially in mitochondria. This creates a vicious cycle, which leads to the accumulation of altered proteins impairing several pathways such as lysosomal-related autophagy and vesicle exocytosis. This contributes to the deposition of misfolded proteins and toxic compounds throughout the cell, leading to neuroinflammation that aggravates neuronal damage7,8,17,21 (Fig. 1).
PRKN variants determine juvenile PD with disease onset typically in the third or fourth decade. At onset, clinical manifestations often include symmetrical foot dystonia.22,23 Depression is quite common in the absence of cognitive impairment.22 Usually, PRKN-PD patients have an excellent response to levodopa and a slower disease progression. Dyskinesias are common at later stages.23
PINK1
PINK1 encodes for ‘Pten-induced putative kinase 1’, a mitochondrial membrane Serine/Threonine kinase. It has a protective activity from stress-induced mitochondrial dysfunction and relevance in mitochondrial quality control, protein clearance and damage-induced mitophagy.24
Biallelic PINK1 variants are associated with early-onset PD and are fully penetrant.24,25 It represents the second most common example of AR early-onset PD.
PINK1 cooperates to correctly translocate proteins between the inner and the outer mitochondrial membrane. This interaction is fundamental for mitochondrial selection and turnover. PINK1 is a sensor for misfolded or damaged proteins, uncoupled respiratory chain complexes and oxidative stress thus adapting the mitochondrial response to cellular stressors. Furthermore, recent studies proved the interactions between PINK1 and other PD-related proteins such as Parkin and LRRK2 (Fig. 1).21,24
PINK1 variants are associated with early-onset PD. Their clinical manifestations are indistinguishable from that of other early-onset PD forms, with lower limb dystonia as a relevant clinical feature. PINK1-PD patients usually display a good response to levodopa and a slow disease progression. Some patients may develop dementia at later stages.26
VPS35
VPS35 encodes for ‘Vacuolar Protein Sorting 35’, a core subunit of a heteropentameric complex called retromer, involved in retrograde transport of proteins from endosome to the trans-Golgi network.27 The retromer acts at different levels, interacting with multiple cellular compartments to manage protein turnover. It has been proposed that variants in VPS35 can affect its protein clearance function and its ability to regulate the formation of lysosomes. Impaired retromer function could alter the turnover of mitochondria-derived vesicles with important impact on mitochondrial viability and dynamics (Fig. 1).28
Vilariño-Güell and colleagues described a family with late-onset AD typical PD carrying the p.D620N VPS35 missense variant.29 This variant, with incomplete penetrance, has been established as an uncommon cause of late-onset AD PD28.
VPS35 and the retromer as a whole seems to play an important role in PD pathogenesis, even though more studies are needed to clarify its exact involvement.
VPS13C
VPS13C encodes for Vacuolar Protein Sorting 13 Homolog C, a protein involved in mitochondrial homeostasis through Pink1/Parkin-mediated mitophagy in response to mitochondrial depolarization as well as in maintenance of mitochondrial transmembrane potential.30 Furthermore, it seems to mediate the transfer of lipids between membranes.31
In 2016, a genome-wide association study (GWAS) found several VPS13C SNPs associated with PD32. Subsequently, biallelic variants of VPS13C were identified in early-onset PD patients with rapid progression, early cognitive decline, dystonic features and diffuse Lewy body pathology.30,33
Genetic risk factors for PD and genotype–phenotype correlations
GBA1
GBA1 encodes for β-Glucosylceramidase (or Glucocerebrosidase), a lysosomal membrane enzyme responsible for the hydrolysis of glucosylceramides and glucosylsphingosines, into free ceramides/sphingosine and glucose. It plays a central role in the homeostasis of complex lipids and in cellular membrane turnover.34
Biallelic mutations in GBA1 are responsible for Gaucher Disease (GD), the most common lysosomal storage disorder. More recently, GD was associated with PD development.35 Several large cohort studies focused either on GD patients and their healthy parents highlighted a link between GBA1 variants and a higher risk of developing PD36 Consequently, studies on PD patients detected heterozygous variants in GBA1 in 5–15% of PD cases, making variants in this gene the most important genetic risk factor for PD (average odds ratio 5.4 from 1.5 to 20).34 Approximately, 300 GBA1 variants have been found (mostly SNV but also insertion/deletion and complex rearrangements with its pseudogene) with diverse frequencies in different ethnic subpopulations. The proximity to the active site is not a reliable predictor of disease severity since disease-causing variants have been identified all over the gene. Furthermore, not only rare variants but even relative common variants (not pathogenic for GD) have been associated with increased risk and different prognosis.34
The best characterized GBA1-associated mechanism of disease is the alteration of lysosomal activity due to the accumulation of the mutated glucocerebrosidase to the outer lysosomal membrane. Variants in GBA1 decrease its activity with different severity leading to deficient autophagic pathways, including macroautophagy, lysosome-mediated and Chaperone-Mediated Autophagy. These altered pathways lead to an imbalance in protein clearance and synthesis that can reinforce α-synuclein and other toxic compounds accumulation within neurons.34 In addition, variants in GBA1 may lead to the production of a misfolded protein which can be retained in the endoplasmic reticulum (ER) and induce ER-stress. In PD patients, an association between ER-stress and an increase in α-synuclein accumulation has been observed.34
It is possible that induced ER-stress and dysfunctional lysosomal autophagy pathways coexist and cause the deposition of toxic and damaged structures, which increase mitochondrial dysfunction and promote neuroinflammation cooperating to self-sustain pathogenicity34 (Fig. 1). The variability of clinical features among GBA1 variants carriers is remarkable; this may be partially explained by the diverse effect of mutations on protein functions.37
The clinical features of GBA1-PD are similar to idiopathic PD38. Nonetheless, GBA1-PD is overall characterized by earlier onset, worse motor impairment, higher risk of cognitive decline and dysautonomia, more rapid progression and decreased survival. Moreover, GBA1 variants are frequently associated with an akinetic-rigid phenotype and the presence of several neuropsychiatric symptoms, such as anxiety, impulsive–compulsive behaviour and hallucinations.39–42
Areas of controversy
Debated genes
During the last 10 years, candidate pathogenic variants of DNAJC13, TMEM230, CHCHD2 and LRP10 have been reported as novel genetic causes of monogenic PD43,44. These genetic associations need confirmation; therefore, the opinion of the authors is that these genes should not be considered for diagnostic purposes yet. Conversely, the inclusion of these genes in PD NGS panels for research purposes is warranted to elucidate their role.
Proposal of genetic testing to PD patients and relatives: why, who and how?
PD patients frequently ask about their relatives’ risk of PD and genetic testing. However, genetic testing is not widely offered yet due to the low clinical utility perceived by clinicians and the assumed high costs of sequencing.45 On the contrary, genetic testing can help the neurologists to give a more accurate prognosis, including disease course and response to treatments. Furthermore, the rise of PD clinical trials enrolling mutation carriers is going to significantly change this outdated view in the next future.14 Moreover, the advent of NGS techniques dramatically increased the feasibility of genetic diagnosis in PD since it allows a rapid cost-effective comprehensive genetic screening. It is important to note that NGS approaches must be combined with dosage analysis techniques (i.e. MLPA, qPCR or specific bioinformatic analyses on raw NGS data) to screen for rearrangements that cannot be revealed by the standard sequencing approaches (i.e. large deletions and duplications).
Nowadays, there are no international guidelines for PD genetic testing. The expert opinion of the authors is that, at least in developed countries, diagnostic genetic testing should be offered to all PD patients with disease onset before 50 years of age, positive familial history, specific ethnic ancestries and/or clinical-radiological features suggestive of a genetic aetiology. This opinion is justified by the higher diagnostic yield of genetic testing in these specific sub-populations. However, as several clinical trials focusing on specific PD genes (i.e. GBA1 and LRRK2) are ongoing, it is reasonable to broaden genetic testing also to late-onset patients in order to allow trial enrolment. Monogenic forms in late-onset PD patients are rare (<5% of patients) and essentially attributable to GBA1 and LRRK2 mutations. Therefore, a more conservative approach can be applied in this population, such as sequencing of specific genes or even testing for single mutations.
Genetic testing of PD patients may have relevant implications for families. For example, genetic diagnosis allows counselling for family planning and informs on disease risk of relatives. Genetic counselling should be modulated on the specific gene involved and family history. Since genetic forms of PD represent about 10–15% of the total and the low penetrance genes (e.g. GBA1 and LRRK2) are the most frequently involved, asymptomatic relatives can be, in most cases, reassured and informed on the low risk of developing PD. Pre-symptomatic testing should be considered with caution and relatives should be carefully counselled about the pros and cons of genetic testing, also in consideration of the psychological burden for the carrier and the present lack of disease-modifying treatments. Predictive genetic testing in PD could be modelled on the example of Huntington disease (HD) in which published guidelines are available.46 However, while HD is an AD monogenic disease with a rather predictable penetrance, PD is a disorder with wide genetic heterogeneity and few data available on the penetrance of many variants. In this sense, PD is more similar to ALS and it could be useful to refer also to the guidelines for genetic testing in motoneuron diseases.47
Moreover, genetic testing in PD should consider the uncertainties about the role of some genetic variants (i.e. variants of unknown significance), the possible presence of variants in more than one gene in the same individual, the incomplete knowledge on genotypic-phenotypic correlation in most PD genes and the phenotypic pleiotropy of some genes. In any case, whenever PD genetic testing is offered (diagnostic or pre-symptomatic), pre- and post-test counselling at a centre with specific expertise is required.
Tailored clinical management
Device-aided therapies in monogenic PD
Device-aided therapies in PD include DBS, continuous apomorphine subcutaneous infusion (CASI) and levodopa/carbidopa intestinal gel infusion (LCIG). They are used in the management of advanced PD when oral pharmacological treatments become less effective or not tolerated by patients. In many cases, it is still unclear whether the genetic background can affect the outcome of these therapies.
To date, only anecdotal reports on CASI and LCIG are available, while most data are about patients receiving DBS surgery.48 DBS is generally associated with positive outcomes in patients with genetic PD. However, some discrepancies have been observed in patients with different genetic forms. For example, PD patients harbouring PRKN variants have sustained improvement of motor function even in long-term follow-up49. LRRK2-PD patients, who frequently undergo DBS surgery because of troublesome dyskinesias, usually display satisfactory outcomes.50 Conversely, although good motor outcomes have been observed in GBA1-PD, there is significant concern on the possible post-operative cognitive deleterious effects in these patients.51 Nonetheless, different types of GBA1 mutations underlie distinct phenotypic profiles. Variants can be classified as mild, severe, complex and risk with patients with severe and complex GBA1-PD had the highest burden of symptoms and a higher risk of hallucinations and cognitive impairment.42 Thus, it is likely that new studies will soon clarify this controversial issue.
Despite genetic testing is not routinely performed before device-aided therapies, a molecular characterization of these patients is critical to collect data and increase our knowledge for future more tailored advanced therapies for specific genetic forms.
Areas timely for developing research
Polygenic Risk Scores in PD risk assessment
Most human traits are influenced by numerous genetic polymorphisms, each with small effects that, along with the environment, contribute to the manifestation of the overall phenotype. In multifactorial diseases such as PD, efforts to quantify the joint effect of common genetic variants and to develop predictive tools, measuring the cumulative genetic load within individuals, facilitate population stratification and identification of high-risk individuals.
An example of such a predictive tool is given by Polygenic Risk Scores (PRS). These are constructed from GWAS prioritized SNPs, weighted by the corresponding effect size estimates and P-values derived from GWAS summary statistics. This allows to capture the cumulative effect of many low to intermediate risk variants in patients’ populations. The aim is to obtain a reliable score capable of predicting both disease risk and continuous clinical outcomes.52
To date, several studies have tried to estimate reliable PRS able to predict differences in PD age at onset (AAO), prognosis, biomarkers and therapeutic responses. Many studies confirmed that higher PRS is significantly associated with earlier AAO tendency.52–54 Different studies tried to establish a PRS association with Levodopa induced dyskinesias55 as well as cognitive impairment.56,57 Moreover, Iwaki and colleagues investigated if the cumulative genetic risk affects the penetrance of the most common PD variants,58 e.g. LRRK2 p.G2019S, resulted in a strong association of high PRS with high variant’s penetrance; GBA variants penetrance have been reported in association with common variants in SNCA and CTSB.59 Unfortunately, to date, none of the studies reached robust results in predicting the probability of developing the disease, or in predicting the disease progression and prognosis, likely because of the still limited available data.
Ultimately, much effort has been made to stratify patients based on their PRS, as well as on RNA expression and biochemical profiles. Examples come from recent studies,60,61 which applied powerful machine learning algorithms to predict PD disease stage, subtypes and relative progression of the disease based on multimodality networks. In particular, genomics, transcriptomics, metabolomics and clinical features have been interconnected to build robust classifiers of disease characteristics and progression, reaching significant results, practically displaying the true aim of such studies, i.e. the potential stratification of patients based on their features, aiming at ameliorating their management. Moreover, this is fundamental to offer the best setup for clinical trials to develop specific therapeutic schemes according to patients’ genetics, avoiding treatment mis-responses and improving medical interventions.
Key Points
In up to 15% of PD patients, a pathogenic (or risk) variant may be detected. Some genetic variants act in a mendelian fashion, while others are risk factors.
Genetic background influences age of onset, disease course, prognosis and therapeutic response.
Genetic testing for PD may be offered in clinical practice to all the patients for its present and future implications. Pre- and post-test counselling should be offered.
Standard guidelines on gene testing in PD patients are missing. The Movement Disorders and Human Genetics scientific societies should address this issue in the near future.
PRS may represent a new tool to genetically stratify the population in terms of disease risk, prognosis and therapeutic outcomes.
Acknowledgements
This work was developed within the framework of the DINOGMI Department of Excellence of MIUR 2018–2022 (legge 232 del 2016).
Contributor Information
L Trevisan, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal and Child Health, University of Genoa, Largo P. Daneo 3, Genova, 16132, Italy; IRCCS Ospedale Policlinico San Martino – SS Centro Tumori Ereditari, Largo R. Benzi 10, Genova, 16132, Italy.
A Gaudio, IRCCS Ospedale Policlinico San Martino- UOC Genetica Medica, Largo R. Benzi 10, Genova, 16132, Italy.
E Monfrini, Dino Ferrari Center, Neuroscience Section, Department of Pathophysiology and Transplantation, University of Milan, Via Francesco Sforza 35, Milan, 20122, Italy; Neurology Unit, Foundation IRCCS Ca’Granda Ospedale Maggiore Policlinico, Via Festa del Perdono 7, Milan, 20122, Italy.
L Avanzino, Department of Experimental Medicine, Section of Human Physiology, University of Genoa, Viale Benedetto XV/3, Genova, 16132, Italy; IRCCS Ospedale Policlinico San Martino, Largo Rosanna Benzi 3, Genova, 16132, Italy.
A Di Fonzo, Dino Ferrari Center, Neuroscience Section, Department of Pathophysiology and Transplantation, University of Milan, Via Francesco Sforza 35, Milan, 20122, Italy; Neurology Unit, Foundation IRCCS Ca’Granda Ospedale Maggiore Policlinico, Via Festa del Perdono 7, Milan, 20122, Italy.
P Mandich, Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal and Child Health, University of Genoa, Largo P. Daneo 3, Genova, 16132, Italy; IRCCS Ospedale Policlinico San Martino- UOC Genetica Medica, Largo R. Benzi 10, Genova, 16132, Italy.
Funding
Italian Ministry of Health (Rete delle Neuroscienze e della Neuroriabilitazione (RIN- RCR-2019-23669119_003 and RCR-2022-23 682 291); Italian Ministry of Health (Ricerca Corrente 2022–2024).
Conflict of interest statement
The authors have no potential conflicts of interest.
Data Availability
No new data were generated or analyzed in support of this research.
References
- 1. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA 2020;323:548–60. 10.1001/jama.2019.22360. [DOI] [PubMed] [Google Scholar]
- 2. Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–4. 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. The Lancet Neurology 2006;5:235–45. 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 4. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015;30:1591–601. 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 5. Isaacson SH, Fisher S, Gupta F, et al. Clinical utility of DaTscan™ imaging in the evaluation of patients with parkinsonism: a US perspective. Expert Rev Neurother 2017;17:219–25. 10.1080/14737175.2017.1256205. [DOI] [PubMed] [Google Scholar]
- 6. Fox SH, Katzenschlager R, Lim SY, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord 2018;33:1248–66. 10.1002/mds.27372. [DOI] [PubMed] [Google Scholar]
- 7. Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry 2020;91:795–808. 10.1136/jnnp-2019-322338. [DOI] [PubMed] [Google Scholar]
- 8. Nishioka K, Imai Y, Yoshino H, et al. Clinical manifestations and molecular backgrounds of Parkinson’s disease regarding genes identified from familial and population studies. Front Neurol 2022;13:764917. 10.3389/fneur.2022.764917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trinh J, Zeldenrust FMJ, Huang J, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 2018;33:1857–70. 10.1002/mds.27527. [DOI] [PubMed] [Google Scholar]
- 10. Steger M, Tonelli F, Ito G, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016;5:e12813. 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J, Chen Y, Pu J. Leucine-rich repeat kinase 2 in Parkinson’s disease: updated from pathogenesis to potential therapeutic target. Eur Neurol 2018;79:256–65. 10.1159/000488938. [DOI] [PubMed] [Google Scholar]
- 12. Monfrini E, Di Fonzo A. Leucine-rich repeat kinase (LRRK2) genetics and Parkinson’s disease. Adv Neurobiol 2017;14:3–30. 10.1007/978-3-319-49969-7_1. [DOI] [PubMed] [Google Scholar]
- 13. Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 2008;7:583–90. 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tolosa E, Vila M, Klein C, et al. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol 2020;16:97–107. 10.1038/s41582-019-0301-2. [DOI] [PubMed] [Google Scholar]
- 15. Bieri G, Brahic M, Bousset L, et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol 2019;137:961–80. 10.1007/s00401-019-01995-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pont-Sunyer C, Iranzo A, Gaig C, et al. Sleep disorders in parkinsonian and nonparkinsonian LRRK2 mutation carriers. PloS One 2015;10:1–20. 10.1371/journal.pone.0132368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshino H, Li Y, Nishioka K, et al. Genotype-phenotype correlation of Parkinson’s disease with PRKN variants. Neurobiol Aging 2022;114:117–28. 10.1016/j.neurobiolaging.2021.12.014. [DOI] [PubMed] [Google Scholar]
- 18. Wasner K, Smajic S, Ghelfi J, et al. Parkin deficiency impairs mitochondrial DNA dynamics and propagates inflammation. Mov Disord 2022;37:1405–15. 10.1002/mds.29025. [DOI] [PubMed] [Google Scholar]
- 19. Reed X, Bandrés-Ciga S, Blauwendraat C, et al. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol Dis 2019;124:230–9. 10.1016/j.nbd.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu W, Huang X, Yoon E, et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease. Brain 2022;145:2077–91. 10.1093/brain/awab456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Borsche M, König IR, Delcambre S, et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020;143:3041–51. 10.1093/brain/awaa246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song J, Shen B, Yang YJ, et al. Non-motor symptoms in Parkinson’s disease patients with Parkin mutations: more depression and less executive dysfunction. J Mol Neurosci 2020;70:246–53. 10.1007/s12031-019-01444-3. [DOI] [PubMed] [Google Scholar]
- 23. Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–7. 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 24. Borsche M, Pereira SL, Klein C, et al. Mitochondria and Parkinson’s disease: clinical, molecular, and translational aspects. J Parkinsons Dis 2021;11:45–60. 10.3233/JPD-201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004;304:1158–60. 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 26. Kasten M, Hartmann C, Hampf J, et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 2018;33:730–41. 10.1002/mds.27352. [DOI] [PubMed] [Google Scholar]
- 27. Williams ET, Chen X, Moore DJ. VPS35, the Retromer complex and Parkinson’s disease. J Parkinsons Dis 2017;7:219–33. 10.3233/JPD-161020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sassone J, Reale C, Dati G, et al. The role of VPS35 in the pathobiology of Parkinson’s disease. Cell Mol Neurobiol 2021;41:199–227. 10.1007/s10571-020-00849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011;89:162–7. 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lesage S, Drouet V, Majounie E, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent Mitophagy. Am J Hum Genet 2016;98:500–13. 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Melia TJ, Reinisch KM. A possible role for VPS13-family proteins in bulk lipid transfer, membrane expansion and organelle biogenesis. J Cell Sci 2022;135:jcs259357. 10.1242/jcs.259357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Cheng L, Li NN, et al. Association of four new candidate genetic variants with Parkinson’s disease in a Han Chinese population. Am J Med Genet B Neuropsychiatr Genet 2016;171:342–7. 10.1002/ajmg.b.32410. [DOI] [PubMed] [Google Scholar]
- 33. Monfrini E, Spagnolo F, Canesi M, et al. VPS13C-associated Parkinson’s disease: two novel cases and review of the literature. Parkinsonism Relat Disord 2022;94:37–9. 10.1016/j.parkreldis.2021.11.031. [DOI] [PubMed] [Google Scholar]
- 34. Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cell 2022;11:1261. 10.3390/cells11081261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 2003;79:104–9. 10.1016/S1096-7192(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 36. Riboldi GM, Di Fonzo AB. GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cell 2019;8:E364. 10.3390/cells8040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jesús S, Huertas I, Bernal-Bernal I, et al. GBA variants influence motor and non-motor features of Parkinson’s disease. PloS One 2016;11:1–17. 10.1371/journal.pone.0167749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013;136:392–9. 10.1093/brain/aws318. [DOI] [PubMed] [Google Scholar]
- 39. Brockmann K, Srulijes K, Pflederer S, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30:407–11. 10.1002/mds.26071. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y, Shu L, Zhou X, et al. A meta-analysis of GBA-related clinical symptoms in Parkinson’s disease. Parkinsons Dis 2018;2018:1–7. 10.1155/2018/3136415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol 2016;80:662–73. 10.1002/ana.24777. [DOI] [PubMed] [Google Scholar]
- 42. Petrucci S, Ginevrino M, Trezzi I, et al. GBA-related Parkinson’s disease: dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord 2020;35:2106–11. 10.1002/mds.28195. [DOI] [PubMed] [Google Scholar]
- 43. Puschmann A. New genes causing hereditary Parkinson's disease or parkinsonism. Curr Neurol Neurosci Rep 2017;17:66. 10.1007/s11910-017-0780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manini A, Straniero L, Monfrini E, et al. Screening of LRP10 mutations in Parkinson's disease patients from Italy. Parkinsonism Relat Disord 2021;89:17–21. 10.1016/j.parkreldis.2021.06.014. [DOI] [PubMed] [Google Scholar]
- 45. Cook L, Schulze J, Kopil C, et al. Genetic testing for Parkinson disease: are we ready? Neurol Clin Pract 2021;11:69–77. 10.1212/CPJ.0000000000000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacLeod R, Tibben A, Frontali M, et al. Recommendations for the predictive genetic test in Huntington’s disease. Clin Genet 2013;83:221–31. 10.1111/j.1399-0004.2012.01900.x. [DOI] [PubMed] [Google Scholar]
- 47. Chiò A, Battistini S, Calvo A, et al. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 2014;85:478–85. 10.1136/jnnp-2013-305546. [DOI] [PubMed] [Google Scholar]
- 48. Chan GH-F. The role of genetic data in selecting device-aided therapies in patients with advanced Parkinson’s disease: a mini-review. Front Aging Neurosci 2022;14:107–10. 10.3389/fnagi.2022.895430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim HJ, Yun JY, Kim YE, et al. Parkin mutation and deep brain stimulation outcome. J Clin Neurosci 2014;21:107–10. 10.1016/j.jocn.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 50. Leaver K, Viser A, Kopell BH, et al. Clinical profiles and outcomes of deep brain stimulation in G2019S LRRK2 Parkinson disease. J Neurosurg 2021;137:1–8. 10.3171/2021.7.JNS21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pal G, Mangone G, Hill EJ, et al. Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann Neurol 2022;91:424–35. 10.1002/ana.26302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dehestani M, Liu H, Gasser T. Polygenic risk scores contribute to personalized medicine of Parkinson’s disease. J Pers Med 2021;11:1030. 10.3390/jpm11101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ibanez L, Dube U, Saef B, et al. Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol 2017;17:198. 10.1186/s12883-017-0978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Escott-Price V, International Parkinson's Disease Genomics Consortium, Nalls MA, et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann Neurol 2015;77:582–91. 10.1002/ana.24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eusebi P, Romoli M, Paoletti FP, et al. Risk factors of levodopa-induced dyskinesia in Parkinson’s disease: results from the PPMI cohort. NPJ Parkinsons Dis 2018;4:33. 10.1038/s41531-018-0069-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paul KC, Schulz J, Bronstein JM, et al. Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol 2018;75:360–6. 10.1001/jamaneurol.2017.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kusters CDJ, Paul KC, Duarte Folle A, et al. Genetic risk scores and hallucinations in patients with Parkinson disease. Neurol Genet 2020;6:e492. 10.1212/NXG.0000000000000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iwaki H, Blauwendraat C, Makarious MB, et al. Penetrance of Parkinson’s disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. Mov Disord 2020;35:774–80. 10.1002/mds.27974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blauwendraat C, Reed X, Krohn L, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020;143:234–48. 10.1093/brain/awz350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Makarious MB et al. Multi-modality machine learning predicting Parkinson’s disease. npj Parkinsons Dis 2022;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dadu A, Satone V, Kaur R, et al. Identification and prediction of Parkinson’s disease subtypes and progression using machine learning in two cohorts. NPJ Parkinsons Dis 2022;8:172. 10.1038/s41531-022-00439-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.