

Abstract
Cardiac fibrosis is a pathological condition that occurs after injury and during aging. Currently, there are limited means to effectively reduce or reverse fibrosis. Key to identifying methods for curbing excess deposition of extracellular matrix is a better understanding of the cardiac fibroblast, the cell responsible for collagen production. In recent years, the diversity and functions of these enigmatic cells have been gradually revealed. In this review, I outline current approaches for identifying and classifying cardiac fibroblasts. An emphasis is placed on new insights into the heterogeneity of these cells as determined by lineage tracing and single-cell sequencing in development, adult, and disease states. These recent advances in our understanding of the fibroblast provide a platform for future development of novel therapeutics to combat cardiac fibrosis.
Keywords: fibroblast, myofibroblast, myocardial infarction, interstitium, adventitia, inflammation
INTRODUCTION
Cardiac fibroblasts are central actors in normal cardiac physiology and cardiovascular disease. They play essential roles in development by depositing collagens and other extracellular matrix (ECM) components and, in adult hearts, fibroblasts are constantly modifying the microenvironment by degrading and depositing ECM (1). Fibroblasts are also responsible for cardiac fibrosis, which is the accumulation of ECM in response to a pathological stimulus. Fibrosis is a common feature of many acute and progressive cardiac diseases. When fibroblasts become activated by injury or inflammation, they exhibit a pronounced upregulation in collagen production, at first aiding to stabilize the heart. While the deposition of ECM may initially strengthen tissue integrity, extensive fibrosis can impair heart function. Persistent fibrosis may ultimately lead to heart failure.
Cardiovascular disease is the leading cause of death in the United States, and annual health care costs for these conditions are over $300 billion (2). The severity of fibrosis is correlated with the progression of heart failure, and fibrosis is a common finding in almost all types of heart disease. Thus, a better understanding of the basal function of these cells, the signaling pathways that control their activation, and the functions of those pathways after injury is warranted. Currently, there are no effective antifibrotic therapies that specifically target the cardiac fibroblast (3). Because identification and tracking of fibroblasts in vivo have been challenging, much of our current knowledge regarding the fibroblast has been based on studies of the myofibroblast [alpha-smooth muscle actin (α-SMA)-positive cells] or in vitro culture of primary cardiac fibroblasts. Now, with advances in technology and development of more precise means for fibroblast identification, we can evaluate in vivo fibroblast function at baseline and after injury.
The purpose of this review is to outline our current understanding of cardiac fibroblast subtypes and describe how these cells potentially contribute to cardiac repair and disease. Three different means of classification are outlined: anatomic location, embryonic origin, and gene expression/function. The reader should keep in mind that the field of cardiac fibroblast biology is rapidly evolving and that these classification schemes are provided as general guidelines when evaluating and identifying cardiac fibroblasts.
MESENCHYMAL CELLS IN THE UNINJURED HEART
Traditionally, a cell has been considered a fibroblast if it produces type I collagen and resides in the connective tissue or interstitium of an organ. Alternatively, fibroblasts have also been identified by their ability to rapidly attach to a culture dish after enzymatic tissue digestion. Fibroblasts constitute only one group of mesenchymal cells that reside within the heart. Perivascular cells with contractile properties, such as vascular smooth muscle cells (VSMCs) and pericytes, are also present, but the total number of these cells is estimated to be less than the number of fibroblasts (4). Distinguishing between mural cells and fibroblasts is usually based on expression of several markers. Cardiac fibroblasts express discoidin domain–containing receptor 2 (DDR2) (5), platelet-derived growth factor receptor alpha (Pdgfrα) (4–8), Tcf21 (4, 8–10), and vimentin (11). Pericytes express NG2 (12), platelet-derived growth factor receptor beta (Pdgfrβ) (12), and Tbx18 (13). VSMCs express α-SMA (14) and Myh11 (15). Some of these markers exhibit overlap between these three cell populations. For example, α-SMA can be found in subsets of pericytes and activated cardiac fibroblasts (12). Often, expression of two or more markers has been used to more confidently identify the cell population of interest. Alternatively, indelible lineage tracers using Cre/loxP technology in mice can also be used (reviewed in 16, 17).
Understanding of cardiac fibroblast biology has increased significantly over the past decade. Mouse lines that permit the genetic manipulation of fibroblasts, the lineage tracing of fibroblasts, and the profiling of fibroblast transcriptomes by microarray or RNA sequencing have provided important insights into the behavior and diversity of cardiac fibroblasts. However, these techniques are often limited because they focus on bulk populations of fibroblasts. Fine details such as genes expressed at low transcript levels or variations due to anatomic location may be lost in these analyses. Recent single-cell analysis of cell populations in the heart has provided a relatively unbiased understanding of the diversity and subsets of cell types. Although the number and properties of these subsets are not always consistent, a growing consensus suggests that most of the major cell types can be subcategorized on the basis of specific patterns of gene expression.
CLASSIFYING FIBROBLAST POPULATIONS IN THE HEART
Cardiac Fibroblast Subtypes by Location
Location may be an underappreciated factor causing differential gene expression by cardiac fibroblasts. There are broadly five cardiac fibroblast locales: adventitia (ventricular and septal), interstitium (ventricular and septal), atrium, annulus fibrosus, and valves (Figure 1). It is plausible that fibroblasts within each of these locations have different gene expression profiles and functions, as there are regional differences in ECM in the heart (18). For example, changes in fibroblast gene expression and proliferation were observed when cardiac fibroblasts were compared between two-dimensional (2D) and three-dimensional (3D) cultures, conditions that illustrate the importance of ECM composition and rigidity in dictating fibroblast gene expression (19, 20). A dramatic and reversible shift in gene expression related to ECM organization and actin cytoskeleton was noted, including upregulation of matrix metalloproteinases (MMPs) and reduced α-SMA expression in fibroblasts cultured in 3D. 3D culture also resulted in reduced expression of genes related to cell division. These data suggest that the surrounding microenvironment, including neighboring cells and ECM, can influence fibroblast gene expression. When considering the known differences in compliance and mechanical strain that exist between the chambers of the heart, cardiac fibroblast location is an important point to consider when describing fibroblast subtypes.
Figure 1.
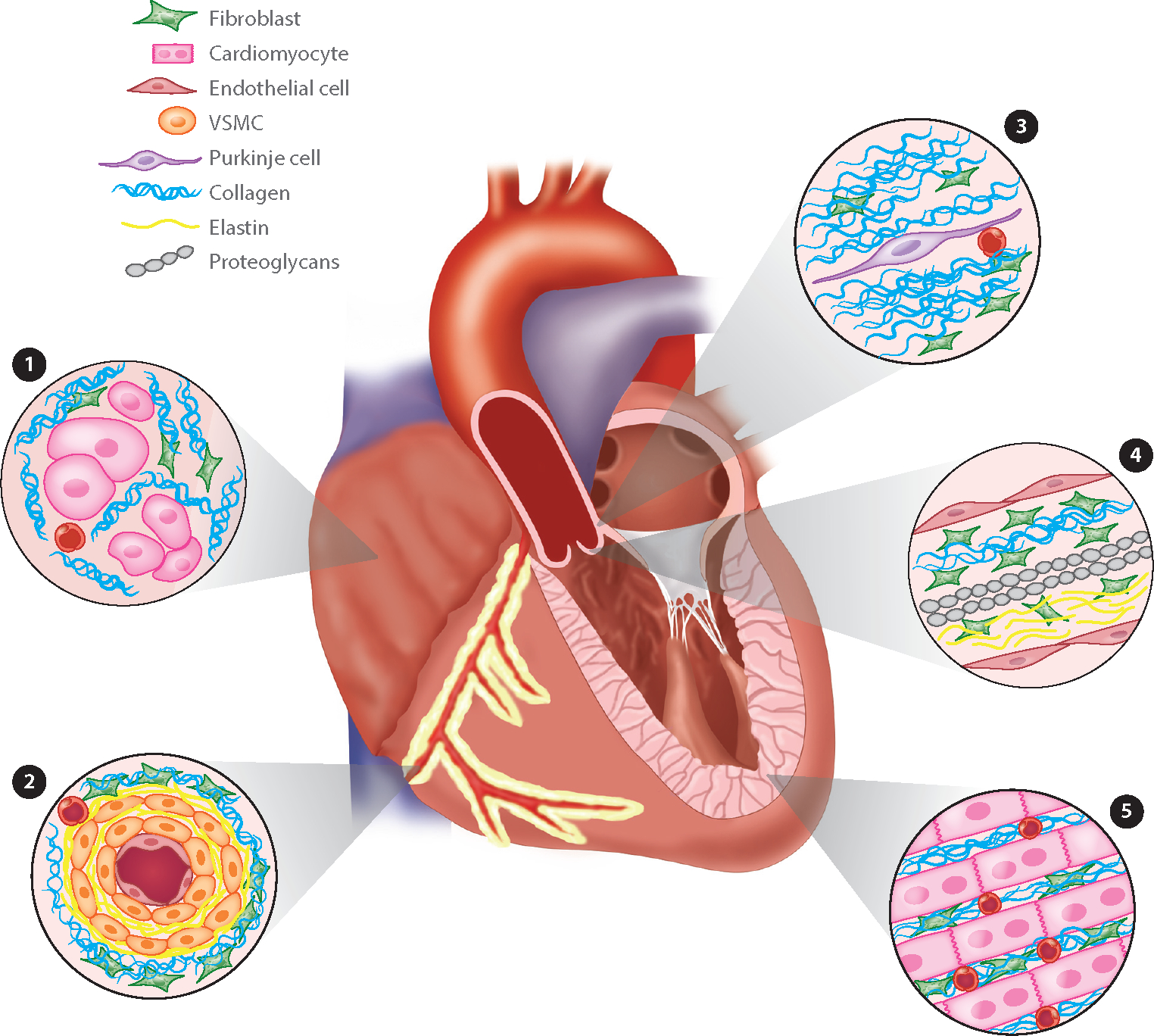
Anatomic location of cardiac fibroblasts. Fibroblasts reside in distinct anatomic locations in the heart, and each population of fibroblasts is likely to express a different transcriptional profile. ➊1 Atrium-transverse section of atria where cardiomyocyte fibers are thinner and less dense compared to the ventricles. The fibroblast to cardiomyocyte ratios in atria have not been clearly documented. ➋2 Adventitia-transverse section through a coronary artery. The number of vascular smooth muscle cells (VSMCs) can vary depending on vessel diameter. Cell types other than fibroblasts have also been noted in the dense collagen surrounding the vessel. ➌3 The annulus fibrosus is a collagen-rich area surrounding the conduction system. Fibroblasts are present in these regions. ➍4 Valves: Fibroblasts reside in three layers (fibrosa, spongiosa, and ventricularis). These are composed of collagen, proteoglycans, and elastin, respectively. ➎5 Interstitium: This longitudinal section of myocardium would be found in the ventricles or ventricular septum.
Interstitial septal and ventricular fibroblasts.
Few studies have compared the expression of interstitial cardiac fibroblasts to other populations of cardiac fibroblasts. If one assumes that the majority of the cardiac fibroblasts are interstitial fibroblasts, then many of the data regarding gene expression and behavior of cardiac fibroblasts are derived from these fibroblasts. Our understanding of cardiac fibroblasts is often based upon comparison of cultured ventricular cardiac fibroblasts from uninjured hearts to cultured cells from injured hearts, and these studies suggest that cells retain some of their transcriptional differences over time. Control fibroblasts are even available commercially, although a recent report suggests that these may also have distinct phenotypes attributable to isolation and culture conditions (21). Because of these hurdles, few studies have focused on how regional differences in microenvironment may affect fibroblast phenotype. It is plausible that fibroblasts may differ not only depending on where they are located (ventricle or septum) but also due to proximity to a blood vessel or nerve fiber. Proteomic data from the human heart support the idea that ECM varies from chamber to chamber (22). Future analyses should include consideration of the specific anatomic sites where fibroblasts reside.
Adventitial septal and ventricular fibroblasts.
Anatomically, the adventitia is the most recognizable population of fibroblasts in the heart. These fibroblasts surround the medial layer of the coronary arteries and are responsible for depositing dense collagen matrix that supports the artery. As vasculature permeates the ventricles and ventricular septum, adventitial fibroblasts can be found throughout these areas in close proximity to blood vessels. While fibroblasts are one of the cellular components surrounding large vessels, other cell types also reside in the same space, including endothelial cells, immune cells, and smooth muscle progenitors (23). A limited number of studies have described baseline gene expression profiles of coronary adventitial fibroblasts in the absence of injury or stress. Several studies have reported a preferential expression of Sonic Hedgehog (Shh) signaling components in adventitial fibroblasts. Patched-1 and Patched-2 are expressed in adventitial fibroblasts of large or medium-sized arteries until postnatal day 10. Subsequently, expression is reduced in the adult (24). Similarly, the Shh responsive transcription factor Gli1 labels a perivascular cell population that resides in the coronary artery adventitia and ventricular interstitium, and some of these cells express α-SMA and collagen after injury (25). Removal of these Gli1-expressing cells using diphtheria toxin resulted in reduced fibrotic remodeling. The authors did not report the extent of Gli1 adventitial expression in the absence of injury. These Gli1-expressing cells were subsequently shown to contribute to vascular calcification and described as progenitors of VSMCs (26). Therefore, it is unclear if these cells can be classified as adventitial fibroblasts. Further studies are required to elucidate the differences between interstitial and adventitial fibroblasts in the heart.
Atrial fibroblasts.
Although fibrosis is associated with persistent atrial fibrillation (27, 28) and, in certain pathological circumstances, atrial fibrosis can exceed that observed in the ventricles (29), we know very little about the differences between atrial and ventricular fibroblasts. Atrial fibroblasts form from the same source as the majority of the ventricular fibroblasts, the epicardium. Despite a similar origin, it is likely that atrial fibroblasts have characteristics that differ from ventricular fibroblasts. The cardiomyocyte organization, tissue stiffness, and the density of collagen matrix in the atria are very different compared to the ventricle. Studies in isolated primary canine fibroblasts revealed that atrial fibroblasts were more proliferative in response to growth factor stimulation and showed differential gene expression compared to ventricular fibroblasts (30).
Annulus fibrosus.
Additional populations of fibroblasts that have distinct anatomic locations are those that constitute the fibrous rings that separate the atria and ventricles, the annulus fibrosus. The atrioventricular conduction system penetrates the annulus fibrosus, and it is thought that this dense matrix provides insulation for normal electrical conduction (31). Although it has a distinct role in heart physiology, little is known regarding any specific gene expression by fibroblasts that reside in these areas.
Cardiac valve fibroblasts.
Because of their involvement in heart valve disease and their segregated anatomic location, the fibroblasts of the valves, also known as valve interstitial cells, are usually not categorized as cardiac fibroblasts. These cells derive from the endocardial cushions, the epicardium, and neural crest and appear to express unique gene sets, perhaps in part due to exposure to mechanical stress. The reader is encouraged to refer to other reviews for further information regarding this population of fibroblasts (32, 33).
Cardiac Fibroblasts Subtypes by Origin
An alternative method for classifying fibroblasts uses their embryonic origin. A majority of the fibroblasts originate from the outer mesothelial covering of the heart, the epicardium, but some of the fibroblasts within the ventricular septum and the left ventricle derive from the endocardium. No significant behavioral differences have been noted between these two populations, but caution must be used when performing lineage tracing and gene deletion based on developmental origin, as the Cre recombinase may not be expressed in both populations.
Epicardial fibroblasts.
The epicardial origin of ventricular cardiac fibroblasts has been appreciated for over 20 years. Using a retrovirus to label the epicardium, it was observed that epicardial cells migrated into the ventricular wall and developed into cells surrounding the coronary arteries as well as into interstitial cells (34). Subsequent to this study, it was determined that a fraction of epicardial cells undergo an epithelial to mesenchymal transition to form a majority of coronary VSMCs and cardiac fibroblasts (35, 36). These epicardial derivatives contribute to the fibroblasts of the ventricles and atria (8, 37), the cardiac annulus fibrosus (38), and atrioventricular valve leaflets (39). Development of epicardial-specific Cre recombinase mouse lines, including Gata5-Cre and Wt1CreERT2 (37), led to the discovery that loss of either Pdgfrα (6) or Tcf21 (8) resulted in an absence of cardiac fibroblasts within the ventricular walls. Thus far, these two genes are the only ones identified that are uniquely required for the development of epicardial fibroblast development. Although both are considered stable markers for cardiac fibroblasts, some studies report changes in transcript levels dependent on the activation state of the fibroblast (10, 40, 41).
Endocardial fibroblasts.
Until 2014, the epicardium was believed to be the sole developmental source of ventricular cardiac fibroblasts, but two independent studies found that some fibroblasts have an endocardial or endothelial origin (5, 7). These Pdgfrα-, Ddr2-, and collagen-expressing fibroblasts were found predominantly in the septum and a region of the left ventricle. Both groups further investigated potential differences between the endocardial lineage and the epicardial lineage. Surprisingly, there were no significant differences in gene expression or proliferation between the two lineages either before or after pressure overload. In fact, the relative contribution of each cell lineage to pressure overload-induced fibrosis was similar to the relative abundance of endocardial-derived cells in the absence of injury (~20%). These studies suggest that the two fibroblast populations converge with regard to gene expression and that the stimulus, not their origin, is the dominant factor controlling their fibrogenic response.
New Fibroblast Subtypes
In addition to segregating cardiac fibroblasts by location and developmental origin, some investigators have begun to use gene expression as a means for discriminating between subtypes. As might be expected, this method of distinction can result in an infinite number of subpopulations, but several consistent profiles have been described by independent laboratories.
Populations revealed by single-cell sequencing analysis.
Single-cell RNA sequencing has provided a powerful view into gene expression of individual cells and reveals new insights into subsets of cell populations. One of the initial studies to characterize mesenchymal populations in the adult murine heart suggested that two populations of Col1a1-expressing fibroblasts exist (42). One population expresses markers that have been reported in the past, including Pdgfrα and Tcf21. A second group expressed Col1a1 transcripts but lacked Pdgfrα and Tcf21. The most distinguishing genes for this subset of fibroblasts were Wnt pathway genes Dkk3, Wif1, Tbx20, and Frzb. Interestingly, another study using Pdgfrα expression to identify cardiac fibroblasts also identified a subset of fibroblasts with a similar transcriptional profile. This population was termed Wntx, and further examination suggested that Wnt pathway–related genes were more highly expressed in this population than the other fibroblast subsets. Periostin expression, usually associated with fibroblast activation, was also present in this population. At baseline and three days post–myocardial infarction, these cells constituted 6% and 10% of the Pdgfrα-expressing fibroblast population, respectively (43).
A second distinction between resting fibroblast populations was Sca1 expression. At baseline, approximately half of the Pdgfrα-expressing fibroblasts had low expression of Sca1, while roughly 30% of the cells expressed high levels of Sca1 (43). Gene ontogeny analysis suggested that the high Sca1 population was enriched for proliferation and stemness genes, including Thy1 and Cd34, while the low Sca1 cells were enriched for cell signaling genes such as Bmp4 and ApoE. Previous reports have suggested that the high Sca1 population contains multipotent progenitor cells that are capable of self-renewal and differentiation into adipocytes, smooth muscle cells, and cardiomyocytes in vitro (44, 45). A similar population of fibroblasts, termed fibro-adipocyte progenitors, or FAPS, has been described previously in heart (46) and skeletal muscle (47, 48). These cells are discussed in more detail in the section titled Mesenchymal Stromal/Stem Cells.
Several additional reports have described fibroblast populations identified by single-cell sequencing during heart development. In the embryonic heart, when fibroblasts were profiled, the defining transcripts were type I collagen, decorin, sox9, Tcf21, and periostin (49–51). It was also noted that transcriptional profiles did not vary dramatically from embryonic into postnatal time points (49), although some differences in embryonic fibroblast markers were noted between human and mouse fibroblast profiles (51). Finally, when the transcriptomes of perinatal heart nuclei were sequenced, two fibroblast populations were described. Neither of these two populations exhibited a Wntx profile as described above for the adult mouse heart, and the populations were distinguished more by different levels of gene expression rather than by unique gene transcripts (52).
Fibroblast Subtypes After Injury
During cardiac stress such as pressure overload or myocardial infarction, a series of cellular responses lead to a process termed cardiac remodeling. Cardiomyocytes send stress signals that trigger inflammatory cell infiltration and fibroblast activation. The term activation in reference to a cardiac fibroblast has a broad definition that includes increased proliferation, matrix deposition, contractile protein expression, and cytokine/growth factor secretion. Because not all fibroblasts express the contractile proteins that define myofibroblasts, identification of fibroblasts after injury has become more complicated than simple expression of α-SMA, but new methods for identifying cells and single-cell sequencing have now provided deeper insights for understanding cardiac fibroblast biology after injury.
Before the advent of single-cell sequencing, identification of fibroblast subsets relied predominantly on examination of primary fibroblast gene expression and behaviors after enzymatic isolation and in vitro culture. These studies provided important insights into general categories of fibroblast activities such as proinflammatory, proangiogenic, and profibrotic. Here, I consider the existing data regarding the subsets of fibroblasts based on their function, but it should be noted that fibroblast function is likely to fall along a spectrum. In addition, some of these studies are based on bulk populations of cells and may be limited by either temporal or spatial constraints.
Proliferating fibroblasts.
There have been several investigations of the proliferation profile of fibroblasts after injury. Two independent reports investigated the resident fibroblast response after myocardial infarction and observed that a strong proliferative burst resulted in 2–3 times more fibroblasts in the infarct region (53, 54). One week after injury, both fibroblast proliferation and expression of α-SMA substantially declined. Similar profiles of proliferation were observed when either pressure overload or isoproterenol were used to induce fibrosis (53).All single-cell sequencing data sets describe a proliferating population of cells, but many of the genes driving the proliferation phenotype vary between studies, with Mki67 being one proliferation gene found in common (10, 43, 54–56). One study suggested that follistatin-like protein 1 is essential for fibroblast expansion, and ventricular rupture was observed when cardiac fibroblasts lacked this growth factor (56). Follistatin-like 1 also promoted cardiac fibroblast activation and protected hearts from rupture (57). Gene expression profiling demonstrated that the proliferating fibroblasts were similar to neonatal fibroblasts (56).
Fibroblasts in inflammation.
Inflammatory cells have an important influence on the activation and matrix production of fibroblasts (58), and studies have indicated that after injury fibroblasts can inform the inflammatory response. For example, apoptotic cell clearance by cardiac fibroblasts has been observed after myocardial infarction. Mechanistically, activated fibroblasts secrete milk fat globule-epidermal growth factor 8 (Mfge8), which enhances apoptotic cell engulfment. Supplementation of Mfge8 resulted in reduced inflammatory signals, whereas loss of Mfge8 leads to extended inflammatory responses (59).
Fibroblasts also secrete a variety of cytokines, growth factors, and chemokines that modulate inflammatory cells. For example, interleukin (IL)-1ra (60), IL-1β (61, 62), IL-6 (61–65), INF-γ (60), TNF-α (61, 62, 65), MCP-1 (62, 64, 65), GM-CSF (66), IL-9 (60), IL-10 (60, 64, 65, 67), IL-11 (55), IL-12 (65), IL-23a (68), CCL5 (68), and TLR4 (69) are upregulated in fibroblasts after in vitro activation or cardiac injury, suggesting that fibroblasts directly control inflammatory cell activity within the infarct region (reviewed in 70). Although expression of these molecules by fibroblasts has been documented by multiple groups, the relative importance of fibroblast expression of these genes compared to other cardiac or infiltrating immune cells is less certain. Most studies have used global knockout or antibody inhibition to address a given inflammatory signaling pathway. Therefore, unless fibroblasts are the only cells that secrete the protein, these studies only indicate the importance of the signaling pathway rather than the specific role of fibroblasts in the inflammatory response.
Fibroblasts in angiogenesis.
Although cardiac fibroblast involvement in angiogenesis has been suggested in vitro and in engineered tissue (71, 72), a limited number of studies have demonstrated angiogenic properties of cardiac fibroblasts either during development or after a cardiac insult. The Wntx cells described above were suggested to have angiogenic potential based on gene expression of paracrine factors that could interact with endothelial cells such as pleiotrophin, myocilin, and Timp3 (43). Although the angiogenic capacity of this population of fibroblasts was not determined biologically in this study, another group also suggested that after myocardial infarction the fibroblast transcription profile shifts from proinflammatory to proangiogenic. Using bulk sequencing to evaluate fibroblast gene expression in mice, fibroblasts isolated from infarct regions 3 days after myocardial infarction had increased expression of angiogenic pathway genes, including Vegfa, and cultured supernatant from fibroblasts isolated 3 days after myocardial infarction stimulated angiogenesis. However, at day 7 after myocardial infarction, inhibitors of angiogenesis were upregulated in fibroblasts (68). Further investigation of fibroblast roles in angiogenesis is necessary to elucidate additional interactions between fibroblasts and endothelial cells.
Fibroblasts and extracellular matrix production.
Historically, the term for an activated fibroblast has been myofibroblast. This cell nomenclature is rooted in the observation that populations of cells within a zone of injury or inflammation establish actin stress fibers that contain α-SMA or SM22. While expression of these contractile proteins is one reliable means to identify myofibroblasts, VSMCs also express these proteins, and distinguishing between the two cell types may be difficult. In addition, recent studies suggest that a more complex spectrum of fibroblast profiles exists than a simple binary state between activated and resting fibroblasts. One gene that is upregulated in activated cardiac fibroblasts is periostin. Periostin is a matricellular protein that is not present in resting or quiescent fibroblasts, but after injury it is rapidly, specifically, and robustly expressed by cardiac fibroblasts in regions of matrix reorganization. A comparison of Tcf21 lineage cells and periostin-expressing cells, either after myocardial infarction or angiotensin infusion, demonstrated that periostin cells represent an activated fibroblast population that derives predominantly from the existing resident fibroblast population (10). This paper also proposed that a percentage of these activated, periostin-expressing cells return to a quiescent state after regression of the fibrotic response.
Pdgfrα-expressing fibroblasts were also examined after myocardial infarction by single-cell sequencing analyses. Four fibroblast subpopulations that were not present in sham-treated hearts emerged. These included a population that expressed cell cycle genes denoting proliferative cells. A second population that clustered with the cycling fibroblasts expressed activation markers such as periostin and α-SMA but no cell cycle genes. These cells were thought to be transitioning toward the proliferative state. Seven days after myocardial infarction two additional populations emerged that were thought to be myofibroblasts. One population was considered antifibrotic while the other population expressed ECM-related genes and was termed profibrotic. Additional analyses with more limited cell numbers did not detect these specific subsets, but similar gene profiles were observed in all of the activated populations (73). In a spontaneous mouse model of fibrosis, single-cell sequencing also identified subpopulations of fibroblasts that could be distinguished as ECM remodeling, Tgfβ1 signaling, Wnt signaling, and proliferation. The genes driving these categories had some overlap with the genes in the previous fibroblast categories identified by other single-cell sequencing described above but did not segregate into the same clusters (55), suggesting that each fibrotic model may have its own temporal and spatial fibroblast gene expression profiles.
Fibroblast expression of matrix-modifying genes can occur weeks after the fibroblast proliferative phase. One profile of expression was described as a specialized matrix-producing cell, a matrifibrocyte. These cells were identified because they persisted after the majority of the activated fibroblasts had either developed a quiescent profile of gene expression or underwent programmed cell death (54). This matrifibrocyte population robustly expressed genes previously associated with bone and cartilage remodeling, including Chad, Cilp2, and Comp. The authors suggested that these specialized fibroblasts are maintained to stabilize the scar after myocardial infarction. Other reports have also found upregulation of some of these genes in fibroblasts after myocardial infarction (43).
Fibroblast senescence.
Recent studies have suggested that a population of cardiac fibroblasts become senescent after injury or during aging. These senescent fibroblasts may secrete inflammatory molecules that modulate innate immunity (74). Senescent myofibroblast cells have been described in both animal models of cardiac fibrosis and human biopsies with cardiac disease, and cardiomyocyte secretion of a senescence-inducing protein, CCN1, resulted in improved heart function after pressure overload (75). Adipose-derived osteopontin has also been suggested to inhibit cardiac fibroblast senescence in aged mice, thereby promoting a fibrotic response. Osteopontin-null animals exhibited reduced fibrosis and increased numbers of senescent fibroblasts (76). After myocardial infarction, p53 was demonstrated to promote senescence of cardiac fibroblasts, thus altering fibroblast ECM and inflammatory regulators (77). The matrifibrocyte, described above, may be considered as a cell exhibiting some characteristics of senescence in that it appears refractory to proliferation upon subsequent insult, though senescence was not investigated. In vitro stimulation of cardiac fibroblasts with palmitate resulted in activation of the NLRP3 inflammasome and senescence as determined by senescence-associated β-galactosidase activity and cell cycle arrest (78). Taken together, these studies suggest that senescent fibroblasts may have differential gene expression compared to other fibroblasts, but further investigation is necessary to determine how these cells contribute to fibrosis.
Other cell types implicated in fibrosis.
There is a current debate regarding the cell population(s) responsible for fibrogenesis. In addition to the resident fibroblast populations described above, multiple cell types have been identified that express collagen or α-SMA after injury.
Extracardiac and Endothelial Cells
Several sources of myofibroblasts have been proposed, including fibrocytes, macrophages, bone marrow–derived cells (79–81), and endothelial cells (82). Multiple lines of evidence now suggest that these sources may not contribute significantly to the matrix deposition in the damaged heart.
Using a variety of experimental heart injury models, researchers investigated the relative contribution of collagen-producing nonresident cells to the collagen-producing resident fibroblasts. These studies have found that in most circumstances the resident fibroblasts comprise the majority of responding fibrogenic cells. Bone marrow chimeras with labeled donor cells were used to determine the number of myofibroblasts derived from circulating and bone marrow–derived cells after myocardial infarction. These studies concluded that few bone marrow cells contributed to the myofibroblast population (59, 83, 84).Even when the animals’ circulatory systems were linked by parabiosis, minimal contribution of the circulating cells was observed after pressure overload (5). Lineage tracing of endothelial cells, VSMCs, and hematopoietic cells also found few fibrogenic or myofibroblast cells derived from these cell populations after myocardial infarction (10, 84) or pressure overload (5, 7). In multiple experimental systems evaluated to date, the resident fibroblast population contributes the majority of the collagen-producing and myofibroblast cells, although all types of cardiac insult have not been examined. It should be remembered that all of these findings were based on mouse studies, and further work is needed to identify the fibrogenic populations in the human heart.
Perivascular Cells
Some studies have suggested that additional mesenchymal populations may be capable of generating ECM. Several groups have described a perivascular, mesenchymal stromal/stem cell that responds to cardiac injury by producing collagen or expressing α-SMA (themyofibroblastmarker). Each study used different means for identifying these cells. For example, a Gli1-positive perivascular cell that develops into a myofibroblast was described. These cells express CD29, Sca1, CD44, and CD105. They constitute 0.07% of the non-cardiomyocytes in the adult, and ablation of these Gli1-expressing cells resulted in reduced fibrosis in response to pressure overload or angiotensin II infusion induced hypertension (25). In skeletal muscle, Nestin- and NG2-positive pericytes formed adipocytes and fibroblasts after injury (85). When the same population was studied in the heart the cells expanded after myocardial infarction but did not express collagen (86). Although relative numbers of perivascular/pericyte populations were not compared to the resident fibroblast cells, these studies demonstrate that a continuum of gene expression may exist between fibroblasts, pericytes, and VSMCs.
Mesenchymal Stromal/Stem Cells
Interestingly, several studies have suggested that a subset of fibroblasts may possess stem cell potential (reviewed in 87). These multipotent cells have been described by multiple names including FAPs, colony-forming unit fibroblasts (CFU-Fs), and cardiac progenitor cells. Identification of this cardiac population varies between studies, but expression of both Sca1/Ly6a and Pdgfrα are consistent markers used by several investigators. Sca1-expressing fibroblasts may secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) and promote heart failure by changing the macrophage response (88). Others have shown that these Sca1 cells can self-renew and are multipotent (44, 45, 89). When cultured in vitro, these cells form adipocytes, VSMCs, fibroblasts, and in some cases cardiomyocytes. The reported abundance of these cells suggests that they are rarer than the Sca1 population described above. The tripotency of these cells in vitro is very reminiscent of that described for mesenchymal stem cells, but a clear in vivo role has not been established. It has been suggested that these cells may contribute to fat accumulation associated with arrhythmogenic cardiomyopathy, as discussed below.
NOVEL ROLES FOR FIBROBLASTS
Fibroblasts Adopting Characteristics of Other Cell Types
I have described the fibroblast phenotypes at rest and after injury, but some studies have suggested that fibroblasts can become other cell types. After cryoinjury in the C3H strain of mice, fibroblasts can express osteogenic genes and contribute to calcification (90). These results are similar to the finding that Gli1-expressing cells in the aorta can also develop into osteoblast-like cells and contribute to aortic calcification. The authors concluded that the adventitial cell was a VSMC progenitor (26). Other studies have suggested that after myocardial infarction fibroblasts have the capacity to become endothelial like and generate 30% of the endothelial cells in newly formed patent vessels (91). This conversion has been challenged by others who found that most nascent endothelial cells derive from existing endothelial cells (92). Consistent with the idea that some fibroblasts exhibit tripartite stem cell behavior, one study using Pdgfrα lineage tracing observed that some of the lineage-traced cells form adipocytes after myocardial infarction and contribute to arrhythmogenic cardiomyopathy (46, 93). However, these results may have technical limitations, as others have demonstrated that epicardial cells can contribute to epicardial adipose tissue (94, 95). Considering the lineage tracing methods that were used, it may be difficult to distinguish between the contribution of an epicardial derived fibroblast and the epicardium (16).
Additional Comparisons
While some criteria for segregating fibroblast phenotypes are outlined above, other fibroblast distinctions have also been reported. For example, one study compared human fetal cardiac fibroblast gene expression to adult human cardiac fibroblasts. They found that a great number of common genes were expressed between these two populations, but several differences were noted. Notch signaling was enriched in the fetal fibroblasts, while IL-6 signaling was enriched in the adult (96). In a forced swim model of exercise, cardiac fibroblasts upregulated antioxidant genes and downregulated ECM gene expression compared to control animals (41). Another study demonstrated by single-cell sequencing that modest sexual dimorphism in cardiac fibroblast gene expression occurs (42), and even mouse strain-specific fibrotic responses have been reported (90, 97).
CONCLUSIONS
Cardiac fibroblasts have been problematic to study because there have been limited robust means for unambiguously identifying these cells. The field of cardiac fibroblast biology is at a turning point where we can begin to appreciate the diverse roles of these cells (Figure 2). Single-cell sequencing analysis has revealed that subpopulations of fibroblasts have heterogeneous gene expression, and these variations should be considered in future studies. Fibroblast subsets are likely to vary depending on the type and extent of the cardiac insult, and a deeper understanding of these differences could provide important insights into the development of therapeutics to combat cardiac fibrosis. Additionally, location within the chamber and proximity to other cell types may be important to consider. Much of the current information regarding cardiac fibroblasts is dependent on animal studies, and we should strive to validate these findings in human cardiac disease if we are to develop strategies for managing pathologic fibrosis.
Figure 2.
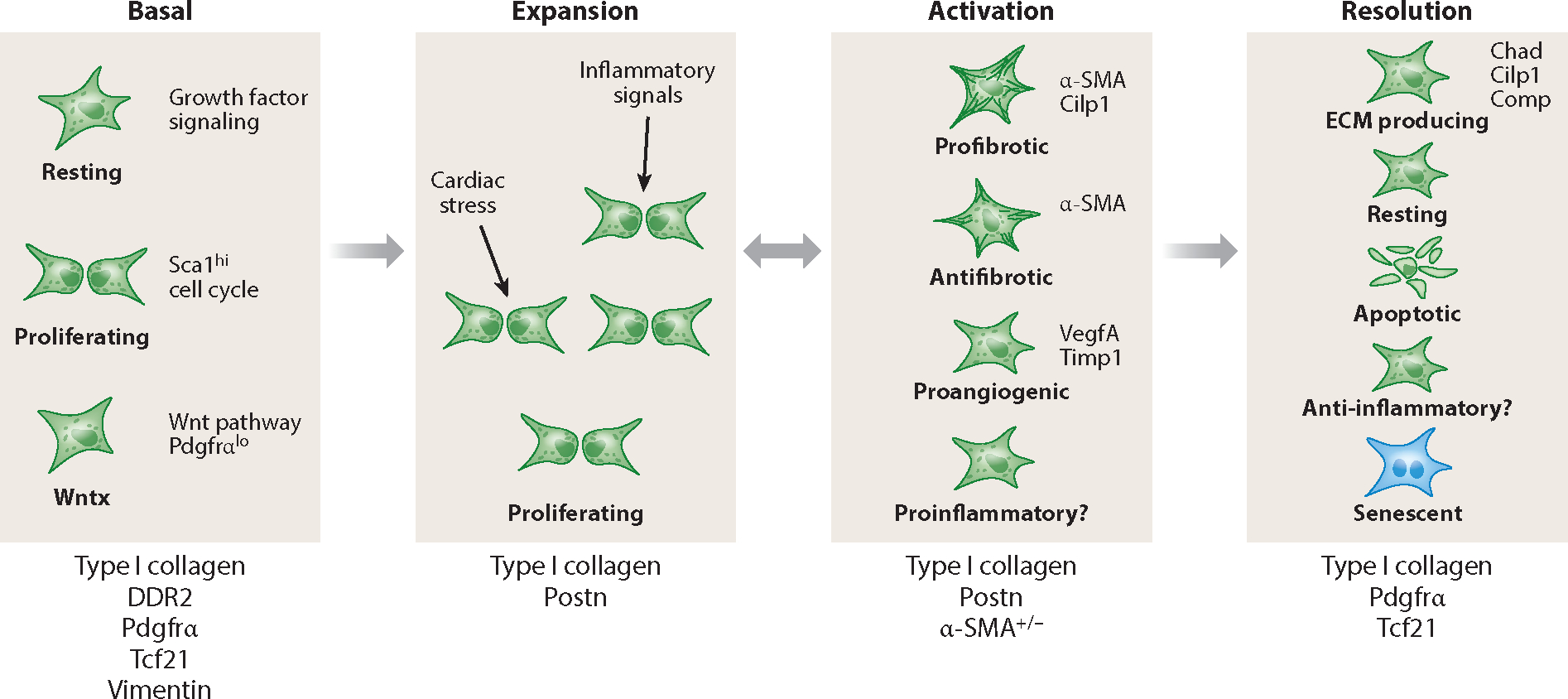
Fibroblast stages at rest and after injury. Several studies have found three subsets of resting cardiac fibroblasts. After injury or infection or during aging, fibroblasts become activated in response to inflammatory cytokines or cardiomyocyte-secreted factors. Fibroblasts enter a proliferative phase, which may overlap with the production of extracellular matrix (ECM). The expanded population of fibroblasts can be divided into at least four different subsets. Myofibroblasts are α-SMA+, while α-SMA− cells may promote angiogenesis. There are multiple proposed fates for the expanded fibroblasts. Some revert to resting fibroblasts, whereas others exhibit a unique and continued ECM production phase. Others either undergo apoptosis or become senescent. Although multiple studies have implicated a role for fibroblasts in regulating inflammation, single-cell sequencing experiments have not described an inflammatory gene expression profile. Below each fibroblast phase are some markers that may be expressed by a majority of cells in that phase.
ACKNOWLEDGMENTS
I would like to thank the Tallquist lab and Ralph Shohet for their thoughtful comments. I would also like to apologize for any research that was omitted due to space limitations. This work was funded by grants from the US National Institutes of Health (HL144067, GM113134, and AHA 17GRNT33660474).
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Eghbali M, Weber KT. 1990. Collagen and the myocardium: fibrillar structure, biosynthesis and degradation in relation to hypertrophy and its regression. Mol. Cell. Biochem. 96:1–14 [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, et al. 2018. Heart disease and stroke statistics—2018 update. A report from the American Heart Association. Circulation 137:e67–492 [DOI] [PubMed] [Google Scholar]
- 3.Gourdie RG, Dimmeler S, Kohl P. 2016. Novel therapeutic strategies targeting fibroblasts and fibrosisin heart disease. Nat. Rev. Drug Discov. 15:620–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, et al. 2016. Revisiting cardiac cellular composition. Circ. Res. 118:400–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, et al.2014. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 115:625–35 [DOI] [PubMed] [Google Scholar]
- 6.Smith CL, Baek ST, Sung CY, Tallquist MD. 2011. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 108:e15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, et al. 2014. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 124:2921–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, et al. 2012. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139:2139–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song K, Nam YJ, Luo X, Qi X, Tan W, et al. 2012. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485:599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, et al. 2016. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT. 1995. Regulation of collagen degradation in the rat myocardium after infarction. J. Mol. Cell. Cardiol. 27:1281–92 [DOI] [PubMed] [Google Scholar]
- 12.Armulik A, Genove G, Betsholtz C. 2011. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21:193–215 [DOI] [PubMed] [Google Scholar]
- 13.Guimarães-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, et al. 2017. Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 20:345–59.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida T, Owens GK. 2005. Molecular determinants of vascular smooth muscle cell diversity. Circ. Res. 96:280–91 [DOI] [PubMed] [Google Scholar]
- 15.Miano JM, Cserjesi P, Ligon KL, Periasamy M, Olson EN. 1994. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ. Res. 75:803–12 [DOI] [PubMed] [Google Scholar]
- 16.Swonger JM, Liu JS, Ivey MJ, Tallquist MD. 2016. Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation 92:66–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuwabara JT, Tallquist MD. 2017. Tracking adventitial fibroblast contribution to disease: a review of current methods to identify resident fibroblasts. Arterioscler. Thromb. Vasc. Biol. 37:1598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camelliti P, Borg TK, Kohl P. 2005. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 65:40–51 [DOI] [PubMed] [Google Scholar]
- 19.Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, et al. 2014. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc. Res. 101:411–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu J, Seldin MM, Fu K, Li S, Lam L, et al. 2018. Topological arrangement of cardiac fibroblasts regulates cellular plasticity. Circ. Res. 123:73–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hookway TA, Matthys OB, Mendoza-Camacho FN, Rains S, Sepulveda JE, et al. 2019. Phenotypic variation between stromal cells differentially impacts engineered cardiac tissue function. Tissue Eng. A 25. 10.1089/ten.tea.2018.0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doll S, Dressen M, Geyer PE, Itzhak DN, Braun C, et al. 2017. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 8:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, et al. 2013. The adventitia: essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 75:23–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, et al. 2008. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. PNAS 105:9349–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, et al. 2015. Perivascular Gli1+progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16:51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, et al. 2016. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell 19:628–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zahid S, Cochet H, Boyle PM, Schwarz EL, Whyte KN, et al. 2016. Patient-derived models link re-entrant driver localization in atrial fibrillation to fibrosis spatial pattern. Cardiovasc. Res. 110:443–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marrouche NF, Wilber D, Hindricks G, Jais P, Akoum N, et al. 2014. Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA 311:498–506 [DOI] [PubMed] [Google Scholar]
- 29.Hanna N, Cardin S, Leung TK, Nattel S. 2004. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc. Res. 63:236–44 [DOI] [PubMed] [Google Scholar]
- 30.Burstein B, Libby E, Calderone A, Nattel S. 2008. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation 117:1630–41 [DOI] [PubMed] [Google Scholar]
- 31.Kolditz DP, Wijffels MC, Blom NA, van der Laarse A, Hahurij ND, et al. 2008. Epicardium-derived cells in development of annulus fibrosis and persistence of accessory pathways. Circulation 117:1508–17 [DOI] [PubMed] [Google Scholar]
- 32.Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. 2017. Developmental mechanisms of aortic valve malformation and disease. Annu. Rev. Physiol. 79:21–41 [DOI] [PubMed] [Google Scholar]
- 33.Liu AC, Joag VR, Gotlieb AI. 2007. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 171:1407–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikawa T, Gourdie RG. 1996. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 174:221–32 [DOI] [PubMed] [Google Scholar]
- 35.Dettman RW, Denetclaw W Jr., Ordahl CP, Bristow J 1998. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193:169–81 [DOI] [PubMed] [Google Scholar]
- 36.Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. 1998. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 82:1043–52 [DOI] [PubMed] [Google Scholar]
- 37.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, et al. 2008. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. 2010. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 338:251–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, et al. 2012. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366:111–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampaio-Pinto V, Rodrigues SC, Laundos TL, Silva ED, Vasques-Novoa F, et al. 2018. Neonatal apex resection triggers cardiomyocyte proliferation, neovascularization and functional recovery despite local fibrosis. Stem Cell Rep. 10:860–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lighthouse JK, Burke RM, Velasquez LS, Dirkx RA Jr., Aiezza A 2nd, et al. 2019. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight 4:e92098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, et al. 2018. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 22:600–10 [DOI] [PubMed] [Google Scholar]
- 43.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, et al. 2019. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 8:e43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chong JJ, Chandrakanthan V, Xaymardan M, Asli NS, Li J, et al. 2011. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 9:527–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noseda M, Harada M, McSweeney S, Leja T, Belian E, et al. 2015. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat. Commun. 6:6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, et al. 2016. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ. Res. 119:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, et al. 2015. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 21:786–94 [DOI] [PubMed] [Google Scholar]
- 48.Joe AW,Yi L, Natarajan A, Le Grand F, So L, et al.2010.Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12:153–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, et al. 2016. Single-cell resolution of temporal gene expression during heart development. Dev. Cell 39:480–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, et al. 2018. Hippo signaling plays an essential rolein cell state transitions during cardiac fibroblast development. Dev. Cell 45:153–69.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui Y, Zheng Y, Liu X, Yan L, Fan X, et al. 2019. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26:1934–50.e5 [DOI] [PubMed] [Google Scholar]
- 52.Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, et al. 2018. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 32:1344–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD. 2018. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 114:161–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, et al. 2018. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 128:2127–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, et al. 2017. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552:110–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kretzschmar K, Post Y, Bannier-Helaouet M, Mattiotti A, Drost J, et al. 2018. Profiling proliferative cells and their progeny in damaged murine hearts. PNAS 115:E12245–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, et al. 2016. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med 8:949–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prabhu SD, Frangogiannis NG. 2016. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119:91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, et al. 2017. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Investig. 127:383–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rossini A, Zacheo A, Mocini D, Totta P, Facchiano A, et al. 2008. HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. J. Mol. Cell. Cardiol. 44:683–93 [DOI] [PubMed] [Google Scholar]
- 61.Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE. 2009. Interleukin-1α stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 297:H1117–27 [DOI] [PubMed] [Google Scholar]
- 62.Tomita K, Takashina M, Mizuno N, Sakata K, Hattori K, et al. 2015. Cardiac fibroblasts: contributory role in septic cardiac dysfunction. J. Surg. Res. 193:874–87 [DOI] [PubMed] [Google Scholar]
- 63.Ma F, Li Y, Jia L, Han Y, Cheng J, et al. 2012. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLOS ONE 7:e35144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, et al. 2007. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am. J. Physiol. Cell Physiol. 292:C1799–808 [DOI] [PubMed] [Google Scholar]
- 65.Humeres C, Vivar R, Boza P, Muñoz C, Bolivar S, et al.2016. Cardiac fibroblast cytokine profiles induced by proinflammatory or profibrotic stimuli promote monocyte recruitment and modulate macrophage M1/M2 balance in vitro. J. Mol. Cell. Cardiol. 101:69–80 [DOI] [PubMed] [Google Scholar]
- 66.Anzai A, Choi JL, He S, Fenn AM, Nairz M, et al. 2017. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 214:3293–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boza P, Ayala P, Vivar R, Humeres C, Cáceres FT, et al. 2016. Expression and function of toll-like receptor 4 and inflammasomes in cardiac fibroblasts and myofibroblasts: IL-1β synthesis, secretion, and degradation. Mol. Immunol. 74:96–105 [DOI] [PubMed] [Google Scholar]
- 68.Mouton AJ, Ma Y, Gonzalez OJR, Daseke MJ 2nd, Flynn ER, et al. 2019. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 114:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muñoz-Rodríguez C, Fernández S, Osorio JM, Olivares F, Anfossi R, et al. 2018. Expression and function of TLR4- induced B1R bradykinin receptor on cardiac fibroblasts. Toxicol. Appl. Pharmacol. 351:46–56 [DOI] [PubMed] [Google Scholar]
- 70.Turner NA. 2016. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 94:189–200 [DOI] [PubMed] [Google Scholar]
- 71.Twardowski RL, Black LD 3rd. 2014. Cardiac fibroblasts support endothelial cell proliferation and sprout formation but not the development of multicellular sprouts in a fibrin gel co-culture model. Ann. Biomed. Eng. 42:1074–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nehls V, Herrmann R, Hühnken M, Palmetshofer A. 1998. Contact-dependent inhibition of angiogenesis by cardiac fibroblasts in three-dimensional fibrin gels in vitro: implications for microvascular network remodeling and coronary collateral formation. Cell Tissue Res. 293:479–88 [DOI] [PubMed] [Google Scholar]
- 73.Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, et al. 2018. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation 138:166–80 [DOI] [PubMed] [Google Scholar]
- 74.Hoare M, Narita M. 2013. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. 15:887–89 [DOI] [PubMed] [Google Scholar]
- 75.Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. 2016. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 67:2018–28 [DOI] [PubMed] [Google Scholar]
- 76.Sawaki D, Czibik G, Pini M, Ternacle J, Suffee N, et al. 2018. Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production. Circulation 138:809–22 [DOI] [PubMed] [Google Scholar]
- 77.Zhu F, Li Y, Zhang J, Piao C, Liu T, et al. 2013. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLOS ONE 8:e74535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sokolova M, Vinge LE, Alfsnes K, Olsen MB, Eide L, et al. 2017. Palmitate promotes inflammatory responses and cellular senescence in cardiac fibroblasts.Biochim.Biophys.Acta Mol.Cell Biol.Lipids 1862:234–45 [DOI] [PubMed] [Google Scholar]
- 79.Mollmann H, Nef HM, Kostin S, von Kalle C, Pilz I, et al. 2006. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 71:661–71 [DOI] [PubMed] [Google Scholar]
- 80.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, et al. 2006. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. PNAS 103:18284–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, et al. 2010. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 49:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, et al. 2007. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13:952–61 [DOI] [PubMed] [Google Scholar]
- 83.Ruiz-Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda G, et al. 2015. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 65:2057–66 [DOI] [PubMed] [Google Scholar]
- 84.Moore-Morris T, Cattaneo P, Guimarães-Camboa N, Bogomolovas J, Cedenilla M, et al. 2018. Infarct fibroblasts do not derive from bone marrow lineages. Circ. Res. 122:583–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. 2013. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am. J. Physiol. Cell Physiol. 305:C1098–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birbrair A, Zhang T, Files DC, Mannava S, Smith T, et al. 2014. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 5:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santini MP, Forte E, Harvey RP, Kovacic JC. 2016. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 143:1242–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen G, Bracamonte-Baran W, Diny NL, Hou X, Talor MV, et al. 2018. Sca-1+ cardiac fibroblasts promote development of heart failure. Eur. J. Immunol. 48:1522–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chong JJ, Reinecke H, Iwata M, Torok-Storb B, Stempien-Otero A, Murry CE. 2013. Progenitor cells identified by PDGFR-α expression in the developing and diseased human heart. Stem Cells Dev. 22:1932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pillai IC, Li S, Romay M, Lam L, Lu Y, et al. 2017. Cardiac fibroblasts adopt osteogenic fates and can be targeted to attenuate pathological heart calcification. Cell Stem Cell 20:218–32.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, et al. 2014. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 514:585–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He L, Huang X, Kanisicak O, Li Y, Wang Y, et al. 2017. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 127:2968–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zangi L, Oliveira MS, Ye LY, Ma Q, Sultana N, et al. 2017. Insulin-like growth factor 1 receptor-dependent pathway drives epicardial adipose tissue formation after myocardial injury. Circulation 135:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamaguchi Y, Cavallero S, Patterson M, Shen H, Xu J, et al. 2015. Adipogenesis and epicardial adipose tissue: a novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. PNAS 112:2070–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Q, Huang X, Oh JH, Lin RZ, Duan S, et al. 2014. Epicardium-to-fat transition in injured heart. Cell Res. 24:1367–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jonsson MKB, Hartman RJG, Ackers-Johnson M, Tan WLW, Lim B, et al. 2016. A transcriptomic and epigenomic comparison of fetal and adult human cardiac fibroblasts reveals novel key transcription factors in adult cardiac fibroblasts. JACC Basic Transl. Sci. 1:590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park S, Ranjbarvaziri S, Lay FD, Zhao P, Miller MJ, et al. 2018. Genetic regulation of fibroblast activation and proliferation in cardiac fibrosis. Circulation 138:1224–35 [DOI] [PMC free article] [PubMed] [Google Scholar]