

Abstract
Many viruses interfere with apoptosis of infected cells, presumably preventing cellular apoptosis as a direct response to viral infection. Since cytotoxic T lymphocytes (CTL) induce apoptosis of infected cells as part of the “lethal hit,” inhibition of apoptosis could represent an effective immune evasion strategy. We report here herpes simplex virus type 1 (HSV-1) interference with CTL-induced apoptosis of infected cells and show that HSV-1 inhibits the nuclear manifestations of apoptosis but not the membrane changes. The HL-60 cell line (human promyelocytic leukemia) undergoes apoptosis in response to many stimuli, including incubation with ethanol. After HSV-1 infection (strains E115 and 17+), ethanol-treated cells did not produce oligonucleosomal DNA fragments characteristic of apoptosis, as assayed by gel electrophoresis and enzyme-linked immunosorbent assay. Inhibition was detected 2 h after infection and increased over time. Importantly, HSV-1-infected cells were resistant to apoptosis induced by antigen-specific CD4+ CTL, despite the fact that CTL recognition and degranulation in response to infected targets remained intact. Unlike HSV-1, HSV-2 (strains 333 and HG52) did not inhibit DNA fragmentation. In contrast to the inhibition of DNA fragmentation by HSV-1, none of the HSV-1 or -2 strains interfered with the ethanol-induced exposure of surface phosphatidylserine characteristic of apoptosis, as determined by annexin V binding. These results demonstrate that genes of HSV-1 inhibit the nuclear manifestations of apoptosis but not the membrane manifestations, suggesting that these may be mediated via separate pathways. They also suggest that HSV-1 inhibition of CTL-induced apoptosis may be an important mechanism of immune evasion.
The herpes simplex viruses (HSV) are thought to have evolved roughly in parallel with the vertebrate immune system (8, 23), and HSV is well adapted for survival in its host. After infection with HSV, a reservoir of latent virus is maintained within the neurons of the dorsal root ganglia. Periodically, the virus reactivates and infectious virions are released; virus replicates in epithelial cells of mucosal surfaces. Host immune responses appear to locally control viral replication (30). While it has long been recognized that patients with impaired T-cell responses exhibit frequent HSV disease, recent studies have indicated that HSV reactivation is frequent even in immunocompetent patients (37). Normal, healthy adults reactivate HSV type 2 (HSV-2) on an average of 10 to 30% of days; suggesting that HSV have likely evolved several novel ways to avoid even normal host defenses. Workers in several laboratories, including ours, have described HSV-mediated downregulation of major histocompatibility complex (MHC) class I expression on fibroblasts and keratinocytes (11, 13, 15). Recently, HSV protein ICP47 has been shown to bind to the TAP-1 protein and prevent the exportation of peptide-laden MHC class I molecules to the surface of cells (7, 10). While downregulation of MHC class I would be expected to affect CD8+ cytotoxic T lymphocytes (CTL) only, HSV also inhibits the killing ability of CD4+ CTL (27), suggesting that additional viral strategies to avoid CTL responses may be operative.
CTL kill nucleated target cells predominantly by inducing apoptosis or programmed cell death (2). CTL induce apoptosis either by using components of their lytic granules (25) or by engagement of the target cell Fas receptor by the Fas ligand on the CTL surface (9). Since induction of apoptosis is critical to the control of viral infections by CTL, inhibition of apoptosis could be an important mechanism of immune evasion. Several viruses are known to produce proteins interfering with apoptosis (34), although many have no effect on apoptosis induced by CTL. Recent reports suggest that HSV may also encode antiapoptotic proteins (14, 17), although their ability to inhibit CTL-induced apoptosis has not been established.
In this report, we demonstrate that laboratory strains of HSV-1, but not HSV-2, inhibit the oligonucleosomal DNA fragmentation characteristic of apoptosis, including apoptosis induced by CTL. In contrast, neither HSV-1 nor HSV-2 had any inhibitory effect on membrane phosphatidylserine (PS) exposure, another characteristic feature of apoptosis. These results suggest that inhibition of CTL-induced apoptosis may be an important mechanism of immune suppression by HSV-1. The results also suggest that the nuclear and membrane manifestations of apoptosis may be differentially regulated.
MATERIALS AND METHODS
Cell lines.
The human promyelocytic leukemia cell line HL-60 was the kind gift of Kindred Ritchie. CTL clones ESL3.326, ESL4.34, and 1A.B.25 were derived from patients with culture-proven HSV-2 infection. ESL3.326 is a CD4+, HLA DQB1*0302-restricted CTL clone which recognizes amino acid residues 433 to 445 of the HSV-2 VP16 protein (16). ESL4.34 is a CD4+, DRB1*0402-restricted CTL clone which recognizes amino acid residues 393 to 405 of the HSV-2 VP16 protein (5). 1A.B.25 is a CD4+, DQB1*0201-restricted CTL clone which recognizes amino acid residues 431 to 440 of the HSV-2 VP16 protein (12). Epstein-Barr virus-immortalized lymphoblastoid cell lines (LCL) Priess (DQ0302), YAR (DR0402), and MAT (DQ0201) were obtained from the VIIIth and Xth International Histocompatibility Panels.
Viruses.
HSV-1 strains E115, 17+, and 90 and HSV-2 strains 333 and HG52 were grown and their titers were determined in Vero cells or human foreskin fibroblasts. The cell lines in which viral stocks were grown were screened for mycoplasma contamination and were negative.
Cytotoxicity assays.
For detection of CTL-induced apoptosis, we used a modification of the JAM assay (22). Target cells consisted of peptide-pulsed, HLA-matched LCL. Target cells were labeled for 4 h with [3H]thymidine at 37°C in growth medium and, at the same time, loaded with 5 μg of the peptide recognized by the CTL clone per ml. After loading and labeling, the cells were washed and either infected with HSV at 10 or 33 PFU/cell or mock infected for 4 h. The target cells were then incubated with CTL for 4 h and aspirated onto fiberglass filters (Unifilter GF/C; Packard Instruments, Meriden, Conn.) by using a Packard Unifilter-96 harvester. Filters were counted in a Packard TopCount scintillation counter, and percent DNA fragmentation was calculated by the following formula: % DNA fragmentation = (S − E)/E, where S is retained DNA (counts) in the absence of treatment (spontaneous) and E is experimentally retained DNA (counts) with treatment.
Agarose gel electrophoresis.
HL-60 cells (2 × 105) were mock treated or treated with increasing concentrations of ethanol. After 4 h, the cells were collected by centrifugation and washed once in phosphate-buffered saline (PBS). The cells were resuspended in 50 μl of lysis buffer (1× Tris-borate-EDTA containing 0.5% Nonidet P-40 [Sigma] and 0.1 mg of RNase A [Sigma] per ml) and incubated for 30 min at 37°C. Proteinase K (Sigma) was added to a final concentration of 1.5 mg/ml, and the sample was incubated at 37°C for 30 min. A 15-μl volume of the specimen was loaded on a dry 2% agarose gel, and 1× Tris-borate-EDTA buffer was added. Horizontal gel electrophoresis was run at 7 V/cm for 90 min, and the gel was stained with SYBR green (FMC BioProducts, Rockland, Maine) and visualized under UV light.
ELISA.
As an alternative method to detect oligonucleosomal DNA fragmentation, the Cell Death Detection ELISA (enzyme-linked immunosorbent assay; Boehringer Mannheim, Indianapolis, Ind.) was used. HL-60 cells (2 × 104 to 2 × 105) were treated with increasing concentrations of ethanol or mock treated. After 4 h, the cells were collected by centrifugation, washed, resuspended in 500 μl of incubation buffer to lyse the cells, and incubated for 30 min at 4°C. The lysate was centrifuged at 15,000 rpm in an Eppendorf 5412 centrifuge (Brinkmann) for 5 min, and 400 μl of the supernatant, containing the fragmented DNA, was removed. The supernatant was diluted with incubation buffer to a final concentration of 105 cell equivalents/ml, and 100 μl was added to microtiter plate wells coated with antihistone antibody. After 90 min of incubation at room temperature, the wells were washed three times with PBS. Peroxidase-conjugated anti-DNA antibody was added to the wells, and they were incubated for 90 min at room temperature. After the wells were washed three times, 100 ml of colorimetric substrate was added and serial readings of optical density at 405 and 490 nm were taken at 5-min intervals.
Flow cytometry for membrane PS exposure.
Fluorescein isothiocyanate (FITC)-annexin V (1 mol/mol) was prepared as previously described (33). HL-60 cells (3 × 104 to 3 × 105) were infected with HSV strains or mock-infected for 4 to 6 h. The cells were then treated with various concentrations of ethanol for 4 h at 37°C. After treatment, the cells were harvested, washed in PBS, and resuspended in 300 μl of annexin labeling buffer (10 mM HEPES-Na [pH 7.4], 136 mM NaCl, 2.7 mM KCl, 2.0 mM MgCl2, 1 mM NaH2PO4, 5 mM glucose, 5 mg of bovine serum albumin per ml, 2.5 mM Ca2+, 10 μg of propidium iodide [PI] per ml, 100 nM FITC-annexin V). After 10 min of incubation at room temperature, paraformaldehyde was added to a final concentration of 1.0% and the cells were incubated for another 15 min at room temperature. The cells were then analyzed by two-color flow cytometry using a Coulter EPICS XL-MCL. Cells showing red fluorescence due to PI uptake were considered to be necrotic; that is, they had lost membrane integrity. Only those cells negative for PI uptake (cells with intact membrane integrity), which also stained with FITC-annexin V, were considered apoptotic.
BLT esterase assay for CTL degranulation.
The N-α-benzyloxycarbonyl-l-lysine thiobenzyl ester (BLT) assay was performed essentially as previously described (32). Briefly, CTL were coincubated with peptide-pulsed and/or HSV-infected target cells for 5 h. A 50-μl volume of the supernatant was removed and frozen at −20°C until the time of assay. Enzyme assays were conducted in 96-well plates at room temperature by addition of an equal volume of 0.1 M Tris (pH 8.1) containing 0.4 mM BLT (Calbiochem, San Diego, Calif.) and 0.4 mM 5′5′-dithiobis(2-nitrobenzoic acid) [Ellman’s Reagent]; Sigma Chemical Co., St. Louis, Mo.). The color change was monitored at 10-min intervals for 60 min at 405 nm by using a Bio-Tek EL310 automated plate reader, and the optical density change per minute was calculated.
RESULTS
Inhibition of DNA fragmentation by HSV-1.
To investigate the effect of HSV infection on the induction of apoptosis in infected cells, we initially used the human promyelocytic leukemia cell line HL-60. This cell line is easily induced to undergo apoptosis by a variety of stimuli. Incubation of HL-60 cells with ethanol for 4 h resulted in oligonucleosomal DNA fragmentation characteristic of apoptosis (Fig. 1, top). DNA fragmentation was most pronounced after treatment with 2.5 to 5% ethanol and was markedly greater than the background DNA fragmentation (0% ethanol) seen in these cultures. To determine whether infection with HSV-1 inhibited DNA fragmentation, HL-60 cells were infected with 10 PFU of HSV-1 strain E115 per cell for 13 h prior to treatment with ethanol. HSV-1-infected cells were resistant to the induction of DNA fragmentation (Fig. 1, bottom). DNA fragmentation after incubation with 2.5 to 5% ethanol was completely inhibited by HSV-1 infection and markedly reduced after treatment with ethanol concentrations as high as 10 to 20%.
FIG. 1.
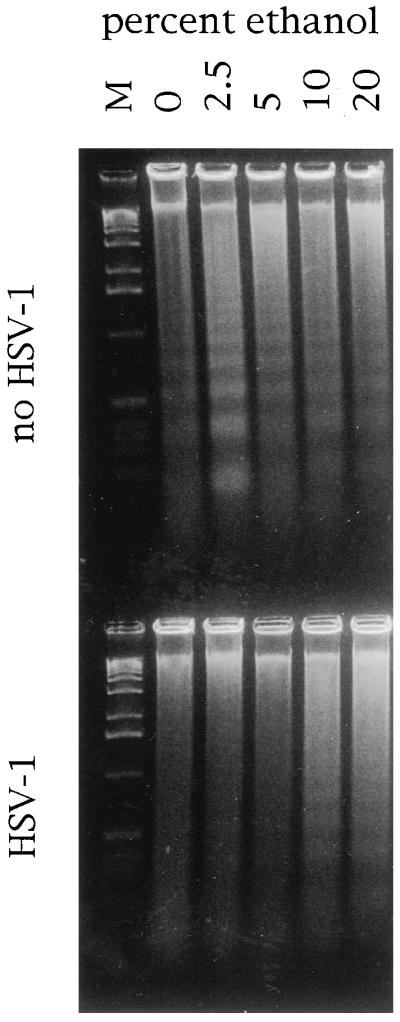
HSV-1 inhibits oligonucleosomal DNA fragmentation. HSV-infected or uninfected HL-60 cells were treated with the indicated concentrations of ethanol and assayed for oligonucleosomal DNA fragmentation by agarose gel electrophoresis. M, DNA molecular size marker.
To confirm these findings, the effect of HSV-1 on DNA fragmentation was assessed by a sandwich ELISA technique. Again, infection of HL-60 cells with HSV-1 strain E115 for 13 h before ethanol treatment almost completely prevented DNA fragmentation (Fig. 2A). To evaluate the kinetics of the HSV-1 effect on DNA fragmentation, the effect of HSV-1 infection 2 h prior to ethanol treatment, as well as that of infection 15 min before ethanol addition, was assessed. Preinfection of HL-60 cells for 2 h prior to addition of ethanol still had a strong inhibitory effect on DNA fragmentation (Fig. 2B). However, when the cells were infected only 15 min before ethanol addition, little or no inhibitory effect was seen (Fig. 2C). Subsequent experiments used a preinfection time of 5 h to allow full expression of the antiapoptotic effect.
FIG. 2.

Inhibition of oligonucleosomal DNA fragmentation by HSV-1 strains E115 and 17+ and lack of inhibition by HSV-2 strain 333. HL-60 cells were infected with HSV-1 strain E115 for 13 h (A), HSV-1 strain E115 for 2 h (B), HSV-1 strain E115 for 15 min (C), HSV-1 strain 17+ for 5 h (D), HSV-2 strain 333 for 5 h (E) prior to apoptosis induction with ethanol (EtOH). Squares, uninfected control cells; diamonds, HSV-infected cells. O.D. 405/490; ratio of optical density at 405 nm to that at 490 nm.
To determine whether the inhibitory effect on DNA fragmentation would also be seen with other HSV-1 strains, HL-60 cells were preinfected with strain 17+ for 5 h prior to ethanol treatment. Inhibition of DNA fragmentation by the 17+ strain of HSV-1 was similar to that seen with strain E115, as determined by sandwich ELISA (Fig. 2D). Surprisingly, HSV-2 strain 333 did not show inhibition of DNA fragmentation (Fig. 2E), nor did a second HSV-2 strain, HG52 (data not shown). Successful HSV-2 infection was confirmed by immunofluorescent staining for HSV-2 proteins, followed by flow cytometry (data not shown).
Lack of inhibition of membrane changes of apoptosis by HSV-1.
Although DNA fragmentation is the most well-described manifestation of apoptosis, apoptotic cells also demonstrate characteristic membrane changes. One of these is the exposure of PS on the outer leaflet of the cell membrane. In the normal, healthy cell, PS is localized almost exclusively to the inner leaflet of the cell membrane. During apoptosis, however, PS equilibrates between the two leaflets (6). PS exposure is thought to play a role in alerting phagocytic cells to the presence of apoptosis (1, 29, 36). HL-60 cells treated with 5% ethanol for 4 h showed a marked increase in PS exposure compared with untreated cells (Fig. 3). To determine whether HSV-1 infection inhibited PS exposure during apoptosis, HL-60 cells were infected with 10 PFU of HSV-1 strain E115 per cell for 5 h prior to incubation with ethanol. In contrast to the marked inhibitory effect of HSV-1 on DNA fragmentation, the virus had no inhibitory effect on PS exposure (Fig. 3). HSV-2 also had no effect on ethanol-induced PS exposure.
FIG. 3.

Neither HSV-1 nor HSV-2 inhibits the membrane PS exposure associated with apoptosis. HL-60 cells were mock infected or infected with HSV-1 strain E115 or HSV-2 strain 333 for 5 h and then mock treated or treated with 5% ethanol. Annexin V-FITC (x axis)-propidium iodide (y axis) labeling, followed by flow cytometry, was performed as described in the text. Quadrants: 2, necrotic cells; 3, nonnecrotic, PS-negative cells; 4, viable, PS-positive cells. The percentage of nonnecrotic, PS-positive cells is shown in quadrant 4. The small decrease in quadrant 4 cells in HSV-1-infected cells seen in this representative experiment was within the range of error of the assay and was not reproducible in multiple repeat experiments.
Inhibition of CTL-induced apoptosis by HSV-1.
CTL kill their targets by inducing them to undergo apoptosis (25). It has been reported that cells undergoing DNA fragmentation no longer support viral replication, suggesting that this may be an important mechanism for CTL control of viral infections (21). We therefore investigated whether HSV-1 could inhibit DNA fragmentation induced by CTL. MHC class II-restricted CTL clones were used for these experiments to avoid confounding effects from MHC class I downregulation by the HSV-1 gene product ICP47.
CTL clones were mixed at various effector-to-target cell ratios with peptide-pulsed, MHC-matched LCL which had been mock infected or infected with HSV 5 h earlier. After 4 h of incubation, the cells were harvested and target cell DNA fragmentation was determined by the JAM assay (22). When target cell line Priess (LCL) was infected with HSV-1 strain E115 at 33 PFU/cell, inhibition of DNA fragmentation induced by clone ESL3.326 was essentially complete (Fig. 4A). With infection by E115 at 10 PFU/cell, the HSV-1 inhibitory effect on DNA fragmentation was approximately 50%, similar to the effect of HSV-1 on ethanol-induced apoptosis (Fig. 4B). In contrast to HSV-1, HSV-2 strain 333 had no inhibitory effect on DNA fragmentation induced by CTL clone ESL3.326 (Fig. 4C).
FIG. 4.

Effects of HSV infection on DNA fragmentation induced by CTL clones ESL3.326 and 1A.B.25. A, clone ESL3.326 versus targets infected with HSV-1 strain E115 at 33 PFU/cell; B, clone ESL3.326 versus targets infected with HSV-1 strain E115 at 10 PFU/cell; C, clone ESL3.326 versus targets infected with HSV-2 strain 333 at 10 PFU/cell; D, clone 1A.B.25 versus targets infected with HSV-1 strain E115 at 10 PFU/cell; E, clone 1A.B.25 versus targets infected with UV-inactivated HSV-1 strain E115 at 10 PFU/cell; F, clone 1A.B.25 versus targets infected with HSV-2 strain 333 at 10 PFU/cell; G, clone 1A.B.25 versus targets infected with HSV-2 strain 333 at 33 PFU/cell. Squares, uninfected control cells; diamonds, HSV-infected cells. E:T ratio, effector-to-target cell ratio.
Although HSV-1 inhibited apoptosis induced by several other stimuli, it remained possible that the apparent inhibitory effect of HSV on CTL-induced apoptosis was due to impairment of CTL recognition of target cells, either by inhibition of antigen presentation by MHC class II, as previously described in neurons (18), or by some other means. However, immunofluorescence staining for MHC class II antigens DP, DQ, and DR showed no effect of HSV infection on MHC class II expression (data not shown). To evaluate antigen presentation to CTL, degranulation of CTL in response to infected versus uninfected target cells was evaluated by the BLT esterase assay. Specific CTL degranulation was equivalent or slightly higher in response to HSV-1-infected cells in comparison with uninfected cells (Fig. 5), confirming that HSV-1 did not inhibit CTL recognition of target cells.
FIG. 5.

HSV infection of targets does not interfere with recognition by CTL. Mock- or HSV-infected, peptide-pulsed LCL targets were incubated with CTL clone 3.326 at a 20:1 effector-to-target cell ratio. Supernatant was collected 5 h later, and CTL degranulation was measured by BLT esterase assay. O.D., optical density.
To confirm the ability of HSV-1 to inhibit DNA fragmentation induced by other CTL clones, a second CTL-target pair was used. Infection of target cell line MAT (LCL) with HSV-1 at 10 PFU/cell resulted in marked inhibition of DNA fragmentation induced by CTL clone 1A.B.25 (Fig. 4D). Incubation of target cells with UV-inactivated HSV-1 had no inhibitory effect (Fig. 4E), confirming that the antiapoptosis function requires active virus. Furthermore, treatment of infected cells with acyclovir, which inhibits viral DNA replication and therefore viral late gene expression, still allowed expression of the antiapoptotic effect (data not shown), suggesting that this effect is mediated by an immediate-early or early gene of HSV-1. As expected, infection with HSV-2 had no inhibitory effect on DNA fragmentation, even at 33 PFU/cell (Fig. 4F and G).
In contrast to the effects of HSV-1 on DNA fragmentation induced by CTL clones ESL3.326 and 1A.B.25, however, HSV-1 had no effect on the DNA fragmentation of target cell line YAR (LCL) induced by clone ESL4.34, another CD4+ CTL clone (Fig. 6A). Again, HSV-2 also had no effect on DNA fragmentation induced by this clone (Fig. 6B). To determine whether the failure to inhibit DNA fragmentation was at the level of the effector or the target cell, the ability of HSV-1 to protect YAR cells from DNA fragmentation induced by other stimuli was assessed. HSV-1 was able to inhibit UV-induced DNA fragmentation of YAR cells (the YAR cell line does not undergo apoptosis in response to ethanol incubation). DNA fragmentation after exposure to UV light (30-W source at 10 cm) was decreased 50 to 100% by infection with HSV-1 strain E115, depending on the conditions of the JAM assay (Fig. 6C). This result suggests that the lack of HSV inhibition of CTL-induced apoptosis may be specific for the mechanism of apoptosis induction used by clone ESL3.326 rather than a feature of the YAR target cells.
FIG. 6.

Lack of inhibition of DNA fragmentation induced in target cell line YAR by CTL clone ESL4.34. Neither HSV-1 strain E115 (10 PFU/cell) (A) nor HSV-2 strain 333 (10 PFU/cell) (B) inhibited fragmentation induced by this clone. HSV-1 strain E115 (10 PFU/cell) (C) inhibited UV-induced DNA fragmentation in YAR cells. Cells were UV irradiated (30-W source at 10 cm) for the indicated times. Squares, uninfected control cells; diamonds, HSV-1 infected cells. E:T ratio, effector-to-target cell ratio.
DISCUSSION
This paper describes two unique features of HSV-1 inhibition of apoptosis. First, we have demonstrated that the HSV-1 antiapoptotic genes can protect the infected cell from DNA fragmentation induced by CTL. Although other viruses have genes which can inhibit apoptosis (reviewed in reference 34), these function mainly to prevent apoptosis as a direct cellular response to viral infection. Few of these proteins have been shown to inhibit apoptosis induced by CTL, possibly because CTL have evolved to bypass many of the proximal triggering events preceding apoptosis (the events typically inhibited by viral antiapoptotic proteins). Instead, CTL can activate caspases (ICE-like proteases) directly via the action of granzyme B (19). The results in this paper demonstrate that HSV-1 can be added to cowpox virus (35) and baculovirus (28) as a virus which inhibits CTL-induced apoptosis. Since CTL are critical for the control of HSV-1 infection (3, 26, 30), these results suggest that inhibition of apoptosis plays an important role in the immune evasion strategy of the virus by preventing CTL-induced cell death until viral replication is complete.
Second, HSV-1 inhibited the DNA fragmentation associated with apoptosis but not the membrane changes such as PS exposure. This implies that the various manifestations of apoptosis, in this case, the nuclear and membrane changes, can be inhibited independently. This also suggests that the HSV-1 antiapoptotic genes may have mechanisms of action distinct from those of other viral inhibitors of apoptosis. For example, another viral inhibitor shown to inhibit CTL-induced apoptosis, CrmA (35), acts by inhibiting the action of caspases. If the HSV-1 antiapoptotic genes also acted to inhibit caspase activity, this would imply that PS exposure is regulated by a mechanism other than the caspase cascade. A recent report showing that PS exposure requires cleavage of ICE-like proteases (caspases) makes this unlikely (20). Alternatively, the HSV-1 inhibitors may act after the caspase cascade, when the nuclear and membrane effector arms of the apoptotic mechanism have separated. Further studies are necessary to establish the exact mechanism of the HSV-1 effect. These studies may provide new and unique insights into the late stages of the apoptotic cascade.
Two recent reports have suggested an antiapoptotic effect of HSV-1, although they did not address the effect of HSV-1 on apoptosis induced by CTL or the effect of HSV-1 infection on PS exposure during apoptosis. Koyama and Miwa (14) reported that HSV-1 infection protected HEp-2 cells from sorbitol-induced apoptosis and that this effect was mediated by an immediate-early (α) or early (β) gene of HSV-1. Similarly, Leopardi and Roizman (17) reported that the HSV-1 protein kinase US3 could protect Vero cells from apoptosis. Our results showing the appearance of the HSV-1 antiapoptotic effect within 2 h of infection support the hypothesis that this function is mediated by immediate-early or early genes of HSV-1. This conclusion is further supported by the observation that treatment of infected cells with acyclovir, which prevents HSV late gene expression, still allows expression of the antiapoptotic effect.
We were somewhat surprised to find that laboratory strains of HSV-2 had no antiapoptotic effect in our system. We are currently testing clinical isolates of both HSV-1 and HSV-2 to determine whether this distinction holds for these strains as well. In either event, the availability of various strains which do and do not express an antiapoptotic function may prove helpful in further defining the viral regulatory regions which inhibit apoptosis. We were also surprised by our observation that HSV-1 inhibited apoptosis induced by only two of the three CTL clones tested. Various T-cell clones may use different mechanisms to induce apoptosis in their target cells (24, 31), so it is possible that HSV-1 is only able to inhibit some CTL killing pathways. Further experiments are required to elucidate the details of this inhibition and also to determine the effect of HSV-1 infection on killing mediated by CD8+ CTL.
Finally, it is interesting to speculate on the evolutionary value of a viral inhibitor of apoptosis which prevents DNA fragmentation but allows membrane PS exposure. DNA fragmentation appears to result in the cessation of viral replication (21), so its inhibition, therefore allowing the infected cell to survive until viral replication is complete, would clearly be advantageous for viral survival. What, then, is the advantage of allowing PS exposure to proceed? One possibility is raised by reports suggesting that PS exposure serves as a signal for phagocytosis (4), particularly by nonprofessional phagocytes. Among the nonprofessional phagocytes are epithelial cells, and it is tempting to speculate that PS exposure may facilitate viral transmission to adjacent, uninfected cells.
ACKNOWLEDGMENTS
This work was supported by a National Foundation for Infectious Diseases/Astra fellowship (to K.R.J.) and NIH grants AI34616 and CA70017 (to D.M.K.).
We thank Don Gibson and Matthew L. Johnson for excellent technical assistance.
REFERENCES
- 1.Bennett M R, Gibson D F, Schwartz S M, Tait J F. Binding and phagocytosis of apoptotic vascular smooth muscle cells is mediated in part by exposure of phosphatidylserine. Circ Res. 1995;77:1136–1142. doi: 10.1161/01.res.77.6.1136. [DOI] [PubMed] [Google Scholar]
- 2.Berke G. The CTL’s kiss of death. Cell. 1995;81:9–12. doi: 10.1016/0092-8674(95)90365-8. [DOI] [PubMed] [Google Scholar]
- 3.Borysiewicz L K, Sissons J G. Cytotoxic T cells and human herpes virus infections. Curr Top Microbiol Immunol. 1994;189:123–150. doi: 10.1007/978-3-642-78530-6_8. [DOI] [PubMed] [Google Scholar]
- 4.Bruckheimer E M, Schroit A J. Membrane phospholipid asymmetry: host response to the externalization of phosphatidylserine. J Leukocyte Biol. 1996;59:784–788. doi: 10.1002/jlb.59.6.784. [DOI] [PubMed] [Google Scholar]
- 5.Doherty D G, Koelle D M, Kwok W W, Masewicz S, Domeier M E, Nepom G T. Allelic variants of MHC class II molecules can act as partial agonists of antigen specific T cell responses. Hum Immunol. 1996;47:149. [Google Scholar]
- 6.Fadok V A, Voelker D R, Campbell P A, Cohen J J, Bratton D L, Henson P M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 7.Fruh K, Ahn K, Djaballah H, Semp’e P, van Endert P M, Tamp’e R, Peterson P A, Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 8.Gentry G A, Rana S, Hutchinson M, Starr P. Evolution of herpes and pox viruses and their hosts: a problem with the molecular clock. Intervirology. 1988;29:277–280. [PubMed] [Google Scholar]
- 9.Golstein P. Fas-based T cell-mediated cytotoxicity. Curr Top Microbiol Immunol. 1995;198:25–37. doi: 10.1007/978-3-642-79414-8_2. [DOI] [PubMed] [Google Scholar]
- 10.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 11.Jennings S R, Rice P L, Kloszewski E D, Anderson R W, Thompson D L, Tevethia S S. Effect of herpes simplex virus types 1 and 2 on surface expression of class I major histocompatibility complex antigens on infected cells. J Virol. 1985;56:757–766. doi: 10.1128/jvi.56.3.757-766.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koelle D M, Johnson M L, Ekstrom A N, Byers P, Kwok W W. Preferential presentation of herpes simplex virus T-cell antigen by HLA DQA1*0501/DQB1*0201 in comparison to HLA DQA1*0201/DQB1*0201. Hum Immunol. 1997;53:195–205. doi: 10.1016/S0198-8859(97)00034-7. [DOI] [PubMed] [Google Scholar]
- 13.Koelle D M, Tigges M A, Burke R L, Symington F W, Riddell S R, Abbo H, Corey L. Herpes simplex virus infection of human fibroblasts and keratinocytes inhibits recognition by cloned CD8+ cytotoxic T lymphocytes. J Clin Invest. 1993;91:961–968. doi: 10.1172/JCI116317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koyama A H, Miwa Y. Suppression of apoptotic DNA fragmentation in herpes simplex virus type 1-infected cells. J Virol. 1997;71:2567–2571. doi: 10.1128/jvi.71.3.2567-2571.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzushima K, Isobe K, Morishima T, Takatsuki A, Nakashima I. Inhibitory effect of herpes simplex virus infection to target cells on recognition of minor histocompatibility antigens by cytotoxic T lymphocytes. J Immunol. 1990;144:4536–4540. [PubMed] [Google Scholar]
- 16.Kwok W W, Domeier M E, Johnson M L, Nepom G T, Koelle D M. HLA-DQB codon 57 is a critical determinant of peptide binding and recognition. J Exp Med. 1996;183:1253–1258. doi: 10.1084/jem.183.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leopardi R, Van Sant C, Roizman B. The herpes simplex virus type 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci USA. 1997;94:7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewandowski G A, Lo D, Bloom F E. Interference with major histocompatibility complex class II-restricted antigen presentation in the brain by herpes simplex virus type 1: a possible mechanism of evasion of the immune response. Proc Natl Acad Sci USA. 1993;90:2005–2009. doi: 10.1073/pnas.90.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin S J, Amarante Mendes G P, Green D R. Cytotoxic lymphocyte killing enters the ice age. Adv Exp Med Biol. 1996;406:29–37. doi: 10.1007/978-1-4899-0274-0_4. [DOI] [PubMed] [Google Scholar]
- 20.Martin S J, Finucane D M, Amarante-Mendes G P, O’Brien G A, Green D R. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- 21.Martz E, Gamble S R. How do CTL control virus infections? Evidence for prelytic halt of herpes simplex. Viral Immunol. 1992;5:81–91. doi: 10.1089/vim.1992.5.81. [DOI] [PubMed] [Google Scholar]
- 22.Matzinger P. The JAM test. A simple assay for DNA fragmentation and cell death. J Immunol Methods. 1991;145:185–192. doi: 10.1016/0022-1759(91)90325-a. [DOI] [PubMed] [Google Scholar]
- 23.McGeoch D J, Cook S, Dolan A, Jamieson F E, Telford E A. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 24.Pham C T N, Ley T J. The role of granzyme B cluster proteases in cell-mediated cytotoxicity. Semin Immunol. 1997;9:127–133. doi: 10.1006/smim.1997.0060. [DOI] [PubMed] [Google Scholar]
- 25.Podack E R. Perforin, killer cells and gene transfer immunotherapy for cancer. Curr Top Microbiol Immunol. 1995;198:121–130. doi: 10.1007/978-3-642-79414-8_7. [DOI] [PubMed] [Google Scholar]
- 26.Posavad C M, Koelle D M, Shaughnessy M F, Corey L. Severe genital herpes infections in HIV-infected individuals with impaired herpes simplex virus-specific CD8+ cytotoxic T lymphocyte responses. Proc Natl Acad Sci USA. 1997;94:10289–10294. doi: 10.1073/pnas.94.19.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Posavad C M, Rosenthal K L. Herpes simplex virus-infected human fibroblasts are resistant to and inhibit cytotoxic T-lymphocyte activity. J Virol. 1992;66:6264–6272. doi: 10.1128/jvi.66.11.6264-6272.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarin A, Williams M S, Alexander Miller M A, Berzofsky J A, Zacharchuk C M, Henkart P A. Target cell lysis by CTL granule exocytosis is independent of ICE/Ced-3 family proteases. Immunity. 1997;6:209–215. doi: 10.1016/s1074-7613(00)80427-6. [DOI] [PubMed] [Google Scholar]
- 29.Schlegel R A, Callahan M, Krahling S, Pradhan D, Williamson P. Mechanisms for recognition and phagocytosis of apoptotic lymphocytes by macrophages. Adv Exp Med Biol. 1996;406:21–28. doi: 10.1007/978-1-4899-0274-0_3. [DOI] [PubMed] [Google Scholar]
- 30.Schmid D S, Rouse B T. The role of T cell immunity in control of herpes simplex virus. Curr Top Microbiol Immunol. 1992;179:57–74. doi: 10.1007/978-3-642-77247-4_4. [DOI] [PubMed] [Google Scholar]
- 31.Stenger S, Mazzaccaro R J, Uyemura K, Cho S, Barnes P F, Rosat J-P, Sette A, Brenner M B, Porcelli S A, Bloom B R, Modlin R L. Differential effects of cytolytic T cell subsets on intracellular infection. Science. 1997;276:1684–1687. doi: 10.1126/science.276.5319.1684. [DOI] [PubMed] [Google Scholar]
- 32.Suhrbier A, Fernan A, Burrows S R, Saul A, Moss D J. BLT esterase activity as an alternative to chromium release in cytotoxic T cell assays. J Immunol Methods. 1991;145:43–53. doi: 10.1016/0022-1759(91)90309-4. [DOI] [PubMed] [Google Scholar]
- 33.Tait J F, Gibson D, Fujikawa K. Phospholipid binding properties of human placental anticoagulant protein-I, a member of the lipocortin family. J Biol Chem. 1989;264:7944–7949. [PubMed] [Google Scholar]
- 34.Teodoro J G, Branton P E. Regulation of apoptosis by viral gene products. J Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tewari M, Telford W G, Miller R A, Dixit V M. CrmA, a poxvirus-encoded serpin, inhibits cytotoxic T-lymphocyte-mediated apoptosis. J Biol Chem. 1995;270:22705–22708. doi: 10.1074/jbc.270.39.22705. [DOI] [PubMed] [Google Scholar]
- 36.Verhoven B, Schlegel R A, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wald A, Zeh J, Selke S, Ashley R L, Corey L. Virologic characteristics of subclinical and symptomatic genital herpes infections. N Engl J Med. 1995;333:770–775. doi: 10.1056/NEJM199509213331205. [DOI] [PubMed] [Google Scholar]