

The assembly of the mammalian brain is a complex process and depends on tightly regulated developmental and genetic programs. The coordinated process of nerve cell (neuron) migration is essential to home neurons into their correct position, which is vital for normal brain development. Disturbance of the neuron migration process at any point of development leads to severe brain malformations resulting in devastating disease. To date, studies have mainly focused on cell-intrinsic gene functions controlling neuronal migration. Therefore, we got very little knowledge about the possible contribution of the cellular surrounding and tissue-wide effects. The scale and nature of such global tissue-wide effects remain completely unclear. We thus established genetic platforms based on mosaic analysis with double markers to visualize and quantify tissue-wide effects in a defined genetic context and at single cell resolution. We found a critical predominant role of the genetic landscape and state of cellular environment affecting overall neuron migration properties. In a broader context, our results suggest that global tissue-wide effects are a major component of the underlying etiology of neurological diseases such as focal malformations of cortical development.
Keywords: cell-autonomous gene function, 4D live-imaging, single-cell genetics, neuronal migration, mosaic analysis with double markers (MADM), cerebral cortex development, non-cell-autonomous effects
ABSTRACT
The mammalian neocortex is composed of diverse neuronal and glial cell classes that broadly arrange in six distinct laminae. Cortical layers emerge during development and defects in the developmental programs that orchestrate cortical lamination are associated with neurodevelopmental diseases. The developmental principle of cortical layer formation depends on concerted radial projection neuron migration, from their birthplace to their final target position. Radial migration occurs in defined sequential steps, regulated by a large array of signaling pathways. However, based on genetic loss-of-function experiments, most studies have thus far focused on the role of cell-autonomous gene function. Yet, cortical neuron migration in situ is a complex process and migrating neurons traverse along diverse cellular compartments and environments. The role of tissue-wide properties and genetic state in radial neuron migration is however not clear. Here we utilized mosaic analysis with double markers (MADM) technology to either sparsely or globally delete gene function, followed by quantitative single-cell phenotyping. The MADM-based gene ablation paradigms in combination with computational modeling demonstrated that global tissue-wide effects predominate cell-autonomous gene function albeit in a gene-specific manner. Our results thus suggest that the genetic landscape in a tissue critically affects the overall migration phenotype of individual cortical projection neurons. In a broader context, our findings imply that global tissue-wide effects represent an essential component of the underlying etiology associated with focal malformations of cortical development in particular, and neurological diseases in general.
INTRODUCTION
The mammalian neocortex, arranged in a six-layered structure, executes essential higher order cognitive brain functions. The laminated organization emerges during embryonic development and instructs the wiring diagram of cortical microcircuits [26, 48]. Deficits in the developmental programs orchestrating cortical layering represent a key underlying mechanism of neurodevelopmental disorders including cortical malformation [22, 30, 38].
A cardinal feature of cortical layering during development is the temporally sequential arrangement of the distinct layers in an ‘inside-out’ fashion [2, 52, 80]. As such, progressively later born, radial glial cell (RGC)-derived, projection neurons migrate out radially past earlier born cohorts to gradually build up the inversely arranged laminae. Seminal live-imaging experiments at single-cell resolution revealed that radial migration of cortical projection neurons occurs in a defined sequence [54, 57, 58, 79]. First, newly-born projection neurons acquire a bipolar morphology and migrate from the ventricular zone (VZ) to the subventricular zone (SVZ) where they adopt a multipolar shape [76]. While in the SVZ, multipolar neurons move slowly tangentially, a process that may be required to explore the extracellular environment for putative polarity-inducing cues [14, 37]. Next, multipolar neurons switch back to a bipolar state with the ventricle-oriented process developing into the axon. Bipolar projection neurons reattach to the radial glial fiber, migrate through the intermediate zone (IZ) and enter the cortical plate (CP) by using a locomotion mode [29, 41]. Once the locomoting neurons reach the most superficial layer, they detach from the radial glial fiber and perform terminal somal translocation to settle in their target position [18, 71, 72].
In the past decades, a large collection of signaling pathways including extracellular cues/receptors, cell adhesion molecules and their receptors, endocytic/exocytic regulators, intracellular signal transduction machinery and transcription factors have been implicated in controlling the discrete sequential steps of radial projection neuron migration in healthy and disease conditions [4, 9, 16, 27, 30, 31, 42, 64]. Thus far, most studies using loss-of-function paradigms focused almost exclusively on the cell-autonomous role of the genes encoding the above regulatory cues. Yet, accumulating evidence suggests that non-cell-autonomous tissue-wide properties could substantially impact and/or contribute to the regulation of radial projection neuron migration [6, 18, 19, 21 24, 25, 28, 31, 32, 55, 68, 82–84]. The nature and dynamics of such global tissue-wide effects remain however unclear.
In our study, we addressed this issue in a quantitative manner at single-cell level and set out to establish experimental genetic paradigms enabling the separation of intrinsic cell-autonomous gene function from the contribution of tissue-wide effects. To this end we utilized mosaic analysis with double markers (MADM) technology [12, 86], which provides a quantitative platform in situ to (i) monitor neuronal migration with exquisite single-cell resolution and (ii) probe the role of the genetic landscape and cellular environment on individual migrating neurons in a defined genetic context. Based on results from holistic single-cell phenotypic analysis upon loss of gene function, we provide evidence that global tissue-wide effects predominate the overall cell-autonomous phenotype of migrating projection neurons.
RESULTS
MADM-based platform to probe cell-autonomous candidate gene function and the contribution of global tissue-wide effects at single-cell level
In order to establish a quantitative assay to dissect cell-autonomous gene function and to measure the contribution of global tissue-wide cues/properties with single-cell resolution, we conceived a platform consisting of three genetic MADM-based paradigms: (i) control-MADM; (ii) GeneX-MADM (mosaic-MADM); and (iii) KO/cKO-GeneX-MADM (KO/cKO-MADM) (Fig. 1A–D and Fig. S1). In control-MADM, mice carrying MADM cassettes on a particular chromosome were combined with Emx1-Cre driver to generate experimental MADM mice with sparse fluorescent labeling in cortical projection neurons. Since these control-MADM mice did not carry any loss-of-function (LOF) allele all cells were considered as ‘control’ with some sparse cells expressing the fluorescent GFP and tdT markers enabling single-cell tracing. In GeneX-MADM with Emx1-Cre, a genetic mutation was coupled to one MADM cassette by meiotic recombination (see Materials and Methods [1, 12] for details) in a way that sparse homozygous mutant cells were always labeled in green (GFP+) and wild-type cells in red (tdT+), respectively. In KO/cKO-GeneX-MADM with Emx1-Cre, the mutant allele of the gene of interest was coupled to both MADM cassettes resulting in full (KO) or conditional (cKO) tissue-specific knockout (Emx1+ projection neuron lineage) with sparse fluorescent labeling in respective experimental MADM mice. The key rationale in our overall assay was based on the following assumptions. First, in sparse genetic mosaic-MADM (where we ablated candidate gene function in just very few GFP+ migrating neurons), the observed phenotype of homozygous mutant cells implied the cell-autonomous gene function when compared to tdT+ control cells. Second, the phenotype of individual mutant GFP+ cells in an all mutant environment (KO/cKO-MADM), of the same candidate gene as above, reflected the combination of (i) cell-autonomous loss of gene function; plus (ii) global community effects in response to the loss of gene function in the vast majority of (if not all) cortical projection neurons across the entire tissue. Third, any quantitatively significant difference in the observed single-cell mutant phenotype in mosaic-MADM when compared to KO/cKO-MADM indicated non-cell-autonomous and thus global tissue-wide effects (Fig. 1D).
Figure 1.

MADM analysis reveals that global tissue-wide effects predominates the cell-autonomous phenotype due to loss of p35/CDK5. (A–D) Experimental MADM paradigm to genetically dissect cell-autonomous gene function and non-cell-autonomous effects. (A) Control (control-MADM: all cells GeneX+/+); (B) sparse genetic mosaic (GeneX-MADM: only green cells are GeneX−/− mutant, red cells are GeneX+/+ in an otherwise heterozygous GeneX+/− environment); (C) global/whole-tissue gene knockout (KO/cKO-GeneX-MADM: all cells mutant); (D) direct phenotypic comparison of mutant cells in GeneX-MADM (mosaic MADM) to mutant cells in KO/cKO-gene-MADM (KO/cKO-MADM). Any significant difference in their respective phenotypes implies non-cell-autonomous effects. (E–H) Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (E) control-MADM (MADM-11GT/TG;Emx1Cre/+); (F) Cdk5r1-MADM (MADM-11GT/TG,Cdk5r1;Emx1Cre/+); and (G) KO-Cdk5r1-MADM (MADM-11GT,Cdk5r1/TG,Cdk5r1;Emx1Cre/+). Relative distribution (%) of MADM-labeled projection neurons is plotted in ten equal zones across the cortical wall. (H) Direct distribution comparison of Cdk5r1−/− mutant cells in Cdk5r1-MADM (grey) versus KO-Cdk5r1-MADM (black) distribution. (I–L) Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (I) control-MADM (MADM-5GT/TG;Emx1Cre/+); (J) Cdk5-MADM (MADM-5GT/TG,Cdk5;Emx1Cre/+); and (K) KO-Cdk5-MADM (MADM-11GT,Cdk5/TG,Cdk5;Emx1Cre/+). Relative distribution (%) of MADM-labeled projection neurons is plotted in 10 equal zones (1–10) across the cortical wall. (L) Direct distribution comparison of Cdk5−/− mutant cells in Cdk5-MADM (grey) versus KO-Cdk5-MADM (black) distribution. Nuclei were stained using DAPI (blue). N = 3 for each genotype with 10 (MADM-11) and 20 (MADM-5) hemispheres analysed. Data indicate mean ± SD, *P < 0.05, **P < 0.01 and ***P < 0.001. Scale bar: 100 μm. Marginal zone (MZ), cortical layers (II–VI), white matter (WM).
Global tissue-wide effects predominates the cell-autonomous phenotype due to loss of p35/CDK5
The role of the p35/CDK5 signaling pathway in cortical projection neuron migration has been studied extensively [13, 23, 25, 29, 40, 59]. Thus with ample information available, based on LOF studies, this pathway provided an ideal case to dissect cell-autonomous requirement and the contribution of tissue-wide effects due to LOF. We first analysed the single-cell phenotype upon ablation of p35 (encoded by Cdk5r1, located on chr.11), the main activator of CDK5. (Fig. S2A). Therefore, we generated control-MADM (MADM-11GT/TG;Emx1Cre/+), Cdk5r1-MADM (MADM-11GT/TG,Cdk5r1;Emx1Cre/+) and KO-Cdk5r1-MADM (MADM-11GT,Cdk5r1/TG,Cdk5r1;Emx1Cre/+). Next we quantified the relative distribution of red and green MADM-labeled cortical projection neurons (terminal position) in somatosensory cortex at postnatal day (P) 21 to assess overall migration capacity in the above MADM paradigms (Fig. 1E–G). In control-MADM red and green (both control) projection neurons distributed similarly but in a defined fashion (relatively higher number in upper layers) across 10 zones as previously reported [32] (Fig. 1E). In Cdk5r1-MADM, the green homozygous Cdk5r1−/− mutant neurons accumulated in lower layers, indicative of the cell-autonomous requirement for radial migration [23, 25], while red control neurons showed a distribution pattern similar to red/green cells in control-MADM (Fig. 1F). In contrast, red and green (both homozygous Cdk5r1−/− mutant) neurons in KO-Cdk5r1-MADM showed similar pattern when compared to each other but clearly different distribution pattern when compared to control and mutant cells in control-MADM and/or Cdk5r1-MADM, respectively (Fig. 1G). Strikingly, the distribution of green homozygous Cdk5r1−/− mutant neurons in Cdk5r1-MADM was significantly distinct when compared to green homozygous Cdk5r1−/− mutant neurons in KO-Cdk5r1-MADM despite that both cell populations had the same genotype (Fig. 1H). Thus, the cell-autonomous phenotype (i.e. terminal location), of Cdk5r1−/− mutant cortical projection neurons, was significantly influenced by the genetic tissue-wide landscape.
To identify the layer specificity of the MADM-labelled neurons in all three genotypes we applied immunohistochemistry for the markers Cux1, FoxP2 and Ctip2 (Fig. S3). While mutant neurons in the Cdk5r1-MADM were mainly distributed in the lower zones of the cortical wall (Fig. S3B), we observed a dispersion of upper layer neurons in the KO-Cdk5r1-MADM (Fig. S3C). Moreover, the lower layer markers revealed an inverted distribution with a dispersion of lower layer neurons in the upper half of the cortical wall in the KO-Cdk5r1-MADM, when compared to control-MADM (Fig. S3E–L). Nevertheless, in the Cdk5r1-MADM, a portion of FoxP2 and Ctip2-positive mutant neurons were ectopically located in the lowest zones (Fig. S3F–S3J).
To corroborate the above finding we established a second MADM platform by using MADM cassettes inserted in chr.5 (i.e. MADM-5) to more directly assay the consequences of Cdk5 (located on chr.5) loss of function (Fig. S2B). We thus generated control-MADM (MADM-5GT/TG;Emx1Cre/+), Cdk5-MADM (MADM-5GT,Cdk5/TG;Emx1Cre/+) and cKO-Cdk5-MADM (MADM-5GT,Cdk5/TG,Cdk5;Emx1Cre/+) and analysed the terminal distribution of red and green cells across the somatosensory cortex at P15 (since cKO-Cdk5-MADM animals tend to die soon thereafter). Again, in control-MADM, red and green cells (both wild-type) distributed similarly when compared to each other, and similarly when compared to red and green cells in control-MADM using MADM-11 (Fig. 1E and I). In mosaic Cdk5-MADM, green mutant Cdk5−/− cells accumulated in lower layers and the white matter (Fig. 1J). The migration phenotype of Cdk5−/− cells in Cdk5-MADM appeared slightly stronger than the one of Cdk5r1−/− cells in Cdk5r1-MADM (Fig. 1F and J). The small phenotypic difference could presumably be due to possible compensation by p39 upon loss of p35 and thus some minor residual CDK5 activity in Cdk5r1−/− cells [40, 75]. In any case, red and green (both homozygous Cdk5−/− mutant) neurons in KO-Cdk5-MADM showed clearly different distribution patterns when compared to control and mutant cells in control-MADM and/or Cdk5-MADM, respectively (Fig. 1I, J and K). Again, the distribution of green, homozygous Cdk5−/− mutant neurons, in Cdk5-MADM, was significantly distinct when compared to green homozygous Cdk5−/− mutant neurons in KO-Cdk5-MADM despite that both cell populations had the same genotype (Fig. 1L). Altogether, our data indicate that the state of the genetic environment contributes critically to the overall phenotype of individual cells.
Developmental progression of tissue-wide effects impacting phenotypic manifestation upon sparse and global KO of p35/CDK5
To determine the emergence of non-cell-autonomous tissue-wide effects influencing radial neuron migration we pursued developmental time course analysis. We utilized the same MADM-based paradigms as described in the above section to visualize the single-cell phenotype upon sparse and global elimination of Cdk5r1 (Fig. 2A–L) and Cdk5 (Fig. 2M–X), respectively. At embryonic day (E) 14 no phenotypic difference—i.e. relative vertical distribution of mutant cells in mosaic versus cKO/KO—in VZ/SVZ and IZ was observed. However, small but significant differences were apparent in the upper zones, corresponding to the emerging CP, in both Cdk5r1 and Cdk5 comparative sparse and global MADM deletion paradigms (Fig. 2D and P). At E16 (Figs 2E–F and 2Q–R) and P0 (Figs 2I–J and 2U–V) time points, based on sparse mosaic MADM paradigms, the cell-autonomous phenotypes demonstrated critical requirement for Cdk5r1/Cdk5 in migrating projection neurons to enter the developing cortical plate. In effect, although mutant cells could migrate from the VZ/SVZ through IZ, Cdk5r1−/− and Cdk5−/− mutant cells accumulated below the CP. The phenotypic manifestation became progressively stronger as the development of the CP proceeded. In contrast, mutant cells upon global Cdk5r1/Cdk5 ablation in KO/cKO appeared to distribute more evenly across the developing cortical wall (Fig. 2G, K, S and W). Thus, the emerging cell-autonomous phenotype upon Cdk5r1/Cdk5 ablation (deficit to enter the developing CP) was significantly affected by the global genetic constitution of the developing cortical wall.
Figure 2.
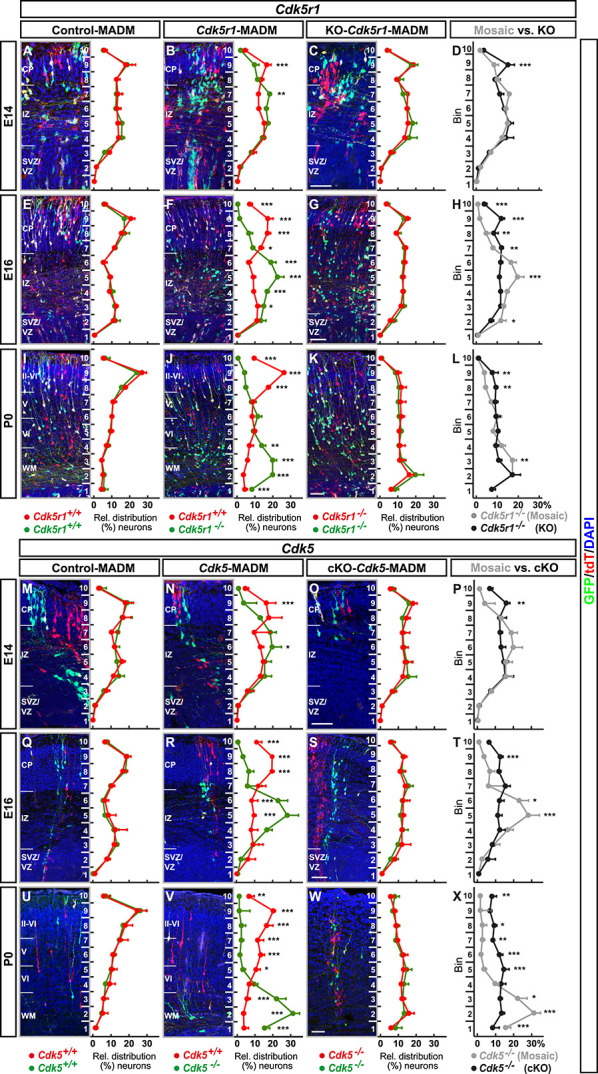
Developmental time course analysis of MADM-based sparse and global KO of p35/CDK5. (A–L) Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (A, E, I) control-MADM (MADM-11GT/TG;Emx1Cre/+); (B, F, J) Cdk5r1-MADM (MADM-11GT/TG,Cdk5r1;Emx1Cre/+); and (C, G, K) KO-Cdk5r1-MADM (MADM-11GT,Cdk5r1/TG,Cdk5r1; Emx1Cre/+) at E14 (A–D), E16 (E–H) and P0 (I–L). Relative distribution (%) of MADM-labeled projection neurons is plotted in ten equal zones across the developing cortical wall. (D, H, L) Direct distribution comparison of Cdk5r1−/− mutant cells at E14 (D), E16 (H) and P0 (L) in Cdk5r1-MADM (grey) versus KO-Cdk5r1-MADM (black) distribution. (M–X) Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (M, Q, U) control-MADM (MADM-11GT/TG;Emx1Cre/+); (N, R, V) Cdk5-MADM (MADM-11GT/TG,Cdk5;Emx1Cre/+); and (O, S, W) KO-Cdk5-MADM (MADM-11GT,Cdk5/TG,Cdk5; Emx1Cre/+) at E14 (M–P), E16 (Q–T) and P0 (U–X). Relative distribution (%) of MADM-labeled projection neurons is plotted in 10 equal zones across the developing cortical wall. (P, T, X) Direct distribution comparison of Cdk5−/− mutant cells at E14 (P), E16 (T), and P0 (X) in Cdk5-MADM (grey) versus KO-Cdk5-MADM (black) distribution. Nuclei were stained using DAPI (blue). N = 3 for each genotype with 10 (MADM-11) and 20 (MADM-5) hemispheres analysed. Data indicate mean ± SD, *P < 0.05, **P < 0.01 and ***P < 0.001. Cortical plate (CP), intermediate zone (IZ), subventricular zone/ventricular zone (SVZ/VZ), cortical layers (II–VI), white matter (WM). Scale bars: 100 μm.
Distinct projection neuron migration dynamics upon sparse and global KO of p35/CDK5
To directly assess cortical projection neuron migration dynamics we measured physical movement in-situ by time-lapse imaging (Fig. 3A). We analysed neuronal migration in embryonic brain slices at E16 when cortical projection neurons were located at all stages in their sequential radial migration trajectory. We capitalized upon the exquisite single-cell resolution in the three—control-MADM (Fig. 3B, Movie S1), Cdk5r1-MADM (Fig. 3C, Movie S2) and KO-Cdk5r1-MADM (Fig. 3D, Movie S3)—experimental paradigms and recorded confocal images at 15 min intervals over an extended period of >15 h. Frames were generated from individual confocal stacks and processed for analysis (see Materials and Methods for details). From all imaging data, we could systematically analyse migration dynamics in movies of at least 12 h (725 min) for all three genetic MADM paradigms. We divided the developing cortical wall into two (lower and upper) zones. The lower zone comprised the VZ/SVZ and IZ, whereas the upper zone corresponded to the emerging CP. To empower our analysis, we employed a semi-automated tracking method to eliminate any experimenter bias (see Materials and Methods). We focused the analysis on the following four populations of MADM-labeled cells: control (red and green neurons in control-MADM), mosaic-control (red control neurons in Cdk5r1-MADM), mosaic-mutant (green Cdk5r1−/− mutant neurons in Cdk5r1-MADM) and KO-mutant (red and green Cdk5r1−/− mutant neurons in KO-Cdk5r1-MADM). First, we evaluated the relative distribution of cells with distinct genotypes across the two (upper and lower) zones at the start (t = 0) and the end (t = 725 min) of the recorded time series (Fig. 3E and F). We noticed that at the start time point roughly 50% of control, mosaic-control and KO-mutant (Cdk5r1−/−) cells were located in the upper and ~ 50% in the lower zone, respectively. In contrast, mosaic-mutant (Cdk5r1−/−) cells in Cdk5r1-MADM failed to distribute evenly between upper and lower zones, and accumulated significantly in the lower zone with much less cells in the upper zone. These data demonstrate that the cell-autonomous Cdk5r1 function, to promote the transition from the IZ into the CP (e.g. Fig. 2B, F and J), is preserved in our in vitro migration assay. At the end time point, there was a trend whereby control and mosaic-control were slightly overrepresented in the upper zone relative to the amount of cells in the lower zone. However, mosaic-mutant neurons still displayed strong bias towards the lower zone, and KO-mutant cells remained at ~50/50 distribution at the end versus start time point.
Figure 3.
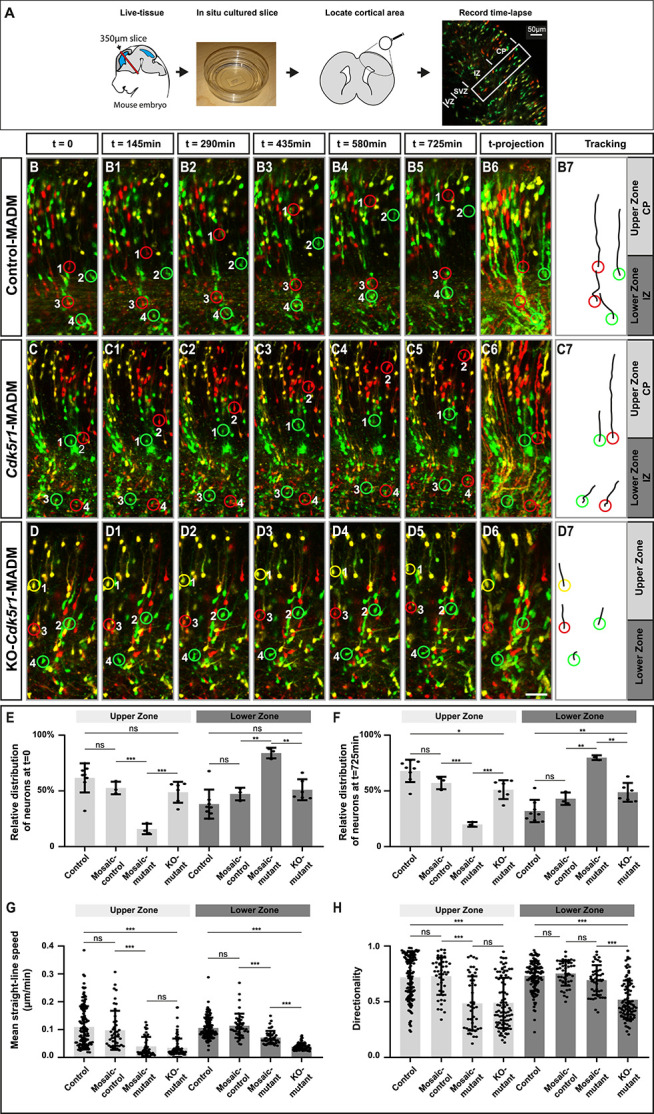
Projection neuron migration dynamics upon sparse and global KO of p35/CDK5. (A) Experimental setup for time-lapse imaging of MADM-labeled neurons in developing somatosensory cortex at E16. (B–D) Time-lapse imaging of (B-B5) control-MADM (MADM-11GT/TG;Emx1Cre/+), (C-C5) Cdk5r1-MADM (MADM-11GT/TG,Cdk5r1;Emx1Cre/+) and (D-D5) KO-Cdk5r1-MADM (MADM-11GT,Cdk5r1/TG,Cdk5r1; Emx1Cre/+). (B6, C6, D6) 12 h time projection of sequential images in IZ (lower zone) and emerging CP (upper zone) at 15 min frame rate. (B7, C7, D7) Tracking trajectories of indicated neurons (red and green rings) in control-MADM (B7), Cdk5r1-MADM (C7) and KO-Cdk5r1-MADM (D7). (E) Relative distribution of neurons at the start of the time-lapse t = 0 for each replicate time-lapse per genotype. (F) Relative distribution of neurons at the end of the time-lapse t = 725 min for each replicate time-lapse per genotype. (G) Mean straight-line speed of the top 15 tracks per replicate time-lapse per genotype. (H) Directionality of the top 15 tracks per replicate time-lapse per genotype. N = 3 videos from >2 independent animals. Data indicate mean ± SD, *P < 0.05, **P < 0.01 and ***P < 0.001. Scale bar: 40 μm.
Next we assessed migration speed (Fig. 3G) and directionality (Fig. 3H). Control and mosaic-control neurons did migrate at equal speeds in the upper and lower zones while mosaic-mutant and KO-mutant migrated at significantly slower speed in both compartments. Interestingly, KO-mutant neurons migrated even significantly slower than mosaic-mutant neurons in the lower zone. While control and mosaic-control neurons showed highly directional (vertical orientation from VZ toward pial surface) migration behavior in both, the upper and lower zones, mosaic-mutant and KO-mutant cells in the upper zone showed significantly less directional movement. In the lower zone, mosaic-mutant cells were not affected in directional movement when compared to control cells but migrating KO-mutant neurons showed significantly less directionality. Altogether, KO-mutant neurons migrated significantly slower and showed significantly less directional movement in the lower zone when compared to mosaic-mutant neurons. Thus, while both populations were Cdk5r1−/−, their environments were distinct (i.e. Cdk5r1−/− only in KO-mutant). We therefore conclude that dynamics and directionality of radially migrating cortical projection neurons is critical dependent on the genetic landscape of the cellular environment.
In silico modelling reveals cell-autonomous force generation and directionality in concert with environmental tissue resistance as minimal parameters instructing cortical projection neuron migration
The above data clearly demonstrated that the interplay of cell-autonomous gene function with non-cell-autonomous tissue-wide properties controls overall efficiency of cortical projection neuron migration. In order to obtain a more quantitative model we set out to define a minimal set of physical parameters sufficient to encompass migration dynamics on a statistical level. We based our approach on the cell tracking data, which we extracted from the time-lapse imaging experiments described above. We first analysed the trajectories of control cells from control-MADM (Fig. 4A), KO-mutant cells from KO-Cdk5r1-MADM (Fig. 4B) and mosaic-control and mosaic-mutant cells from Cdk5r1-MADM (Fig. 4C) and plotted the overall experimental velocity distribution of each cell population as a reference (Fig. 4D). More specifically, the temporal change in position resulted in a distribution of velocities (i.e. normalized velocity) throughout the developing cortical wall ranging from the ventricle to the pia (y axis in Fig. 4D). For control, mosaic-control and to some extent mosaic-mutant (cells in CP considered as ‘escapers’ being able to cross the IZ/CP border despite loss of Cdk5r1), neurons the distribution showed strong velocity peak within the lower half of the tissue (lower zone) followed by a sharp decrease in velocity around the border of the upper zone. However, KO-mutant cells did not display such characteristics but rather showed a more or less even distribution throughout the tissue. The velocity distributions and thus migration dynamics at the IZ-CP (lower–upper zone) border with a positional change in velocity implied a change in tissue architecture and suggested a difference in ‘stiffness’ between the two compartments. Indeed, atomic force microscopy measurements (i.e. Young’s moduli E as proxy for stiffness or resistance) previously indicated distinct stiffness of individual cortical compartments at different developmental stages [35].
Figure 4.
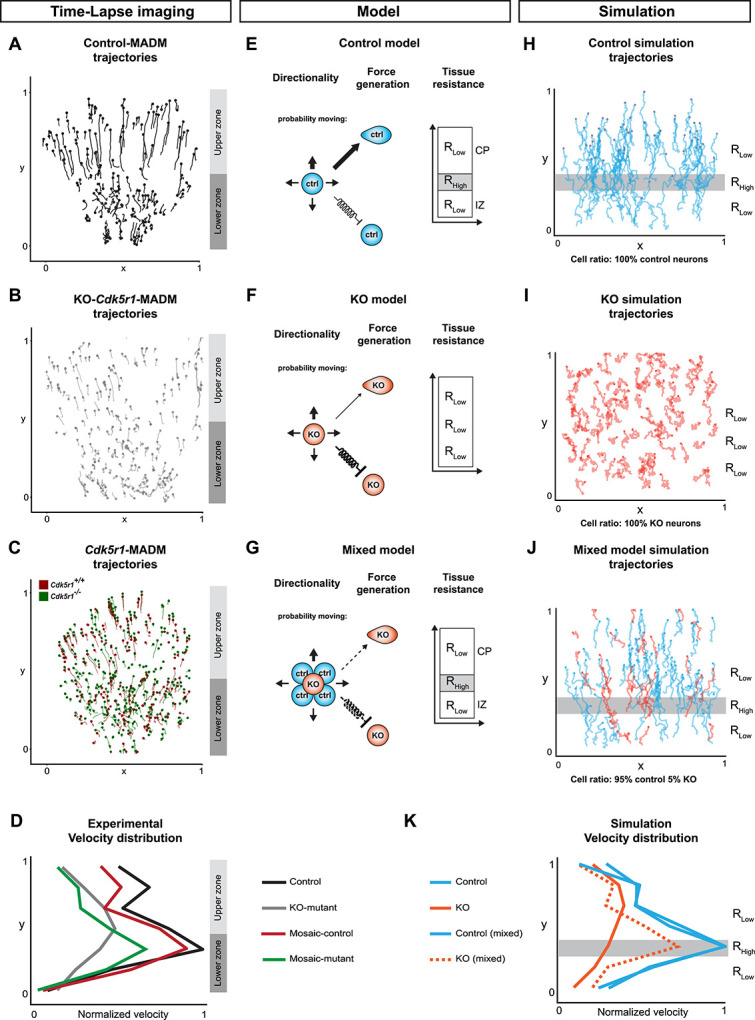
In silico modelling of neuronal migration dynamics upon MADM-based Cdk5r1 ablation. (A–C) Representative migration trajectories in (A) control-MADM, (B) KO-Cdk5r1-MADM and (C) Cdk5r1-MADM. (D) Normalized velocity distributions from experimental data in control-MADM (n = 4), KO-Cdk5r1-MADM (n = 3) and Cdk5r1-MADM (n = 3). (E) Model of neuron migration in control environment with corresponding resistance zones. The thickness of the arrows indicates the probability to move in any direction. The random walk bias of cells in form of directionality and force generation when cells move (arrow) and with force conservation included as a spring constant when cells do not move (spring arrow). The directionality bias is defined as 65% in pial-direction for control. (F) Model of neuron migration in global Cdk5r1 KO environment with a single resistance zone. The thickness of the arrows indicates the probability to move in any direction, here the directionality bias is defined as 51% in pial-direction. The random walk bias of cells in form of directionality and force generation when cells move (arrow) and with force conservation included as a spring constant when cells do not move (spring arrow). (G) Mixed model indicating cross interactions of Cdk5r1−/− mutant and control cells, respectively. The directionality bias is defined in pial-direction as a function of N_ctrl/N_Mut with a minimum at 51% and a maximum at 65%. The thickness of the arrows indicates the probability of the mutant neuron to move in any direction. The random walk bias of cells in form of directionality and force generation when cells move (arrow) and with force conservation included as a spring constant when cells do not move (spring arrow). (H–I) Simulation of migration trajectories in (H) control model, (I) global Cdk5r1 KO model and (J) mixed model (95% Ctrl, 5% KO). Resistant zones are indicated accordingly. (K) Normalized velocity distributions of simulation trajectories.
Next, from the experimental data we extracted two cell-intrinsic parameters, directionality (ρ) and force generation (α), and inferred one extrinsic parameter defining the environmental tissue resistance (Ri) (Fig. 4E, see also Materials and Methods). For the latter, in a reductionist model, we assumed that IZ and CP are relatively homogenous and that overall stiffness can be attributed to differences in physical pore-size distribution. In other words, when a cell migrates through a porous environment it will have to squeeze through different pores in order to advance. Thus, the overall resistance that the cells are experiencing decreases with increasing pore size. The magnitude of the resistance fields Ri is defined as a free parameter of our system. As described above, in control conditions, migrating cells at the IZ-CP border seem to struggle in order to squeeze through. In contrast, due to virtual inexistence of physiological layer structure in KO-Cdk5r1-MADM the migration dynamics appear independent of the location and thus of the Rtissue. To integrate the two described environments in the model, we designed an environment with three compartments and two different resistances (Rlow and Rhigh). In the control environment, the layers were defined by RControl,tissue = R1,R2,R3 for R2 > R1 ~ R3, with R2 roughly corresponding to the IZ-CP border (Fig. 5E and Table S2) while in KO-Cdk5r1-MADM environment we set RKO,tissue = RKO with magnitude on the order of R1,R3 (Fig. 5F and Table S2). The tissue resistance parameters in combination with directional bias ρ and force generation α allowed us to model cells as persistent random walkers (Fig. S4A, see also Materials and Methods for details).
Figure 5.
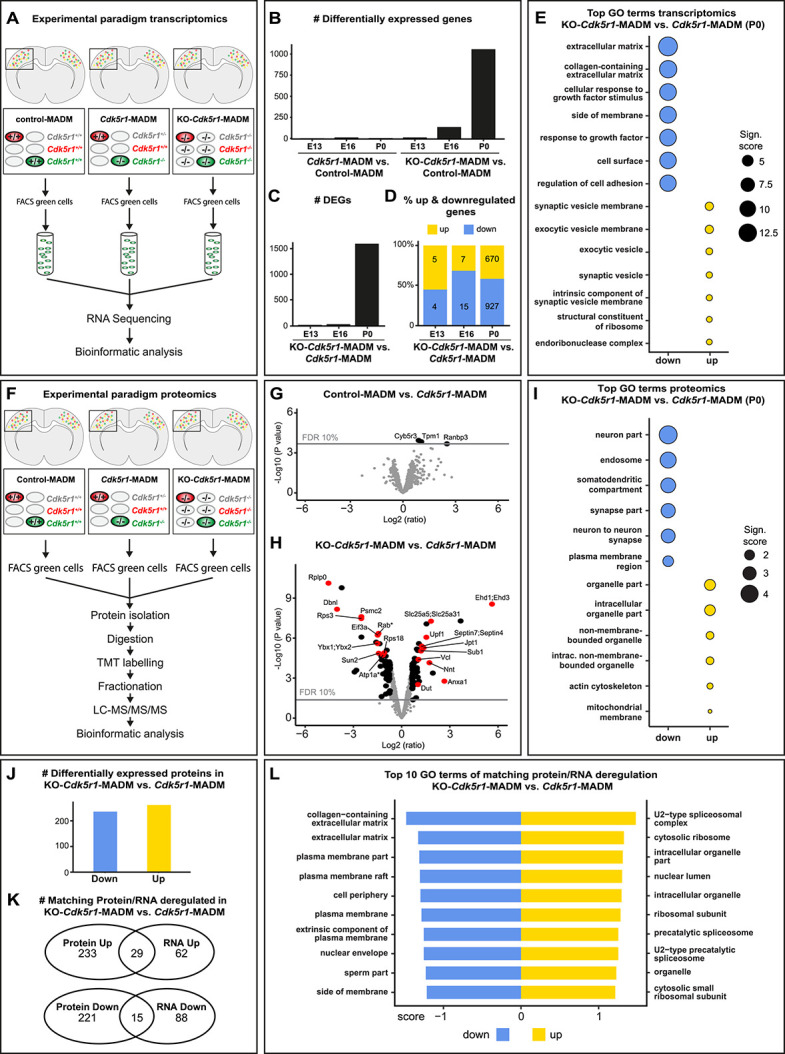
Gene and protein expression in Cdk5r1−/− mutant cells upon sparse and global KO. (A) Experimental paradigm and pipelines for gene expression profiling in control-MADM (left), Cdk5r1-MADM (middle) and KO-Cdk5r1-MADM (right). (B) Number of differentially expressed genes (DEGs) in Cdk5r1-MADM and KO-Cdk5r1-MADM versus control at E13, E16 and P0. (C) Number of DEGs in KO-Cdk5r1-MADM versus Cdk5r1-MADM at E13, E16 and P0. (D) Percentage of up- and downregulated genes in KO-Cdk5r1-MADM versus Cdk5r1-MADM at E13, E16 and P0. (E) Top GO terms associated with genes in (C and D) at P0. Note that GO term enrichments for upregulated genes are non-significant. (F) Experimental paradigm and pipelines for proteome profiling in control-MADM (left), Cdk5r1-MADM (middle) and KO-Cdk5r1-MADM (right). (G) Volcano plot showing deregulated proteins in control-MADM versus Cdk5r1-MADM comparison at P0. Note that only three proteins were significantly upregulated. (H) Volcano plot showing deregulated proteins KO-Cdk5r1-MADM versus Cdk5r1-MADM comparison at P0. Asterisks indicate that Rab and Atp1a protein groups consist of several isoforms not listed in the figure. (I) Top enriched GO terms associated with genes encoding the proteins as shown in (H). (J) Number of genes associated with differentially expressed proteins in KO-Cdk5r1-MADM versus Cdk5r1-MADM. Note that criteria for significant differential expression were relaxed compared to (H). (K) Venn diagrams indicating the overlap of deregulated genes in transcriptomic and proteomic datasets in KO-Cdk5r1-MADM versus Cdk5r1-MADM. (L) Top 10 GO-terms associated with gene sets that are up- and downregulated in both (transcriptomic and proteomic) data sets (overlap in K).
Based on our model, we next simulated control neuron migration (Fig. 4H) and could very closely mimic the experimentally observed migration behavior and velocity distribution (Fig. 4A, D, H and K). Likewise, the simulation of KO-mutant cells (Fig. 4I) in a uniformly low resistance environment RKO,tissue (with reduced directionality parameter ρ and force scaling parameter α, respectively) matched our observations in experimental data from KO-Cdk5r1-MADM paradigm (Fig. 4B, D, I and K).
Next we utilized a ‘mixed model’ where we introduced KO-mutant cells into a surrounding of control cells with according control tissue resistance environment (Fig. 4G). Here we introduced a linear coupling of directionality ρ and force generation coefficient α with the ratio of control to KO-mutant cells (Fig. 4J, Fig. S4B). The simulation conducted with the mixed model mimicked the dynamics and distributions we detected in Cdk5r1-MADM where mutant neurons appeared more dynamic than in KO-Cdk5r1-MADM (Fig. 4J–K). We could model the emergence of the non-cell-autonomous effects with increasing ratio of KO-mutant cells to control (Fig. S4C). The more KO-mutant cells present in the simulation, the more uniform the velocity distribution appeared across the vertical axis of the cortical wall. At a ratio of 4/96% (KO-mutant to control), the KO-mutant cells showed velocity distribution like mosaic-mutant cells and control cells (similar to the observation in the experimental data). In contrast, at a ratio of 10/90%, the KO-mutant cells displayed velocity distribution as observed in KO-Cdk5r1-MADM. Moreover, in our model, we found that the transition between the two distributions appeared abruptly between 5% and 6% abundance of KO-mutant cells. In summary, our in silico model can provide an accurate quantitative replicate of the migration behavior and dynamics as measured in situ. Our data further demonstrate that the relative amount of Cdk5r1−/− mutant cells and thus the overall genetic landscape within the cortical tissue critically impacts on the migration dynamics of individual cells.
Distinct deregulation of gene expression in Cdk5r1 −/− mutant cells upon sparse and global KO
Our data so far indicate distinct single-cell in vivo phenotypes in Cdk5r1−/− mutant cells depending on the genetic state of the environment. By using in silico modelling, we also showed that the tissue resistance (in combination with cell-intrinsic directionality and force generation capability) along the migration path represented a most critical physical parameter. To obtain a hint on molecular correlates mirroring the distinct observed phenotypes, we next pursued transcriptome analysis. We specifically isolated green GFP+ MADM-labeled cortical projection neurons by FACS [45, 46] in control-MADM (Cdk5r1+/+), Cdk5r1-MADM (Cdk5r1−/−) and KO-Cdk5r1-MADM (Cdk5r1−/−) at E13, E16 and P0, respectively (Fig. 5A). We performed RNA-sequencing of small bulk samples using SMARTer technology, followed by bioinformatics analysis (see Materials and Methods). First we analysed Cdk5r1 expression and found that in both (sparse and global KO) deletion paradigms, the Cdk5r1 expression level was close to zero while in control (Cdk5r1+/+) cells substantial Cdk5r1 expression was evident (Fig. S5). These data validated the MADM-based Cdk5r1 ablation paradigms. We next analysed differentially expressed genes (DEGs) in Cdk5r1-MADM and KO-Cdk5r1-MADM in comparison to control-MADM (Fig. 5B). At P0, we identified three significant DEGs in Cdk5r1-MADM, which was in stark contrast to the KO-Cdk5r1-MADM cells where we identified 1056 DEGs (padj<0.05, DESeq2). Thus depending on the state of the genetic environment (global Cdk5r1 KO or not), three orders of magnitude higher number of DEGs was observed in individual Cdk5r1−/− mutant cells. Next, we directly compared the difference in DEGs between Cdk5r1−/− mutant cells in sparse versus global KO. We observed a progressive increase of DEGs during development with slightly more genes showing downregulation (Fig. 5C–D). To obtain more insight into the biological DEG functions we performed gene ontology (GO) enrichment analysis at P0 on the 670 up- and 927 downregulated genes (Fig. 5E). The top enriched GO terms for the downregulated genes (blue) were highly significant (padj<2.45x10−9, hypergeometric test) and associated with extracellular matrix (ECM), cell membrane and cell adhesion. GO term enrichment in the upregulated (yellow) genes was not significant (padj>0.5) and associated with synaptic terms (Fig. 5E). In summary, transcriptome analysis identified downregulation of ECM, membrane associated and cell adhesion genes as major classes of DEGs in Cdk5r1−/− cells in global KO-Cdk5r1-MADM but not sparse mosaic Cdk5r1-MADM.
Deregulation of ECM and cell adhesion proteins upon global but not sparse Cdk5r1 −/− KO
Next, we determined putative differences in Cdk5r1−/− mutant cells upon global versus sparse KO at the proteomic level. We thus isolated the green GFP+ MADM-labeled cortical projection neurons by FACS [45, 46] in control-MADM (Cdk5r1+/+), Cdk5r1-MADM (Cdk5r1−/−) and KO-Cdk5r1-MADM (Cdk5r1−/−) (Fig. 5F), followed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) (Fig. 5F, see also Materials and Methods). First we compared the proteome of Cdk5r1-MADM mutant neurons to control-MADM cells whereby we identified (only) three significantly deregulated proteins (Fig. 5G). However, when mutant cells in Cdk5r1-MADM (Cdk5r1−/−) were compared with mutant cells in KO-Cdk5r1-MADM (Cdk5r1−/−), we identified 59 and 61 significantly up- and downregulated proteins, respectively (Fig. 5H). GO-term enrichment analysis (of the genes associated with the above differentially expressed proteins) indicated top GO terms associated with membrane and neuron–neuron contact among the downregulated group, thus corroborating our findings from the transcriptome analysis. GO-terms for the upregulated group were mainly associated with intracellular processes such as organelles (Fig. 5I). Having proteomic and transcriptomic information at hand, we next analysed putative overlap in the two data sets. Since the direct correlation between transcriptome and proteome was complex, we did lower the thresholds for differential expression (see Materials and Methods). We focused the analysis on 1060 gene annotations, which were informative in both data sets, and identified 262 downregulated and 236 upregulated DEGs from the proteomics data set (Fig. 5J). Strikingly, 29 upregulated and 15 downregulated genes were common to both transcriptomic and proteomic analyses (Fig. 5K). GO-term enrichment analysis identified ECM and membrane-associated GO terms among the down regulated gene group and GO terms related to cell-intrinsic entities among the upregulated gene group (Fig. 5L). In summary, the above analysis revealed significant downregulation of mRNAs and proteins related to membrane, cell adhesion and ECM, specifically in Cdk5r1−/− mutant cells upon global tissue-wide but not sparse KO.
Global tissue-wide effects predominates the cell-autonomous phenotype due to loss of Dab1
Global elimination of p35/CDK5 signaling in cortical projection neurons results in dominant tissue-wide effects that affect the individual cell-autonomous phenotype. To evaluate whether such observation is specific to p35/CDK5 signaling pathway we next investigated the consequences of ablation of Reelin/DAB1 signaling (which operates in parallel to CDK5 [7, 39, 44, 59]). We therefore generated MADM-based Dab1 (located on chr.4) ablation paradigms using MADM-4 platform (Fig. S2C). We analysed radial neuron migration in control-MADM (MADM-4GT/TG;Emx1Cre/+), Dab1-MADM (MADM-4GT/TG,Dab1;Emx1Cre/+) and KO-Dab1-MADM (MADM-4GT,Dab1/TG,Dab1;Emx1Cre/+) at E16, P0 and P21 in somatosensory cortex (Figs 6A–D and S6). In Dab1-MADM the majority of Dab1−/− mutant neurons were positioned in a biased manner, reflecting the cell-autonomous Dab1 function [18, 24, 68] within lower zones in the distribution charts (i.e. below CP during earlier development and lower cortical layers at later stages). Conversely, Dab1−/− mutant neurons in KO-Dab1-MADM showed much more even distribution throughout the cortical wall (Figs 6A–D and S6).
Figure 6.

MADM-based analysis of Dab1 and Cdk5r1/Dab1 epistasis. (A–D) Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (a) control-MADM (MADM-4GT/TG;Emx1Cre/+); (F) Dab1-MADM (MADM-4GT/TG,Dab1;Emx1Cre/+); and (G) KO-Dab1-MADM (MADM-4GT,Dab1/TG,Dab1;Emx1Cre/+). Relative distribution (%) of MADM-labeled projection neurons is plotted in 10 equal zones across the cortical wall. (H) Direct distribution comparison of Dab1−/− mutant cells in Dab1-MADM (grey) versus KO-Dab1-MADM (black) distribution. (E–H) Cdk5r1/Dab1 epistasis in MADM-11 labeling background. Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (E) KO-Dab1; MADM-11 (MADM-11GT/TG;Dab1−/−;Emx1Cre/+), (F) Cdk5r1-MADM-11;KO-Dab1 (MADM-11GT/TG,Cdk5r1;Dab1−/−;Emx1Cre/+) and (G) double-KO-Cdk5r1-MADM-11;KO-Dab1 (MADM-11GT,Cdk5r1/TG,Cdk5r1;Dab1−/−;Emx1Cre/+). Relative distribution (%) of MADM-labeled projection neurons is plotted in 10 equal zones across the cortical wall. (H) Direct distribution comparison of Cdk5r1−/− mutant cells upon sparse (grey) and global (black) KO in Dab1−/− mutant background. (I–L) Dab1/Cdk5r1 epistasis in MADM-4 labeling background. Analysis of green (GFP+) and red (tdT+) MADM-labeled projection neurons in (I) KO-Cdk5r1; MADM-4 (MADM-4GT/TG;Cdk5−/−;Emx1Cre/+), (J) Dab1-MADM-4;KO-Cdk5r1 (MADM-4GT/TG,Dab1;Cdk5r1−/−;Emx1Cre/+) and (G) double-KO Dab1-MADM-4;KO-Cdk5r1 (MADM-4GT,Dab1/TG,Dab1;Cdk5r1−/−;Emx1Cre/+). Relative distribution (%) of MADM-labeled projection neurons is plotted in 10 equal zones across the cortical wall. (H) Direct distribution comparison of Dab1−/− mutant cells upon sparse (grey) and global (black) KO in Cdk5r1−/− mutant background. All analysis was carried out at P21. N = 3 for each genotype. From each animal 10 (MADM-11) or 20 (MADM-4) hemispheres were analysed. Nuclei were stained using DAPI (blue). Data indicate mean ± SD, *P < 0.05, **P < 0.01 and ***P < 0.001. Marginal zone (MZ), superplate (SPP), white matter (WM). Scale bar: 100 μm.
Tissue-wide non-cell-autonomous effects impacting neuronal migration are specific for distinct signaling pathways
Global, but not sparse KO, of Dab1 results in tissue-wide effects affecting the single-cell Dab1−/− mutant phenotype in quite a similar manner to the above p35/CDK5 findings. Whether the observed tissue-wide effects exhibit ‘gene’ specificity or may dominate over distinct signaling pathways was however not clear. We thus conceived epistasis experiments (Fig. S7) to test possible pathway exclusivity of tissue-wide properties upon global gene KO. First, we utilized MADM-11 platform to generate control, global and sparse Cdk5r1 deletion paradigms in a full Dab1−/− KO background (Fig. 6F–H). We thus tested whether tissue-wide effects due to global Dab1 ablation may affect or interfere with the cell-autonomous Cdk5r1−/− mutant phenotype (i.e. accumulation of mutant cells in lower layers and the white matter as described in Fig. 1F). We however observed a similar cell-autonomous Cdk5r1−/− mutant phenotype (upon sparse mosaic Cdk5r1 deletion) in Dab1−/− background (Fig. 6F) as in Dab1+/+ background (i.e. in Cdk5r1-MADM context). Next we reversed the genetic background conditions and utilized MADM-4 platform to generate control, global and sparse Dab1 deletion paradigms in a full Cdk5r1−/− KO background (Fig. 6I–L). Again, the cell-autonomous Dab1−/− mutant phenotype upon sparse deletion was highly similar in Cdk5r1−/− KO (Fig. 6J) when compared to Dab1-MADM in Cdk5r1+/+ background (Fig. 6B). In contrast to the above findings, concomitant global KO of both Cdk5r1 and Dab1 resulted in a relatively uniform distribution of double mutant cells across the cortical wall (Fig. 6G and K), comparable to the distribution in individual KO of Dab1 (Fig. 6E) or Cdk5r1 (Fig. 6I), respectively. In summary, tissue-wide non-cell-autonomous effects predominates the cell-autonomous phenotype albeit in a gene-specific manner.
Global KO of both Cdk5r1 and Dab1 triggers deregulation of genes associated with ECM and cell adhesion
The phenotypic manifestation of global tissue-wide effects appeared very similar for Cdk5r1 and Dab1 because global ablation of either gene resulted in relatively uniform distribution of mutant neurons across the vertical axis of the cortical wall. We therefore asked next whether we might find overlap in deregulated gene expression upon global Cdk5r1 and Dab1 LOF or not. To this end, we applied the identical approach as described above and FACS-isolated green GFP+ MADM-labeled cells (using MADM-11 platform) from control-MADM (MADM-11GT/TG;Emx1Cre/+) and MADM;KO-Dab1 (MADM-11GT/TG;Dab1−/−;Emx1Cre/+) at P0 (Fig. 7A). We complemented this dataset with control-MADM (MADM-11GT/TG;Emx1Cre/+) and KO-Cdk5r1-MADM (MADM-11GT,Cdk5r1/TG,Cdk5r1;Emx1Cre/+) data as described before (Fig. 5). We performed DEG analysis on the combined data set (see also Materials and Methods). Dab1 expression level was close to zero in MADM;KO-Dab1 but readily detectable in all other samples, validating the MADM;KO-Dab1 ablation paradigm (Fig. S8A).
Figure 7.
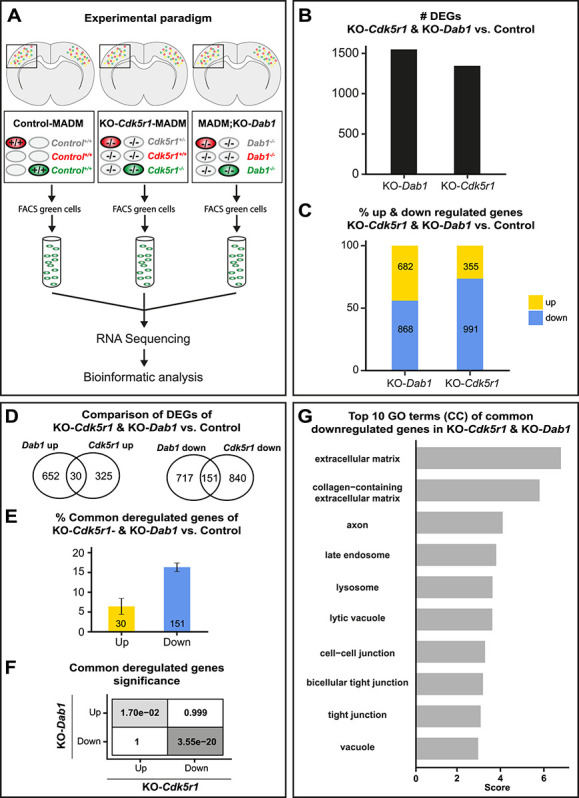
Gene expression upon combined global KO of Cdk5r1 and Dab1. (A) Experimental paradigm and pipelines for gene expression profiling in control-MADM (left), KO-Cdk5r1-MADM (middle; KO-Cdk5r1) and MADM;KO-Dab1 (right; KO-Dab1) at P0. (B) Number of differentially expressed genes (DEGs) in KO-Cdk5r1 and KO-Dab1 versus control. (C) Percentage of up- and downregulated genes in KO-Cdk5r1 and KO-Dab1 versus control. (D) Venn diagrams showing common up- and down-regulated genes in KO-Cdk5r1 and KO-Dab1 versus control. (E) Percentage of common up- and downregulated genes in KO-Cdk5r1 and KO-Dab1 versus control. (F) Significance of all pairwise overlaps of DEGs shown in (D). (G) Top 10 GO terms of commonly downregulated genes in KO-Cdkr5r1 and KO-Dab1, according to overlap shown in (D, right). Commonly upregulated genes did not yield any significant GO term enrichment.
Next, we analysed DEGs in KO-Cdk5r1-MADM and MADM;KO-Dab1, relative to control, and compared the data to each other. Upon global ablation of either, Cdk5r1 or Dab1, we found >1000 deregulated genes in both mutants (Fig. 7B) with the majority of DEGs being downregulated (Fig. 7C). We next analysed the overlap of DEGs in Cdk5r1 and Dab1 KOs and found both common and non-common deregulated genes (Fig. 7D). The majority of the commonly deregulated genes were downregulated (Fig. 7E) and the overlap of both, up- and downregulation was significant (Fig. 7F). Among the upregulated genes we did not obtain any significantly enriched GO-terms although the non-common deregulated genes showed overlap of many of their respective GO-terms between KO-Cdk5r1-MADM and MADM;KO-Dab1 (Fig. S8B). In contrast, among the commonly downregulated genes, we found significant GO-terms associated with cell–cell and cell–matrix interaction (Fig. 7G). Taken together, the above analysis suggested that cell-adhesion and ECM represent major elements in tissue-wide effects upon global KO of genes encoding components of the p35/CDK5 and Reelin/DAB1 signaling pathways.
DISCUSSION
Radial migration of cortical projection neurons has been studied extensively, and a rich catalogue of regulatory signaling cues and pathways have been compiled. The systematic study of the cell-autonomous functions of the genes encoding the signaling cascades has revealed an extensive genetic framework. However, the nature and relative contributions of global tissue-wide effects, in regulating radial migration in the developing neocortex, remained unclear. To this end, we established genetic paradigms enabling the dissection of cell-autonomous gene function and non-cell-autonomous effects. Our data revealed that the genetic landscape of the cellular environment, surrounding migrating neurons, fundamentally affects the individual single-cell phenotype (Fig. 8).
Figure 8.
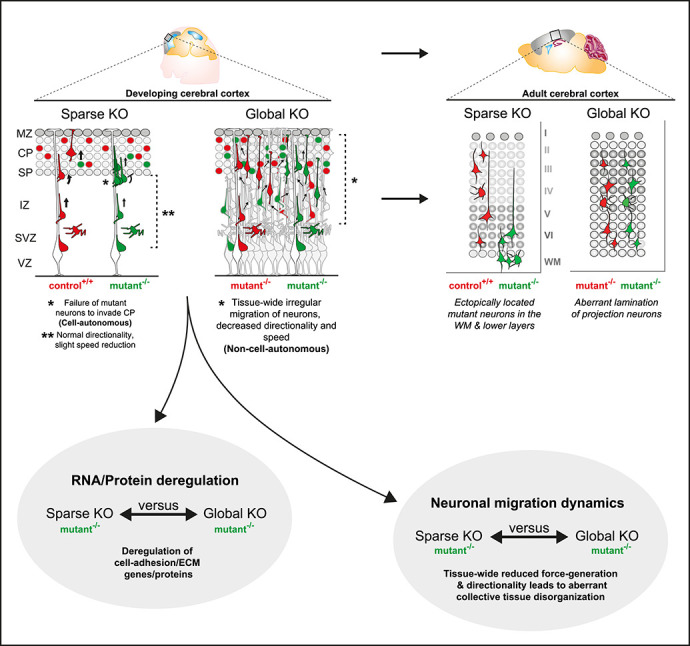
Interplay of cell-autonomous and global tissue-wide properties in cortical projection neuron migration. Schematic illustrating the MADM-based subtractive phenotypic analysis of sparse genetic mosaics (control background) and global knockout (cKO/KO) (mutant background), both coupled with fluorescent MADM-labeling of homozygous mutant and control neurons. Such assay enabled the high-resolution analysis of projection neuron migration dynamics in distinct genetic environments with concomitant isolation of genomic and proteomic profiles. In combination with computational modeling, we utilized these experimental paradigms to visualize non-cell-autonomous effects in radial neuron migration at single-cell resolution. In sparse KO, mutant neurons migrated more dynamically and expressed cell adhesion molecules similar like in control. However, in global KO, we observed that cell adhesion molecules were significantly downregulated. Mutant neurons in global KO also showed much more severe migration phenotype resulting in drastic disorganization of the mature cortical wall.
We focused our analysis mainly on the well-characterized p35/CDK5 pathway providing a defined genetic framework and context. We could show that sparse p35/CDK5 ablation results in a highly specific phenotype that we attributed to the loss of cell-autonomous gene function. Individual p35/Cdk5−/− mutant cortical projection neurons in sparse genetic mosaic conditions failed to pass the border between the IZ and the emerging CP and progressively accumulated below the CP and the white matter. However, radial migration from the VZ and through the IZ occurred, albeit at lower speed. In contrast, single p35/Cdk5−/− mutant neurons in a global homozygous p35/Cdk5−/− mutant KO environment migrated even slower in the VZ/IZ and showed skewed directionality. The perhaps more surprising finding was that in the above full KO condition, p35/Cdk5−/− mutant neurons distributed relatively evenly across the entire cortical wall. Thus, the cell-autonomous phenotype as observed in sparse KO scenario seemed wiped out. There are a few (not mutually exclusive) possibilities that could explain the distinct phenotypic observations. First, the IZ-CP border lost its property as a physical gatekeeper upon global loss of p35/Cdk5. Such scenario actually formed an important basis of our modeling approach where we assumed uniform tissue stiffness/resistance that radially migrating neurons encounter upon global p35 ablation. Indeed, the simulation of such conditions revealed very similar migration trajectories of p35/Cdk5−/− mutant cells as observed by live imaging in experimental global p35/Cdk5 KO conditions. Second, p35/Cdk5−/− mutant neurons in sparse genetic mosaic lost an essential intrinsic property required for transiting the IZ-CP border, reflecting the cell-autonomous p35/Cdk5 gene functions. One important p35/CDK5 downstream signaling hub is NDEL1, which is phosphorylated at specific sites by CDK5 [56, 69]. Interestingly, sparse mosaic deletion of Ndel1 results in a congeneric phenotype, to the sparse p35/Cdk5−/− condition, whereby Ndel1−/− mutant neurons fail to invade the CP and progressively accumulate below the CP and later in the white matter [32]. However, tissue-wide Ndel1 KO results in severely disorganized neocortex with seemingly immobile Ndel1−/− mutant neurons [84]. Thus, sparse loss of any component of the p35/CDK5/NDEL1 signaling hub leads to the inability of individual radially migrating neurons to enter the target area, the emerging CP, while global KO results in predominant cortical tissue disorganization with slower or even immobile projection neurons. The sparse ablation of p35/CDK5/NDEL1 might lead to deficits in LIS1/Dynein-mediated force generation [8, 36, 51, 65, 81], necessary to overcome the increased physical tissue resistance at the IZ/CP border, although the precise biochemical mechanism remains to be clarified in future studies.
The observation that global KO, of the same signaling component as in sparse KO, results in predominant tissue-wide effects appears to exhibit pathway specificity. As such, sparse elimination of Cdk5r1 gene function in a Dab1−/− global KO background (which also shows severe disorganization of the cortical wall) showed a similar cell-autonomous phenotype (failure to enter CP) like in Dab1+/+ background conditions. Thus, the cell-autonomous Cdk5r1−/− phenotype was preserved regardless of the global Dab1 genotype. It will be revealing to investigate the generality of the above findings in future experiments including systematic assessment of potential epistatic interactions of mutations in distinct genes that lead to cortical malformation.
Cdk5r1 and Dab1 knockout mouse models show a difference in their neuronal layering distribution with Dab1 mutants presenting inverted layering [68]. In contrast the Cdk5r1 mutants show intermingled upper and lower layer neurons [11, 23] (Fig. S3). As the cell-autonomous p35−/− phenotype was preserved in the global Dab1 environment, we suggest that the p35−/− phenotype arises due to the inability to surpass previously born neurons rather than due to a deficit in crossing the subplate. Along this line, a recent study showed that the density of deep layer Dab1 deficient neurons prevented superficial layer neurons from entering the cortical plate [83]. Difference in tissue resistance/stiffness is an attractive explanation for the accumulation of the Cdk5r1−/− mutant cells in the Dab1−/− global KO background. For example, if the Dab1−/− KO environment is stiffer than the Cdk5r1−/− KO environment, passing cells will face a higher tissue resistance and their migration will be impaired. So far, biophysical measurement and data of tissue resistance in brain tissue are very limited. Future studies investigating such properties will undoubtedly provide important insights into our understanding of the gene mutations affecting neuronal migration.
What are the underlying characteristics of global tissue-wide effects affecting radial projection neuron migration? It is clear that any tissue consists of a complex extracellular environment. Thus, individual cells are exposed to many extrinsic elements including (i) secreted factors acting locally, globally or even systemically; (ii) the ECM; and (iii) neighboring cells mediating cell–cell interactions through receptors and/or direct physical stimuli [28]. While secreted factors mostly signal to other cells (besides autocrine signaling), and thus mainly act non-cell-autonomously, how cell-intrinsic signaling molecules could act at the global tissue-wide level is not yet clear. Our MADM data are based on the genetic deletion of the intracellular CDK5 kinase or DAB1 adapter protein function. In both instances, the cell-autonomous migration phenotype upon sparse deletion differed from the one upon global KO. In order to get insights at the molecular level, we pursued transcriptome/proteome analysis and found that upon sparse deletion of for instance p35, very few genes and proteins were deregulated in mutant cells. In stark contrast, global KO led to a high number of deregulated proteins and genes. A sizeable fraction of differentially expressed genes even correlated significantly with deregulation of their encoded proteins. The top GO terms associated with deregulated genes/proteins included ECM, receptors and cell adhesion. These results indicate that at the global tissue-wide level the ECM and cell adhesion landscape was drastically changed in global KO condition when compared to wildtype. Although p35/CDK5 and DAB1 signal in parallel, deregulated ECM and cell adhesion was a common denominator upon global KO. Our data are in agreement with earlier studies that showed N-cadherin dependent signaling to be relevant for both p35/CDK5 [42] and DAB1 [18, 71, 72] pathways, respectively. Interestingly, transcriptomic analysis in whole cortex KO mouse models for Ndel1, Lis1 and Ywhae, all acting in the LIS1/Dynein signaling pathway and to some extent downstream of p35/CDK5, have also revealed altered cell adhesion and cytoskeleton organization pathways [63].
Migrating neurons can exert positive and negative interaction on each other depending on the cellular environment and their genetic constitution [19, 21, 28]. In collective cell migration for instance, cell–cell interaction balancing adhesion and repulsion is a key mechanism [73]. Besides, direct physical interactions might be critical whereby for example less agile mutant cells are passively pushed, pulled or simply piggyback onto more dynamic cells.
To better comprehend tissue-wide effects on neuronal migration, it will be important to investigate the migration behavior in combination with transcriptomic and proteomic profiles of rescued ‘wild type’ neurons in a global KO environment. A few studies have observed a rescue of migration by re-expression of the mutant gene in a subset of neurons in a global-KO of the same gene [23, 68, 74]. Examples include chimeric mice consisting of Dab1 wild-type and Dab1 mutant cells. In such Dab1-KO environment Dab1 wild-type cells were capable of migrating radially but failed to rescue the inversion of cortical layers of the Dab1-KO. Interestingly, Dab1 mutant cells did not impose a mutant phenotype on the wild-type cells [24]. Moreover, induction of Dab1 expression in a subset of superficial layer neurons in Dab1-deficient mice using in-utero electroporation [55] or lentivirus vectors [68] rescued the migration phenotype of the transfected cells. Taken together, these findings suggest that the effect of a mutant-environment on rescued wild type cells is minimal although subtle changes in transcriptomics/proteomics cannot be excluded.
Our data indicate that ECM and/or cell–cell interaction are perturbed in Dab1-KO. Since sparse re-expression of Dab1 rescues the migration capability [74] we propose that impaired tissue-wide ECM/cell–cell interaction did not predominantly impair the cell-autonomous migration capability of rescued cells. However, the single-cell Dab1-KO phenotype likely precipitated due to the interaction of impaired ECM/cell–cell environment (tissue-wide effect) with the Dab1 deficient cell. Such scenario would be supported by our finding that Dab1 deficient cells in a phenotypically normal Dab1 environment (normal ECM/cell–cell interaction) showed a significantly different migration phenotype compared to Dab1 deficient cells in a global Dab1 KO environment.
Radial glia cells play an important role in neuronal migration and therefore the mutant radial glia could potentially contribute to the migration defect. However, multiple lines of evidence argue against this possibility. In the case of RGCs exhibiting a dominant effect on migrating neurons, phenotypes would appear throughout the cortical wall. In contrast to such expectation, we found that mutant neurons in mosaic-MADM and KO-MADM did migrate through the IZ and sometimes beyond into the CP. In addition, migration defects were strongest in the outer zones of the cortical wall. Studies re-expressing either Cdk5r1 or Dab1 specifically in a subset of neurons in a global-KO environment observed a ‘rescue’ of the targeted neurons in their ability to migrate, which suggest no substantial direct contribution of either Cdk5r1 or Dab1 mutant radial glia on the migrating neurons [23, 68, 74]. Detailed morphological analysis found no differences between wild-type and p35 KO RGPs [11].
Labelling efficiency did vary between the different MADMs [12] dependent on the chromosome and insertion. However, the phenotypes observed between Cdk5r1, Cdk5 and Dab1 did show a similar cell-autonomous versus non-cell-autonomous phenotype regardless of the MADM labelling efficiency. As such, the distribution pattern of the labeled neurons was highly similar when comparing the different control-MADM scenarios. Furthermore, only slight differences were observed between e.g. Cdk5r1 (MADM-11) and Cdk5 (MADM-5) for all MADM genotypes analysed. These differences were most likely due to individual gene (loss of) function rather than a reflection of the efficiency of the MADM reporter. However, we cannot completely exclude local effects due to more or less mutant cells in distinct MADM reporters.
MADM relies on interchromosomal recombination and differential segregation of recombinant chromosomes during mitotic cell division to generate homozygous mutant cells for a particular gene of interest. Thus, mutant cells that are generated during the MADM process could in principle inherit certain amounts of the corresponding protein during cell division, a process called protein perdurance. We cannot completely exclude a possible effect of protein perdurance in the interpretation of the phenotypes described in the above mosaic sparse deletion paradigms. However, we consider the effect to be minimal at best for the following reasons. The Emx1-Cre driver is active from E9 onward when neural stem cells in the mouse embryonic forebrain still divide in an expanding proliferative manner with relatively short cell-cycle length of ~8 h [77]. Thus, before the generation of postmitotic projection neurons, every proliferative stem cell division will dilute protein perdurance. Assuming that half the amount of protein from the mother cell was inherited into the daughter cells, after five cell cycles, <4% of protein would be left in the cells if the protein were infinitely stable. Yet, the p35 protein encoded by Cdk5r1, is a very short lived protein with a half-life of 20-30 min in vivo [60] and DAB1 has a half-life of ~12 h [3]. Hence, it is unlikely that a biologically relevant amount of p35 or DAB1 protein was present in a substantial number of mutant migrating cells in any of the mosaic MADM scenarios.
Our findings put a new perspective on the clinical symptoms and/or appearance of disease-causing mutations affecting radial migration in particular, but also mutations causing cortical malformation in general. For instance in focal malformations of cortical development (FMCD) a small fraction of mutant cells can disrupt neighboring cells and even large areas thereby affecting overall cortical architecture [5, 47, 62, 66]. In order to obtain a better understanding of FMCD disease etiology, it will thus be essential to rigorously scrutinize the contribution of not only cell-autonomous loss of gene function but also tissue-wide and systemic effectors. Interestingly, lissencephaly with cerebellar hypoplasia has been attributed to individuals with mutations in CDK5 or RELN [33, 48]. The common clinical appearance in such patients could (at least in part) emerge from tissue-wide effects alike the ones we observed in our MADM-based analysis of Cdk5r1/CDK5 and Dab1, respectively.
CONCLUSION
Our study provides quantitative evidence, in a correlative manner, that global tissue-wide effects play essential roles in the control of radial projection neuron migration in the developing cortex. Based on defined genetic conditions in combination with single-cell tracing, we could show that sparse mosaic ablation of gene function results in highly specific migration phenotypes. In contrast, in global KO, individual migrating neurons exhibit distinct deficits that result from predominant tissue-wide effects. Altogether, cortical projection neuron migration is tightly regulated by intrinsic gene function and depending on the cellular and genetic landscape of the overall surrounding tissue.
MATERIALS AND METHODS
Mouse lines
All mouse colonies were maintained in accordance with protocols approved by the institutional animal care and use committee, institutional ethics committee and the preclinical core facility at Institute of Science and Technology Austria. Experiments were performed under a license approved by the Austrian Federal Ministry of Science and Research following the Austrian and EU animal laws (license numbers: BMWF-66.018/0007-II/3b/2012 and BMWFW-66.018/0006-WF/V/3b/2017).
Mice with specific pathogen-free status according to FELASA recommendations [50] were bred and maintained in experimental rodent facilities (room temperature 21 ± 1°C [mean ± SEM]; relative humidity 40%–55%; photoperiod 12 L:12D). Food (V1126, Ssniff Spezialitäten GmbH, Soest, Germany) and tap water were available ad libitum.
Mouse lines with MADM cassettes inserted on chr.4, chr.5 and chr.11 [12, 32] [MADM-4-GT, MADM-4-TG, MADM-5-GT, MADM-5-TG, MADM-11-GT (JAX stock # 013749), MADM-11-TG (JAX stock # 013751)], Cdk5r1 [11] (JAX stock # 004163), Cdk5-flox [67] (JAX stock # 014156), Dab1 [34] (JAX stock # 003581); Emx1-Cre [20] (JAX stock # 005628) were previously described. We have not observed any influence of sex on the results in our study, and all experiments and analyses were carried out using animals of both sexes. Phenotypic time course analysis of Dab1-MADM-4, Cdk5-MADM-5, Cdk5r1-MADM-11 in combination with Emx1-Cre was performed at E14, E16, P0 and P21. For sequencing and/or proteomics experiments, MADM-11 animals were used in combination with Emx1-Cre and were analysed at E13, E16 and P0. Genetic epistasis experiments of Cdk5r1 and Dab1 on MADM-4 and MADM-11 in combination with Emx1-Cre were all performed at P21.
Isolation of fixed tissue
Tissues from postnatal time points (P15/P21) were collected by cardiac perfusion. Mice were deeply anesthetized through injection of a ketamine/xylazine/acepromazine solution (65 mg, 13 mg, and 2 mg/kg body weight, respectively) and unresponsiveness was confirmed through pinching the paw. The diaphragm of the mouse was opened from the abdominal side to expose the heart. Cardiac perfusion was performed with phosphate buffered saline (PBS) followed immediately by ice-cold 4% PFA prepared in PB buffer (Sigma-Aldrich). Brains were removed and further fixed in 4% PFA for 24 h at 4°C to ensure complete fixation. Brains were cryopreserved with 30% sucrose (Sigma-Aldrich) solution in PBS for ~48 h. Brains were then embedded in Tissue-Tek O.C.T. (Sakura). For adult time points, 45 μm coronal sections were collected in 24 multi-well dishes (Greiner Bio-one) and stored at −20°C in antifreeze solution (30% v/v ethylene glycol, 30% v/v glycerol, 10% v/v 0.244 M PO4 buffer) until used. Tissue from embryonic time points (E14/E16) and postnatal day zero (P0) was directly transferred into 4% PFA and kept at least 24 h at 4°C. Cryopreservation and embedding were done as described for adult brains. For embryonic and early postnatal brains, 25 μm cryosections were directly mounted onto Superfrost glass-slides (Thermo Fisher Scientific).
Immunohistochemistry
Brain sections were mounted onto Superfrost glass-slides (Thermo Fisher Scientific) and let to dry, followed by three wash steps each of 5 min with PBS. Tissue sections were blocked for 30 min in a buffer solution containing 5% normal donkey serum (Thermo Fisher Scientific) and 0.5% Triton X-100 in PBS. Primary antibodies for GFP (1:500 Chick, Aves Labs Inc.) and RFP (1:500 Rabbit, MBL) were mixed in the blocking buffer and incubated on the tissue for at least 12 h at 4°C. Sections were washed 3 times for 5 min each with PBS with triton (PBT) (0.5% Triton X-100 in PBS) and incubated with the corresponding secondary antibodies Alexa Fluor 488 (1:500 Anti-Chicken IgG, Jackson ImmunoResearch Labs) and Cy3 (1:500 Anti-Rabbit IgG, Jackson ImmunoResearch Labs) diluted in PBT for 1 h. Sections were then washed two times with PBT and once with PBS each for 5 min. Finally, nuclear staining was done using 10 min of incubation with PBS containing 2.5% DAPI (Thermo Fisher Scientific). Sections were embedded in mounting medium containing 1,4-diazabicyclooctane (DABCO; Roth) and Mowiol 4-88 (Roth) and stored at 4°C. For the neuronal layer identity staining, primary antibodies for Cux1 (1:100, Goat, Santa Cruz), FoxP2 (1:300, Goat, Santa Cruz) and Ctip2 (1:400, Rat, Abcam) were used with primary antibodies for GFP (1:500, Chick, Aves Labs Inc.) and RFP (1:500, Rabbit, MBL) in the same manner as described above. Alexa Flour 647 (1:500 Anti-Goat IgG or 1:500, Anti-Rat IgG,Jackson ImmunoResearch Labs) were used for Goat or Rat originating primary antibodies. For primary antibodies requiring antigen retrieval, the tissue was processed by incubating in sodium citrate buffer (10 mM Sodium Citrate, 0.05% Tween 20, pH 6.0) at 80°C for 10 min prior to immunostaining. Once cooled to room temperature, sections were washed with PBS and standard immunostaining protocol as described above was performed.
Imaging of fixed brain tissue
Histological brain sections were imaged using confocal microscopy (Zeiss inverted LSM800) or epifluorescence microscopy (Olympus VS120 Slide scanner). Confocal images were recorded on a Zeiss LSM 800 laser-scanning confocal microscope mounted with a plan-apochromat 10x/0.45, WD = 2.1 mm objective. Excitation/emission wavelengths were 488/509 nm (EGFP), 554/581 nm (tdTomato), and 353/465 nm (DAPI). Z-series images were collected on a PC running ZEN 2.6 software (Zeiss). Image series were Z-projected, stitched and contrast-enhanced using ZEN 2.6 software (Zeiss). Slidescanner images were recorded with a 10x/NA 0.4 objective. Excitation/emission wavelengths were 485/518 nm (FITCH), 560/580 nm (Cy3), and 387/455 nm (DAPI). Slide scanner images were processed using ImageJ software [70].
Analysis of relative distribution of MADM-Labeled neurons
Images were imported into ImageJ software [70] and MADM-labeled neurons were manually quantified based on the respective fluorescent marker expression and their relative position, which was calculated with respect to the bottom of the ventricle and the pial surface (for details see http://github.com/hippenmeyerlab/cell2layer). The analysis script used computes the relative and absolute distances of each manually marked neuron to its boundaries (ventricular surface and pia) provided manually as two segmented lines. For each neuron, the shortest distance to the two-layer boundaries is computed, resulting in two distances d1 and d2. The normalized (relative) distance is computed by:
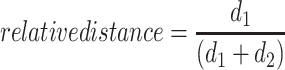 |
Statistical analyses were done with GraphPad Prism 8.0.1, applying an arcsin conversion of relative percentages, a two-way analysis of variance (ANOVA) and a Tukey post hoc test.
Slice culture and time-lapse imaging
Embryos were collected at E16 and stored in ice-cold PBS during genotyping. Immediately after genotyping, MADM labeled embryonic brains were dissected and mounted in 4% low-melting agarose (Fisher BioReagents). A total of 300 μm coronal slices were prepared in oxygenated ice-cold artificial cerebrospinal fluid using a vibratome (Leica VT 1200S). Thereafter, slices were placed on Milicell culture inserts (Millipore) in 6-well glass-bottom dishes (MatTek) containing culture medium (1% 100X N2 supplement (Gibco), 1% Penicillin–Streptomycin (Gibco) in transparent F12/DMEM (Gibco)) and incubated (37°C, 5% CO2) for at least 45 min prior to imaging acquisition. To reduce the evaporation of media during imaging, a FoilCover lid (Pecon) was applied on top of the glass-bottom dishes during time-lapse imaging. A time-lapse of minimum 15 h with a frame rate of 15 ± min was recorded unidirectionally at seven Z-positions with 5 μm spacing using confocal microscopy [Zeiss LSM800, Plan-Apochromat 10×/0.45, WD = 2.1 mm objective, equipped with a heating chamber and stage-top incubator chamber and gas mixed (Ibidi) (37°C, 5% CO2)]. Excitation/emission wavelengths were 488/509 nm (EGFP) and 554/581 nm (tdTomato). Time-lapse images were collected on a PC running Zeiss ZEN Blue software. Time-lapse image series were Z-projected, time-stitched using ZEN blue software.
Correction of non-linear local drift in time-lapse images
To correct any local tissue drift in the original 3D multi-channel movies, we developed the Python package undrift. First, dense optical flow from successive image pairs is estimated with the Farnebäck method [17] using the OpenCV library (version 3.3.1). For movies with more than one input channel, we used the averaged channel intensities before estimating the optical flow for each pixel. Input parameters for the Farnebäck method were set as follows: the number of image pyramid levels to 3, the averaging window size to 512 × 512 (px), the size of the pixel neighborhood used to find polynomial expansions to 5 px, the standard deviation of the Gaussian that is used to smooth derivatives to 0.4 px and the number of iterations per pyramid level to 3. Parameters were optimized to capture the movement of single cells and the locally coherent drift of tissue regions (if present). Then, the pairwise optical flow fields were smoothed (locally weighted averaged) with a spatio-temporal Gaussian (σ_t = 1 px and σ_xy = 25.6 px) using the scikit-image library (0.16.2). The strong spatial smoothing effectively removes movement on a small scale (single cells), whereas spatially coherent optical flow on a bigger scale (tissue drift) is maintained in the output. The smoothed pairwise optical flow fields are integrated over time to obtain an optical flow field relative to the reference frame (first time-point) using cubic spline interpolation and the Python SciPy library (version 1.4.1). New movies are rendered by artificially unwarping this integrated flow field on the original movie channels starting from the reference frame. For more information see http://github.com/hippenmeyerlab/undrift.
Analysis of neuronal trajectories
Neurons were tracked semi-automatically with the ImageJ plugin TrackMate [78] using the LoG detector (estimated blob diameter: 10.0micron, threshold: 2.0, Median filter: enabled, sub-pixel localization: enabled) and the linear motion LAP tracker (initial search radius: 15, max search radius: 15, max frame gap: 2) for each channel. Tracks were manually curated to ensure correct tracking of neurons. Only red and green neuronal tracks were included in the analysis, all yellow neurons were excluded in the analysis. Tissue compartments (upper/lower zone) were drawn manually. All parameters were extracted in a .csv file for analysis. For each neuron, we first determined if it was located in the located in the upper/lower zone in the first frame or last frame. In Fig. 3E–F: each cell track was grouped into upper zone if in any frame the cell was positioned in the upper zone area. The cell track was grouped into lower zone if the cell was in no frame placed in the upper zone area. An arcsin conversion was performed of relative percentages for statistical calculations. Figures 3G–H: we extracted the x/y coordinates of cells based on their position (upper zone, lower zone). The resulting cells were re-grouped into cell tracks and for each track we calculated mean straight-line speed and directionality as follows.
Distance of one cell between two frames as:
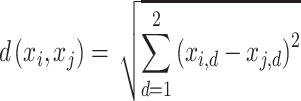 |
Sum of all distances (total distance traveled) with N being the number of frames a cell was tracked in:
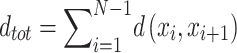 |
Net distance traveled:
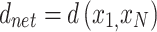 |
Net time traveled:
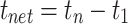 |
Mean straight line speed:
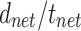 |
Directionality (meandering) index:
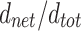 |
Note that the time between frames can differ for each cell (e.g. if a cell was not identified in one frame) and that the total time a cell was tracked can also differ between cells. Therefore, to assure the same frame rate for the analysis, we calculated the velocities in micrometer per minute. Note that a single track can be divided into sub-tracks if crossing the middle line. These tracks are ‘crossing’ and make up on average ~6% of all tracks. For Fig. 3E–H, we ranked each cell track from each video based on dnet and plotted the indicated values for the top 15 cells for each video. Statistical analyses were done with GraphPad Prism 8.0.1, applying a two-way ANOVA and a Tukey post hoc test.
Formulation of computational migration model
The formulated 2D migration model is evaluated via a Python 3.6 script. The force generation mechanism is based on a random walk model with directional bias on a single cell of unit mass [10]. We include an alteration from [10] by generating a 2D force 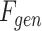 in an intermediate step instead of directly calculating a displacement for each cell. The total force acting on a given cell at each time instance is given by:
in an intermediate step instead of directly calculating a displacement for each cell. The total force acting on a given cell at each time instance is given by:
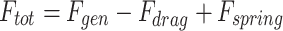 |
For each timestep 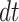 the internal molecular machinery of a cell generated force
the internal molecular machinery of a cell generated force 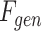 . If this force is insufficiently high to overcome the subsequently resulting drag force
. If this force is insufficiently high to overcome the subsequently resulting drag force 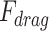 it is stored in form of a spring-like force
it is stored in form of a spring-like force 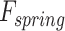 for the next timestep. Whereas the drag force is defined as:
for the next timestep. Whereas the drag force is defined as:
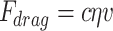 |
Here, 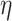 denotes the dynamic viscosity and
denotes the dynamic viscosity and 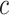 is a parameter dependent on the cell shape, which we consider a spherical particle of unit radius. In our model both parameters are considered to be locally constant and can therefore be unified as a resistance parameter
is a parameter dependent on the cell shape, which we consider a spherical particle of unit radius. In our model both parameters are considered to be locally constant and can therefore be unified as a resistance parameter 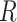 . Similar models have been previously described for 3D cell migration in extra cellular matrices [85]. We include the cell intrinsic difference between control and KO-Cdk5r1-MADM in the form of a directionality parameter
. Similar models have been previously described for 3D cell migration in extra cellular matrices [85]. We include the cell intrinsic difference between control and KO-Cdk5r1-MADM in the form of a directionality parameter 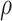 and a force scaling parameter
and a force scaling parameter 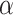 [10]. Here, a value of
[10]. Here, a value of 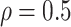 corresponds to a pure random walk model, which KO-Cdk5r1-MADM neurons tend towards. However, control neurons are closer to
corresponds to a pure random walk model, which KO-Cdk5r1-MADM neurons tend towards. However, control neurons are closer to 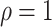 , indicating a high directionality. The force scaling parameter
, indicating a high directionality. The force scaling parameter 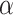 controls the magnitude of the generated force and henceforth velocity magnitudes. The non-cell-autonomous effect is included by introducing a linear coupling of directionality and force generation coefficient with the ratio
controls the magnitude of the generated force and henceforth velocity magnitudes. The non-cell-autonomous effect is included by introducing a linear coupling of directionality and force generation coefficient with the ratio 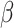 of control cells
of control cells 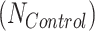 to mutant cells
to mutant cells 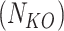 . Boundary values for
. Boundary values for 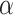 and
and 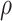 at
at 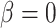 and
and 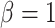 correspond to values from experimental data for KO-Cdk5r1-MADM and control, respectively. For more details of our model and parameters see Table 1 below and Fig. S4.
correspond to values from experimental data for KO-Cdk5r1-MADM and control, respectively. For more details of our model and parameters see Table 1 below and Fig. S4.
Table 1.
Modelling parameters

|
Preparation of cell suspension and FACS
Preparation of cell suspension for cell sorting was prepared as previously described [46] for E13, E16 and P0 time points for RNA-seq and P0 for proteomics. From the overall MADM samples, GFP+ cells were collected for each genotype. For RNA-sequencing, 2000 cells per replicate (1 replicate=1 cerebral cortex) were sorted directly into a custom-made lysis buffer (30 nM TRIS pH 8, 10 nM EDTA pH 8, 1% SDS and 200 μg/μl Proteinase K). For proteomics, 10 000 cells per replicate (1 replicate = 1 cerebral cortex) were prepared as described above except no serum was added to the media and an extra wash step with DMD/F12 wash was carried out. Samples for proteomics were sorted directly into 50 μl lysis buffer (LYSE-NHS, Preomics iST-NHS kit).
RNA extraction of MADM samples for RNA sequencing
Directly after cell sorting using FACS, samples were incubated for 30 min at 37°C. Total volume was filled to 250 μl using RNase-free H2O (Thermo Fisher Scientific) followed by the addition of 750 μl Trizol LS (Thermo Fisher Scientific). Samples were mixed by inversion (five times). After a 5 min incubation step at RT, the entire solution was transferred into a MaXtract tube (QIAGEN). A total of 200 μl chloroform (Sigma-Aldrich) was added, followed by three times 5 s vortexing and 2 min incubation at RT. Samples were centrifuged for 2 min at 12 000 rpm at 18°C. The supernatant was transferred to a new tube and isopropanol (Sigma-Aldrich) was added in a 1:1 ratio. For better visibility of the RNA pellet 1 μl GlycoBlue (Thermo Fisher Scientific) was added and the entire solution was mixed by vortexing (3× 5 s). Samples were left for precipitation o/n at −20°C. After precipitation samples were centrifuged for 20 min with 14 000 rpm at 4°C. The supernatant was removed and the RNA pellet was washed with 70% ethanol, followed by a 5 min centrifugation step (14 000 rpm at 4°C). The RNA pellet was resuspended in 12.5 μl RNase-free H2O. RNA quality was analysed using RNA 6000 Pico kit (Agilent) following the manufacturer’s instructions. The RNA samples were stored at −80°C until further use. RNA sequencing was performed by VBCF GmbH on Illumina platforms.
Statistical analysis of RNA-Seq
Read processing, alignment and annotations are described elsewhere [45]. STAR alignment parameters: clip5pNbases 3, outFilterMultimapNmax 1, outSAMtype BAM SortedByCoordinate and quantMode GeneCounts. Downstream analyses were performed in R (v3.6.1). Read coverages of the deleted Cdk5r1 region (chr11:80477417–80478722, mm10) and the deleted Dab1 region (chr4:104605298–104605437, mm10) were calculated using bedtools intersect with the split option on the aligned bam file produced by STAR. These read counts were added to the count tables produced by STAR with the gene name Cdk5r1_del and Dab1_del, respectively.
For Fig. 5A–E, we analysed 81 samples and removed 9 samples with a low percentage of uniquely aligned reads (<50%) or due to their position on the PCA plot. Statistics on differential expression between all pairs of genotypes were calculated with DESeq2 (v1.26.0) using contrasts for each developmental time point separately. To reduce noise, only genes with an average read coverage of >10 were used in the analyses. We used an adjusted P-value (padj) cutoff of 0.05 for DEGs for all analyses in Fig. 5A–E. Up- and down-regulated genes were determined by a log2 fold-change of >0 or < 0, respectively. For Gene Ontology term enrichment, we used the enrichGO function from the clusterProfiler package (v3.14.0) with parameters: universe = [all informative genes in the respective comparison], ont = ‘ALL’, pool = T, readable = T, OrgDb = org.Mm.eg.db (v3.10.0), minGSSize = 20, maxGSSize = 500, P-value cutoff = 0.1, qvalueCutoff = 0.2. For the GO term plot in Fig. 5E, we focused on P0 timepoint and first removed GO terms that are not related to neuronal development by removing GO terms with blood, vascu or angio in their description. Then we calculated the negative log10 of the uncorrected P-value for the remaining GO terms (score). Finally, we ranked GO terms by the score and plotted the score of the top 10 GO terms focusing on the ontology BP.
For Fig. 7, we analysed 38 samples consisting of 22 samples already used for Fig. 5A–E (control, KO-Cdk5r1-MADM, P0 time point) and 16 new samples (control, Dab1-KO). We removed three samples due to their position on a PCA plot. Statistics on differential expression were calculated as for Fig. 5A–E using genes with an average read coverage over all samples >20. For all analyses in Fig. 7, we used an adjusted P-value (padj) cutoff of 0.05 and an absolute log2 fold-change >0.35 to define DEGs. Figure 7E: We calculated the % common up- and down-regulated genes relative to all DEG in KO-Dab1/control and KO-Cdk5r1-MADM/control, respectively, and plotted the mean of these two values. Figure 7F: significance of the overlap between DEG groups was calculated using newGOM from package GeneOverlap (v1.22.0) using the number of informative genes in this comparison as genome.size. We used different gene groups for further analysis: Common_up defines genes that are common up-regulated DEGs (intersection Fig. 7D left), Common_down defines genes that are common down-regulated DEGs (intersection Fig. 7D right), Dab1_spec defines genes that are DEG in Dab1-KO/control but not in KO-Cdk5r1-MADM/control (Dab1 up, Dab1 down in Fig. 7D) and Cdk5r1_spec defines genes that are DEG in KO-Cdk5r1-MADM/control but do not overlap (Cdk5r1 up, Cdk5r1 down Fig. 7D). GO term enrichment for the respective gene group was performed using enrichGO with parameters: enrichGO(OrgDb = org.Mm.eg.db, readable = T, pool = T, maxGSSize = 900, minGSSize = 100, P-value cutoff = 0.05, qvalueCutoff = 0.1, separately for different GO ontologies. For Fig. 7G, we calculated a score as in Fig. 7E and plotted the top 10 GO terms from cellular components (CC) ontology without prior filtering. For Fig. S8 we used the used top 50 GO terms (ranked by adjusted P-value) from Dab1_spec and Cdk5r1_spec analysis and calculated all pairwise semantic similarity goSim from GOSemSim (v2.12.1) package with parameters: measure = ‘Jiang’. We plotted the resulting similarity matrix using the pheatmap package.
Sample processing for proteomics
Samples were divided into two batches, each containing three control samples and five Cdk5r1-MADM or KO-Cdk5r1-MADM samples. Batch 1, processed on day 1, contained green cells from two individual litters and corresponded to control versus KO-Cdk5r1-MADM comparison; batch 2, processed on day 2, contains green from three individual litters and was used for the control versus Cdk5r1-MADM comparison. Each litter contained both control and either Cdk5r1-MADM or KO-Cdk5r1-MADM. Protein extraction, tryptic digestion and peptides cleanup were performed using a TMT-labeling compatible variant of the in-Stage Tips method (iST-NHS-12x kit, Preomics). Briefly, immediately after cell-sorting, collected cells were supplemented with 50 μl LYSE-NHS buffer, boiled for 10 min, then processed according to the manufacturer’s protocol with the following minor modifications: sonication was skipped (the number of cells was low enough that DNA would not be an issue, and this would reduce the chance of proteins loss or samples contamination due to having to use a probe sonicator); digestion was performed overnight. Prior to TMT labeling, small aliquots of each of the 16 samples were taken and mixed to generate a mixed reference sample. Individual samples were then labeled with TMT-10plex (lot # UL291039, ThermoFisher Scientific), splitting the contents of each TMT vial to label one sample of each batch. Individual samples were combined into two TMT-labeled samples, each containing the eight samples from one batch plus a mixed reference sample. Combined samples were then loaded onto the iST-NHS kit’s cartridges in several steps, washed as per the manufacturer’s protocol, eluted and dried in a speedvac. Since phospho-peptides were of interest, although the amount of material as determined by a Pierce Quantitative Colorimetric Peptide Assay (Thermo Scientific) was low (~100 μg/sample), the samples were subjected to phospho-peptides enrichment (MagReSyn Ti-IMAC beads, ReSyn Biosciences) according to manufacturer’s protocol but scaling down the amount of beads, then the flow-throughs were fractionated into eight fractions using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (ThermoFisher Scientific).
LC–MS/MS analysis
Samples were dried, redissolved in 0.1% TFA and analysed by LC–MS/MS on an Ultimate HPLC (ThermoFisher Scientific) coupled to a Q-Exactive HF (ThermoFisher Scientific). Each sample was concentrated over an Acclaim PepMap C18 pre-column (5 μm particle size, 0.3 mm ID × 5 mm length, ThermoFisher Scientific) then bound to a 50 cm EasySpray C18 analytical column (2 μm particle size, 75 μm ID × 500 mm length, ThermoFisher Scientific) and eluted over the following 90 min gradient: solvent A, water +0.1% formic acid; solvent B, 80% acetonitrile in water +0.08% formic acid; constant 300 nl/min flow; B percentage: start, 2%; 70 min, 31%; 90 min, 44%. Mass spectra were acquired in positive mode with a data-dependent acquisition method: FWHM 20s, lock mass 445.12003 m/z; MS1: profile mode, 120 000 resolution, AGC target 3e6, 50 ms maximum IT, 380 to 1500 m/z; MS2: top 20, centroid mode, 1 microscan, 60 000 resolution, AGC target 1e5, 100 ms maximum IT, 0.7 m/z isolation window (no offset), 100 m/z fixed first mass, NCE 32, excluding charges 1 and 8 or higher, 60s dynamic exclusion.
Statistical analysis of proteomics
Raw files were searched in MaxQuant 1.6.14.0 against the Mus musculus reference proteome from UniProtKB. Fixed cysteine modification was set to H11OC6N. Variable modifications were Oxidation (M), Acetyl (Protein N-term), Deamidation (NQ), Gln- > pyro-Glu and Phospho (STY). Match between runs and second peptides search were active. All FDRs were set to 1%. MaxQuant results were further processed in R using in-house scripts, starting from evidence (PSM) tables. Briefly, potential contaminants, reverse database hits or evidences with null intensity values were excluded. Evidence reporter intensities were scaled to integrated feature intensity, normalized using the Levenberg–Marquardt procedure row-wise, then assembled into peptidoforms (post-translationally modified peptides), summing up intensities per sample. Peptidoform reporter intensities were corrected for TMT lot label impurity values, median normalized, log-transformed, subjected to variance stabilizing normalization, re-normalized using the Levenberg–Marquardt procedure row-wise, then corrected for TMT batch effect using the Internal Reference Scaling method. Ratios to the average of all either control or Cdk5r1-MADM samples (two parallel analyses) were calculated, then protein groups were inferred from peptidoforms. Because the focus was on discovering and quantifying as many protein groups as possible from limiting sample amounts, all groups including those with just one peptide were retained. Protein groups were quantified by averaging the intensity profile of matching peptidoforms (excluding phospho-peptides and counterparts), weighted by the inverse of individual posterior error probabilities, then these values were averaged per sample and P-values calculated using a moderated t-test and an F-test (limma package). Significance thresholds were calculated using the Benjamini Hochberg procedure for 10%, 20% and 30% FDR. In addition, protein groups with an absolute log2 ratio smaller than 95% of individual to average reference log2 ratios were excluded. Results were saved as an EXCEL file and used for downstream analyses. Figures 5G–H: we plotted the Moderated.t-test:.-log10(P-value) against the Ratio:.log2.-.Mean for the respective comparison. All genes that were marked ‘up, FDR = 10%’ and ‘down, FDR = 10%’ were labeled in the volcano plot. For Fig. 5I: we used all gene names linked to peptide groups for subsequent analyses. We defined significant DEGs as genes marked ‘up, FDR = 10%’, ‘down, FDR = 10%’ and calculated GO term enrichments using clusterProfiler (v3.14.3) with parameters: universe = [all genes in ‘Genes’], OrgDb = org.Mm.eg.db (v3.10.0), ont = ‘CC’, P-value cutoff = 0.1, minGSSize = 50, maxGSSize = 2000, pool = F, readable = T. We plotted selected terms of the top 15 GO terms, ranked by P-value. Figures 5J and K: comparison to RNA-Seq: we defined RNA-Seq DEGs by using statistics calculated in Fig. 5B and extracting genes with an adjusted P-value of <0.1 and a log2 fold-change >0 (RNA Up) or <0 (RNA Down). DEGs based on proteomics were defined as having a ‘+’ in the ‘Significant:.FDR = 10%.-.full-KO’ column and ‘Ratio:.log2.-.Mean.-.full-KO’ <0 (Protein Down) or >0 (Protein Up). Note that we only used genes that were informative on both RNA-Seq and Proteomics for this analysis. Figure 5L: for GO term analysis, we used gene sets commonly up- and down-regulated in both RNA-Seq and proteomics (intersection Fig. 5K). GO term enrichment was calculated using enrichGO with parameters: universe = [all genes informative in RNA-Seq and proteomics], OrgDb = org.Mm.eg.db (v3.10.0), ont = ‘CC’, P-value cutoff = 0.9, minGSSize = 10, maxGSSize = 2000, pool = F, readable = T. We calculated a score as described before with a negative prefix for down regulated genes and plotted the top 10 GO terms, ranked by P-value.
Transcriptomics raw data
The raw sequencing data used in this publication were deposited in NCBI’s Gene Expression Omnibus [15] and be accessible through GEO Series accession number GSE200604 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200604).
Proteomics raw data
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [61] partner repository with the dataset identifier PXD023222.
Code for analysis of relative distribution of MADM-Labeled neurons
The Python (version > = 3.6) package cell2layer (version 0.2) for analysing the relative and absolute distances of each manually marked neuron to its layer boundaries along with documentation is available at http://github.com/hippenmeyerlab/cell2layer as open-source software under the GNU General Public License v3.0.
Code for correction of non-linear local drift in time-lapse images
The Python (version > = 3.6) package undrift (version 0.2) for correcting non-linear, local tissue drift along with documentation is available at http://github.com/hippenmeyerlab/undrift as open-source software under the GNU General Public License v3.0.
Supplementary Material
Contributor Information
Andi H Hansen, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Florian M Pauler, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Michael Riedl, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Carmen Streicher, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Anna Heger, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Susanne Laukoter, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Christoph Sommer, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Armel Nicolas, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Björn Hof, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
Li Huei Tsai, Picower Institute for Learning and Memory, MIT, Cambridge, MA 02139, USA.
Thomas Rülicke, Department of Biomedical Sciences, University of Veterinary Medicine Vienna, 1210 Vienna, Austria.
Simon Hippenmeyer, Institute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria.
SUPPLEMENTARY MATERIAL
Supplementary material are available at Oxford Open Neuroscience online.
STUDY FUNDING
A.H.H. was a recipient of a DOC Fellowship (24812) of the Austrian Academy of Sciences. This work also received support from IST Austria institutional funds; the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA grant agreement No 618444 to S.H.
APC FUNDING
APC funding was obtained by IST Austria institutional funds.
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
S.H. and A.H.H. conceived the research. A.H.H., C.St., and A.H generated all experimental MADM tissue. A.H.H., C.St., T.R, and S.L., performed all the experiments except LCMS which was performed by A.N. A.H.H., F.M.P, and C.So. analyzed imaging data. F.M.P. performed computational and bioinformatic analysis of RNA-seq and proteomics with inputs from A.H.H. and S.H. M.R. and A.H.H. established the computational model with input from B.H and S.H. L.H.T provided critical resources. S.H. and A.H.H. wrote the manuscript with input from F.M.P. and M.R. All authors edited and proofread the manuscript.
DATA AVAILABILITY
All data have been presented in Figures and Supplemental Figures. Original images will be made available upon request. Raw sequencing/proteomics data, sample lists and code will be accessible as detailed.
ACKNOWLEDGMENTS
We thank A. Sommer and C. Czepe (VBCF GmbH, NGS Unit), L. Andersen, J. Sonntag and J. Renno for technical support and/or initial experiments; M. Sixt, J. Nimpf and all members of the Hippenmeyer lab for discussion. This research was supported by the Scientific Service Units of IST Austria through resources provided by the Imaging and Optics Facility, Lab Support Facility and Preclinical Facility.
References
- 1.Amberg N, Hippenmeyer S. Genetic mosaic dissection of candidate genes in mice using mosaic analysis with double markers. STAR Protoc. 2021;2: 100939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–8 [DOI] [PubMed] [Google Scholar]
- 3.Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol. Cell. Biol. 2003;23:9293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43 [DOI] [PubMed] [Google Scholar]
- 5.Baek ST, Copeland B, Yun E-J et al. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. 2015;21:1445–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van den Berghe V, Stappers E, Seuntjens E. How cell-autonomous is neuronal migration in the forebrain? Molecular cross-talk at the cell membrane. Neurosci. 2014;20:571–5 [DOI] [PubMed] [Google Scholar]
- 7.Bock HH, May P. Canonical and non-canonical Reelin signaling. Front Cell Neurosci. 2016;10:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradshaw NJ, Hayashi MAF. NDE1 and NDEL1 from genes to (mal)functions: parallel but distinct roles impacting on neurodevelopmental disorders and psychiatric illness. Cell Mol Life Sci. 2017;74:1191–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchsbaum IY, Cappello S. Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development. 2019;146: [DOI] [PubMed] [Google Scholar]
- 10.Caffrey JR, Hughes BD, Britto JM, Landman KA. An in silico agent-based model demonstrates Reelin function in directing lamination of neurons during cortical development. Plos One. 2014;9:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chae T, Kwon YT, Bronson R et al. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42 [DOI] [PubMed] [Google Scholar]
- 12.Contreras X, Amberg N, Davaatseren A et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 2021;35:109274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delalle I, Bhide PG, Caviness VS, Tsai LH. Temporal and spatial patterns of expression of p35, a regulatory subunit of cyclin-dependent kinase 5, in the nervous system of the mouse. J Neurocytol. 1997;26:283–96 [DOI] [PubMed] [Google Scholar]
- 14.Dimidschstein J, Passante L, Dufour A et al. Ephrin-B1 controls the columnar distribution of cortical pyramidal neurons by restricting their tangential migration. Neuron. 2013;79:1123–35 [DOI] [PubMed] [Google Scholar]
- 15.Edgar R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evsyukova I, Plestant C, Anton ES. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol. 2013;29:299–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farnebäck G. Two-frame motion estimation based on polynomial expansion. Lect Notes Comput Sci (Including Subser Lect Notes Artif Intell Lect Notes Bioinformatics). 2003;2749:363–70 [Google Scholar]
- 18.Franco SJ, Martinez-Garay I, Gil-Sanz C et al. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorelik A, Sapir T, Woodruff TM, Reiner O. Serping1/C1 inhibitor affects cortical development in a cell autonomous and non-cell autonomous manner. Front Cell Neurosci. 2017;11:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorski JA, Talley T, Qiu M et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenman R, Gorelik A, Sapir T et al. Non-cell autonomous and non-catalytic activities of ATX in the developing brain. Front Neurosci. 2015;9:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerrini R, Parrini E. Neuronal migration disorders. Neurobiol Dis. 2010;38:154–66 [DOI] [PubMed] [Google Scholar]
- 23.Gupta A, Sanada K, Miyamoto DT et al. Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat Neurosci. 2003;6:1284–91 [DOI] [PubMed] [Google Scholar]
- 24.Hammond V, Howell B, Godinho L, Tan SS. Disabled-1 functions cell autonomously during radial migration and cortical layering of pyramidal neurons. J Neurosci. 2001;21:8798–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammond V, Tsai LH, Tan SS. Control of cortical neuron migration and layering: cell and non cell-autonomous effects of p35. J Neurosci. 2004;24:576–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanganu-Opatz IL, Butt SJB, Hippenmeyer S et al. The logic of developing neocortical circuits in health and disease. J Neurosci. 2021;41:813–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen AH, Duellberg C, Mieck C et al. Cell polarity in cerebral cortex development—cellular architecture shaped by biochemical networks. Front Cell Neurosci. 2017;11:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen AH, Hippenmeyer S. Non-cell-autonomous mechanisms in radial projection neuron migration in the developing cerebral cortex. Front Cell Dev Biol. 2020;8: 574382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatanaka Y, Hisanaga S-I, Heizmann CW, Murakami F. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J Comp Neurol. 2004;479:1–14 [DOI] [PubMed] [Google Scholar]
- 30.Heng JIT, Chariot A, Nguyen L. Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci. 2010;33:38–47 [DOI] [PubMed] [Google Scholar]
- 31.Hippenmeyer S. Molecular pathways controlling the sequential steps of cortical projection neuron migration. Cell Mol Control Neuron Migrat. 2014;800:1–24 [DOI] [PubMed] [Google Scholar]
- 32.Hippenmeyer S, Youn YH, Moon HM et al. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong SE, Shugart YY, Huang DT et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–6 [DOI] [PubMed] [Google Scholar]
- 34.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–7 [DOI] [PubMed] [Google Scholar]
- 35.Iwashita M, Kataoka N, Toida K, Kosodo Y. Systematic profiling of spatiotemporal tissue and cellular stiffness in the developing brain. Development. 2014;141:3793–8 [DOI] [PubMed] [Google Scholar]
- 36.Jheng G-W, Hur SS, Chang C-M et al. Lis1 dysfunction leads to traction force reduction and cytoskeletal disorganization during cell migration. Biochem Biophys Res Commun. 2018;497:869–75 [DOI] [PubMed] [Google Scholar]
- 37.Jossin Y. Molecular mechanisms of cell polarity in a range of model systems and in migrating neurons. Mol Cell Neurosci. 2020;106: 103503 [DOI] [PubMed] [Google Scholar]
- 38.Juric-Sekhar G, Hevner RF. Malformations of cerebral cortex development: molecules and mechanisms. Annu Rev Pathol. 2019;14:293–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keshvara L, Magdaleno S, Benhayon D et al. Cyclin-dependent kinase 5 phosphorylates disabled 1 independently of Reelin signaling. J Neurosci. 2002;22:4869–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ko J, Humbert S, Bronson RT et al. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci. 2001;21:6758–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–9 [DOI] [PubMed] [Google Scholar]
- 42.Kwan KY, Sestan N, Anton ES. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development. 2012;139:1535–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwon YT, Gupta A, Zhou Y et al. Regulation of N-cadherin-mediated adhesion by the p35-Cdk5 kinase. Curr Biol. 2000;10:363–72 [DOI] [PubMed] [Google Scholar]
- 44.Kwon YT, Tsai LH. A novel disruption of cortical development in p35(−/−) mice distinct from reeler. J Comp Neurol. 1998;395:510–22 [DOI] [PubMed] [Google Scholar]
- 45.Laukoter S, Amberg N, Pauler FM, Hippenmeyer S. Generation and isolation of single cells from mouse brain with mosaic analysis with double markers-induced uniparental chromosome disomy. STAR Protoc. 2020a;1: 100215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laukoter S, Beattie R, Pauler FM et al. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat Commun. 2020b;11:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JH, Huynh M, Silhavy JL et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lodato S, Arlotta P. Generating neuronal diversity in the mammalian cerebral cortex. Annu Rev Cell Dev Biol. 2015;31:699–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magen D, Ofir A, Berger L et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5. Hum Genet. 2015;134:305–14 [DOI] [PubMed] [Google Scholar]
- 50.Mähler (Convenor M, Berard M, Feinstein R et al. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab Anim. 2014;48:178–92 [DOI] [PubMed] [Google Scholar]
- 51.Marín O, Valiente M, Ge X, Tsai LH. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol. 2010;2:1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–8 [DOI] [PubMed] [Google Scholar]
- 53.Morimura T, Ogawa M et al. Relative importance of the tyrosine phosphorylation sites of Disabled-1 to the transmission of Reelin signaling. Brain Res. 2009;1304:26–37 [DOI] [PubMed] [Google Scholar]
- 54.Nadarajah B, Brunstrom JE, Grutzendler J et al. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–50 [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa N, Plestant C, Yabuno-Nakagawa K et al. Memo1-mediated tiling of radial glial cells facilitates cerebral cortical development. Neuron. 2019;103:836–852.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niethammer M, Smith DS, Ayala R et al. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711 [DOI] [PubMed] [Google Scholar]
- 57.Noctor SC, Flint AC, Weissman TA et al. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–20 [DOI] [PubMed] [Google Scholar]
- 58.Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–44 [DOI] [PubMed] [Google Scholar]
- 59.Ohshima T. Neuronal migration and protein kinases. Front Neurosci. 2015;8:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patrick GN, Zhou P, Kwon YT et al. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–64 [DOI] [PubMed] [Google Scholar]
- 61.Perez-Riverol Y, Csordas A, Bai J et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poduri A, Evrony GD, Cai X et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pramparo T, Libiger O, Jain S et al. Global developmental gene expression and pathway analysis of normal brain development and mouse models of human neuronal migration defects. PLoS Genet. 2011;7: e1001331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiner O, Parichha A, Sapir T. Modeling human neuronal migration deficits in 3D. Curr Opin Neurobiol. 2021;66:30–6 [DOI] [PubMed] [Google Scholar]
- 65.Reiner O, Sapir T. LIS1 functions in normal development and disease. Curr Opin Neurobiol. 2013;23:951–6 [DOI] [PubMed] [Google Scholar]
- 66.Rivière JB, Mirzaa GM, O’Roak BJ et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Samuels BA, Hsueh Y-P, Shu T et al. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56:823–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211 [DOI] [PubMed] [Google Scholar]
- 69.Sasaki S, Shionoya A, Ishida M et al. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–96 [DOI] [PubMed] [Google Scholar]
- 70.Schindelin J, Arganda-Carreras I, Frise E et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sekine K, Honda T, Kawauchi T et al. The outermost region of the developing cortical plate is crucial for both the switch of the radial migration mode and the Dab1-dependent “inside-out” lamination in the neocortex. Rapid Commun. 2011;31:9426–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sekine K, Kawauchi T, Kubo K et al. Reelin controls neuronal positioning by promoting cell-matrix adhesion via inside-out activation of integrin α5β1. Neuron. 2012;76:353–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shellard A, Mayor R. Rules of collective migration: from the wildebeest to the neural crest. Decennial index. 2020;375: 20190387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simo S, Jossin Y, Cooper JA. Cullin 5 regulates cortical layering by modulating the speed and duration of Dab1-dependent neuronal migration. J Neurosci. 2010;30:5668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Su SC, Tsai L-H. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–91 [DOI] [PubMed] [Google Scholar]
- 76.Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi T, Nowakowski R, Caviness V. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15:6046–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tinevez J-Y, Perry N, Schindelin J et al. TrackMate: an open and extensible platform for single-particle tracking. Methods. 2017;115:80–90 [DOI] [PubMed] [Google Scholar]
- 79.Tsai J, Vallee RB. Stem Cell Migration. Totowa, NJ: Humana Press, 2011 [Google Scholar]
- 80.Valiente M, Marín O. Neuronal migration mechanisms in development and disease. Curr Opin Neurobiol. 2010;20:68–78 [DOI] [PubMed] [Google Scholar]
- 81.Vallee RB, Seale GE, Tsai J-W. Emerging roles for myosin II and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol. 2009;19:347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang H, Jensen P, Goldowitz D. The community effect and Purkinje cell migration in the cerebellar cortex: analysis of scrambler chimeric mice. J Neurosci. 2002;22:464–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoshinaga S, Honda T, Kubo K, Nakajima K. Dab1-deficient deep layer neurons prevent Dab1-deficient superficial layer neurons from entering the cortical plate. Neurosci Res. 2022; 10.1016/j.neures.2022.03.011 [DOI] [PubMed] [Google Scholar]
- 84.Youn YH, Pramparo T, Hirotsune S, Wynshaw-Boris A. Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J Neurosci. 2009;29:15520–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zaman MH, Kamm RD, Matsudaira P, Lauffenburger DA. Computational model for cell migration in three-dimensional matrices. Biophys J. 2005;89:1389–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zong H, Espinosa JS, Su HH et al. Mosaic analysis with double markers in mice. Cell. 2005;121:479–92 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data have been presented in Figures and Supplemental Figures. Original images will be made available upon request. Raw sequencing/proteomics data, sample lists and code will be accessible as detailed.