

Abstract
The prediction of molecular interactions is vital for drug discovery. Existing methods often focus on individual prediction tasks and overlook the relationships between them. Additionally, certain tasks encounter limitations due to insufficient data availability, resulting in limited performance. To overcome these limitations, we propose KGE-UNIT, a unified framework that combines knowledge graph embedding (KGE) and multi-task learning, for simultaneous prediction of drug–target interactions (DTIs) and drug–drug interactions (DDIs) and enhancing the performance of each task, even when data availability is limited. Via KGE, we extract heterogeneous features from the drug knowledge graph to enhance the structural features of drug and protein nodes, thereby improving the quality of features. Additionally, employing multi-task learning, we introduce an innovative predictor that comprises the task-aware Convolutional Neural Network-based (CNN-based) encoder and the task-aware attention decoder which can fuse better multimodal features, capture the contextual interactions of molecular tasks and enhance task awareness, leading to improved performance. Experiments on two imbalanced datasets for DTIs and DDIs demonstrate the superiority of KGE-UNIT, achieving high area under the receiver operating characteristics curves (AUROCs) (0.942, 0.987) and area under the precision-recall curve ( AUPRs) (0.930, 0.980) for DTIs and high AUROCs (0.975, 0.989) and AUPRs (0.966, 0.988) for DDIs. Notably, on the LUO dataset where the data were more limited, KGE-UNIT exhibited a more pronounced improvement, with increases of 4.32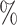 in AUROC and 3.56
in AUROC and 3.56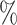 in AUPR for DTIs and 6.56
in AUPR for DTIs and 6.56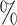 in AUROC and 8.17
in AUROC and 8.17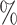 in AUPR for DDIs. The scalability of KGE-UNIT is demonstrated through its extension to protein–protein interactions prediction, ablation studies and case studies further validate its effectiveness.
in AUPR for DDIs. The scalability of KGE-UNIT is demonstrated through its extension to protein–protein interactions prediction, ablation studies and case studies further validate its effectiveness.
Keywords: molecular interactions prediction, KGE, multi-task learning, DDIs, DTIs
INTRODUCTION
Drug discovery and development is a lengthy and costly process [1]. Various studies focus on understanding molecular interactions and associations, such as DDIs, DTIs and protein–protein interactions (PPIs). These interactions play a crucial role in downstream applications, including drug repurposing, prediction of drug side effects and drug discovery.
In recent years, a series of computational methods have been proven effective for a specific task of predicting molecular interactions. These methods can be broadly categorized into three groups: similarity-based methods, matrix factorization-based methods and network-based methods. Similarity-based methods assume that if two pairs of nodes are similar, their interactions will also be similar [2, 3]. However, these methods overlook crucial features and information, such as molecular structure, functionality and metabolic pathways. Matrix factorization-based methods involve the decomposition and reconstruction of an adjacency matrix to infer novel molecular interactions. These methods utilize mathematical techniques to factorize the matrix into lower-dimensional representations, which capture latent features and relationships between molecules. Examples of matrix factorization-based methods include neighborhood regularized logistic matrix factorization [4], semi-nonnegative matrix factorization [5] and manifold regularized matrix factorization [6]. Matrix factorization-based methods often assume that the molecular interaction data can be represented by a low-rank matrix, which may not accurately capture the complex and heterogeneous nature of molecular interactions. Furthermore, these methods primarily rely on the inherent structure of the interaction matrix, neglecting other relevant features such as molecular descriptors, chemical structures or biological pathways.
Network-based methods have emerged as effective approaches for downstream link prediction based on constructed networks. GCN-DTI [7] was the first approach to introduce graph neural network (GNN) into the field of DTI prediction. And more and more methods, GNN [8, 9] and other variants, such as graph convolutional network (GCN) [10] and graph attention network (GAT) [11, 12], have been demonstrated relatively better performance in predicting molecular interactions. In addition, network-based methods have been further developed by heterogeneous data from multiple omics resources, such as genomics, proteomics and metabolomics. These networks consist of multiple types of nodes (such as drug, protein and disease) and edges (such as interaction indicators and similarity scores of nodes). For example, Luo et al. [13] proposed DTINet, which constructs heterogeneous networks to integrate drug- and protein-related information. Wan et al. [14] introduced NeoDTI to learn low-dimensional feature representations of drugs and proteins from heterogeneous data, enhancing the prediction performance of DTIs. Lin et al. [15] mined and fused multi-source features of heterogeneous data using a multi-network structure to facilitate DDI prediction. Furthermore, studies utilizing knowledge graphs (KGs) have achieved groundbreaking results in predicting molecular interactions [8, 16]. For instance, Mohamed et al. [17] developed TriModel, which employs knowledge graph embedding (KGE) techniques to learn vector representations of drugs and proteins, enabling the discovery of unknown DTIs. Lin et al. [18] proposed KGNN, an end-to-end framework that captures drug information and its potential neighborhood entities to predict DDIs. Both TriModel and KGNN demonstrate the effectiveness of KG in molecular interaction prediction.
There exists a certain degree of interconnectedness among molecular relationship prediction tasks. For instance, the mechanism of action for drugs often involves specific interactions with protein targets, and the interaction between drugs and protein targets can be influenced by other drugs or proteins. This implies the presence of correlations and mutual influences between drug–drug interactions (DDIs) and drug–protein target interactions [19]. However, the current focus of research in molecular relationship prediction tasks is predominantly on individual tasks, often disregarding the potential relationships and interactions between them. Furthermore, given the limited number of known molecular interactions in comparison to the vast number of potential interactions, certain methods may encounter limitations in predicting specific tasks due to insufficient data. To overcome the challenge of data scarcity, leveraging information and knowledge from other labeled tasks has emerged as a feasible solution. Multi-task learning takes advantage of the high correlation present in labeled data through implicit data augmentation and has shown superiority over traditional single-task learning in drug discovery [20, 21]. DeepDISOBind [22] models the interaction between DNA, RNA and proteins to simultaneously predict these three types of interactions, achieving better results than single-task models. Given the potential relationships among various molecular relationships and node data, multi-task learning can effectively leverage correlations between different molecular relationship tasks and improve model performance. KG-MTL [23] joint preserves the semantic relations of drug entities and the neighbor structures of the compound, resulting in improved predictions of drug–target and compound–protein interactions. However, the implementation of multi-task learning is challenging and requires careful consideration of the correlation between tasks [24].
Overall, the existing methods primarily focus on individual tasks without considering the potential correlation information among different molecular interactions prediction tasks. Additionally, certain tasks encounter limitations due to insufficient data availability, thereby impeding the algorithms’ performance. In the study, to overcome the abovementioned drawbacks, we present a pioneering approach that combines a large-scale KG and multi-task learning, named KGE-UNIT, for joint prediction of multiple types of molecular interactions. Based KGE, we extract heterogeneous features from the drug KG to supplement the structural features of drug and protein nodes, thereby improving the quality of features. Based multi-task learning, we propose an effective and novel predictor consisting of the task-aware CNN-based encoder and the task-aware attention decoder. This predictor has the capability to simultaneously predict multiple types of molecular interactions, and it can improve the performance of each respective task even under conditions of limited data availability. The encoder is responsible for fusing multimodal features, while the attention decoder is designed to capture the interacting context of molecular interactions tasks by the task-interacted attention block, impose constraints specific to each task and enhance task awareness by the task-aware attention block, ultimately leading to improve their respective performance. The ablation study proves that the performance enhancement of single tasks, particularly in data-scarce tasks, by multimodal features and multi-task learning. Furthermore, relevant experiments show that KGE-UNIT outperforms some state-of-the-art methods for both DDIs prediction and DTIs prediction. The main code can be accessed through https://github.com/zcc1203/KGE-UNIT.
METHODS AND MATERIALS
In the section, we first formulate the joint multiple types of interactions prediction problem (refer Section 2.1). And, we introduce the proposed KGE-UNIT in detail (refer Section 2.2).
Problem formulation
Knowledge graph. We consider a KG that provides heterogeneous information of drug–target and drug–drug pairs, denoted by 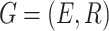 , where E (resp., R) is the set of entities (resp., relations). The KG stores information as triplets that represent interactions between two entities. A triplet can be denoted by
, where E (resp., R) is the set of entities (resp., relations). The KG stores information as triplets that represent interactions between two entities. A triplet can be denoted by 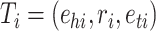 , where
, where 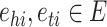 and
and 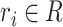 . Problem definition. To facilitate the understanding of our proposed methods, in this study, we chose two representative interaction prediction tasks: DTIs and DDIs prediction. We formulate these tasks as binary classification problems and aim to estimate interaction probabilities
. Problem definition. To facilitate the understanding of our proposed methods, in this study, we chose two representative interaction prediction tasks: DTIs and DDIs prediction. We formulate these tasks as binary classification problems and aim to estimate interaction probabilities 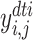 of a drug–target pair
of a drug–target pair 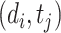 and
and 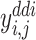 of a drug–drug pair
of a drug–drug pair 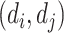 . Given the SMILES sequences
. Given the SMILES sequences 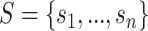 for n drugs, protein sequences
for n drugs, protein sequences 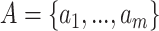 for m proteins and the KG G, the goal is to learn a multi-task prediction function
for m proteins and the KG G, the goal is to learn a multi-task prediction function 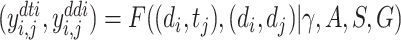 , where
, where 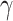 denotes the model parameters.
denotes the model parameters.
Workflow of KGE-UNIT
The workflow of KGE-UNIT (Figure 1) includes three main parts: multi-modal feature extraction through KGE and CNNs, integration and encoding of features with a CNN-based encoder and task-aware feature decoding with a task-aware attention decoder.
Figure 1.

The structure of KGE-UNIT.
KG construction and heterogeneous features extractor
Molecular interaction prediction involves multiple dimensions of information, including the structure, function and interactions of molecules. Building a KG allows for the integration of multi-source data, enabling a better capture of complex associations between molecules. In the study, we employed the integration of heterogeneous data from genomics, proteomics and metabolomics to construct a KG, in which biomedical concepts are represented as nodes and interactions/associations (such as DTIs, DDIs and drug–disease interactions) are represented as edges. For instance, the triple representation <DB15035, DTI, P04626> in the KG illustrates the interaction between drug DB15035 and protein P04626. Therefore, we can obtain the KG triples with plenty of information, including the topological structure and semantic relations.
After constructing the KG, we employed KGE models to learn the topological structures and semantic relation of all entities and relations. In this study, we tried many KGE methods to test the performance of heterogeneous features extractor, and finally, we utilized ConvE [25] as the KGE model of choice. ConvE is a KGE model in which the interactions between input entities and relationships are modeled by convolutional and fully connected layers. And ConvE has consistently proven to be a powerful baseline model on the drug discovery in prior studies [26]. Compared with other KGE models, ConvE exhibits a superior suitability for tasks involving the prediction of molecular interactions. This heightened suitability is chiefly attributed to its use of convolution to enhance feature learning capability.
Structural features extractor
The structural features extractor is designed to extract the structural characteristics of both drug and protein entities from drug SMILES structures and protein sequences. To accomplish this, the drug SMILES structures are transformed into molecule graphs using RDKit [27], which represents atoms as nodes and chemical bonds as edges. Additionally, the protein sequences are transformed into high-dimensional feature vectors through the Composition, Transition and Distribution method [28]. Given the high dimensionality and potential for noise in these feature vectors, we apply Principal Component Analysis to reduce the dimensions while preserving the essential information contained within the relevant entity features. Then, we employ task-aware CNN-based encoders to extract local chemical contexts and molecular structures from the drug and protein structures, respectively. CNN have demonstrated remarkable success and have become popular in a wide range of bioinformatics tasks. Deepconv-dti [29] utilizes CNNs to extract local features of protein sequences for the purpose of DTI prediction. MDeePred [30] employs CNNs to encode target proteins and generate multi-channel fusion features for the prediction of drug–target binding affinity. CNNs extract features through local connections, enabling them to integrate both local and global features, thereby improving the quality of the features in the context of molecular relationship prediction. The forward propagation of convolutional layer l is calculated as follows:
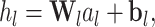 |
(1) |
where 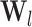 and
and 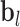 are the weight matrix and bias vector for
are the weight matrix and bias vector for 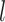 th layer,
th layer, 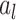 and
and 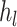 are the input and output of the forward propagation process, respectively.
are the input and output of the forward propagation process, respectively.
Joint multi-task predictor
To unit multiple molecular interactions tasks, we have developed a novel encoder–decoder predictor in KGE-UNIT. The task-aware CNN-based encoder is designed to fuse heterogeneous and structural features. The task-aware attention decoder is composed of task-interacted attention blocks, task-aware attention blocks and task-specific prediction heads as shown in Figure 2. The task-interacted attention block is utilized to capture the interactive impacts between different molecular interactions. And the task-aware attention block is proposed to optimize their representations for individual tasks in the context of DDIs, DTIs and other relevant interactions. Both blocks consist of an Multi-head self attention (MHSA) [31] and a multi-layer perceptron (MLP). MHSA have been proven effective in multi-task learning [32], and we have chosen it as a crucial component of our predictor due to the following advantages; MHSA has the capability to incorporate both local and global information, thereby facilitating the generation of feature representations that are more comprehensive and enriched in content. The independent learning ability of each attention head enables them to attend to various positions in the input sequence, effectively capturing the feature information associated with amino acid residues and atoms present in the target and drug molecules. By conducting scaled dot-product attention computations in parallel, MHSA operates on three key entities, namely the query, key, and value, which are all represented as vectorized forms. MHSA can be calculated as
Figure 2.

The structure of the joint multi-task predictor in KGE-UNIT.
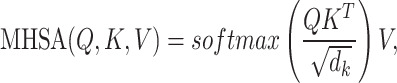 |
(2) |
where Q, K and V are the query, key and value matrices, respectively.
Task-interacted Attention Block
The goal of the task-interacted attention block is to exploit the interacting infomation of different molecular interactions tasks. As shown in Figure 2, we integrated heterogeneous and structural features by the CNN-based encoder for each task as follows:
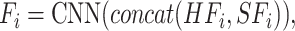 |
(3) |
where 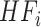 ,
, 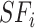 are heterogeneous and structural features of
are heterogeneous and structural features of 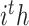 task,
task, 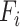 is mixed features. CNN represents the CNN-based encoder, including convolutional layers, batch normalization layers and activation layers. Specifically, we employed the LeakyReLU [33] function as the non-linearity activation. The mixed features
is mixed features. CNN represents the CNN-based encoder, including convolutional layers, batch normalization layers and activation layers. Specifically, we employed the LeakyReLU [33] function as the non-linearity activation. The mixed features 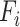 serve as input to the task interaction block. First, we concatenate these features as follows:
serve as input to the task interaction block. First, we concatenate these features as follows:
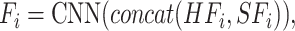 |
(4) |
where N is the number of tasks, concat(*) is the concatenate operator. Then, 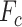 serves as the query, key and value of MHSA in task interaction block as follows:
serves as the query, key and value of MHSA in task interaction block as follows:
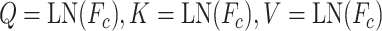 |
(5) |
 |
(6) |
where Q, K and V are query, key and value entities. LN is Layer Normalization [34], MLP is a multi-layer perceptron, which includes a fully connected layer and batch normalization layer.
Task-aware Attention Block
Task-aware attention blocks are used to learn the representation of DDIs and DTIs tasks while considering their interacting contexts. They can comprehend the interactions of tasks and dynamically assign different attention to each specific task. Each task is processed by its corresponding task-aware attention block. These two blocks possess a similar structure but differ in the entities of query, key and value. In task-specific attention blocks, the output of the task-interacted attention block is employed as the key and value, while the output of CNN-based encoder serves as the query as follows:
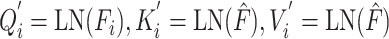 |
(7) |
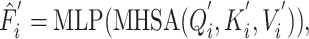 |
(8) |
where 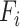 denotes mixed features, which are the outputs of the
denotes mixed features, which are the outputs of the  CNN-based encoder.
CNN-based encoder. 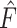 denotes the output of the task-interacted attention block, and
denotes the output of the task-interacted attention block, and 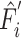 denotes task-interacted features, which are the outputs of the
denotes task-interacted features, which are the outputs of the 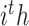 task-aware attention block. Then, we integrated task-interacted features and mixed features to get task-aware features as follows:
task-aware attention block. Then, we integrated task-interacted features and mixed features to get task-aware features as follows:
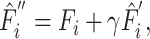 |
(9) |
where 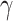 is a hyper parameter.
is a hyper parameter.
Task-specific Prediction Head
After the extraction of task-aware features, we employ task-specific prediction heads to make predictions for molecular interactions, which are treated as binary classifications. Each task is processed by its corresponding task-specific prediction head. A task-specific prediction head consists of fully connected layers, batch normalization layers and activation layers, defined as follows:
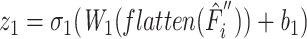 |
(10) |
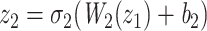 |
(11) |
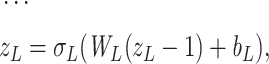 |
(12) |
where L is the number of fully connected layers. And flatten(*) is flatten operator. 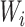 ,
, 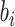 , and
, and 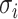 are the weight matrix, bias vector and activation function for
are the weight matrix, bias vector and activation function for 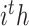 layer. In the study,
layer. In the study, 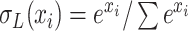 is the softmax function.
is the softmax function. 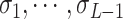 are LeakyReLU function.
are LeakyReLU function.
Multi-task loss optimization
Prediction DTIs and DDIs are all treated as binary classification tasks. Given the DTI and DDI pairs in the training process, our optimization goal is to minimize the cross-entropy loss as follows:
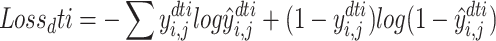 |
(13) |
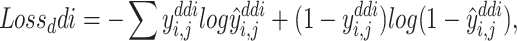 |
(14) |
where 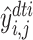 (resp.,
(resp.,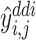 ) is the prediction of DTI pair (
) is the prediction of DTI pair (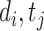 )(resp., DDI pair (
)(resp., DDI pair (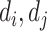 )).
)). 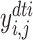 (resp.,
(resp., 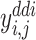 ) is the true label of DTI pair (
) is the true label of DTI pair (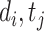 ) (resp., DDI pair (
) (resp., DDI pair (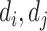 )). The performance of a multi-task model is sensitive to the choice of loss weight in the training process, as it determines the relative importance of each task on the joint loss. To balance loss contribution for multiple tasks, we employ weight
)). The performance of a multi-task model is sensitive to the choice of loss weight in the training process, as it determines the relative importance of each task on the joint loss. To balance loss contribution for multiple tasks, we employ weight 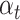 to determine the loss contribution for the task
to determine the loss contribution for the task 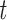 . We generate a weighted sum of task-specific losses,
. We generate a weighted sum of task-specific losses, 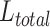 , thereby ensuring the accurate evaluation of each task’s relative importance
, thereby ensuring the accurate evaluation of each task’s relative importance
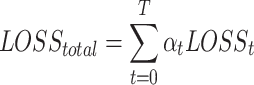 |
(15) |
where 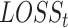 is a loss function for task t.
is a loss function for task t.
For specific parameter settings of KGE-UNIT, please refer to Supplementary Section 1 Parameters settings (see Supplementary Data available online at http://bib.oxfordjournals.org).
Evaluation metrics
In this study, the area under the receiver operating characteristics curve (AUROC) and the area under the precision-recall curve (AUPR) are adopted to evaluate each method’s performance.
RESULTS
Datasets
In the study, we use two benchmark datasets, Luo’s dataset [13] and BioKG [35], to compare KGE-UNIT with other state-of-the-art methods for DDI and DTI predictions. Luo’s dataset contains 12 015 types of nodes and 1 895 445 types of edges, and the BioKG utilized in the study comprises of 105 524 unique nodes and 2 043 846 unique edges. (See Supplementary Section 2 Datasets, see Supplementary Data available online at http://bib.oxfordjournals.org).
The distribution of edge categories in both datasets is somewhat imbalanced. In Luo’s dataset, the number of DDIs is five times greater than that of DTIs, while in BioKG, the number of DDIs is nearly 50 times greater than DTIs. Moreover, the volume of DTI data is significantly insufficient in comparison to that of DDIs.
We consider the known DTIs and DDIs as positive samples and choose non-existing DTIs and DDIs as negative samples to create the experimental dataset. Futhermore, our experiments are divided into two scenarios: the warm start and the cold start for drugs. In the scenario of warm start, we apply 10-fold cross-validation (10-CV) and split all DTIs and DDIs into 10 subsets. Then, we randomly select negative samples to maintain a 1:1 ratio between positive and negative samples. In the scenario of cold start for drugs, we employ 10-CV on drugs. We randomly divide the drugs into 10 subsets and utilize one of them for testing drugs.
Evaluations on DTIs prediction task
We compared our proposed KGE-UNIT with the existing methods: SVM, DNN, TransE [36], DistMult [37], ConvKB [38], RGCN [39], ConvE [25], CrossE [40], RotatE [41], DistMA [42], MuRE [43], AutoSF [44], BoxE [45], PairRE [46], DeepDTI [47] and KGE_NFM [48] (See Supplementary Section 3 Baselines, see Supplementary Data available online at http://bib.oxfordjournals.org) on the DTIs prediction task. In order to eliminate potential accuracy differences caused by different implementation frameworks, we conducted experiments on all KGE models using the PyKEEN [49] framework. As shown in Table 1, KGE-UNIT demonstrates a clear superiority over all other state-of-the-art methods. Specifically, on the Luo’s dataset, KGE-UNIT achieves a significant improvement of at least 4.32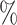 in AUROC score and 3.56
in AUROC score and 3.56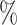 in AUPR score. Similarly, on the BioKG dataset, it achieves a minimum improvement of 1.44
in AUPR score. Similarly, on the BioKG dataset, it achieves a minimum improvement of 1.44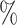 in AUROC and 1.03
in AUROC and 1.03 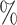 in AUPR. These results indicate that, firstly, all methods perform better on the BioKG dataset compared with the LUO dataset. This is because the BioKG dataset contains richer data information, including drug–target interactions (DTIs) and the heterogeneous information composed of all types of drug-related entities. Secondly, among the 12 tested heterogeneous data-driven KGE models, BoxE and RotatE achieved the best performance, while DistMult and TransE exhibited the poorest performance. This is primarily attributed to the limited capacity of DistMult and TransE in capturing semantic information from the drug KG. Similar conclusions have also been demonstrated in [26, 50]. Furthermore, compared with feature-based methods (e.g. SVM, DNN, DeepDTI) and heterogeneous data driven methods (e.g. BoxE, RotatE), under using the same heterogeneous data-driven methods, methods which integrate multiple types of features (e.g. KGE_NFM, KGE-UNIT) outperform bettaer. Furthermore, in tasks with relatively limited data, KGE-UNIT not only considers the features of the target entities but also extensively explores the potentially useful features from other nodes related to the task. This leads to a substantial improvement in algorithm performance.
in AUPR. These results indicate that, firstly, all methods perform better on the BioKG dataset compared with the LUO dataset. This is because the BioKG dataset contains richer data information, including drug–target interactions (DTIs) and the heterogeneous information composed of all types of drug-related entities. Secondly, among the 12 tested heterogeneous data-driven KGE models, BoxE and RotatE achieved the best performance, while DistMult and TransE exhibited the poorest performance. This is primarily attributed to the limited capacity of DistMult and TransE in capturing semantic information from the drug KG. Similar conclusions have also been demonstrated in [26, 50]. Furthermore, compared with feature-based methods (e.g. SVM, DNN, DeepDTI) and heterogeneous data driven methods (e.g. BoxE, RotatE), under using the same heterogeneous data-driven methods, methods which integrate multiple types of features (e.g. KGE_NFM, KGE-UNIT) outperform bettaer. Furthermore, in tasks with relatively limited data, KGE-UNIT not only considers the features of the target entities but also extensively explores the potentially useful features from other nodes related to the task. This leads to a substantial improvement in algorithm performance.
Table 1.
DTIs Prediction Results on Luo’s Dataset and BioKG Dataset in the scenario of the warm start.
| Luo | BioKG | |||
|---|---|---|---|---|
| Methods | AUROC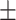 std std |
AUPR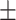 std std |
AUROC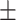 std std |
AUPR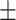 std std |
| SVM | 0.654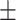 0.037 0.037 |
0.739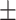 0.045 0.045 |
0.730 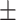 0.041 0.041 |
0.803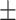 0.021 0.021 |
| DNN | 0.882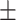 0.031 0.031 |
0.881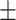 0.026 0.026 |
0.934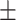 0.018 0.018 |
0.933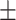 0.014 0.014 |
| TransE | 0.731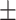 0.042 0.042 |
0.725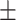 0.033 0.033 |
0.756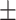 0.011 0.011 |
0.799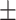 0.018 0.018 |
| DistMult | 0.752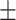 0.025 0.025 |
0.745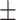 0.038 0.038 |
0.903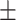 0.049 0.049 |
0.889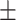 0.031 0.031 |
| ConvKB | 0.823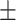 0.010 0.010 |
0.826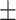 0.036 0.036 |
0.911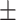 0.029 0.029 |
0.913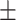 0.015 0.015 |
| RGCN | 0.842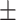 0.032 0.032 |
0.848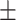 0.045 0.045 |
0.915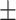 0.017 0.017 |
0.902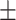 0.026 0.026 |
| ConvE | 0.795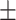 0.033 0.033 |
0.828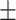 0.041 0.041 |
0.922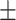 0.032 0.032 |
0.925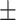 0.029 0.029 |
| CrossE | 0.862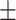 0.015 0.015 |
0.874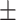 0.027 0.027 |
0.931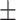 0.041 0.041 |
0.928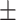 0.039 0.039 |
| RotatE | 0.869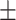 0.025 0.025 |
0.872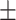 0.013 0.013 |
0.946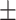 0.019 0.019 |
0.952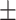 0.032 0.032 |
| DistMA | 0.837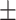 0.028 0.028 |
0.838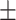 0.019 0.019 |
0.928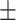 0.025 0.025 |
0.930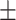 0.041 0.041 |
| MuRE | 0.839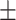 0.028 0.028 |
0.839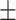 0.053 0.053 |
0.931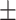 0.052 0.052 |
0.937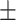 0.015 0.015 |
| AutoSF | 0.733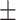 0.040 0.040 |
0.731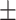 0.058 0.058 |
0.877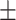 0.034 0.034 |
0.885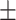 0.049 0.049 |
| BoxE | 0.857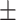 0.054 0.054 |
0.843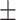 0.030 0.030 |
0.913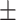 0.047 0.047 |
0.916 0.026 0.026 |
| PairRE | 0.831 0.017 0.017 |
0.828 0.044 0.044 |
0.915 0.027 0.027 |
0.911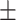 0.062 0.062 |
| DeepDTI | 0.859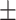 0.039 0.039 |
0.840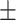 0.017 0.017 |
0.973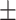 0.024 0.024 |
0.970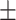 0.036 0.036 |
| KGE_NFM | 0.903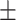 0.026 0.026 |
0.898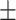 0.019 0.019 |
0.946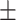 0.016 0.016 |
0.946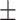 0.028 0.028 |
| KGE-UNIT | 0.942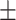 0.025 0.025 |
0.930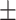 0.034 0.034 |
0.987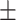 0.035 0.035 |
0.980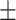 0.042 0.042 |
Evaluations on DDIs prediction task
We compared our proposed KGE-UNIT with the existing methods: SVM, DNN, TransE, DistMult, ConvKB, RGCN, ConvE, CrossE, RotatE, DistMA, MuRE, AutoSF, BoxE, PairRE, DeepDDI [51] and KGNN [18] (See Supplementary Section 3 Baselines, see Supplementary Data available online at http://bib.oxfordjournals.org) on the DDIs prediction task. In the BioKG dataset, there is a total of 1 334 085 DDIs information, accounting for 65.27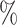 of all the relationship information. However, in the Luo dataset, there are 10 036 DDIs, which represent a mere 0.53
of all the relationship information. However, in the Luo dataset, there are 10 036 DDIs, which represent a mere 0.53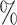 of the total. It represents a substantial disparity in the distribution between the two datasets. From the results of DDIs prediction shown in Table 2, it can be observed that on the Luo’s dataset, KGE-UNIT (AUROC= 0.975, AUPR=0.966) outperforms other methods, exhibiting at least 6.56
of the total. It represents a substantial disparity in the distribution between the two datasets. From the results of DDIs prediction shown in Table 2, it can be observed that on the Luo’s dataset, KGE-UNIT (AUROC= 0.975, AUPR=0.966) outperforms other methods, exhibiting at least 6.56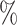 in AUROC and 8.17
in AUROC and 8.17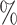 in AUPR. On the BioKG dataset, due to the larger volume of the dataset, all methods perform better, especially the heterogeneous data-driven methods, and KGE-UNIT achieved the best performance. The results indicate that based on the ability to effectively integrate and learn from multiple types of features, KGE-UNIT showcases exceptional performance even in scenarios with limited data availability.
in AUPR. On the BioKG dataset, due to the larger volume of the dataset, all methods perform better, especially the heterogeneous data-driven methods, and KGE-UNIT achieved the best performance. The results indicate that based on the ability to effectively integrate and learn from multiple types of features, KGE-UNIT showcases exceptional performance even in scenarios with limited data availability.
Table 2.
DDIs Prediction Results on Luo’s Dataset and BioKG Dataset in the scenario of the warm start.
| Luo | BioKG | |||
|---|---|---|---|---|
| Methods | AUROC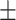 std std |
AUPR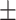 std std |
AUROC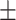 std std |
AUPR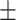 std std |
| SVM | 0.700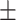 0.018 0.018 |
0.774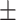 0.015 0.015 |
0.761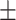 0.041 0.041 |
0.785 0.044 0.044 |
| DNN | 0.902 0.022 0.022 |
0.889 0.035 0.035 |
0.967 0.034 0.034 |
0.964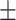 0.020 0.020 |
| TransE | 0.725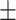 0.016 0.016 |
0.715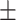 0.032 0.032 |
0.915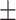 0.038 0.038 |
0.906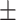 0.032 0.032 |
| DistMult | 0.741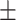 0.016 0.016 |
0.736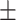 0.019 0.019 |
0.908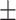 0.053 0.053 |
0.910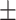 0.068 0.068 |
| ConvKB | 0.866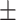 0.023 0.023 |
0.853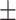 0.034 0.034 |
0.947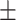 0.018 0.018 |
0.944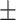 0.017 0.017 |
| RGCN | 0.853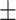 0.013 0.013 |
0.848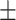 0.020 0.020 |
0.938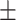 0.038 0.038 |
0.943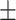 0.054 0.054 |
| ConvE | 0.822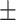 0.035 0.035 |
0.831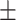 0.019 0.019 |
0.929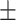 0.033 0.033 |
0.928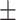 0.068 0.068 |
| CrossE | 0.894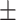 0.016 0.016 |
0.892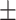 0.015 0.015 |
0.948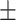 0.031 0.031 |
0.945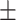 0.044 0.044 |
| RotatE | 0.885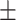 0.018 0.018 |
0.879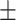 0.021 0.021 |
0.943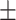 0.042 0.042 |
0.947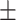 0.016 0.016 |
| DistMA | 0.842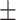 0.031 0.031 |
0.823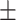 0.036 0.036 |
0.936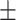 0.031 0.031 |
0.941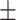 0.036 0.036 |
| MuRE | 0.848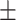 0.017 0.017 |
0.836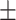 0.033 0.033 |
0.946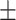 0.038 0.038 |
0.942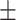 0.064 0.064 |
| AutoSF | 0.755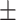 0.015 0.015 |
0.762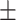 0.028 0.028 |
0.929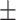 0.033 0.033 |
0.925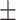 0.027 0.027 |
| BoxE | 0.902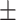 0.017 0.017 |
0.898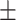 0.053 0.053 |
0.945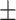 0.027 0.027 |
0.936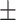 0.022 0.022 |
| PairRE | 0.879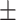 0.018 0.018 |
0.863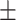 0.036 0.036 |
0.932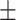 0.029 0.029 |
0.921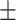 0.051 0.051 |
| DeepDDI | 0.915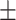 0.030 0.030 |
0.893 0.024 0.024 |
0.972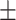 0.025 0.025 |
0.968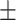 0.047 0.047 |
| KGNN | 0.812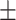 0.040 0.040 |
0.769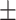 0.023 0.023 |
0.947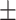 0.038 0.038 |
0.944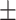 0.024 0.024 |
| KGE-UNIT | 0.975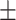 0.024 0.024 |
0.966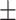 0.032 0.032 |
0.989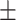 0.027 0.027 |
0.988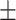 0.035 0.035 |
Influence of KGE methods on KGE-UNIT
To further illustrate the impact of different heterogeneous feature extraction methods on KGE-UNIT, we conducted experiments by varying the KGE methods within the KGE-UNIT framework. The results of DTI and DDI predictions on Luo’s dataset are shown in Figure 3 (more in Supplementary Table 4, see Supplementary Data available online at http://bib.oxfordjournals.org), respectively. From the results, it can be observed that compared with using KGE methods individually, KGE-UNIT consistently exhibits improved performance, with the most significant improvement observed for DistMult. Specifically, ConvE-UNIT achieves the highest performance, surpassing the second-ranked DistMult-UNIT. Notably, the results of DistMult-UNIT and ConvE-UNIT are similar. Compared with the third-ranked RotatE-UNIT, in DTI tasks, ConvE-UNIT achieves an improvement of 0.85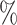 in AUROC and 0.97
in AUROC and 0.97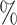 in AUPR, while in DDI tasks, it achieves an improvement of 3.39
in AUPR, while in DDI tasks, it achieves an improvement of 3.39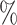 in AUROC and 4.78
in AUROC and 4.78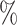 in AUPR. This suggests that the performance of KGE-UNIT is not solely correlated with the performance of KGE methods. Other factors to consider include the integration of multi-modal features and the capability of extracting features of the joint multi-task predictor in KGE-UNIT.
in AUPR. This suggests that the performance of KGE-UNIT is not solely correlated with the performance of KGE methods. Other factors to consider include the integration of multi-modal features and the capability of extracting features of the joint multi-task predictor in KGE-UNIT.
Figure 3.
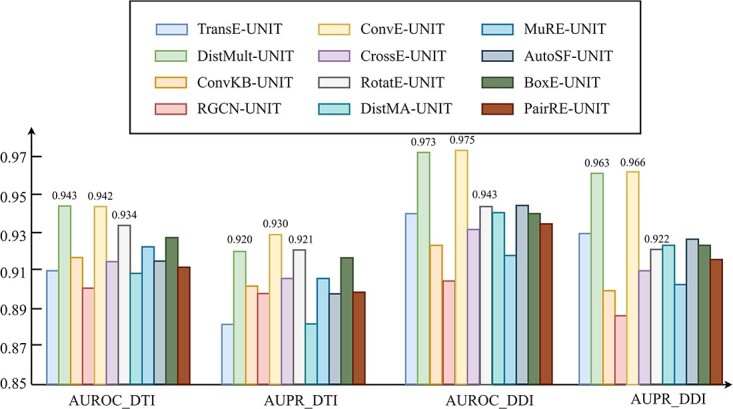
Influence of KGE methods on KGE-UNIT on DTIs and DDIs Prediction.
Evaluations in the scenario of the cold start for drugs
Then, we discussed the performance of KGE-UNIT in the scenario of the cold start drugs. As described in 3.1 Datasets, we applied 10-cv on drugs, randomly divided the drugs into 10 subsets. The results on Luo’s dataset are shown in Table 3. In the cold start scenario for drugs, we observed that KGE-UNIT performed best in both DTIs and DDIs prediction tasks. For the DTI task, it achieved an AUROC of 0.956 and an AUPR of 0.947. In the DDI task, it attained an AUROC of 0.935 and an AUPR of 0.939. And in the cold start scenario, the performance reduction of the KGE models is more significant. Overall, methods that incorporate multi-modal features, such as KGE-UNIT and KGE_NFM, demonstrate better predictive performance for cold start drug scenarios.
Table 3.
DTIs and DDIs Prediction Results on Luo’s Dataset in the scenario of the cold start for drugs.
| DTIs | DDIs | |||
|---|---|---|---|---|
| Methods | AUROC std std |
AUPR std std |
AUROC std std |
AUPR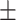 std std |
| TransE | 0.640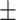 0.062 0.062 |
0.635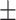 0.057 0.057 |
0.603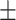 0.072 0.072 |
0.599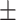 0.088 0.088 |
| DistMult | 0.617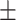 0.069 0.069 |
0.623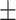 0.043 0.043 |
0.621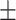 0.057 0.057 |
0.622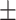 0.082 0.082 |
| ConvKB | 0.690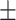 0.075 0.075 |
0.701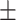 0.051 0.051 |
0.611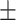 0.064 0.064 |
0.620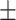 0.059 0.059 |
| RGCN | 0.682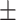 0.072 0.072 |
0.705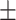 0.049 0.049 |
0.616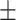 0.072 0.072 |
0.626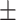 0.051 0.051 |
| ConvE | 0.692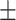 0.061 0.061 |
0.687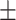 0.046 0.046 |
0.621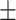 0.039 0.039 |
0.639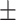 0.051 0.051 |
| CrossE | 0.749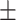 0.053 0.053 |
0.762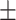 0.049 0.049 |
0.635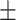 0.074 0.074 |
0.639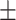 0.069 0.069 |
| RotatE | 0.721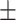 0.064 0.064 |
0.727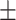 0.071 0.071 |
0.661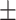 0.085 0.085 |
0.642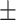 0.077 0.077 |
| DistMA | 0.655 0.039 0.039 |
0.659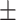 0.058 0.058 |
0.592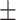 0.074 0.074 |
0.597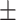 0.088 0.088 |
| MuRE | 0.723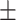 0.058 0.058 |
0.736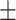 0.041 0.041 |
0.650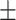 0.075 0.075 |
0.646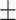 0.089 0.089 |
| AutoSF | 0.637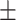 0.057 0.057 |
0.651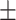 0.042 0.042 |
0.593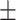 0.089 0.089 |
0.600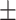 0.073 0.073 |
| BoxE | 0.742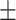 0.049 0.049 |
0.763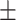 0.057 0.057 |
0.603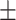 0.062 0.062 |
0.611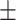 0.059 0.059 |
| PairRE | 0.717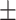 0.043 0.043 |
0.727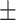 0.038 0.038 |
0.652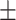 0.041 0.041 |
0.669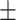 0.074 0.074 |
| DeepDTI/DeepDDI | 0.793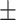 0.068 0.068 |
0.825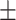 0.053 0.053 |
0.825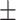 0.081 0.081 |
0.829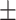 0.077 0.077 |
| KGE_NFM/KGNN | 0.903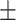 0.041 0.041 |
0.909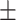 0.033 0.033 |
0.713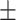 0.086 0.086 |
0.701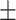 0.098 0.098 |
| KGE-UNIT | 0.956 0.055 0.055 |
0.947 0.066 0.066 |
0.935 0.127 0.127 |
0.939 0.112 0.112 |
Ablation study
To explore how the heterogeneous features, structural features and multi-task joint structure improve the performance of KGE-UNIT, we conduct the ablation study on the following variants:
KGE-UNIT without structural features and joint multi-task predictor (w/o SF +MP) is the variant of KGE-UNIT where we only used ConvE to predict DTIs and DDIs, respectively. It should be noted that the result is slightly different from Tables 1 and 2, because the result of ConvE on Table 1 (resp., Table 2) is got by training all DTI (resp., DDI) samples.
KGE-UNIT without structural features (w/o SF) is the variant of KGE-UNIT where we only applied the heterogeneous features of drug–drug pairs and drug–target pairs learnt from the KG as input of the predictor.
KGE-UNIT without heterogeneous features (w/o HF) is the variant of KGE-UNIT where we only used the structure features learnt from drug and protein structure as input of the predictor.
KGE-UNIT without DTI task (w/o DTI) is the variant of KGE-UNIT that is designed as a single-task method specifically focused on the DDI task.
KGE-UNIT without DDI task (w/o DDI) is the variant of KGE-UNIT that is designed as a single-task method specifically focused on the DTI task.
The ablation study results on Luo Dataset are shown in Figure 4. It is evident that the KGE-UNIT, which incorporates all modules, achieves the best performance. And both heterogeneous features and structural features contribute to the prediction of molecular interactions, with each providing certain benefits. However, compared with heterogeneous features, structural features have a more significant impact. Furthermore, compared with the DDIs task, in the DTIs task, using multi-task learning has a more significant performance improvement than single task learning. To further elucidate the role of each module, we extracted the learned representations of each module from the trained KGE-UNIT model and projected them into a two-dimensional space using t-SNE [52]. We present the visualization results on the Luo’s test set in Figure 5. It can be observed that KGE methods, despite being trained, exhibit limited capability in discriminating molecular pairs. In contrast, KGE-UNIT effectively distinguishes interacting and non-interacting pairs by integrating multiple features through the CNN-based encoder and the task-aware attention decoder, further enhancing the discriminative ability of KGE-UNIT.
Figure 4.

Results of ablation study.
Figure 5.

The learned representations of each module from KGE-UNIT on the Luo’s test set.
Scalability discussion
To further demonstrate the effectiveness and scalability of KGE-UNIT, we extended the prediction module to support DTIs, DDIs and PPIs prediction tasks. The KGE-UNIT structure is shown in Figure 6. The corresponding results on Luo’s dataset are presented in Table 4. The results prove that based on the rich multiple types of features and the fusion of CNN and MHSA in the predictor, KGE-UNIT maintains good performance even when extended to more tasks.
Figure 6.

The structure of joint multi-task predictor for DTI, DDI and PPI prediction tasks.
Table 4.
The results of KGE-UNIT for DTI, DDI and PPI prediction tasks.
| Tasks | AUROC std std |
AUPR std std |
|---|---|---|
| DTI | 0.903 0.027 0.027 |
0.916 0.015 0.015 |
| DDI | 0.935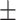 0.036 0.036 |
0.941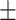 0.032 0.032 |
| PPI | 0.892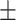 0.019 0.019 |
0.887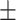 0.013 0.013 |
Case study
Next, we conducted case studies using KGE-UNIT to validate its effectiveness. We identified several DDIs and DTIs that are not present in the DrugBank database [53], and we obtained some supporting evidence from other studies.
Drug–target interactions
(i) Mitochondrial acyl-CoA dehydrogenase 9 (ACAD9) deficiency was demonstrated to improve with the use of coenzyme Q10 [54].
(ii) Acetaminophen was found that had the potential to upregulate the expression of myeloperoxidase [55].
(iii) The expression of ABCB1 was found to reduce the cellular accumulation of rivaroxaban, thereby confirming the role of ABCB1 in the active efflux of rivaroxaban [56].
Drug–drug interactions
(i) Zelavespib (PU-H71) was shown to synergize with bortezomib and significantly inhibits the growth of Ewing sarcoma [57].
(ii) Rivaroxaban may potentially cause bleeding in patients due to gastroduodenal ulcers [58], while Dihydroxyaluminum sodium carbonate is commonly used to treat conditions such as peptic ulcers.
(iii) Garozzo et al. [59] found that N-acetytcysteine (NAC) and oseltamivir significantly improve the therapeutic efficacy against influenza virus.
The above case studies illustrate the ability of KGE-UNIT to identify new DTIs and DDIs. Therefore, KGE-UNIT has a positive impact on the design and development process of new drugs.
DISCUSSION
The prediction of molecular interactions, such as DTIs and DDIs prediction, plays a crucial role in various aspects of drug discovery. While several methodologies have been proposed to uncover molecular interactions within specific domains, most existing methods tend to focus on individual prediction tasks with limited consideration for diverse task features. Simultaneously, many tasks face bottlenecks due to the impact of data scarcity.
In the study, we propose KGE-UNIT, a novel method that combining both merits of KGE and multi-task learning for multiple types of molecular interactions prediction. By leveraging superior multimodal features and employing a predictor that effectively mines task interaction information and emphasizes task-specific features, this method enhances the performance of individual tasks.
In conclusion, the results of two public available datasets with varying distributions of molecular relationships demonstrate that KGE-UNIT outperforms both state-of-the-art DTIs and DDIs prediction algorithms. Moreover, the framework of KGE-UNIT is easily extensible to solving more molecular interaction prediction problems simultaneously. Furthermore, the results obtained by KGE-UNIT in discovering unknown DTIs and DDIs are supported by existing literature. This not only demonstrates the reliability of our findings but also highlights the effectiveness of KGE-UNIT in identifying real-world drug interactions.
In summary, KGE-UNIT offers improved performance and scalability compared with existing methods. This research opens up possibilities for further advancements in multi-task learning and the application of unified frameworks in the field of molecular interaction prediction. In the future, we will take more attention on further improvements of the prediction ability of this framework and explore the scalability on other downstream tasks.
We summarized the limitations and future improvements of KGE-UNIT. First, our KGE-UNIT framework has not yet considered the incorporation of 3D structures of drugs and proteins. Models based on 3D structures should ideally be trained on highly reliable datasets containing measured ligand-receptor affinities and co-crystal structures of ligands and proteins. However, these datasets are relatively scarce due to their high costs and the need for experimental validation of structural information. Therefore, one direction that requires consideration that how to fully leverage the advantages of 3D structural features when dealing with limited data volume. Second, in the section Ablation Study, we have discussed the importance of structural features within the KGE-UNIT framework. Nevertheless, it is worth noting that not all nodes related to drugs can offer structural features, especially for those newly discovered. The absence of structural features can impact the performance of KGE-UNIT, and this limitation is inherent to the approach. Therefore, one direction that requires consideration that how to integrate more multimodality features and more kinds of associations expand heterogeneous data to enhance feature quality. Last, a common challenge in KGE is the necessity to retrain the model when introducing new entities or relations, which consumes substantial time and computational resources. This challenge also applies to our model. Therefore, one direction that requires consideration that how to learn feature representations by pretrained models and reduce training process.
Overall, there are still some related works to be done in future. (i) Using pre-trained models and transfer learning to optimize the feature extraction process and quality. (ii) Integrating various forms of structural features to enhance stability, such as sequence strings, molecular graphs and 3D structures. And constructing a larger-scale KG to improve the quality of heterogeneous information. (iii) Further optimizing the KGE-UNIT framework and apply it to a broader range of multi-task drug relationship predictions.
Key Points
We present a unified framework, which combines KG embedding and multi-task learning, named KGE-UNIT, for joint prediction of DTIs and DDIs. KGE-UNIT enables simultaneous prediction of multiple types of molecular interactions and enhances the performance of each task, even when data availability is limited.
Through KGE, KGE-UNIT can extract heterogeneous features from the drug KG to enhance the structural features of drug and protein nodes, which ultimately leads to an improvement in the quality of the features.
Based on multi-task learning, in KGE-UNIT, a novel and effective encoder–decoder predictor (i.e. task-aware CNN-based encoder and task-aware attention decoder) is proposed to fuse better multimodal features, capture the contextual interactions of molecular tasks and enhance task awareness, leading to improved performance of all tasks.
Supplementary Material
Author Biographies
Chengcheng Zhang is a PHD student in Department of Computer Science at Harbin Institute of Technology, China. Her expertise is bioinformatics.
Tianyi Zang is a professor in the School of Computer Science and Technology at Harbin Institute of Technology (HIT), China. Before joining HIT in 2009, he was a research fellow at the Department of Computer Science at University of Oxford, UK. His current research is concerned with biomedical bigdata computing and algorithms, deep-learning algorithms for network data, intelligent recommendation algorithms, and modeling and analysis methods for complex systems.
Tianyi Zhao is a professor in school of Medicine and Heath at Harbin Institute of Technology and Harbin Institute of Technology Zhengzhou Research Institute. His expertise is bioinformatics and machine learning.
Contributor Information
Chengcheng Zhang, Department of Computer Science, Harbin Institute of Technology, Harbin, 150001, China.
Tianyi Zang, Department of Computer Science, Harbin Institute of Technology, Harbin, 150001, China.
Tianyi Zhao, School of Medicine and Health, Harbin Institute of Technology, Harbin, 150001, China.
FUNDING
National Natural Science Foundation of China (62076082, 62102116); National Key Research and Development Project (2016YFC0901605); Interdisciplinary Research Foundation of HIT.
DATA AVAILABILITY
The source data and code are available at https://github.com/zcc1203/KGE-UNIT.
References
- 1. Mohs RC, Greig NH. Drug discovery and development: role of basic biological research. Alzheimer’s Dement: Transl Res Clin Interv 2017;3(4):651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kastrin A, Ferk P, Leskošek B. Predicting potential drug-drug interactions on topological and semantic similarity features using statistical learning. PLoS One 2018;13(5):e0196865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi J-Y, Gao K, Shang X-Q, Yiu S-M. LCM-DS: a novel approach of predicting drug-drug interactions for new drugs via Dempster-Shafer theory of evidence. In 2016 IEEE international conference on bioinformatics and biomedicine (BIBM). Shenzhen, China: IEEE, 2016.
- 4. Liu Y, Min W, Miao C, et al. Neighborhood regularized logistic matrix factorization for drug-target interaction prediction. PLoS Comput Biol 2016;12(2):e1004760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hui Y, Mao K-T, Shi J-Y, et al. Predicting and understanding comprehensive drug-drug interactions via semi-nonnegative matrix factorization. BMC Syst Biol 2018;12(1):101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang W, Chen Y, Li D, Yue X. Manifold regularized matrix factorization for drug-drug interaction prediction. J Biomed Inform 2018;88:90–7. [DOI] [PubMed] [Google Scholar]
- 7. Zhao T, Yang H, Valsdottir LR, et al. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief Bioinform 2021;22(2):2141–50. [DOI] [PubMed] [Google Scholar]
- 8. Yi H-C, You Z-H, Huang D-S, Kwoh CK. Graph representation learning in bioinformatics: trends, methods and applications. Brief Bioinform 2022;23(1):bbab340. [DOI] [PubMed] [Google Scholar]
- 9. Yue X, Wang Z, Huang J, et al. Graph embedding on biomedical networks: methods, applications and evaluations. Bioinformatics 2020;36(4):1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng J, Wang Y, Guan J, et al. An end-to-end heterogeneous graph representation learning-based framework for drug–target interaction prediction. Brief Bioinform 2021;22. [DOI] [PubMed] [Google Scholar]
- 11. Shao K, Zhang Y, Wen Y, et al. Dti-heta: prediction of drug–target interactions based on gcn and gat on heterogeneous graph. Brief Bioinform 2022;23(3):bbac109. [DOI] [PubMed] [Google Scholar]
- 12. Cheng Z, Zhao Q, Li Y, Wang J. Iifdti: predicting drug–target interactions through interactive and independent features based on attention mechanism. Bioinformatics 2022;38(17):4153–61. [DOI] [PubMed] [Google Scholar]
- 13. Luo Y, Zhao X, Zhou J, et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun 2017;8(1):573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wan F, Hong L, Xiao A, et al. Neodti: neural integration of neighbor information from a heterogeneous network for discovering new drug–target interactions. Bioinformatics 2019;35(1): 104–11. [DOI] [PubMed] [Google Scholar]
- 15. Lin S, Wang Y, Zhang L, et al. Mdf-sa-ddi: predicting drug–drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism. Brief Bioinform 2022;23(1):bbab421. [DOI] [PubMed] [Google Scholar]
- 16. Zeng X, Xinqi T, Liu Y, et al. Toward better drug discovery with knowledge graph. Curr Opin Struct Biol 2022;72:114–26. [DOI] [PubMed] [Google Scholar]
- 17. Mohamed SK, Nováček V, Nounu A. Discovering protein drug targets using knowledge graph embeddings. Bioinformatics 2020;36(2):603–10. [DOI] [PubMed] [Google Scholar]
- 18. Lin X, et al. KGNN: Knowledge Graph Neural Network for Drug-Drug Interaction Prediction. in IJCAI. Yokohama, Japan, 2020. [Google Scholar]
- 19. Chen S, Li T, Yang L, et al. Artificial intelligence-driven prediction of multiple drug interactions. Brief Bioinform 2022;23(6):bbac427. [DOI] [PubMed] [Google Scholar]
- 20. Liu K, Sun X, Jia L, et al. Chemi-net: a molecular graph convolutional network for accurate drug property prediction. Int J Mol Sci 2019;20(14):3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li S, Wan F, Shu H, et al. Monn: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Systems 2020;10(4):308–322.e11. [Google Scholar]
- 22. Zhang F, Zhao B, Shi W, et al. Deepdisobind: accurate prediction of rna-, dna-and protein-binding intrinsically disordered residues with deep multi-task learning. Brief Bioinform 2022;23(1):bbab521. [DOI] [PubMed] [Google Scholar]
- 23. Ma T, Lin X, Bosheng Song SY, Philip, and Xiangxiang Zeng. Kg-mtl: knowledge graph enhanced multi-task learning for molecular interaction. IEEE Trans Knowl Data Eng 2022;1–12. [Google Scholar]
- 24. Yuting X, Ma J, Liaw A, et al. Demystifying multitask deep neural networks for quantitative structure–activity relationships. J Chem Inf Model 2017;57(10):2490–504. [DOI] [PubMed] [Google Scholar]
- 25. Dettmers T, Minervini P, Stenetorp P, Riedel S. Convolutional 2d knowledge graph embeddings. in Proceedings of the AAAI conference on artificial intelligence. New Orleans, Louisiana, USA, 2018.
- 26. Rivas-Barragan D, Domingo-Fernández D, Gadiya Y, Healey D. Ensembles of knowledge graph embedding models improve predictions for drug discovery. Brief Bioinform 2022;23(6):bbac481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landrum G. Rdkit: open-source cheminformatics software. 2016;149(150):650. URL https://github.com/rdkit/rdkit. [Google Scholar]
- 28. Dubchak I, Muchnik I, Holbrook SR, Kim S-H. Prediction of protein folding class using global description of amino acid sequence. Proc Natl Acad Sci U S A 1995;92(19):8700–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee I, Keum J, Nam H. Deepconv-dti: prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol 2019;15(6):e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rifaioglu AS, Atalay RC, Cansen Kahraman D, et al. Mdeepred: novel multi-channel protein featurization for deep learning-based binding affinity prediction in drug discovery. Bioinformatics 2021;37(5):693–704. [DOI] [PubMed] [Google Scholar]
- 31. Vaswani A, et al. Attention is All you Need. in Neural Information Processing Systems. Long Beach, California, USA: Curran Associates, Inc., 2017. [Google Scholar]
- 32. Xu Y, Yang Y, Zhang L. DeMT: Deformable mixer transformer for multi-task learning of dense prediction. In Proceedings of the AAAI conference on artificial intelligence. Washington DC, USA: AAAI Press, 2023.
- 33. Maas AL, Hannun AY, Ng AY. Rectifier nonlinearities improve neural network acoustic models. in Proc icml. Atlanta, GA, USA: Curran Associates, Inc., 2013.
- 34. Ba JL, Kiros JR, Hinton GE. Layer normalization. Stat 2016;1050:21. [Google Scholar]
- 35. Walsh B, Mohamed SK, Nováček V. Biokg: A knowledge graph for relational learning on biological data. In Proceedings of the 29th ACM International Conference on Information & Knowledge Management. New York, NY, USA: Association for Computing Machinery, 2020.
- 36. Bordes A, Usunier N, Garcia-Duran A, et al. Translating embeddings for modeling multi-relational data. Adv Neural Inf Process 2013;26:2787–95. [Google Scholar]
- 37. Yang B, et al. Embedding Entities and Relations for Learning and Inference in Knowledge Bases. In Proceedings of the International Conference on Learning Representations (ICLR) 2015. San Diego, CA, USA, 2015. [Google Scholar]
- 38. Dai Quoc Nguyen TDN, Nguyen DQ, Phung D. A novel embedding model for knowledge base completion based on convolutional neural network. In Proceedings of NAACL-HLT. 2018, 327–33. [Google Scholar]
- 39. Schlichtkrull M, Kipf TN, Bloem P, et al. Modeling relational data with graph convolutional networks. In: The Semantic Web: 15th International Conference, ESWC 2018, Heraklion, Crete, Greece, June 3–7, 2018, Proceedings 15, pages 593–607. Springer, 2018. [Google Scholar]
- 40. Zhang W, et al. Interaction embeddings for prediction and explanation in knowledge graphs. In Proceedings of the twelfth ACM international conference on web search and data mining. Melbourne, VIC, Australia: Association for Computing Machinery, 2019.
- 41. Sun Z, Deng Z-H, Nie J-Y, Tang J. RotatE: Knowledge graph embedding by relational rotation in complex space. In International Conference on Learning Representations. New Orleans, LA, USA, 2018. [Google Scholar]
- 42. Shi X, Xiao Y. Modeling multi-mapping relations for precise cross-lingual entity alignment. In Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP). Hong Kong, China: Association for Computational Linguistics, 2019.
- 43. Balazevic I, Allen C, Hospedales T. Multi-relational poincaré graph embeddings. In: Wallach H, et al. (eds.) Advances in Neural Information Processing Systems. Vancouver, BC, Canada: NeurIPS, 2019. [Google Scholar]
- 44. Zhang Y, Yao Q, Dai W, Chen L. AutoSF: Searching scoring functions for knowledge graph embedding. In 2020 IEEE 36th International Conference on Data Engineering (ICDE). Dallas, Texas USA: IEEE, 2020.
- 45. Abboud R, Ceylan I, Lukasiewicz T, Salvatori T. Boxe: a box embedding model for knowledge base completion. Advances in Neural Information Processing Systems 2020;33:9649–61. [Google Scholar]
- 46. Chao L, He J, Wang T, Chu W. PairRE: Knowledge Graph Embeddings via Paired Relation Vectors. In Proceedings of the 59th Annual Meeting of the Association for Computational Linguistics and the 11th International Joint Conference on Natural Language Processing (Volume 1: Long Papers). Online: Association for Computational Linguistics, 2021. [Google Scholar]
- 47. Wen M, Zhang Z, Niu S, et al. Deep-learning-based drug–target interaction prediction. J Proteome Res 2017;16(4):1401–9. [DOI] [PubMed] [Google Scholar]
- 48. Ye Q, Hsieh C-Y, Ziyi Yang Y, et al. A unified drug–target interaction prediction framework based on knowledge graph and recommendation system. Nat Commun 2021;12(1):6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ali M, Berrendorf M, Hoyt CT, et al. Pykeen 1.0: a python library for training and evaluating knowledge graph embeddings. J Mach Learn Res 2021;22(1):3723–8. [Google Scholar]
- 50. Bonner S, Barrett IP, Ye C, et al. Understanding the performance of knowledge graph embeddings in drug discovery. Artif Intell Life Sci 2022;2:100036. [Google Scholar]
- 51. Ryu JY, Kim HU, Lee SY. Deep learning improves prediction of drug–drug and drug–food interactions. Proc Natl Acad Sci 2018;115(18):E4304–E4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van der Maaten L, Hinton G. Visualizing data using t-sne. J Mach Learn Res 2008;9(11):2579–2605. [Google Scholar]
- 53. Wishart DS, Feunang YD, Guo AC, et al. Drugbank 5.0: a major update to the drugbank database for 2018. Nucleic Acids Res 2018;46(D1):D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kadoya T, Sakakibara A, Kitayama K, et al. Successful treatment of infantile-onset acad9-related cardiomyopathy with a combination of sodium pyruvate, beta-blocker, and coenzyme q10. J Pediatr Endocrinol Metab 2019;32(10):1181–5. [DOI] [PubMed] [Google Scholar]
- 55. Zheng Z, Sheng Y, Bing L, Ji L. The therapeutic detoxification of chlorogenic acid against acetaminophen-induced liver injury by ameliorating hepatic inflammation. Chem Biol Interact 2015;238:93–101. [DOI] [PubMed] [Google Scholar]
- 56. Sennesael A-L, Panin N, Vancraeynest C, et al. Effect of abcb1 genetic polymorphisms on the transport of rivaroxaban in hek293 recombinant cell lines. Sci Rep 2018;8(1):10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ambati SR, Lopes EC, Kosugi K, et al. Pre-clinical efficacy of pu-h71, a novel hsp90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol Oncol 2014;8(2):323–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Samuel ZG. Thromboembolism prophylaxis for patients discharged from the hospital: easier said than done. Journal of the American College of Cardiology 2020;75:3148–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garozzo A, Tempera G, Ungheri D, et al. N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int J Immunopathol Pharmacol 2007;20(2):349–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data and code are available at https://github.com/zcc1203/KGE-UNIT.