

Abstract
Alpha-diversity indices are an essential tool for describing and comparing biodiversity. Microbial ecologists apply indices originally intended for, or adopted by, macroecology to address questions relating to taxonomy (conserved marker) and function (metagenome-based data). In this Perspective piece, I begin by discussing the nature and mathematical quirks important for interpreting routinely employed alpha-diversity indices. Secondly, I propose a metagenomic alpha-diversity index (MD) that measures the (dis)similarity of protein-encoding genes within a community. MD has defined limits, whereby a community comprised mostly of similar, poorly diverse protein-encoding genes pulls the index to the lower limit, while a community rich in divergent homologs and unique genes drives it toward the upper limit. With data acquired from an in silico and three in situ metagenome studies, I derive MD and typical alpha-diversity indices applied to taxonomic (ribosomal rRNA) and functional (all protein-encoding) genes, and discuss their relationships with each other. Not all alpha-diversity indices detect biological trends, and taxonomic does not necessarily follow functional biodiversity. Throughout, I explain that protein Richness and MD provide complementary and easily interpreted information, while probability-based indices do not. Finally, considerations regarding the unique nature of microbial metagenomic data and its relevance for describing functional biodiversity are discussed.
Keywords: biodiversity ecosystem function, microbial ecology, statistical ecology, Theoretical ecology
A proposed metagenomic diversity index provides simple and easily intrepretable information to link microbial functional potential and ecosystem processes.
Introduction
As microbial ecologists, we are interested in how microorganisms shape the world around us. No taxon single-handedly drives any given biochemical process in isolation, and so when we wish to understand how a process functions, we must consider taxa at the scale of the communities they exist in. Ultimately, we seek to explain how each respective taxon within a community contributes toward (or hinders) a given process of interest, succinctly termed as the biodiversity-ecosystem function (BEF) relationship (Manning et al. 2018). Typically, successful enquiries consider three avenues of investigation in combination: (i) the biodiversity of a community (alpha-diversity); (ii) the composition of that community (beta-diversity); and (iii) what makes them differ (determined via differential abundances, biochemical analyses etc). For example, we may be interested in how antibiotics can inadvertently disrupt the typical function of the gut microbiome. After seven days of clindamycin application, many commensal Bacteroidota within an individual's gut are driven to extinction (decreased alpha-diversity), a small number of Bacteroidota taxa fill the newly unoccupied niches (shifted beta-diversity), because they are antibiotic-resistant (increased abundance of antibiotic-resistance genes) (Jernberg et al. 2007). Thus, we can describe the impact of antibiotics on the gut microbiome, understand the consequences of local extinction (i.e. severely reduced alpha-diversity), and even make some predictions. For example, we could expect that the long-term persistence (over two years) of relatively poorly diverse communities that host elevated antibiotic-resistance genes may increase the risk of resistant pathogens becoming established (Macfarlane 2014).
This Perspective piece will focus on the first avenue of investigation, i.e. alpha-diversity, specifically within the context of deriving measures of functional biodiversity from metagenomic data. Issues regarding the application of historical alpha-diversity indices from macroecology will be discussed. Finally, a simple index to derive the biodiversity of a set of protein-encoding genes is proposed and its application demonstrated. The examples here show how alpha-diversity indices are an essential tool for explaining how communities respond to environmental stressors, changing conditions or develop over time. The value of these indices goes beyond acting as explanatory tools, though, and ultimately have the potential to act as quantitative predictors of (un)desirable ecosystem functions (Petchey and Gaston 2006), e.g. an x increase in rhizosphere functional biodiversity is associated with a y increase in plant growth.
An abridged discussion of alpha-diversity indices
Most alpha-diversity indices are univariate metrics that measure specific qualities of the ranked Species Abundance Distribution (SAD), which represents one of the fundamental means by which ecologists consider a community (McGill et al. 2007). Each index has its own nuances and quirks that must be taken into consideration when comparing between communities.
The simplest and most intuitive is the Richness of taxa present (TR), which represents the sum of unique taxa within a community. It is expressed as the length of the SAD. This metric is unbounded and can, theoretically, vary from one to an infinite number of taxa. However, TR is the metric most sensitive to sampling depth (i.e. total number of observations per sample) and sampling scale (Gotelli and Colwell 2001). Care must therefore be taken when defining what is meant by a ‘community’ in the context of the experiment, e.g. the community of Prokaryotes living within a 2 mg soil aggregate versus all Prokaryotes present within 500 mg of a composite soil (Szoboszlay and Tebbe 2021). Clearly TR will be higher in the latter. In the same fashion, it makes little sense to compare TR between environmental samples of similar properties where sampling depth differs greatly, e.g. a sample with 103 amplicon sequences versus a sample with 105.
Evenness is a measure of equality between taxon abundances, and is reflective of the slope of the SAD. Take, for example, Simpson's Diversity index as a measure of Evenness, as:
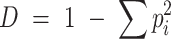 |
Where pi is the proportional abundance of the ith taxon. (Please note that this is a simplified expression of Simpson's original index (Simpson 1949)). Specifically, D asks ‘If I choose two individuals from a community at random, what is the likelihood that these belong to different taxa?’ D is bound between 0 and 1–1/TR, with a value approaching 0 indicating an uneven community where there is a high chance that the two selected individuals share the same taxonomy. It follows logically that a high probability of selecting individuals from the same taxon means the community has a low biodiversity. As D approaches 1–1/TR, this represents a perfectly even community where all taxa are equally abundant and there is a high probability of randomly selecting individuals from distinct taxa. In microbial ecology, we could expect nutrient-rich environments dominated by a small number of fast-growing, copiotrophic taxa or environments subject to extremes in temperature, pH etc. to have a D approaching 0. Unlike TR, D is a proportional metric, and is therefore less biased by sampling depth than TR. Although D was conceived to handle communities of infinite population sizes (Simpson 1949) in practice, the TR of microbial communities is much greater than for plants or animals, and ‘large’ numbers can render D difficult to interpret (described further below).
A third example that is widespread in microbial ecology is the Shannon diversity index, as:
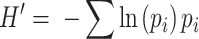 |
Where, as above, pi is the proportional abundance of the ith taxon. H′ was not originally intended for ecological applications, yet somewhat like D, it asks a probability-based question: ‘How likely is it that the next individual in a sequence belongs to the same taxon as the current individual?’ As H′ increases, it is less likely that these two individuals belong to the same taxon. Unlike D, H′ is not strictly proportional, and so it is relatively more sensitive to sampling constraints. Furthermore, H' acts in a highly non-linear fashion, meaning that it increases rapidly in poorly diverse communities and slowly in more complex communities. In regard to the SAD, H' functions as an intermediary between Richness and Evenness. As they measure different, but unified, aspects of the SAD, it is possible to derive them as extensions of each other (Hill 1973).
With the advent of metagenomics, alpha-diversity indices were quickly applied to discrete counts of protein-encoding genes in order to quantify the functional biodiversity of communities. The Richness of protein-encoding genes (PR) can correlate strongly and positively with TR across temperature gradients (Ruhl et al. 2022), aridity gradients (Song et al. 2019), and with seasonality (Galand et al. 2018). Similarly, the H′ of protein-encoding genes can correlate positively with taxonomic-based H′ (Fierer et al. 2013). Alpha-diversity measures of protein-encoding genes have also been used as one of our three pillars of investigating ecosystem processes. Such studies support general concepts such as: functional potential of host-associated microbiomes change over host developmental life-stages (e.g. early versus late growth stages of Arabidopsis rhizosphere (Chaparro et al. 2014)); greater functional potential conveys benefits for host physiology (e.g. corals become more resistant to bleaching (Cardenas et al. 2022)); and increased functional potential is linked to higher rates of certain ecosystem processes (e.g. increased greenhouse gas emissions from peatlands (Pavia et al. 2023)). Tracking alpha-diversity changes also shows that it is possible to restore lost functional potential in disturbed ecosystems, e.g. re-vegetation of deforested landscapes (Guo et al. 2018). These are fundamentally important basic questions toward understanding BEF relationships. However, sequencing data also allows us to consider underlying genetic relationships between taxa, e.g. alpha- and beta-diversity metrics that compare (dis)similarity between taxa that share a single conserved genetic marker (Faith 1992, Lozupone et al. 2007), and we should therefore not feel limited to treating genes simply as discrete counts in a series, nor to only employ indices that ask fairly abstract probability-based questions.
Imagine a forest
Where every tree represents a unique protein-encoding gene, e.g. pyruvate kinase, ammonia mono-oxygenase, predicted but functionally unknown proteins, and so on. Some of these trees will have long branches that spread far from the trunk, ending in many individual leaves. These branch lengths represent the dissimilarity in the gene between taxa (the leaves of the branch) that encode for the same gene (at the end of different, but connected, branches). We could speculate that these are the most interesting trees in this forest as they represent homologous genes that share a common ancestor, yet have diverged over time, and while the protein's key function is shared, they may perform optimally under different niches, e.g. low-affinity versus high-affinity particulate methane mono-oxygenase. Other trees may be very large, yet ‘stumpy’ in terms of their branch lengths. These would represent highly-conserved homologous genes that are unlikely subject to (or direct contributors toward) niche differentiation between taxa, e.g. glutamate synthase. Some short trees are more akin to shrubs—these have relatively few leaves (i.e. fewer taxa in the community encode for these proteins), yet may still carry out key functions, e.g. nitrogenase. In this analogy, the genetic diversity inherent within certain communities will give rise to dense, broad-branched leafy forests whereas others will be more like an arid shrubland. This is not to say that the genetic diversity in the imaginary arid shrubland is unimportant for that given ecosystem (Shade 2017), but one can reasonably expect a greater potential for unique functionality under more variable conditions in the forest. Our aim is to quantify this in a meaningful manner.
Let us ask ‘What is the biodiversity amongst a set of observed protein-encoding genes?’ We have a set number of observations (N) that could be entire coding sequences from a collection of genomes, or predicted protein-encoding genes from a metagenome, depending on what is being analysed. These are the leaves in our forest. There are also a set number of unique protein-encoding genes, the protein Richness (PR), to which N are distributed amongst, acting as the trees that support each leaf. Each protein-encoding gene (leaf) that belongs to a P (tree) also differs from the other leaves, calculated as a % of dissimilarity in sequence identity (d) (pair-wise branch length). Therefore, the biodiversity within the ith P is simply a ratio of the sum of pair-wise dissimilarities (di) to the number of pair-wise combinations amongst the protein-encoding genes in the ith P (ci). This is summed to give the biodiversity across all P:
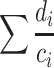 |
It should be noted that this ratio is compatible with gene clustering algorithms that report pair-wise (dis)similarities between a representative gene and all others within the homolog, including a self-comparison, and therefore in these cases ci will always be at least 1 (see Supplementary Fig. S1 for a conceptual visualization of this). This simplistic ratio will, however, lose information from so-called Orphan proteins that are only detected once (i.e. singletons). The di of a protein-encoding gene observed only once will be 0, and so it will not contribute to the biodiversity sum. As these Orphans are protein-encoding genes that may be rare (yet potentially interesting!) within the community, or our sequencing depth may simply not be deep enough to observe its homologs, we still wish to retain information from their detection. To save the Orphans, we adjust the biodiversity ratio as so:
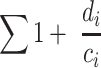 |
Such a value is inherently tied to PR, however, and as described above, such alpha-diversity indices are sensitive to sampling depth and scale. To improve comparability between samples (i.e. communities) we weight the overall value by our total observations N. This has the added benefit of creating upper and lower boundaries on the index. Our metagenomic alpha-diversity index (MD) is thus:
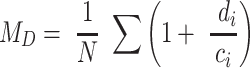 |
MD increases for communities with diverse functional gene homologs associated with either completely unique and/or dissimilar protein-encoding genes. Conversely, communities dominated by protein-encoding genes that are highly similar will yield a low MD. Somewhat similar to D, MD is bound between a theoretical lower limit of no biodiversity among protein-encoding genes, 1/N, and a theoretical upper limit of ‘perfect’ biodiversity where each protein-encoding gene is absolutely unique, 1 (please consult the supplementary material for a simplified mathematical proof).
Let us consider a simple example. Imagine three in silico ‘communities’ as: (i) varying Escherichia coli strains; (ii) commensal host-associated human gut taxa (Bacteroides thetaiotaomicron, Bacteroides fragilis, Faecalibacterium prausnitzii, Clostridium butyricum, Lactobacillus acidophilus, Bifidobacterium lactis) (Newton et al. 2013); and (iii) a phototrophic biological soil crust (BSC) of free-living taxa (Microcoleus vaginatus, Stenotrophomonas maltophila, Pelomonas saccharophila, Azotobacter beijerinckii, Lactiplantibacillus plantarum, Methylobacterium aerolatum) (Couradeau et al. 2019) (Table 1; Table S1 for genome source information). Each community has six distinct taxa (TR). Amino acid sequences of protein-encoding genes among genomes, N, were clustered dependent on shared kmers, and pairwise dissimilarity between clustered homologs calculated, with MMSeqs2 (Steinegger and Söding 2017) (although other pairwise comparative methods could be employed, such as all-vs-all BLAST (Price et al. 2008) or mapping predicted protein-encoding genes back to custom databases (Galand et al. 2018)). The lower cut-off E value of homologs clustered by MMSeqs2 was ca. 10−4, which equates to a false discovery rate of incorrectly assigning a protein-encoding gene to a group of homologs as roughly 10−4 (Steinegger and Söding 2017). A minimum sequence identity cut-off was not imposed. PR, H′ and D were calculated from the proportional sizes of clustered homologs. The log10 P dissimilarity and MD are also reported. The indices PR, H′ and MD show expected trends of E. coli < Human Gut < BSC. As mentioned above, D suffers from the ‘large’ numbers of PR here.
Table 1.
Alpha-diversity index comparisons within three simplistic, in silico communities. TR = taxonomic Richness; N = total number of protein-encoding genes compared within the in silico community; PR, H′, D, Log10 P dissimilarity and MD = respectively as Richness, Shannon, Simpson Evenness, protein dissimilarity and metagenomic diversity indices derived from protein-encoding genes within the community.
| Community | TR | N | PR | H′ | D | Log10 P dissimilarity | MD |
|---|---|---|---|---|---|---|---|
| E. coli | 6 | 28 029 | 5 485 | 8.25 | 0.999 | 3.77 | 0.21 |
| Human Gut | 6 | 20 007 | 10 456 | 8.87 | 0.999 | 4.06 | 0.57 |
| Biological Soil Crust | 6 | 26 814 | 15 318 | 9.29 | 0.999 | 4.22 | 0.62 |
While PR, H′ and MD all indicate that BSC has the greatest functional potential, I argue that the value of MD lies in its interpretability. Rather than asking an abstract, probability-based question, it specifically asks how much diversity exists amongst the observed protein-encoding genes. It is immediately apparent from the MD approaching 0 that the protein-encoding genes in the E. coli group (0.21) are highly similar to each other relative to the gut and BSC groups, i.e. there is high redundancy, poor biodiversity and ultimately lower potential for varied functionality.
As the theoretical upper limit of 1 indicates that every protein-encoding gene is absolutely unique, and the lower limit is effectively 0, the BSC MD of 0.62 indicates that most of the protein-encoding genes in this group are either divergent within/between, or are completely unique to, these six taxa. PR is also quite simple to interpret, e.g. there are 3 x more unique protein-encoding genes in the BSC group than the E. coli group. Indeed, while PR and MD provide distinct information, they have a complementary interpretation—the ca. 3 x more unique protein-encoding genes in BSC versus E. coli also equates to ca. 3 x more genetic dissimilarity amongst these genes. In isolation, though, PR cannot provide information regarding genetic dissimilarity and/or potential functional redundancy among the six taxa. For example, while the PR of the BSC group is ca. 50% greater than the gut taxa, MD is only marginally higher in the BSC group, and this implies a relatively greater overlap in general functionality amongst these six free-living taxa.
In contrast, H′ seems to suggest that the functional biodiversity among six E. coli strains (8.25) is not that dissimilar from the two groups comprised of distinct prokaryotes (8.87 and 9.29). Due to the highly non-linear nature of H′, one cannot interpret this difference as ca. 10% greater diversity in the BSC versus E. coli groups. H′ can only tell us that diversity in BSC is higher than E. coli.
But what about metagenomes?
The following three examples of measuring functional biodiversity are from metagenomes. For specific methods of how metagenomic data was processed, please refer to the Supplementary Methods.
The first example considers changes in taxonomic and functional biodiversity across a steep temperature gradient within a geothermal hotspring (Ruhl et al. 2022). The original study found that both TR (as operational taxonomic units) and PR (as Pfam annotated protein-encoding genes) decreased as temperature increased. The bioinformatic approaches used here differed, e.g. all protein-encoding genes were analysed and not only those that could be assigned a functional annotation. Even so, the same strong trends unifying both taxonomic and functional biodiversity are clear (Fig. 1). Ruhl et al., concluded that the rapid decrease in taxonomic and functional biodiversity across the gradient was a consequence of heat-stress selecting for relatively simple communities of thermophilic taxa. Additionally, the thermophilic taxa were also predicted to have on average smaller genomes than mesophiles, further contributing to the decreased functional biodiversity. From the analyses performed here (Fig. 1), this strong temperature-dependent trend was apparent regardless of how taxonomic biodiversity (TR, H′, D) or functional biodiversity (PR, H′, MD) was considered. Regardless, comparing PR between the coldest and hottest communities, we see that the mesophilic community has ca. 10 000 more unique protein-encoding genes, equivalent to a ca. 20% increase in Richness. Similarly, MD shows that there is a ca. 15% increase in the genetic diversity with these additional 10 000 unique protein-encoding genes present. However, as above with the E. coli example, an increase in H' from 10.77 (hottest) to 10.98 (coldest), or roughly a 1% increase, does not tell us anything about the underlying relationship between temperature and functional diversity here, other than that mesophilic communities are more diverse than thermophilic.
Figure 1.

Example one, geothermal hotspring temperature biodiversity gradient. Both taxonomic (ribosomal rRNA gene) and functional (all protein-encoding genes) biodiversity decreases with increasing temperature as stress selects for few heat-adapted taxa with relatively limited functionality. Linear regression slopes for each index are shown (n = 1 per temperature point). TR = taxonomic-marker derived Richness, H′ = Shannon, D = Simpson Evenness, PR = protein Richness, MD = metagenomic diversity derived from genetic (dis)similarity of all protein-encoding genes.
Example number two comes from observations during a natural, annual event: how a dramatic increase in summer daylight hours gives rise to a bloom of life in the pelagic Arctic Ocean (Puente-Sanchez et al. 2022). Samples were taken in March, April, May and June. In early spring a 2 m thick ice-sheet covered the sea, no photosynthetically active radiation (PAR) could reach the pelagic communities and integrated chlorophyll a was < 2 mg m−2. Over time as the season transitioned into summer, ice-melt was prolific, sea ice was breaking apart and sufficient PAR had led to > 200 mg m−2 integrated chlorophyll a. Neither taxonomic nor functional alpha-diversity indices were reported in this study, however the seasonal change was marked by strong compositional shifts, with photosynthetic Pro- and Eukaryotes, heterotrophic Bacteroidota and Pseudomonadota (formerly Proteobacteria) blooming in summer, and a concurrent relative decrease in Thermoproteota (formerly Thaumarchaeota), Planctomycetota and Verrucomicrobiota (Puente-Sanchez et al. 2022). Derivation of alpha-diversity indices here showed that overall taxonomic biodiversity decreased in June photosynthetic communities (Fig. 2). Despite a lower taxonomic biodiversity, the June communities had a greater functional biodiversity, likely driven by an enrichment in functional potential (and the great repertoire of associated genetic machinery) of photosynthetic microorganisms, e.g. Photosystem I, Photosystem II, carboxysome, Calvin-Benson-Bassham cycle etc. (Rubin et al. 2015). Previous studies of ecological succession in oceanic diatom blooms have also demonstrated that, while a relatively small subset of Bacteroidota and Pseudomonadota heterotrophs are enriched alongside photoautotrophs, these taxa possess diverse carbohydrate active enzymes and broad oligomer and monomer substrate preferences that target diatom and cyanobacterial exopolysaccharides (Teeling et al. 2012, Zheng et al. 2019). It is therefore worth emphasising that trends in taxonomic and functional biodiversity are not necessarily linked—having more unique heterotrophs in March and April, as per the 16S rRNA gene, does not necessarily mean that their genomes host a greater diversity, or functional potential, of homologous protein-encoding genes relative to the photosynthetically-active community and its associated specialist heterotrophs. As with previous examples, PR and MD give complementary information—June communities have ca. 6 000 more unique protein-encoding genes with a ca. 7% greater genetic diversity amongst them. H′, of course, can only tell us that the June communities are more functionally biodiverse than those in March.
Figure 2.

Example two, seasonal comparison of Arctic Ocean communities as they transition from spring into summer. Increased daylight during the summer month of June drives a decrease in taxonomic (ribosomal rRNA gene) biodiversity as communities become dominated by photosynthetic organisms and a subset of specialist heterotrophs. However, functional (all protein-encoding genes) biodiversity is greater in the photosynthetic communities. Results of significance testing with gamma-distributed general linear models are shown where April, May or June differed from March. n = 3 for March, n = 1 for April, n = 2 for May and n = 2 for June. (*) P < 0.05 (**) P = 0.001 (***) P < 0.001. TR = taxonomic-marker derived Richness, H′ = Shannon, D = Simpson Evenness, PR = protein Richness, MD = metagenomic diversity derived from genetic (dis)similarity of all protein-encoding genes.
The third and final example involves the successional development of soil microbial (and plant) communities after volcanic eruptions at the Llaima volcano in Chile (Hernández et al. 2020a,b). Lava flow had essentially created new substrate for colonisation at distinct geographical sites around the volcano, allowing for a successional time gradient for comparisons across ca. 50, 250 and 350 years. At the time of sampling, the ‘early’ successional stage was colonised by lichen-prokaryote symbiotic communities, while the intermediate and latter stages were colonised by understory plants. Hernández et al. 2020a,b show that as soils developed, overall TR (as operational taxonomic units) increased, with the early stage strongly dominated by ‘simplistic’ communities of autotrophic archaeal ammonia oxidisers, Cyanobacteriota, nitrogen, hydrogen and carbon monoxide-fixing Chloroflexota that transitioned to the more ‘typical’ soil communities dominated by highly diverse heterotrophic Pseudomonadota, Acidobacteriota and Actinobacteriota. Here, a significant increase in TR was noted at the intermediate successional stage, however community Evenness (D) actually decreased by the late successional stage, as communities shifted from primarily autotrophic to heterotroph-dominated soil assemblages (Fig. 3). In terms of functional biodiversity, both PR and MD identified a decreased functional potential after 371 years of ecological succession. Therefore, while taxonomic alpha-diversity indices gave somewhat inconsistent results for overall biodiversity (increased Richness yet decreased Evenness) the functional genetic information showed a consistent trend in that functional biodiversity decreased as the niche-differentiated autotrophic communities were replaced by heterotrophs that shared relatively similar functions for nutrient acquisition and metabolism of plant-derived organic substrates.
Figure 3.

Example three, successional development of soil communities after volcanic eruptions. Taxonomic Richness peaks at a mid-successional stage. Taxonomic Evenness, protein Richness and genetic functional biodiversity as MD ultimately decrease as niche-differentiated autotrophic communities are replaced by soil organoheterotrophs after 371 years. Results of significance testing with gaussian-distributed general linear models are shown where mid or late successional stages differed from the earliest sampled stage. (*) P < 0.05 (**) P = 0.001 (***) P < 0.001. n = 3 per successional stage. TR = taxonomic-marker derived Richness, H′ = Shannon, D = Simpson Evenness, PR = protein Richness, MD = metagenomic diversity derived from genetic (dis)similarity of all protein-encoding genes.
Some technical considerations
While MD seeks to measure the functional biodiversity of a community from a different angle (i.e. genetic dissimilarity) than pre-existing alpha-diversity indices, it remains constrained by data quality and processing. Larger contigs will improve gene prediction and clustering, potentially yielding ‘more accurate’ alpha-diversity indices, and so direct comparisons between MD values should share assembly software, parameters etc. Similarly, the parameters used to cluster protein-encoding genes as homologs must also be consistent (who should be considered as belonging to a leaf on the same tree?) as these cutoffs are essential to how the pair-wise genetic dissimilarities are calculated. Secondly, due to redundancy in the genetic code, amino acid sequences are better reflective of actual protein function than nucleic acid sequences (Wang et al. 2013). Therefore, I suggest that amino acid sequences should preferentially be analysed when the overall goal is to investigate meaningful relationships in how functional biodiversity may inform actual BEF relationships. Thirdly, while MD is less sensitive to N (i.e. total observations) than PR or H′, it is not a perfectly proportional metric bounded between 0 and 1, and so rarefying or randomly subsampling to a shared N will improve comparability between samples within similar ecosystems. Interestingly, unlike PR and H′, MD actually has a negative relationship with increasing N, which shows that it is redundancy/high similarity between genes that primarily drives MD downwards. Thus, a sample with N = 2 M may be resequencing/re-observing the same genes over and over, which will lower MD relative to the same sample with N = 1 M. Finally, deriving MD from host-associated communities may prove tricky—any ‘contaminant’ host genetic material that is sequenced alongside its microbiota will affect how MD is calculated. While pre-existing pipelines remove human-associated genetic material (Uritskiy et al. 2018) this would not be sufficient for deriving MD from, for example, a root endophyte community.
A future for microbial diversity metrics
The suggested MD is by no means meant to replace pre-existing alpha-diversity metrics, nor will it be the last proposed metric. However, going into the future, the following points are worth considering. As most protein-encoding genes from environmental sources cannot currently be annotated (Nayfach et al. 2021), functional biodiversity studies should not be limited to only analysing the relatively small fraction of genes that can currently be annotated. Galand et al. (2018) demonstrate this point very well. There are many ways one can compare the (dis)similarity of protein-encoding genes without resorting to annotation, for example local alignment-based (Schloss and Handelsman 2008) or kmer-based techniques (Steinegger and Söding 2017). Furthermore, the leap between determining a relationship between functional biodiversity and measurable ecosystem function is vast (Petchey and Gaston 2006) and fraught with many conflicting concepts. When only a subset of taxa are active at any one time (Shi et al. 2011), and indeed a lot of environmental DNA comes from necromass (Carini et al. 2017) and a non-negligible amount of genetic material may represent ‘pseudo-genes’ (Goodhead and Darby 2015), it is understandable to question the usefulness of deriving an alpha-diversity index for functional biodiversity. Care must also be taken not to conflate concepts of ‘functional biodiversity’, i.e. as described throughout this Perspective piece, with ‘functional traits’ (Escalas et al. 2019), which are an emergent property from collections of specific genes and/or gene variants, e.g. methanogenesis, maximum growth rate, copiotrophic or stress tolerant life-strategies etc. (Krause et al. 2014, Malik et al. 2020, Westoby et al. 2021). Once again, alpha-diversity indices provide information on only one piece of our biological puzzles, and must be considered within a greater context and systems-based understanding when investigating why communities ‘are as they are and do what they do’. Here they serve a useful explanatory purpose as a quantifiable summary of functional potential, with the benefit of MD lying in its simple interpretability, comparability between communities as a bounded metric, and that it tries to more directly address the gap between microbial metagenomic information and the overall BEF. Despite the abovementioned hurdles that must be considered and/or overcome, sensible, quantitative metrics that explain BEF relationships are a worthy goal to strive toward as they have the potential to model and predict (un)desirable ecosystem functions in the world around us.
Supplementary Material
Acknowledgements
I would like to express my thanks toward the anonymous reviewers for truly constructive input, Dr. Brandon Seah for enlightening conversations and suggestions for recent (and ever-faster) bioinformatic tools, and for sowing the seeds of the protein-forest metaphor. I would also like to express heart-felt thanks toward Prof. Dr. Christoph Tebbe, also for enlightening conversations, and for eternal support and mentorship.
Conflict of interest
I declare that I have no conflicting interests associated with thinking about alpha-diversity indices.
Funding
I would like to thank the Deutsche Forschungsgemeinschaft (DFG) for financial support under project number 522758166.
Data Availability
All (meta)genomes analysed in this study are publically available (Tables S1, S2, S3 and S4 for details). Python code to apply MMseqs2 and to derive MD from clustered amino-acid sequences of protein-encoding genes is available at: github.com/DamienFinn/MD/blob/main/MD.py. This code also includes several parameters that can be adjusted by users, including changing E value and sequence identity thresholds, and setting a user defined N for random sampling without replacement from clustered protein-encoding genes. Please note that this code begins to run quite slowly where observations are greater than 0.5 M. I do not have a computer science background, and interpretation-based computing languages are the best I can do—more clever individuals will be needed to write these functions in a faster language.
References
- Cardenas A, Raina JB, Pogoreutz C et al. Greater functional diversity and redundancy of coral endolithic microbiomes align with lower coral bleaching susceptibility. ISME J. 2022;16:2406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carini P, Marsden PJ, Leff JW et al. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol. 2017;2:16242. [DOI] [PubMed] [Google Scholar]
- Chaparro JM, Badri DV, Vivanco JM. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014;8:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couradeau E, Giraldo Silva A, de Martini F et al. Spatial segregation of the biological soil crust microbiome around its foundational cyanobacterium, microcoleus vaginatus, and the formation of a nitrogen-fixing cyanosphere. Microbiome. 2019;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalas A, Hale L, Voordeckers JW et al. Microbial functional diversity: from concepts to applications. Ecol Evol. 2019;9:12000–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- Fierer N, Ladau J, Clemente JC et al. Reconstructing the microbial diversity and function of pre-agricultural Tallgrass prairie soils in the United States. Science. 2013;342:621–4. [DOI] [PubMed] [Google Scholar]
- Galand PE, Pereira O, Hochart C et al. A strong link between marine microbial community composition and function challenges the idea of functional redundancy. ISME J. 2018;12:2470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodhead I, Darby AC. Taking the pseudo out of pseudogenes. Curr Opin Microbiol. 2015;23:102–9. [DOI] [PubMed] [Google Scholar]
- Gotelli NJ, Colwell RK. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett. 2001;4:379–91. [Google Scholar]
- Guo Y, Chen X, Wu Y et al. Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci Total Environ. 2018;635:598–606. [DOI] [PubMed] [Google Scholar]
- Hernández M, Calabi M, Conrad R et al. Analysis of the microbial communities in soils of different ages following volcanic eruptions. Pedosphere. 2020a;30:126–34. [Google Scholar]
- Hernández M, Vera-Gargallo B, Calabi-Floody M et al. Reconstructing genomes of carbon monoxide oxidisers in volcanic deposits including members of the Class Ktedonobacteria. Microorganisms. 2020b;8:1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology. 1973;54:427–32. [Google Scholar]
- Jernberg C, Löfmark S, Edlund C et al. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56–66. [DOI] [PubMed] [Google Scholar]
- Krause S, Le Roux X, Niklaus PA et al. Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front Microbiol. 2014;5:251. 10.3389/fmicb.2014.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST et al. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microb. 2007;73:1576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane S. Antibiotic treatments and microbes in the gut. Environ Microbiol. 2014;16:919–24. [DOI] [PubMed] [Google Scholar]
- Malik AA, Martiny JBH, Brodie EL et al. Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. ISME J. 2020;14:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning P, van der Plas F, Soliveres S et al. Redefining ecosystem multifunctionality. Nat Ecol Evol. 2018;2:427–36. [DOI] [PubMed] [Google Scholar]
- McGill BJ, Etienne RS, Gray JS et al. Species abundance distributions: moving beyond single prediction theories to integration within an ecological framework. Ecol Lett. 2007;10:995–1015. [DOI] [PubMed] [Google Scholar]
- Nayfach S, Roux S, Seshadri R et al. A genomic catalog of Earth's microbiomes. Nat Biotechnol. 2021;39:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton DF, Macfarlane S, Macfarlane GT. Effects of antibiotics on bacterial species composition and metabolic activities in chemostats containing defined populations of human gut microorganisms. Antimicrob Agents Chemother. 2013;57:2016–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavia MJ, Finn D, Macedo-Tafur F et al. Genes and genome-resolved metagenomics reveal the microbial functional make up of Amazon peatlands under geochemical gradients. Environ Microbiol. 2023;25:2388–403. [DOI] [PubMed] [Google Scholar]
- Petchey OL, Gaston KJ. Functional diversity: back to basics and looking forward. Ecol Lett. 2006;9:741–58. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastBLAST: homology relationships for millions of proteins. PLoS One. 2008;3:e3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente-Sanchez F, Macias L, Campbell KL et al. 2022; Bacterioplankton taxa compete for iron along the early spring-summer transition in the Arctic Ocean. Biorxiv. 10.1101/2022.02.07.479392 [DOI] [PMC free article] [PubMed]
- Rubin BE, Wetmore KM, Price MN et al. The essential gene set of a photosynthetic organism. P Natl Acad Sci USA. 2015;112:E6634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl IA, Sheremet A, Smirnova AV et al. Microbial functional diversity correlates with species diversity along a temperature gradient. Msystems. 2022;7:e00991–00921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. A statistical toolbox for metagenomics: assessing functional diversity in microbial communities. BMC Bioinf. 2008;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade A. Diversity is the question, not the answer. ISME J. 2017;11:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Tyson GW, Eppley JM et al. Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 2011;5:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EH. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- Song HK, Shi Y, Yang T et al. Environmental filtering of bacterial functional diversity along an aridity gradient. Sci Rep. 2019;9:866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35:1026–8. [DOI] [PubMed] [Google Scholar]
- Szoboszlay M, Tebbe CC. Hidden heterogeneity and co-occurrence networks of soil prokaryotic communities revealed at the scale of individual soil aggregates. MicrobiologyOpen. 2021;10:e1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling H, Fuchs BM, Becher D et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–11. [DOI] [PubMed] [Google Scholar]
- Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Quensen JF, Fish JA et al. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio. 2013;4:e00592–00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westoby M, Nielsen DA, Gillings MR et al. Strategic traits of bacteria and archaea vary widely within substrate-use groups. FEMS Microbiol Ecol. 2021;97:fiab142. [DOI] [PubMed] [Google Scholar]
- Zheng Q, lu J, Wang Y et al. Genomic reconstructions and potential metabolic strategies of generalist and specialist heterotrophic bacteria associated with an estuary synechococcus culture. FEMS Microbiol Ecol. 2019;95:fiz017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All (meta)genomes analysed in this study are publically available (Tables S1, S2, S3 and S4 for details). Python code to apply MMseqs2 and to derive MD from clustered amino-acid sequences of protein-encoding genes is available at: github.com/DamienFinn/MD/blob/main/MD.py. This code also includes several parameters that can be adjusted by users, including changing E value and sequence identity thresholds, and setting a user defined N for random sampling without replacement from clustered protein-encoding genes. Please note that this code begins to run quite slowly where observations are greater than 0.5 M. I do not have a computer science background, and interpretation-based computing languages are the best I can do—more clever individuals will be needed to write these functions in a faster language.