

Abstract
Aortic aneurysm (AA) and dissection (AD) are aortic diseases caused primarily by medial layer degeneration and perivascular inflammation. They are lethal when the rupture happens. Vascular smooth muscle cells (SMCs) play critical roles in the pathogenesis of medial degeneration, characterized by SMC loss and elastin fiber degradation. Many molecular pathways, including cyclic nucleotide signaling, have been reported in regulating vascular SMC functions, matrix remodeling, and vascular structure integrity. Intracellular cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) are second messengers that mediate intracellular signaling transduction through activating effectors, such as protein kinase A (PKA) and PKG, respectively. cAMP and cGMP are synthesized by adenylyl cyclase (AC) and guanylyl cyclase (GC), respectively, and degraded by cyclic nucleotide phosphodiesterases (PDEs). In this review, we will discuss the roles and mechanisms of cAMP/cGMP signaling and PDEs in AA/AD formation and progression and the potential of PDE inhibitors in AA/AD, whether they are beneficial or detrimental. We also performed database analysis and summarized the results showing PDEs with significant expression changes under AA/AD, which should provide rationales for future research on PDEs in AA/AD.
Keywords: Aortic aneurysm, Aortic dissection, cAMP, cGMP, PDE, GPCR
1. Introduction
AA and AD are aortic diseases that could happen in every anatomic segment of the aorta, including the ascending aorta, aorta arch, descending thoracic aorta, and abdominal aorta [1, 2]. Based on their geometric positions, AA is classified into thoracic aortic aneurysm (TAA), abdominal aortic aneurysm (AAA), and thoracoabdominal aortic aneurysm (TAAA) when a TAA extends into the abdominal aorta [2, 3]. AA is defined when the diameter of the artery reaches 1.5 times increase compared to the normal diameter; however, the threshold is 1.25 times increase when the aneurysm occurs in the aortic root and ascending thoracic aorta according to the 2022 guideline [2]. Most of the AA is asymptomatic but lethal when ruptured. The thoracic aorta AA (such as TAA or TAAA) is mostly caused by genetic factors and is inheritable [2, 4]. Marfan syndrome (MFS) is the most common genetic disorder that causes TAA [4, 5]. Those patients carry a mutation in FBN1, which encodes Fibrillin-1[5]. Fibrillin-1 is a kind of microfibril that plays an important role in maintaining tissue integrity [6, 7]. It provides structural support for the elastic fiber formation [6]. It also regulates the bioavailability of TGFβ superfamily [8]. The mutation in Fibrillin-1 leads to a disorder of the fibrillar elastic matrix in the vascular wall [5]. It weakens the vascular wall, resulting in a high risk of AA or AD. Besides, Loeys-Dietz syndrome (mutation in TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD2, SMAD3)[9, 10], vascular Ehlers-Danlos syndrome (mutation in COL3A1, COL1A2)[9], and many other syndromes could also lead to TAA [4, 11]. For the aneurysm initiated from the abdominal aorta, the risk factors are mostly hypertension, atherosclerosis, smoke, and gender [1, 2]. Treatments for AA include medical therapy, open surgery for a prosthetic graft replacement, or endovascular repair with a stent graft fixed on a non-aneurysmal segment [1, 2, 12]. The management strategy varies based on the risk factor, the impaired anatomic segment level, the histopathology, and the condition of rupture/unruptured, which is well described in the 2022 guideline [1, 2, 12].
AD occurs when the aortic wall is torn, and the aortic lumen is separated into two channels [3]. Patients with AD present with an acute pain [1–3]. Occasionally, AD has a noticeable bulging in the aorta. AD has a high risk of rupture because of the breach in the aortic wall. For patients with acute AD in the ascending aorta, the overall early mortality within 48 h is 5.8% (including 91.4% subjects in surgery group and 8.6% in medication group) [13]. The 48h-mortality in the medical treatment group is 23.8%, while the surgery group is 4.4% [13]. After the first 48h and surgery, patients still face high risks of complications, such as cardiovascular diseases, acute renal failure, and stroke [14, 15]. A nationwide cohort study of the Danish population showed that 30-day mortality for patients with ascending AD is 22%; 5-year mortality is 57% [15]. AD is classified into four types: classic dissection, incomplete dissection, intramural hematoma, and penetrating aortic ulcer. The definition is based on the pathophysiological changes, such as fake lumen, thrombosis, and ulcer formation, which could be detected by CT and ultrasonic examination [3]. According to the Stanford classification, the AD with the ascending aorta involvement is type A, and without ascending aorta involvement is type B. The non-A/non-B dissection only impairs the aortic arch. Management with medical therapy, endovascular repair, or open surgery depends on the AD types, conditions, and risk factors [2]. AA can lead to AD when the dilated aorta wall cannot stand the flow of pressure and the blood flow into the vascular wall [2]. AD may cause a pseudoaneurysm due to the bulge of the hematoma [15].
2. Animal models to study AA and AD
Many rodent animal models have been developed to study AA and AD experimentally in laboratories. Different models induce AA/AD with distinct mechanisms and unique pathophysiological features. Among them, systemic Ang II infusion or local vascular damage by elastase or CaCl2 are the most commonly used methods. It is noteworthy that none of the rodent AA models are exactly identical to human AAs. Different models resemble unique human features. Therefore, it might be more appropriate to use different animal models to confirm findings. Table 1 summarizes the features of each animal model.
Table 1.
Animal models of AA/AD.
| Models | Species | Pathological processes | Time | Aortic Segment | Acceleration | Advantages | Disadvantage |
|---|---|---|---|---|---|---|---|
| Fbn1 C1039G/+ | Mice | Spontaneous dilatation, aneurysm, dissection, and rupture. | 6–12 months | Ascending, arch, and descending aorta | AngII infusion | Mimic human MFS development. Best model to study MFS and TAA/TAD. | Progress slowly |
| AngII infusion | Mice | Dilatation, aneurysm, dissection, and rupture. | 2–4 weeks | All segments | Hyperlipidmia via HFD, ApoE−/−, Ldlr−/−, or AAV-PCSK9-mut | Mimic the pathological feature in human AA/AD. Severe enough to induce AD in aorta. | Affected by cardiac function, renal function, aorta function and lipid metabolism. Transgenic mice with double knockout are hard to generate. |
| Elastase (perivascular application) | Mice Rats | Dilatation and aneurysm. | 2–4 weeks | Infra-renal aorta | BAPN intake | Form severe aneurysm. Not affected by other system. | Limit to infra-renal segment. Surgery is difficult. |
| CaCl2 (perivascular application) | Mice Rats | Dilatation, aneurysm, and calcification. | 6–10 weeks | Infra-renal aorta | Additional PBS application | Form calcification. Not affected by other system. | Limit to infra-renal segment. Surgery is difficult. Progress slowly. |
| TAC | Mice | Dilatation, aneurysm | 1–3 weeks | Ascending aorta and right carotid artery | NR | A pressure overload model affecting aorta function and renal function. | Limit to ascending aorta. Mild progress (mostly aorta dilatation). |
AngII, angiotensin II; HFD, high fat diet; BAPN, β-Aminopropionitrile; CaCl2, Calcium chloride; TAC, transverse aortic constriction. NR, not reported.
2.1. Fbn mutation
FBN1 missense mutation is the main cause of MFN in patients. To mimic this pathophysiology in human, transgenic mice with Fbn1 mutation is generated. In 1997, the first Fbn1 mutation mouse line was created and named as mgΔ [16]. In this strain, 19–24 extrons of Fbn1 were replaced by a neomycin-resistance expression cassette, leading to 272 residues missing in Fbn1 [16]. FBN1 mutation in humans was thought to be completely penetrated; however, no obvious phenotype was found in heterozygote mice. The homozygote mice had a 90% decrease in Fbn1 transcription level and died in two weeks due to the defect in the cardiovascular system [16, 17]. In 2001, another Fbn1 mutation strain, mgΔloxPneo, was developed. mgΔloxPneo was modified from mgΔ. It deleted the 19–24 extrons by the Cre-loxP system. In addition to the phenotype found in mgΔ, mgΔloxPneo also showed a variation in phenotype among various strains with different background (e.g., C57BL/6 and 129Sv), indicating an epigenomic contribution in the MFS development [18]. Nowadays, the most commonly used animal model of MFS is Fbn1C1039G/+ that was developped in 2004. Fbn1C1039G/+ is a heterozygous knock-in mouse line carrying a missense mutant Fbn1(Cys1039Gly). This strain develops mild MFS, but mimics the classic MFS (Cys1039Tyr) in human[19, 20]. In this model, mice start to have aortic dilatation after 6-month normal feeding, and the mice have a high risk of AD and rupture after 12-month normal feeding [19]. These mice could have a normal life span, but the MFS development can be accelerated by the Angiotensin II (Ang II) infusion [21].
2.2. Ang II infusion
Ang II is the most commonly used reagent to induce aneurysms [22]. Ang II infusion increases blood pressure, which is a high risk of aneurysm development in humans; however, Ang II-induced aneurysm does not completely depend on blood pressure increase [22, 23]. The doses of Ang II infusion vary from 1000ng/kg/min to 2500ng/kg/min. Ang II infusion causes many vascular changes, including AA, AD, and chronic inflammation [24, 25]. After Ang II infusion, dissection (usually in the ascending aorta) happens in the early stage (first two weeks), and AA, particularly AAA in the suprarenal aorta, happens later. Ang II triggers chronic inflammation, which lasts the entire process [24]. With low doses of AngII infusion, such as 1000ng/kg/min, aortic dilatation can be found in all segments; however, AA mainly happens in the abdominal segment [23]. Higher doses of the Ang II infusion, such as 2000 ng/kg/min, are seen as a model for sporadic AA/AD [26]. The incidence of AA/AD in the ascending or descending aorta increases with higher doses than lower ones of the Ang II infusion [26]. There are many ways to modify the Ang II-infusion model to increase the incidence, most of which are by inducing dyslipidemia. The reported methods include a high-fat diet (HFD) feeding [26], ApoE−/− or Ldlr−/−background with HFD [27, 28], and expressing mutant proprotein convertase subtilisin/kexin type 9 (PCSK9) via adeno-associate virus (AAV) combined with HFD [29], which are common used methods. Those hypercholesterolemic mice have a 3–4-fold higher AA/AD incidence than Ang II-infused normocholesterolemic mice [22, 24]. Although the modified Ang II-infusion animal models mimic risks of AA/AD in patients (hypertension and hyperlipidemia), some features of AA/AD are different from humans. For example, Ang II-induced AAA is suprarenal, while human AAA is infrarenal. In addition, the phenotype of this model is modified by other factors, such as cardiac function, renal function, blood pressure, lipid levels, and gender [30]. Thus, genetically engineered mice with cell/tissue-specific changes in gene expression have often been used to evaluate the specific contributions of this model.
2.3. Elastase application
Intraluminal infusion or perivascular application of elastase directly degrades the elastin in the aorta and induces local vascular enlargement and AA. Due to the technique challenge of intraluminal infusion, perivascular application has become more popular. The aneurysm incidence of perivascular application with elastase reaches 70% 14 days after surgery [31]. This method can be modified with additional β-Aminopropionitrile (BAPN) oral administration. BAPN is a lysyl oxidase inhibitor that blocks the cross-links with collagen and elastin [32]. BAPN/elastase model increases the incidence (reaches 93%), the severity of aneurysm, and perivascular inflammatory cell infiltration compared with elastase application alone [32]. Perivascular elastase application can be used in mice or rats. Since elastase is painted on the adventitia of the infrarenal segment during the surgery, the AAA is limited to the area receiving the elastase. The pathology feature in the painted area is mostly AA but not AD. Since this model is a local reaction, the contributions from other organs are limited. However, the surgery procedure is more challenging.
2.4. Calcium chloride (CaCl2) application
Like the perivascular elastase application animal model, the CaCl2 application also directly damages the elastin by painting on the adventitia of the infrarenal segment. Mechanismically, CaCl2 penetrates the aortic wall, damaging the elastin and causing perivascular inflammation and calcification [33]. The incidence of aneurysm in mice with a C57BL/6 background is about 50%, and it takes 6–10 weeks to form an aneurysm [34]. One way to accelerate this process is additional PBS application after the CaCl2 treatment [35]. A mixture of CaCl2 and PBS forms CaPO4, the main component of calcium deposition in the aorta, making the calcification faster [35]. The CaCl2 model possesses a pathological feature of calcium deposition, making it an excellent model to investigate calcification development in animal models; however, CaCl2 damage does not mimic the pathology of human AA/AD.
2.5. Transverse aortic constriction (TAC)
The TAC model is commonly seen in studying pressure overload-induced heart failure. The blood flow load is increased before the ligation area by ligating the transverse aorta. Therefore, in addition to heart failure, the proximal aorta (ascending aorta and right carotid artery) is also affected. The aortic wall thickening and dilatation start one week after TAC. Aortic root diameter is around 21.5 % increase, and 23% increase in ascending aorta one week after TAC [36, 37], and the diameter of the right carotid artery (RCA) is increased by 35% three weeks after TAC [38]. Based on the diameter increase level, the pathological feature could be hard to reach the definition of aneurysm (1.5 times increase).
3. Pathogenesis of AA and AD
A normal aorta wall comprises 3 layers, intima, media, and adventitia. Genetic background or risk factors can weaken the aorta wall. When the aorta wall is weak and cannot stand the blood flow pressure, the aorta wall can be pushed to dilate and form the AA. If there is a breach in the aortic wall, the blood can flood into the media of the aortic wall, forming AD. The media layer degeneration plays a vital role during AA and AD. SMC is the major cell type of the medial layer of the aorta and is important for maintaining homeostasis and the structural integrity of the aorta. Vascular SMC actively regulates contraction/relaxation in response to the pulsatile and high-pressure blood flow in the aortas. Vascular SMC is also the major source of elastin that provides elasticity to the aorta.
The process of medial degeneration includes vascular SMC phenotype switching, senescence, death, vascular inflammation, and oxidative stress [39]. SMCs can undergo phenotype changes, e.g., from a contractile phenotype to a synthetic one, which leads to a low capacity to regulate vascular contraction/relaxation. Increasing evidence has suggested that vascular SMC contractile dysfunction contributes to TAA and AD. For example, human genetics studies have revealed that genetic mutations in SMC contractile genes, such as ACTA2 (encoding SMC-specific alpha-actin, aSMA) [40], MYH11 (encoding SMC-specific myosin heavy chain, SM-MHC) [41], and MYLK (encoding myosin light chain kinase, MLK) [42], cause familial TAA or AD. It has been reported that anti-hypertensive drugs, such as calcium channel blockers, accelerate TAA in a MFS mouse model and increase the risk of AD in MFS patients [43]. SMC relaxant hydrolazine also increases the risk of AD in turkeys [44]. Moreover, it has been shown that there is a decrease in maximum contraction in vascular SMCs from ≈30% sporadic AAA patients compared to normal SMCs [45], suggesting that impaired vascular SMC contractility may also play a role in AAA pathogenesis.
Additionally, synthetic SMCs gain the ability for pro-mitosis, pro-inflammation, pro-senescence, pro-death, etc., which weaken the structural stability of the aortic wall. SMC senescence causes cell cycle arrest and secretion of pro-inflammatory cytokines [46]. Apoptosis, pyroptosis, and ferroptosis are ways that cause SMC loss in the aortic wall. Vascular SMC death decreases cell numbers and reduces the capability to synthesize elastic matrix proteins. The medial layer becomes thinner and loses vascular integrity, which is crucial for aortic dilatation and rupture. Matrix metalloproteinase (MMP) production is another way that destroys vascular integrity. MMPs can be released from both vascular SMCs and inflammatory cells [7, 26]. MMPs degrade elastin and collagen, causing an extracellular matrix (ECM) degradation [7, 26]. Besides, oxidative stress from vascular SMCs and inflammatory cells is another way that causes vascular SMC dysfunction, death, vascular inflammation, and ECM degradation.
Immune cells also regulate VSMC function during AA/AD development. In the early stage, innate immune cells are the predominant cell type, including neutrophils, monocytes, macrophages, eosinophils, mast cells, and dendritic cells [47]. In the late stage, adaptive immune response participated by B cells and T cells could be seen [48]. Immune cells are recruited from circulating blood or infiltrated from perivascular adipose tissue (PVAT) [49]. Roles of perivascular immune cells could be summarized as: a) directly impairing VSMC, b) amplifying immune response, and c) maintaining homeostasis. MMPs, ROS, and pro-inflammatory cytokines (such as TNF-α) induce VSMC death and phenotype switch directly. Many immune cell types can secrete these factors. IL-4, IL-6, IL-10, etc, mediate pro-inflammatory effects by interacting with other immune cells and amplifying inflammatory status. Treg, eosinophils, and some subtypes of macrophages are recognized as beneficial immune cells [50–52]. During AA/AD development, they are recruited around the aorta, exerting a protective role by suppressing inflammation or maintaining VSMC homeostasis. Many signaling pathways have been reported for regulating vascular SMC pathogenesis and imflammatory status in AA/AD, which has been well reviewed [7, 12, 26, 39, 53]. This review focuses on studies about the roles and mechanisms of cyclic nucleotide signaling and PDEs in AA/AD.
4. Mechanism of cyclic nucleotide-mediated regulation of vascular SMC functions
4.1. cAMP signaling
Intracellular cAMP can be generated by membrane adenylyl cyclases (ACs) upon the activation of stimulatory G-protein (Gs) by stimulation of G-protein-coupled receptors (GPCRs) [54]. Knockout of Gs in SMCs caused SMC phenotype switching from contractile to synthetic SMC [55]. SMC-specific knockout of Gs in mice exhibited lower cAMP levels in the aorta, SMC phenotype switch in vivo, and aggravated AA in AngII-infused mouse model [55].
Several GsPCRs have been studied in AA or AD, including the adenosine type 2 receptors (A2Rs), beta-adrenergic receptor (β-AR), and prostaglandin receptors [56]. There are two A2Rs, A2aR and A2bR. Both are coupled to Gs and involved in cAMP production after stimulation by endogenous agonist adenosine. In human AA tissues, A2aR expression was increased [57]. In the intraluminal elastase-infused mouse aneurysm model, A2aR knockout or A2aR antagonism aggravated AAA formation, while A2aR agonism alleviated AAA formation [57]. In cultured vascular SMCs, A2aR agonism attenuated MMP2, MMP9, and VCAM1 expression [57]. Even though A2aR plays a beneficial role in vascular SMCs, the effects of A2aR antagonist/agonist treatment on AAA are more likely contributed by cAMP-mediated regulation of T cell activation and macrophage retention [57]. Based on the online database (proteinatlas.org), A2aR is indeed highly expressed in immune cells, such as T cells, B cells, and macrophages. This is consistent with the predominant contribution of immune cells to AAA formation in A2aR−/− or A2aR antagonist-treated mice. Immune cell infiltration is observed in the perivascular tissue of an aneurysm. The increased A2aR expression observed in aneurysm tissue might be contributed by A2aR expression in infiltrated immune cells. A2bR activation by adenosine also increases cAMP and is also present in vascular SMCs [58, 59]. However, the role of A2bR in AA/AD has not been reported.
β-ARs, including β1-AR and β2-AR, are activated upon the stimulation of catecholamines, leading to cAMP production [60]. The functions of β-ARs in SMC relaxation, proliferation, and apoptosis have been reported decades ago [61, 62]. Antagonizing β-ARs with β-blockers effectively treats many cardiovascular diseases, such as hypertension [60]. In a cohort study, long-term use of β-blockers decreases the risk of all-cause mortality compared with other antihypertension agents [63]. Another study found long-term use of β-blockers is beneficial to the progression of aortic dilatation and improved the outcomes in Type B dissection patients [64]; however, a recently published meta-analysis showed that β-blockers do not affect the aneurysm growth [65]. There is still no experimental evidence to show the role of β-ARs in AA/AD in animal models.
Prostaglandin E2 receptor 2 (EP2), EP4, prostaglandin D2 receptor 1 (DP1), and prostacyclin receptor (IP receptors) are different prostaglandin receptors coupled with Gs. They promote cAMP elevation by ligands such as prostaglandin E2 (PGE2) for EP2/4, PGD2 for DP1, and PGI2 for the IP receptor. The functions of EP2 are mostly focused on the cerebral aneurysm. EP2 in endothelial cells and macrophages activates NF-κB signaling, promoting aneurysm formation in the brain [66, 67]. Compared with EP2, EP4 has a predominant role in promoting AAA development. EP4 overexpressed mice have a more severe AAA phenotype, while EP4 heterozygous knockout mice or EP4 antagonist treatment alleviated AAA [68, 69]. PGE2 stimulation of EP4, but not EP2, mediates IL-6 expression in macrophages and vascular SMCs via increasing cAMP [68, 70]. VSMC-derived IL-6 promotes the inflammatory status by recruiting more monocytes and macrophages. The recruited monocytes and macrophages produce more PGE2, which constitutes a positive feedback loop [68]. DP1, together with DP2, are PGD2 receptors. DP1 knockout mice or antagonist treatment attenuated AAA formation, probably through decreasing inflammation, as DP1 and DP2 are mainly expressed in immune cells [71]. These findings indicate that stimulating different GsPCRs may have distinct functions in AA/AD, although they all elevate intracellular cAMP levels (Figure 1).
Figure 1.
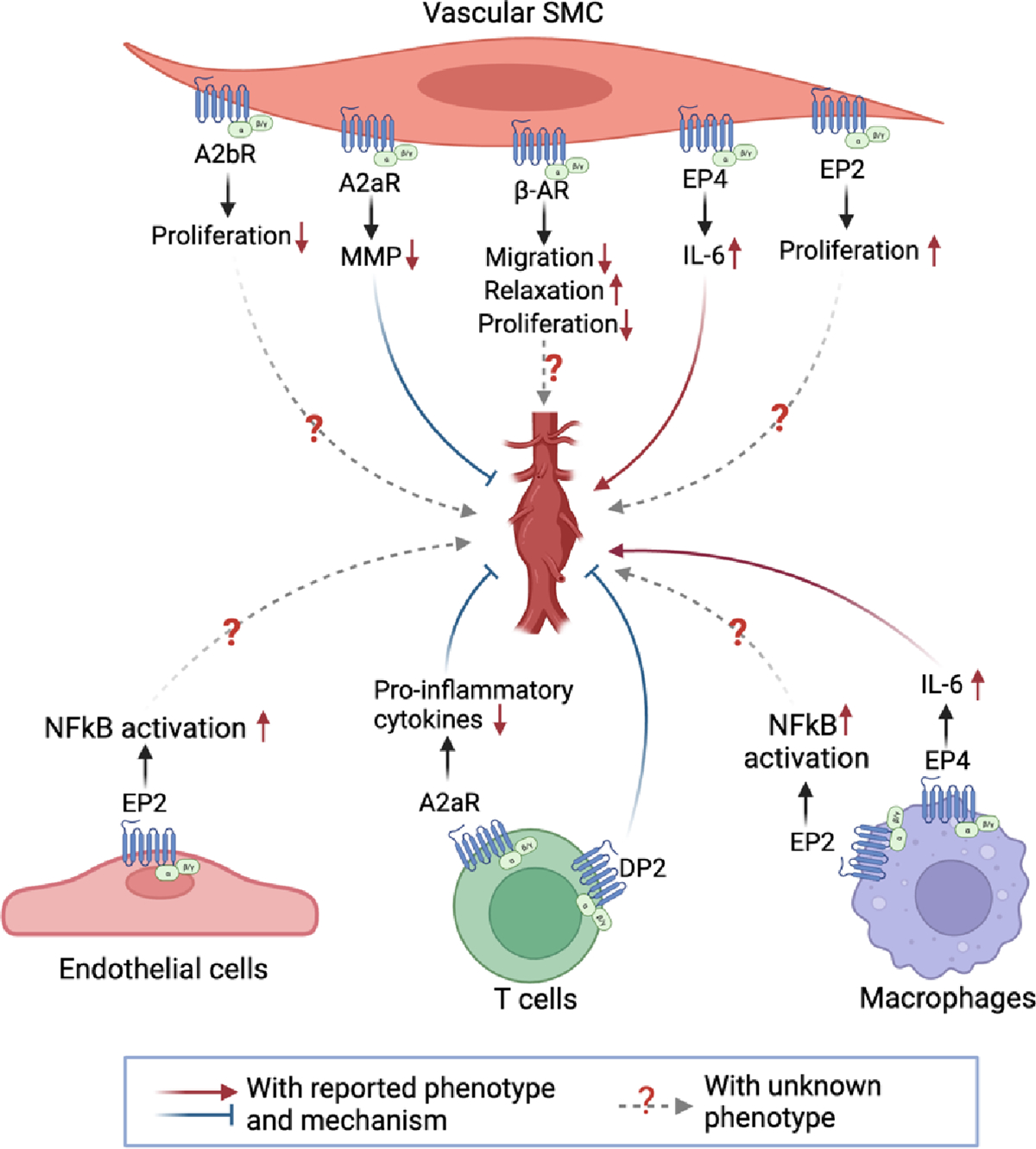
The functions of GPCR in aortic aneurysm development. A2aR alleviates AAA development by suppressing MMP production in vascular SMCs and pro-inflammatory cytokine production in T cells. EP4 promotes AAA development through mediating IL-6 expression in both vascular SMC and macrophages. DP2 promotes AAA development mainly through mediating T-cell activation. A2bR suppresses proliferation in vascular SMC. β-AR inhibits vascular SMC migration, relaxation, and proliferation. EP2 promotes vascular SMC proliferation and activates NF-kB signaling in endothelial cells and macrophages. However, the roles of A2bR, β-AR, and EP2 in AAA development remain unknown.
4.2. cGMP signaling
cGMP can be generated through stimulating soluble guanylate cyclases (sGCs) by NO or CO or stimulating particulate guanylate cyclases (pGCs) by natriuretic peptides (such as ANP or BNP). sGC is a heterodimer composed of α and β subunits. α and β subunits have similar domain structures, but dimerization is needed for sGC enzyme activity [72]. Both α and β subunits contain an amino-terminus heme domain (binds NO), a PAS domain (for dimerization), a coiled-coil domain, and a cyclase domain [72]. Each α and β subunit have different isoforms, e.g., α1and α2 for the α subunit and β1and β2 for the β unit [72]. sGCs with different subunit compositions may have different enzyme activity and tissue distribution. The most common dimer of sGC in the aorta is α1/β1[73]. Decreased sGC activity is found in human AA tissues [73]. One explanation is the switch of the α1 subunit isoform. In normal aorta, the α1 subunit is constituted with isoform A (α1A, non-functional) and isoform B (α1B, functional); however, in AA, the proportion of functional α1B is largely decreased, and the non-functional α1A becomes the predominant component of α1 subunit [73]. This isoform switch results in sGC inactivation [73]. However, another study showed increased sGC activity in TAA of human and animal MFS, and inhibition of sGC/cGMP signaling by sGC inhibitor ODG reverts TAA in the MFS animal model [74]. Supraphysiological NO levels and increased plasma cGMP levels were also found in humans and mice with MFS [74]. Moreover, inducible NO synthase (iNOS) expression, which catalyzes NO production, is found to be increased in aneurysmal tissues of AAA patients and Elastase-induced rat models [75, 76]. These results suggest an activated sGC/cGMP signaling in aneurysm development. Thus, the regulation and function of sGC/cGMP signaling in AA/AD remain controversial. This may be related to the complexity of NO-mediated regulation of vascular SMC functions. For example, the effect of NO on vascular relaxation could be mediated by the canonical NO/sGC/cGMP signaling or by a non-canonical pathway that depends on superoxide anion (O2−) but not the sGC/cGMP signaling [55]. Although the sGC subunit isoform switch occurred in AA, how the nonfunctional sGC isoform contributes to functional changes of sGC in vascular SMCs remains to be further investigated.
PKG is a predominant effector molecule that mediates the cGMP effects in various cell types. PKG is encoded by two genes, PRKG1 and PRKG2. PKG1 (encoded by PRKG1) is the predominant one expressed in vascular SMCs. A previous study reported increased PKG activity in the aorta from human and mouse MFS. Silencing PKG1 by shRNA attenuated TAA in MFS animal model [74]. A Sanger sequencing study identified a PRKG1 variant among familial TAA patients. PRKG1 c.530G>A (p.Arg177Gln) altered the high-affinity cGMP binding site within the regulatory domain, which results in a cGMP-independent constitutively activation [77]. Transgenic mice carrying the PRKG1R177Q mutation exhibited increased PKG activity, age-dependent aortic dilation, increased SMC apoptosis, and elastin fiber break [78]. These findings suggest that PKG1 overactivation promotes TAA.
4.3. Cyclic nucleotide phosphodiesterases (PDEs)
Cyclic nucleotide phosphodiesterases (PDEs) are important in modulating cyclic nucleotide signaling through hydrolyzing cAMP and/or cGMP. The PDE superfamily comprises 11 families with different genes and many transcript variants. Each PDE has a unique substrate specificity, kinetic property, and tissue-specific expression. Generally, the protein structure of PDE includes a regulatory region (R region) in the N-terminus and a catalytic region (C region) in the C-terminus. R region contains protein binding sites, such as Ca2+/Calmodulin binding sites in PDE1. These protein binding sites decide the PDE activation, subcellular location, and many other functions. C region contains cAMP or cGMP hydrolyzing sites, determining the affinity and dynamics for hydrolyzing cAMP/cGMP. Different PDEs show distinct substrate specificities, either specific for cAMP (e.g., PDE4), specific for cGMP (e.g., PDE5), or for both cAMP and cGMP (e.g., PDE1). The substrate specificities of different PDEs have been reviewed in many review articles [79]. Herein, we focus on PDEs’ expression, function, and mechanism in AA/AD (Figure 2).
Figure 2.
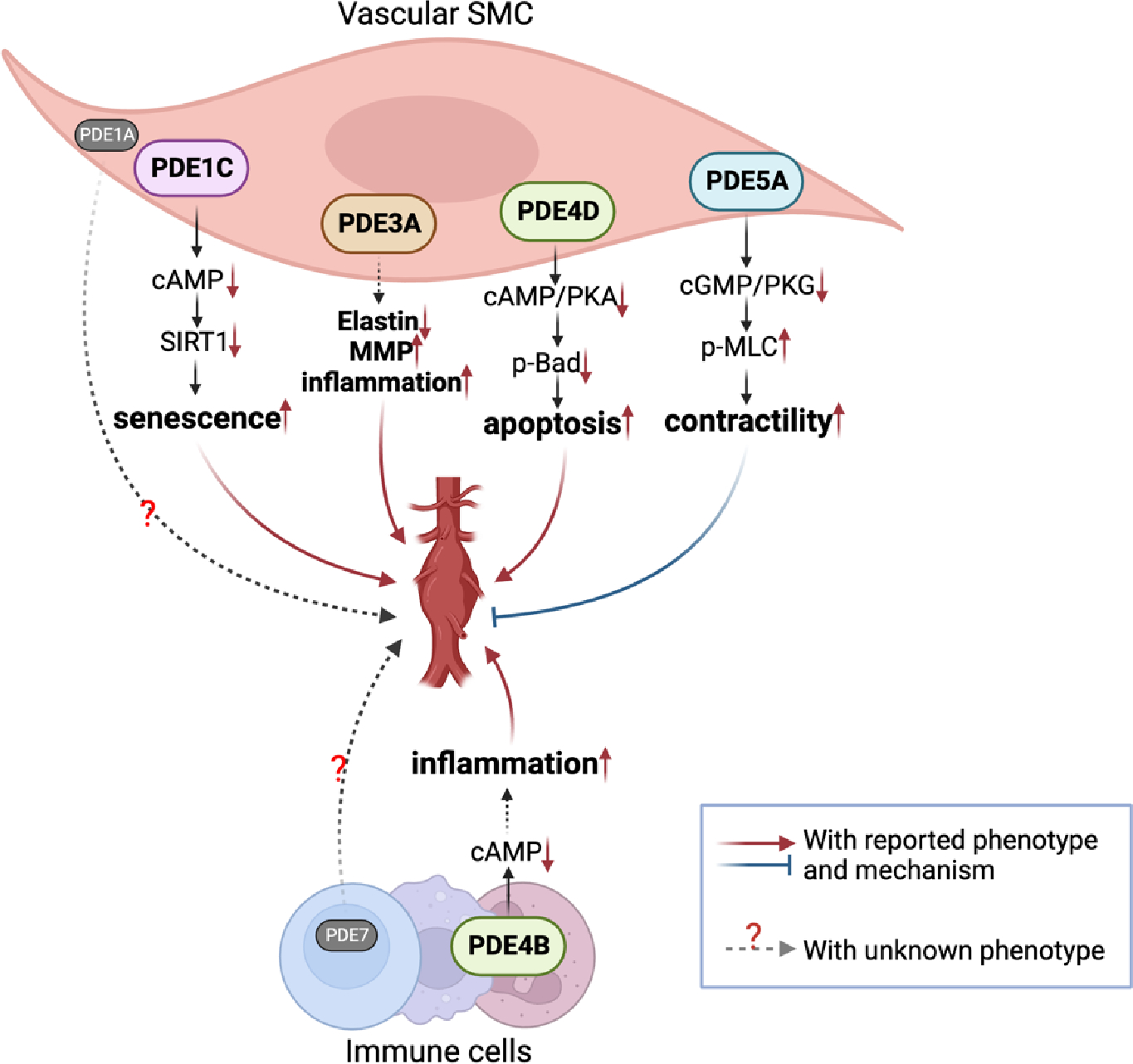
The role of different PDEs in aortic aneurysm/dissection development. PDE1C contributes to AAA by antagonizing cAMP mediated SIRT1 activation and accelerating vascular SMC senescence. PDE3A contributes to AAA development by promoting elastin degradation, MMP production, and inflammation. PDE4B is mainly expressed in immune cells. PDE4B mediates perivascular inflammation by antagonizing the anti-inflammatory cAMP-signaling in immune cells. PDE4D contributes to AAA by antagonizing the cAMP/PKA/p-BAD signaling pathway and mediating vascular SMC apoptosis. PDE5A inactivation contributes to AAA may be due to promoting the cGMP/PKG/MLCP pathway and vascular SMC relaxation. The vascular contractile function is important for minimizing vessel wall stress. PDE1A is mainly expressed in vascular SMC. PDE7 is mainly expressed in immune cells, such as T cells. The roles of PDE1A and PDE7 in AAA development remains unknown. Bad, BCL2-antagonist of cell death; MLC, myosin light chain.
4.3.1. PDE1
PDE1 family members are encoded by three genes, PDE1A, PDE1B, and PDE1C [80]. PDE1 is featured by the Ca2+/Calmodulin (CAM) binding sites in the regulatory region, thus activated upon Ca2+ elevation and referred to as Ca2+/CAM-stimulated PDE [80, 81]. Different PDE1 subtypes have distinct substrate affinities: PDE1A and 1B have much higher affinities for cGMP than cAMP, while PDE1C has similar high affinities for cAMP and cGMP. In vasculature, the function of PDE1 appears to be mostly focused on vascular SMCs and myofibroblasts [82, 83]. Under normal conditions, PDE1A is the predominant PDE1 member in vascular SMCs [84–86]. In contractile SMCs, PDE1A is expressed in the cytosol, primarily responsible for regulating SMC contractility. This is consistent with the findings that PDE1A contributes to blood pressure regulation [87, 88]. However, in synthetic SMCs, PDE1A is expressed mainly in the nucleus, which is primarily responsible for regulating the SMC growth [89]. Human genetic studies identified that PDE1A SNP is associated with diastolic blood pressure and carotid intima-media thickness [90, 91]. The expression and function of PDE1A in AA/AD remain unknown.
The reports of PDE1B in vascular tissues are limited. One study showed that PDE1B expression was detected in monkey vascular SMCs but not in human or rodent vascular SMCs [92]. Although limited PDE1B is detected in human and rodent vascular SMCs, PDE1B expresses and functions in immune cells, such as macrophages and lymphocytes [82]. As chronic inflammation is critical for AA/AD, the role of PDE1B in AA/AD warrants further study. PDE1C expression is almost undetectable in normal vasculature, but drastically upregulated in synthetic SMCs under the culture or vascular lesions [84, 85].
The expression and function of PDE1C in AAA have been explored recently [85]. For example, PDE1C expression is induced during AAA development, primarily in synthetic SMC-like cells of human and mouse AAA tissues. No significant PDE1C expression was detected in normal aortic tissues. PDE1C knockout attenuated AAA formation in ApoE−/− mice with AngII-infusion and C57BL/6J mice treated with BAPN and periaortic elastase [85]. Aortic SMCs from PDE1C knockout mice exhibited longer maintaining time under the culture in vitro and lower levels of senescent markers than SMCs from PDE1C wild-type mice [85]. PDE1C inactivation attenuated aortic SMC senescence in human and mouse aortic SMCs. This is consistent with a previous report that PDE1 plays a role in vascular aging [91]. PDE1C hydrolyzes both cAMP and cGMP; however, the conventional downstream effectors, such as PKA, EPAC, and PKG, are not involved in PDE1C regulation of senescence in vascular SMCs [85]. Interestingly, PDE1C regulates vascular SMC senescence via cAMP-mediated activation of SIRT1, a longevity gene that encodes a NAD-dependent protein deacetylase [85]. cAMP can directly bind to and activate SIRT1 [93, 94]. PDE1C suppresses the SIRT1 activation by decreasing cAMP [85]. A pan PDE1 inhibitor, IC86340, suppressed VSMC senescence and attenuated aneurysm development in mice [85]. The nutraceutical reagent, vinpocetine, acting as a PDE1 inhibitor, attenuated experimental AAA induced by BANP and the periaortic elastase treatment in mice [95]. These results suggest that pharmacological inhibition of all PDE1 isozymes may elicit a net beneficial effect against AAA. ITI-214 is a newly developed pan PDE1 inhibitor that has beneficial effects on memory performance and heart failure in clinical and animal studies [96, 97]. Thus, PDE1 inhibition may represent a new therapeutic strategy in treating AA/AD.
4.3.2. PDE3
PDE3 family members are encoded by 2 genes, PDE3A and PDE3B. PDE3A is highly enriched in heart, vascular, and placental smooth muscle, corpus cavernosum smooth muscle, and platelets, while PDE3B is highly enriched in white and brown adipose tissues. Although PDE3A and PDE3B have different structures in R regions, their C regions are similar [98]. PDE3 hydrolyzes both cAMP and cGMP, with much higher Vmax for cAMP than for cGMP. Thus, cGMP inhibits cAMP hydrolysis by PDE3, and PDE3 is called a cGMP-inhibited cAMP PDE [99, 100]. PDE3A has three splicing variants, PDE3A1, PDE3A2 and PDE3A3. They have the same structures in the C regions but differ in the N-terminal structures [98, 101]. PDE3A2 is the predominant variant in vascular SMCs and platelets, while PDE3A1 is mainly in cardiomyocytes and PDE3A3 in the placenta [101].
The function of PDE3 in AA/AD has been investigated in various animal models using PDE3 inhibitors, such as cilostazol [102, 103] or K-134 [104]. Cilostazol, a pan PDE3 inhibitor, is clinically used to treat intermittent claudication by dilating the vessel and avoiding platelet aggregation [105]. In an intraluminal elastase-infusion rat AAA model and an AngII/ApoE−/− mouse AAA model, cilostazol decreased the AAA development, accompanied by decreased elastin degradation, MMP activity, chronic inflammation, and ROS production in the AAA tissues [102, 103]. K-134, another pan PDE3 inhibitor, exhibits higher properties in blocking antiplatelet activity than cilostazol. In a hypoperfusion-induced aneurysm rat model, K-134 significantly decreased the AAA diameter and rupture, improving the survival rate [104]. Similar results were also found in the intraluminal elastase-infusion rat AAA model and AngII/ApoE−/− mouse AAA model with K-134 [104]. K-134 treatment decreased medial disruption, macrophage infiltration, ROS generation, MMP activity, and AAA formation [104].
Despite the protective effects of PDE3 inhibition against AAA, the PDE3 subtype that is mainly responsible for the effects of PDE3 inhibition on AAA remains unknown. A previous study using PDE3A and PDE3B knockout mice has defined that PDE3A is predominantly responsible for the effects of cilostazol on the cardiovascular system, such as the cardiac contractile function, heart rate, and platelet function [106]. PDE3A is reported to involve blood pressure regulation [107]. Gain-of-function mutations in the N-terminus (T445N) or C region (R862C) increase the catalytic activity of PDE3A [101, 107]. T445N mutation only happens in PDE3A1 and PDE3A2, but not PDE3A3 due to their structure differences in the N-terminus [101]. These gain-of-function mutations increase the risk of mendelian hypertension and brachydactyly type E (HTNB) in patients [101, 108]. In the hypertension animal model, rats carrying this mutation exhibited more severe hypertension [107]. Vascular SMCs with this mutation had lower cAMP content, reduced PKA activity, and increased MLC phosphorylation, thus leading to SMC constriction [101, 108]. PDE3A/cAMP/PKA also regulates the cell cycle through P53/P21 [101, 107, 109]. A gain-of-function mutation of PDE3A results in vascular SMC proliferation and vascular hyperplasia. The gain-of-function PDE3A mutation causes hypertension but is protective from cardiac damage [98, 110]. Therefore, developing novel strategies for cell type-specific targeting of PDE3A is important.
4.3.3. PDE4
PDE4 is a cAMP-specific PDE. PDE4 activity is one of the predominant cAMP-hydrolyzing PDE activities in vascular SMCs [111]. For example, PDE4 inhibitor Ro 20-1724 decreased 40% of PDE activity in vascular SMCs [111]. PDE4 family members are encoded by 4 genes, PDE4A, PDE4B, PDE4C, and PDE4D. The compositions and relative abundances of different PDE4 subtypes varied with cell types. In mouse vascular SMCs, the expression of PDE4 subtypes: PDE4B>PDE4D>PDE4A>PDE4C; however, in human vascular SMCs, PDE4A and PDE4B expression levels are almost equivalent [112, 113].
PDE4B and PDE4D have been respectively studied in AAA. In human and mouse AAA, PDE4B expression is increased in inflammatory cells [114]. Blocking PDE4B by pan PDE4 inhibitor, Rolipram, decreased the AAA incidence, aortic diameter, and rupture. Rolipram also decreases immune cell infiltration, such as macrophages, lymphocytes, and neutrophils in AAA tissues. PDE4D expression is also increased in mouse AAA tissues, particularly in aortic SMCs [112]. SMC-specific PDE4D knockout decreased aortic diameter and elastin degradation in mouse AAA [112]. PDE4D promoted aortic SMC apoptosis since PDE4D deficiency suppressed aortic SMC apoptosis [112]. PDE4D inactivation elevated the cAMP level and activated the PKA, subsequently attenuating the vascular SMC apoptosis [112]. PKA can suppress cell apoptosis by phosphorylating BCL2-antagonist of cell death (Bad). Indeed, increased PKA activity by PDE4D inhibition or knockdown elevated Bad phosphorylation, one of the mechanisms by which PDE4D regulates vascular SMC viability [112].
A synergistic effect between PDE3 and PDE4 inhibition has been reported in vascular SMCs. For example, for PDGFBB-induced vascular SMC migration, a PDE3 inhibitor alone did not significantly alter the cell migration; however, when combined with a PDE4 inhibitor, the inhibitive effect was augmented [111]. An additive effect between PDE3 and PDE4 inhibition is also documented in treating chronic obstructive pulmonary disease (COPD). PDE4 inhibition with roflumilast suppressed the inflammation, and PDE3/PDE4 dual-inhibition showed an additive effect on the anti-inflammation [115]. Thus, the combined PDE3 and PDE4 inhibition may have synergistic or additive effects on AAA, which deserves further investigation.
4.3.4. PDE5
PDE5 family members hydrolyze cGMP specifically, encoded by the PDE5A gene with 3 splice variants [79]. PDE5 is highly expressed in contractile SMCs, negatively regulating the NO-cGMP-PKG signaling pathway and SMC relaxation. PDE5 inhibitors are clinically used to treat erectile dysfunction and pulmonary hypertension by relaxing blood vessels in the corresponding tissues [116].
A series of cases reported AD after using PDE5 inhibitor sildenafil or tadalafil in patients [117–121], which is summarized in Table 2. The most common syndrome is sudden chest pain about 2 hours after sildenafil or tadalafil use, accompanied by fainting in some cases (Table 2). The dissection mostly happens in the thoracic aorta, such as ascending or descending aorta, and an intima tear was detected. The PDE5 inhibitor-caused AD could happen at any age, with or without any medical history, medicine history, or drug abuse. Those patients received management in line with the guidelines then, but it has a high mortality among them even though they received surgery replacement right after the syndrome onset. Besides AD, PDE5 inhibitors caused arterial dissection in the cerebral artery and vertebral artery [103, 104], which could also be lethal. These cases strongly suggest the relationship between PDE5 and dissection. The affected areas in MFS or TAA patients have lower PDE5A expression than the control aorta [122].
Table 2.
Reported cases of sildenafil/tadalafil abuse causing aortic dissection.
| Patients | Syndrome | Time and PDE5i dosage | Long-term PDE5i usage | Medical history | Medication history | Risk factors | Sexual intercourse (Y/N) | Diagnosis | Management and follow-up | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 28-year-old man | Sudden chest pain and fainting | 2 h after the use of 50 mg sildenafil | 1 year | No history of cardiac problems | No history of medication | Heavy smoker | N | Ascending aortic dissection | Bentall procedure during operation. Patient recovered. | Aortic dissection due to sildenafil abuse. Interact Cardiovasc Thorac Surg, 2009 |
| 42-year-old man | Pain in his right groin and leg. onset of atypical chest pain radiating to the back | 1 h after the use of 50mg sildenafil | Not mentioned | Hypertension | Losartan | Heavy smoker, cocaine abuse | Y | Descending aortic dissection with an intimal tear in aortic arch. | Nitroglycerin, labetalol, metoprolol, captopril, and ranitidine and closely monitored but died 12 days after admission | Acute aortic dissection after cocaine and sildenafil abuse. J Emerg Med. 2001 |
| 61-year-old man | Abdominal pain, diaphoresis, vomiting, diarrhea and chest pain | 30 minutes after ingestion of 50 mg of sildenafil. | 2 years | Arterial hypertension, venous insufficiency, and atrial fibrillation | Digitoxin, isosorbid-5-mononitrate, allopurinol, ranitidine, flavonoid, diosmin and phenprocoumon | Not mentioned | N | Ascending aortic dissection with an intimal tear above the aortic valve. | An aortic prosthesis was implanted. Patients died 4 days after surgery. | Aortic dissection after sildenafil-induced erection. South Med J, 2006 |
| 63-year-old man | Chest pain, excessive sweating, nausea, and collapsing | 1h after 20mg tadalafil | The second time | Controlled hypertension and erectile dysfunction but no evident cardiac problems | β-blocker | No history of drug abuse. | Y | Descending aortic dissection | Endoprosthesis and recovered. | Type B aortic dissection after the use of tadalafil. Ann Thorac Surg, 2012. |
| 63-year-old male | Died with bruise on the chest area. No apparent symptoms before death. | 45 ng/ml Sildenafil in blood | Not regularly | Hypertension | Not mentioned | Not mentioned | Mot mentioned | Ascending aortic dissection. | Not applied | 2021 Aortic Dissection After Sildenafil Use, A Rare Cooccurence, Case Report of An Autopsy |
| 66-year-old man | Chest pain, fatigue and suddenly collapsed. | 30–60 min after 50–100 mg oral dose of sildenafil | More than 1 year | No history of cardiac problems | No medication history | not mentioned | Y | Ascending and descending aortic dissection | Not applied before death. | Post-coital death in chronic sildenafil abuser. J Geriatr Cardiol, 2020. |
| 45-year-old man | Headache and left-sided weakness | 2 h after the use of 100 mg sildenafil | 1 year | Hypertension and diabetes mellitus | No medication history | Nonsmoker | N | Dissection at the right anterior cerebral artery | Antiplatelet and prophylactic antiepileptic medication. Patient died 22 days after treatment. | Anterior Cerebral Artery Dissection Due to Sildenafil Use. Noro Psikiyatr Ars. 2017 |
| 49-year-old man | Hemiparesthesia of the right side, neck pain and ataxia of the right arm | 300 mg sildenafil | 2 years | No medical history | Not mentioned | Smoker | N | Dissection of the right vertebral artery | Heparin intravenously for 6 days, then medication was switched to clopidogrel. Dissection is resolved. | Vertebral artery dissection associated with sildenafil abuse. Journal of Clinical Neuroscience.2013 |
In a periaortic elastase-induced mouse AAA model, PDE5A expression was reduced in the medial areas of AAA tissues [123]. Sildenafil treatment aggravated the elastase-induced AAA [123], consistent with the phenotype observed in patients with PDE5 inhibitors. An increased cGMP content, PKG activity, and decreased MLC phosphorylation were observed in the medial layers of sildenafil-treated aortas [123], which may reflect a decreased aortic contractile function. Although the animal study with sildenafil and the reported clinical cases strongly suggest a role of loss-of-PDE5A function in AA/AD, further studies with genetically engineered PDE5A mice may provide better evidence for the role and mechanistic insight of PDE5A in AA/AD.
5. Conclusion and perspective
This review discussed the clinical reports and/or experimental studies of cAMP/cGMP signaling regulators/modulators in AA/AD, including Gs, PKG1, PDE3 (probably PDE3A), PDE4B, PDE4D, PDE5A (Figure 2), and their inhibiors (Table 3). AA/AD are multifactorial diseases that involve various cell types (e.g., immune cells, vascular SMCs, etc.), pathogenic mechanisms (e.g., inflammation, cell death, matrix remodeling, etc.), and risk factors (e.g., hyperlipidemia, hypertension, ROS, smoking, etc.). cAMP and cGMP exist and function in all cell types and are modulated by different PDEs. How dysregulation of the cyclic nucleotide signaling contributes to AA/AD is far from completely understood. The studies of PDEs mostly focus on AAA in animal models. Their roles in other parts of AA and AD remain to be investigated due to the different mechanistic actions of different AAs. Many other PDEs are expressed in cells critical for AA/AD, and their roles deserve to be studied. Several previously published studies reported AA’s single-cell RNA sequence (scRNA-seq) data in different species. For example, Yanming et al. reported the scRNA-seq data of human TAD samples [124]. Through re-analyzing these data, we found that several PDEs are changed in TAD samples. For example, besides PDE3A and PDE5A, PDE1A is another PDE isozyme highly enriched in contractile vascular SMCs (Figure 3A), and PDE1A expression is reduced in SMCs of TAD (Figure 3B). Thus, the role of PDE1A in AA/AD is currently studied in the authors’ lab. In addition, PDE7A, a cAMP-hydrolyzing PDE [79], is upregulated in immune cells of human TAD (Figure 3C). It is known that PDE7A is mainly enriched in immune cells, such as T cells, which are the largest immune cell population in the TAD samples [124]. PDE7A shows a strong relationship with T cell activation. For example, increasing PDE7A expression is found in activated T cells [125]; PDE7A knockdown or inhibition in T cells affects T cell activation [125, 126]. Given the role of T cells, it warrants further investigation into the role of PDE7 in AA/AD. PDEs are important pharmacological targets for various diseases, and several PDE inhibitors are currently used as therapeutic reagents. Understanding the roles of PDEs in AA/AD may facilitate the development of PDE-inhibitor replacement therapy in AA/AD or predict the side effects of AA/AD when using PDE inhibitors for treating other diseases.
Table 3.
PDE inhibitors in AA/AD development.
| Inhibitor name | PDE selectivity | Effect on AA/AD | Mechanism | Reported indications |
|---|---|---|---|---|
| Vinpocetine | PDE1 | Ameliorates AA in elastase-induced mouse model. | Suppressed elastin degradation, media smooth muscle cell depletion, collagen fibers remodeling and macrophage infiltration. | Cerebrovascular disorders, cognitive impairment, dietary supplement. |
| Cilostazol | PDE3 | Ameliorates AA in elastase-infusion rat model and an AngII/ApoE−/− mouse model | Decreased elastin degradation, MMP activity, chronic inflammation, and ROS production. | Intermittent claudication |
| K-134, Phase II | PDE3 | Ameliorates AA elastase-infusion rat model and AngII/ApoE−/− mouse model. | Decreased medial disruption, macrophage infiltration, ROS generation, MMP activity. | Inhibit platelet aggregation and thrombus formation |
| Rolipram, Phase II | PDE4 | Ameliorates AA in AngII/ApoE−/− mouse model. | Decreased immune cell infiltration. Suppressed SMC apoptosis via the cAMP/PKA/pBad axis. | Major depressive disorder, Huntington’s disease, multiple sclerosis |
| Sildenafil | PDE5 | Aggravates AA in elastase-induced mouse model. | Induced vascular relaxation through cGMP/PKG/pMLC axis in SMCs. | Erectile dysfunction, pulmonary arterial hypertension |
Figure 3.
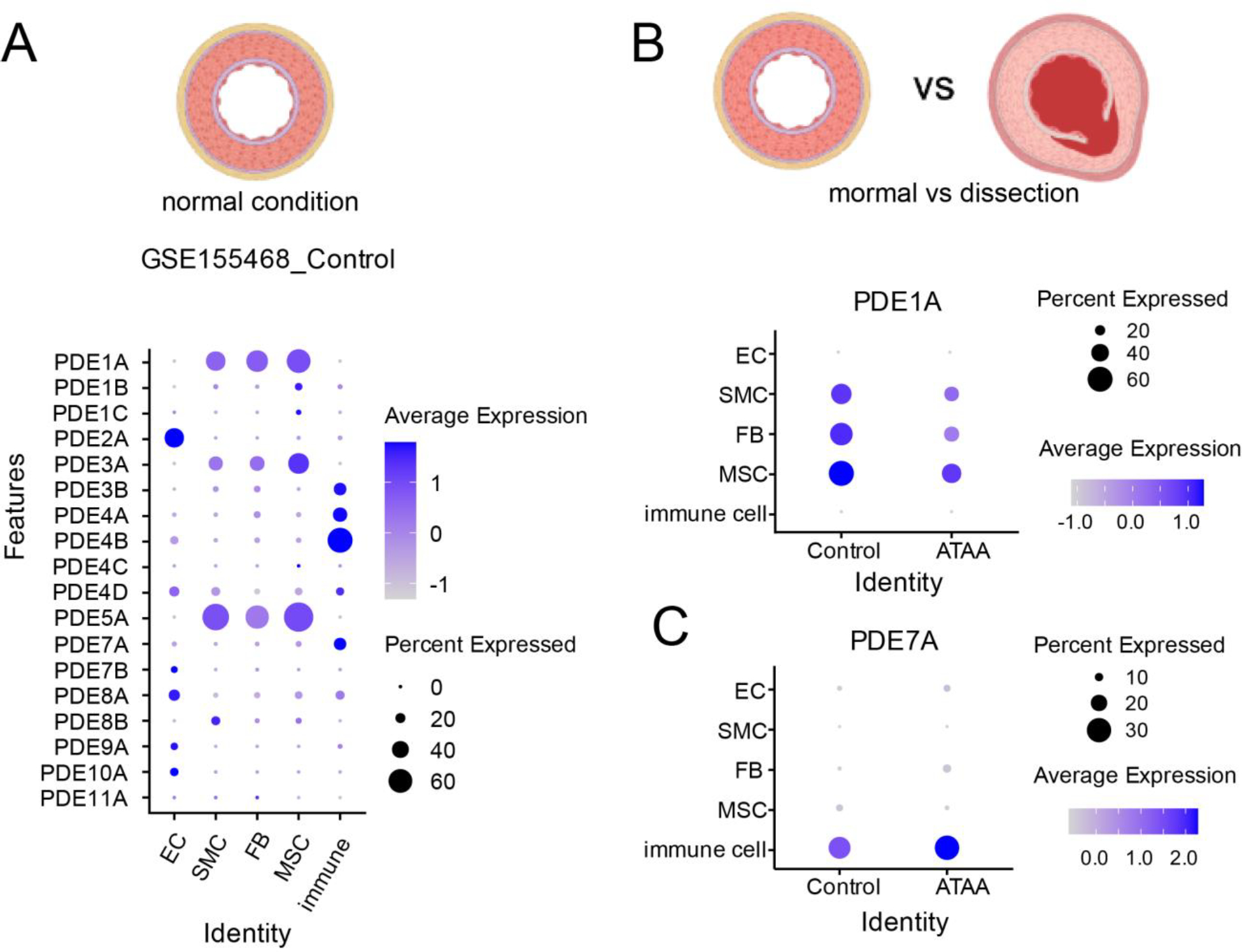
Sc-RNA seq reveals different PDE expression in the aorta (re-analyzed from GSE155468). A. PDE expression in the normal aorta of ECs, SMCs, fibroblast FBs, and immune cells. B. PDE1A expression in normal condition (control) and ascending thoracic aortic aneurysm (ATAA). C. PDE7A expression in normal condition (control) and ascending thoracic aortic aneurysm (ATAA). EC, endothelial cell; FB, fibroblast; MSC, mesenchymal stem cell; SMC, smooth muscle cell.
Acknowledgements
Figure 1 and 2 were created with BioRender.com.
Funding
This work was financially supported by the National Institute of Health HL154318, HL162259, and HL170024-01 (to C.Y.).
Non-standard Abbreviations and Acronyms
- AA
aortic aneurysm
- AD
aortic dissection
- SMC
smooth muscle cell
- cAMP
intracellular cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- PKA
protein kinase A
- PKG
protein kinase G
- AC
adenylyl cyclase
- GC
guanylyl cyclase
- PDE
phosphodiesterase
- MFS
Marfan syndrome
- AngII
Angiotensin II
- BAPN
β-Aminopropionitrile
- MMP
matrix metalloproteinase
- ECM
extracellular matrix
- GPCR
G-protein-coupled receptor
- Gs
stimulatory G-protein
- NO
nitric oxide
Footnotes
Declaration of Competing interest
The authors declared no conflict of interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Ting Shu: Conceptualization, Writing-original draft, Writing – review&editing, Visualization. YitianZhou: Software, Visualization. Chen Yan: Conceptualization, Writing – review&editing, Supervision, Funding acquisition.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
ScRNA-seq data can be accessed at GSE155468.
References
- 1.Writing Committee M, et al. , 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol, 2022. 80(24): p. e223–e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isselbacher EM, et al. , 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation, 2022. 146(24): p. e334–e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vilacosta I, et al. , Acute Aortic Syndrome Revisited: JACC State-of-the-Art Review. J Am Coll Cardiol, 2021. 78(21): p. 2106–2125. [DOI] [PubMed] [Google Scholar]
- 4.Krywanczyk A, et al. , Thoracic Aortic Aneurysm and Dissection: Review and Recommendations for Evaluation. Am J Forensic Med Pathol, 2023. [DOI] [PubMed] [Google Scholar]
- 5.Zeigler SM, Sloan B, and Jones JA, Pathophysiology and Pathogenesis of Marfan Syndrome. Adv Exp Med Biol, 2021. 1348: p. 185–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakai LY, et al. , FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene, 2016. 591(1): p. 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen YH, et al. , Aortic Aneurysms and Dissections Series. Arterioscler Thromb Vasc Biol, 2020. 40(3): p. e37–e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhry SS, et al. , Fibrillin-1 regulates the bioavailability of TGFbeta1. J Cell Biol, 2007. 176(3): p. 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asta L, et al. , Genetic Basis, New Diagnostic Approaches, and Updated Therapeutic Strategies of the Syndromic Aortic Diseases: Marfan, Loeys-Dietz, and Vascular Ehlers-Danlos Syndrome. Int J Environ Res Public Health, 2023. 20(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velchev JD, et al. , Loeys-Dietz Syndrome. Adv Exp Med Biol, 2021. 1348: p. 251–264. [DOI] [PubMed] [Google Scholar]
- 11.Downey RT and Aron RA, Thoracic and Thoracoabdominal Aneurysms: Etiology, Epidemiology, and Natural History. Anesthesiol Clin, 2022. 40(4): p. 671–683. [DOI] [PubMed] [Google Scholar]
- 12.Gao J, et al. , The mechanism and therapy of aortic aneurysms. Signal Transduct Target Ther, 2023. 8(1): p. 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris KM, et al. , Early Mortality in Type A Acute Aortic Dissection: Insights From the International Registry of Acute Aortic Dissection. JAMA Cardiol, 2022. 7(10): p. 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lasica RM, et al. , Early and Late Mortality Predictors in Patients with Acute Aortic Dissection Type B. Cardiol Res Pract, 2022. 2022: p. 7869356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obel LM, et al. , Clinical Characteristics, Incidences, and Mortality Rates for Type A and B Aortic Dissections: A Nationwide Danish Population-Based Cohort Study From 1996 to 2016. Circulation, 2022. 146(25): p. 1903–1917. [DOI] [PubMed] [Google Scholar]
- 16.Pereira L, et al. , Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet, 1997. 17(2): p. 218–22. [DOI] [PubMed] [Google Scholar]
- 17.Bunton TE, et al. , Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res, 2001. 88(1): p. 37–43. [DOI] [PubMed] [Google Scholar]
- 18.Lima BL, et al. , A new mouse model for marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS One, 2010. 5(11): p. e14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Judge DP, et al. , Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest, 2004. 114(2): p. 172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campens L, et al. , Intrinsic cardiomyopathy in Marfan syndrome: results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr Res, 2015. 78(3): p. 256–63. [DOI] [PubMed] [Google Scholar]
- 21.Cavanaugh NB, et al. , A Novel Murine Model of Marfan Syndrome Accelerates Aortopathy and Cardiomyopathy. Ann Thorac Surg, 2017. 104(2): p. 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawada H, et al. , Twenty Years of Studying AngII (Angiotensin II)-Induced Abdominal Aortic Pathologies in Mice: Continuing Questions and Challenges to Provide Insight Into the Human Disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 2022. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassis LA, et al. , ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol, 2009. 296(5): p. H1660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daugherty A, Cassis LA, and Lu H, Complex pathologies of angiotensin II-induced abdominal aortic aneurysms. J Zhejiang Univ Sci B, 2011. 12(8): p. 624–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trachet B, et al. , Angiotensin II infusion into ApoE−/− mice: a model for aortic dissection rather than abdominal aortic aneurysm? Cardiovasc Res, 2017. 113(10): p. 1230–1242. [DOI] [PubMed] [Google Scholar]
- 26.Luo W, et al. , Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation, 2020. 141(1): p. 42–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rateri DL, et al. , Prolonged infusion of angiotensin II in apoE(−/−) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol, 2011. 179(3): p. 1542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, et al. , Hypercholesterolemia Accelerates Both the Initiation and Progression of Angiotensin II-induced Abdominal Aortic Aneurysms. Ann Vasc Med Res, 2020. 6(2). [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, et al. , Hypercholesterolemia Induced by a PCSK9 Gain-of-Function Mutation Augments Angiotensin II-Induced Abdominal Aortic Aneurysms in C57BL/6 Mice-Brief Report. Arterioscler Thromb Vasc Biol, 2016. 36(9): p. 1753–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alsiraj Y, et al. , Sex Chromosome Complement Defines Diffuse Versus Focal Angiotensin II-Induced Aortic Pathology. Arterioscler Thromb Vasc Biol, 2018. 38(1): p. 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue C, et al. , Mouse Abdominal Aortic Aneurysm Model Induced by Perivascular Application of Elastase. J Vis Exp, 2022(180). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu G, et al. , A novel chronic advanced stage abdominal aortic aneurysm murine model. J Vasc Surg, 2017. 66(1): p. 232–242 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berman AG, et al. , Experimental aortic aneurysm severity and growth depend on topical elastase concentration and lysyl oxidase inhibition. Sci Rep, 2022. 12(1): p. 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Krishna S, and Golledge J, The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis, 2013. 226(1): p. 29–39. [DOI] [PubMed] [Google Scholar]
- 35.Yamanouchi D, et al. , Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg, 2012. 56(2): p. 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei H, et al. , Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-beta signaling. Proc Natl Acad Sci U S A, 2012. 109(14): p. E841–50. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Kuang SQ, et al. , Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol, 2013. 33(9): p. 2172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Da X, et al. , AGGF1 therapy inhibits thoracic aortic aneurysms by enhancing integrin alpha7-mediated inhibition of TGF-beta1 maturation and ERK1/2 signaling. Nat Commun, 2023. 14(1): p. 2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu H, et al. , Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J Am Heart Assoc, 2021. 10(24): p. e023601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo DC, et al. , Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet, 2007. 39(12): p. 1488–93. [DOI] [PubMed] [Google Scholar]
- 41.Zhu L, et al. , Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet, 2006. 38(3): p. 343–9. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, et al. , Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet, 2010. 87(5): p. 701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyle JJ, et al. , A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson CF and Taylor WJ, Effect of hydralazine on aortic rupture induced by B-aminopropionitrile in turkeys. Circulation, 1982. 65(4): p. 704–8. [DOI] [PubMed] [Google Scholar]
- 45.Bogunovic N, et al. , Impaired smooth muscle cell contractility as a novel concept of abdominal aortic aneurysm pathophysiology. Sci Rep, 2019. 9(1): p. 6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang D, et al. , Cellular senescence and abdominal aortic aneurysm: From pathogenesis to therapeutics. Front Cardiovasc Med, 2022. 9: p. 999465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H, et al. , Single-Cell RNA Sequencing Reveals Heterogeneity of Vascular Cells in Early Stage Murine Abdominal Aortic Aneurysm-Brief Report. Arterioscler Thromb Vasc Biol, 2021. 41(3): p. 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marquez-Sanchez AC and Koltsova EK, Immune and inflammatory mechanisms of abdominal aortic aneurysm. Front Immunol, 2022. 13: p. 989933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye T, et al. , Relationships Between Perivascular Adipose Tissue and Abdominal Aortic Aneurysms. Front Endocrinol (Lausanne), 2021. 12: p. 704845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong W, Tian Y, and Li L, T cells in abdominal aortic aneurysm: immunomodulation and clinical application. Front Immunol, 2023. 14: p. 1240132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu CL, et al. , Eosinophils Protect Mice From Angiotensin-II Perfusion-Induced Abdominal Aortic Aneurysm. Circ Res, 2021. 128(2): p. 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raffort J, et al. , Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol, 2017. 14(8): p. 457–471. [DOI] [PubMed] [Google Scholar]
- 53.Nienaber CA, et al. , Aortic dissection. Nat Rev Dis Primers, 2016. 2: p. 16053. [DOI] [PubMed] [Google Scholar]
- 54.Kamato D, et al. , Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Galpha/q,11. Front Cardiovasc Med, 2015. 2: p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin X, et al. , Smooth muscle-specific Gsalpha deletion exaggerates angiotensin II-induced abdominal aortic aneurysm formation in mice in vivo. J Mol Cell Cardiol, 2019. 132: p. 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Billington CK and Penn RB, Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res, 2003. 4(1): p. 2. [PMC free article] [PubMed] [Google Scholar]
- 57.Bhamidipati CM, et al. , Adenosine 2A receptor modulates inflammation and phenotype in experimental abdominal aortic aneurysms. FASEB J, 2013. 27(6): p. 2122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dubey RK, et al. , Adenosine inhibits growth of human aortic smooth muscle cells via A2B receptors. Hypertension, 1998. 31(1 Pt 2): p. 516–21. [DOI] [PubMed] [Google Scholar]
- 59.Dubey RK, et al. , Adenosine, Via A(2B) Receptors, Inhibits Human (P-SMC) Progenitor Smooth Muscle Cell Growth. Hypertension, 2020. 75(1): p. 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frishman WH, Cardiology patient page. Beta-adrenergic blockers. Circulation, 2003. 107(18): p. e117–9. [DOI] [PubMed] [Google Scholar]
- 61.Werstiuk ES and Lee RM, Vascular beta-adrenoceptor function in hypertension and in ageing. Can J Physiol Pharmacol, 2000. 78(6): p. 433–52. [PubMed] [Google Scholar]
- 62.Johnson R, et al. , Regulation of human vascular smooth muscle cell migration by beta-adrenergic receptors. Am Surg, 2006. 72(1): p. 51–4. [PubMed] [Google Scholar]
- 63.Sahil F, et al. , Association Between Long-Term Use of Non-steroidal Anti-inflammatory Drugs and Hyperkalemia in Diabetic Patients. Cureus, 2021. 13(6): p. e15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Genoni M, et al. , Chronic beta-blocker therapy improves outcome and reduces treatment costs in chronic type B aortic dissection. Eur J Cardiothorac Surg, 2001. 19(5): p. 606–10. [DOI] [PubMed] [Google Scholar]
- 65.Siordia JA, Beta-Blockers and Abdominal Aortic Aneurysm Growth: A Systematic Review and Meta-Analysis. Curr Cardiol Rev, 2021. 17(4): p. e230421187502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aoki T, et al. , Prostaglandin E2-EP2-NF-kappaB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci Signal, 2017. 10(465). [DOI] [PubMed] [Google Scholar]
- 67.Aoki T, et al. , PGE(2)-EP(2) signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-kappaB. Br J Pharmacol, 2011. 163(6): p. 1237–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hiromi T, et al. , Excessive EP4 Signaling in Smooth Muscle Cells Induces Abdominal Aortic Aneurysm by Amplifying Inflammation. Arterioscler Thromb Vasc Biol, 2020. 40(6): p. 1559–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao RY, et al. , Prostaglandin receptor EP4 in abdominal aortic aneurysms. Am J Pathol, 2012. 181(1): p. 313–21. [DOI] [PubMed] [Google Scholar]
- 70.Bayston T, et al. , Prostaglandin E2 receptors in abdominal aortic aneurysm and human aortic smooth muscle cells. J Vasc Surg, 2003. 38(2): p. 354–9. [DOI] [PubMed] [Google Scholar]
- 71.Weintraub NL, et al. , Role of prostaglandin D2 receptors in the pathogenesis of abdominal aortic aneurysm formation. Clin Sci (Lond), 2022. 136(5): p. 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Montfort WR, Wales JA, and Weichsel A, Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxid Redox Signal, 2017. 26(3): p. 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin E, et al. , Alternative splicing impairs soluble guanylyl cyclase function in aortic aneurysm. Am J Physiol Heart Circ Physiol, 2014. 307(11): p. H1565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de la Fuente-Alonso A, et al. , Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO. Nat Commun, 2021. 12(1): p. 2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang J, et al. , Inducible nitric oxide synthase is present in human abdominal aortic aneurysm and promotes oxidative vascular injury. J Vasc Surg, 2003. 38(2): p. 360–7. [DOI] [PubMed] [Google Scholar]
- 76.Sigala F, et al. , Relationship between iNOS expression and aortic cell proliferation and apoptosis in an elastase-induced model of aorta aneurysm and the effect of 1400 W administration. Surgery, 2005. 137(4): p. 447–56. [DOI] [PubMed] [Google Scholar]
- 77.Guo DC, et al. , Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet, 2013. 93(2): p. 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwaerzer GK, et al. , Aortic pathology from protein kinase G activation is prevented by an antioxidant vitamin B(12) analog. Nat Commun, 2019. 10(1): p. 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keravis T and Lugnier C, Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol, 2012. 165(5): p. 1288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Francis SH, Corbin JD, and Bischoff E, Cyclic GMP-hydrolyzing phosphodiesterases. Handb Exp Pharmacol, 2009(191): p. 367–408. [DOI] [PubMed] [Google Scholar]
- 81.Samidurai A, et al. , Role of phosphodiesterase 1 in the pathophysiology of diseases and potential therapeutic opportunities. Pharmacol Ther, 2021. 226: p. 107858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan S and Yan C, PDE1 isozymes, key regulators of pathological vascular remodeling. Curr Opin Pharmacol, 2011. 11(6): p. 720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roks AJM, Phosphodiesterase-1 in the cardiovascular system. Cell Signal, 2022. 92: p. 110251. [DOI] [PubMed] [Google Scholar]
- 84.Cai Y, et al. , Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res, 2015. 116(7): p. 1120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang C, et al. , Cyclic nucleotide phosphodiesterase 1C contributes to abdominal aortic aneurysm. Proc Natl Acad Sci U S A, 2021. 118(31). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rybalkin SD, et al. , Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res, 2002. 90(2): p. 151–7. [DOI] [PubMed] [Google Scholar]
- 87.Wang X, et al. , Generation and phenotypic characterization of Pde1a mutant mice. PLoS One, 2017. 12(7): p. e0181087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laursen M, et al. , Novel selective PDE type 1 inhibitors cause vasodilatation and lower blood pressure in rats. Br J Pharmacol, 2017. 174(15): p. 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagel DJ, et al. , Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res, 2006. 98(6): p. 777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tam CHT, et al. , Identification of a Common Variant for Coronary Heart Disease at PDE1A Contributes to Individualized Treatment Goals and Risk Stratification of Cardiovascular Complications in Chinese Patients With Type 2 Diabetes. Diabetes Care, 2023. 46(6): p. 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bautista Nino PK, et al. , Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin Sci (Lond), 2015. 129(12): p. 1061–75. [DOI] [PubMed] [Google Scholar]
- 92.Rybalkin SD, et al. , Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J Clin Invest, 1997. 100(10): p. 2611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gerhart-Hines Z, et al. , The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell, 2011. 44(6): p. 851–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iside C, et al. , SIRT1 Activation by Natural Phytochemicals: An Overview. Front Pharmacol, 2020. 11: p. 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang C, et al. , Vinpocetine protects against the development of experimental abdominal aortic aneurysms. Clin Sci (Lond), 2020. 134(22): p. 2959–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gilotra NA, et al. , Acute Hemodynamic Effects and Tolerability of Phosphodiesterase-1 Inhibition With ITI-214 in Human Systolic Heart Failure. Circ Heart Fail, 2021. 14(9): p. e008236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Snyder GL, et al. , Preclinical profile of ITI-214, an inhibitor of phosphodiesterase 1, for enhancement of memory performance in rats. Psychopharmacology (Berl), 2016. 233(17): p. 3113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Movsesian M, Ahmad F, and Hirsch E, Functions of PDE3 Isoforms in Cardiac Muscle. J Cardiovasc Dev Dis, 2018. 5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peiro AM, et al. , Genetic variation in phosphodiesterase (PDE) 7B in chronic lymphocytic leukemia: overview of genetic variants of cyclic nucleotide PDEs in human disease. J Hum Genet, 2011. 56(9): p. 676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Degerman E, Belfrage P, and Manganiello VC, Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J Biol Chem, 1997. 272(11): p. 6823–6. [DOI] [PubMed] [Google Scholar]
- 101.Ercu M, Walter S, and Klussmann E, Mutations in Phosphodiesterase 3A (PDE3A) Cause Hypertension Without Cardiac Damage. Hypertension, 2023. 80(6): p. 1171–1179. [DOI] [PubMed] [Google Scholar]
- 102.Zhang Q, et al. , Suppression of experimental abdominal aortic aneurysm in a rat model by the phosphodiesterase 3 inhibitor cilostazol. J Surg Res, 2011. 167(2): p. e385–93. [DOI] [PubMed] [Google Scholar]
- 103.Umebayashi R, et al. , Cilostazol Attenuates Angiotensin II-Induced Abdominal Aortic Aneurysms but Not Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol, 2018. 38(4): p. 903–912. [DOI] [PubMed] [Google Scholar]
- 104.Unno N, et al. , K-134, a phosphodiesterase 3 inhibitor, reduces vascular inflammation and hypoxia, and prevents rupture of experimental abdominal aortic aneurysms. JVS Vasc Sci, 2020. 1: p. 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rogers KC, Oliphant CS, and Finks SW, Clinical efficacy and safety of cilostazol: a critical review of the literature. Drugs, 2015. 75(4): p. 377–95. [DOI] [PubMed] [Google Scholar]
- 106.Sun B, et al. , Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal, 2007. 19(8): p. 1765–71. [DOI] [PubMed] [Google Scholar]
- 107.Ercu M, et al. , Phosphodiesterase 3A and Arterial Hypertension. Circulation, 2020. 142(2): p. 133–149. [DOI] [PubMed] [Google Scholar]
- 108.Maass PG, et al. , PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet, 2015. 47(6): p. 647–53. [DOI] [PubMed] [Google Scholar]
- 109.Begum N, Hockman S, and Manganiello VC, Phosphodiesterase 3A (PDE3A) deletion suppresses proliferation of cultured murine vascular smooth muscle cells (VSMCs) via inhibition of mitogen-activated protein kinase (MAPK) signaling and alterations in critical cell cycle regulatory proteins. J Biol Chem, 2011. 286(29): p. 26238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yan C, Miller CL, and Abe J, Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res, 2007. 100(4): p. 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Palmer D, Tsoi K, and Maurice DH, Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ Res, 1998. 82(8): p. 852–61. [DOI] [PubMed] [Google Scholar]
- 112.Gao R, et al. , Phosphodiesterase 4D contributes to angiotensin II-induced abdominal aortic aneurysm through smooth muscle cell apoptosis. Exp Mol Med, 2022. 54(8): p. 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fan T, et al. , Phosphodiesterase 4D promotes angiotensin II-induced hypertension in mice via smooth muscle cell contraction. Commun Biol, 2022. 5(1): p. 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Varona S, et al. , Rolipram Prevents the Formation of Abdominal Aortic Aneurysm (AAA) in Mice: PDE4B as a Target in AAA. Antioxidants (Basel), 2021. 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Banner KH and Press NJ, Dual PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br J Pharmacol, 2009. 157(6): p. 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Corbin JD, Francis SH, and Webb DJ, Phosphodiesterase type 5 as a pharmacologic target in erectile dysfunction. Urology, 2002. 60(2 Suppl 2): p. 4–11. [DOI] [PubMed] [Google Scholar]
- 117.Nachtnebel A, et al. , Aortic dissection after sildenafil-induced erection. South Med J, 2006. 99(10): p. 1151–2. [DOI] [PubMed] [Google Scholar]
- 118.Lameijer CM, et al. , Type B aortic dissection after the use of tadalafil. Ann Thorac Surg, 2012. 93(2): p. 651–3. [DOI] [PubMed] [Google Scholar]
- 119.Famularo G, et al. , Acute aortic dissection after cocaine and sildenafil abuse. J Emerg Med, 2001. 21(1): p. 78–9. [DOI] [PubMed] [Google Scholar]
- 120.Tiryakioglu SK, et al. , Aortic dissection due to sildenafil abuse. Interact Cardiovasc Thorac Surg, 2009. 9(1): p. 141–3. [DOI] [PubMed] [Google Scholar]
- 121.D’Errico S, Bonuccelli D, and Neri M, Post-coital death in chronic sildenafil abuser. J Geriatr Cardiol, 2020. 17(3): p. 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cesarini V, et al. , Regulation of PDE5 expression in human aorta and thoracic aortic aneurysms. Sci Rep, 2019. 9(1): p. 12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang C, et al. , Sildenafil (Viagra) Aggravates the Development of Experimental Abdominal Aortic Aneurysm. J Am Heart Assoc, 2022. 11(2): p. e023053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li Y, et al. , Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation, 2020. 142(14): p. 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Szczypka M, Role of Phosphodiesterase 7 (PDE7) in T Cell Activity. Effects of Selective PDE7 Inhibitors and Dual PDE4/7 Inhibitors on T Cell Functions. Int J Mol Sci, 2020. 21(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang G, et al. , Phosphodiesterase 7A-deficient mice have functional T cells. J Immunol, 2003. 171(12): p. 6414–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ScRNA-seq data can be accessed at GSE155468.