

Abstract
Adipsin is an adipokine predominantly synthesized in adipose tissues and released into circulation. It is also known as complement factor-D (CFD), acting as the rate-limiting factor in the alternative complement pathway and exerting essential functions on the activation of complement system. The deficiency of CFD in humans is a very rare condition. However, complement overactivation has been implicated in the etiology of numerous disorders, including cardiovascular disease (CVD). Increased circulating level of adipsin has been reported to promote vascular derangements, systemic inflammation, and endothelial dysfunction. Prospective and case-control studies showed that this adipokine is directly associated with all-cause death and rehospitalization in patients with coronary artery disease. Adipsin has also been implicated in pulmonary arterial hypertension, abdominal aortic aneurysm, pre-eclampsia, and type-2 diabetes which is a major risk factor for CVD. Importantly, serum adipsin has been recognized as a unique prognostic marker for assessing cardiovascular diseases. At present, there is paucity of experimental evidence about the precise role of adipsin in the etiology of CVD. However, this mini review provides some insight on the contribution of adipsin in the pathogenesis of CVD and highlights its role on endothelial, smooth muscle and immune cells that mediate cardiovascular functions.
Keywords: Adipsin, Cardiovascular disease, Complement factor-D, Endothelial cells, Smooth muscle cells
Graphical Abstract
1.0. Introduction
Adipsin, a serine-protease homolog, is an adipokine produced exclusively in adipose tissues and released into circulation (1). The concentration of adipsin in plasma typically ranges from 1 to 2 μg/mL in healthy adults, where it is filtered through the glomerular capillaries, reabsorbed by the renal tubules, and rapidly catabolized within cells. However, its concentration may vary with disease condition involving renal injury, causing approximately a 10-fold increase in adipsin level due to reduced glomerular filtration (2). Adipsin concentrations are not different between healthy men and women (3). While a high adipsin concentration may correlate positively with body weight, a low plasma concentration has been reported in diabetic animals and humans (4). There is no comprehensive study that compares adipsin levels among different ethnic groups. Also, there is paucity of data to support that adipsin concentration varies with genetic polymorphism. Adipsin is also known as complement factor-D (CFD), exerting significant influence on the complement system by acting as a rate-limiting component in the activation of alternative complement pathway (5). Once released into circulation, CFD is rendered inactive by self-inhibitory loop because there is no specific endogenous inhibitor. Thus, a well-coordinated control is crucial to inhibit inappropriate cleavage of the endogenous biomolecules aside its specific substrate. However, this protein is activated during immune suppression of infectious substance (6).
Functionally, adipsin plays a crucial role in the activation of complement system, which represents a major constituent of the innate immunity. This adipokine initiates and amplifies the alternative complement pathway by catalyzing the cleavage of its specific substrate, factor-B, to synthesize complement factor-3. Absence of CFD has been shown to cause inactivity of the alternative pathway (7, 8). Moreover, adipsin contributes to the complement amplification loop that mediates responses from the lectin (LP) and classical complement pathway (CP) activation (9). Human adipsin deficiency is a very rare condition, and patients tend to have normal phenotype but prone to infection due to reduced CFD level and defective alternative pathway activation (10).
Adipsin may also exert detrimental effects on cardiovascular function. Numerous experimental evidences have shown that complement system activation enhances leukocyte accumulation in extracellular matrix (ECM), facilitates endothelial cell (EC) activation, stimulates pro-inflammatory mediators from smooth muscle cells thereby promoting cardiovascular derangements (11, 12). Interestingly, increased adipsin concentration correlates positively with all-cause death, rehospitalization, incidences of myocardial infarction and has been recognized as a unique prognostic marker in coronary artery disease (13). Also, increased circulating adipsin is directly associated with increased carotid intima-media thickness (CIMT) and metabolic disturbances (14). In this mini review, we search through literatures to provide insight on the contributions of adipsin in the pathogenesis of cardiovascular diseases, especially its roles on endothelial, smooth muscle and immune cells that significantly impact the cardiovascular system.
2.0. Adipsin in alternative pathway and complement system activation in cardiovascular diseases.
The alternative pathway is constantly active due to self-activation, and it plays a critical role in complement response amplification independent of the classic pathway as well as maintaining low-level activity of the complement system (15). CFD regulates complement system by promoting the activation of alternative pathway (AP). Studies have shown that CFD is a protease specific to the AP and that a small amount of CFD is sufficient to elicit an appreciable time-dependent increase in AP activity (16). On the other hand, studies using animal models have shown that CFD deficient mice with lipodystrophy exhibit a reduced AP activity, while antibody to CFD suppress AP activation without affecting the classical pathway (17).
CFD functions not only as the rate-limiting protease in the production of C3 convertase required for the AP activation, but also is important in the proliferation of complement system through the amplification loop of the AP (Figure 1). Although selective inhibition of the AP by CFD inhibitor results in significant inhibition of CP-mediated terminal complement activation (9), both the CP and LP uses the AP amplification loop to elicit their responses. Thus, CFD can eventually amplify the output of all the three pathways via a positive feedback mechanism due to the enhanced production of C3-convertase required for the breakdown of complement factor-5, a major effector protein in the complement system, with resultant production of C5a (anaphylatoxin) and C5b, a membrane attack complex that destroys pathogens and promotes cell lysis (18, 19). Amplification of the complement pathways is associated with continuous production of C3a and C5a that trigger more cleavage of proteolytic enzymes as well as other cellular biomolecules, which promote inflammation as well as immune cell and phagocyte infiltration that further cause activation of the complement system (20, 21), consequently contributing to cardiovascular diseases.
Fig. 1.

Overview of the complement system activated by the classical, lectin and alternative pathways. Factor-D (FD) acts as an important protease in AP activation resulting in the generation of C3 convertase (C3bBb) that cleaves C3 into C3a (anaphylatoxin) and C3b. Also. FD contributes significantly to AP amplification, in which multiple C3s are cleaved into C3b to produce extra C3 convertase (C3bBb) to generate even more C3 that is utilized by the CP and LP to elicit their responses. This strong amplification loop leads to full complement system activation. C3b attaches to the membrane-bound C3bBb complex to produce C5 convertase (C3bBb3b) that cleaves C5 into C5a (anaphylatoxin) and C5b, which assemble membrane attack complex (MAC) and further disrupt and lyse pathogens.
The CODAM study conducted by Hertle, Arts (22) shows that the AP can be spontaneously activated in blood vessels and is associated with cardiovascular derangements. The AP can also be activated and dysregulated without terminal complement activation in patients with heart failure, owing to an increased level of CFD (23, 24). In addition, the complement system promotes the formation of atherosclerotic plaques, which narrows the vascular lumen and causes ischemia or rupture with resultant thrombosis that enhances the development of acute coronary disease (25, 26).
3.0. Adipsin and cardiovascular diseases: insight from epidemiological studies
The circulating level of Adipsin has been correlated with the incidence of CVD, making this protease an important biomarker for assessing CVD and potential therapeutic target in this disorder. Ohtsuki, Satoh (13) have shown that Adipsin is a novel biomarker in the diagnosis of coronary artery disease (CAD) and significantly associated with all-cause death and rehospitalization in these patients. An increased level of adipsin is reported in adults with growth hormone deficiency complicated with dysfunctional glucolipid metabolism (27). Abnormal lipid metabolism is a significant risk factor for CAD, suggesting that adipsin can be an early biomarker of CVD. Coronary angiography assessment using optical coherence tomography suggests that serum adipsin is an accurate biomarker for evaluating thin-cap fibroatheroma (TCFA), and this protease is positively associated with plaque vulnerability in patients with CAD (28).
Transcriptomic analyses indicate that an increased expression of adipsin in systemic sclerosis is associated with pulmonary arterial hypertension (PAH) (29). EC Fli1 (Friend leukemia virus integration 1) deficiency partially upregulates adipsin expression and contributes to the development of PAH (30). A predominant increase in C3a and CFD has been reported in the human right ventricle, and CFD deficiency in animal model of pulmonary artery constriction (PAC) reduces right ventricular remodeling and fibrosis, while pharmacological inhibition of C3a receptor significantly improves right ventricular function (31). Additionally, an increased circulating adipsin level positively correlates with increased carotid intima-media thickness (14). In patients with polycystic ovarian syndrome (PCOS), the increased level of circulating adipsin is positively associated with body mass index (BMI) and insulin resistance and is an independent predictor of carotid intima-media thickness (14). Moreover, the increased plasma and urine level of adipsin in healthy pregnant women at the third trimester is hypothesized to be an important indicator of preeclampsia, a pathological condition characterized by increased blood pressure (32, 33).
Adipsin promotes vascular endothelial dysfunction by activating the AP and thus increasing the production of complement proteins such as C3a and C5a (34, 35), which justifies the potential to use circulating adipsin level as a novel biomarker for CVD. Activation of the AP is longitudinally correlated with negative cardiovascular outcomes, such as ischemia and infarction (22). Population-based cohort study shows significant correlation of adipsin and complement protein-3 (C3) with arterial stiffness although it is related to type-2 diabetes and metabolic dysfunctions Jin, Reesink (36).
4.0. Adipsin in cardiovascular disease: insight from experimental studies
Adipose tissues, especially the epicardial and perivascular adipose tissues (PVAT) surrounding the heart and blood vessels, have significant influence on the vascular system due to their anatomical location as well as synthesis and release of adipokines including adipsin into the circulation (37, 38). These adipokines mediate vascular remodeling, compromise EC integrity, promote smooth muscle cell phenotypic switch, oxidative stress, macrophage infiltration and cellular inflammation that contribute to the pathogenesis of cardiovascular dysfunctions (39, 40).
Adipose tissue-derived adipsin promotes risk factors associated with CVD. Genomic analyses of epicardial adipose tissue in patients with atrial fibrillation show that adipsin is abundantly expressed in this tissue (41), which could be responsible for the involvement of epicardial adipose tissue (EAT) in cardiac diseases (42, 43). A recent study finds that EAT-derived adipsin exacerbates cardiovascular injury by promoting cardiomyocyte death following myocardial infarction (44). An increased adipsin level activates Poly ADP-ribosepolymerase-1 (PARP-1) activity, which depletes NAD level with resultant decrease in Sir2alpha deacetylase activity in cardiomyocytes. However, another study shows that adipsin mediates iron homeostasis by upregulating ferritin heavy chain (FTH) and downregulating transferrin receptor (TFRC) to mitigate cardiac remodeling after myocardial infarction (45). In this study, both co-immunoprecipitation assay and immunofluorescence staining show a significant interaction and colocalization of adipsin with iron homeostasis related protein-2 (IRP2) in an in-vitro hypoxia model. Thus, IRP2, an important regulator of iron homeostasis, is proposed as a downstream mediator for the cardioprotective effect by adipsin. On the other hand, Shahini, Michelsen (24) have reported that an increase in adipsin level exacerbates systemic inflammation, cardiac dysfunction (especially worsen diastolic function), and neurohormonal deterioration in patients with chronic heart failure. Despite the conflicting reports from recent studies, the mechanistic role of adipsin in the pathogenesis of cardiovascular disease needs to be further explored, especially the roles of cellular mediators (such as endothelial, smooth muscle, and immune cells) in cardiac functions.
4.1. Effect of adipsin on ECs
Vascular ECs mediate the complex interplay between plasma and vascular tissue, especially the VSMCs, thereby maintaining VSMC contractile phenotype and enhancing optimal vascular functions (such as vascular tone) vital to cardiovascular health (46, 47). Studies have shown that complement activation can increase the expression of inflammatory cytokines and promote immune cell attachment to ECs via chemotaxis, which further inhibits endothelial nitric oxide production and initiate or exacerbate vascular dysfunction (12, 48). This implies that adipsin may be involved in endothelial damage with resultant cardiovascular dysfunction, including PAH.
Recently, Jin, Eussen (49) conducted the Maastricht study involving 2947 participants and reported that circulating adipsin is significantly associated with endothelial dysfunction and low-grade inflammation, which contribute to the progression of cardiovascular injury. In addition, studies elucidating the interplay between obesity and vascular function show that the increased circulating adipsin significantly reduces endothelium-induced vasodilation and exacerbates endothelin-1-dependent vasoconstriction in humans, suggesting that adipsin may aggravate cardiovascular derangement by compromising endothelial functions (50).
Structural analyses show that the endothelium is endowed with complement inhibitory proteins such as Decay accelerating factors (DAF) and other membrane-bound inhibitors including membrane cofactor protein CD46 that helps prevent the interaction between excessive complement activation in the circulation and VSMCs (51). Importantly, theses membrane-bound inhibitors protect endothelium against constant low level AP activation mediated by adipsin (52), suggesting that adipsin may aggravate complement-mediated vascular injury by promoting the AP activation.
4.2. Effect of Adipsin on VSMCs
The medial layer of the blood vessel wall is comprised mainly of VSMCs that control vascular tone and blood pressure. VSMCs can modify their functions and morphology in relation to different stimuli, allowing mature VSMCs to change from a contractile to a synthetic state, a phenomenon known as “phenotype modulation”. This phenotypic modulation contributes to the etiology of several cardiovascular dysfunctions (53). Although, there is a lack of literature on the direct role of adipsin on VSMC phenotype switch, its downstream C3 protein has been reported to exert significant effect on VSMCs phenotype. PVAT-derived C3 due to the abundance of adipsin in this tissue promotes differentiation and migration of adventitial fibroblasts by activating c-Jun N-terminal kinase pathway, thereby contributing to adventitial remodeling and myofibroblast aggregation (54). The adventitial fibroblasts can migrate into the medial layer to induce VSMC phenotype switch via TGF-β1 pathway, promoting revascularization (55, 56). Notably, C3 directly increases synthetic phenotype, promotes VSMC growth, as well as mediates the increased neointimal formation caused by C-reactive protein in spontaneously hypertensive rats and mouse carotid wire injury model (57, 58). Furthermore, increased serum level of adipsin is observed in patients with abdominal aortic aneurysm, suggesting that adipsin may promote VSMC phenotypic modulation during the aneurysm development (59).
4.3. Effect of Adipsin on immune cells
Although the immune system functions as an inherent defense against infection or injury, immune cell (especially T-cells and macrophages) activation has been implicated as a pathogenic mediator of cardiovascular diseases. These cells are often recruited from circulation into vascular beds by activated ECs and later differentiated into unique effector cells that contribute to the development of CVD (60, 61).
Macrophages participate in dynamic interactions among different vascular cells and mediate VSMC phenotype modulation (62). Evaluation of the interaction between macrophages and adipocytes shows that both cells express adipsin that promotes preadipocyte differentiation into macrophages (63). Adipsin deficiency inhibits the recruitment of monocytes, neutrophils, and macrophages into the synovial fluid in serum-injected K/BxN mouse model of inflammatory arthritis (64). In addition, a reduction in atherosclerotic plaque area and macrophage accumulation is reported in ApoE−/−/AdipsinTg mice after high fat diet feeding for 3 months. Furthermore, adipsin overexpression in oxLDL-stimulated macrophages attenuates lipid uptake by inhibiting CD36/PPARγ levels (65), implicating that adipsin is a unique CVD risk factor. These experimental studies suggest that adipsin may either modulate or enhance macrophage phenotype or function in a disease-dependent manner.
T-cells may infiltrate into myocardium or heart valves to compromise cardiac function with resultant scarring (66). Various cytokines such as leptin, visfatin, adiponectin, resistin from adipose tissues may regulate T-cell activation and differentiation (67). DAF-deficiency in mice activates complement proteins with subsequent amplification of T-cell activity. This enhanced T-cell activity is associated with the AP activation in T-cells, which further promotes the production of local anaphylatoxin (68, 69). Although there is paucity of data on the direct contribution of adipsin to T-cell polarization, literatures have shown that adipsin may influence complement system activation implicated in T-cell immunity. Thus, adipsin may enhance T-cell polarization in the myocardium indirectly, which further regulates cardiac remodeling and the progression of CVD.
5.0. Conclusion
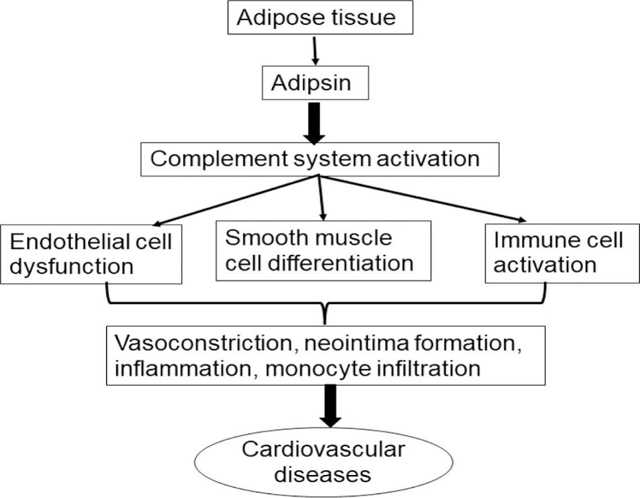
Adipose tissues synthesize and release adipokines, including adipsin, into the circulation, which contributes to the pathogenesis of CVD. Although the correlation between adipsin and CVD, such as coronary artery disease, heart failure, pulmonary arterial hypertension, and pre-eclampsia, has been well documented, the precise mechanisms underlying adipsin functions in the pathogenesis of CVD are yet to be determined. Thus, comprehensive studies using in vitro and in vivo experimental paradigms are needed to explore the specific contributions of this adipokine to the development/progression of CVD, especially its effects on cellular mediators, i.e., immune cells, VSMCs, and ECs (Figure 2).
Fig. 2.

Schematic effect of adipsin on cellular mediators of cardiovascular diseases.
6.0. Future perspective
Insights from epidemiological and experimental studies have shown that adipsin contributes to the development and progression of CVD. However, future research should be geared toward unraveling the underlying mechanisms by which this adipokine promotes cellular derangement associated with CVD. Basic and clinical researchers may employ different approaches, such as genetic modifications or pharmacological interventions with adipsin agonists or antagonists, to evaluate the specific roles of adipsin in these disorders. The results from these studies will help address the paucity of information regarding any confounding variables such as lifestyle, gender, age, and comorbidities that may influence the roles of adipsin in the etiology of CVD. While some adipokines, like adiponectin, omentin, and apelin, exert anti-inflammatory and cardioprotective effects (70, 71), others adipokines, such as visfatin, leptin, resistin, and lipocalin-2, promote inflammation, insulin resistance, EC dysfunction and VSMC apoptosis (72–74). Notably, adipsin may compromise EC function, promote neointimal formation and recruit immune cells into different vascular beds in the setting of CVD. Thus, there may be interplays between adipsin and other adipokines in the development of CVD, which could also be an important subject for future inquiry. Although increased adipsin level has been identified as a unique biomarker in patients with coronary artery disease, atherosclerosis, and correlates positively with ischemic events (13, 75), the combination of adipsin with other biomarkers such as brain natriuretic peptide (BNP) or high-sensitive C-reactive protein (hs-CRP) may be a more valuable diagnostic tool, likely better than adipsin alone, and thus may improve the clinical practice for diagnosing CVD. Furthermore, pharmacological interventions targeting circulating adipsin may inhibit the activation of complement system and further improve management of risk factors associated with CVD. Therefore, unravelling the detailed contributions of adipsin in the etiology of CVD remain an active area of research, which is crucial for revealing the complex interplays between adipsin and other adipokines and addressing confounding variables. The outcomes of these studies could lead to significant discoveries of the therapeutic and diagnostic values of adipsin in the clinical management of CVD.
Funding statement
This work was supported by grants from National Institutes of Health (HL117247, HL119053, and HL147313) and Department of Veterans Affairs Merit Review Awards (I01 BX006161).
Abbreviations
- CFD
Complement factor-D
- ECM
extracellular matrix
- CIMT
carotid intima-media thickness
- AP
Alternative pathway
- CP
Classical pathway
- LP
Lectin pathway
- FD
Factor D
- C3
Complement factor-3
- C5
Complement factor-5
- MASPs
Manna-binding lectin-associated serine protease
- CVD
Cardiovascular disease
- CAD
coronary artery disease
- PVAT
Perivascular adipose tissue
- EAT
Epicardial adipose tissue
- VSMC
Vascular smooth muscle cell
- ECs
Endothelial cells
- TGF-β1
Transforming Growth factor-beta1
- ApoE
Apolipoprotein-E
- OxLDL
Oxidized low-density lipoprotein
- CD36
Cluster of Differentiation 36
- PPARγ
Peroxisome Proliferated activated receptor-γ
- DAF
Decay accelerating factor
Footnotes
CRediT authorship and contribution statement
AD drafted and edited the manuscript; SYC revised the manuscript.
Declaration of competing interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237(4813):402–5: doi: 10.1126/science.3299705. [DOI] [PubMed] [Google Scholar]
- 2.Pascual M, Paccaud JP, Macon K, Volanakis JE, Schifferli JA. Complement activation by the alternative pathway is modified in renal failure: the role of factor D. Clin Nephrol. 1989;32(4):185–93: [PubMed] [Google Scholar]
- 3.Milek M, Moulla Y, Kern M, Stroh C, Dietrich A, Schön MR, et al. Adipsin Serum Concentrations and Adipose Tissue Expression in People with Obesity and Type 2 Diabetes. Int J Mol Sci. 2022;23(4): doi: 10.3390/ijms23042222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tafere GG, Wondafrash DZ, Zewdie KA, Assefa BT, Ayza MA. Plasma Adipsin as a Biomarker and Its Implication in Type 2 Diabetes Mellitus. Diabetes Metab Syndr Obes. 2020;13:1855–61: doi: 10.2147/dmso.S253967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Y, Ma M, Ippolito GC, Schroeder HW Jr., Carroll, Volanakis. Complement activation in factor D-deficient mice. Proc Natl Acad Sci U S A. 2001;98(25):14577–82: doi: 10.1073/pnas.261428398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jing H, Macon KJ, Moore D, DeLucas LJ, Volanakis JE, Narayana SV. Structural basis of profactor D activation: from a highly flexible zymogen to a novel self-inhibited serine protease, complement factor D. Embo j. 1999;18(4):804–14: doi: 10.1093/emboj/18.4.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesavre PH, Müller-Eberhard HJ. Mechanism of action of factor D of the alternative complement pathway. J Exp Med. 1978;148(6):1498–509: doi: 10.1084/jem.148.6.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sprong T, Roos D, Weemaes C, Neeleman C, Geesing CL, Mollnes TE, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. 2006;107(12):4865–70: doi: 10.1182/blood-2005-07-2820. [DOI] [PubMed] [Google Scholar]
- 9.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138(3):439–46: doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shariat M, Heydrzadeh M, Abolhassani H, Bemanian MH, Yazdani R. Chapter 9 - Complement deficiencies. In: Aghamohammadi A, Abolhassani H, Rezaei N, Yazdani R, editors. Inborn Errors of Immunity: Academic Press; 2021. p. 291–315. [Google Scholar]
- 11.Carter AM. Complement activation: an emerging player in the pathogenesis of cardiovascular disease. Scientifica (Cairo). 2012;2012:402783: doi: 10.6064/2012/402783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruan C-C, Gao P-J. Role of Complement-Related Inflammation and Vascular Dysfunction in Hypertension. Hypertension. 2019;73(5):965–71: doi:doi: 10.1161/HYPERTENSIONAHA.118.11210. [DOI] [PubMed] [Google Scholar]
- 13.Ohtsuki T, Satoh K, Shimizu T, Ikeda S, Kikuchi N, Satoh T, et al. Identification of Adipsin as a Novel Prognostic Biomarker in Patients With Coronary Artery Disease. J Am Heart Assoc. 2019;8(23):e013716: doi: 10.1161/jaha.119.013716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gursoy Calan O, Calan M, Yesil Senses P, Unal Kocabas G, Ozden E, Sari KR, et al. Increased adipsin is associated with carotid intima media thickness and metabolic disturbances in polycystic ovary syndrome. Clin Endocrinol (Oxf). 2016;85(6):910–7: doi: 10.1111/cen.13157. [DOI] [PubMed] [Google Scholar]
- 15.Łukawska E, Polcyn-Adamczak M, Niemir ZI. The role of the alternative pathway of complement activation in glomerular diseases. Clin Exp Med. 2018;18(3):297–318: doi: 10.1007/s10238-018-0491-8. [DOI] [PubMed] [Google Scholar]
- 16.Langereis JD, van der Molen RG, de Kat Angelino C, Henriet SS, de Jonge MI, Joosten I, et al. Complement factor D haplodeficiency is associated with a reduced complement activation speed and diminished bacterial killing. Clin Transl Immunology. 2021;10(4):e1256: doi: 10.1002/cti2.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Hutson I, Akk AM, Mascharak S, Pham CTN, Hourcade DE, et al. Contribution of Adipose-Derived Factor D/Adipsin to Complement Alternative Pathway Activation: Lessons from Lipodystrophy. J Immunol. 2018;200(8):2786–97: doi: 10.4049/jimmunol.1701668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyer DS, Schmidt-Erfurth U, van Lookeren Campagne M, Henry EC, Brittain C. THE PATHOPHYSIOLOGY OF GEOGRAPHIC ATROPHY SECONDARY TO AGE-RELATED MACULAR DEGENERATION AND THE COMPLEMENT PATHWAY AS A THERAPEUTIC TARGET. Retina. 2017;37(5):819–35: doi: 10.1097/iae.0000000000001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harboe M, Mollnes TE. The alternative complement pathway revisited. Journal of Cellular and Molecular Medicine. 2008;12(4):1074–84: doi: 10.1111/j.1582-4934.2008.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Frontiers in Immunology. 2015;6: doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholz W, McClurg MR, Cardenas GJ, Smith M, Noonan DJ, Hugli TE, et al. C5a-mediated release of interleukin 6 by human monocytes. Clin Immunol Immunopathol. 1990;57(2):297–307: doi: 10.1016/0090-1229(90)90043-p. [DOI] [PubMed] [Google Scholar]
- 22.Hertle E, Arts IC, van der Kallen CJ, Feskens EJ, Schalkwijk CG, Stehouwer CD, et al. The alternative complement pathway is longitudinally associated with adverse cardiovascular outcomes. The CODAM study. Thromb Haemost. 2016;115(2):446–57: doi: 10.1160/th15-05-0439. [DOI] [PubMed] [Google Scholar]
- 23.Holt MF, Michelsen AE, Shahini N, Bjørkelund E, Bendz CH, Massey RJ, et al. The Alternative Complement Pathway Is Activated Without a Corresponding Terminal Pathway Activation in Patients With Heart Failure. Front Immunol. 2021;12:800978: doi: 10.3389/fimmu.2021.800978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, et al. The alternative complement pathway is dysregulated in patients with chronic heart failure. Scientific Reports. 2017;7(1):42532: doi: 10.1038/srep42532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torzewski M, Bhakdi S. Complement and atherosclerosis—united to the point of no return? Clinical Biochemistry. 2013;46(1):20–5: doi: 10.1016/j.clinbiochem.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Kiss MG, Binder CJ. The multifaceted impact of complement on atherosclerosis. Atherosclerosis. 2022;351:29–40: doi: 10.1016/j.atherosclerosis.2022.03.014. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Zheng X, Xie X, Qian W, Zhang L, Ren W. CORRELATION OF INCREASED SERUM ADIPSIN WITH INCREASED CARDIOVASCULAR RISKS IN ADULT PATIENTS WITH GROWTH HORMONE DEFICIENCY. Endocr Pract. 2019;25(5):446–53: doi: 10.4158/ep-2018-0541. [DOI] [PubMed] [Google Scholar]
- 28.Sun R, Qiao Y, Yan G, Wang D, Zuo W, Ji Z, et al. Association between serum adipsin and plaque vulnerability determined by optical coherence tomography in patients with coronary artery disease. J Thorac Dis. 2021;13(4):2414–25: doi: 10.21037/jtd-21-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korman BD, Marangoni RG, Hinchcliff M, Shah SJ, Carns M, Hoffmann A, et al. Brief Report: Association of Elevated Adipsin Levels With Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis & rheumatology (Hoboken, N.J.). 2017;69(10):2062–8: doi: 10.1002/art.40193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyagawa T, Taniguchi T, Saigusa R, Fukayama M, Takahashi T, Yamashita T, et al. Fli1 deficiency induces endothelial adipsin expression, contributing to the onset of pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford). 2020;59(8):2005–15: doi: 10.1093/rheumatology/kez517. [DOI] [PubMed] [Google Scholar]
- 31.Ito S, Hashimoto H, Yamakawa H, Kusumoto D, Akiba Y, Nakamura T, et al. The complement C3-complement factor D-C3a receptor signalling axis regulates cardiac remodelling in right ventricular failure. Nat Commun. 2022;13(1):5409: doi: 10.1038/s41467-022-33152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Luo X, Xu Q, Yu H, Gao L, Zhou R, et al. Adipsin of the Alternative Complement Pathway Is a Potential Predictor for Preeclampsia in Early Pregnancy. Front Immunol. 2021;12:702385: doi: 10.3389/fimmu.2021.702385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poveda NE, Garcés MF, Ruiz-Linares CE, Varón D, Valderrama S, Sanchez E, et al. Serum Adipsin Levels throughout Normal Pregnancy and Preeclampsia. Sci Rep. 2016;6:20073: doi: 10.1038/srep20073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shivshankar P, Li Y-D, Mueller-Ortiz SL, Wetsel RA. In response to complement anaphylatoxin peptides C3a and C5a, human vascular endothelial cells migrate and mediate the activation of B-cells and polarization of T-cells. The FASEB Journal. 2020;34(6):7540–60: doi: 10.1096/fj.201902397R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, et al. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. Journal of Neuroinflammation. 2016;13(1):23: doi: 10.1186/s12974-016-0485-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin S, Reesink KD, Kroon AA, de Galan B, van der Kallen CJH, Wesselius A, et al. Complement factors D and C3 cross-sectionally associate with arterial stiffness, but not independently of metabolic risk factors: The Maastricht Study. J Hypertens. 2022;40(11):2161–70: doi: 10.1097/hjh.0000000000003237. [DOI] [PubMed] [Google Scholar]
- 37.Britton KA, Fox CS. Perivascular adipose tissue and vascular disease. Clin Lipidol. 2011;6(1):79–91: doi: 10.2217/clp.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oikonomou EK, Antoniades C. The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol. 2019;16(2):83–99: doi: 10.1038/s41569-018-0097-6. [DOI] [PubMed] [Google Scholar]
- 39.Akoumianakis I, Tarun A, Antoniades C. Perivascular adipose tissue as a regulator of vascular disease pathogenesis: identifying novel therapeutic targets. Br J Pharmacol. 2017;174(20):3411–24: doi: 10.1111/bph.13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lau WB, Ohashi K, Wang Y, Ogawa H, Murohara T, Ma XL, et al. Role of Adipokines in Cardiovascular Disease. Circ J. 2017;81(7):920–8: doi: 10.1253/circj.CJ-17-0458. [DOI] [PubMed] [Google Scholar]
- 41.Zhao L, Ma Z, Guo Z, Zheng M, Li K, Yang X. Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. Biomedicine & Pharmacotherapy. 2020;121:109634: doi: 10.1016/j.biopha.2019.109634. [DOI] [PubMed] [Google Scholar]
- 42.Ansaldo AM, Montecucco F, Sahebkar A, Dallegri F, Carbone F. Epicardial adipose tissue and cardiovascular diseases. Int J Cardiol. 2019;278:254–60: doi: 10.1016/j.ijcard.2018.09.089. [DOI] [PubMed] [Google Scholar]
- 43.Song Y, Song F, Wu C, Hong YX, Li G. The roles of epicardial adipose tissue in heart failure. Heart Fail Rev. 2022;27(1):369–77: doi: 10.1007/s10741-020-09997-x. [DOI] [PubMed] [Google Scholar]
- 44.Hao S, Zhang J, Pei Y, Guo L, Liang Z. Complement factor D derived from epicardial adipose tissue participates in cardiomyocyte apoptosis after myocardial infarction by mediating PARP-1 activity. Cell Signal. 2022:110518: doi: 10.1016/j.cellsig.2022.110518. [DOI] [PubMed] [Google Scholar]
- 45.Man W, Song X, Xiong Z, Gu J, Lin J, Gu X, et al. Exosomes derived from pericardial adipose tissues attenuate cardiac remodeling following myocardial infarction by Adipsin-regulated iron homeostasis. Frontiers in Cardiovascular Medicine. 2022;9: doi: 10.3389/fcvm.2022.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Méndez-Barbero N, Gutiérrez-Muñoz C, Blanco-Colio LM. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int J Mol Sci. 2021;22(14): doi: 10.3390/ijms22147284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lilly B. We have contact: endothelial cell-smooth muscle cell interactions. Physiology (Bethesda). 2014;29(4):234–41: doi: 10.1152/physiol.00047.2013. [DOI] [PubMed] [Google Scholar]
- 48.Brunn GJ, Saadi S, Platt JL. Differential Regulation of Endothelial Cell Activation by Complement and Interleukin 1α. Circulation Research. 2006;98(6):793–800: doi:doi: 10.1161/01.RES.0000216071.87981.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin S, Eussen S, Schalkwijk CG, Stehouwer CDA, van Greevenbroek MMJ. Plasma factor D is cross-sectionally associated with low-grade inflammation, endothelial dysfunction and cardiovascular disease: The Maastricht study. Atherosclerosis. 2023;377:60–7: doi: 10.1016/j.atherosclerosis.2023.06.079. [DOI] [PubMed] [Google Scholar]
- 50.Schinzari F, Tesauro M, Campia U, Cardillo C. Dysregulated adipokine secretory profile is associated with endothelial dysfunction in human obesity. European Heart Journal. 2020;41(Supplement_2): doi: 10.1093/ehjci/ehaa946.3032. [DOI] [Google Scholar]
- 51.Mason JC, Yarwood H, Sugars K, Morgan BP, Davies KA, Haskard DO. Induction of decay-accelerating factor by cytokines or the membrane-attack complex protects vascular endothelial cells against complement deposition. Blood. 1999;94(5):1673–82: [PubMed] [Google Scholar]
- 52.Sohn JH, Kaplan HJ, Suk HJ, Bora PS, Bora NS. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest Ophthalmol Vis Sci. 2000;41(11):3492–502: [PMC free article] [PubMed] [Google Scholar]
- 53.Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, et al. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc Sci. 2021;2:79–94: doi: 10.1016/j.jvssci.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruan C-C, Zhu D-L, Chen Q-Z, Chen J, Guo S-J, Li X-D, et al. Perivascular Adipose Tissue–Derived Complement 3 Is Required for Adventitial Fibroblast Functions and Adventitial Remodeling in Deoxycorticosterone Acetate–Salt Hypertensive Rats. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(12):2568–74: doi:doi: 10.1161/ATVBAHA.110.215525. [DOI] [PubMed] [Google Scholar]
- 55.Han X, Wu A, Wang J, Chang H, Zhao Y, Zhang Y, et al. Activation and Migration of Adventitial Fibroblasts Contributes to Vascular Remodeling. The Anatomical Record. 2018;301(7):1216–23: doi: 10.1002/ar.23793. [DOI] [PubMed] [Google Scholar]
- 56.Chen P-Y, Qin L, Li G, Tellides G, Simons M. Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFβ)-dependent smooth muscle cell phenotype modulation. Scientific Reports. 2016;6(1):33407: doi: 10.1038/srep33407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hage FG, Oparil S, Xing D, Chen YF, McCrory MA, Szalai AJ. C-reactive protein-mediated vascular injury requires complement. Arterioscler Thromb Vasc Biol. 2010;30(6):1189–95: doi: 10.1161/atvbaha.110.205377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin ZH, Fukuda N, Jin XQ, Yao EH, Ueno T, Endo M, et al. Complement 3 is involved in the synthetic phenotype and exaggerated growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2004;44(1):42–7: doi: 10.1161/01.HYP.0000129540.83284.ca. [DOI] [PubMed] [Google Scholar]
- 59.Feridooni T, Zamzam A, Popkov M, Syed MH, Djahanpour N, Wheatcroft M, et al. Plasma complement component C2: a potential biomarker for predicting abdominal aortic aneurysm related complications. Scientific Reports. 2022;12(1):21252: doi: 10.1038/s41598-022-24698-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simons KH, de Jong A, Jukema JW, de Vries MR, Arens R, Quax PHA. T cell co-stimulation and co-inhibition in cardiovascular disease: a double-edged sword. Nat Rev Cardiol. 2019;16(6):325–43: doi: 10.1038/s41569-019-0164-7. [DOI] [PubMed] [Google Scholar]
- 61.Reali E, Ferrando-Martinez S, Catalfamo M. Editorial: The Interplay Between Immune Activation and Cardiovascular Disease During Infection, Autoimmunity and Aging: The Role of T Cells. Frontiers in Immunology. 2021;12: doi: 10.3389/fimmu.2021.719517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butoi E, Gan AM, Tucureanu MM, Stan D, Macarie RD, Constantinescu C, et al. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim Biophys Acta. 2016;1863(7 Pt A):1568–78: doi: 10.1016/j.bbamcr.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Chazenbalk G, Bertolotto C, Heneidi S, Jumabay M, Trivax B, Aronowitz J, et al. Novel Pathway of Adipogenesis through Cross-Talk between Adipose Tissue Macrophages, Adipose Stem Cells and Adipocytes: Evidence of Cell Plasticity. PLOS ONE. 2011;6(3):e17834: doi: 10.1371/journal.pone.0017834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Zou W, Brestoff JR, Rohatgi N, Wu X, Atkinson JP, et al. Fat-Produced Adipsin Regulates Inflammatory Arthritis. Cell Rep. 2019;27(10):2809–16.e3: doi: 10.1016/j.celrep.2019.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duan Y, Zhang X, Zhang X, Lin J, Shu X, Man W, et al. Inhibition of macrophage-derived foam cells by Adipsin attenuates progression of atherosclerosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2022;1868(12):166533: doi: 10.1016/j.bbadis.2022.166533. [DOI] [PubMed] [Google Scholar]
- 66.Cunningham MW. T cell mimicry in inflammatory heart disease. Mol Immunol. 2004;40(14–15):1121–7: doi: 10.1016/j.molimm.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 67.Song J, Deng T. The Adipocyte and Adaptive Immunity. Frontiers in Immunology. 2020;11: doi: 10.3389/fimmu.2020.593058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwan WH, van der Touw W, Heeger PS. Complement regulation of T cell immunity. Immunol Res. 2012;54(1–3):247–53: doi: 10.1007/s12026-012-8327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunkelberger JR, Song W-C. Complement and its role in innate and adaptive immune responses. Cell Research. 2010;20(1):34–50: doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 70.Peng J, Chen Q, Wu C. The role of adiponectin in cardiovascular disease. Cardiovascular Pathology. 2023;64:107514: doi: 10.1016/j.carpath.2022.107514. [DOI] [PubMed] [Google Scholar]
- 71.Tan Y-L, Zheng X-L, Tang C-K. The protective functions of omentin in cardiovascular diseases. Clinica Chimica Acta. 2015;448:98–106: doi: 10.1016/j.cca.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 72.Askin L, Abus S, Tanriverdi O. Resistin and Cardiovascular Disease: A Review of the Current Literature Regarding Clinical and Pathological Relationships. Curr Cardiol Rev. 2022;18(1):e290721195114: doi: 10.2174/1573403×17666210729101120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dakroub A, Nasser SA, Kobeissy F, Yassine HM, Orekhov A, Sharifi-Rad J, et al. Visfatin: An emerging adipocytokine bridging the gap in the evolution of cardiovascular diseases. Journal of Cellular Physiology. 2021;236(9):6282–96: doi: 10.1002/jcp.30345. [DOI] [PubMed] [Google Scholar]
- 74.Kang K-W, Ok M, Lee S-K. Leptin as a Key between Obesity and Cardiovascular Disease. Journal of Obesity & Metabolic Syndrome. 2020;29(4):248–59: doi: 10.7570/jomes20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Askin L, Askin H, Abus S, Hidayet S. Adipsin as a novel prognostic biomarker for cardiovascular diseases. Cor Vasa. 2022;66:34–7: [Google Scholar]