

SUMMARY
Elevated interleukin (IL)-1β levels, NLRP3 inflammasome activity, and systemic inflammation are hallmarks of chronic metabolic inflammatory syndromes, but the mechanistic basis for this is unclear. Here, we show that levels of plasma IL-1β are lower in fasting compared to fed subjects, while the lipid arachidonic acid (AA) is elevated. Lipid profiling of NLRP3-stimulated mouse macrophages shows enhanced AA production and an NLRP3-dependent eicosanoid signature. Inhibition of cyclooxygenase by nonsteroidal anti-inflammatory drugs decreases eicosanoid, but not AA, production. It also reduces both IL-1β and IL-18 production in response to NLRP3 activation. AA inhibits NLRP3 inflammasome activity in human and mouse macrophages. Mechanistically, AA inhibits phospholipase C activity to reduce JNK1 stimulation and hence NLRP3 activity. These data show that AA is an important physiological regulator of the NLRP3 inflammasome and explains why fasting reduces systemic inflammation and also suggests a mechanism to explain how nonsteroidal anti-inflammatory drugs work.
In brief
By studying how arachidonic acid (AA) impacts the inflammasome, Pereira et al. demonstrate that AA inhibits NLRP3-mediated IL-1β production by blocking the activities of phospholipase C and the downstream protein kinases PKD and JNK. This provides a mechanistic basis for the inverse relationship between serum levels of AA and IL-1β.
Graphical Abstract
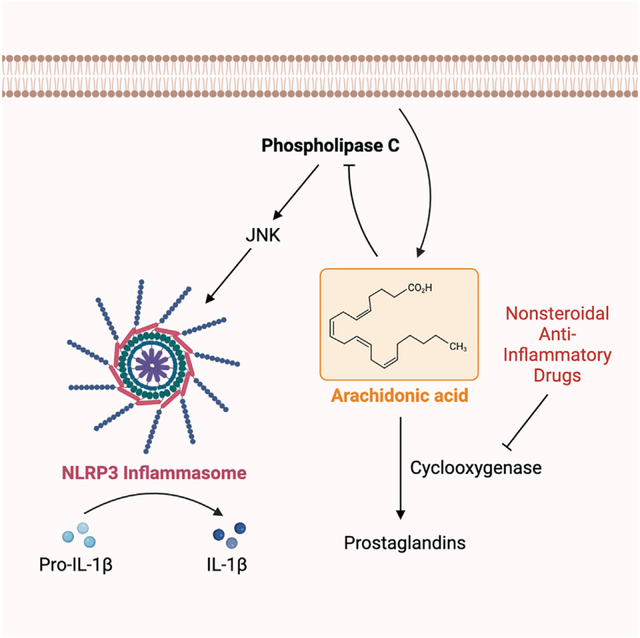
INTRODUCTION
The consumption of a Western, high-calorie diet (WD) is associated with a chronic metabolic inflammatory syndrome (metaflammation), which underpins many prevalent noncommunicable diseases.1 How this complex process, whereby tissue-specific and systemic immune responses are integrated alongside metabolic regulation, is governed remains poorly understood. Fasting leads to suppression of metabolic inflammation and is characterized by a drop in serum pro-inflammatory cytokines, particularly interleukin-1β (IL-1β), which is closely associated with insulin regulation and blood glucose levels.2–4
One emerging regulator of metaflammation is the inflammasome induced by nucleotide-binding and oligomerization domain-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) activation.1 Inflammasomes are multi-protein signaling platforms that consist of a receptor (usually an NLR), an adaptor (apoptosis-associated speck-like protein containing a caspase-recruitment domain, ASC), and an effector (caspase-1), which jointly process IL-1β and IL-18 to their bioactive forms and cleave the cell death effector gasdermin D (GSDMD) to drive pyroptosis.5 Fasting regulates NLRP3 activity,2 but the mechanisms that regulate this process are not well understood. Oxidized low-density lipoprotein (LDL) and cholesterol crystals trigger activation of NLRP3 in macrophages when the cellular capacity for metabolizing cholesterol is exceeded, but if the cell remains capable of processing cholesterol, then anti-inflammatory responses are induced.1 Diets rich in saturated fatty acids, such as palmitic acid (PA) or stearic acid, also trigger NLRP3 inflammatory activity.1 In mice, the systemic inflammation is seen when Ldlr−/− are fed a WD and requires NLRP3-mediated trained immunity, which is reversed when the mice are placed on a normal chow diet.6
Activation of the NLRP3 inflammasome is a tightly regulated two-step process.7 First transcription of pro-IL-1β and NLRP3 is induced by Toll-like receptor (TLR) activators, such as lipopolysaccharide (LPS), or pro-inflammatory cytokines, such as tumor necrosis factor α. This is followed by an activation phase, for which many post-translational regulatory steps are important, one of which is phosphorylation of both NLRP3 and ASC.7 Phosphorylation of ASC by spleen tyrosine kinase and c-Jun N-terminal kinase (JNK) leads to the formation of the ASC signaling scaffold and to inflammasome activation.8 The regulation of NLRP3 by phosphorylation is more complex, with the pyrin domain of this protein being the phospho-regulatory target.9 We have shown that biphasic JNK1 phosphorylation of NLRP3 in response to mitochondrial reactive oxygen species leads to inflammasome activation,10 but phosphorylation of NLRP3 at serine 58 and JNK2 activity both prevent activation of the NLRP3 inflammasome.11 This suggests there is a complex network of phosphorylation events that regulate NLRP3.
NLRP3 is increasingly recognized as being interlinked with lipid metabolism.12–14 Eicosanoids, which are derived from oxidation of arachidonic acid (AA), comprise a complex family of signaling lipids with important roles in regulating inflammation.15 Among them, prostaglandins (PGs), such as PGE2, formed by cyclooxygenase (COX) metabolism of AA, may regulate NLRP3 activity, although the precise role for this lipid as an activator or inhibitor of inflammasome activity is controversial.16 The nonsteroidal anti-inflammatory drugs (NSAIDs) are COX inhibitors, yet despite these medicines being in clinical use for decades, how they achieve their anti-inflammatory effects is still not fully understood.17
Here, we show that in fasting subjects where serum IL-1β is suppressed, AA is elevated, which is reversed upon refeeding. NLRP3 stimulation induced lipid production, including AA and eicosanoids, from macrophages, but only AA profoundly inhibited this inflammasome by suppressing phospholipase C (PLC) and JNK activity. NSAID treatment of macrophages suppressed NLRP3 activity and prostanoid production, but AA levels remained unchanged. These data show that AA is an important physiological regulator of the NLRP3 inflammasome and provide a mechanism explaining why fasting reduces systemic inflammation.
RESULTS
Fasting human subjects have elevated AA levels
The emerging importance of lipids in regulating NLRP3 activity12 led us to investigate the lipidomic profile in fasted compared to fed individuals. Serum samples were taken from a cohort of volunteers. In this study, 21 volunteers consumed a baseline 500 kcal meal, fasted for 24 h, and then consumed a 500 kcal refeed meal. In peripheral blood mononuclear cells (PBMCs) from these volunteers, IL-1β levels were elevated 3 h after refeeding (Figure 1A). Plasma AA was elevated in the volunteers during fasting but reduced upon refeeding (Figure 1B).
Figure 1. Fasting human volunteers have elevated arachidonic acid (AA) levels.
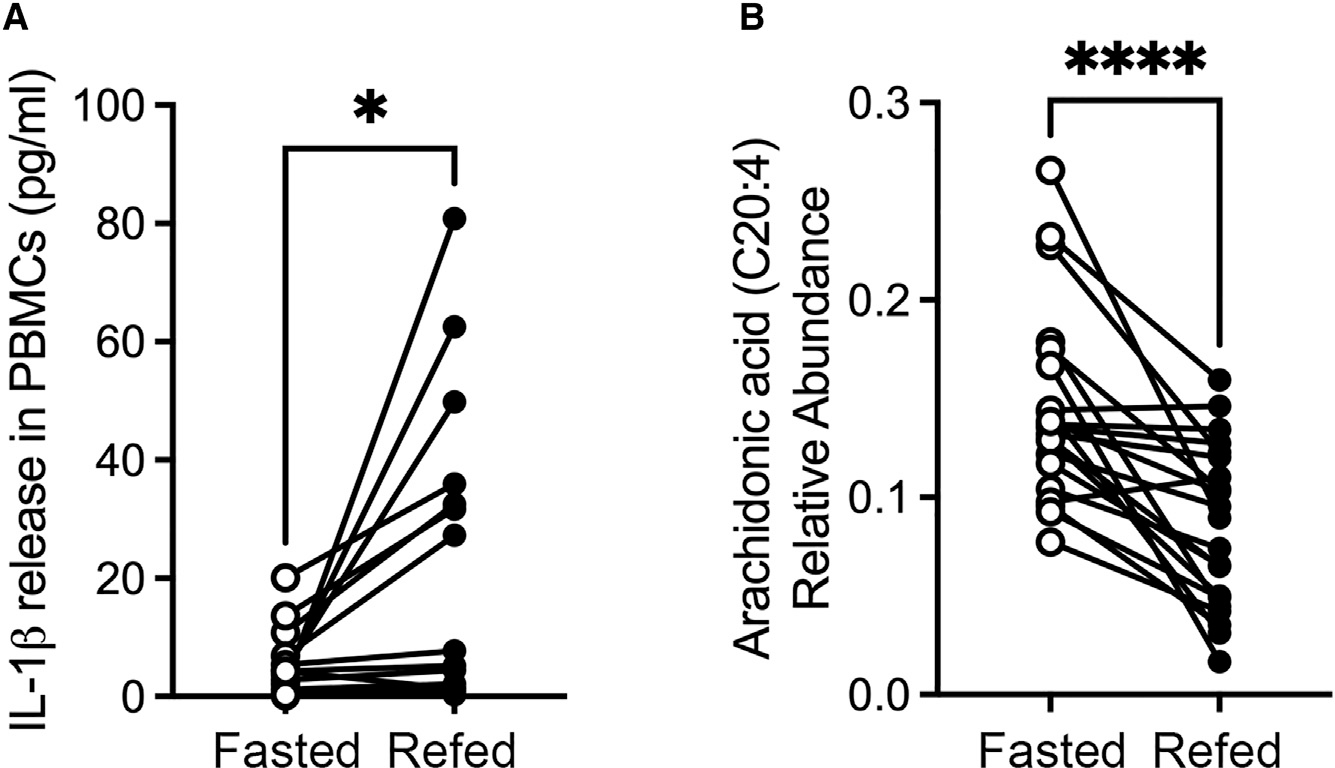
(A) Peripheral blood mononuclear cells were isolated from the volunteers and IL-1β production measured by ELISA before and after refeeding (n = 16).
(B) Plasma AA was measured in the same volunteers before and after fasting (n = 21).
Canonical NLRP3 inflammasome activation leads to AA production
The elevation of AA production coupled to the suppression of IL-1bβ production in fasted subjects led us to investigate how AA and its eicosanoid metabolites are synthesized upon NLRP3 stimulation with nigericin in macrophages. Wild-type (WT) and Nlrp3−/− bone-marrow-derived macrophages (BMDMs) were primed with LPS (3 h, 200 ng/mL) and stimulated with nigericin (10 μM, 1 h). As expected, IL-1β production only occurred in nigericin-treated WT BMDMs (Figure S1). The culture supernatants were collected and lipids extracted and submitted to reverse-phase liquid chromatography/drift tube ion mobility-mass spectrometry.18 Identification was carried out using an internal database and LIPIDMAPS19,20 to match exact mass and collision cross-sections derived from the ion mobility measurements and then subjected to analysis.21,22 Heatmaps and principal-component analysis of the samples revealed clustering between primed WT and Nlrp3−/− BMDMs, suggesting that the lipid compositions of these cell populations are similar. Upon nigericin stimulation, however, WT and Nlrp3−/− cells formed two separate clusters, establishing an NLRP3-dependent lipid signature (Figures 2A and 2B). The complete list of lipids and their abundance can be found in the Table S1.
Figure 2. Canonical NLRP3 inflammasomes leads to eicosanoid production.
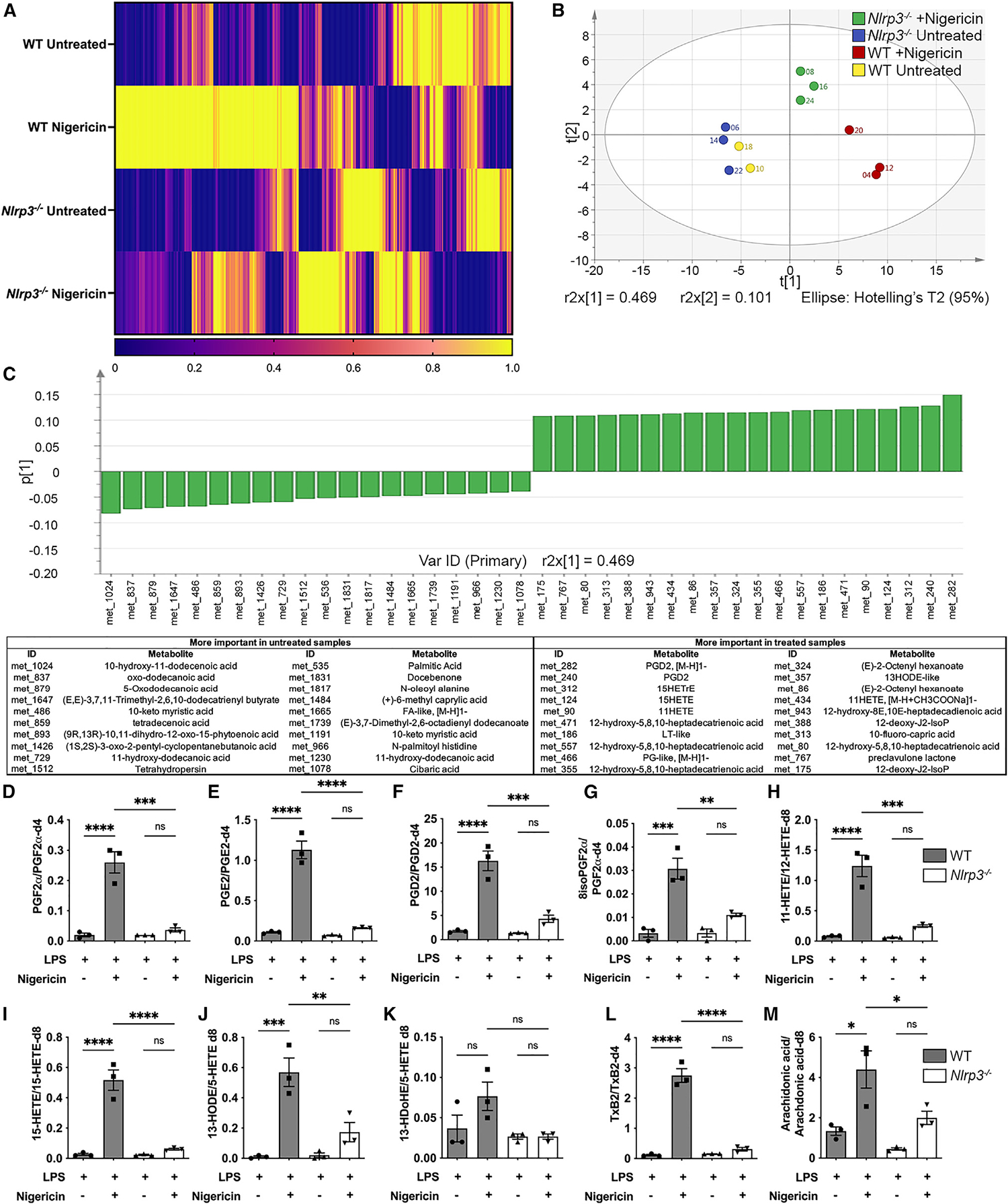
(A) Heatmap analysis of lipids present in culture supernatants of LPS-primed (200 ng/mL for 3 h) WT and Nlrp3−/− BMDMs stimulated with nigericin (10 μM, 1 h), identified by liquid chromatography/drift tube ion mobility-mass spectrometry (LC/DTIM-MS).
(B) Principal-component analysis of the samples used in this study.
(C) Lipids that contribute the most to the differences between nigericin-treated and untreated groups.
(D–M) Quantification by LC/DTIM-MS of eicosanoids PGF2α (D), PGE2 (E), PGD2 (F), 8-iso-PGF2α (G), 11-HETE (H), 15-HETE (I), 13-hydroxydocosahexaenoate (J), 13-hydroxyoctadecadienoic acid (K), TxB2 (L), and AA (M), present in culture supernatants of LPS-primed (200 ng/mL for 3 h) WT and Nlrp3−/− BMDMs stimulated with nigericin (10 μM, 1 h).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way analysis of variance with Tukey’s multiple comparison test). Data are from three independent experiments (mean and SEM).
Eicosanoids, including PG PGD2 and 11-hydroxyeicosatetraenoic acid (11-HETE), were among the species identified as more prevalent following NLRP3 activation (Figure 2C). We therefore quantified a variety of eicosanoids using their respective retention time and exact mass. Very similar patterns were observed regarding molecules such as PGF2α, PGE2, PGD2, 8isoPGF2α, 11-HETE, 15-HETE, 13-hydroxyoctadecadienoic acid (but not 13-hydroxydocosahexaenoate), thromboxane B2 (TxB2), and the eicosanoid precursor AA, with enhanced production in response to NLRP3 stimulation (Figures 2D–2M).
AA inhibits the NLRP3 inflammasome
We hypothesized that the inverse relationship between IL-1β and AA seen in the volunteer samples might be linked to an effect of AA, or its associated eicosanoid metabolites, inhibiting NLRP3 inflammasome activity. In our lipidomic analysis, stimulation of LPS-primed WT BMDMs with nigericin elicited a substantial increase in the production of AA release, in addition to eicosanoid production (Figure 2). We therefore compared the effects of AA with a number of eicosanoids on NLRP3 activation. Stimulation of LPS-primed BMDMs with nigericin in the presence of exogenous AA (40 μM) inhibited NLRP3-induced IL-1β production (Figure 3A). This inhibition was sustained when AA was present both during LPS priming and nigericin stimulation (Figure 3B). Titration analysis revealed that AA did not inhibit nigericin-stimulated, NLRP3-induced lytic cell death in BMDMs despite reducing IL-1β production even at low AA concentrations (Figures 3C and 3D). Similar results were found when NLRP3 was stimulated in BMDMs using ATP (Figures 3E and 3F).
Figure 3. AA inhibits the NLRP3 inflammasome.
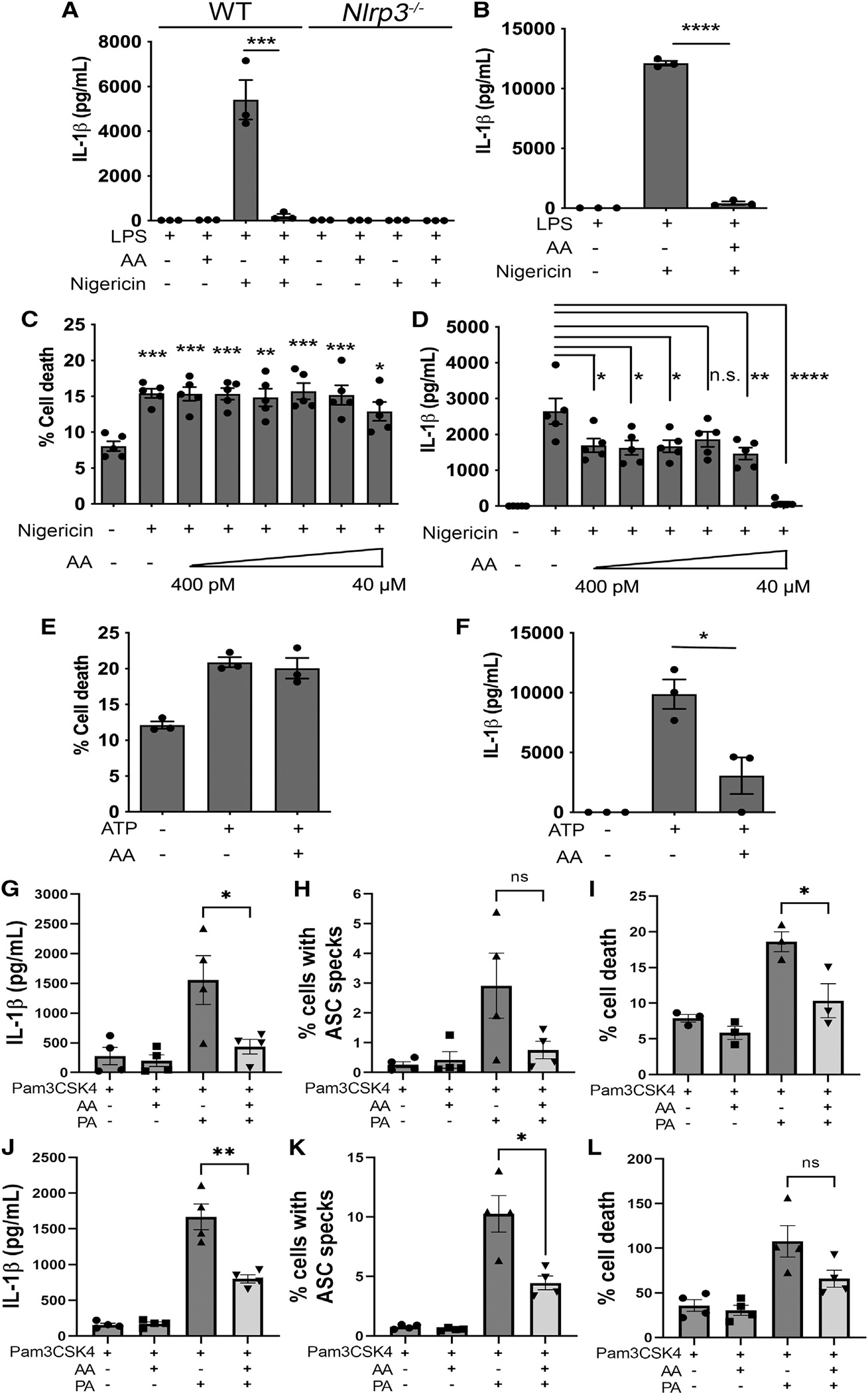
(A and B) IL-1β production in LPS-primed BMDMs after 1 h stimulation with 10 μM nigericin with or without 40 μM AA added after priming (A) or during priming (B).
(C-F) Cellular viability (C and E) and IL-1β (D and F) produced by LPS-primed WT BMDMs after stimulation with nigericin (10 μM, 1 h) (C and D) or ATP (5 mM, 30 min) (E and F) in presence of 40 μM AA.
(G-I) Cellular viability (G), ASC speck quantification (H), and IL-1β (I) produced by WT BMDMs primed with Pam3CSK4 (200 ng/mL, 4 h) after stimulation with palmitic acid (PA; 1 mM, 16 h) in presence of 40 μM AA.
(J-L) Cellular viability (J), ASC speck quantification (K), and IL-1β (L) produced by THP-1 cells (differentiated with PMA, 200 ng/mL for 24 h followed by 24 h washout) primed with Pam3CSK4 (200 ng/mL, 4 h) after stimulation with PA (500 μM, 24 h) in presence of 40 μM AA.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (one-way analysis of variance with Tukey’s multiple comparison test or Student’s unpaired t test). n.s., not significant. Data are from at least three independent experiments (mean and SEM).
Serum PA is elevated in metabolic syndrome.23 We therefore used this lipid to stimulate NLRP3 in the human THP-1 monocyte/macrophage cells differentiated with PMA to a macrophage-like phenotype. Cells were primed with TLR1/2 agonist PAM3CSK4 (200 ng/mL, 4 h), followed by NLRP3 stimulation with nigericin or PA.24 We measured IL-1β production but also immunolabeled the inflammasome adaptor protein ASC to determine the efficiency of ASC speck formation within these cells. NLRP3 activation recruits ASC prior to binding to, and activating, caspase-1.5 If AA inhibits NLRP3, then ASC speck formation, as well as IL-1β production, is expected to be reduced. AA inhibited IL-1β secretion and ASC speck formation but also lytic cell death in human THP-1 cells (Figures 3G–3I and S2). The inhibitory effect of AA was particularly noticeable when PA was used as the NLRP3 trigger, suggesting a potentially important role for AA in regulating lipid-driven inflammation (Figures 3J–3L). AA dose-dependently inhibited NLRP3-induced IL-1β production, ASC speck formation, and cell death after activation by PA in the concentration range of 10–40 μM (Figure S2).
We then explored the possibility that the effect of AA on the NLRP3 inflammasome could be mediated by its metabolites. Specifically, we stimulated LPS-primed (3 h, 200 ng/mL) WT and Nlrp3−/− BMDMs for 1 h with 1 μM freshly prepared eicosanoids/stable eicosanoid analogs (PGE1 analog misoprostol, PGD2, PGE2, PGF2α, and PGI2). Stimulation with these eicosanoids failed to induce cell death or IL-1β production (Figures 4A and 4B), suggesting that these individual eicosanoids do not induce inflammasome activation. The possibility remained, however, that individual eicosanoids could modify NLRP3 activity. To test this idea, LPS-primed (3 h, 200 ng/mL) WT and Nlrp3−/− BMDMs were simultaneously stimulated with nigericin (10 μM, 1 h) and 1 μM freshly prepared eicosanoids. No statistically significant differences in cell death or IL-1β production were observed in comparison to nigericin-only controls (Figures 4C and 4D). Additional titration experiments were then performed in LPS-primed (3 h, 200 ng/mL) WT BMDMs stimulated with nigericin (10 μM for 1 h) in the presence of increasing concentrations of PGE2 or PGF2α (100 pM–10 μM). Again, no effect was seen on cell death (Figures 4E and 4G). Independent of concentration, PGE2 also did not significantly impact IL-1β production, although a trend toward lower IL-1β production at low PGE2 concentrations was observed, similar to previously reported data.24 Concentrations of PGF2α below 1 μM induced a significant decrease in IL-1β production (Figures 4F and 4H). Stimulation with increasing concentrations of PGE2 during LPS priming revealed an increase in IL-1β upon nigericin stimulation (in this experiment, PGE2 was present both during LPS priming and nigericin stimulation) (Figure S3). These results illustrated a complex regulatory network of individual eicosanoids upon inflammasome activation that is heavily influenced by experimental design and thus probably explains the apparent inconsistencies in the literature. It is possible that the net effect of the various COX-derived eicosanoids reflects a regulatory network on NLRP3 inflammasome activity consistent with an emerging role for lipids as modulators of the activity of this inflammasome. Collectively, however, our data uncover that AA itself is a potent inhibitor of the NLRP3 inflammasome.
Figure 4. Individual eicosanoids have little effect on NLRP3 activity.
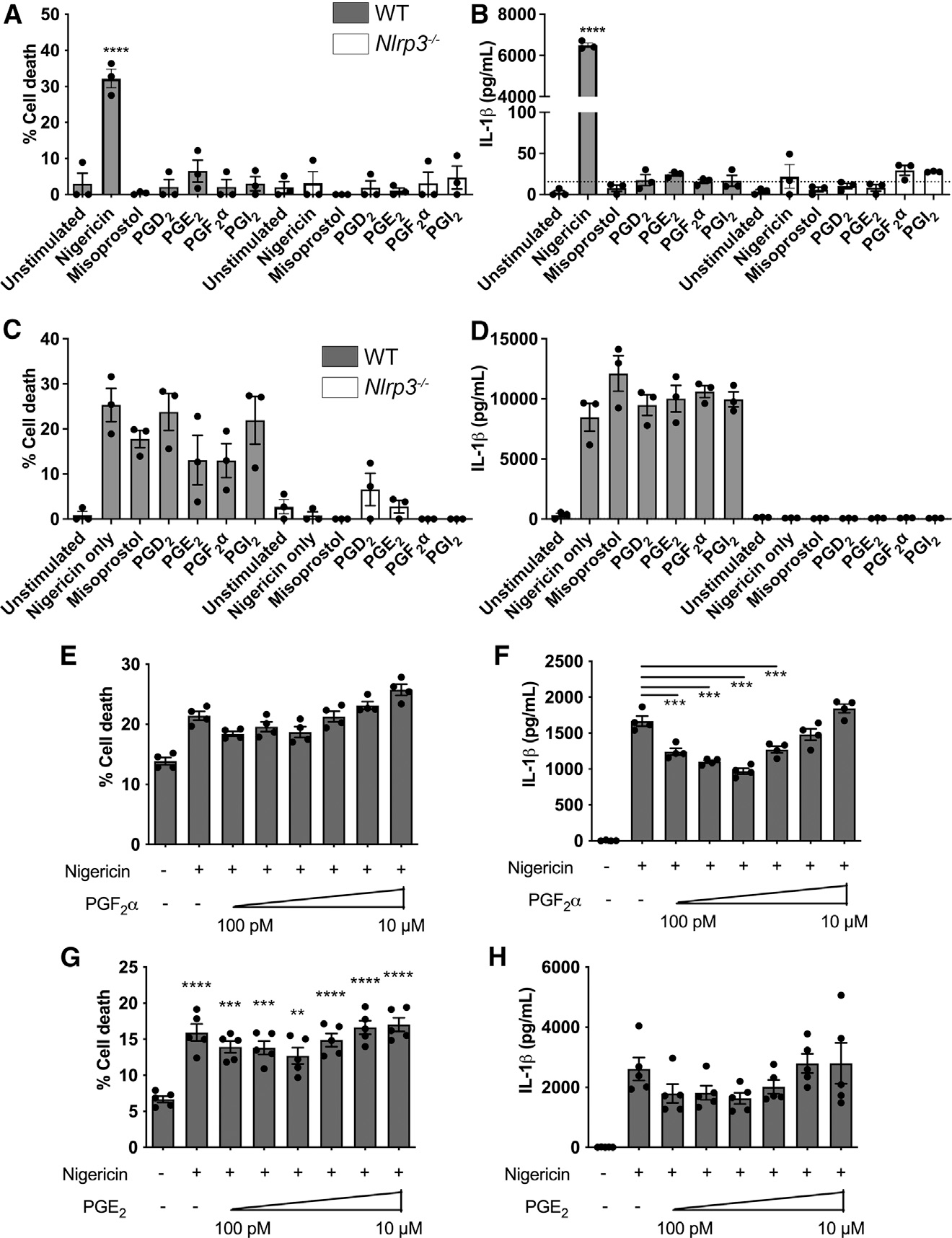
(A-D) Cell death (A and C), and IL-1β (B and D) produced by LPS-primed (200 ng/mL for 3 h) WT and Nlrp3−/− BMDMs in response to 1 h stimulation with 1 μM misoprostol, PGD2, PGE2, PGF2α, and PGI2.
(E-H) Cell death (E and G) and IL-1β (F and H) produced by LPS-primed WT BMDMs in response to 1 h stimulation with 10 mM nigericin in presence of increasing concentrations of PGF2α (E and F) and PGE2 (G and H).
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 in comparison to untreated controls unless indicated otherwise (one-way analysis of variance with Tukey’s multiple comparison test). Dashed line represents the assay detection limit. Data are from three (A-D) or four (E-H) independent experiments (mean and SEM).
Inhibition of AA processing by COX selectively regulates canonical NLRP3 activity
AA is metabolized to eicosanoids, such as PGs, through the activity of COXs (COX-1 and COX-2). Inhibition of COX activity, by NSAIDs, suppresses eicosanoid production and enhances the availability of AA for processing through alternative metabolic pathways. We therefore investigated whether NSAID inhibition of AA metabolism through COX would also lead to NLRP3 inflammasome inhibition. Our lipidomic analysis showed that the COX-2 inhibitor celecoxib, while markedly reducing the production of eicosanoids such as PGF2aα, PGE2, PGD2, 11-HETE, 15-HETE, and TxB2 in response to nigericin stimulation, did not significantly suppress AA release (Figure S4). In previous work, COX inhibitors have been used to show that PGE2 stimulates IL-1β production via increasing il1b transcription25,26 or suppresses IL-1β production either due transcriptional or post-transcriptional effects.16,24 These inconsistencies could be due to differences in experimental design, as LPS priming of macrophages and COX inhibition were performed differently in each study, and COX metabolites can influence signaling pathways involved in pro-IL-1β expression.27,28 To circumvent this problem, we performed LPS stimulation and COX inhibition in conditions that induced similar levels of pro-IL-1β expression but with COX-2, NLRP3, and caspase-1 expressed at similar levels under all experimental conditions (Figures S5A–S5E). Using this system, LPS-primed WT and Nlrp3−/− BMDMs were stimulated with the canonical NLRP3 stimulants nigericin or ATP in the presence or absence of COX-1/-2 inhibitors. COX inhibitors alone, in the absence of nigericin or ATP, did not induce inflammasome activity (Figures S5F–S5G). Nlrp3−/− BMDMs, as expected, responded poorly to nigericin across all conditions, whereas WT BMDMs showed substantial IL-1β and IL-18 production upon treatment with either nigericin or ATP. In each case, we saw striking inhibition of both IL-1β and IL-18 production in the presence of COX inhibitors (Figures 5A–5E). The nonselective COX-1 and COX-2 inhibitor indomethacin and the COX-2 selective inhibitor celecoxib had similar effects on NLRP3 inflammasome activity. Collectively, our data support a model whereby COX inhibitors block NLRP3-induced inflammasome activity, most likely through the actions of AA.
Figure 5. Cyclooxygenase (COX) activity regulates NLRP3 activity but not NLRC4.
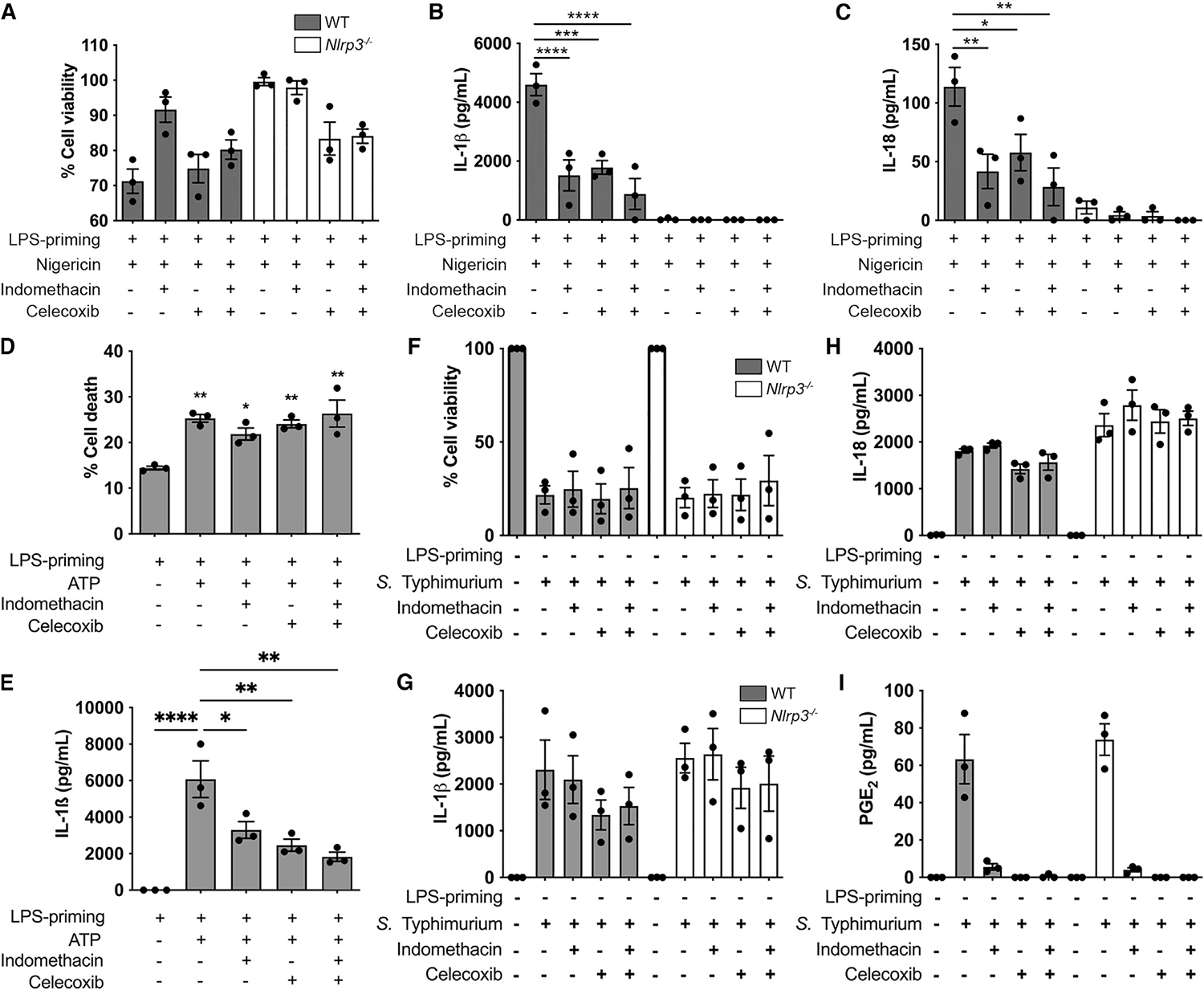
(A-H) Cellular viability (A and D), IL-1β (B and E), and IL-18 (C) produced by LPS-primed (200 ng/mL for 3 h) WT and Nlrp3−/− BMDMs in response to NLRP3 stimulant nigericin (10 μM, 1 h) or ATP (5 mM, 30 min) in presence of COX inhibitors indometacin (100 μM) or celecoxib (10 μM).
(F-I) Cellular viability (F), IL-1β (G), IL-18 (H), and PGE2 (I) produced by unprimed WT and Nlrp3−/− BMDMs in response to S. Typhimurium infection (MOI 10, 2 h) in presence of COX inhibitors.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (one-way analysis of variance with Tukey’s multiple comparison test). Data are from three independent experiments (mean and SEM).
NLRC4 stimulation triggers production of eicosanoids such as PGE2 both in vivo and in macrophages in vitro.18,29 To determine whether COX inhibition also regulates NLRC4 activation, we infected BMDMs with Salmonella Typhimurium (MOI 10) for 2 h in the presence of indomethacin or celecoxib. Under these conditions, S. Typhimurium activates primarily the NLRC4 inflammasome.30 During Salmonella infection of BMDMs, and similar to previous studies, COX-1/-2 inhibition blocked PGE2 production as expected but had no effect on NLRC4 inflammasome activity (Figures 5F–5I). These data suggest that COX inhibitors suppress NLRP3-induced inflammasome activity but do not impact the NLRC4 inflammasome.
AA inhibits PLC activity and suppresses downstream protein kinases (PKs)
Our lipidomic analysis suggested that COX inhibition had little impact on AA abundance in macrophages, but both COX inhibition and AA addition during nigericin stimulation led to decreased IL-1β production. AA is released from the membrane predominantly by the activity of PLA2 but also by the action of PLC.31 NLRP3 is highly regulated by post-translational modifications (PTMs) including, for example, phosphorylation and ubiquitination.7 PLC activity can regulate NLRP3 PTM through JNK1-induced phosphorylation7,. We hypothesized that the presence of a “static” AA pool, for example in the presence of COX inhibitors, might trigger a negative feedback loop driven by classical product inhibition32 of PLC shutting off downstream PKs and hence NLRP3 activation.
To explore this idea, we investigated whether inhibition of PLA2 or PLC affects NLRP3-dependent IL-1β production and cell death. The PLA2 inhibitors ASB14780 and NAAA had no impact on NLRP3-induced IL-1β production, suggesting that PLA2 is not involved in regulating NLRP3 activation in response to nigericin (Figures 6A and 6B). Inhibition of PLC with U-73122, however, revealed a dose-dependent decrease in IL-1β production (Figure 6B). Cell death was increased at 10 μM inhibitor (Figure 6A), but this effect is due to toxicity unrelated to the inflammasome (Figure S6). Titration of the PLC inhibitor showed a dose-dependent decrease in nigericin-induced IL-1β production without affecting cell death (Figures 6C and 6D). Similarly, stimulation of the NLRP3 inflammasome with ATP was also regulated by PLC but not by PLA2 (Figures 6E and 6F).
Figure 6. PLC is required for canonical NLRP3 activity.
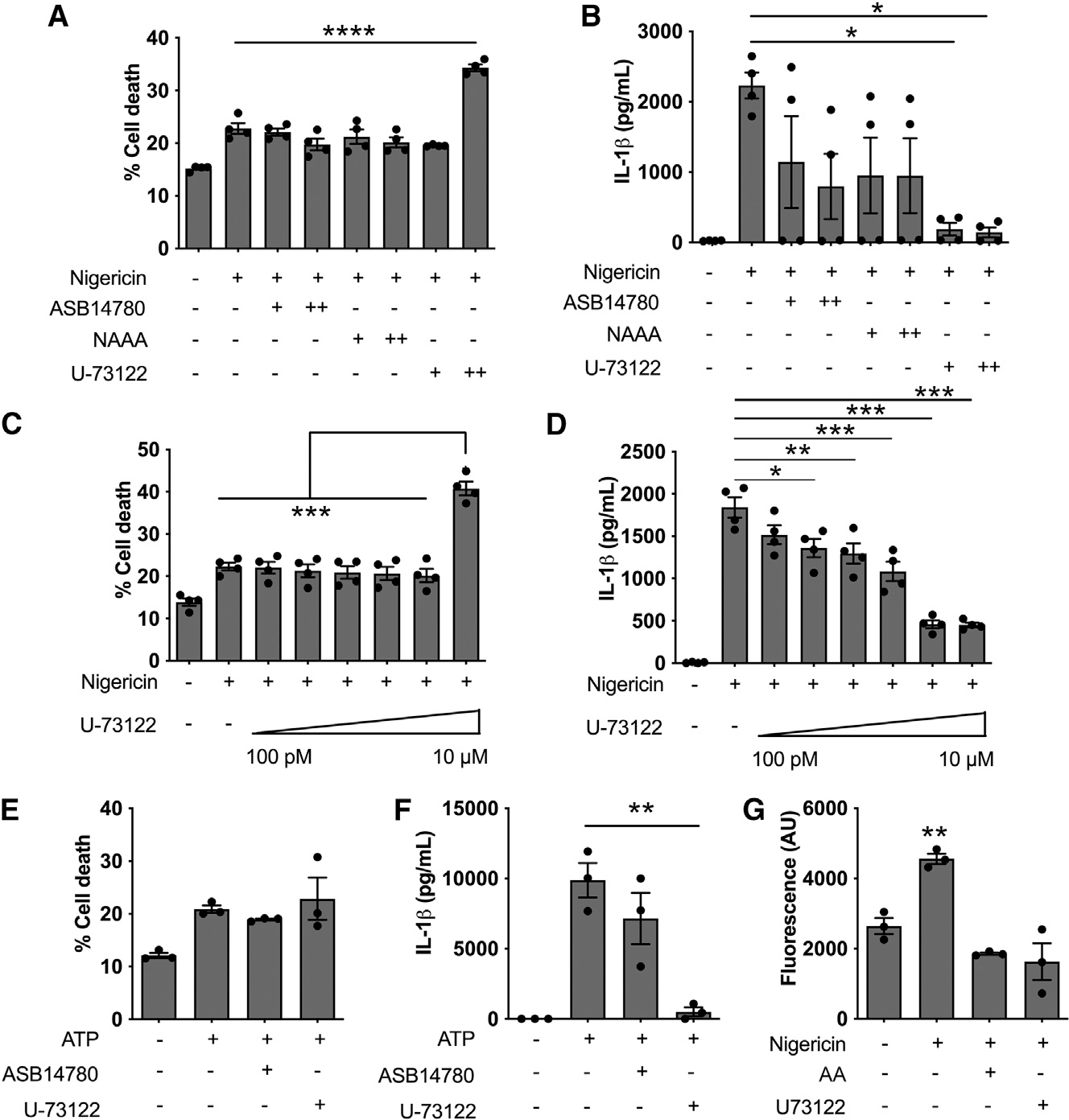
(A and B) Cell death (A) and IL-1β production (B) in LPS-primed (200 ng/mL for 3 h) WT BMDMs in response to 1 h stimulation with 10 mM nigericin in presence of 1 (+) or 10 μM (++)PLA2 inhibitors ASB1414780 and NAAA or PLC inhibitor U-73122. (C and D) Cell death (C) and IL-1β (D) produced by LPS-primed WT BMDMs stimulated for 1 h with 10 mM nigericin in presence of increasing concentrations of PLC inhibitor U-73122.
(E and F) Cell death (E) and IL-1β (F) produced by LPS-primed WT BMDMs stimulated for 30 min with 5 mM ATP in presence of 1 mM ASB14780 (PLA2 inhibitor) or 1 μM U-73122 (PLC inhibitor).
(G) PLC activity of LPS-primed WT BMDMs stimulated with 10 μM nigericin for 1 h in presence of U-73122 or 40 μM AA.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (one-way analysis of variance with Tukey’s multiple comparison test or Student’s unpaired t test). Data are from three (E–G) or four (A-D) independent experiments (mean and SEM).
Since product inhibition is a well-recognized negative feedback mechanism to control metabolic pathways,32 we next investigated whether AA can inhibit PLC activity. Some evidence for this to occur already exists,33 but whether a negative AA feedback loop operates in the context of inflammasome activation is unknown. To investigate this, we quantified PLC activity in protein extracts from LPS-primed WT BMDMs treated with nigericin (10 μM, 1 h) in the presence of AA or the PLC inhibitor (U-73122) as a control. Upon nigericin treatment, PLC activity increases significantly. Unsurprisingly, U-73122 suppressed PLC activity in response to nigericin, but AA also reduced nigericin-stimulated PLC activity (Figure 6G). These data suggested that activation of the canonical NLRP3 inflammasome is modified by PLC, but not PLA2, and that AA, by a product inhibition negative feedback loop, inhibits PLC activity.
PLC stimulates the activation of PKs such as PKC, PKD, and JNK1,31,34,35 which, in turn, upregulates NLRP3 activation.8,10,36,37 We therefore tested whether AA could suppress NLRP3-induced JNK phosphorylation. In THP-1 cells stimulated with nigericin, inhibition of either PKD or JNK led to decreased NLRP3 activation (Figures 7A and 7B). Accordingly, the presence of AA inhibited PKC, PKD, and JNK activation, as suggested by decreased phosphorylation (Figures 7C and 7D). The bile salt lithocholic acid (LCA) also inhibits NLRP3 inflammasome activation.38 LCA, however, does not affect PKC, PKD, and JNK phosphorylation, suggesting that AA and bile salts inhibit the NLRP3 inflammasome by distinct mechanisms (Figures 7A–7D).
Figure 7. AA inhibits PKC, PKD, and JNK.
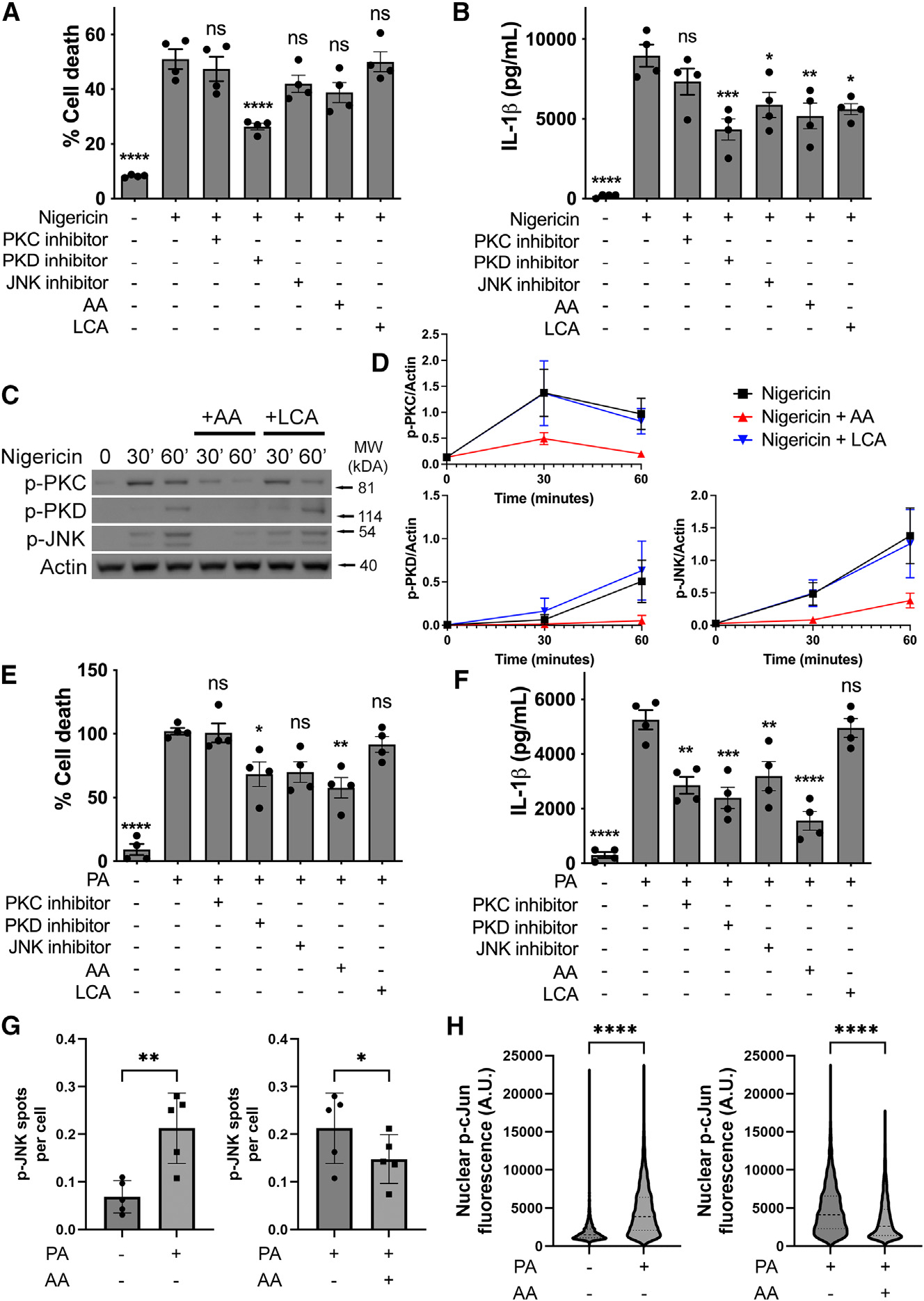
(A and B) Cell death (A) and IL-1β production (B) in THP-1 cells primed with LPS (200 ng/mL, 3 h) followed by stimulation with nigericin (10 μM, 1 h) in presence of PKC inhibitor (sotrastaurin, 10 μM), PKD inhibitor (CRT006601, 10 μM), JNK inhibitor (SP600125, 10 μM), AA (40 μM), or LCA (30 μM).
(C and D) Immunoblots (C) and densitometric quantification (D) of THP-1 cells primed with LPS (200 ng/mL, 3 h) and stimulated with nigericin (10 μM) in presence of AA (40 μM) or LCA (30 μM) for up to 60 min.
(E and F) Cell death (E) and IL-1β production (F) in THP-1 cells primed with Pam3CSK4 (200 ng/mL, 4 h) followed by stimulation with PA (500 μM, 16 h) in presence of PKC inhibitor (sotrastaurin, 10 μM), PKD inhibitor (CRT006601, 10 μM), JNK inhibitor (SP600125, 10 μM), AA (40 μM), or LCA (30 μM).
(G and H) Quantification of p-JNK (G) and nuclear p-c-Jun (H) in THP-1 cells primed with Pam3CSK4 (200 ng/mL, 4 h) after stimulation with PA (500 μM, 24 h) in presence of 40 μM AA.
THP-1 cells were differentiated with PMA (200 ng/mL) for 24 h followed by 24 h washout prior to the experiments. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 in comparison to nigericin-treated cells (A and B) or PA-treated cells (E and F) unless indicated otherwise (one-way analysis of variance with Tukey’s multiple comparison test). n.s., not significant. Data are from three (C and D), four (A, B, E, and F), or five (G and H) independent experiments (mean and SEM).
Similarly, in PA-stimulated THP-1 cells, inhibition of PKC, PKD, and JNK led to impaired NLRP3 activation (Figures 7E and 7F). In response to PA, JNK was phosphorylated and activated as demonstrated by phosphorylation of the JNK target c-Jun. AA suppressed both PA-induced JNK phosphorylation and c-Jun phosphorylation (Figures 7G and 7H). These data identified that AA drives a negative feedback loop, via product inhibition, that suppresses NLRP3 activation through JNK phosphorylation. This provides a mechanistic basis explaining how NLRP3 activity may be regulated by AA either in response to NSAID (COX) inhibition or fasting.
DISCUSSION
Here, we show that AA inhibits the NLRP3 inflammasome, through a PLC-JNK-dependent mechanism, to supress IL-1β production under physiological conditions. This provides a mechanistic basis for how dietary manipulation such as fasting influences the inflammatory state and is likely to be critical for reducing the metaflammation underpinning many diseases induced by the WD. Fasting can differentially regulate NLRP3 activity,2 and elevated IL-1β is observed after feeding,39,40 but the mechanism for this is unclear. It is increasingly clear that lipids are important regulators of NLRP3 activity,12,13 but how this occurs is still poorly understood. Here, we show that in a fasting subject cohort where IL-1β is suppressed, AA is elevated, and this effect is reversed upon feeding. While we do not have direct in vivo evidence that AA suppresses IL-1β production, our data highlighted the possibility that AA, or one of its metabolites, might be an NLRP3 regulator. Unexpectedly, we showed in vitro that AA itself inhibits activation of the NLRP3 inflammasome, while AA metabolites do not. COX inhibitors, which decrease the production of AA eicosanoid metabolites, but not AA itself, also decrease NLRP3 activity, providing further evidence that the NLRP3-inhibitory effect is a direct result of AA rather than a metabolite.
We hypothesized that AA-driven NLRP3 inhibition might occur by product inhibition of the enzymes that liberate AA from the plasma membrane, which are PLA2- and, to a lesser extent, PLC dependent. Our data show that AA inhibits PLC activity, supporting our hypothesis that a product inhibition negative feedback loop occurs.33 Links between PLC and canonical inflammasome activity were reported,41,42 but how precisely PLC activates the NLRP3 inflammasome remains to be fully elucidated. Here, we hypothesized that PLC regulates NLRP3 activity via PKC, PKD, and JNK. PLC drives the generation of the second messenger diacylglycerol, which activates PKC and PKD.31,34,35 The PLC/PKC axis activates JNK1, which we, and others, have shown is important in activating NLRP3.8,10 Additionally, PKD directly phosphorylates NLRP3, enhancing its activity.37 Our data supported this hypothesis, as PLC inhibitors, as well as AA, inhibit JNK1 activation, evidence that the PLC-JNK axis is an important regulator of NLRP3 activity.
We also observed that while AA inhibited NLRP3-mediated IL-1β production, its effect on cell death was marginal. Recently published work has shown that dendritic cells release IL-1β in the absence of cell death upon stimulation with oxidized phospholipids (OxPaPC).43 The mechanisms underpinning this are due to GSDMD-perforated plasma membranes being repaired by triggering the assembly of the endosomal sorting complexes required for transport III (ESCRT III) machinery at the plasma membrane, where it removes the GSDMD pores by shedding them into vesicles, thus preventing GSDMD-mediated pyroptosis.44 We speculate that a similar GSDMD pore repair process may explain our observations, although the relationships between AA, ESCRT III, and NLRP3 remain to be elucidated.
Our work demonstrates a mechanism by which the NSAID COX inhibitors have broad anti-inflammatory effects: through AA inhibition of NLRP3 activity. A number of papers have investigated how COX-derived eicosanoids might interface with inflammasomes. Eicosanoid regulation of IL-1β has, for the most part, focused on transcriptional events.16,25,26 Here, COX inhibition decreased NLRP3-mediated IL-1β and IL-18 production. Our analysis indicated that individual eicosanoids had some minor regulatory effects on canonical NLRP3 activation. Eicosanoids such as PGE2 and PGF2α showed some decrease in the production of IL-1β in LPS-primed BMDMs stimulated with nigericin similar to data from a previous report where PGE2 inhibits the inflammasome via PKA and phosphorylation of NLRP3.23 Others report that PGE2 stimulates IL-1β production in response to Tytius serralatus venom via PKA activation.45 Our data show that COX inhibition decreases the production of PGE2 (and other eicosanoids) but not AA, suggesting that this lipid, rather than an eicosanoid, is regulating NLRP3 activation. The apparent discrepancies in the data around eicosanoid regulation of NLRP3 inflammasome activity suggest there is a complex lipid regulatory network that may have a concentration dependency.
Our data also support an important metabolic loop whereby fasting elevates AA, which feeds back on to the inflammasome, suppressing IL-1β. Elegant data suggest that in adults, IL-1β production is produced as a physiological response to food.3 Our data here show that AA is enhanced in adults upon fasting, when IL-1β is suppressed. This suggests that rather than IL-1β always being involved in pathological responses, in response to feeding, there is a novel, physiological regulatory loop between IL-1β, NLRP3, and AA. Since we have shown that NLRP3 inflammasome activation yields an increase in AA, this regulatory loop may prevent excessive inflammasome activation under normal physiologic conditions. We hypothesize that this differs in the context of the WD, where the NLRP3 inflammasome is chronically activated, and hence IL-1β is contributing to the pathology of long-term inflammation. How AA is regulated in these people is unclear, but it is tempting to speculate that the success of intermittent fasting diets may involve fasting-triggered AA production to suppress inflammasome activity, thereby reducing metaflammation associated with WD metabolic syndrome.
In conclusion, we provide data to suggest that AA is an important physiological inhibitor of the NLRP3 inflammasome. The elevation of AA in plasma lipids from fasting volunteers provides a mechanism to explain the drop in IL-1β production from PBMCs from these people and, potentially, one way in which fasting has beneficial anti-inflammatory effects. Our data also suggest a mechanism by which NSAIDs are anti-inflammatory and identify that AA may have a previously unappreciated role as a primary signaling lipid.
Limitations of the study
Our study demonstrates a mechanism for AA-mediated inflammasome inhibition in vitro. While our in vivo data from human volunteers observed elevated AA and reduced IL-1β, in agreement with our in vitro data, this evidence is indirect. Further studies are required to unambiguously link AA to reduced inflammation in vivo and validate the mechanisms we proposed in this article.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and resource requests can be directed to and will be fulfilled by the lead contact Clare E. Bryant (ceb27@cam.ac.uk).
Materials availability
Mouse lines and cell lines used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The uncropped immunoblots, quantitative and lipidomic data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit pAb anti-Caspase-1 p10 | Santa Cruz | sc-514; RRID AB_2068895 |
| Goat pAb anti-IL-1 beta/IL-1F2 | R&D | AF-401; RRID AB_416684 |
| Mouse mAb anti-Actin | Abcam | AB3280; RRID AB_303668 |
| Rat mAb anti-NLRP3 | R&D | MAB7578; RRID AB_2889405 |
| Goat pAb anti-COX2 | R&D | AF4198; RRID AB_2229909 |
| Rabbit pAb anti-Goat IgG Peroxidase | Santa Cruz | sc-2922; RRID AB_656965 |
| Horse pAb anti-mouse IgG Peroxidase | Cell Signaling | 7076; RRID AB_330924 |
| Goat pAb anti-Rabbit IgG (whole molecule) Peroxidase | Sigma-Aldrich | 7077; RRID AB_10694715 |
| Rabbit pAb anti-phospho-PKC (pan) | Cell Signaling | 9371; RRID AB_2168219 |
| Rabbit pAb anti-phospho-PKD | Cell Signaling | 2051; RRID AB_330841 |
| Rabbit pAb anti-phospho-JNK | Cell Signaling | 4668; RRID AB_823588 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Salmonella enterica Typhimurium SL1344 | Hoiseth and Stocker46 | N/A |
|
| ||
| Biological samples | ||
|
| ||
| Human blood samples | From subject cohort – ClinicalTrials.gov ID NCT02719899 | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetic Acid | Millipore Sigma | 5330010050 |
| Arachidonic Acid | Tocris | 2756 |
| Arachidonic Acid | Sigma-Aldrich | 10931 |
| ASB-14780 | Sigma-Aldrich | SML1913 |
| ATP | Sigma-Aldrich | A1852 |
| Celecoxib | Millipore Sigma | SML3031 |
| Chloroform | Millipore Sigma | 288306 |
| CRT0066101 | Tocris | 4975 |
| Deutered COX and LOX LC-MS Mixture | Cayman Chemical | 19228 |
| Dulbecco’s Modification of Eagle’s Medium | Corning | 10–013-CV |
| Dulbecco’s Phosphate-Buffered Saline | Corning | 21–031-CV |
| Gentamicin | Millipore Sigma | G1397 |
| Halt Phosphatase Inhibitor Cocktail (100x) | Thermo-Fisher | 1862495 |
| Halt Protease Inhibitor Cocktail (100x) | Thermo-Fisher | 87786 |
| Hexane | Millipore Sigma | 1043911000 |
| HyClone Fetal Bovine Serum | Fisher Scientific | SH3008803 |
| Indomethacin | Millipore Sigma | I7378 |
| Isopropanol | Millipore Sigma | 1027811000 |
| L-Glutamine | Millipore Sigma | G7513 |
| LB Broth (Miller) | Sigma-Aldrich | L3522 |
| LB Broth with Agar (Miller) | Sigma-Aldrich | L3147 |
| Lithocholic Acid | Millipore Sigma | L6250 |
| Methanol | Millipore Sigma | 1.06035 |
| Misoprostol | Tocris | 2297 |
| N-(p-Amylcinnamoyl)anthranilic acid | Sigma-Aldrich | A8486 |
| Nigericin | Sigma-Aldrich | N7143 |
| NP-40 (Nonidet P-40 Substitute) | Boston Bioproducts | #P-877 |
| Palmitic Acid | Millipore Sigma | P0500 |
| Pam3CSK4 | Invivogen | tlrl-pms |
| Penicilin Streptomycin Solution, 100x | Corning | 30–002-CI |
| Pierce™ Lane Marker Reducing Sample Buffer | Thermo-Fisher | 39000 |
| Prostaglandin D2 | Cayman Chemical | 12010 |
| Prostaglandin E2 | Tocris | 2296 |
| Prostaglandin F2alpha | Tocris | 4214 |
| Prostaglandin I2 | Tocris | 2989 |
| Sotrastaurim | Abcam | ab219867 |
| SP600125 | Tocris | 1496 |
| TMB Substrate Reagent Set | BD | 555214 |
| Ultrapure LPS, E. coli O111:B4 | Invivogen | tlrl-3pelps |
| U-73122 | Sigma-Aldrich | 662035 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CytoTox 96(R) Non-Radioactive Cytotoxicity Assay | Promega | G1780 |
| Clarity Max Western ECL Substrate | Bio-Rad | 1705062 |
| EnzChek(TM) Direct Phospholipase C Assay Kit | Invitrogen | E10215 |
| Human IL-1Beta/IL-1F2 DuoSet ELISA | R&D Systems | DY201 |
| Mouse IL-1Beta/IL-1F2 DuoSet ELISA | R&D Systems | DY401 |
| Mouse IL-18 Elisa kit | MBL International | 7625 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 |
| PGE2 ELISA kit | Enzo Life Sciences | ADI-900–001 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data | This paper | Mendeley Data: https://doi.org/10.17632/vtpjtgm2tw.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| THP-1 | ATCC | ATCC TIB-202 |
|
| ||
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles Rivers | N/A |
| Mouse: C57BL/6 NLRP3 Knockout | Millenium Pharmaceuticals | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | Graphpad Software | www.graphpad.com |
| ImageJ | NIH | imagej.nih.gov/ij/ |
| SIMCA | Sartorius | www.sartorius.com |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
WT C57BL/6 mice were obtained from Charles River, UK. Nlrp3−/− mice on a C57BL/6 background were produced by Millenium Pharmaceuticals and obtained from Kate Fitzgerald (University of Massachusetts). All animals were housed in a pathogen-free facility and all work involving live animals complied with the University of Cambridge Ethics Committee regulations under Home Office Project License number 80/2572. This study used male and female mice in similar proportions, all ranging from 8 to 12-weeks old.
Human studies
The fasting and refeeding clinical study in healthy volunteers was approved by the US National Institutes of Health Intramural Institutional Review Board (ClinicalTrials Identifier No. NCT02719899).47 In the first visit, volunteers were screened in the ambulatory clinic and signed informed consent for the protocol before enrolling in the study. In a second visit, blood was drawn from overnight-fasted participants to establish the baseline immune response. Next, overnight-fasted participants consumed a 500-calorie meal before 8 a.m. and fasted for 24h with unrestricted water intake. After 24h, blood was draw (fasted samples), the participants ate a 500-calorie meal and after 3h, blood was draw once again (refed samples).47 The PBMCs were extracted following the 24h fast and 3h after refeeding and incubated with ATP (3 mM) for 30 min, prior to the measurement of IL-1β release by ELISA assays.2 Serum from these same samples were employed to measure circulating arachidonic acid levels.
METHOD DETAILS
Lipid extraction and mass spectrometry
Lipid extraction and mass spectrometry extractions were performed as follows18: 2.5 mL of isopropanol/hexane/acetic acid solution (20:30:2, v/v/v) at 4°C and 10 ng of COX and LOX LC/MS mixture (CAY19228–1, Cayman Chemical) were added to 1 mL supernatants from stimulated or stimulated BMDMs and kept on ice for 10 min. Samples were then vortexed for 15 s, 2.5 mL of hexane (4°C) was added and the solution vortexed for another 15 s. The samples were subjected to centrifugation (5 min, 4°C, 900 G), the organic layer collected. The aqueous fraction was re-extracted by addition of 3.75 mL chloroform/methanol (1:2, v/v), vortexed for 15 s and 1.25 mL of chloroform was added. Samples were then vortexed for 15 s, 1.25 mL of water was added, vortexed again, and centrifuged (5 min, 4°C, 900 G). The organic layer was combined with the organic layer from the first extraction, dried under nitrogen, and analyzed by liquid chromatography/drift tube ion mobility coupled with high resolution mass spectrometry (LC/DTIM-MS) (Agilent 6560 IM QTOF MS (Agilent Technologies) with a reverse-phase ACQUITY CSH C18 column (Waters Company). Annotation and identification of lipids was performed with an adaption of the KniMet pipeline22 with combined annotation comprising the LIPID MAPS Structure Database and an internal lipid library.19
Cell isolation and culture
C57BL/6 mice were killed by cervical dislocation, ethanol 70% was sprayed for sterilization and the skin around the leg was removed. Afterward, the leg was removed and placed in DMEM (Dulbecco’s modified Eagle’s medium) on ice. In a laminar flow cabinet, the muscle was removed, the tibia and femur were separated at the knee ligament, and the proximal and distal epiphysis removed. For bone marrow derived macrophages (BMDM) culture, bone marrow was flushed out using BMDM growth media (DMEM supplemented with 10% HyClone (Thermo Fisher Scientific), 20% L929 conditioned media and 5 mM L-Glutamine (Sigma)), and the collected cells were centrifuged at 300 × G for 10 min at 15°C and resuspended in BMDM growth media. The cells were then cultured at 37°C under 5% CO2 atmospheric conditions, with the addition of equal volume of the appropriate media after 2 days in culture and completely replenishing the media after 4 days in culture. Every experiment was conducted using cells with 7–9 days in culture.
L929 conditioned media was prepared by growing L929 cells to confluence for 2 weeks, in RPMI 1640 (Sigma) supplemented with 10% Hyclone and 5 mM L-Glutamine. The culture supernatant was collected and sterilized by filtration in 0.22 μm filters (Milipore).
Cell stimulation and infection
Salmonella Typhimurium strain SL134446 were cultivated to log phase by pre-culturing the bacteria for 17.5 h in 5 mL LB broth (Sigma) at 37°C and 200 rpm, followed by a 1 in 10 dilution of the pre-culture in LB broth and further culture for 2 h. The bacteria were then centrifuged for 10 min at 4.300 × G and washed in BMDM growth media and allowed to infect cells at the indicated multiplicity of infection (MOI) for an hour at 37°C and 5% CO2. For the 2 and 6 h timepoints, the infection was followed by washing and incubation with media containing 50 μg/mL gentamicin (Sigma) for another hour. For the 6 h timepoint, media was replaced by supplemented DMEM containing 10 μg/mL gentamicin and incubated at 37°C for another 4 h. Culture supernatants were collected for every timepoint for cytokine quantitation.
Selected experiments required priming with LPS or Pam3CSK4. This was performed by incubation of cells in growth media containing 200 ng/mL ultrapure LPS from Escherichia coli O111:B4 (Invivogen) for 3 h or 200 ng/mL Pam3CSK4 (Invivogen) for 4 h at 37°C and 5% CO2, followed by successive washing in media alone. Before priming, COX inhibition experiments included a pre-incubation step of 30 min with indomethacin 100 mM (Sigma) or celecoxib 10 μM (Sigma). COX inhibitors were also present at these concentrations during LPS priming and stimulations.
For stimulation experiments, the LPS-primed cells were incubated with Nigericin 10 μM (Sigma) for 1 h or ATP 5 mM (Sigma) for 30 min. In selected assays, nigericin 10 μM and prostaglandins (obtained from Tocris) at 1 μM (unless otherwise stated) were added simultaneously and incubated for 1 h. A pre-incubation step of 30 min was performed after priming in experiments containing arachidonic acid 40 μM (Tocris), U-73122 1 μM (Sigma), ASB14780 1 μM (Sigma), N-(p-Amylcinnamoyl)anthranilic acid 1 μM (Sigma), sotrastaurin 10 μM (abcam), CRT0066101 10 μM (Tocris), SP600125 10 μM (Tocris), LCA 30 μM (Sigma).
Cellular viability assays
BMDM cytotoxicity was measure using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). Briefly, after cellular infection and stimulation as indicated above, the adhered cells were washed three times in non-supplemented DMEM, incubated with 40 μL per well of Triton X-100 0.5% for 15 min at 4°C. The cells were then scrapped from the wells and 10 μL of each well was transferred to another plate containing 105 μL Triton X-100 1.2% per well and incubated at 37°C for an hour, diluted in PBS if necessary and the CytoTox reagent was used as described by the manufacturer. Cellular viability was then calculated in relation to the uninfected control containing 200.000 cells (100% viability). To quantify cell death, the supernatants of stimulated cells were collected and LDH activity was measured using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega), in relation to a 100% cell lysis control done by lysing 200.000 cells with the lysis reagent present in the CytoTox kit.
Cytokine quantification
Secreted cytokines were quantified by enzyme linked immunosorbent assay (ELISA) using the experiments supernatants after appropriate dilution in growth media. All cytokines were measured according to the manufacturer’s instructions. For IL-1β the kit OptEIA Mouse IL-1β Set (BD Biosciences) and Human IL-1β DuoSet (R&D Systems) were used. For IL-18 the kit mouse IL-18 ELISA (MBL International) was used.
Immunoblots
After stimulation, cells were lysed for 10 min in ice using buffer containing 10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM NaVO4, 20 mM PMSF, Phosphatase inhibitor cocktail 3 (1 in 100 dilution, Sigma) and Protease inhibitor cocktail (1 in 100 dilution, P8340, Sigma). Protein levels were quantified using Pierce BCA Protein Assay Kit (Life Technologies) and adjusted to 500 μg/mL for immunoblotting. The samples were then incubated for 5 min at 100°C with Pierce Lane Marker Reducing Sample Buffer (Life Technologies). Gels were loaded with 20 μL of the sample per lane, with a final protein mass of 10 μg.
Immunoblots were probed using the following primary antibodies: caspase-1 p10 (mouse) (sc-514, Santa Cruz) 1 in 500; IL-1β (goat) (AF-401, R&D Systems) 1 in 1000; β-Actin (mouse) (AB3280, ABCAM) 1 in 2500; NLRP3 (rat) (MAB7578-SP, R&D Systems) 1 in 2000; COX2 (goat) (AF4198, R&D Systems). The secondary antibodies used were: anti-goat IgG-HRP (sc-2922, Santa Cruz) 1 in 5000; anti-mouse IgG-HRP (7076, Cell Signaling) 1 in 6000; anti-rabbit IgG-HRP (A24537, Thermo Scientific) 1 in 6000 as appropriate; anti-rat IgG-HRP (7077S, Cell Signaling) 1 in 5000.
PLC activity assay
10.000.000 BMDMs were plated in petri dishes overnight and primed with LPS as described above. The cells were then pre-incubated with arachidonic acid or U-73122 as described above and next stimulated with nigericin 10 μM for 1 h with or without inhibitors. The cells were then collected and lysed for 10 min in ice using buffer containing 10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM NaVO4, 20 mM PMSF, Phosphatase inhibitor cocktail 3 (1 in 100 dilution, Sigma) and Protease inhibitor cocktail (1 in 100 dilution, P8340, Sigma). PLC activity was then measured with EnzChek Direct Phospholipase C Assay Kit (Thermo) as indicated by the manufacturer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was done using the software Prism 6.0 (GraphPad Software) as indicated in each individual experiment. In summary, statistical difference between two groups was determined using unpaired t test, differences between multiple groups were determined using one-way analysis of variance (ANOVA) with Tukey’s post-test. In this work, a p value below 0.05 was considered significant. Analysis of metabolomic data was done using SIMCA 15 (Sartorius).
Supplementary Material
Highlights.
In fasting compared to fed subjects, plasma IL-1β is lower and arachidonic acid (AA) is higher
Exogenous AA impairs NLRP3 inflammasome activity in human and mouse macrophages
AA inhibits phospholipase C and reduces JNK stimulation and hence NLRP3 activity
ACKNOWLEDGMENTS
This work was supported by a Wellcome Trust Investigator award (108045/Z/15/Z) and MRC project grant (MR/X000826/1) to C.E.B. We would like to thank Stephen Webster and Panagiotis Tourlomousis for their help and guidance with the experimental planning and for providing critical resources for this research. The studies in the initial human protocol were supported by funding from the US NHLBI Division of Intramural Research (ZIA-HL005199) to M.N.S. Work in the J.L.G. lab is supported by the Medical Research Council (MR/P011705/1). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any author-accepted manuscript version arising from this submission.
Footnotes
DECLARATION OF INTERESTS
C.E.B. is a co-founder of Danger Bio, on the scientific advisory board of Nodthera and Related Sciences, and is a consultant for Janssen.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113700.
REFERENCES
- 1.Christ A, Lauterbach M, and Latz E (2019). Western Diet and the Immune System: An Inflammatory Connection. Immunity 51, 794–811. [DOI] [PubMed] [Google Scholar]
- 2.Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, Waclawiw MA, Han K, Pelletier M, Sauve AA, et al. (2015). Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Invest. 125, 4592–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiedemann SJ, Trimigliozzi K, Dror E, Meier DT, Molina-Tijeras JA, Rachid L, Le Foll C, Magnan C, Schulze F, Stawiski M, et al. (2022). The cephalic phase of insulin release is modulated by IL-1βeta. Cell Metab. 34, 991–1003.e6. [DOI] [PubMed] [Google Scholar]
- 4.Misaki Y, Miyauchi R, Mochizuki K, Takabe S, Shimada M, Ichikawa Y, and Goda T (2010). Plasma interleukin-1beta concentrations are closely associated with fasting blood glucose levels in healthy and pre-clinical middle-aged nonoverweight and overweight Japanese men. Metabolism 59, 1465–1471. [DOI] [PubMed] [Google Scholar]
- 5.Swanson KV, Deng M, and Ting JPY (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christ A, Gu€nther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, et al. (2018). Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, and Núñez G (2023). The NLRP3 inflammasome: activation and regulation. Trends Biochem. Sci. 48, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, and Mitsuyama M (2013). Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stutz A, Kolbe C-C, Stahl R, Horvath GL, Franklin BS, van Ray O, Brinkschulte R, Geyer M, Meissner F, and Latz E (2017). NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 214, 1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradfield CJ, Liang JJ, Ernst O, John SP, Sun J, Ganesan S, de Jesus AA, Bryant CE, Goldbach-Mansky R, and Fraser IDC (2023). Biphasic JNK signaling reveals distinct MAP3K complexes licensing inflammasome formation and pyroptosis. Cell Death Differ. 30, 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Q, Kuang H, Chen C, Yan J, Do-Umehara HC, Liu XY, Dada L, Ridge KM, Chandel NS, and Liu J (2015). The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat. Immunol. 16, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kagan JC (2023). Excess lipids on endosomes dictates NLRP3 localization and inflammasome activation. Nat. Immunol. 24, 3–4. [DOI] [PubMed] [Google Scholar]
- 13.Liang JJ, Fraser IDC, and Bryant CE (2021). Lipid regulation of NLRP3 inflammasome activity through organelle stress. Trends Immunol. 42, 807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Venditti R, Ran L, Liu Z, Vivot K, Schürmann A, Bonifacino JS, De Matteis MA, and Ricci R (2023). Distinct changes in endosomal composition promote NLRP3 inflammasome activation. Nat. Immunol. 24, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- 16.Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL, Chae JJ, and Shelhamer JH (2015). Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macro-phages. J. Immunol. 194, 5472–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenstein A, Hilliard BK, Pope SD, Zhang C, Taskar P, Waizman DA, Israni-Winger K, Tian H, Luan HH, and Wang A (2022). Activation of the transcription factor NRF2 mediates the anti-inflammatory properties of a subset of over-the-counter and prescription NSAIDs. Immunity 55, 1082–1095.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz C, Liggi S, Mocciaro G, Jung S, Induruwa I, Pereira M, Bryant CE, Meckelmann SW, O’Donnell VB, Farndale RW, et al. (2019). A Comprehensive UHPLC Ion Mobility Quadrupole Time-of-Flight Method for Profiling and Quantification of Eicosanoids, Other Oxylipins, and Fatty Acids. Anal. Chem. 91, 8025–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CRH, Shimizu T, Spener F, van Meer G, Wakelam MJO, and Dennis EA (2009). Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahy E, Sud M, Cotter D, and Subramaniam S (2007). LIPID MAPS online tools for lipid research. Nucleic Acids Res. 35, W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinz C, Liggi S, and Griffin JL (2018). The potential of Ion Mobility Mass Spectrometry for high-throughput and high-resolution lipidomics. Curr. Opin. Chem. Biol. 42, 42–50. [DOI] [PubMed] [Google Scholar]
- 22.Liggi S, Hinz C, Hall Z, Santoru ML, Poddighe S, Fjeldsted J, Atzori L, and Griffin JL (2018). KniMet: a pipeline for the processing of chromatography-mass spectrometry metabolomics data. Metabolomics 14, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bermúdez-Cardona J, and Velásquez-Rodríguez C (2016). Profile of Free Fatty Acids and Fractions of Phospholipids, Cholesterol Esters and Triglycerides in Serum of Obese Youth with and without Metabolic Syndrome. Nutrients 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortimer L, Moreau F, MacDonald JA, and Chadee K (2016). NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 17, 1176–1186. [DOI] [PubMed] [Google Scholar]
- 25.Hua KF, Chou JC, Ka SM, Tasi YL, Chen A, Wu SH, Chiu HW, Wong WT, Wang YF, Tsai CL, et al. (2015). Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1βeta production. J. Cell. Physiol. 230, 863–874. [DOI] [PubMed] [Google Scholar]
- 26.Zas1ona Z, Pålsson-McDermott EM, Menon D, Haneklaus M, Flis E, Prendeville H, Corcoran SE, Peters-Golden M, and O’Neill LAJ. (2017). The Induction of Pro-IL-1βeta by Lipopolysaccharide Requires Endogenous Prostaglandin E(2) Production. J. Immunol. 198, 3558–3564. [DOI] [PubMed] [Google Scholar]
- 27.Poligone B, and Baldwin AS (2001). Positive and negative regulation of NF-kappaB by COX-2: roles of different prostaglandins. J. Biol. Chem. 276, 38658–38664. [DOI] [PubMed] [Google Scholar]
- 28.Tang T, Scambler TE, Smallie T, Cunliffe HE, Ross EA, Rosner DR, O’Neil JD, and Clark AR (2017). Macrophage responses to lipopolysaccharide are modulated by a feedback loop involving prostaglandin E(2), dual specificity phosphatase 1 and tristetraprolin. Sci. Rep. 7, 4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, and Vance RE (2012). Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, and Bryant CE (2014). Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 111, 7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadamur G, and Ross EM (2013). Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154. [DOI] [PubMed] [Google Scholar]
- 32.Walter C, and Frieden E (1963). The Prevalence and Significance of the Product Inhibition of Enzymes. Adv. Enzymol. Relat. Subj. Biochem. 25, 167–274. [DOI] [PubMed] [Google Scholar]
- 33.Sumida C, Graber R, and Nunez E (1993). Role of fatty acids in signal transduction: modulators and messengers. Prostaglandins Leukot. Essent. Fatty Acids 48, 117–122. [DOI] [PubMed] [Google Scholar]
- 34.Mérida I, Arranz-Nicolás J, Rodríguez-Rodríguez C, and Ávila-Flores A (2019). Diacylglycerol kinase control of protein kinase C. Biochem. J. 476, 1205–1219. [DOI] [PubMed] [Google Scholar]
- 35.Wang QJ (2006). PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 27, 317–323. [DOI] [PubMed] [Google Scholar]
- 36.Shio MT, Christian JG, Jung JY, Chang K-P, and Olivier M (2015). PKC/ROS-Mediated NLRP3 Inflammasome Activation Is Attenuated by Leishmania Zinc-Metalloprotease during Infection. PLoS Negl. Trop. Dis. 9, e0003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, Mihlan M, Liu Y, Puig Gá mez M, Goginashvili A, et al. (2017). Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 214, 2671–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo C, Xie S, Chi Z, Ke Y, Lu L, Guo C, Xie S, Chi Z, Zhang J, Liu Y, et al. (2016). Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome Article Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 45, 802–816. [DOI] [PubMed] [Google Scholar]
- 39.Dror E, Dalmas E, Meier DT, Wueest S, Thé venet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, et al. (2017). Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 18, 283–292. [DOI] [PubMed] [Google Scholar]
- 40.Hepprich M, Wiedemann SJ, Schelker BL, Trinh B, Stärkle A, Geigges M, Löliger J, Böni-Schnetzler M, Rudofsky G, and Donath MY (2020). Postprandial Hypoglycemia in Patients after Gastric Bypass Surgery Is Mediated by Glucose-Induced IL-1β. Cell Metab. 31, 699–709.e5. [DOI] [PubMed] [Google Scholar]
- 41.Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, et al. (2018). Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negash AA, Olson RM, Griffin S, and Gale M (2019). Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 15, e1007593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, and Kagan JC (2016). An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, and Broz P (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- 45.Zoccal KF, Sorgi CA, Hori JI, Paula-Silva FWG, Arantes EC, Serezani CH, Zamboni DS, and Faccioli LH (2016). Opposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortality. Nat. Commun. 7, 10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoiseth SK, and Stocker BA (1981). Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239. [DOI] [PubMed] [Google Scholar]
- 47.Han K, Singh K, Rodman MJ, Hassanzadeh S, Wu K, Nguyen A, Huffstutler RD, Seifuddin F, Dagur PK, Saxena A, et al. (2021). Fasting-induced FOXO4 blunts human CD4+ T helper cell responsiveness. Nat. Metab. 3, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The uncropped immunoblots, quantitative and lipidomic data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit pAb anti-Caspase-1 p10 | Santa Cruz | sc-514; RRID AB_2068895 |
| Goat pAb anti-IL-1 beta/IL-1F2 | R&D | AF-401; RRID AB_416684 |
| Mouse mAb anti-Actin | Abcam | AB3280; RRID AB_303668 |
| Rat mAb anti-NLRP3 | R&D | MAB7578; RRID AB_2889405 |
| Goat pAb anti-COX2 | R&D | AF4198; RRID AB_2229909 |
| Rabbit pAb anti-Goat IgG Peroxidase | Santa Cruz | sc-2922; RRID AB_656965 |
| Horse pAb anti-mouse IgG Peroxidase | Cell Signaling | 7076; RRID AB_330924 |
| Goat pAb anti-Rabbit IgG (whole molecule) Peroxidase | Sigma-Aldrich | 7077; RRID AB_10694715 |
| Rabbit pAb anti-phospho-PKC (pan) | Cell Signaling | 9371; RRID AB_2168219 |
| Rabbit pAb anti-phospho-PKD | Cell Signaling | 2051; RRID AB_330841 |
| Rabbit pAb anti-phospho-JNK | Cell Signaling | 4668; RRID AB_823588 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Salmonella enterica Typhimurium SL1344 | Hoiseth and Stocker46 | N/A |
|
| ||
| Biological samples | ||
|
| ||
| Human blood samples | From subject cohort – ClinicalTrials.gov ID NCT02719899 | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetic Acid | Millipore Sigma | 5330010050 |
| Arachidonic Acid | Tocris | 2756 |
| Arachidonic Acid | Sigma-Aldrich | 10931 |
| ASB-14780 | Sigma-Aldrich | SML1913 |
| ATP | Sigma-Aldrich | A1852 |
| Celecoxib | Millipore Sigma | SML3031 |
| Chloroform | Millipore Sigma | 288306 |
| CRT0066101 | Tocris | 4975 |
| Deutered COX and LOX LC-MS Mixture | Cayman Chemical | 19228 |
| Dulbecco’s Modification of Eagle’s Medium | Corning | 10–013-CV |
| Dulbecco’s Phosphate-Buffered Saline | Corning | 21–031-CV |
| Gentamicin | Millipore Sigma | G1397 |
| Halt Phosphatase Inhibitor Cocktail (100x) | Thermo-Fisher | 1862495 |
| Halt Protease Inhibitor Cocktail (100x) | Thermo-Fisher | 87786 |
| Hexane | Millipore Sigma | 1043911000 |
| HyClone Fetal Bovine Serum | Fisher Scientific | SH3008803 |
| Indomethacin | Millipore Sigma | I7378 |
| Isopropanol | Millipore Sigma | 1027811000 |
| L-Glutamine | Millipore Sigma | G7513 |
| LB Broth (Miller) | Sigma-Aldrich | L3522 |
| LB Broth with Agar (Miller) | Sigma-Aldrich | L3147 |
| Lithocholic Acid | Millipore Sigma | L6250 |
| Methanol | Millipore Sigma | 1.06035 |
| Misoprostol | Tocris | 2297 |
| N-(p-Amylcinnamoyl)anthranilic acid | Sigma-Aldrich | A8486 |
| Nigericin | Sigma-Aldrich | N7143 |
| NP-40 (Nonidet P-40 Substitute) | Boston Bioproducts | #P-877 |
| Palmitic Acid | Millipore Sigma | P0500 |
| Pam3CSK4 | Invivogen | tlrl-pms |
| Penicilin Streptomycin Solution, 100x | Corning | 30–002-CI |
| Pierce™ Lane Marker Reducing Sample Buffer | Thermo-Fisher | 39000 |
| Prostaglandin D2 | Cayman Chemical | 12010 |
| Prostaglandin E2 | Tocris | 2296 |
| Prostaglandin F2alpha | Tocris | 4214 |
| Prostaglandin I2 | Tocris | 2989 |
| Sotrastaurim | Abcam | ab219867 |
| SP600125 | Tocris | 1496 |
| TMB Substrate Reagent Set | BD | 555214 |
| Ultrapure LPS, E. coli O111:B4 | Invivogen | tlrl-3pelps |
| U-73122 | Sigma-Aldrich | 662035 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CytoTox 96(R) Non-Radioactive Cytotoxicity Assay | Promega | G1780 |
| Clarity Max Western ECL Substrate | Bio-Rad | 1705062 |
| EnzChek(TM) Direct Phospholipase C Assay Kit | Invitrogen | E10215 |
| Human IL-1Beta/IL-1F2 DuoSet ELISA | R&D Systems | DY201 |
| Mouse IL-1Beta/IL-1F2 DuoSet ELISA | R&D Systems | DY401 |
| Mouse IL-18 Elisa kit | MBL International | 7625 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 |
| PGE2 ELISA kit | Enzo Life Sciences | ADI-900–001 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data | This paper | Mendeley Data: https://doi.org/10.17632/vtpjtgm2tw.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| THP-1 | ATCC | ATCC TIB-202 |
|
| ||
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles Rivers | N/A |
| Mouse: C57BL/6 NLRP3 Knockout | Millenium Pharmaceuticals | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | Graphpad Software | www.graphpad.com |
| ImageJ | NIH | imagej.nih.gov/ij/ |
| SIMCA | Sartorius | www.sartorius.com |