

SUMMARY
Predicting the risk of cancer mutations is critical for early detection and prevention, but differences in allelic severity of human carriers confound risk predictions. Here, we elucidate protein folding as a cellular mechanism driving differences in mutation severity of tumor suppressor BRCA1. Using a high-throughput protein-protein interaction assay, we show that protein-folding chaperone binding patterns predict the pathogenicity of variants in the BRCA1 C-terminal (BRCT) domain. HSP70 selectively binds 94% of pathogenic BRCA1-BRCT variants, most of which engage HSP70 more than HSP90. Remarkably, the magnitude of HSP70 binding linearly correlates with loss of folding and function. We identify a prevalent class of human hypomorphic BRCA1 variants that bind moderately to chaperones and retain partial folding and function. Furthermore, chaperone binding signifies greater mutation penetrance and earlier cancer onset in the clinic. Our findings demonstrate the utility of chaperones as quantitative cellular biosensors of variant folding, phenotypic severity, and cancer risk.
Graphical Abstract

In brief
Gracia et al. show that the binding pattern of protein-folding chaperones to BRCA1 variant proteins predicts structure-function relationships and clinical outcomes. They find that the degree of HSP70 binding to variants correlates with age of cancer onset and identify a special class of hypomorphic BRCA1 mutations prevalent within human populations.
INTRODUCTION
Understanding how changes in genotype alter phenotypes is paramount to predicting cancer risk. However, the number of variants of uncertain clinical significance and discrepancies among commercial variant classification laboratories confound cancer risk predictions.1–4 Furthermore, features of human heredity remain difficult to integrate into clinical practice, including reduced penetrance (likelihood of disease among individuals with similar genotypes), variable expressivity (differences in disease severity among patients with similar genotypes), and hypomorphism (partial loss of gene function).5–8 Indeed, the percentage of genetic variants in the ClinVar database currently classified as a practice guideline is miniscule (0.03%).9
Protein folding links the effect of coding mutations to disease because pathogenic mutations frequently disrupt protein structure. A fundamental aspect of protein structures are domains, which fold into specific 3D structures encoded by the primary amino acid sequence.10–13 Folded domains tend to be only marginally more stable than unfolded domains,14,15 so small changes in DNA sequence can disrupt structure and function. Indeed, pathogenic missense mutations cluster within globular protein domains.13,16 Moreover, the marginal thermodynamic stability of domain structures can sensitize proteins to disease-associated environmental factors.17–20 Hence, changes in protein folding may explain differences in the functional severity, penetrance, and expressivity of human mutations.
Several variant effect predictors (VEPs) can infer the pathogenicity of coding mutations.21–24 Modern VEPs combine co-variation analyses and machine learning to predict variant effects.25–30 However, VEPs vary substantially in their accuracy and require experimental validation in a relevant system that can account for the crowdedness of the intracellular milieu, where most disease-associated proteins are found.31,32 Thus, a generalizable approach to measure changes in the folding of protein variants in a cellular context would greatly aid efforts to classify the clinical significance and disease risk of genetic variations.
Protein-folding chaperones (used interchangeably with “chaperones” herein) are highly conserved and abundant proteins that assist in the folding, maturation, localization, and degradation of proteins within cells.33–35 Two of the most abundant cytosolic chaperones, HSP70 and HSP90, fold a large fraction of the human proteome.36,37 Early in the folding cycle, HSP70 binds to stretches of amino acids that are highly hydrophobic and generally found in the core of globular domains.37 In contrast, HSP90 preferentially binds partially folded native-like proteins later in the folding cycle.38–40 Folded proteins are then released from HSP70 and HSP90 to recycle chaperones for additional rounds of chaperoning. The unique interaction specificities of HSP70 and HSP90 have proven useful for evaluating the impact of genetic and environmental factors on protein folding.41–44 Previously, we used HSP70 and HSP90 chaperone interactions to characterize disease-associated coding mutations in FANCA.19 Because hydrophobic polypeptides are exposed at early stages of folding, we hypothesized that HSP70 preferentially binds protein variants populating unfolded conformations that are functionally compromised. Consistent with this hypothesis, cells carrying FANCA (Fanconi Anemia complementation group A) protein variants that preferentially interact with HSP70 develop severe cellular phenotypes.19 Whether chaperone binding patterns can predict cancer risk of individuals carrying mutations in a cancer driver gene remains to be determined.
The tumor suppressor BRCA1 is essential to life.45 Thus, heterozygous individuals carrying a pathogenic BRCA1 mutation are ~5- to 10-fold more likely to develop breast, ovarian, pancreatic, and prostate cancers.5,46,47 The encoded BRCA1 protein suppresses tumor formation by regulating DNA damage checkpoint activation and assisting in repair of double-strand breaks in homology-directed repair (HDR).48–51 Thus, predicting the outcome of BRCA1 protein variants is critical to cancer risk management, diagnosis, and treatment. However, BRCA1 mutations have been associated with diverse functional consequences including separation of function,52,53 hypomorphic,54–56 and dominant-negative effects,57,58 obscuring variant predictions in the clinic. Investigators have applied many independent high-throughput approaches to evaluate the effects of BRCA1 variations on protein function in vitro,59–63 but whether these data can integrate clinical features of human heredity or predict mutation-associated cancer risk remains unclear.
Here, we use a high-throughput, quantitative protein-chaperone interaction platform (LUMIER) to test whether chaperone binding patterns predict structure-function relationships of BRCA1 variants and the clinical manifestations of BRCA1 mutations in carriers. We find that 94% of pathogenic variants in the BRCT domain of BRCA1 bind to HSP70 in cells. We show that increased chaperone binding reflects specific structural and functional disruptions induced by each variant. Specifically, the degree of chaperone binding is proportional to the structural and functional severity of the corresponding BRCA1 allele. We identify a class of BRCA1 variants that exhibit an intermediate chaperone interaction pattern, retain partial BRCA1 structure and function, and manifest hypomorphic functional consequences. Furthermore, patients carrying germline BRCA1 variants that bind HSP70 manifest greater disease penetrance and earlier age of cancer onset. Patients carrying hypomorphic chaperone-engaged variants exhibit delayed cancer onset as compared to strong HSP70-bound variants, suggesting more variable clinical expression for partially folded variants.
RESULTS
Most pathogenic BRCA1-BRCT variants bind chaperones
A large percentage of the BRCA1 missense variants in ClinVar are of uncertain or conflicting clinical significance (Figure 1A). 93% of pathogenic missense mutations are within the N-terminal RING or C-terminal BRCT structured domains (Figure 1B). We expressed the BRCA1-RING and BRCA1-BRCT domains fused to a 33FLAG tag and measured chaperone interactions in living cells using a high-throughput co-immunoprecipitation (coIP) assay (Figures 1C and 1D).41,64 We used NanoLuc luciferase-tagged chaperones because they produced higher signal-to-noise ratios as compared to the Renilla luciferase system (Figure S1A). We validated NanoLuc chaperone binding patterns using a panel of established HSP70 and HSP90 clients and co-chaperones and observed chaperone binding patterns consistent with prior reports19 (Figure S1B). Unlike control proteins, the wild-type BRCA1-BRCT domain did not bind to either chaperone significantly (Figure S1C). These results suggest that the wild-type BRCA1-BRCT domain is well folded in cells. In contrast, the wild-type BRCA1-RING domain strongly interacted with HSP70 and did not bind HSP90 (Figure S1D), suggesting that the BRCA1-RING domain does not fold independently in cells and likely relies on heterodimerization with the BARD1-RING for folding.49,63,65
Figure 1. Most pathogenic BRCA1-BRCT variants bind chaperones.
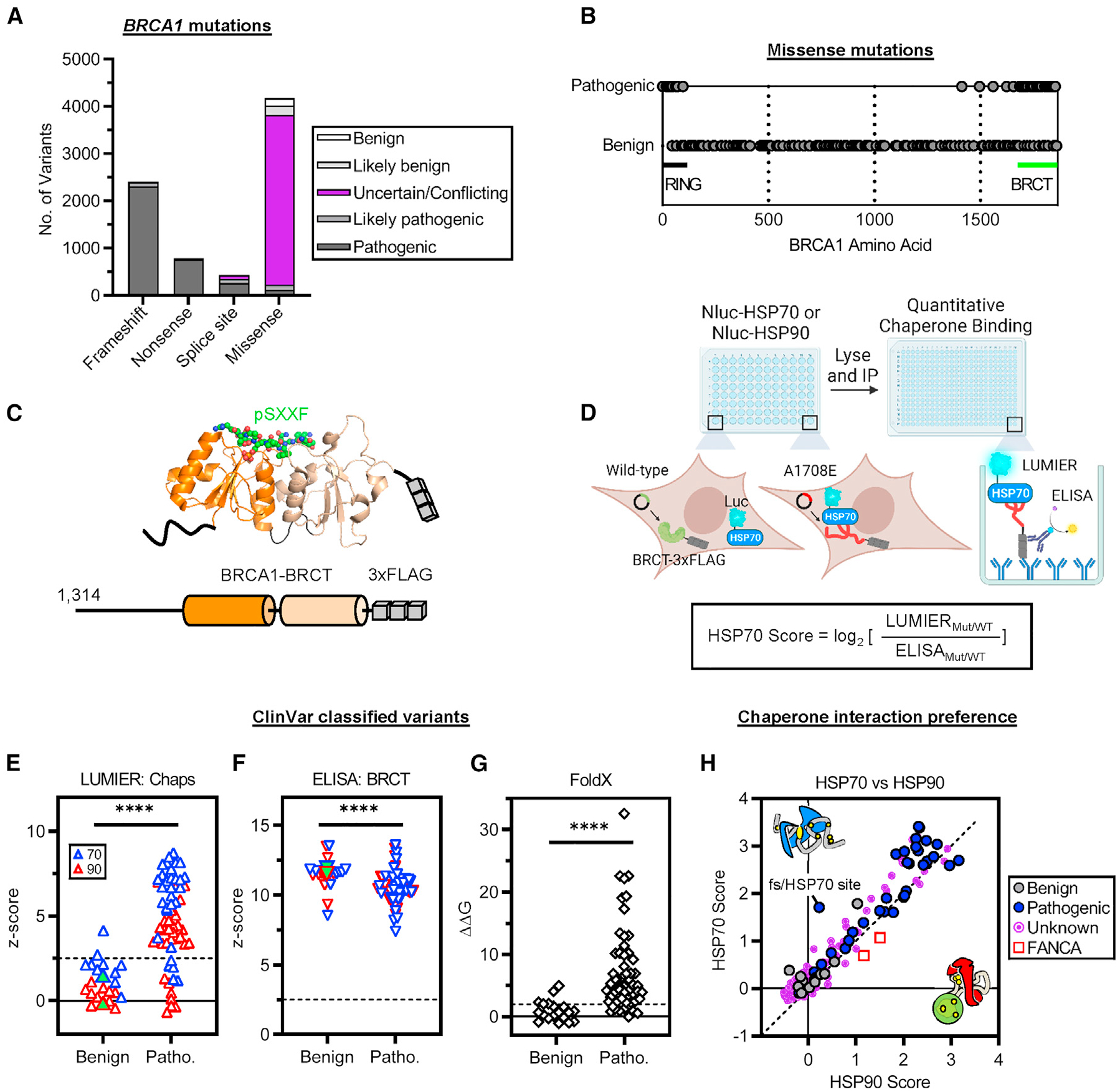
(A and B) BRCA1 mutations cataloged in ClinVar (accessed April 27, 2023).9
(A) Clinical significance of BRCA1 mutations grouped by mutation type.
(B) Location of pathogenic missense mutations cataloged in ClinVar with at least a “two-gold-star” review status (n = 285).
(C) C-terminal BRCA1 3×FLAG-tagged truncation construct used for LUMIER. The two BRCT subdomains are colored (orange and wheat). pSXXF denotes the BRCT binding phosphopeptide (BACH1). Protein Data Bank (PDB): 1T29.
(D) LUMIER with BACON (bait control) approach to quantify chaperone interactions in HEK293T cells. Diagram was created with BioRender.
(E) Chaperone binding to benign and pathogenic BRCA1-BRCT variants in ClinVar (including “likely” classifications). Z scores were calculated by averaging non-transfected wells to illustrate the significance of the data. Dashed line indicates the threshold for a statistically significant signal (Z score > 2.5). Wild-type BRCA1-BRCT values are shown as green-filled triangles. Chaps, chaperones; Patho., pathogenic.
(F) BRCA1-BRCT variant levels after pull-down detected by ELISA.
(G) Variant ΔΔG values (kcal/mol) predicted by FoldX relative to the wild-type value (ΔΔG = 0). Dashed line indicates the threshold for structure disruption (ΔΔG > 2).
(H) HSP70/HSP90 interaction preferences. Diagonal shows the identity line. The unknown group includes variants annotated as having uncertain significance, no significance provided, conflicting interpretations, or “one-gold-star” review status. FANCA variants and a frameshifted (fs) variant encoding an additional out-of-frame HSP70 site shown for comparison.
Statistical significance was determined using two-tailed Mann-Whitney t test (E–G). ****p ≤ 0.0001. Data are presented as mean values from at least two independent experiments.
We cloned a library of clinical variants in the BRCA1-BRCT domain and in the BRCA1-RING domain and control variants not part of either domain and measured the binding of each variant to HSP70 and HSP90. Control variants had no effect on chaperone binding, except for a frameshifting variant predicted to introduce a new HSP70 binding site (P1575L fs*25; Table S1, ChaperISM DnaK variant predictions). In contrast, variants in the BRCA1-BRCT domain exhibited dramatic increases in chaperone binding that were highly reproducible (Figure S1E). Unlike BRCA1-BRCT variants, BRCA1-RING variants did not significantly alter chaperone binding, consistent with the wild-type BRCA1-RING domain not folding properly in our assay (Figure S1D). Thus, we focused on disease-associated BRCA1-BRCT variants in determining chaperone interactions.
24 out of 31 pathogenic BRCA1-BRCT variants significantly interacted with HSP70 and HSP90, whereas 9 out of 11 benign variants did not interact with either chaperone (Z score > 2.5; Figure 1E). Because chaperones are involved in the degradation of misfolded proteins,66,67 we tested if increased chaperone binding correlates with changes in protein abundance. We used a quantitative enzyme-linked immunosorbent assay (ELISA) to quantify BRCA1-BRCT variant pull-down for benign and pathogenic variants (Figure S1F). We observed that pathogenic BRCA1-BRCT variants were pulled down ~1.3-fold less (ΔZ scores ~1) than benign variants and the wild-type control (p ≤ 0.0001; Figure 1F; Table S1), suggesting that pathogenic variants were more prone to misfolding and degradation within the cell. Indeed, we observed increased binding to endogenous chaperones and decreased protein abundance for pathogenic BRCA1-BRCT variants using conventional coIP and western blotting (Figure S1G). These interactions were reproduced in three different BRCT-containing BRCA1 fragments (Figure S1H), suggesting that chaperone binding reflects disruption of the BRCT domain structure. In silico predictions of variant stability using FoldX21 showed that 86% of pathogenic BRCT missense variants from ClinVar increased the Gibbs free energy of folding as compared to the wild type (Figure 1G; Table S2). Taken together, these results show that most pathogenic BRCA1-BRCT missense variants bind chaperones and are unstable in cells.
Next, we evaluated the HSP70/HSP90 chaperone binding preference of pathogenic BRCA1-BRCT variants by generating normalized HSP interaction scores (Figure 1D). 94% of the pathogenic variants bound to HSP70 at least 1.5-fold more than the wild-type domain (HSP70 score ≥ 0.5, Figure 1H). Moreover, pathogenic BRCA1-BRCT variants were predominantly engaged by HSP70 over HSP90 (~71%), especially when compared with HSP90-engaged FANCA variants that retain protein function.19 In addition, ~29% of BRCA1-BRCT variants of unknown significance bound to either chaperone (HSP score ≥ 0.5; Figure 1H), suggesting that chaperone interaction patterns can help assign clinical significance to human BRCA1-BRCT variants. Notably, chaperone binding varied drastically among BRCA1-BRCT variants (1.5- to 11-fold increased binding relative to wild type). The broad diversity of chaperone binding patterns suggests that chaperones detect different types of BRCA1-BRCT folding variants.
Chaperone biosensors detect a diversity of protein-folding variants
Chaperone binding to BRCA1-BRCT variants appears to reflect changes in domain thermodynamic stability. However, HSP70 binding to variants may reflect an increased number of HSP70 binding motifs created by the mutation.37 We tested this using ChaperISM68 to identify HSP70 binding motifs in the wild-type and variant protein sequences. We observed no significant difference between variants predicted to have an increased number of DnaK binding sites (bacterial HSP70) compared to variants with no change in or fewer DnaK sites (Figures S2A and S2B; Table S1). These results suggest that increased HSP70 binding to BRCA1-BRCT variants reflects decreased domain stability. To test this, we compared chaperone binding with a protease sensitivity assay described previously.69–71 We grouped BRCA1-BRCT variants exhibiting increased sensitivity to protease cleavage and compared them with variants resistant to protease cleavage. In line with our findings described above, increased HSP70 binding and decreased amounts of BRCA1-BRCT were pulled down (ELISA) for variants sensitive to protease digestion (Figure S2C).
Collectively, our data suggest that increased chaperone binding to BRCA1-BRCT variants generally reflects changes in the thermodynamic stability of each protein variant. To further validate this conjecture, we rationally designed BRCA1-BRCT variants (n = 49) to disrupt fundamental elements of protein structure and measured chaperone binding (Table S1). For this, we focused on secondary structure elements, hydrophobic interactions, and side-chain hydrogen bonds.
We targeted secondary structure elements observed in the BRCA1-BRCT crystal structure. Proline variants are typically not tolerated in α-helix secondary structures.72 Indeed, FoldX predicts strong structural disruptions when helical residues are mutated to proline as compared to alanine (Figure S2D). Additionally, proline is the most frequent pathogenic mutation reported for BRCA1-BRCT α-helical residues in ClinVar (5 out of 19; Table S2). We observed increased chaperone binding to the BRCA1-BRCT when prolines were swapped in at α-helices (V1665P, Y1666P, T1720P, and D1840P; Figure 2A). Similar results were obtained for charged variants in the BRCT β-sheets (V1687E and I1807E; Figure 2A). Notably, variants designed to disrupt α-helices or β-sheets were engaged predominantly to HSP70 (e.g., I1807E bound 2.1-fold more to HSP70 than to HSP90).
Figure 2. Chaperone biosensors detect a diversity of protein-folding variants.

(A) Variants targeting secondary structure elements in the BRCA1-BRCT domain.
(B) Y1845 variants that disrupt or support hydrophobic interactions. FoldX ΔΔG predictions for each variant are shown.
(C) T1685 variants that disrupt or support side-chain hydrogen bonding. Two different T1685S codons were tested because these codons previously exhibited different functional effects.60
(D and E) Chaperone binding predictions to the BRCA1-BRCT variant library. The “disruptor” bin includes variants that introduce prolines in α-helices; truncations within the BRCT domain; buried variants that introduce a charge, decrease hydrophobicity, or create a steric clash; and charged variants that mutate residues in hydrophobic networks or disrupt side-chain hydrogen bonding. The “no effect” bin includes variants that fully delete the BRCT domain; variants outside the BRCT domain; buried variants that retain hydrophobicity; isosteric or quasi-isosteric variants; variants that maintain side-chain hydrogen bonds; and other surface variants not included prior. Dashed line indicates the cutoff for binding (HSP70 score > 0.5). (E) excluded variants previously characterized using protease sensitivity.71
Statistical significance was determined using a two-tailed Mann-Whitney t test. ****p ≤ 0.0001. Data are presented as mean ± standard deviation values from at least two independent experiments.
Hydrophobic interactions stabilize the core of folded domains.73 We rationally designed 11 variants to target hydrophobic interactions. Replacing buried residues in the hydrophobic core with charged amino acids increased chaperone binding (Y1845D, Y1845E, M1783R, and M1783K; Figures 2B and S2E). In contrast, biochemically compatible variants did not increase chaperone binding (Y1845F and Y1845W) or weakly increased it (M1783A). Overall, we observed greater chaperone binding to variants in our library that mutate buried residues than to variants that mutate surface residues (Figures S2F).
Next, we tested if chaperone interactions can detect disruptions to hydrogen-bonding networks. Surface residues of the BRCA1-BRCT were targeted to avoid compromising other structural elements. T1685 forms side-chain hydrogen bonds with neighboring residues (Figure 2C). Two pathogenic variants in ClinVar block these hydrogen bonds (T1685A and T1685I), and both variants were predominantly engaged by HSP70. We tested if hydrogen bonds formed by the hydroxyl side chain of T1685 stabilize the structure by engineering a BRCA1-BRCT variant that preserves hydrogen bonding. Indeed, mutating T1685 to serine (T1685S) did not increase chaperone binding in two codon backgrounds. Hence, the hydroxyl side chain of T1685 coordinates local hydrogen bonds that stabilize the BRCA1-BRCT structure. Furthermore, 9 out of 12 additional surface variants targeting side-chain hydrogen bonds showed increased chaperone binding (Figure S2G; Table S1). These results suggest that side-chain hydrogen bonds are surface determinants of BRCA1-BRCT folding.
There were examples where the effect of mutation on chaperone binding was difficult to explain quantitatively. For example, we mutated charged residues at the termini of α-helix 1 to disrupt a potential helix dipole moment that supports protein structure.74,75 Chaperone binding was specifically increased to E1661K but not to other charge-swapping variants (E1660K, E1661K, R1670E, and K1671E; Figure S2H). However, residue E1661 also forms hydrogen bonds via its side chain with K1690, T1658, and the pSXXF. Disruption of T1658 hydrogen bonds (T1658A, T1658N, and T1658S) did not increase chaperone binding (Figure S2H), suggesting minimal impact on BRCA1-BRCT folding. Notably, a variant that disrupts E1661 hydrogen bonds and does not introduce a charge swap (E1661A) bound HSP70 ~2-fold less than E1661K, suggesting that the E1661K variant disrupts the BRCA1-BRCT structure via a complex mechanism. Although the mechanism of BRCA1-BRCT structure disruption by E1661K is unclear, the increased chaperone binding to the variant suggests it is clinically relevant. Indeed, E1661K has been observed in a human patient with cancer (skin cancer, non-melanoma), and the variant is annotated with conflicting interpretations of pathogenicity in ClinVar (Table S1).
Another set of variants that were difficult to interpret involved residues between the BRCA1-BRCT tandem subdomains. Two pathogenic variants in our library (A1708E and G1706E) introduced a charge within this groove, and both variants bound strongly to chaperones (Figure S2I). Interestingly, eight additional variants in this region increased chaperone binding to different degrees. Variants that preserve hydrophobicity or accommodate the steric constraints of the pocket interacted with chaperones only moderately (L1705I, L1705V, and G1706A) as compared to variants that introduce a charge (L1705K, L1705R, L1705D, A1708R, and F1704D). Notably, 3 of these variants were predominantly engaged by HSP90 (F1704D, G1706E, and A1708E), suggesting structural changes different from variants that selectively engaged HSP70. Hence, chaperones bind BRCA1-BRCT variants in different ways that reflect the structural effect of the specific mutation.
In summary, the results described in this section showed that chaperones bind BRCA1-BRCT variants that disrupt protein structure. We assessed the accuracy of chaperone binding as a detector of structure disruption using criteria to classify “disruptor” and “no effect” variants (Table S1; STAR Methods, selection and characterization of variants). For rationally designed variants, we correctly assigned 44 out of 49 variants, amounting to a total accuracy rate of 90% (Figure 2D). Using the same criteria for variants of unknown clinical significance in ClinVar, we accurately assigned 88 out of 109 variants, amounting to a total accuracy rate of 81% (Figure 2E). In conclusion, increased chaperone binding can help identify BRCA1-BRCT variants that disrupt domain structure, even when the mechanism of disruption is ambiguous.
The degree of chaperone binding is proportional to the severity of BRCA1 mutation
The large variation of increased chaperone binding to BRCA1-BRCT variants compelled us to examine whether the degree of chaperone binding reflects fractional loss of BRCA1-BRCT structure. We quantified the maximal increase in chaperone binding to BRCA1-BRCT variants by generating 13 double variants. The inclusion of single variants that do not bind chaperones did not change binding (e.g., F1695L/M1775K compared to M1775K; Figure S3A). Combining two variants that both interacted moderately with HSP70 increased chaperone binding to the double variant (S1655F/M1775K, ~4.3-fold increased binding relative to single variants). In contrast, combining two single variants that interacted strongly with HSP70 only weakly increased binding, indicating an upper limit on HSP70 binding to BRCA1-BRCT variants (C1697R/D1840P and V1687E/G1788V; ~1.5-fold increased binding relative to single variants; Figure S3A). These results suggest that maximal chaperone binding in our assay reflects complete structural disruption of the BRCA1-BRCT domain.
Below the upper limit of chaperone binding, we observed a positive correlation between chaperone binding to each BRCA1-BRCT missense variant and the predicted increase in Gibbs free energy of folding in silico (R2 = 0.51; Figure 3A). This correlation was reproduced when using empirically derived stability measures for 18 recombinant BRCA1-BRCT variants purified from Escherichia coli (R2 = 0.55; Figure 3B).76 In addition, 10 variants that were insoluble in E. coli interacted strongly with HSP70. We observed similar results when evaluating HSP90 binding scores to BRCA1-BRCT variants (Figures S3B and S3C). These data suggest that strong HSP70 binding to variants reflects a severely disrupted BRCA1-BRCT domain, whereas intermediate chaperone binding to BRCA1-BRCT variants generally reflects partial structure disruption.
Figure 3. The degree of chaperone binding is proportional to the severity of BRCA1 mutation.

(A and B) Correlation of HSP70 binding with BRCA1-BRCT variant stability. Horizontal dashed line is the average of four double variants that combine strongly chaperone-bound single variants. Vertical dashed line reflects the upper limit of stability measurements owing to variant insolubility. The correlations were fit to a linear regression within the linear regime (FoldX: < 10 kcal/mol, empirical: < 5 kcal/mol). G1788V was not fit because the stability measurement for this variant was previously reported as contradictory.76
(C and D) HSP70 binding binned according to the functional effect class measured using aggregation/degradation or transcriptional activation assays.77
(E) Linear correlation of HSP70 binding and variant levels (ELISA) at different concentrations of cell lysates pulled down. Dashed lines indicate the average LUMIER and ELISA signals observed under our standard assay conditions for variants that bound strongly to HSP70.
(F and G) BRCA1 variant function in HAP160 (cell fitness) or HeLa62 (HDR) cells binned according to the degree of HSP70 binding.
(H) Integrated functional data from neXtProt binned by the magnitude of HSP70 binding. Percentages calculated using the percentage of variants associated with each HSP70 binding magnitude and phenotype intensity.77 Func., functional.
Statistical significance was determined using Kruskal-Wallis ANOVA test (C, D, F, and G) or chi-squared test (H). ****p ≤ 0.0001, **p ≤ 0.01, and *p ≤ 0.05. Data are presented as mean ± standard deviation values from at least two independent experiments.
To test if the degree of chaperone binding is proportional to the functional (phenotypic) severity of the corresponding BRCA1 protein variant, we collected BRCA1 functional data from the neXtProt server.77 This database catalogs the functional severity of BRCA1 variants measured in many different assays and laboratories as a qualitative “phenotypic intensity” with four classifications: functional, mild, moderate, and severe (573 unique entries for variants in our library; Table S3A). Using these phenotypic intensity classes, we observed progressive increases in HSP70 binding that paralleled increasing aggregation or degradation propensity of the variants (Figure 3C). Importantly, we observed the same trend in HSP70 binding when comparing the phenotypic intensity with the BRCA1-BRCT variant effect on transcriptional activation, cell localization, and cell viability (Figures 3D, S3D, and S3E). These relationships cannot be explained by assay saturation because HSP70 binding for each mutant was well within the linear regime (Figures 3E and S3F). Taken together, these results suggest that the degree of chaperone binding to BRCA1-BRCT variants reflects the functional severity of BRCA1 mutants.
To further examine this relationship, we collected functional data from two deep mutational scanning cell-based assays (Table S3B). We compared the degree of HSP70 binding with growth defect scores induced by BRCA1 missense mutations at the endogenous BRCA1 gene locus in HAP1 cells.60 We observed functional defects that increased progressively with the magnitude of HSP70 binding (Figure 3F). Moreover, BRCA1-BRCT variants that bound moderately to HSP70 conferred intermediate effects on the growth phenotype. We observed a similar relationship when investigating the effect of BRCA1 mutation on HDR efficiency in HeLa cells (Figure 3G)62 or when binning BRCA1-BRCT variant data using FoldX predictions (Figures S3G and S3H). Furthermore, there was a strong positive correlation between the functional defect induced by BRCA1 mutations and the magnitude of HSP70 binding to the encoded variants (HAP1: R2 = 0.52, HeLa: R2 = 0.49; Figures S3I and S3J). These data further suggest that the degree of chaperone binding to BRCA1-BRCT domain variants is proportional to the phenotypic severity of the full-length BRCA1 variant in cells.
However, chaperone binding was not increased for all deleterious BRCA1-BRCT variants. 25% of the variants that induced growth defects in HAP1 cells did not bind HSP70 (35% in the HeLa HDR assay). We hypothesized that these mutants impair BRCA1 function in a manner independent of structure disruption (Figures S3I and S3J). Indeed, we found four missense variants that compromised splice sites.78 In addition, six variants may interfere with pSXXF binding to the BRCT domain because these variants occurred at residues protected from solvent when the pSXXF was present in the crystal structure (“pSXXF protected”). Five additional surface variants were not pSXXF protected, suggesting that the variant disrupts function through a mechanism independent of pSXXF binding. Together, these variants were designated as “contact” because the defect in BRCA1-BRCT function is likely attributable to the loss of protein-partner binding, not domain structure disruption (15% of functionally impaired variants in HAP1 and 19% in HeLa; Figure S3K). Notably, we also observed moderate chaperone binding to certain variants that mutate these contact residues (e.g., S1655F, R1699W, M1775K, and M1775R), suggesting these variants partially disrupt domain folding, in agreement with previous reports.71,76,79 Thus, integrating chaperone interactions with functional data can help distinguish true contact mutations from complex mutations that disrupt both protein-partner interactions and domain structure.
Taken together, the findings described in this section highlight the ability of chaperones to gauge the functional severity of structure-disrupting BRCA1 mutations in cells. After aggregating all functional data from the neXtProt server, we found that 68% of variants that bind weakly to HSP70 are functional and 39% of those that bind moderately confer mild or moderate defects, whereas 65% of variants that bind strongly exhibit severe functional defects (Figure 3H; Table S3). We conclude that the degree of chaperone binding to BRCA1-BRCT variants is generally proportional to the phenotypic severity of the corresponding BRCA1 mutation.
Chaperones identify diverse classes of pathogenic BRCA1 variations in humans
To ascertain the prevalence of BRCA1 variants that bind moderately to HSP70 in humans, we compared chaperone binding across diverse clinical and population studies. BRCA1 missense variants that bind moderately to chaperones are abundant and equally prevalent in patients with cancer and the general population (Figure 4A). After combining missense variants from ClinVar (unknown clinical significance only), The Cancer Genome Atlas (TCGA),80,81 and the gnomAD population database,3 we observed that 18% of the variants moderately bound HSP70 and 12% strongly bound HSP70. Variants that bound moderately to HSP70 were also prevalent in all subpopulations cataloged in gnomAD (Figure S4A). In addition, moderate HSP70 interactors were the most abundant group of hypomorphic BRCA1-BRCT missense variants observed in cell-based deep mutational scans (Figure 4B; Table S3B).60,62 Furthermore, variants that bound moderately were slightly more abundant than variants that bound strongly to HSP70 for unknown variants in ClinVar (Figure S4B). Thus, variants exhibiting moderate HSP70 binding are abundant and relevant alleles in humans.
Figure 4. Chaperones identify diverse classes of pathogenic BRCA1 variations in humans.

(A) HSP70 interaction scores for natural human variants observed in patients with cancer (ClinVar9 and cBioPortal/TCGA80,81) or the general population (gno-mAD3). Unknown (unk.) includes variants annotated as having uncertain significance, no significance provided, conflicting interpretations, or one-gold-star review status.
(B) The functional severity60,62 of natural BRCA1-BRCT variants grouped by the degree of HSP70 binding. LoF, loss of function.
(C) HSP70 binding to natural BRCA1-BRCT human variants binned by the predicted effect on domain structure. Variants were ranked using an informed hierarchical approach (see STAR Methods). Moderate HSP70-bound variants are colored magenta and overlaid onto the BRCT crystal structure. An example long-range interaction that coordinates the two BRCT subdomains is indicated with an arrow. Struct., structure.
(D and E) ROC curves using pathogenicity annotated in ClinVar (D) or mode phenotypic intensity annotated in neXtProt (E) as the target datasets. The neXtProt curves designate mild, moderate, and ambiguous (multimodal) variants as pathogenic.
(F) The observed AUCs when mild, moderate, and ambiguous variants in the neXtProt target dataset were designated benign or pathogenic.
Statistical significance was determined using Kruskal-Wallis ANOVA test (A and C) or chi-squared test (B). ****p ≤ 0.0001 and **p ≤ 0.01. ns, not significant. Data are presented as mean values from at least two independent experiments.
We hypothesized that BRCA1 variations that increased chaperone binding are subject to purifying selection in humans because increased chaperone binding correlates with loss of protein function. To test this, we compared allele frequencies in gnomAD3 for variants that increased chaperone binding with those that maintain wild-type-like chaperone interactions. Notably, variants that bound moderately or strongly to chaperones were 23-fold less abundant than variants that did not bind chaperones (Figure S4C), suggesting that both variant groups are subject to purifying selection. However, these differences in allele frequencies were not significant, likely owing to the abundance of rare BRCA1 variants in the gnomAD database or the possible influence of unaccounted risk factors. In contrast, variants observed in tumors that bound chaperones have allelic frequencies similar to those that do not bind chaperones, suggesting reduced purifying selection of mutants in tumors (Figure S4D).
To identify structural mechanisms driving partial folding of human hypomorphic variants, we investigated variants moderately bound by HSP70 to search for structural commonalities. We hierarchically grouped missense BRCA1 variants into structural effect classes based on functional data, structural features, and stability predictions (see STAR Methods). For example, known protease-sensitive mutants were ranked as “structural disruption” followed by “pSXXF/contact” because these variants can be confidently assigned. We compared structural effect classes with variants expected to severely disrupt the BRCA1-BRCT domain structure (Table S3B). We observed weak and moderate HSP70 binding for variants expected to accommodate the structure, variants within the hydrophobic pSXXF groove, and variants that disrupt long-range interactions (Figures 4C and S4E). Interestingly, variants that interacted moderately with HSP70 were abundant in all BRCT structural effect classes we analyzed and found throughout the domain. Hence, hypomorphic structure-disrupting variations in humans may be inherent to protein-structured domains.
To compare the accuracy of chaperone interactions with other indicators of pathogenicity, we used receiver operating characteristic (ROC) curves (Table S3B). HSP70 binding classified ClinVar variants with near-perfect accuracy (Figures 4D and S4F; area under the curve [AUC] 0.94). Similar results were obtained using HAP1 cells60 (AUC, 0.98). Moreover, next-generation VEPs27–30 performed equally well (AlphaMissense AUC 0.95) and correlated well with HSP70 binding (Figures S4G–S4J). Notably, the performance of pathogenicity algorithms decreased when partial loss-of-function BRCA1 variants were included using integrated neXtProt data (Figure 4E). In contrast, HSP70 binding outperformed all other pathogenicity metrics when mild, moderate, and ambiguous variants were assigned as pathogenic (AUC 0.95). Indeed, chaperone binding performed well regardless of how mild, moderate, and ambiguous variants were assigned in the target dataset (Figure 4F). We did not compare HAP1 function scores to neXtProt data to avoid the confounding effects of splice variants. In summary, these results suggest that quantitative chaperone interactions can be used to identify pathogenic variants and hypomorphic alleles in humans.
Chaperone binding patterns stratify cancer risk and variant expressivity of human BRCA1 carriers
To determine if chaperone binding can elucidate the cancer risk of BRCA1 mutation carriers, we collected familial penetrance likelihood data for heritable BRCA1 variants from the Leiden Open Variation Database.82 Breast cancer risk was markedly higher in carriers of BRCA1 mutations that moderately or strongly increased HSP70 binding to the BRCA1-BRCT domain (Figures 5A and S5A–S5C; Table S4). Notably, four highly penetrant variants that moderately bound HSP70 mutated residues near the pSXXF binding site (D1692H, R1699W, M1775K, and M1775R), suggesting that pSXXF cleft disruption drives increased penetrance in carriers. In contrast, two highly penetrant variants not in proximity to the pSXXF cleft were moderately bound by HSP70 (V1736A and M1783T; Table S1), suggesting that partial disruption of the BRCT domain structure drives increased penetrance. Hence, BRCA1-BRCT variants that compromise domain folding, even partially, can increase the likelihood of cancer development.
Figure 5. Chaperone binding patterns stratify cancer risk and variant expressivity of human BRCA1 carriers.

(A) BRCA1 mutation penetrance likelihood binned by the magnitude of HSP70 binding. Data shown were obtained from the Leiden Open Variation Database (LOVD) (accessed April 24, 2023).82
(B–D) Age of first cancer diagnosis for patients carrying BRCA1 mutations binned by mutation type (B), missense mutations in domains (C), and the degree of HSP70 binding (D).
Statistical significance was determined using two-tailed (A) or one-tailed (B–D) Mann-Whitney t test. ****p ≤ 0.0001, **p ≤ 0.01, and *p ≤ 0.05. The number of variants in each bin is shown in parentheses. Data are presented as mean values from at least two independent experiments.
To test if chaperone interactions predict mutation expressivity in the clinic, we evaluated an independent cohort of 1,106 BRCA1 carriers who underwent clinical genetic testing at The University of Texas MD Anderson Cancer Center (Table S5).83,84 Breast cancer developed in 71% of the individuals in this cohort, and the remainder were negative at the time of examination (Figures S5D and S5E). Patient survival was not significantly different when binning BRCA1 carriers based on HSP70 binding (p = 0.20; Figure S5F). The lack of significance is likely due to the small number of deaths that have occurred since the inception of this prospective cohort (~79% survival for patients carrying variants that bound HSP70). To improve statistical power, we tested the relationship between increased chaperone binding and age of disease onset, which was collected for each patient with cancer at the time of diagnosis.
We hypothesized that individuals who developed cancer earlier in life carry a BRCA1 variant that is more functionally compromised. Indeed, patients carrying nonsense BRCA1 mutations developed cancer earlier than those carrying missense mutations (Figure 5B). In addition, missense mutations within the BRCT and RING domains resulted in earlier age of onset (Figure 5C). Strikingly, patients with breast cancer carrying BRCA1-BRCT variants that bound strongly to HSP70 were diagnosed 12 years earlier (median age of onset) than those carrying weakly HSP70-bound mutants (Figure 5D). Interestingly, BRCA1-BRCT variants that moderately bound HSP70 were associated with delayed cancer onset (median, 8 years) as compared to strongly HSP70-bound variants. FoldX predictions, HAP1 functional data, and next-generation VEPs27–30 also showed a delay in disease onset for patients carrying variants that partially disrupt structure or function (Figures S5G–S5L). These results demonstrate that hypomorphic, moderately HSP70-bound variants are an important, phenotypically distinct class of pathogenic BRCA1 variations in humans. We conclude that the degree of chaperone binding can be used to detect the penetrance and expressivity of cancer variations in humans, enabling stratification of mutation-associated cancer risk in BRCA1 carriers.
DISCUSSION
A major barrier to increasing the clinical relevance of genetic variations is linking the molecular mechanisms of mutations to their clinical manifestations. Here, we present a high-throughput approach to precisely quantify structure-function relationships based on physical interactions between protein variants and ancient solutions to the protein-folding problem, protein-folding chaperones. This approach delineated loss of protein folding as the cellular mechanism driving loss of function and its corresponding effect on cancer risk for mutations in the BRCA1-BRCT domain.
HSP70 and HSP90 recognize non-native polypeptides during folding.34,35 We hypothesized that chaperones can be used as biosensors to infer the structural and phenotypic effects of mutations, thus bypassing the need for fully understanding structure-function relationships in the cell. Chaperone interactions facilitated classification of otherwise indistinguishable BRCA1 mutations into discrete functional groups based on the pattern of chaperone binding to the encoded variant. Severely compromised BRCA1 variants bound strongly to chaperones, and BRCA1’s tumor-suppressive function was impaired through full-length BRCA1 degradation. Even the absence of chaperone binding to variants was informative when compared with available functional data, revealing true contact mutants that disrupt protein-partner interactions without affecting BRCA1-BRCT domain structure. This is important considering that different variations of the same residue can manifest different diseases and clinical severities. Indeed, one such natural BRCA1 variant, R1699Q, does not bind chaperones and confers intermediate cancer risk and Fanconi anemia-like symptoms.85–88 However, a different variant at the same position, R1699W, binds moderately to chaperones and partially folds.76 Predicting the clinical manifestations of such similar protein-coding variations may be achievable using protein-folding chaperone interactions in cells.
Importantly, the degree of increased chaperone binding is proportional to the structural and functional defect induced by mutation. Variants exhibiting moderately increased chaperone binding were a signature of hypomorphic allelic severity. These mutants partially fold and function, thereby leading to partial or mild defects in function and intermediate phenotypic effects. BRCA1 hypomorphs have been identified and suggested to play a role in cancer predisposition (germline variants) and PARP inhibitor resistance (somatic mutations).54,85,91 However, a majority of known hypomorphs arise due to separation-of-function activity. In contrast, we find that partial loss of BRCT folding results in hypomorphic clinical outcomes in cells and humans. We show that these BRCA1-BRCT hypomorphs predis-pose individuals to breast cancer with high-penetrance yet variable disease expressivity, as reflected in the age of onset.
The data presented herein may have broad applicability for cancer therapy. Determining whether HSP70-engaged BRCA1 variants hypersensitize tumors to genotoxic agents warrants investigation. Moreover, it would be interesting to see if variants that partially disrupt BRCA1-BRCT domain folding are buffered by protein-folding chaperones, imparting increased sensitivity to environmental stressors for individuals carrying these variants in their genome. Indeed, R1699W binds to HSP90 and has been previously characterized as a temperature-sensitive mutant.89,90 Hence, establishing a framework to accurately predict the effects of these BRCA1 variations on protein folding using chaperone interactions would further improve risk stratification in carriers and therapeutic options for patients. We propose parsing variations in disease-relevant genes by chaperone binding as a powerful strategy with broad applicability for risk stratification and precision medicine applications.
Limitations of the study
A limitation of this study is that the target variant dataset must map to globular domains to allow for the interrogation of changes in chaperone binding across different variants. Fortunately, more than 60% of residues in the human proteome are found in globular domains.13 Another limitation exists in the detection threshold of our assay. For example, we did not detect chaperone binding to the wild-type BRCA1-BRCT, but there may be a rare populated state of chaperone-bound molecules or transient chaperone interactions that we cannot detect. For this reason, stabilizing the wild-type BRCA1-BRCT by mutation is not measurable directly in our assay. Instead, our approach can be used to identify compensatory mutations that manifest reduced or wild-type-like chaperone binding in the context of a destabilizing mutation that increases chaperone binding.19 Indeed, there are instances where decreases in chaperone binding to variants are informative, such as upon binding of a co-factor or compound that induces protein stabilization42 or a mutation that changes an intrinsically disordered protein into a well-structured protein. In summary, our study highlights the utility of chaperone binding patterns as predictors of mutation severity and cancer risk in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information or reagents may be directed to the lead contact, Georgios I. Karras (gkarras@mdanderson.org).
Materials availability
Plasmids generated in this study will be made available upon request by contacting the lead author.
Data and code availability
Chaperone interaction data generated in this study have been made publicly available through Mendeley. https://data.mendeley.com/drafts/rmj7fwm6n7
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
Mammalian cells
Stable HEK293T cell lines expressing N-terminally tagged NanoLuc-chaperones (HSP70/HSPA8 or HSP90/HSP90AB1) were maintained in 10-cm petri dishes (Corning, 08–772–22) at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (Corning, 10–013-CV) supplemented with 10% fetal bovine serum (Sigma-Aldrich, F2442) and Pen-Strep (Gibco, 15140–122). Cells were collected using treatment with Accumax (Innovative Cell Technologies, AM-105) at 37°C for 15–20 min and dilution in media. Cell lines were routinely tested for mycoplasma contamination (Lonza, LT07–318) and validated using STR fingerprinting by the MD Anderson Cytogenetics and Cell Authentication Core.
Patient data curation
Patients were identified in a prospectively maintained database after approval by the MD Anderson institutional review board. Informed consent was obtained from all human subjects. Germline BRCA1 genetic test results from 1996 to 2023 and age of cancer diagnosis were included in this study. De-identified patient data were curated to exclude ambiguous clinical data from the study. Genomic alterations reported by clinicians were cross-referenced to the human BRCA1 sequence taken from NCBI to validate mutations (GenBank: NM_007294.4). Genomic alterations in BRCA1 were then categorized as frameshift, gene deletion, in-frame deletion, intronic, large deletion, large duplication, large insertion, missense, nonsense, splice site, synonymous, or undeterminable (15 of 1,121 individuals (1.3%) had undeterminable alterations and were filtered out). Kaplan-Meier survivor curves were generated using artificial censorship at 20 years to observe patient survival probability on a reasonable timescale after cancer diagnosis. For patients that had developed multiple cancers at different times, the lowest age was used as the age of first cancer diagnosis. When comparing age of first cancer diagnosis among groups in the cohort, one-tailed Mann-Whitney (unpaired nonparametric) t-tests were used to test for significantly decreased age of cancer onset. Most of the patients in this cohort were White female (~49%) given that breast cancer is the predominant diagnosis among female individuals with BRCA1 mutations and most relevant to their health. In our analysis using HSP70 binding and age of first cancer diagnosis, the racial diversity (~2% Native American, ~2% Other, ~4% Asian, ~23% African American, ~30% Hispanic, ~39% White) was much greater than that in the entire cohort, suggesting that race did not substantially influence our chaperone binding age of onset results.
METHOD DETAILS
Plasmids and cloning
Plasmids containing human BRCA1 cDNA were a gift from Junjie Chen (Addgene, Plasmid #99394). The BRCT and RING variant libraries were cloned by amplification of the C-terminal (residues 1,314–1,863) or N-terminal (residues 1–324) region of BRCA1 using primers (Sigma-Aldrich) designed with attB handles compatible with Gateway cloning. PCR products were purified using agarose gel extraction (Takara Bio, 740609), and BP clonase (Invitrogen, 11789) was used to insert BRCT fragments into the entry vector pDONR221. Next, two-step PCR-mediated site-directed mutagenesis was performed using Phusion High-Fidelity DNA Polymerase (New England BioLabs, M0530), and primers containing the mutation of interest were used to generate mutant BRCA1 fragments flanked by attL sequences for LR clonase reactions (Invitrogen, 11791). Inserts were cloned into pcDNA3.1 expression vectors encoding tags (N-terminal, 3xFLAG-V5; C-terminal, V5–3xFLAG) and then propagated using transformation into DH5α bacteria and isolation of single colonies on LB plates supplemented with ampicillin. Plasmids were mini-prepped (Promega, A1222) and validated using restriction enzyme digestion and Sanger sequencing.
LUMIER with BACON
LUMIER with bait control (BACON) was carried out as described previously using automated liquid handling robots (Biotek, EL406; TECAN, Freedom EVO).41,64 Each experiment included eight replicates of wild-type BRCA1-BRCT and four replicates of the RING wild-type plasmid DNA as well as controls to assess reproducibility. Plasmid DNA (700 ng) was arrayed in round-bottom 96-well plates (Corning, 353227) and mixed with 200 μL of PEI MAX (Polysciences, 24765) with OptiMEM (Gibco, 319850) at a ratio of 1:100 (10 μg/mL final). This DNA transfection mix was then evenly distributed to four flat-bottom 96-well plates (two per HSP70 or HSP90 cell line; Corning, 3598) and incubated at room temperature for 20 min. Reverse transfection was then performed by adding 100 μL of chaperone luciferase-fused stable cells to the DNA transfection mix in flat-bottom 96-well plates at a density of 30,000–50,000 cells per well in duplicate plates. Cells were grown for 2 days, washed three times with PBS using an automated Biotek plate washer and incubated with lysis buffer (50 mM HEPES-KOH, pH 8.0, 150 mM NaCl, 10 mM MgCl2, 20 mM NaMo, 0.7% Triton X-100, 5% glycerol) supplemented with protease inhibitors (Leupeptin, Sigma-Aldrich, 11034626001; Aprotinin, Santa Cruz Biotechnology, sc3595; Pepstatin A, VWR, 97064–248; PMSF, Sigma-Aldrich, 7110), phosphatase inhibitors (Sodium Orthovanadate, New England BioLabs, P0758; Sodium Fluoride, Sigma-Aldrich, S7920), RNAse A (Fisher Scientific, EN0531) and Benzonase (MilliporeSigma, 70664) for 5 min at room temperature. Lysates were transferred to 384-well plates pre-coated in-house with anti-FLAG antibodies (Sigma-Aldrich, F1804). To test different concentrations of cell lysates pulled-down, lysates were diluted with non-transfected cell extracts to minimize effects on protein folding related to molecular crowding. Lysates were incubated at 4°C for 3 h with gentle rocking and then washed six times in 4°C HENG buffer (50 mM HEPES-KOH, pH 8.0, 150 mM NaCl, 2 mM EDTA, pH 8.0, 20 mM NaMo, 0.5% Triton X-100, 5% glycerol). 20 μL of luciferase substrate mix was diluted 200x for NanoLuc substrate (Promega, N11) and 100x dilution for RenillaLuc substrate (Promega, E28) using a reaction buffer (20 mM Tris HCl, pH 7.5, 1 mM EDTA, 150 mM KCl, 0.5% Tergitol NP9) and added to all wells. Luminescence readings were taken using a 400 to 700-nm filter equipped to an EnVision Plate Reader (PerkinElmer). For BACON, the luciferase reagent was dumped into a sink, and 30 μL of anti-FLAG-coupled horseradish peroxidase (abcam, ab1238) diluted at 1:10,000 in an ELISA buffer (PBS, 5% Tween 20, 1% goat serum (Sigma-Aldrich, G9023) was added to all wells, and incubated for 90 min at room temperature with gentle rocking. Wells were washed six times with PBST (0.05% Tween 20), 30 μL of ELISA Supersignal ELISA chemiluminescent substrate (Thermo Scientific, 370) was added and the absorbance was read.
Co-IP
Co-IP was carried out using agarose-linked anti-FLAG beads (Sigma-Aldrich, F2426). 400,000 parental HEK293T cells were seeded in 6-well plates (Corning, 3506) on Day 0. On Day 1, forward transfection was performed by mixing 3,200 ng of DNA packaged with 400 μL of OptiMEM combined with 8 μL of Lipofectamine2000 following incubation times recommended by the manufacturer (Invitrogen, 11668). On day 3, cells were lysed on ice for 20 min with ice-cold LUMIER lysis buffer. Samples were then centrifuged at 14,000xg for 10 min at 4°C, and a soluble fraction was mixed with pre-washed anti-FLAG beads and gently mixed for 3 h at 4°C. Beads were then washed four times with ice-cold lysis buffer and proteins were eluted using 150 μg of 3xFLAG peptide competitor (Biomatik, 56305) and gentle rocking for 30 min at room temperature. Eluates were then collected and mixed 1:1 with a urea/SDS protein sample buffer followed by 65°C incubation for 5 min.
Western blotting
Denatured protein samples were loaded on pre-cast 4–12% SDS-PAGE gels (Invitrogen, WG1402) and resolved for 1.5 h at 160 V in SDS-MOPS buffer (Invitrogen, NP0001). Proteins were transferred to PVDF membranes (Bio-Rad Laboratories, 1620177) with wet transfer using tris-glycine buffer supplemented with 10% fresh methanol. Membranes were blocked in 5% milk (Lab Scientific, M0841) prepared in PBST (0.05% Tween 20) and incubated with corresponding antibodies described in key resources table. Membranes were washed three times with PBST for 5 min each before incubation with anti-mouse secondary antibodies (Sigma-Aldrich, F1804) for at least 60 min, and then washed again before development using horseradish peroxidase substrate (Millipore, WBKLS0) and visualized using a Chemidoc MP imaging system (Bio-Rad Laboratories).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG (M2) | Sigma Aldrich | Cat. #: F1804; RRID:AB_262044 |
| Anti-DDDDK-HRP | abcam | Cat. #: ab1238; RRID:AB_299061 |
| Anti-HSP90 (F-8) | Santa Cruz | Cat. #: sc-13119; RRID:AB_675659 |
| Anti-HSP70/HSC70 (N27F3–4) | StressMarq Biosciences | Cat. #: SMC-104B; RRID:AB_2820319 |
| EZview Red ANTI-FLAG agarose beads | Sigma Aldrich | Cat. #: F2426; RRID:AB_2616449 |
| Chemicals, peptides, and recombinant proteins | ||
| MDYKDHDGDYKDHDIDYKDDDDK (3x-FLAG) | Biomatik | Cat. #: 56305, custom peptide synthesis |
| LR Clonase II Enzyme Mix | Invitrogen | Cat. #: 11791100 |
| BP Clonase II Enzyme Mix | Invitrogen | Cat. #: 11789020 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat. #: M0530L |
| Critical commercial assays | ||
| Renilla Luciferase Assay System | Promega | Cat. #: E2820 |
| Nano-Glo Luciferase Assay System | Promega | Cat. #: N1120 |
| SuperSignal ELISA Pico Chemiluminescent Substrate | Thermo Scientific | Cat. #: 37069 |
| PureYield Plasmid Miniprep System | Promega | Cat. #: A1222 |
| NucleoSpin Gel and PCR Clean-Up | Takara Bio | Cat. #: 740609.250 |
| Immobilon Western Chemiluminescent HRP Substrate | Millipore Sigma | Cat. #: WBKLS0500 |
| Deposited data | ||
| Mutant chaperone interaction and computational data | This study | https://data.mendeley.com/datasets/rmj7fwm6n7/1 |
| Experimental models: Cell lines | ||
| Rluc-HSPA8 | Taipale et al.,41 2012 | R156 |
| Rluc-HSP90AB1 | Taipale et al.,41 2012 | R166 |
| Nluc-HSPA8 | This paper | N156 |
| Nluc-HSP90AB1 | This paper | N166 |
| Recombinant DNA | ||
| pDONR221–1314-BRCA1-STOP | This paper | pKlab182 |
| pDONR221-ATG-1528-BRCA1 | This paper | pKlab183 |
| pDONR221-ATG-1314-BRCA1 | This paper | pKlab184 |
| pcDNA3.1–1314-BRCA1-WT-V5–3xFLAG | This paper | pKlab187 |
| pcDNA3.1–1314-BRCA1–V5–3xFLAG variant library (for details see Table S1) | This paper | BRCT variant library |
In silico predictions of variant effects
DnaK (bacterial HSP70) binding sites were predicted using ChaperISM.68 The algorithm was run using qualitative mode prediction, and the number of binding sites predicted for variants was compared with predictions of the wild-type sequence. Variants were then grouped into three bins: unchanged (number of binding sites “stayed same”), more sites (“more binding sites” or “existing site became larger”), and fewer sites (“less binding sites” or “existing site became smaller”). Variants that resulted in out of frame translated polypeptides were designated as “new out of frame sites” or “no new out of frame sites” depending on ChaperISM prediction of the frameshifted variant. The number of DnaK sites predicted for these variants could not be directly compared with the wild-type because the protein sequence length was different. DnaK binding sites were also predicted using the LIMBO algorithm,92 and no significant difference in chaperone binding was observed when comparing variants in DnaK predicted sites with variants not within predicted sites.
FoldX (version 5.0)21 was used to build variant models and calculate the stability of BRCT variants relative to the wild-type (PDB: 1t29). First, the wild-type structure was repaired using “RepairPDB”. Next, variant models were built using “BuildModel” with the NumberofRuns parameter set to five to derive averages and standard deviations. ΔΔG values relative to the corresponding wild-type model generated for each variant were extracted. Variants at G1738 were not shown in the figures owing to abnormally high ΔΔG values arising from modeling errors. Unless otherwise noted, all FoldX data shown were obtained from predictions made without the pSXXF.
Mutation pathogenicity predictions were collected using PolyPhen223 with the HumDiv classifier model, GRCh37/hg19 genome assembly and canonical transcript settings. SIFT93 predictions were made using the UniProt-SwissProt_2010_09 database, a median conservation score of 3.00 and sequences with more than 90% identity removed from predictions. LIST-S294 was used to generate mutation pathogenicity predictions enhanced by taxonomy-based sorting using the full-length BRCA1 sequence. SuSPect95 variant pathogenicity predictions were obtained using the full-length BRCA1 sequence. ESM1b,30 AlphaMissense,29 EVE27 and VARITY28 variant predictions were obtained from their corresponding webservers.
Secondary structure predictions were obtained using the STRIDE web portal96 and the BRCT crystal structure repaired by FoldX (PDB: 1t29). B-factors for each amino acid were extracted using the swift.cmbi web portal, again using the crystal structure repaired by FoldX. PONDR97 was used to calculate residue disorder using the full-length BRCA1 sequence and the VL-XT predictor. SASA values were collected from the GetArea webserver98 using PDB files repaired by FoldX with the radius of water probe set to 1.4Å and without gradient calculations. SABLE99 was used to predict solvent accessibility and secondary structure using the full-length BRCA1 sequence with the predictor type set to include Approximator (Exposure Pred). The secondary structure of the BRCA1 sequence was also predicted by JPRED4100 using the 1,314–1,863 BRCA1 sequence. For sequence-based secondary structure predictions (SABLE and JPRED4), α-helix and β-sheet predictions were summed to generate a parameter that reflects the probability of a secondary structure at a given residue.
Selection and characterization of variants
BRCA1 variants were extracted from ClinVar and curated to filter for single missense mutations. Variants were called pathogenic or benign including “likely” classifications if the review status was at least ‘two-gold-stars’. Variants we classified as “unknown” included entries in ClinVar annotated as uncertain significance, conflicting interpretations, not provided or ‘one-gold-star’ review status. All ClinVar BRCT missense variants were tested using FoldX and variants were selected to represent benign and pathogenic variant groups. 11 out of 19 benign and 31 out of 56 pathogenic BRCA1-BRCT missense mutants from ClinVar were cloned. This included a majority of the BRCA1-BRCT missense variants from ClinVar (56%); thus, our variant library reflects the entire BRCT ClinVar missense mutant dataset. Additionally, variants were extracted from cBioPortal using the “curated set of non-redundant studies” dataset. BRCA1 variants were extracted for 13 cancer types in TCGA (KICH, KIRP, COAD, BRCA, LUAD, LUSC, PAAD, PRAD, LIHC, HNSC, BLCA, KIRC and READ). gnomAD variants were extracted from the v2.1.1 dataset. Variants extracted from cBioPortal, TCGA and gnomAD were filtered using FoldX to select for mutations predicted to either disrupt or have no effect on protein structure.
To test if chaperones detect changes in thermodynamic stability, a subset of ClinVar variants (n = 44) were compared with previously published protease sensitivity data.71 A protease-sensitive variant group was generated using a < 30% protease resistance cutoff (relative to wild-type). These data were compared with a control group of variants that behaved similar to the wild-type in all assays (> 70% cutoff). Variants that exhibited partial sensitivity to protease digestion or ambiguous functional effects were not included in these comparisons. 49 variants were rationally designed using BRCA1-BRCT crystal structures to disrupt or support protein folding (interactions we investigated were validated in five BRCA1-BRCT crystal structures). ProteinTools101 was run using the BRCA1-BRCT PDB repaired by FoldX without the pSXXF to identify residues involved in hydrophobic networks, side chain hydrogen bond networks (including salt bridges) and long-range interactions (BRCT structure contact map). Residues in the crystal structure were defined as buried (SASA < 10%), intermediate (10% < SASA < 30%), or surface (SASA >= 30%). Surface folding determinants were defined as surface or intermediate variants (SASA > 10%) that disrupted side chain hydrogen bonds and bound to HSP70 (HSP70 score > 0.5), excluding proline variants introduced within α-helices which are expected to disrupt α-helix secondary structures.
Variants were predicted to bind (disruptor) or not bind (no effect) to chaperones based on criteria that considered the biochemical properties of the wild-type BRCA1-BRCT residue and variant. Variants predicted to disrupt structure and bind chaperones included 1) prolines in α-helices, 2) truncations within the BRCT domain (e.g., D1778G fs*27), 3) buried variants (SASA < 10%) that introduced charged side chains, 4) buried variants that decrease hydrophobicity (e.g., V1713A), 5) buried variants that introduced steric clashes (e.g., M1783W), 6) hydrophobic network residues mutated to charged variants and 7) disruptions to side chain hydrogen bonding. Variants predicted to have no effect on chaperone binding were 1) complete BRCT deletions and mutations outside the BRCT domain (e.g., P1575L fs*25), 2) hydrophobic swaps at buried positions (e.g., V1719L), 3) isosteric and quasi-isosteric swaps at buried positions (e.g., D1692N), 4) variants that maintain wild-type side chain hydrogen bonds and 5) all remaining surface variants (SASA > 10%). Variants were considered to decrease hydrophobicity if they lost at least two carbon-containing side chain groups. Buried variants were considered to create a steric clash when the side chain was increased by at least two functional groups. Variants were called isosteric if the mutant structure was the same as the wild-type residue (e.g., aspartate mutated to asparagine). Quasi-isosteric variants differed by one functional group.
BRCA1 mutation data were collected from high-throughput variant screens measured in HAP160 or HeLa62 cell systems. For HDR assays taken from HeLa cells, average BRCA1 siRNA function scores were used. False-negative variants were defined as variants that do not bind HSP70 (interaction score < 0.5) and exhibit intermediate or complete loss of function in either HAP1 cell fitness or HeLa HDR assays. Contact mutants were false-negative variants that were either pSXXF protected or surface as follows. Residues designated as “pSXXF protected” were identified using ΔSASA values after comparing SASA in the presence and absence of the pSXXF (ΔSASA > 5%). Surface BRCT residues, excluding those near the pSXXF, were identified using SASA values calculated in the presence of the pSXXF (SASA >= 30%). Also, a large collection of BRCA1 functional data was extracted from the neXtProt server.77 Each functional assay in this server classifies the effect of BRCA1 variants relative to wild-type as “phenotypic intensity” (none, mild, moderate or severe effect). The functional assays were grouped into eight assay groups: 1) aggregation/degradation, 2) cell localization, 3) cell viability, 4) DNA repair, 5) genome stability, 6) genotoxic stress, 7) protein-partner interaction and 8) transcriptional activation. In our analysis, variants with multiple entries corresponding to the same functional assay group were included because they represent a unique measurement from an independent publication for those variants. A stacked bar graph was generated by collecting all phenotypic intensity counts for variants in our library, splitting variants into bins according to the degree of HSP70 binding and calculating the percentage of variants associated with each phenotypic intensity.
Human alleles were classified as functional, hypomorphic, or severe using HAP1 cell fitness and HDR cell-based variant data60,62 considering the following possibilities: 1) functional in both assays, 2) functional in one assay with no data available for the second assay, 3) functional in one assay and intermediate in the second assay, 4) functional in one assay and loss of function in the second assay, 5) intermediate in both assays, 6) intermediate in one assay and no data available for the second assay, 7) intermediate in one assay and loss of function in the second assay, 8) loss of function in one assay and no data available in the second assay and 9) loss of function in both assays. Groups 1 and 2 were designated “functional”, groups 3–6 were designated “hypomorphic” and groups 7–9 were designated “loss of function”. The structural features of hypomorphic BRCA1 variants that bound moderately to chaperones were compared by binning variants into structural effect classes. Because some variants can fit into more than one class (e.g., A1708P introduces a proline in an alpha helix but also disrupts inter-subdomain interactions) a hierarchical variant assignment protocol was developed to avoid redundancies. Variants were grouped into structural classes in the following order: 1) protease-sensitive, 2) pSXXF protected (defined above) and other surface contact mutants, 3) helix-breaking prolines/charged variants in β-sheets/buried charged variants/buried loss of hydrophobicity, 4) inter-subdomain interactions, 5) disrupted hydrogen bonding, 6) buried hydrophobic swap/quasi-isosteric/isosteric swaps, 7) FoldX ΔΔG values greater than 5 kcal/mol, 8) non-BRCT and 9) other. Groups 1, 3 and 7 were combined into a “structural disruption” bin, groups 4 and 5 were combined into a “long-range” bin and group 6 was denoted “structural fit”. Residues participating in long-range interactions were defined as being at least 50 amino acids apart in primary sequence and within 5Å in the BRCT contact map.
QUANTIFICATION AND STATISTICAL ANALYSIS
Chaperone interaction scores
Raw data were log2–transformed and background subtracted separately using the average of non-transfected wells taken from each individual experiment. Stable HSP70 and HSP90 cell line values were background subtracted separately owing to differences in the luminescence background signal of each cell line. Luminescence and ELISA z-scores were generated by normalizing log2-transformed values to the log2 average and standard deviation of all non-transfected wells across all experiments. Next, ELISA z-scores were compared to filter out data when the amount of bait protein purified varied from the median value for all experiments (cutoff Z score < −2.5 or Z score > 2.5; ~1–2% of wells excluded). After these exclusions, log2 background subtracted values were averaged across all experiments to generate log2 SNRs. Variant chaperone interaction scores were generated by subtracting the luminescence log2 SNR by ELISA log2 SNR to normalize for slight differences in the amount of BRCA1-BRCT variants after pull-down. Variant normalized values were then subtracted from the average of all wild-type normalized values to generate HSP interaction scores.
To determine chaperone interaction preference (HSP70 vs. HSP90), the normalized chaperone interaction scores were converted to interaction z-scores by normalizing the overall HSP scores to the standard deviation for all HSP70 and HSP90 interaction scores. First, variants that did not bind to either chaperone (HSP interaction Z score < 2.5) were identified. Second, variants that only bound to HSP70 or HSP90 significantly were designated HSP70>HSP90 and HSP90RHSP70, respectively. Third, for variants that exhibited increased binding to both HSP70 and HSP90, the ΔHSP interaction Z score (70 minus 90) was used to designate variants as HSP70>HSP90 (ΔHSP Z score > 1) or HSP90≥HSP70 (ΔHSP Z score < 1).
BRCA1 missense variant data were binned according to the magnitude of chaperone binding to test if variants that bound HSP70 to a moderate degree reflected partial loss-of-function BRCA1 variants. Frameshifting and truncating variants were excluded because the degree of chaperone binding cannot be directly compared with the wild-type BRCA1-BRCT owing to a change in the length of the protein variants. In addition to the binning of BRCA1-BRCT variants shown in the main text, additional grouping strategies were used to test if the moderate chaperone binding variant group was apparent for different binning approaches. Despite small differences in the significance, all of our conclusions were robust to independent binning strategies, including binning according to HSP90 interaction scores (HSP90 < 0.5, weak; 0.5 < HSP90 < 2, moderate; HSP90 > 2, strong), HSP70 interaction quartiles (Q1/Q2, weak; Q3, moderate; Q4, strong), luminescence signal z-scores (Z score < 2.5, weak; 2.5 < Z score < 5, moderate; Z score > 5, strong), hybrid quartile and z-scores (Q1/Q2, Z score < 2.5, weak; Q3, Z score > 2.5, moderate; Q4, strong) and various HSP70 interaction score cutoffs (HSP70 < 1, weak; 1 < HSP70 < 2, moderate; HSP70 > 2, strong or HSP70 < 1.3, weak; 1.3 < HSP70 < 2.7, moderate; HSP70 > 2.7, strong).
ROC analysis
ROC curves were generated using the ROCR package in RStudio. SASA and FoldX predictions without the pSXXF were used. FoldX predictions for variants at G1738 were set to 20 kcal/mol to account for abnormally high ΔΔG values arising from modeling errors (this improved FoldX parameter predictions by preventing outlier-driven scaling bias). For the ELISA parameter, a minimum threshold was set also to prevent outlier-driven scaling bias (this improved ELISA parameter predictions). SASA (GetArea and SABLE), B-factor values, secondary structure predictions (both JPRED and SABLE) and disorder predictions (PONDR) were based on the wild-type BRCT residue and did not take into account the variants. The data were trimmed so that all variants had predictions available for all parameters tested (ClinVar target dataset, n = 47; neXtProt target dataset, n = 74). For ROC curve analysis using ClinVar as the ground truth, ‘one-gold-star’ review status entries were considered, including “likely” classifications to avoid data trimming. For HAP1 ROC curves using ClinVar as the target dataset, a separate run was performed to avoid further trimming of the dataset (ClinVar target, n = 44). For ROC curve analysis using aggregated data from the neXtProt server as the ground truth, the mode functional consequence for each mutant was used to designate variants in the target dataset as benign or pathogenic. For variants with a mode severity of mild, moderate, or multi-modal (“ambiguous”), multiple ROC curves were generated, whereby mild/moderate/ambiguous variants were systematically designated either benign or pathogenic. The HAP1 parameter was not run against the neXtProt target dataset owing to large discrepancies arising from splice site variants that were largely undetected by neXtProt assays that use BRCA1 cDNA.
Statistical analysis
Statistical analyses are described at the end of each figure legend. Analyses were performed using the GraphPad Prism software package. Significance was defined as p ≤ 0.05 (indicated by a single *).
Supplementary Material
Highlights.
Chaperones detect an abundance of pathogenic folding variants of BRCA1-BRCT
The degree of chaperone binding reflects structural and phenotypic severity
Chaperones identify hypomorphic mutations and contact variants
Chaperone interactions indicate the penetrance and expressivity of BRCA1 alleles
ACKNOWLEDGMENTS
We thank Junjie Chen for providing plasmids encoding wild-type BRCA1 and for guidance with BRCA1 fragment expression. We thank Mikko Taipale for providing HEK293T cell lines stably expressing N-terminally NanoLuc-tagged HSP70 (HSPA8) and HSP90 (HSP90AB1) and Susan Lindquist for pcDNA3.1 destination vectors. We thank Natalia Condic for outstanding technical assistance; Nikitha Kota for help with ChaperISM predictions; Wenyi Wang and Shaolong Cao for help retrieving TCGA data; Jay Shendure and Greg Findlay for help accessing their HAP1 data; and Richard Wood, John Tainer, Dan Dickinson, Swathi Arur, Pierre McCrea, Junjie Chen, Daniel Herschlag, and Donald Norwood at MD Anderson Editing Services Research Medical Library for feedback on our data and the manuscript. Tumor allele frequencies are in whole based upon data generated by TCGA Research Network (https://www.cancer.gov/tcga). G.I.K. is a CPRIT Scholar in Cancer Research. Research reported in this publication was supported by the CPRIT under award number RR180005 and by the National Cancer Institute of the National Institutes of Health under award numbers F32CA253780 to B.G. and K22CA222938 to G.I.K., and from the Joe W. and Dorothy Dorsett Brown Foundation and the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113803.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Ready K, Gutierrez-Barrera AM, Amos C, Meric-Bernstam F, Lu K, Hortobagyi G, and Arun B (2011). Cancer risk management decisions of women with BRCA1 or BRCA2 variants of uncertain significance. Breast J. 17, 210–212. 10.1111/j.1524-4741.2010.01055.x. [DOI] [PubMed] [Google Scholar]
- 2.Balmaña J, Digiovanni L, Gaddam P, Walsh MF, Joseph V, Stadler ZK, Nathanson KL, Garber JE, Couch FJ, Offit K, et al. (2016). Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol 34, 4071–4078. 10.1200/JCO.2016.68.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makhnoon S, Chen M, Levin B, Ensinger M, Mattie KD, Grana G, Shete S, Arun BK, and Peterson SK (2022). Use of breast surveillance between women with pathogenic variants and variants of uncertain significance in breast cancer susceptibility genes. Cancer 128, 3709–3717. 10.1002/cncr.34429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King MC, Marks JH, and Mandell JB; New York Breast Cancer Study Group (2003). Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646. 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, and Parmigiani G (2007). Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol 25, 1329–1333. 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niroula A, and Vihinen M (2017). Predicting Severity of Disease-Causing Variants. Hum. Mutat 38, 357–364. 10.1002/humu.23173. [DOI] [PubMed] [Google Scholar]
- 8.Kingdom R, and Wright CF (2022). Incomplete Penetrance and Variable Expressivity: From Clinical Studies to Population Cohorts. Front. Genet 13, 920390. 10.3389/fgene.2022.920390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsden RL, Lewis TA, and Orengo CA (2007). Towards a comprehensive structural coverage of completed genomes: a structural genomics viewpoint. BMC Bioinf. 8, 86. 10.1186/1471-2105-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drew K, Winters P, Butterfoss GL, Berstis V, Uplinger K, Armstrong J, Riffle M, Schweighofer E, Bovermann B, Goodlett DR, et al. (2011). The Proteome Folding Project: proteome-scale prediction of structure and function. Genome Res. 21, 1981–1994. 10.1101/gr.121475.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A,Bridgland A, Cowie A, Meyer C, Laydon A, et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaeffer RD, Zhang J, Kinch LN, Pei J, Cong Q, and Grishin NV (2023). Classification of domains in predicted structures of the human proteome. Proc. Natl. Acad. Sci. USA 120, e2214069120. 10.1073/pnas.2214069120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dill KA, and Chan HS (1997). From Levinthal to pathways to funnels. Nat. Struct. Biol 4, 10–19. [DOI] [PubMed] [Google Scholar]
- 15.Fersht A (1998). Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (W.H. Freeman; ). [Google Scholar]
- 16.Yang F, Petsalaki E, Rolland T, Hill DE, Vidal M, and Roth FP (2015). Protein domain-level landscape of cancer-type-specific somatic mutations. PLoS Comput. Biol 11, e1004147. 10.1371/journal.pcbi.1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vermeulen W, Rademakers S, Jaspers NG, Appeldoorn E, Raams A, Klein B, Kleijer WJ, Hansen LK, and Hoeijmakers JH (2001). A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat. Genet 27, 299–303. 10.1038/85864. [DOI] [PubMed] [Google Scholar]
- 18.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karras GI, Yi S, Sahni N, Fischer M, Xie J, Vidal M, D’Andrea AD, Whitesell L, and Lindquist S (2017). HSP90 Shapes the Consequences of Human Genetic Variation. Cell 168, 856–866.e12. 10.1016/j.cell.2017.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, Constantinou S, Renaudin X, Lee M, Aebersold R, and Venkitaraman AR (2017). A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 169, 1105–1118.e15. 10.1016/j.cell.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, and Serrano L (2005). The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, and Thomas A (2006). Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet 43, 295–305. 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315. 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy MD, Hamre J, Klimov DK, and Jafri MS (2021). Predicting Genetic Variation Severity Using Machine Learning to Interpret Molecular Simulations. Biophys. J 120, 189–204. 10.1016/j.bpj.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, Gal Y, and Marks DS (2021). Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95. 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Liu H, Li R, Sun S, Weile J, and Roth FP (2021). Improved pathogenicity prediction for rare human missense variants. Am. J. Hum. Genet 108, 2389. 10.1016/j.ajhg.2021.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, Pritzel A, Wong LH, Zielinski M, Sargeant T, et al. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492. 10.1126/science.adg7492. [DOI] [PubMed] [Google Scholar]
- 30.Brandes N, Goldman G, Wang CH, Ye CJ, and Ntranos V (2023). Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet 55, 1512–1522. 10.1038/s41588-023-01465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martelotto LG, Ng CK, De Filippo MR, Zhang Y, Piscuoglio S, Lim RS, Shen R, Norton L, Reis-Filho JS, and Weigelt B (2014). Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 15, 484. 10.1186/s13059-014-0484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pak MA, Markhieva KA, Novikova MS, Petrov DS, Vorobyev IS, Maksimova ES, Kondrashov FA, and Ivankov DN (2023). Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS One 18, e0282689. 10.1371/journal.pone.0282689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feder ME, and Hofmann GE (1999). Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu. Rev. Physiol 61, 243–282. 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- 34.Hartl FU, and Hayer-Hartl M (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858. 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 35.Hartl FU, Bracher A, and Hayer-Hartl M (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 36.Schopf FH, Biebl MM, and Buchner J (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol 18, 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 37.Rosenzweig R, Nillegoda NB, Mayer MP, and Bukau B (2019). The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol 20, 665–680. 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- 38.Morán Luengo T, Mayer MP, and Rüdiger SGD (2019). The Hsp70-Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 29, 164–177. 10.1016/j.tcb.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Noddings CM, Wang RYR, Johnson JL, and Agard DA (2022). Structure of Hsp90-p23-GR reveals the Hsp90 client-remodelling mechanism. Nature 601, 465–469. 10.1038/s41586-021-04236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang RYR, Noddings CM, Kirschke E, Myasnikov AG, Johnson JL, and Agard DA (2022). Structure of Hsp90-Hsp70-Hop-GR reveals the Hsp90 client-loading mechanism. Nature 601, 460–464. 10.1038/s41586-021-04252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, and Lindquist S (2012). Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001. 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taipale M, Krykbaeva I, Whitesell L, Santagata S, Zhang J, Liu Q, Gray NS, and Lindquist S (2013). Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nat. Biotechnol 31, 630–637. 10.1038/nbt.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, and Lindquist S (2014). A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448. 10.1016/j.cell.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, et al. (2015). Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660. 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, and Lu LY (2020). BRCA1 and homologous recombination: implications from mouse embryonic development. Cell Biosci. 10, 49. 10.1186/s13578-020-00412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark SL, Rodriguez AM, Snyder RR, Hankins GDV, and Boehning D (2012). Structure-Function Of The Tumor Suppressor BRCA1. Comput. Struct. Biotechnol. J 1, e201204005. 10.5936/csbj.201204005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ragupathi A, Singh M, Perez AM, and Zhang D (2023). Targeting the BRCA1/2 deficient cancer with PARP inhibitors: Clinical outcomes and mechanistic insights. Front. Cell Dev. Biol 11, 1133472. 10.3389/fcell.2023.1133472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou BB, and Elledge SJ (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 49.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, and Ohta T (2001). The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem 276, 14537–14540. 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 50.Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, and Smerdon SJ (2004). Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat. Struct. Mol. Biol 11, 512–518. 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 51.Varma AK, Brown RS, Birrane G, and Ladias JAA (2005). Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry 44, 10941–10946. 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 52.Nacson J, Di Marcantonio D, Wang Y, Bernhardy AJ, Clausen E, Hua X, Cai KQ, Martinez E, Feng W, Callén E, et al. (2020). BRCA1 Mutational Complementation Induces Synthetic Viability. Mol. Cell 78, 951–959.e6. 10.1016/j.molcel.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krais JJ, Glass DJ, Chudoba I, Wang Y, Feng W, Simpson D, Patel P, Liu Z, Neumann-Domer R, Betsch RG, et al. (2023). Genetic separation of Brca1 functions reveal mutation-dependent Polq vulnerabilities. Nat. Commun 14, 7714. 10.1038/s41467-023-43446-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Bernhardy AJ, Nacson J, Krais JJ, Tan YF, Nicolas E, Radke MR, Handorf E, Llop-Guevara A, Balmaña J, et al. (2019). BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun 10, 5661. 10.1038/s41467-019-13530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. (2018). BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 24, 3513–3527.e7. 10.1016/j.celrep.2018.08.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Kim HH, George E, Swisher EM, Simpkins F, and Johnson N (2016). RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Invest 126, 3145–3157. 10.1172/JCI87033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaclová T, Woods NT, Megías D, Gomez-Lopez S, Setién F, García Bueno JM, Macías JA, Barroso A, Urioste M, Esteller M, et al. (2016). Germline missense pathogenic variants in the BRCA1 BRCT domain, p.Gly1706Glu and p.Ala1708Glu, increase cellular sensitivity to PARP inhibitor olaparib by a dominant negative effect. Hum. Mol. Genet 25, 5287–5299. 10.1093/hmg/ddw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sylvain V, Lafarge S, and Bignon YJ (2002). Dominant-negative activity of a Brca1 truncation mutant: effects on proliferation, tumorigenicity in vivo, and chemosensitivity in a mouse ovarian cancer cell line. Int. J. Oncol 20, 845–853. [PubMed] [Google Scholar]
- 59.Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, and Fields S (2015). Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 200, 413–422. 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernandes VC, Golubeva VA, Di Pietro G, Shields C, Amankwah K, Nepomuceno TC, de Gregoriis G, Abreu RBV, Harro C, Gomes TT, et al. (2019). Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: Implications for clinical annotation. J. Biol. Chem 294, 5980–5992. 10.1074/jbc.RA118.005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adamovich AI, Diabate M, Banerjee T, Nagy G, Smith N, Duncan K, Mendoza Mendoza E, Prida G, Freitas MA, Starita LM, and Parvin JD (2022). The functional impact of BRCA1 BRCT domain variants using multiplexed DNA double-strand break repair assays. Am. J. Hum. Genet 109, 618–630. 10.1016/j.ajhg.2022.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clark KA, Paquette A, Tao K, Bell R, Boyle JL, Rosenthal J, Snow AK, Stark AW, Thompson BA, Unger J, et al. (2022). Comprehensive evaluation and efficient classification of BRCA1 RING domain missense substitutions. Am. J. Hum. Genet 109, 1153–1174. 10.1016/j.ajhg.2022.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taipale M (2018). Quantitative Profiling of Chaperone/Client Interactions with LUMIER Assay. Methods Mol. Biol 1709, 47–58. 10.1007/978-1-4939-7477-1_4. [DOI] [PubMed] [Google Scholar]
- 65.Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, Fukuda M, Ohta T, and Klevit R (2003). Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 100, 5646–5651. 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meacham GC, Patterson C, Zhang W, Younger JM, and Cyr DM (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol 3, 100–105. 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 67.Quintana-Gallardo L, Martín-Benito J, Marcilla M, Espadas G, Sabidó E, and Valpuesta JM (2019). The cochaperone CHIP marks Hsp70- and Hsp90-bound substrates for degradation through a very flexible mechanism. Sci. Rep 9, 5102. 10.1038/s41598-019-41060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gutierres MBB, Bonorino CBC, and Rigo MM (2020). ChaperISM: improved chaperone binding prediction using position-independent scoring matrices. Bioinformatics 36, 735–741. 10.1093/bioinformatics/btz670. [DOI] [PubMed] [Google Scholar]
- 69.Williams RS, Chasman DI, Hau DD, Hui B, Lau AY, and Glover JNM (2003). Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. J. Biol. Chem 278, 53007–53016. 10.1074/jbc.M310182200. [DOI] [PubMed] [Google Scholar]
- 70.Williams RS, Lee MS, Hau DD, and Glover JNM (2004). Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat. Struct. Mol. Biol 11, 519–525. 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 71.Lee MS, Green R, Marsillac SM, Coquelle N, Williams RS, Yeung T, Foo D, Hau DD, Hui B, Monteiro ANA, and Glover JNM (2010). Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 70, 4880–4890. 10.1158/0008-5472.CAN-09-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barlow DJ, and Thornton JM (1988). Helix geometry in proteins. J. Mol. Biol 201, 601–619. 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
- 73.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, and Zehfus MH (1985). Hydrophobicity of amino acid residues in globular proteins. Science 229, 834–838. 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 74.Shoemaker KR, Kim PS, York EJ, Stewart JM, and Baldwin RL (1987). Tests of the helix dipole model for stabilization of alpha-helices. Nature 326, 563–567. 10.1038/326563a0. [DOI] [PubMed] [Google Scholar]
- 75.Nicholson H, Anderson DE, Dao-pin S, and Matthews BW (1991). Analysis of the interaction between charged side chains and the alpha-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry 30, 9816–9828. 10.1021/bi00105a002. [DOI] [PubMed] [Google Scholar]
- 76.Rowling PJE, Cook R, and Itzhaki LS (2010). Toward classification of BRCA1 missense variants using a biophysical approach. J. Biol. Chem 285, 20080–20087. 10.1074/jbc.M109.088922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cusin I, Teixeira D, Zahn-Zabal M, Rech de Laval V, Gleizes A, Viassolo V, Chappuis PO, Hutter P, Bairoch A, and Gaudet P (2018). A new bioinformatics tool to help assess the significance of BRCA1 variants. Hum. Genom 12, 36. 10.1186/s40246-018-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ahlborn LB, Dandanell M, Steffensen AY, Jønson L, Nielsen FC, and Hansen TVO (2015). Splicing analysis of 14 BRCA1 missense variants classifies nine variants as pathogenic. Breast Cancer Res. Treat 150, 289–298. 10.1007/s10549-015-3313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coquelle N, Green R, and Glover JNM (2011). Impact of BRCA1 BRCT domain missense substitutions on phosphopeptide recognition. Biochemistry 50, 4579–4589. 10.1021/bi2003795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, and den Dunnen JT (2011). LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat 32, 557–563. 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 83.Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN, and Arun BK (2008). Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol 26, 4282–4288. 10.1200/JCO.2008.16.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanford RA, Song J, Gutierrez-Barrera AM, Profato J, Woodson A, Litton JK, Bedrosian I, Albarracin CT, Valero V, and Arun B (2015). High incidence of germline BRCA mutation in patients with ER low-positive/PR low-positive/HER-2 neu negative tumors. Cancer 121, 3422–3427. 10.1002/cncr.29572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keupp K, Hampp S, Hübbel A, Maringa M, Kostezka S, Rhiem K, Waha A, Wappenschmidt B, Pujol R, Surrallés J, et al. (2019). Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol. Genet. Genomic Med 7, e863. 10.1002/mgg3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, Pieterse M, Wientjens E, Seibler J, Hogervorst FBL, and Jonkers J (2013). A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 3, 1142–1155. 10.1158/2159-8290.CD-13-0094. [DOI] [PubMed] [Google Scholar]
- 87.Moghadasi S, Meeks HD, Vreeswijk MP, Janssen LA, Borg Å, Ehrencrona H, Paulsson-Karlsson Y, Wappenschmidt B, Engel C, Gehrig A, et al. (2018). The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J. Med. Genet 55, 15–20. 10.1136/jmedgenet-2017-104560. [DOI] [PubMed] [Google Scholar]
- 88.Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, Pettigrew C, kConFab; Van Asperen CJ, Ausems MGEM, et al. (2012). BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J. Med. Genet 49, 525–532. 10.1136/jmedgenet-2012-101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carvalho MA, Billack B, Chan E, Worley T, Cayanan C, and Monteiro ANA (2002). Mutations in the BRCT domain confer temperature sensitivity to BRCA1 in transcription activation. Cancer Biol. Ther 1, 502–508. 10.4161/cbt.1.5.165. [DOI] [PubMed] [Google Scholar]
- 90.Worley T, Vallon-Christersson J, Billack B, Borg A, and Monteiro ANA (2002). A naturally occurring allele of BRCA1 coding for a temperature-sensitive mutant protein. Cancer Biol. Ther 1, 497–501. 10.4161/cbt.1.5.164. [DOI] [PubMed] [Google Scholar]
- 91.Krais JJ, and Johnson N (2020). BRCA1 Mutations in Cancer: Coordinating Deficiencies in Homologous Recombination with Tumorigenesis. Cancer Res. 80, 4601–4609. 10.1158/0008-5472.CAN-20-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Van Durme J, Maurer-Stroh S, Gallardo R, Wilkinson H, Rousseau F, and Schymkowitz J (2009). Accurate prediction of DnaK-peptide binding via homology modelling and experimental data. PLoS Comput. Biol 5, e1000475. 10.1371/journal.pcbi.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ng PC, and Henikoff S (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malhis N, Jacobson M, Jones SJM, and Gsponer J (2020). LIST-S2: taxonomy based sorting of deleterious missense mutations across species. Nucleic Acids Res. 48, W154–W161. 10.1093/nar/gkaa288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yates CM, Filippis I, Kelley LA, and Sternberg MJE (2014). SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J. Mol. Biol 426, 2692–2701. 10.1016/j.jmb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heinig M, and Frishman D (2004). STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32, W500–W502. 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, and Dunker AK (2003). Predicting intrinsic disorder from amino acid sequence. Proteins 53 (Suppl 6), 566–572. 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 98.Hayryan S, Hu CK, Skrivánek J, Hayryane E, and Pokorný I (2005). A new analytical method for computing solvent-accessible surface area of macromolecules and its gradients. J. Comput. Chem 26, 334–343. 10.1002/jcc.20125. [DOI] [PubMed] [Google Scholar]
- 99.Adamczak R, Porollo A, and Meller J (2004). Accurate prediction of solvent accessibility using neural networks-based regression. Proteins 56, 753–767. 10.1002/prot.20176. [DOI] [PubMed] [Google Scholar]
- 100.Drozdetskiy A, Cole C, Procter J, and Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferruz N, Schmidt S, and Höcker B (2021). ProteinTools: a toolkit to analyze protein structures. Nucleic Acids Res. 49, W559–W566. 10.1093/nar/gkab375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Chaperone interaction data generated in this study have been made publicly available through Mendeley. https://data.mendeley.com/drafts/rmj7fwm6n7
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.