

Abstract

Polyethylene deconstruction to reusable smaller molecules is hindered by the chemical inertness of its hydrocarbon chains. Pyrolysis and related approaches commonly require high temperatures, are energy-intensive, and yield mixtures of multiple classes of compounds. Selective cleavage reactions under mild conditions (<ca. 200 °C) are key to improve the efficacy of chemical recycling and upcycling approaches. These can be enabled by introduction of low densities of predetermined breaking points in the polyethylene chains during the step-growth or chain-growth synthetic construction of designed-for-recycling polyethylene-type materials. Alternatively, they can be accomplished by postpolymerization functionalization of postconsumer polyethylene waste via dehydrogenation and follow-up reactions or through oxidation to long-chain dicarboxylates. Deconstruction of litter under environmental conditions via the aforementioned break points can alleviate plastics’ persistency, as a backstop to closed-loop recycling.
1. Introduction
Plastics are a key component of virtually any technology today. A myriad of applications in, e.g., the fields of construction, transportation, communication, water supply, or health care rely on the specific performance of polymer materials. The demand for plastic materials is ever increasing due to technological advances and an increasing world population with its desire for a high quality of life.1 This consumes enormous feedstock resources, as plastics are hardly reused after their useful service life. Currently, a large part of the plastic waste generated is placed in landfills, and a substantial portion is released to the environment in an uncontrolled way.2 Incineration with energy recovery essentially is a method of waste disposal, recovering a part of the caloric value of the plastic at the most.3,4 While the handling and legislation concerning plastic waste varies strongly between countries, a true recycling in the sense of a circular economy remains to be established anywhere. A closed loop that recycles polymers into an identical or similar-value application as in the preceding life cycle today is reality for only a small fraction of the plastic waste collected in advanced waste management systems.5 While such a truly circular approach will not be applicable and sensible for all types of waste streams, it is also evident that plastic waste represents a valuable resource that is not used efficiently today and overall plastics technology and economy could be designed in a more resource-efficient manner.6
Polyethylene is the largest produced synthetic polymer, with an annual production of more than 100 million tons. Polyethylene alone accounts for roughly one-third of the overall plastic production.2 Compared to other plastics, the emission of greenhouse gases per mass of polyethylene produced is comparatively low. Still, an entire cradle-to-grave life cycle, starting from crude oil and ending with incineration with energy recovery, produces 4.4 kg of CO2 equivalents per kg of polyethylene.7 The different types of polyethylene vary in their microstructure as a result of the production process, that is, catalytic insertion or free-radical chain growth, respectively, and the presence or absence of comonomers. High-density polyethylene (HDPE), low-density polyethylene (LDPE), and linear low-density polyethylene (LLDPE) each come in numerous different grades. Further variants comprise waxes and ultrahigh molecular weight polyethylene (UHMWPE), smaller in volume but still amounting to production volumes of millions of tons.8,9 Applications of polyethylenes range from food packaging films, detergent or milk bottles, mulch films, textiles, and high-performance fibers and ropes to fresh water pipes and orthopedic bearings to name only a few examples. Consequently, service life cycles range from short-term use (e.g., LDPE single-use packaging) to >50 years (e.g., HDPE pipes).
The mechanical recycling, requiring material that is washed and sorted in single-polymer streams, of PE is comparatively well-established vs most other plastics (with the exception of poly(ethylene terephthalate), PET). Yet, a closed-loop recycling to produce material suited for the same application is hardly practiced for collected polymer waste (Figure 1).10,11 One reason is that repeated processing and use lead to a deterioration of polymer properties. Further, the existence of many different grades complicates mechanical recycling. For example, PE molecular weight distributions are quite specifically defined for given processing methods and applications, with different molecular weight fractions serving different essential functions (Figure 2). Further, different PE products contain different stabilizers, processing aids, etc., in variable amounts.12 To achieve a sustainable plastic economy within the planetary boundaries, additional chemical recycling processes of existing polymers need to be established, and polymers specifically designed to enable such recycling are necessary.10,13−15
Figure 1.

Fate of collected plastic waste in Western Europe.2
Figure 2.

Schematic representation of a bimodal molecular weight distribution of polyethylene, and role of different molecular weight regimes in processing and materials properties (Reprinted with permission from Tailor-Made Polymers Via Immobilization of Alpha-Olefin Polymerization Catalyst2008, 1–42. Copyright © 2008, Wiley-VCH).12
The mechanical strength of polyethylene arises from crystalline order due to noncovalent (van der Waals) interactions between adjacent segments in all-trans conformation of hydrocarbon chains (Figure 3). This is particularly pronounced for HDPE, as it is composed of linear chains devoid of branches that would disturb this crystalline packing.9
Figure 3.
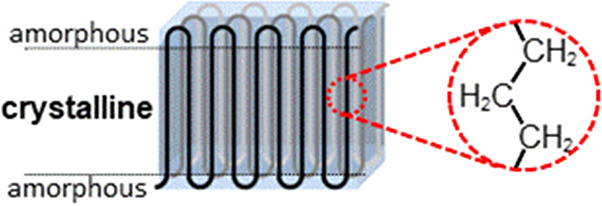
Polyethylene crystallinity arises from van der Waals interactions between adjacent stretched hydrocarbon segments, illustrated here for a folded chain crystallite.
The chemically inert nature and C–C bond uniformity of the hydrocarbon chains hinders their breakdown back to the ethylene monomer. The formation of olefinic monomers from the saturated chains is kinetically hindered and thermodynamically disfavored.16 Thus, pyrolysis to liquid hydrocarbons and their steam cracking to generate olefins in both steps require high temperatures of up to 800 °C and are energy-consuming. Ethylene yields are limited in practice to around 10%.4,17 Catalytic cracking of polyethylene is of interest to reduce the temperatures required in particular (to around 300 °C).18 By contrast, the concepts for chemical recycling of PE-type polymers reviewed here operate at mild conditions (≤ca. 200 °C) and ideally avoid highly endothermic reactions (Figure 4).
Figure 4.

Access to PE-type materials from polyethylene waste, natural oil, and petroleum- or renewable-based ethylene. Chemical recycling is enabled via low densities of in-chain functional groups in a PE chain, and biodegradation acts as a backstop that prevents environmental accumulation of mismanaged plastic waste.
Notably, this inert nature of polyethylene in conjunction with its hydrophobic, apolar, and crystalline nature also impedes degradation of material lost into the environment. Polyolefins can therefore persist for many decades or even centuries.19 While polymer deconstruction is considered here primarily within the context of recycling, the underlying principles of polymer design to enable such deconstruction also can facilitate the breakdown of plastic litter and prevent their accumulation; therefore, these complementary processes will also be discussed. The scope of “polyethylene-type” polymers is guided by the criterion of a hydrocarbon-dominated polymer crystallinity (cf. Figure 3) as a universal characteristic feature found in the various types of traditional polyethylenes. Typically, this is reflected in an orthorhombic crystal structure.
2. Synthesis
One approach to establish a circular plastics economy is the synthesis of polymers already tailored for deconstruction. Accordingly, functional groups as predetermined breaking points can be introduced into the polymer chain. A low density of such in-chain functional groups can enable different deconstruction pathways while not compromising the highly crystalline nature and desirable materials properties of PE. The inclusion of such predesigned break points can be achieved during chain-growth polymerization of olefins with functional comonomers. Alternatively, polyethylene-like chains with in-chain functional groups can also be generated by step-growth polycondensation reactions of long-chain difunctional monomers or by related ring-opening polymerizations of macrocycles.
2.1. Chain-Growth Polymers
Polyethylenes are produced industrially by catalytic (HDPE and LLDPE) or free-radical (LDPE) chain-growth polymerization. An inclusion of in-chain heteroatom-containing functional groups that could enable deconstruction has been a long-sought goal. However, traditional olefin polymerization catalysts are extremely sensitive to any polar reagents or impurities as a result of the high oxophilicity of their early transition-metal (d0) active sites.20 One approach to circumvent this limitation is the introduction of low densities of double bonds into polyolefin chains by copolymerization with, e.g., butadiene. Deconstruction via these double bonds commonly requires several reaction steps (cf. Chapter 3). A direct introduction of heteroatom-containing in-chain functional groups is enabled by recently discovered functional group tolerant catalysts or by free-radical polymerization, which will be discussed in more detail throughout this chapter.
Postpolymerization backbone oxidation reactions can also afford desired in-chain functional groups such as ketone or hydroxy groups. These postpolymerization reactions, however, can suffer from drawbacks such as uncontrolled chain cleavage and selectivity, if promoted in a free-radical fashion.21,22 Substantial efforts led to the development of more controlled, transition-metal catalyzed C–H functionalization.23−27 The introduction of keto and hydroxyl groups via this method can enhance materials properties, for example, enabling better adhesion to polar surfaces.26 Despite significant advances, the functional group selectivity remains a main challenge for postpolymerization modification of polyethylenes, and usually mixtures of ketones and hydroxyl groups are obtained from Ni- or Ru-catalyzed C–H functionalization.25,26 The synthesis of exclusively keto-containing polymers via this pathway was also reported but requires an additional oxidation step of such mixed “oxo-polyethylenes” by Cp*Ir-catalyzed transfer dehydrogenation using acetone as oxidant.28 Even though a high selectivity for keto groups (>99%) can be achieved initially with Cu catalysts and benzaldehyde as a reagent, the C–H oxidation results in a significant molecular weight decrease vs the original material.27 Thus, the oxidation of polyethylenes may unfold its potential rather in PE waste upcycling (cf. Section 3.2.2) or modification of materials properties than in the tailored synthesis of polymers designed for deconstruction.
The incorporation of carbon monoxide (CO) in high-pressure (∼1000 atm) free-radical ethylene polymerization to generate keto-modified LDPE was reported early on.29−31 Due to the relatively higher stability of the acyl radical formed after a CO-incorporation event, the chain-growth rate is decreased compared to ethylene homopolymerization.30,32 Like in ethylene homopolymerization, branched materials are obtained due to backbiting of the growing polyethylene radicals on the chain. In addition, branches adjacent to the carbonyl groups are prominent due to the propensity of H-abstraction from the α-methylene groups, −CH2C(=O)- (Figure 5).32 Recent laboratory-scale studies revealed that in reaction media that are inert to radical transfer reactions, namely, dimethyl carbonate33 or water, the (co)polymerization can be performed under comparatively moderate pressures of <350 atm to yield thermoplastic processable materials or film-forming aqueous dispersions, respectively.32 Notwithstanding the lowered growth rate by incorporation of CO, the copolymerization is practical for the industrial generation of LDPE materials. Keto-modified LDPEs with typically ca. 1 mol % of incorporated in-chain keto groups (Mn ∼ 5 × 104 g mol–1; Đ ∼ 4) have been employed since the 1970s as photodegradable beverage six-pack slings, to enhance the degradation of littered material.34−36
Figure 5.

LDPE-like materials containing in-chain keto and ester groups from controlled free-radical terpolymerization of ethylene with CO and 2-methylene-1,3-dioxepane.37
In the context of recent studies of controlled free-radical copolymerization, the incorporation of keto as well as ester groups into polyethylene chains has been demonstrated.37 In the organometallic-mediated radical polymerization (OMRP) with cobalt(acetyl acetonate) complexes, degrees of branching are comparatively low due to the underlying reversible trapping of radicals (ca. 7/1000 carbon atoms). Notably, CO incorporations are much enhanced compared to reference uncontrolled copolymerizations. This is a result of the dual function of CO as comonomer and coordinating Lewis base. By terpolymerization with a cyclic ketene acetal, 2-methylene-1,3-dioxepane, lightly branched polyethylenes with in-chain keto as well as in-chain ester groups are accessible (∼2 mol % each, Figure 5).37 This is enabled by the propensity of the cyclic ketene acetal to undergo ring opening upon addition of a growing polymeryl radical to the double bond, to yield a new primary alkyl radical.38 The molecular weights reported so far (Mn ∼ 6 × 103 g mol–1; Đ ∼ 2) call for improvement if applications as thermoplastics are targeted.37
In catalytic copolymerizations to linear polyethylenes, traditional early transition-metal catalysts are deactivated by carbon monoxide. Quenching by CO is in fact an established method to determine the number of active sites.39 Late transition-metal catalysts are not subject to such limitation, as evidenced by various large-scale industrial carbonylation processes for the synthesis of small molecules based on cobalt, nickel, rhodium, or iridium catalysts.40 In catalytic olefin-CO copolymerizations, the strong relative binding of CO and low barrier for CO insertion favors the formation of perfectly alternating polyketones.41−47 Alternating olefin-CO copolymers based on ethylene and ca. 5 mol % of propylene had been commercialized as high melting engineering thermoplastics (Tm ∼ 230 °C) by Shell under the trade name “Carilon”48 and have more recently been revived by Hyosung as “Poketone”.49 The polymerization process is based on cationic Pd(II) diphosphine complexes.42−44
Overcoming the preference for CO incorporation is a key to enable catalytic polymerization to polyethylene-type materials with low densities of in-chain keto groups (Figure 6). Nonalternating ethylene CO copolymerizations are favored by a neutral charge of the catalyst that decreases the relative binding affinity of CO vs ethylene compared to cationic counterparts.47,50−52 Indeed, an inclusion of multiple ethylene-based repeat units in addition to alternating motifs was first observed for neutral Pd(II) phosphinosulfonate catalysts (Figure 7).52 Further studies reported materials with likely low keto incorporations; however, these were low molecular weight brittle polymers or oligomers which impeded studies of mechanical properties.53
Figure 6.

Routes to HDPE-like materials with in-chain keto and ester groups accessible by catalytic polymerizations. The postpolymerization oxidation route of polyethylene materials to similar materials is shown in comparison.
Figure 7.

Reported catalysts for the nonalternating copolymerization of ethylene and CO.52,60,66,69,70
Neutral Ni(II) phosphinophenolate catalysts are prominently employed in the Shell Higher Olefin process for ethylene oligomerization to linear 1-olefins. This is based on an effective competition of chain transfer with chain growth, with typical ratios of vgrowth vs vtransfer of five to ten.54−56 Before this established picture, the finding of Shimizu et al. that bulky substituted phosphinophenolate catalysts polymerize ethylene to high molecular weight of Mn > 105 g mol–1 was a breakthrough.57,58 This development was taken further with catalytic polymerizations in which chain transfer is virtually completely suppressed, resulting in a living character and the formation of ultrahigh molecular weights of Mn 3 × 106 g mol–1, in the form of aqueous dispersions.59 Exposure of state-of-the-art catalysts to ethylene-CO mixtures with high olefin/CO ratios affords high molecular weight linear polyethylenes with exclusively nonalternating ketone motifs (“keto-PEs”), preferentially in the form of isolated carbonyls.60−62 Beneficially, the presence of CO increases molecular weights compared to ethylene homopolymerizations, likely due to a further suppression of β-hydride elimination (to Mn 2 × 105 g mol–1; Mw 4 × 105 g mol–1).60 At the target incorporations of ca. 0.3 to 3 mol % keto units these do not disturb the polyethylene crystalline structure, nor is the melting point impacted significantly60 as the incorporation of keto-groups into the crystalline lattice of polyethylene is associated with a rather low energy penalty.63−65
Consequently, these keto-PEs are on par with commercial HDPE concerning their tensile properties, as determined on injection-molded specimens. At the same time they are photodegradable (cf. section 3.1).60 For the synthesis of these materials, the dosing of high ethylene/CO ratios was achieved by premixing in a high-pressure syringe pump60 or by mass flow meters with a customized control system.61 Notably, both setups allow for the convenient switching between natural isotopic abundance CO and isotopically pure 13CO, the latter facilitating exhaustive microstructure analysis by 13C NMR spectroscopy analysis and also monitoring of photodegradation (cf. Chapter 3). In parallel studies, Nozaki et al. demonstrated the formation of high molecular weight (Mn 6 × 104 g mol–1) linear polyethylenes with an excellent ratio of isolated in-chain carbonyls (99%) employing their (dimenthylphosphino)sulfonate Pd(II) catalyst. Metal carbonyls like [Fe(CO)5] were elegantly used as a source of the keto groups.66 As an alternative way of generating low concentrations of CO for nonalternating copolymerization, the electroreduction67 or photoreduction68 of carbon dioxide in a separate compartment of a polymerization reactor has also been reported.
Nonalternating ethylene-CO copolymerizations have also been demonstrated more recently with cationic Pd(II) catalysts but at still relatively high keto incorporations that also reflect in significantly higher melting points compared to polyethylene.69,70 Further development and studies of these catalysts, as well as their Ni(II) analogues, yielded keto-PEs with low CO incorporation and high molecular weights (Mn up to 105 g mol–1).71
From a mechanistic point of view the ability of the aforementioned neutral catalysts to generate keto-PE arises from the barrier of the nonalternating pathway of chain growth being competitive to that of alternating chain growth (Figure 8).45,46,62 DFT studies starting from a five-membered chelate formed by CO insertion revealed that Ni-phosphinophenolate catalysts indeed have a ΔΔG‡alt-nonalt similar to Pd-phosphinosulfonate complexes despite differing in the nature of the rate-determining steps for both pathways.62 In the phosphinosulfonate Pd(II) system, the decisive steps are CO insertion for the alternating and ethylene insertion for the nonalternating pathway. The key steps in Ni-phosphinophenolate systems are ethylene coordination to open a six-membered chelate for the alternating, and metal–alkyl cis–trans isomerization for the nonalternating pathways. These pathways are further governed by different parameters: alternating chain growth is mostly influenced by the electronic properties of the catalyst and nonalternating chain growth by catalyst sterics, respectively.62
Figure 8.

Mechanistic steps and activation barriers of nonalternating vs alternating ethylene/CO copolymerization for Pd-phosphinosulfonate (red) and Ni-phosphinophenolate catalysts (blue) as calculated by DFT.62
Despite conveniently introducing in-chain carbonyl groups and imparting photodegradability, the keto-PE materials obtained from ethylene/CO copolymerization are not inherently susceptible to direct chemical deconstruction by simple reactions, such as solvolysis of ester groups. Nevertheless, the in-chain keto groups can provide an array of potential chemical modifications72−74 and can serve as a platform to make polyolefins more prone for subsequent chemical deconstruction75 as demonstrated recently by Baeyer–Villiger oxidation of keto-PEs to polyethylenes containing a combination of in-chain keto and ester groups.76 While HDPE-like materials properties were retained in these keto-ester-PEs, the combination of functional groups allowed for photolytic degradation via the keto groups as well as chemical deconstruction of the ester groups by methanolysis.76
In-chain ester groups can theoretically be included via catalytic chain-growth copolymerization of carbon dioxide (CO2) and ethylene (Figure 6). The utilization of CO2 as a comonomer with olefins has long been discussed77−80 but suffers from thermodynamic and kinetic limitations. Thermodynamic calculations by Miller77 and DFT studies by Nozaki78 concluded that alternating ethylene/CO2 copolymerization is thermodynamically strongly disfavored. Nonalternating copolymerization with an excess ethylene incorporation (ethylene/CO2 > 2.4), however, was found to be, in principle, thermodynamically accessible,77,80 and a generic catalytic cycle following the coordination–insertion mechanism was proposed by Müller et al.79 Nevertheless, the substantially lower activation barrier for the alternative “escape route” via ethylene homopolymerization compared to CO2 copolymerization78,79 prevents incorporation of CO2 into the polymer chain, and effective kinetic pathways are yet to be found. In ethylene polymerizations by cationic Pd(II)81 or neutral Ni(II) salicylaldiminato catalysts82−84 in supercritical carbon dioxide (scCO2) as a reaction medium, the scCO2 was found to be entirely inert. In contrast, direct catalytic conversion of ethylene and other olefins with one equivalent of CO2 to acrylates is well-documented, but this is enabled by the formation of acrylic acid salts as a driving force.85,86 These significant, if not prohibitive, hurdles for a direct in-chain CO2 incorporation can be circumvented by reaction with 1,3-butadiene to an unstable lactone intermediate. Even though polymers containing up to 30% CO2 are formed, most of these polymers contain the ester groups not as main chain links, and they do not possess PE-like chain microstructures or properties.78,80,87,88 Only recently, Tonks and co-workers reported a well-defined polyester formed via a CO2-butadiene pathway.89 The materials obtained by ring-opening polymerization (ROP) of the formed lactone intermediate, however, possessed properties of an amorphous polyester (with Tg ∼ −40 °C) rather than resembling semicrystalline polyethylene-type materials.
Nevertheless, in addition to the aforementioned free-radical polymerization approach, catalytic approaches to directly incorporate in-chain ester groups in polyethylene chains during polymerization have also been reported. Hydroesterificative polymerization of a linear, long-chain ω-unsaturated alcohol with CO can yield a linear polyester (Figure 9a, right pathway) with Mn up to 1.7 × 104 g mol–1 and Tm > 70 °C.90,91 Here, the formed cobalt- or palladium-acyl intermediate after insertion of CO is trapped by the alcohol group of the monomer to form the respective (poly)ester.92 Monomer extension of the ω-unsaturated alcohol by etherification expands the scope of accessible microstructures.93 Since hydroesterification and CO/alkene copolymerization94 share the same metal-acyl intermediate95 (Figure 9c), the pathways of hydroesterification and CO/alkene copolymerization can compete, if suitable cationic Pd-bisphosphine catalysts are chosen and a polyketoester with tunable ratios of in-chain ester and keto groups can be obtained (Figure 9b).96 As the formation of keto repeat units goes along with formation of a branch, branched microstructures are obtained: short-chain branches are formed by the nonfunctionalized comonomer (e.g., 1-hexene) and long-chain branches originate from the functional comonomer which is incorporated as keto repeat unit and then further reacted on the hydroxyl group. These branched copolymers exhibited only rather low molecular weights with high dispersities (Mn (1.5–25) × 103 g mol–1, Đ 2.4–10.2) and low melting points Tm 13–57 °C.96
Figure 9.

(a) Concept of hydroesterificative polymerization to linear polyesters90,91 and competitive alkene copolymerization.94 (b) Formation of branched polyketoesters by combining compatible pathways of linear hydroesterificative and carbonylative alkene polymerization.96 (c) Catalytic cycles of hydroesterificative and carbonylative alkene polymerization with common acyl intermediate, which allows for switching of the polymerization pathways (Figure 9c: Reprinted with permission from ACS Catal. 2022, 12, 14629–14636. Copyright © 2022, American Chemical Society).96
An alternative to provide in-chain groups that can eventually serve for deconstruction (cf. Chapter 3.2.1) is the introduction of unsaturation during polymer synthesis. This can be achieved by catalytic insertion copolymerization of butadiene, or ring-opening metathesis polymerization (ROMP) of cycloolefins (Figure 10, c). These all-hydrocarbon comonomers do not require catalysts tolerant toward heteroatom-containing monomers, and consequently also very oxophilic catalysts can be employed.
Figure 10.

General overview of different approaches to polyethylene with a low degree of in-chain unsaturation via chain-growth polymerization and post-polymerization functionalization.
Incorporation of butadiene comonomer in a 1,4-fashion in catalytic copolymerization of ethylene97−101 (or propylene102−105) can yield in-chain double bonds, with the degree of unsaturation in the polyolefin chain being controlled by monomer ratios. Terpolymerization with other α-olefins can lead to unsaturated, LLDPE-like structures.99,106,107 A variety of catalysts have been reported for ethylene butadiene copolymerization.100 Most commonly, lanthanide catalysts based on Nd97,106,108−110 or Sc101,109,111 are used for copolymerization of ethylene and butadiene. But also group IV catalysts based on titanocenes,98,112 zirconocenes,103−105,113,114 or hafnium102,113 are suitable. Even cobalt-based late transition-metal catalysts were reported.115 However, the activity of most catalysts is hampered by butadiene, and other microstructure motifs commonly occur in addition to the desired in-chain olefins. Conventional Ziegler catalysts mostly yield alternating copolymers or multiblock copolymers100,106 and therefore are not ideal for the incorporation of a low density of unsaturation. The 1,2-insertion of butadiene results in side-chain olefins. Their in situ cyclization by reinsertion has been widely reported and is used in the synthesis of ethylene–butadiene elastomers.100,106,116 Selectivity for 1,4-insertion over 1,2-insertion is required for incorporation of a low density of in-chain unsaturation.
Several catalysts have been reported to yield good 1,4-selectivity while also enabling low amounts of butadiene in the polymer without forming block-like or alternating sequences under appropriate copolymerization conditions. These are usually: low butadiene feed-ratios, suitable activator species, and activator-catalyst ratio.98,101−104,112,115,117 Silica-supported CpTiCl3 activated with triisobutyl or trioctyl aluminum can yield ethylene–butadiene copolymers with random distribution of olefin groups between 0.5 and 2.5 mol % and high molecular weights of up to 1.1 × 105 g mol–1.98 Postmetallocene single-site catalysts like [Me2Si(NtBu)(Me4Cp)]TiCl2 can also give high molecular weight copolymers (∼3 × 105 g mol–1) with adjustable degrees of in-chain unsaturation between 0.5 and 16 mol %. Especially at lower butadiene ratios, 1,4-insertion prevails (>70%) over 1,2-vinylic structures even at 16 mol % butadiene incorporation.112 Bis(arylimino)pyridyl cobalt(II) catalysts can yield ethylene-rich copolymers with prevailing 1,4-incorporation (70–97% vs 1,2) when activated with modified methylaluminoxane (MMAO), even though only low molecular weights (<1.4 × 104 g mol–1) were obtained.115 Melting points of these copolymers with low degrees of in-chain unsaturation of around approximately 100–120 °C agree with the expected behavior of a polyethylene with occasional defects in the form of unsaturation within the chain.98,112,115 Further, tetradentate [OSSO]-bis(phenolato)Ti(IV)117 and half-sandwich thiophene-fused cyclopentadienyl Sc(III) catalysts101 have also been reported for ethylene–butadiene copolymerization with exclusively 1,4-selectivity but favored incorporation of butadiene in alternating blocks, thus lacking truly polyethylene-like material properties.101,117
In addition to ethylene as comonomer, the copolymerization of butadiene and propylene with good 1,4-selectivity and low degrees of in-chain unsaturation to semicrystalline polypropylene-like polymers with high molecular weights (>5 × 104 g mol–1) and melting points >100 °C was reported for ansa-bis(indenyl)zirconocene catalysts103−105 and bridged biphenylphenoxide Hf(IV) catalysts.102
Recently, also the incorporation of “blocked” in-chain olefin groups by Pd-catalyzed copolymerization of ethylene with oxa-norbornadienes was reported. These can subsequently be unblocked by retro Diels–Alder cleavage upon heating in solution or polymer melt, resulting in unsaturated HDPE chains with up to 2.2 mol % in-chain olefins, while high molecular weights are obtained after unblocking (Mn ∼ 3 × 104 g mol–1).118
Aliphatic polymers with in-chain double bonds can also be accessed by ROMP of cyclic monomers.119−122 However, the degree of unsaturation is usually high enough that the products do not resemble polyethylenes. For example, eight-membered ring cyclooctene monomers are efficiently polymerized by ROMP, to yield products with a double bond for every sixth methylene carbon of the chain.123,124 Larger ring-size monomers on the other hand are often not readily available and require multistep syntheses, and the driving force for their ROMP is limited.121,123 The high density of internal double bonds also leads to relatively short-chain monomers when deconstructed to α,ω-telechelics, as compared to the longer-chain α,ω-telechelics accessible from olefin-butadiene copolymers. Alternatively, an unsaturated polymer obtained from ROMP can be subjected to partial hydrogenation to reduce the degree of unsaturation and therefore better resemble thermal and solid-state properties of polyethylene, as demonstrated for the ROMP of cyclopentene.125 The obtained polymer could be partially hydrogenated by employing variable amounts of p-toluenesulfonyhydrazide to polyethylenes with tailored degrees of unsaturation.125 Similarly, partial hydrogenation of amorphous and highly unsaturated 1,4-polybutadiene homopolymer with [(Ph3P)3RhCl] can give polyethylene-like polymers with varying degrees of unsaturation between 2.6 and 16 mol %, with crystallinity and melting points strongly dependent on the amount of residual in-chain olefins (Tm: 78–118 °C).126
Another possibility arising from ROMP is the direct synthesis of long-chain α,ω-telechelic molecules. Instead of cleaving an unsaturated polymer by oxidation or metathesis, the ROMP is carried out in the presence of a functionalized acyclic alkene, which acts as chain transfer agent (CTA).127 An approach to access such α,ω-telechelics has been explored extensively by Grubbs and Hillmyer.124,128−134 Carboxyl-functionalized telechelic long-chain monomers can be precisely synthesized by employing an unsaturated diacid, such as maleic acid.132 For the synthesis of hydroxyl-terminated telechelic molecules, diacetyl alkenes are used as CTA in ROMP and converted to the free alcohol by hydrolysis.129,131,133 Recently, Hillmyer and co-workers reported the direct synthesis of a hydroxyl-terminated α,ω-telechelic molecule with 7-hexadecene-1,16-diol as CTA.134 Hydrogenation of the unsaturated α,ω-telechelic molecules leads to saturated PE-like segments,132−134 which can be used as long-chain monomers for polycondensation reactions to recyclable PE-like polymers. Particularly, long-chain α,ω-hydroxy or carboxy telechelic molecules offer potential in view of application as long-chain building blocks toward recyclable polyethylene-like polymers (Figure 11), as demonstrated for linear, HDPE-type135,136 as well as branched, therefore rather LLDPE-like building blocks.136
Figure 11.

Synthesis concept of long-chain α,ω-carboxy telechelics from CTA-ROMP as PE-like building blocks.132,135
2.2. Step-Growth Polymers
An alternative to construction of polyethylene-type materials with in-chain functional groups by chain-growth copolymerizations is the synthesis of such materials via polymerization reactions of these very functional groups (Figure 4, right). An illustrative example is the A2 + B2 polyesterification of long-chain dicarboxylic acids, A-(CH2)n-A (A = carboxylate) with long-chain diols B-(CH2)m-B (B = hydroxyl group). This concept requires straightforward access to suitable long-chain monomers, which is provided by state-of-the-art catalytic conversions of suitable long-chain substrates, primarily unsaturated fatty acids.
2.2.1. Access to Monomers
Fatty acids offer themselves as starting materials for the synthesis of long-chain difunctional monomers as they already provide long, linear methylene sequences endowed with a terminal carboxylate group.
Self-metathesis of unsaturated fatty acids or their ester analogs yields a stoichiometric mixture of internally unsaturated alkenes and α,ω-dicarboxylates (Figure 12a). Catalytic double-bond hydrogenation of the latter affords the target long-chain saturated dicarboxylate monomers, e.g., C18 octadecanedioate from oleate feedstocks or C26 from erucates. Molecular catalysts, like Grubbs II ruthenium alkylidene or molybdenum catalysts, are suitable for olefin metathesis of plant oil feedstocks including extensively produced crops like soy bean or palm oil to generate the α,ω-dicarboxylates.137,138 Elevance (now Wilmar) has commercialized olefin metathesis of fatty acids in a large-scale biorefinery in Indonesia since 2013, and offers C18 dicarboxylates commercially, in a quality suitable for polycondensation.139,140
Figure 12.

Schematic diagram for the synthesis of long-chain monomers from fatty acids via self-metathesis, isomerizing alkoxycarbonylation, and ω-oxidation.
Isomerizing alkoxycarbonylation (Figure 12c) of unsaturated fatty acid esters generates a second ester group, at the other terminus of the fatty acid substrate, from reaction of the internal double bond with carbon monoxide and methanol or another alcohol.141,142 In order to generate the new ester group not at the original double bond position in the center of the fatty acid chain but at the terminus, catalysts are required that “walk” back and forth along the fatty acid chain easily and selectively undergo ester formation only when at the terminal position.143 This can be achieved with Pd(II) catalysts with bulky substituted diphosphines like the commercially available 1,2-bis(di-tert-butylphosphino)xylene.144,145 By this kinetic control, selectivities up to 95% at virtually complete conversions have been achieved.146 Unlike olefin metathesis, the entire fatty acid substrate is incorporated in the product. As the second carbonyl group originates from the carbon monoxide substrate, odd-numbered products are formed, e.g., C19 from high oleic sunflower oil or C23 from erucic feedstock.147 Formates such as methyl formate can act as CO surrogates in this process to produce CO in situ, alleviating the need for a CO supply infrastructure.148,149 Isomerizing carbonylation has been demonstrated on a laboratory scale (up to ca. 0.3 kg), yielding long-chain diesters or dicarboxylic acids (from hydroxycarbonylation with water) in polycondensation-grade quality.141
The aforementioned reactions yield long-chain difunctional monomers of similar or equal chain length as the fatty acid substrate. Ultra long-chain difunctional linear products can be generated through “chain doubling” by combination of dynamic catalytic isomerizing crystallization with olefin metathesis. This yields, for example, a C48 linear diester as polyethylene-like telechelic from erucate.150
In addition to the aforementioned carboxylates, long-chain diols are also attractive monomers for the generation of polyethylene-type materials. They are accessible from the α,ω-diesters via reduction with metal hydrides or ruthenium-catalyzed reduction with molecular hydrogen.141,151,152
As an alternative to the catalytic upgrading of oils, biotechnological transformation of saturated or unsaturated fatty acids, or alkanes, allows for an oxidation of the terminal methyl carbon. The ω-oxidation of C14 to C22 fatty acids was enabled by the selective blocking of specific parts of the β-oxidation pathway of different yeast strains.153 The target monomers could be generated in concentrations of up to 160 g L–1 with high substrate conversion efficiency. The need for additional nutrients and a rather complex workup procedure of fermentation broths are limitations of this process. ω-Oxidation was developed in the 1990s by Henkel (now Emery Oleochemicals) to produce C18 diacid, among others, on a pilot scale.154 Today, Cathay Industrial Biotech155 also offers C14 and higher chain length dicarboxylic acids sourced from fermentation.
By blocking the β-oxidation pathway even further upstream, unsymmetrical ω-hydroxy fatty acids are also accessible.156 These can be polymerized via step-growth AB polycondensation to linear polyhydroxyalkanoates (PHAs).157
AB-type long-chain polyesters can also be obtained by ROP of large ring-size lactones.158−160 Pentadecalactone is the most studied large ring lactone used for ROP. It is used in the fragrance industry and produced in a five-step synthesis industrially from cyclododecanone.161 This route comprises (i) the addition of allylic alcohol, followed by (ii) a ring-closure, (iii) oxidation with H2O2, (iv) decomposition of the hydroperoxide to an ester, and finally (v) hydrogenation. Though ROP is a chain-growth-type reaction, extensive transesterification can occur during the entropy-driven ROP of large rings with both enzymes and synthetic small-molecule catalysts. This results in product microstructures similar to those from traditional step-growth polyesterification in terms of molecular weight distributions (Đ ∼ 2) and random monomer sequences in case of ring-opening copolymerizations of several lactones.162 Given the similarities of these linear AB-type long-chain polyesters to A2B2 polyesters and the effort required for the synthesis of large ring lactones,163,164 their utility as monomers compared to long-chain dicarboxylates and diols appears limited.
2.2.2. Long-Chain Polycondensates
Polycondensation of the aforementioned long-chain monomers can yield linear aliphatic chains with different in-chain functional groups, polyesters being a prominent example (Figure 13). In these step-growth reactions, high functional group conversions and sufficiently pure monomers are a prerequisite for achieving reasonable molecular weights. In polymerization protocols employing a long-chain dicarboxylic acid or ester in combination with a short-chain diol, the latter is used in excess and removed during the polycondensation along with the more volatile condensate (alcohol or water) liberated from esterification (e.g., final conditions 200 °C, vacuum of 0.1 mbar).165−167 By contrast, nonvolatile diols need to be employed in an exact stoichiometric ratio in the initial reaction mixture. On the other hand, the removal of the volatile condensates is less demanding compared to the removal of the excess short-chain diol.64,168,169
Figure 13.

(a) Polymer synthesis of polyesters and polycarbonates via polycondensation. (b) Comparison of differential scanning calorimetry (DSC) traces of polyesters with different chain lengths (C18, C26, C48) between functional groups. (c) Comparison of DSC traces of polycarbonates with different chain lengths (C18, C26, C48) between functional groups.150,168,172,173
Notwithstanding, both approaches can yield polyesters with molecular weights in the range of typical commercial polyethylenes, as illustrated in Figure 14b for polyester-18,18 vs an injection molding grade HDPE. The low density of in-chain ester groups does not disturb crystallization of the chains in an orthorhombic, HDPE-like crystal structure (Figure 14a). Consequently, the modulus, ductility, and toughness of injection-molded specimens are similar to the HDPE reference material (Figure 14c,d).168 While the ester groups are preferentially located in the amorphous phase, they can also be incorporated into the polyethylene crystal (Figure 14e).170,171 This goes along with an energy penalty, which results in a reduced melting point compared to linear polyethylene, the extent depending on the ester group density (e.g., Tm’s of PE-18,18: 99 °C; PE-26,26: 113 °C; PE-48,48: 120 °C vs HDPE: 135 °C; Figure 13b).64
Figure 14.

(a) WAXS of PC-18, PE-18,18, and HDPE, reflexes correspond to the orthorhombic unit cell. (b) SEC traces of PE-18,18, PC-18 in comparison to commercial HDPE. (c) Stress–strain curves of PE-18,18, PC-18, and HDPE. (d) Comparison of Young’s moduli and stress at yield values for PE-18,18, PC-18, and HDPE. (e) Schematic representation of the solid-state structure of HDPE (top) and PE-like polymer (bottom) crystallites (Adapted with permission from Nature2021, 590, 423–437. Copyright © 2021, Springer Nature).168
Polyesters from A2 + B2 polycondensation feature precise methylene spacings between the ester groups as given by the choice of monomers. This enables the formation of layers of ester groups in the crystal, subject to dipole interactions between adjacent segments (Figure 15b). For energetically favorable combinations of methylene spacings, for example, combination of even-numbered dicarboxylates with even-number diols, the dipoles of carbonyl groups in adjacent chain segments in the crystal align in opposite directions, effectively canceling the local polarization. The resulting favorable interactions in part alleviate the energy penalty caused by inclusion of ester groups into the crystalline lattice. This compensation is then reflected in an increased melting point compared to a random arrangement of the functional groups or less suitable combinations of methylene spacings. Consequently, pronounced odd–even effects are observed in A2B2 polyesters.64
Figure 15.

(a) Melting points of randomly long-spaced polyketones (red),63 polyesters (green),175 and polycarbonates (blue)176 vs their density of functional groups (reprinted with permission from ACS Macro Lett. 2015, 4, 704–707. Copyright © 2015, American Chemical Society). (b) Molecular and supramolecular structure of PE-22,4 according to SAXS and NMR spectroscopy (reprinted with permission from Macromolecules2007, 40, 8714–8725. Copyright © 2007, American Chemical Society).170
Also, long-chain polyesters from mixtures of dicarboxylate monomers of variable length can adopt polyethylene-like structures. Odd–even effects are absent in these polyesters as the irregular spacing of ester groups in the chain hinders favorable dipole alignment, and their melting points are similar to those of the “mismatched” single-length monomer polyesters from odd-numbered dicarboxylates. The Tm of these “multiplechain length” polyesters increase with a higher center of the monomer-chain length distribution, regardless of the center of the distribution being even or odd. Such polyesters from dicarboxylate mixtures are also of interest as the latter can be potentially sourced from low-value biomass or plastic waste (section 3.2.2).174
For a combination of an (even-numbered) long-chain dicarboxylate, with the particularly short C2 ethylene glycol, relatively high melting points are observed which may be due to the entire ethylene glycol monomer unit acting as single defect in the crystal (Tm PE-2,18: 96 °C; Tm PE-3,18: 82 °C; Tm PE-4,18: 86 °C; Tm PE-18,18: 99 °C).167 At the same time, ethylene glycol is available on a large scale and at lower cost than long-chain diols, and an increased density of ester bonds leads to further desirable properties, such as an increased environmental degradability (section 3.1).165,167,174
The relationship between randomly placed ester, carbonate, and keto group density on polymer properties has been mapped out using hydrogenated model polymers from ADMET. This enables a convenient variation of the functional group density via the ratio of a functional group-containing and a pure hydrocarbon α,ω-diene comonomer (Figure 15a).63,171,175,176 A decrease in polymer Tm’s with increasing ester, carbonate, and keto density and with decreasing dipole moment of the functional group from keto to ester to carbonate is instructive.
Studies on copolymers from varying ratios of pentadecalactone (PDL) and caprolactone (CL) revealed further correlations with the crystal structure of the polyesters. Despite a lowering of the melting point and enthalpy of fusion with an increased random incorporation of ester groups (via a higher ratio of CL), the crystallinity of the polymers and also the crystal thickness remained constant over a range of compositions. However, the increasingly irregular stacking of the ester groups resulted in an increased mobility of the chains in the crystallites and lower energy barriers for their deformation, reflected by a significant lowering of the stress at yield for a polymer with equal molar ratios of PDL and CL.162
Analogous to PE, the crystallinity of the PE-type polymers can be influenced by the introduction of side chains. Duchateau et al. attached C5– or C6–OH chains via radical thiol–ene chemistry onto unsaturated macrolactones. The polymerization of these branched monomers yielded an LLDPE-type material with depressed crystallinity and melting points and LLDPE-like mechanical properties.177
Barrier properties against water or gases such as oxygen are relevant among others in food packaging applications. The water and gas barrier properties strongly depend on the hydrophobicity and the crystallinity of the material, respectively. Consequently, the gas or oxygen barrier properties of crystalline polyesters like PE-2,16 are favorably similar to HDPE, whereas the water barrier is significantly lower. However, the water barrier is still significantly improved compared to poly(butylene adipate terephthalate), an important commercial biodegradable polyester.165,178 While the hydrophobicity of PE is beneficial for barrier properties, it also leads to a low compatibility and thus low adhesion with most other materials. The in-chain ester groups slightly increase the surface free energy of the material,167 enabling the adhesion of hydrophilic ink after printing on PE-18,18 films compared to HDPE films. This effect can be enhanced by the introduction of small amounts of ionic groups.179
In addition to the polyesters outlined, long-chain polycondensates with numerous other in-chain functional groups have been studied, often motivated by the possibility for deconstruction in recycling or biodegradation.
During the polycondensation of long-chain diols to polycarbonates, diethyl carbonate as a reagent can increase the reactivity of end groups in the critical late stages of the reaction by release of CO2 and ethylene to more reactive −OH groups, which has proved beneficial to building up molecular weight. So obtained polycarbonate-18 (Mn = 90 kg mol–1, Mw = 300 kg mol–1) exhibits a PE-like solid-state structure and mechanical properties (Young’s modulus, tensile strength, and ductility) comparable to commercial HDPE (Figure 14). The inclusion of the carbonate groups into PE crystallites goes along with a larger energy penalty compared to polyesters (Figure 15a), which results in somewhat lower melting points compared to polyesters (cf. PC-18: 88 °C vs PE-18,18: 99 °C).168,176 Notwithstanding, with sufficiently long methylene sequences also objects from aliphatic polycarbonate material can be made that are not distorted at boiling water temperatures (PC-48: Tm 113 °C).168
Long-chain aliphatic polyamides (PAs) have also been extensively studied; the motivation for these studies was the lower water uptake, higher dimensional stability, and lower melting points of these PAs compared to traditional short-chain polyamides like Nylon-6,6 (Tm PA-6,6: 265 °C,180Tm PA-18,18: 163 °C181). However, hydrogen bonding between the amide groups dominates their properties, and only for very low amide group densities a polyethylene-like structure emerges.64,149,182,183
Numerous hydrolytically labile groups have been investigated as part of polymers with long methylene sequences between these functional groups. However, the size and conformational preferences of the explored acetal,176 orthoester,184 Vitamin C,185 and numerous phosphorus-containing groups (H-phosphonates,186,187 phenylphosphonates,186,187 phosphoesters,188−190 or pyrophosphates191) impair the formation of a typical orthorhombic polyethylene crystal structure and in this sense compromise a “polyethylene-type” nature of these polymers unless very long spacers (>ca. C40) between the functional groups are employed. Apart from a few notable exceptions,186,188 mostly brittle materials were reported which may in part also be due to the practical difficulties in determining the tensile properties of rapidly hydrolyzing materials. Much of the understanding of these polymers was enabled by ADMET polymerization of corresponding monomers, developed by Wurm et al.185,188−191 However, long-chain poly(H-phosphonates) could also be obtained by straightforward base-catalyzed polycondensation of (ultra)long-chain diols with dimethylphosphonate,186 and polyacetals were generated by “acetal metathesis”192 from long-chain diacetals.176,193 Notably, as blend components such long-chain polymers can be incorporated into a polyethylene-like morphology, likely by cocrystallization, and enhance hydrolytic degradability (Section 3.1).
3. Deconstruction
Deconstruction of PE-type materials can proceed via predetermined breaking points incorporated during its chain-growth or step-growth synthesis, as outlined in the previous sections. This enables a closed-loop chemical recycling to the monomers and, in principle, can alleviate the problematic persistence of mismanaged plastic waste by enabling environmental deconstruction through biodegradation (cf. section 3.1). When such breaking points are not present, waste PE may also be recycled by reaction sequences, often starting with dehydrogenation or oxidation (cf. section 3.2).
3.1. Via Predetermined Breaking Points
Deconstruction to their monomer building blocks via solvolysis was demonstrated for the PE-type polymers PE-18,18 or PC-18 (Figure 13) with methanol or ethanol at 120 to 150 °C, optionally accelerated by KOH as catalyst. The C18 diester and C18 diol monomers could be recovered in virtually quantitative yield and high purity from mixtures of the respective polymers with polypropylene and HDPE, also separating additives like dyes or reinforcing fibers. Recycled polymer generated from the recovered monomers possesses the same mechanical properties as the virgin polymer, demonstrating the feasibility of a closed-loop recycling of HDPE-like materials (Figure 16).168
Figure 16.

Chemical recycling of PC-18 (Adapted with permission from Nature2021, 590, 423–437. Copyright © 2021, Springer Nature).168
The similar physical properties such as melting temperature and solubility of the long-chain diol and long-chain diester lead to crystallization of a uniform monomer mixture during recycling of the polyester. The stoichiometric monomer mixture recovered is suitable directly for further polymerization. However, a straightforward separation of the two monomers, if desired, appears challenging.168 This could be overcome by short–long chain polyesters, like PE-2,18, whose monomers substantially differ in their physical properties. The isolation of these monomers after solvolysis can be achieved by selective crystallization of the long-chain diacid, and it is further aided by the volatility of the ethylene glycol component.167
The depolymerization and repolymerization of PE-type polyesters was found to be robust, as functional groups such as ionic groups179 and thioether groups194 or furan-containing monomers195 did not interfere with the depolymerization process. Even mixtures of diacids could be recovered upon depolymerization in the same ratio as used in the initial polycondensation.174 Also, telechelics with high molar masses in the range of several kg mol–1 could be de- and repolymerized as described above.102,136
The industrial feasibility of solvolysis processes is underlined by the industrial methodology developed for chemical recycling of poly(ethylene terephthalate) (PET). Glycolysis, hydrolysis, or methanolysis at 180 to 250 °C yields terephthalic acid or its esters that can serve as monomers for the polycondensation to PET. Polymer produced in this way meets the stringent requirements of, e.g., food contact materials.196 Life-cycle assessments (LCAs) show that the carbon footprint and environmental impact of mechanically and chemically recycled PET is significantly lower than that of PET from nonrecycled resources and, also, comparable to the footprint of energy recovery by incineration.3,197 The main contributors to the life cycle impacts of chemical recycling of PET are the high energy input for depolymerization caused by the high melting point and glass transition temperature of PET, the shredding to flakes, and the use of sodium hydroxide as catalyst.197,198 By comparison, the lower melting point of polyethylene-type materials is reflected in milder conditions required for solvolysis (120 to 150 °C). In fact, polyethylene-like polyesters could be deconstructed completely to pure monomers in the presence of PET, the latter remaining intact at the lower operating temperatures.167,168,199
The chemical recycling of PE-type polymers by solvolysis extends to further in-chain functional groups. Johnson and Johnson recently reported recycling of a material with bifunctional silyl ether groups in a polyethylene chain (generated through a ROMP copolymerization followed by hydrogenation) by reaction with an alcohol.200
Although not exactly addressing a polyethylene-like material, it is notable that Greiner and Rist also demonstrated the chemical recycling of a long-chain polyamide, PA-6,19, by hydrolysis.149 The monomers could be recovered in a quality suitable for repolymerization into a material with mechanical properties akin to the virgin material.
In general terms, an alternative to chemical recycling via solvolysis that also relies on largely thermoneutral reactions is polymer-cyclic monomer equilibria. The entropy-driven deconstruction of polyamide-6 to ε-caprolactam is applied industrially.201 Materials that can be recycled via cyclic acetals and in fact possess tensile properties similar to polyethylene were reported by Coates et al.202,203 Thermally stable, processable α-disubstituted polybutyrate can also be recycled via a four-membered lactone.204 For the polyethylene-type materials reviewed here with low densities of functional groups in the chain and consequently relatively long methylene segments between functional groups, this would translate to the formation of relatively large rings. The temperature dependency of such polymer-cyclic monomer equilibria is generally less favorable, which also complicates the synthesis of large ring monomers (Section 2.2).
In the processing of postconsumer plastic waste streams, sorting errors205 and virtually inseparable multilayer multicomponent materials206 can lead to mixed material streams, in particular destructive for chlorinated polymers.207 Most polymers, even if relatively similar in their chemical structure, are immiscible.208 Such contaminations will result in phase separation during the processing of mechanically recycled polymers. This can result in poor mechanical properties, as prominently exemplified by PE and iPP mixtures.209 Compatibilization by small amounts of block copolymers can alleviate this problem.210
Consequently, polyethylene is immiscible with PE-like polyesters such as PE-19,19, PE-23,23,169 and PE-15,211 as evidenced by observation of the unaltered melting points of the individual constituents by DSC of blends. However, a good compatibility between HDPE or LDPE and PE-15 was demonstrated by Duchateau et al. using scanning electron microscopy, transmission electron microscopy, and X-ray studies. Epitaxial crystallization from the PE-15 lamellae onto existing HDPE and LDPE lamellae resulted in a fractured surface of the domains, indicating a strong adhesion between the polyester and PE domains. Consequently, clear, ductile films could be prepared from blends of LDPE and PE-15 that crystallized in shish-kebab morphology at higher ratios of PE-15.211 The mechanical properties of HDPE were not adversely affected by blending with PE-18,18.168 This suggests that a conceivable contamination of polyethylene waste streams with long-chain polyesters during mechanical recycling would not be detrimental. A complete separation of blends of HDPE and a PE-like polyester was also feasible via a selective solvolysis of the latter’s in-chain functionalities, as was shown for HDPE and PE-18,18.168
In principle, in-chain ester or other hydrolyzable groups in a polyethylene chain can also enable biodegradation. This can be of interest for particular applications that inherently require biodegradability like compostable packaging or mulch films. More universally, it may provide a backstop for plastic waste littered to the natural environment instead of being delivered to its proper end of life receiving environment. This might alleviate the problematic environmental persistency of polyolefins.19 Note that while clear standards for compostability exist, there is no definition of what constitutes a material nonpersisting in the environment.
The rate-limiting step of biodegradation is usually the hydrolytic breakdown of the polymer chains by enzymes.212,213 Compared to less crystalline polyesters from short-chain monomers, enzymatic or abiotic hydrolysis of HDPE-like materials is expected to be hindered by their crystalline and hydrophobic nature. Indeed, no degradation of PE-15 was observed upon exposure to a buffered lipase solution for 100 days.214 PE-18,18 was found to be unaffected by a one-year exposure to dilute hydrochloric acid as observed by monitoring of sample mass and molecular weight via NMR spectroscopy and SEC (size exclusion chromatography).168 In contrast, the short–long-type polyester PE-2,18 was found to be degraded by naturally occurring enzymes to the monomers at 37 °C in vitro and is fully compostable under industrial composting conditions (Figure 17).167 The melting points of polyesters are generally accepted as an indicative factor for degradability.215,216 The accelerated degradation of PE-2,18 compared to PE-18,18, despite both polymers exhibiting nearly the same melting point, may be due to the increased ester-bond density or the higher solubility of the monomers of PE-2,18 compared to the water-insoluble C18 diol formed upon hydrolysis of PE-18,18.
Figure 17.

Mineralization of PE-2,18 and cellulose based on CO2 evolution under industrial composting conditions at 58 °C following ISO 14855 (reprinted with permission from Angew. Chem., Int. Ed. 2023, 62, e202213438. Copyright © 2023, Wiley-VCH Verlag GmbH & Co. KGaA).167
More hydrolytically labile in-chain groups in long-chain poly-H-phosphonates,186 polyorthoesters,184 polyphosphoesters,189 polyphosphoorthoesters,217 and polypyrophosphates191 can strongly enhance degradation. For example, poly-H-phosphonate-19 degrades completely to the C19 diol and phosphoric acid in 2 days upon exposure to water or moist air. However, the listed functional groups disturb crystallization and hinder adoption of a polyethylene-like solid-state structure of the neat polymer. Notwithstanding, their long-chain structure renders poly-H-phosphonates compatible with PE-18,18 in blends. Upon immersion of these HDPE-like blends in water, the minor hydrolytically labile component degrades completely over the course of months and the liberated phosphoric acid also initiates hydrolysis of the PE-18,18 matrix, resulting in embrittlement and significant molecular weight decrease.218
Photolytically cleavable breaking points are another approach toward nonpersistent polymers. Exposure of PE-like keto-polyethylenes from catalytic and free-radical ethylene-CO copolymerization to (simulated) sunlight resulted in a clear onset of degradation, as evidenced by embrittlement, mass loss, and substantial decrease of molecular weight.32,34,35,60,66 The amount of keto groups decreases due to Norrish-type chain scission,219,220 yet ca. half of the originally present in-chain keto groups remain in those experiments and can promote further degradation. In addition, new carbonyl groups form by accelerated hydrocarbon oxidation in these keto-PEs, which was not observed for an HDPE reference sample within the same period of time.60
3.2. Post-Use Functionalization
Well over a billion metric tons of chemically inert PE materials have already exceeded their use lifetime, and of the ca. 100 million tons of virgin PE produced annually only a small portion is mechanically recycled.221 Thus, waste PE is an abundant feedstock for recycling by chemical methods and also for conversion to other valuable products and chemical intermediates. Substantial efforts for conversion of PE waste into fuel-like hydrocarbon mixtures by hydrogenolytic cracking of the hydrocarbon backbone into smaller fragments have been reported recently.222−228 Similarly, alkane cross metathesis between long- and short-chain alkanes can yield fuel or wax-like products.229,230 Here, combination of alkane dehydrogenation and olefin metathesis leads to the desired linear alkane mixtures, either by a tandem catalysis with an Ir-catalyzed dehydrogenation and a Re2O7 olefin metathesis catalyst229 or by a single-site, W-catalyst immobilized on silica, which can catalyze both reactions.230 All these approaches of hydrogenolytic cracking or alkane metathesis, however, lead to hydrocarbon mixtures, usually suitable for fuel or naphtha applications and therefore limited potential implementation into a circular plastic economy, where a direct reuse of deconstructed intermediates is targeted.
3.2.1. Via Unsaturation
On the other hand, the introduction of in-chain double bonds by dehydrogenation can give a valuable platform for more selective chemical deconstruction of waste PE. The obtained in-chain double bonds in these polymers obtained by dehydrogenation of PE chains or also from previous introduction during polymer synthesis (chapter 2.1) can be used as a platform to further chemically modify and eventually deconstruct the polymer (Figure 18). The latter can be achieved by ethenolysis to α,ω-divinyl telechelics104,105,113,126,231−234 or cross metathesis with various functional molecules to form long-chain α,ω-telechelic molecules, for example, long-chain diesters or diols.98,102,110,113,118,235,236 These can be used as (macro)monomers for polyester or polycarbonate synthesis, thus possibly reintroducing PE waste to a circular economy. For example, Boisson and co-workers synthesized long-chain α,ω-carboxylic acids by oxidation with KMnO498 and long-chain α,ω-diols by metathetical depolymerization110 from ethylene–butadiene copolymers. Coates et al. applied these concepts to unsaturated propylene-butadiene copolymers, which were cleaved via cross metathesis with acrylates and subsequently repolymerized by polycondensation. The so-obtained ester-linked polypropylene showed properties similar to LLDPE and could be recycled chemically.102 Further, the cross metathesis of unsaturated polyethylenes obtained from dehydrogenation237 or “blocked” olefins118 with acrylates could yield long-chain ester monomers, which were repolymerized by polycondensation to HDPE-like materials (Figure 19).118,237
Figure 18.

Deconstruction of polyethylene materials via in-chain unsaturation.
Figure 19.

Transforming waste polyethylene to recyclable materials with HDPE-like mechanical properties via consecutive dehydrogenation and cross metathesis (Adapted from J. Am. Chem. Soc. 2022, 144, 51, 23280–23285. Copyright © 2022, American Chemical Society).237
The postuse dehydrogenation of PE chains is usually carried out using Ir-based pincer-type catalysts229,237−245 which can lead to a random introduction of unsaturation of up to 3.2 mol %.241 These catalysts operate at ca. 200 °C, which facilitates the conversion of the challenging polymeric substrate. Polyethylene is hardly soluble in any solvent at ambient conditions, which can be overcome by high temperatures, above the melting point of polyethylene. The length of the obtained α,ω-telechelic segments after ethenolysis or cross metathesis can be controlled by the amount of unsaturation/dehydrogenation, and segment lengths between approximately 1 and 4 kg mol–1 have been reported.237 As an alternative to direct catalytic dehydrogenation, the reaction of HDPE with Br2 and subsequent elimination of HBr to unsaturated PE chains was reported. Ethenolysis of these unsaturated PE chains resulted in α,ω-divinyl-oligomers with a length of 0.7–0.9 kg mol–1.232
The even further deconstruction of PE waste to small platform molecules like ethylene or propylene could enable complete circularity without limiting the products to α,ω-telechelic macromonomers for polymer synthesis only. The chemical recycling of PE back to its ethylene monomer suffers from high energy demands and low monomer selectivity (under 10% effective yields as ethylene).4,17 Recently, Hartwig241 and Scott and Guironnet240,246 independently reported the breakdown of waste PE to propylene in highly efficient yields of up to 80%,241 by using a combination of Ir-catalyzed dehydrogenation and subsequent tandem-catalytic isomerizing ethenolysis with Pd-isomerization and Hoveyda-Grubbs241/Ultracat240 metathesis catalysts (Figure 20). The driving force for this process, which comprises largely energetically neutral conversions, is the utilization of ethylene as a reagent, which also is the origin of two-thirds of the carbon atoms of the formed propylene molecules. Challenges to overcome are the limited temperature stability, and consequently productivity, of the isomerization and (particularly) the soluble, molecular metathesis catalyst (ca. 80 turnovers), with the reaction temperature being dictated by the need to solubilize the polyethylene substrate rather than optimum conditions of the catalysts. To this end, heterogeneous catalysts are potentially more viable in terms of applicability.240
Figure 20.

(a) Concept of transforming waste polyethylene to propylene as a valuable chemical building block (Reprinted with permission from J. Am. Chem. Soc. 2022, 144, 18526–18531. Copyright © 2022, American Chemical Society).240 (b) Converting waste polyethylene to propylene by dehydrogenation and subsequent tandem isomerizing ethenolysis (Reprinted with permission from Science2022, 377, 1561–1566. Copyright © 2022, The American Association for the Advancement of Science).241
3.2.2. Oxidation
A valorization of HDPE waste via oxidation has been suggested already in the very early days of polyolefin technology (Figure 21). Notably, α,ω-dicarboxylic acids (DCAs) can be produced from HDPE by a variety of such oxidative processes (Figure 22). These products may serve as useful intermediates for consumer products, polymers,174,247 or as biological feedstocks.248,249
Figure 21.

Header of H. Alter’s 1960 article proposing waste PE as a valuable resource (reprinted with permission from Ind. Eng. Chem. 1960, 52, 121–124. Copyright 2022, American Chemical Society).250
Figure 22.

Recent developments and applications of catalytic oxidation processes for the conversion of high-density polyethylene to mixtures of dicarboxylic acids.
Oxidation of PE by hot fuming nitric acid was first developed as a means to isolate crystalline lamellae for the purpose of PE structural elucidation.251 Under longer reaction times, PE can be further oxidized, yielding long-chain nitro-carboxylic acids which can be subsequently defunctionalized to give paraffins252 or, selectively, DCAs.253 The oxidation of PE using nitric acid receives ongoing attention to obtain DCAs as high-value chemicals,254 for instance, as monomers to produce polyester acrylates.255 With this process, recoveries of C3–C9 DCAs up to 35 mol % have been achieved after reacting PE with nitric acid (≤7 wt. eq ) for 3 h at 180 °C (Figure 22a).255,256
PE can also be oxidized with only gaseous reactants (i.e., nitric oxide/oxygen), also selectively yielding short-chain (C4–C7) DCAs in recoveries up to 73 wt % after 16 h reaction at 170 °C.257 Mild conditions (i.e., room temperature) can also be employed for PE oxidation via sulfonation and subsequent grafting of Fe(III) to PE, enabling Fenton degradation yielding C4 DCA and a range of (mono)carboxylic acids.258 Later, metal-catalyzed autoxidation of PE, inspired by the industrial process of p-xylene oxidation to yield terephthalic acid,259 was developed as a preferrable method for the formation of functionalized compounds from polyethylene, especially DCAs.250 As such, the state-of-the-art is a cobalt/manganese cocatalyst system with bromide acting as initiator. More recently, N-hydroxyimides such as N-hydroxyphthalimide (NHPI) have been explored as a less corrosive alternative to bromide.260 The utilization of ≥10 wt % NHPI with a cocatalyst system of Co(II) and Mn(II) (added at 10 wt % each compared to PE) under O2 pressure and temperatures of 150–160 °C has been shown to produce DCAs in the range of C4–C20 with 20–40% yields (Figure 22b).249,261 Broader distributions of DCAs (up to C34) produced without the application of metal catalysts have been observed; however, further oxidized keto-DCAs are produced at the same time.262 Furthermore, different conditions (such as oxidation of aqueous PE dispersions)263 or catalyst systems (such as Ru(III)/TiO2)264 have been applied to yield dicarboxylic acid oils (i.e., long dicarboxylate chains with Mw in the range of 500–2000 g mol–1) (Figure 22c). Recent work has also explored the initial feasibility of the deconstruction of HDPE to low molecular weight, oxidized products such as short-chain ketones and carboxylic acids by applying oxidases isolated from wax moth larvae Galleria mellonella(265) or from Rhodococcus spp. bacteria.266
While the oxidation of PE to DCAs has been well-studied, the purification and isolation and the utilization of the resulting products is relatively underexplored to date. Polyesters generated from C4 to C13 DCA mixtures (from nitric acid oxidation of PE) and 1,4-butandiol or 1,6-hexandiol, respectively, were employed as macrodiols in the production of thermoplastic polyurethane elastomers.267 Note this does not require as high a DCA monomer purity as a generation of high molecular weight thermoplastic linear polyesters. Carboxylates from PE oxidation have also been studied as surfactants.268
Another key application for multiple chain length DCAs is their direct utilization as biological feedstocks. The bioconversion of PE waste products to PHA was first investigated using PE pyrolysis waxes as feedstocks,269 obtained via oxygenation (and ozonation),270 to convert low molecular weight PE to paraffins. The shift to transition-metal-catalyzed oxidation yielding a fatty acid mixture, rather than paraffin waxes, was shown to improve their bioconversion efficiency due to improved solubility.248 In fact, transition-metal catalysts have long been of interest for the process of paraffin oxidation.271 This oxidation strategy has been applied directly to both isolated PE (with DCA yields up to ca. 50 mol %) and PE as part of a simulated mixed-waste stream with other polymers such as polystyrene and PET to produce DCAs.249,261 DCAs could then be used as sole carbon source for bioengineered strains of Pseudomonas putida, a Gram-negative soil bacterium, for production of PHAs,249 or Aspergillus nidulans, a filamentous fungi, to produce fungal secondary metabolites, which can be used in medical applications.261 In the latter case, as short-chain DCAs (<C10) were found to be toxic to select fungal species, they were separated from long-chain DCAs (≥C10) using a pH-controlled liquid–liquid extraction.261 Notably, recent applications of oxidized PE waste to natural microbial consortia, sampled from plastic recycling plants, demonstrated the possibility to convert these products to wax esters as a valorization product, without the need for specific bioengineering.272
It is notable that the chemistry underlying the targeted catalytic oxidation processes has similarities to pro-oxidant additives to PE, used with the aim of catalyzing their inherent oxidation in the environment (so-called “oxo-degradable PEs”). For example, organic salts of Co(III), Mn(II), Fe(III), and Ni(II) have been applied to PE formulations for single-use products.273 Other additives used in PE consumer products such as TiO2 also inadvertently show photooxidative activity.274 While enhanced oxidation of PE results in or accelerates molecular weight decreases and fragmentation, the evidence of subsequent complete biodegradability of such materials remains unproven.273 For this and other reasons, “oxo-degradable” plastics have been banned for single-use products by the European Union.275 Further assessments of the environmental biodegradability of such materials would require long-term tracking of mineralization of PE-carbon to CO2 or assimilation of PE-carbon into microbial biomass, which could be unequivocally verified using stable-isotope labeled materials.276
4. Conclusions
Compared to unselective breakdown of polyolefin chains by pyrolysis or catalytic cracking at high temperatures to hydrocarbon mixtures, a deconstruction via specific break points can enable a selective chemical recycling to monomers that can be repolymerized to polyolefin-type polymers. Key steps of this deconstruction, as well as synthesis of dedicated monomers and the corresponding construction of polymers already designed for recycling, rely on advanced catalytic methods. Many of the specific catalytic conversions reviewed here can be considered a proof of principle in the sense that further refinements are necessary to take them from the laboratory to a commercial scale. Catalyst performance in terms of activity and particularly stability calls for improvements. Prominently, in the deconstruction of waste polyethylene, elevated reaction temperatures are dictated by the need to operate in the polyolefin melt or in solution. This requires stable catalysts for the reactions of a given deconstruction scheme, like polyethylene dehydrogenation, olefin metathesis, and isomerization. Development of heterogeneous catalysts may provide solutions here. In polyethylene oxidation, achieving sufficient selectivity to dicarboxylic acids (or other products) while maintaining a straightforward approach in terms of oxidant used and number of reaction or purification steps appears to be the key challenge.
That being said, it is encouraging that the general types of reactions employed in the approaches to construction of monomers and polymers and the polymer deconstruction reviewed here, namely, carbonylation, olefin isomerization, olefin cross metathesis, fermentative ω-oxidation, olefin insertion polymerization, ring-opening metathesis polymerization, alkane dehydrogenation or oxidation, polyesterification, and solvolysis are provenly feasible on a (very) large scale.
The utility as materials of the reviewed polymers designed for chemical recyclability and biodegradability has been underlined by demonstration of essential critical features, again on a laboratory or small pilot scale. Long-chain polyesters are melt-processable by injection molding, film extrusion, fiber spinning, or also fused filament printing. They possess desirable tensile properties and barrier properties. Expansion to a larger scale along with in-depth studies and development of specific applications that optionally can comprise recycling is an exciting perspective.
Concerning the environmental deconstruction of polyethylene-type materials via in-chain functional groups, mapping out the impact of ester and other groups on (bio)degradation rates and mechanisms in different environments like soil or marine conditions is imperative.
Acknowledgments
The authors thank the ERC (Advanced Grant DEEPCAT, No. 832480) for ongoing financial support of our studies of degradable polyethylenes.
Glossary
Abbreviations
- ΔΔG‡alt-nonalt
difference in Gibbs free activation energies between alternating and nonalternating chain growth
- ADMET
acyclic diene metathesis
- atm
atmosphere
- CL
caprolactone
- CTA
chain transfer agent
- DCA
dicarboxylic acid
- DFT
density functional theory
- DSC
differential scanning calorimetry
- Cp
cyclopentadienyl
- Cp*
1,2,3,4,5-pentamethylcyclopentadienyl
- HDPE
high-density polyethylene
- iPP
isotactic polypropylene
- LCA
life-cycle assessment
- LDPE
low-density polyethylene
- LLDPE
linear low-density polyethylene
- MMAO
modified methylaluminoxane
- Mn
number-average molecular mass
- Mw
weight-average molecular mass
- NHPI
N-hydroxyimides such as N-hydroxyphthalimide
- NMR
nuclear magnetic resonance
- OMRP
organometallic-mediated radical polymerization
- PA-X,Y
polyamide derived from Cx diamine and Cy diacid or diester
- PC-X
Polycarbonate derived from Cx diol
- PDL
pentadecalactone
- PE
polyethylene
- PE-X,Y
polyester derived from Cx diol and Cy diacid or diester
- PE-Z
linear AB-type polyester with z carbon atoms in the repeat unit
- PET
polyethylene terephthalate
- PHA
polyhydroxyalkanoate
- ROMP
ring-opening metathesis polymerization
- ROP
ring opening polymerization
- r.t.
room temperature
- SAXS
small-angle X-ray scattering
- scCO2
supercritical CO2
- SEC
size exclusion chromatography
- Tg
glass transition temperature
- Tm
melting temperature
- UHMWPE
ultrahigh molecular weight polyethylene
- WAXS
wide-angle X-ray scattering
- wt %
weight%
Biographies
Simon T. Schwab studied chemistry at the University of Freiburg, Germany, including research stays with Prof. Howdle at the University of Nottingham, UK, and at BASF’s Catalyst Division in Iselin, NJ, USA. After concluding his studies with a master’s thesis in Prof. Mülhaupt’s group on the design of biobased monomers for stereolithography in 2020, he started working on his PhD with Prof. Mecking at the University of Konstanz on biodegradable and recyclable polyesters mimicking commodity plastics in their mechanical properties.
Maximilian Baur studied chemistry at the University of Konstanz, Germany. After an intermediate research stay with tesa SE, Hamburg, Germany, he joined the Chair of Chemical Materials Science as PhD student under the supervision of Prof. Stefan Mecking at the end of 2019. During his PhD studies, he worked on developing novel and nonpersistent polyethylene-like polymers. His research focused mainly on catalytic copolymerization of ethylene as an approach to such materials.
Taylor F. Nelson received his Ph.D. degree from ETH Zurich in 2019 under the supervision of M. Sander and K. McNeill on the topic of biodegradation of polyesters in agricultural soils. After holding a postdoctoral position at Woods Hole Oceanographic Institution (Massachusetts, USA) on the topic of marine degradation of consumer plastic goods, he is currently a Postdoctoral Research Fellow of the Alexander von Humboldt Foundation in the group of S. Mecking at the University of Konstanz. Currently, his research is focused on the synthesis of polyethylene-type polyesters from novel feedstocks and their environmental biodegradation.
Prof. Stefan Mecking studied chemistry at RWTH Aachen (1986-1992) and obtained his PhD degree with W. Keim (1992-1994), working on carbonylation. Following a postdoctoral stay at University of North Carolina Chapel Hill as a Feodor-Lynen-Fellow of the Alexander von Humboldt-Foundation with M. Brookhart (1994-1996), an industrial assignment with Hoechst Corporate Research in Frankfurt (1996-1998) and as a group leader at the University of Freiburg (1998-2004), in 2004 he was appointed full professor of Chemical Materials Science at the University of Konstanz. He has received national and international awards including the BASF Catalysis Award and the Otto-Roelen-Medal and was an elected visiting fellow of Trinity College, Oxford in 2019. His research, published in 270 papers in refereed journals and 40 patents, focuses on functional group tolerant polymerization catalysis and sustainable materials, including their degradation behavior and recycling. His research on degradable polyolefins enabled by catalysis is supported by an ERC Advanced Grant.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
This paper was originally published ASAP on February 26, 2024. Figure 20 was corrected, and the revised version reposted on February 29, 2024.
Special Issue
Published as part of Chemical Reviewsvirtual special issue “The Future of Plastics Sustainability”.
References
- Policy Scenarios to 2060. Global Plastics Outlook ;OECD, 2022. 10.1787/aa1edf33-en [DOI]
- Plastics - the Facts 2020. Plastics Europe. Online at https://plasticseurope.org/knowledge-hub/plastics-the-facts-2020/ (accessed 2023-10-28).
- Vollmer I.; Jenks M. J. F.; Roelands M. C. P.; White R. J.; van Harmelen T.; de Wild P.; van der Laan G. P.; Meirer F.; Keurentjes J. T. F.; Weckhuysen B. M. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem., Int. Ed. 2020, 59, 15402–15423. 10.1002/anie.201915651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmeister C. G.; Mülhaupt R. Closing the Carbon Loop in the Circular Plastics Economy. Macromol. Rapid Commun. 2022, 43, e2200247 10.1002/marc.202200247. [DOI] [PubMed] [Google Scholar]
- Vogt B. D.; Stokes K. K.; Kumar S. K. Why is Recycling of Postconsumer Plastics so Challenging?. ACS Appl. Polym. Mater. 2021, 3, 4325–4346. 10.1021/acsapm.1c00648. [DOI] [Google Scholar]
- Zibunas C.; Meys R.; Kätelhön A.; Bardow A. Cost-Optimal Pathways towards Net-Zero Chemicals and Plastics Based on a Circular Carbon Economy. Comput. Chem. Eng. 2022, 162, 107798. 10.1016/j.compchemeng.2022.107798. [DOI] [Google Scholar]
- Zheng J.; Suh S. Strategies to Reduce the Global Carbon Footprint of Plastics. Nat. Clim. Chang. 2019, 9, 374–378. 10.1038/s41558-019-0459-z. [DOI] [Google Scholar]
- Stürzel M.; Mihan S.; Mülhaupt R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. 10.1021/acs.chemrev.5b00310. [DOI] [PubMed] [Google Scholar]
- Jeremic D.Polyethylene. In Ullmann’s Encyclopedia of Industrial Chemistry ,7th ed.; Wiley-VCH, 2014. [Google Scholar]
- Bachmann M.; Zibunas C.; Hartmann J.; Tulus V.; Suh S.; Guillén-Gosálbez G.; Bardow A. Towards Circular Plastics within Planetary Boundaries. Nat. Sustain 2023, 6, 599–610. 10.1038/s41893-022-01054-9. [DOI] [Google Scholar]
- Ragaert K.; Delva L.; van Geem K. Mechanical and Chemical Recycling of Solid Plastic Waste. Waste Manage. 2017, 69, 24–58. 10.1016/j.wasman.2017.07.044. [DOI] [PubMed] [Google Scholar]
- Severn J. R.; Chadwick J. C.. Tailor-Made Polymers Via Immobilization of Alpha-Olefin Polymerization Catalyst ;Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Meys R.; Kätelhön A.; Bachmann M.; Winter B.; Zibunas C.; Suh S.; Bardow A. Achieving Net-Zero Greenhouse Gas Emission Plastics by a Circular Carbon Economy. Science 2021, 374, 71–76. 10.1126/science.abg9853. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Romain C.; Williams C. K. Sustainable Polymers from Renewable Resources. Nature 2016, 540, 354–362. 10.1038/nature21001. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Fevre M.; Jones G. O.; Waymouth R. M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2018, 118, 839–885. 10.1021/acs.chemrev.7b00329. [DOI] [PubMed] [Google Scholar]
- Coates G. W.; Getzler Y. D. Y. L. Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy. Nat. Rev. Mater. 2020, 5, 501–516. 10.1038/s41578-020-0190-4. [DOI] [Google Scholar]
- Lopez G.; Artetxe M.; Amutio M.; Bilbao J.; Olazar M. Thermochemical Routes for the Valorization of Waste Polyolefinic Plastics to Produce Fuels and Chemicals. A Review. Renew. Sust. Energy Rev. 2017, 73, 346–368. 10.1016/j.rser.2017.01.142. [DOI] [Google Scholar]
- Galadima A.; Masudi A.; Muraza O. Towards Low-Temperature Catalysts for Sustainable Fuel from Plastic: A Review. J. Environ. Chem. Eng. 2021, 9, 106655. 10.1016/j.jece.2021.106655. [DOI] [Google Scholar]
- Chamas A.; Moon H.; Zheng J.; Qiu Y.; Tabassum T.; Jang J. H.; Abu-Omar M.; Scott S. L.; Suh S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. 10.1021/acssuschemeng.9b06635. [DOI] [Google Scholar]
- Nakamura A.; Ito S.; Nozaki K. Coordination-Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. 10.1021/cr900079r. [DOI] [PubMed] [Google Scholar]
- Moad G. The Synthesis of Polyolefin Graft Copolymers by Reactive Extrusion. Prog. Polym. Sci. 1999, 24, 81–142. 10.1016/S0079-6700(98)00017-3. [DOI] [Google Scholar]
- Gloor P.; Tang Y.; Kostanska A.; Hamielec A. Chemical Modification of Polyolefins by Free Radical Mechanisms: A Modelling and Experimental Study of Simultaneous Random Scission, Branching and Crosslinking. Polymer 1994, 35, 1012–1030. 10.1016/0032-3861(94)90946-6. [DOI] [Google Scholar]
- Boaen N. K.; Hillmyer M. A. Post-Polymerization Functionalization of Polyolefins. Chem. Soc. Rev. 2005, 34, 267–275. 10.1039/b311405h. [DOI] [PubMed] [Google Scholar]
- Williamson J. B.; Lewis S. E.; Johnson R. R.; Manning I. M.; Leibfarth F. A. C-H Functionalization of Commodity Polymers. Angew. Chem., Int. Ed. 2019, 58, 8654–8668. 10.1002/anie.201810970. [DOI] [PubMed] [Google Scholar]
- Bunescu A.; Lee S.; Li Q.; Hartwig J. F. Catalytic Hydroxylation of Polyethylenes. ACS Cent. Sci. 2017, 3, 895–903. 10.1021/acscentsci.7b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Malollari K. G.; Uliana A.; Sanchez D.; Messersmith P. B.; Hartwig J. F. Selective, Catalytic Oxidations of C-H Bonds in Polyethylenes Produce Functional Materials with Enhanced Adhesion. Chem 2021, 7, 137–145. 10.1016/j.chempr.2020.11.020. [DOI] [Google Scholar]
- Teo J. Y. Q.; Yeung C. W. S.; Tan T. T. Y.; Loh W. W.; Loh X. J.; Lim J. Y. C. Benzaldehyde-Mediated Selective Aerobic Polyethylene Functionalisation with Isolated Backbone Ketones. Green Chem. 2022, 24, 6287–6294. 10.1039/D2GC02502G. [DOI] [Google Scholar]
- Shi J. X.; Ciccia N. R.; Pal S.; Kim D. D.; Brunn J. N.; Lizandara-Pueyo C.; Ernst M.; Haydl A. M.; Messersmith P. B.; Helms B. A.; et al. Chemical Modification of Oxidized Polyethylene Enables Access to Functional Polyethylenes with Greater Reuse. J. Am. Chem. Soc. 2023, 145, 21527–21537. 10.1021/jacs.3c07186. [DOI] [PubMed] [Google Scholar]
- Brubaker M. M.; Coffman D. D.; Hoehn H. H. Synthesis and Characterization of Ethylene/Carbon Monoxide Copolymers, A New Class of Polyketones. J. Am. Chem. Soc. 1952, 74, 1509–1515. 10.1021/ja01126a047. [DOI] [Google Scholar]
- Coffman D. D.; Pinkney P. S.; Wall F. T.; Wood W. H.; Young H. S. Compositional Relationships in the Copolymerization of Ethylene with Carbon Monoxide. J. Am. Chem. Soc. 1952, 74, 3391–3393. 10.1021/ja01133a051. [DOI] [Google Scholar]
- Buback M.; Tups H. High-Pressure Copolymerization of C2H4 and CO. Physica B+C 1986, 139–140, 626–628. 10.1016/0378-4363(86)90663-7. [DOI] [Google Scholar]
- Morgen T. O.; Baur M.; Göttker-Schnetmann I.; Mecking S. Photodegradable Branched Polyethylenes from Carbon Monoxide Copolymerization under Benign Conditions. Nat. Commun. 2020, 11, 3693. 10.1038/s41467-020-17542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau E.; Broyer J.-P.; Boisson C.; Spitz R.; Monteil V. Unusual Activation by Solvent of the Ethylene Free Radical Polymerization. Polym. Chem. 2011, 2, 2328. 10.1039/c1py00200g. [DOI] [Google Scholar]
- Andrady A. L. Weathering of Polyethylene (LDPE) and Enhanced Photodegradable Polyethylene in the Marine Environment. J. Appl. Polym. Sci. 1990, 39, 363–370. 10.1002/app.1990.070390213. [DOI] [Google Scholar]
- Andrady A. L.Weatherability of Enhanced Degradable Plastics ;United States Environmental Protection Agency, 1993.
- Guillet J. E.; Huber H. X.; Scott J. A.. Synthesis and Applications of Photodegradable Poly(Ethylene Terephthalate). In Degradable Polymers, recycling, and Plastics Waste Management ,1st ed.; Albertsson A.-C., Huang S. J., Eds.; CRC Press: Boca Raton, FL, 1995; pp 253–264. [Google Scholar]
- Fuchs A.; Mecking S. Controlled Cobalt-Mediated Free-Radical Co- and Terpolymerization of Carbon Monoxide. J. Am. Chem. Soc. 2022, 144, 15879–15884. 10.1021/jacs.2c07200. [DOI] [PubMed] [Google Scholar]
- Agarwal S. Chemistry, Chances and Limitations of the Radical Ring-Opening Polymerization of Cyclic Ketene Acetals for the Synthesis of Degradable Polyesters. Polym. Chem. 2010, 1, 953. 10.1039/c0py00040j. [DOI] [Google Scholar]
- Piovano A.; Zarupski J.; Groppo E. Disclosing the Interaction between Carbon Monoxide and Alkylated Ti3+ Species: a Direct Insight into Ziegler-Natta Catalysis. J. Phys. Chem. Lett. 2020, 11, 5632–5637. 10.1021/acs.jpclett.0c01665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertleff W.; Roeper M.; Sava X.. Carbonylation. Ullmann’s Encyclopedia of Industrial Chemistry, 7 ed.; Wiley-VCH, 2012. [Google Scholar]
- Rix F. C.; Brookhart M.; White P. S. Mechanistic Studies of the Palladium(II)-Catalyzed Copolymerization of Ethylene with Carbon Monoxide. J. Am. Chem. Soc. 1996, 118, 4746–4764. 10.1021/ja953276t. [DOI] [Google Scholar]
- Shultz C. S.; Ledford J.; DeSimone J. M.; Brookhart M. Kinetic Studies of Migratory Insertion Reactions at the (1,3-Bis(diphenylphosphino)propane)Pd(II) Center and Their Relationship to the Alternating Copolymerization of Ethylene and Carbon Monoxide. J. Am. Chem. Soc. 2000, 122, 6351–6356. 10.1021/ja994251n. [DOI] [Google Scholar]
- Drent E.; van Broekhoven J. A. M.; Doyle M. J. Efficient Palladium Catalysts for the Copolymerization of Carbon Monoxide with Olefins to Produce Perfectly Alternating Polyketones. J. Organomet. Chem. 1991, 417, 235–251. 10.1016/0022-328X(91)80176-K. [DOI] [Google Scholar]
- Drent E.; Budzelaar P. H. M. Palladium-Catalyzed Alternating Copolymerization of Alkenes and Carbon Monoxide. Chem. Rev. 1996, 96, 663–682. 10.1021/cr940282j. [DOI] [PubMed] [Google Scholar]
- Haras A.; Michalak A.; Rieger B.; Ziegler T. Theoretical Analysis of Factors Controlling the Nonalternating CO/C2H4 Copolymerization. J. Am. Chem. Soc. 2005, 127, 8765–8774. 10.1021/ja050861d. [DOI] [PubMed] [Google Scholar]
- Haras A.; Michalak A.; Rieger B.; Ziegler T. Comparative Study on Catalytic Systems for the Alternating and Nonalternating CO/Ethene Copolymerization. Organometallics 2006, 25, 946–953. 10.1021/om050846h. [DOI] [Google Scholar]
- Newsham D. K.; Borkar S.; Sen A.; Conner D. M.; Goodall B. L. Inhibitory Role of Carbon Monoxide in Palladium(II)-Catalyzed Nonalternating Ethene/Carbon Monoxide Copolymerizations and the Synthesis of Polyethene-block-poly(ethene-alt-carbon monoxide). Organometallics 2007, 26, 3636–3638. 10.1021/om700523m. [DOI] [Google Scholar]
- Vernyi B.Shell Launches CARILON Line of Aliphatic Polyketone TPS. Plastics News, 1996. [Google Scholar]
- Jung M.-h.Hyosung to Unveil POKETONE at Global Markets. Business Korea, 2016. [Google Scholar]
- Luo R.; Newsham D. K.; Sen A. Palladium-Catalyzed Nonalternating Copolymerization of Ethene and Carbon Monoxide: Scope and Mechanism. Organometallics 2009, 28, 6994–7000. 10.1021/om9008235. [DOI] [Google Scholar]
- Bettucci L.; Bianchini C.; Claver C.; Suarez E. J. G.; Ruiz A.; Meli A.; Oberhauser W. Ligand Effects in the Non-Alternating CO-Ethylene Copolymerization by Palladium(ii) Catalysis. Dalton Trans. 2007, 5590–5602. 10.1039/b711280g. [DOI] [PubMed] [Google Scholar]
- Drent E.; van Dijk R.; van Ginkel R.; van Oort B.; Pugh R. I. The First Example of Palladium Catalysed Non-Perfectly Alternating Copolymerisation of Ethene and Carbon Monoxide. Chem. Commun. 2002, 964–965. 10.1039/b111629k. [DOI] [PubMed] [Google Scholar]
- Soomro S. S.; Cozzula D.; Leitner W.; Vogt H.; Müller T. E. The Microstructure and Melt Properties of CO-Ethylene Copolymers with Remarkably Low CO Content. Polym. Chem. 2014, 5, 3831–3837. 10.1039/C3PY01637D. [DOI] [Google Scholar]
- Keim W.; Kowaldt F. H.; Goddard R.; Krüger C. Novel Coordination of (Benzoylmethylene)triphenylphosphorane in a Nickel Oligomerization Catalyst. Angew. Chem., Int. Ed. 1978, 17, 466–467. 10.1002/anie.197804661. [DOI] [Google Scholar]
- Keim W. Oligomerization of Ethylene to α-Olefins: Discovery and Development of the Shell Higher Olefin Process (SHOP). Angew. Chem., Int. Ed. 2013, 52, 12492–12496. 10.1002/anie.201305308. [DOI] [PubMed] [Google Scholar]
- Mu H.; Pan L.; Song D.; Li Y. Neutral Nickel Catalysts for Olefin Homo- and Copolymerization: Relationships between Catalyst Structures and Catalytic Properties. Chem. Rev. 2015, 115, 12091–12137. 10.1021/cr500370f. [DOI] [PubMed] [Google Scholar]
- Shimizu F.; Xin S.; Tanna A.; Goromaru S.; Matsubara K.. Metal Complex and Method for Producing α-Olefin Polymer and Method for Producing α-Olefin/(Meth) Acrylate Copolymer Using the Same: U.S. Patent US8618319B2, 2013.
- Xin B. S.; Sato N.; Tanna A.; Oishi Y.; Konishi Y.; Shimizu F. Nickel Catalyzed Copolymerization of Ethylene and Alkyl Acrylates. J. Am. Chem. Soc. 2017, 139, 3611–3614. 10.1021/jacs.6b13051. [DOI] [PubMed] [Google Scholar]
- Lin F.; Morgen T. O.; Mecking S. Living Aqueous Microemulsion Polymerization of Ethylene with Robust Ni(II) Phosphinophenolato Catalysts. J. Am. Chem. Soc. 2021, 143, 20605–20608. 10.1021/jacs.1c10488. [DOI] [PubMed] [Google Scholar]
- Baur M.; Lin F.; Morgen T. O.; Odenwald L.; Mecking S. Polyethylene Materials with In-Chain Ketones from Nonalternating Catalytic Copolymerization. Science 2021, 374, 604–607. 10.1126/science.abi8183. [DOI] [PubMed] [Google Scholar]
- Baur M.; Mecking S. Polyethylenes with Combined In-Chain and Side-Chain Functional Groups from Catalytic Terpolymerization of Carbon Monoxide and Acrylate. ACS Macro Lett. 2022, 11, 1207–1211. 10.1021/acsmacrolett.2c00459. [DOI] [PubMed] [Google Scholar]
- Voccia M.; Odenwald L.; Baur M.; Lin F.; Falivene L.; Mecking S.; Caporaso L. Mechanistic Insights into Ni(II)-Catalyzed Nonalternating Ethylene-Carbon Monoxide Copolymerization. J. Am. Chem. Soc. 2022, 144, 15111–15117. 10.1021/jacs.2c04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmann P.; Wimmer F. P.; Mecking S. Long-Spaced Polyketones from ADMET Copolymerizations as Ideal Models for Ethylene/CO Copolymers. ACS Macro Lett. 2015, 4, 704–707. 10.1021/acsmacrolett.5b00324. [DOI] [PubMed] [Google Scholar]
- Stempfle F.; Ortmann P.; Mecking S. Long-Chain Aliphatic Polymers To Bridge the Gap between Semicrystalline Polyolefins and Traditional Polycondensates. Chem. Rev. 2016, 116, 4597–4641. 10.1021/acs.chemrev.5b00705. [DOI] [PubMed] [Google Scholar]
- Sanchez I. C.; Eby R. K. Thermodynamics and Crystallization of Random Copolymers. Macromolecules 1975, 8, 638–641. 10.1021/ma60047a012. [DOI] [Google Scholar]
- Tang S.; Seidel F. W.; Nozaki K. High Density Polyethylenes Bearing Isolated In-Chain Carbonyls. Angew. Chem., Int. Ed. 2021, 133, 26710–26714. 10.1002/ange.202110957. [DOI] [PubMed] [Google Scholar]
- Dodge H. M.; Natinsky B. S.; Jolly B. J.; Zhang H.; Mu Y.; Chapp S. M.; Tran T. V.; Diaconescu P. L.; Do L. H.; Wang D.; et al. Polyketones from Carbon Dioxide and Ethylene by Integrating Electrochemical and Organometallic Catalysis. ACS Catal. 2023, 13, 4053–4059. 10.1021/acscatal.3c00769. [DOI] [Google Scholar]
- Zhou F.; Dai H.; Tang S.; Zhou Y. Synthesis of Polyethylenes with In-Chain Isolated Carbonyls from CO2 and Ethylene via a Tandem Photoreduction/Polymerization Protocol. CCS Chem. 2023, 1–9. 10.31635/ccschem.023.202303275. [DOI] [Google Scholar]
- Chen S.-Y.; Pan R.-C.; Chen M.; Liu Y.; Chen C.; Lu X.-B. Synthesis of Nonalternating Polyketones Using Cationic Diphosphazane Monoxide-Palladium Complexes. J. Am. Chem. Soc. 2021, 143, 10743–10750. 10.1021/jacs.1c04964. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Gaire S.; Ziegler C. J.; Jia L. Nickel Catalysts for Non-Alternating CO-Ethylene Copolymerization. ChemCatChem. 2022, 14, e202200974 10.1002/cctc.202200974. [DOI] [Google Scholar]
- Chen S.-Y.; Song Y.-H.; Jiao S.; Zou C.; Li S.-H.; Chen C.; Lu X.-B.; Liu Y. Carbonyl Functionalized Polyethylene Materials via Ni- and Pd-diphosphazane Monoxide Catalyzed Nonalternating Copolymerization. J. Catal. 2023, 417, 334–340. 10.1016/j.jcat.2022.12.020. [DOI] [Google Scholar]
- Kosaka N.; Hiyama T.; Nozaki K. Baeyer-Villiger Oxidation of an Optically Active 1,4-Polyketone. Macromolecules 2004, 37, 4484–4487. 10.1021/ma0359638. [DOI] [Google Scholar]
- Liguori A.; Hakkarainen M. Designed from Biobased Materials for Recycling: Imine-Based Covalent Adaptable Networks. Macromol. Rapid Commun. 2022, 43, e2100816 10.1002/marc.202100816. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Takahashi K.; Hayashi S.; Suzuki M.; Fujikake N.; Kasuya K.-I.; Zhou J.; Nakagawa S.; Yoshie N.; Li C., et al. Synthesis of Novel Polymers with Biodegradability from Propylene and Carbon Monoxide ChemRxiv 2023. 10.26434/chemrxiv-2023-wj0p6. [DOI] [PubMed]
- Sobkowicz M. J. Polymer Design for the Circular Economy. Science 2021, 374, 540. 10.1126/science.abm2306. [DOI] [PubMed] [Google Scholar]
- Baur M.; Mast N. K.; Brahm J. P.; Habé R.; Morgen T. O.; Mecking S. High-Density Polyethylene with In-Chain Photolyzable and Hydrolyzable Groups Enabling Recycling and Degradation. Angew. Chem., Int. Ed. Engl. 2023, 62, e202310990 10.1002/anie.202310990. [DOI] [PubMed] [Google Scholar]
- Price C. J.; Reich B. J. E.; Miller S. A. Thermodynamic and Kinetic Considerations in the Copolymerization of Ethylene and Carbon Dioxide. Macromolecules 2006, 39, 2751–2756. 10.1021/ma052697k. [DOI] [Google Scholar]
- Nakano R.; Ito S.; Nozaki K. Copolymerization of Carbon Dioxide and Butadiene via a Lactone Intermediate. Nat. Chem. 2014, 6, 325–331. 10.1038/nchem.1882. [DOI] [PubMed] [Google Scholar]
- Moha V.; Cozzula D.; Hölscher M.; Leitner W.; Müller T. E. A DFT Study on the Co-polymerization of CO2 and Ethylene: Feasibility Analysis for the Direct Synthesis of Polyethylene Esters. ChemSusChem 2016, 9, 1614–1622. 10.1002/cssc.201501615. [DOI] [PubMed] [Google Scholar]
- Tang S.; Nozaki K. Advances in the Synthesis of Copolymers from Carbon Dioxide, Dienes, and Olefins. Acc. Chem. Res. 2022, 55, 1524–1532. 10.1021/acs.accounts.2c00162. [DOI] [PubMed] [Google Scholar]
- de Vries T. J.; Kemmere M. F.; Keurentjes J. T. F. Characterization of Polyethylenes Produced in Supercritical Carbon Dioxide by a Late-Transition-Metal Catalyst. Macromolecules 2004, 37, 4241–4246. 10.1021/ma035347b. [DOI] [Google Scholar]
- Guironnet D.; Friedberger T.; Mecking S. Ethylene Polymerization in Supercritical Carbon Dioxide with Binuclear Nickel(II) Catalysts. Dalton Trans. 2009, 8929–8934. 10.1039/b912883b. [DOI] [PubMed] [Google Scholar]
- Guironnet D.; Göttker-Schnetmann I.; Mecking S. Catalytic Polymerization in Dense CO2 to Controlled Microstructure Polyethylenes. Macromolecules 2009, 42, 8157–8164. 10.1021/ma901397q. [DOI] [Google Scholar]
- Bastero A.; Franciò G.; Leitner W.; Mecking S. Catalytic Ethylene Polymerisation in Carbon Dioxide as a Reaction Medium with Soluble Nickel(II) Catalysts. Chemistry 2006, 12, 6110–6116. 10.1002/chem.200600499. [DOI] [PubMed] [Google Scholar]
- Huguet N.; Jevtovikj I.; Gordillo A.; Lejkowski M. L.; Lindner R.; Bru M.; Khalimon A. Y.; Rominger F.; Schunk S. A.; Hofmann P.; et al. Nickel-catalyzed Direct Carboxylation of Olefins with CO2: One-Pot Synthesis of α,β-Unsaturated Carboxylic Acid Salts. Chem.-Eur. J. 2014, 20, 16858–16862. 10.1002/chem.201405528. [DOI] [PubMed] [Google Scholar]
- Hendriksen C.; Pidko E. A.; Yang G.; Schäffner B.; Vogt D. Catalytic Formation of Acrylate from Carbon Dioxide and Ethene. Chem.-Eur. J. 2014, 20, 12037–12040. 10.1002/chem.201404082. [DOI] [PubMed] [Google Scholar]
- Tang S.; Zhao Y.; Nozaki K. Accessing Divergent Main-Chain-Functionalized Polyethylenes via Copolymerization of Ethylene with a CO2/Butadiene-Derived Lactone. J. Am. Chem. Soc. 2021, 143, 17953–17957. 10.1021/jacs.1c08578. [DOI] [PubMed] [Google Scholar]
- Garcia Espinosa L. D.; Williams-Pavlantos K.; Turney K. M.; Wesdemiotis C.; Eagan J. M. Degradable Polymer Structures from Carbon Dioxide and Butadiene. ACS Macro Lett. 2021, 10, 1254–1259. 10.1021/acsmacrolett.1c00523. [DOI] [PubMed] [Google Scholar]
- Rapagnani R. M.; Dunscomb R. J.; Fresh A. A.; Tonks I. A. Tunable and Recyclable Polyesters from CO2 and Butadiene. Nat. Chem. 2022, 14, 877–883. 10.1038/s41557-022-00969-2. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Mecking S. A Synthetic Polyester from Plant Oil Feedstock by Functionalizing Polymerization. Angew. Chem., Int. Ed. 2019, 58, 3346–3350. 10.1002/anie.201810914. [DOI] [PubMed] [Google Scholar]
- Quinzler D.; Mecking S. Renewable Resource-Based Poly(Dodecyloate) by Carbonylation Polymerization. Chem. Commun. 2009, 5400–5402. 10.1039/b912294j. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M.; Zuideveld M. A.; Swennenhuis B. H. G.; Freixa Z.; Kamer P. C. J.; Goubitz K.; Fraanje J.; Lutz M.; Spek A. L. Alcoholysis of Acylpalladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: The Importance of Cis-Coordinating Phosphines. J. Am. Chem. Soc. 2003, 125, 5523–5539. 10.1021/ja029341y. [DOI] [PubMed] [Google Scholar]
- Sachs J. D.; Tonks I. A. Synthesis of Poly(Ester-Ether) Polymers via Hydroesterificative Polymerization of α,ω-Enol Ethers. Macromolecules 2022, 55, 9520–9526. 10.1021/acs.macromol.2c01935. [DOI] [Google Scholar]
- Kacker S.; Jiang Z.; Sen A. Alternating Copolymers of Functional Alkenes with Carbon Monoxide. Macromolecules 1996, 29, 5852–5858. 10.1021/ma960255q. [DOI] [Google Scholar]
- Yee G. M.; Wang T.; Hillmyer M. A.; Tonks I. A. Mechanistic Study of Palladium-Catalyzed Hydroesterificative Copolymerization of Vinyl Benzyl Alcohol and CO. Organometallics 2019, 38, 1778–1786. 10.1021/acs.organomet.9b00091. [DOI] [Google Scholar]
- Lo S.-Y.; Folster C. P.; Harkins R. P.; Anderson R. J.; Lien Y.-L.; Chiu H.-C.; Carpenter A. E.; Tonks I. A. Carbonylative Co- and Terpolymerizations of 10-Undecen-1-ol: A Route to Polyketoesters with Tunable Compositions. ACS Catal. 2022, 12, 14629–14636. 10.1021/acscatal.2c03984. [DOI] [Google Scholar]
- Barbotin F.; Monteil V.; Llauro M.-F.; Boisson C.; Spitz R. First Synthesis of Poly(ethene- co −1,3-butadiene) with Neodymocene Catalysts. Macromolecules 2000, 33, 8521–8523. 10.1021/ma0012043. [DOI] [Google Scholar]
- Shiono T.; Yoshino O.; Ikeda T. Synthesis and Oxidative Degradation of Poly(Ethene-ran-1,3-Butadiene). Macromol. Rapid Commun. 2000, 21, 1297–1301. 10.1002/1521-3927(20001201)21:18<1297::AID-MARC1297>3.0.CO;2-1. [DOI] [Google Scholar]
- Adnan Akram M.; Liu X.; Fu Z.; Fan Z. Ethylene-Butadiene Copolymerization and Ethylene-1-Hexene-Butadiene Terpolymerization with a MgCl2 -Supported Ziegler-Natta Catalyst: Polymer Structure and Active Centers. ChemistrySelect 2021, 6, 8288–8298. 10.1002/slct.202101552. [DOI] [Google Scholar]
- Belaid I.; Monteil V.; Boisson C.. Copolymerization of Ethylene with Conjugated Dienes. In Handbook of Transition Metal Polymerization Catalysts ;Hoff R., Hoff R. E., Mathers R. T., Eds.; Wiley: Hoboken, NJ, 2010; pp 661–692. [Google Scholar]
- Wu C.; Liu B.; Lin F.; Wang M.; Cui D. cis-1,4-Selective Copolymerization of Ethylene and Butadiene: A Compromise between Two Mechanisms. Angew. Chem., Int. Ed. 2017, 56, 6975–6979. 10.1002/anie.201702128. [DOI] [PubMed] [Google Scholar]
- Kocen A. L.; Cui S.; Lin T.-W.; LaPointe A. M.; Coates G. W. Chemically Recyclable Ester-Linked Polypropylene. J. Am. Chem. Soc. 2022, 144, 12613–12618. 10.1021/jacs.2c04499. [DOI] [PubMed] [Google Scholar]
- Ishihara T.; Ban H. T.; Hagihara H.; Shiono T. Additive Effects of Alkylaluminium Compounds on Propylene-1,3-Butadiene Copolymerization Using Isospecific Zirconocene Catalysts. J. Organomet. Chem. 2010, 695, 1694–1699. 10.1016/j.jorganchem.2010.04.003. [DOI] [Google Scholar]
- Ishihara T.; Ban H. T.; Hagihara H.; Shiono T. Copolymerization of Propylene with 1,3-Butadiene Using Isospecific Zirconocene Catalysts. J. Polym. Sci. A-2 Polym. Phys. 2007, 45, 5731–5740. 10.1002/pola.22321. [DOI] [Google Scholar]
- Ishihara T.; Shiono T. Synthesis of Poly(propylene- ran −1,3-butadiene) and Its Metathesis Degradation with Ethylene. Macromolecules 2003, 36, 9675–9677. 10.1021/ma0349480. [DOI] [Google Scholar]
- Boisson C.; Monteil V.; Thuilliez J.; Spitz R.; Monnet C.; Llauro M.-F.; Barbotin F.; Robert P. Advances and Limits in Copolymerization of Olefins with Conjugated Dienes. Macromol. Symp. 2005, 226, 17–24. 10.1002/masy.200550802. [DOI] [Google Scholar]
- Gerum W.; Höhne G. W. H.; Wilke W.; Arnold M.; Wegner T. Hydrogenated Butadiene/Ethene/1-Olefin Terpolymers as Model Substances for Short-Chain Branched Polyethylene. Macromol. Chem. Phys. 1996, 197, 1691–1712. 10.1002/macp.1996.021970511. [DOI] [Google Scholar]
- Boisson C.; Monteil V.; Ribour D.; Spitz R.; Barbotin F. Lanthanidocene Catalysts for the Homo- and Copolymerization of Ethylene with Butadiene. Macromol. Chem. Phys. 2003, 204, 1747–1754. 10.1002/macp.200350037. [DOI] [Google Scholar]
- Woodman T. J.; Sarazin Y.; Fink G.; Hauschild K.; Bochmann M. Heterogenized “Ligand-Free” Lanthanide Catalysts for the Homo- and Copolymerization of Ethylene and 1,3-Butadiene. Macromolecules 2005, 38, 3060–3067. 10.1021/ma047454r. [DOI] [Google Scholar]
- Lucas F.; Peruch F.; Carlotti S.; Deffieux A.; Leblanc A.; Boisson C. Synthesis of Dihydroxy Poly(Ethylene-co-Butadiene) via Metathetical Depolymerization: Kinetic and Mechanistic Aspects. Polymer 2008, 49, 4935–4941. 10.1016/j.polymer.2008.09.012. [DOI] [Google Scholar]
- Zhang K.; Dou Y.; Jiang Y.; Zhang Z.; Li S.; Cui D. High-Performance Elastomer from Trans-1,4 Copolymerization of Ethylene and Butadiene. Macromolecules 2021, 54, 9445–9451. 10.1021/acs.macromol.1c01384. [DOI] [Google Scholar]
- Kaminsky W.; Hinrichs B. Ethene-Butadiene Copolymers by Single-Site Catalysts. Macromol. Symp. 2003, 195, 39–44. 10.1002/masy.200390143. [DOI] [Google Scholar]
- Arriola D. J.; Briggs J. R.; Timmers F. J.; Wagner N. L.; Wenzel T. T.. Production of Telechelic Compounds by Metathesis Depolymerization: US7956132B2, 2011.
- Napoli M.; Pragliola S.; Costabile C.; Longo P. New Group IV Metallocene Systems Active in the Copolymerization of α-Olefins and Conjugated Dienes. Macromol. Chem. Phys. 2006, 207, 304–309. 10.1002/macp.200500457. [DOI] [Google Scholar]
- Železník O.; Merna J. Bis(Arylimino)Pyridyl Cobalt Catalysts for Copolymerization of Buta-1,3-Diene with Ethene. Polymer 2019, 175, 195–205. 10.1016/j.polymer.2019.05.025. [DOI] [Google Scholar]
- Llauro M. F.; Monnet C.; Barbotin F.; Monteil V.; Spitz R.; Boisson C. Investigation of Ethylene/Butadiene Copolymers Microstructure by 1H and 13C NMR. Macromolecules 2001, 34, 6304–6311. 10.1021/ma010421g. [DOI] [Google Scholar]
- Capacchione C.; Avagliano A.; Proto A. Ethylene-Butadiene Copolymerization Promoted by Titanium Complex Containing a Tetradentate [OSSO]-Type Bis(phenolato) Ligand. Macromolecules 2008, 41, 4573–4575. 10.1021/ma800864y. [DOI] [Google Scholar]
- Parke S. M.; Lopez J. C.; Cui S.; LaPointe A. M.; Coates G. W. Polyethylene Incorporating Diels-Alder Comonomers: A ″Trojan Horse″ Strategy for Chemically Recyclable Polyolefins. Angew. Chem., Int. Ed. 2023, 62, e202301927 10.1002/anie.202301927. [DOI] [PubMed] [Google Scholar]
- Walker R.; Conrad R. M.; Grubbs R. H. The Living ROMP of trans-Cyclooctene. Macromolecules 2009, 42, 599–605. 10.1021/ma801693q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock R. R. Living Ring-Opening Metathesis Polymerization Catalyzed by Well-Characterized Transition-Metal Alkylidene Complexes. Acc. Chem. Res. 1990, 23, 158–165. 10.1021/ar00173a007. [DOI] [Google Scholar]
- Cho I. Ring-Opening Polymerizations for the Synthesis of Sequence-Controlled Copolymers. Macromol. Symp. 2003, 195, 89–94. 10.1002/masy.200390149. [DOI] [Google Scholar]
- Trnka T. M.; Grubbs R. H. The Development of L2X2Ru = CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- Hlil A. R.; Balogh J.; Moncho S.; Su H.-L.; Tuba R.; Brothers E. N.; Al-Hashimi M.; Bazzi H. S. Ring Opening Metathesis Polymerization (ROMP) of Five- to Eight-Membered Cyclic Olefins: Computational, Thermodynamic, and Experimental Approach. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 3137–3145. 10.1002/pola.28695. [DOI] [Google Scholar]
- Martinez H.; Ren N.; Matta M. E.; Hillmyer M. A. Ring-Opening Metathesis Polymerization of 8-Membered Cyclic Olefins. Polym. Chem. 2014, 5, 3507. 10.1039/c3py01787g. [DOI] [Google Scholar]
- Naga N.; Arai R.; Kikuchi G.; Toyota A.; Noguchi K.; Sone M.; Shirae F.; Gotoh T.; Kurosu H. Crystalline Structure of Polyethylene Containing Vinylene Units in the Main Chain. Polymer 2011, 52, 4857–4866. 10.1016/j.polymer.2011.08.058. [DOI] [Google Scholar]
- Shiono T.; Naga N.; Soga K. Synthesis of α,ω-Divinylpolyethylene-Like Polymers from cis-1,4-Polybutadiene Using Partial Hydrogenation and Metathesis Degradation with Ethylene. Makromol. Chem., Rapid Commun. 1993, 14, 323–327. 10.1002/marc.1993.030140601. [DOI] [Google Scholar]
- Yan T.; Guironnet D. Synthesis of Telechelic Polyolefins. Polym. Chem. 2021, 12, 5126–5138. 10.1039/D1PY00819F. [DOI] [Google Scholar]
- Hillmyer M. A.; Grubbs R. H. Chain Transfer in the Ring-Opening Metathesis Polymerization of Cyclooctadiene Using Discrete Metal Alkylidenes. Macromolecules 1995, 28, 8662–8667. 10.1021/ma00129a027. [DOI] [Google Scholar]
- Hillmyer M. A.; Grubbs R. H. Preparation of Hydroxytelechelic Poly(Butadiene) via Ring-Opening Metathesis Polymerization Employing a Well-Defined Metathesis Catalyst. Macromolecules 1993, 26, 872–874. 10.1021/ma00056a051. [DOI] [Google Scholar]
- Hillmyer M. A.; Nguyen S. T.; Grubbs R. H. Utility of a Ruthenium Metathesis Catalyst for the Preparation of End-Functionalized Polybutadiene. Macromolecules 1997, 30, 718–721. 10.1021/ma961316n. [DOI] [Google Scholar]
- Bielawski C. W.; Scherman O. A.; Grubbs R. H. Highly Efficient Syntheses of Acetoxy- and Hydroxy-Terminated Telechelic Poly(Butadiene)s Using Ruthenium Catalysts Containing N -Heterocyclic Ligands. Polymer 2001, 42, 4939–4945. 10.1016/S0032-3861(00)00504-8. [DOI] [Google Scholar]
- Pitet L. M.; Hillmyer M. A. Carboxy-Telechelic Polyolefins by ROMP Using Maleic Acid as a Chain Transfer Agent. Macromolecules 2011, 44, 2378–2381. 10.1021/ma102975r. [DOI] [Google Scholar]
- Wang Y.; Hillmyer M. A. Hydroxy-Telechelic Poly(Ethylene-co-Isobutylene) as a Soft Segment for Thermoplastic Polyurethanes. Polym. Chem. 2015, 6, 6806–6811. 10.1039/C5PY00990A. [DOI] [Google Scholar]
- Sample C. S.; Kellstedt E. A.; Hillmyer M. A. Tandem ROMP/Hydrogenation Approach to Hydroxy-Telechelic Linear Polyethylene. ACS Macro Lett. 2022, 11, 608–614. 10.1021/acsmacrolett.2c00144. [DOI] [PubMed] [Google Scholar]
- Arrington A. S.; Brown J. R.; Win M. S.; Winey K. I.; Long T. E. Melt Polycondensation of Carboxytelechelic Polyethylene for the Design of Degradable Segmented Copolyester Polyolefins. Polym. Chem. 2022, 13, 3116–3125. 10.1039/D2PY00394E. [DOI] [Google Scholar]
- Zhao Y.; Rettner E. M.; Harry K. L.; Hu Z.; Miscall J.; Rorrer N. A.; Miyake G. M. Chemically Recyclable Polyolefin-Like Multiblock Polymers. Science 2023, 382, 310–314. 10.1126/science.adh3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikkali S.; Mecking S. Refining of Plant Oils to Chemicals by Olefin Metathesis. Angew. Chem., Int. Ed. 2012, 51, 5802–5808. 10.1002/anie.201107645. [DOI] [PubMed] [Google Scholar]
- Vilela C.; Silvestre A. J. D.; Meier M. A. R. Plant Oil-Based Long-Chain C 26 Monomers and Their Polymers. Macromol. Chem. Phys. 2012, 213, 2220–2227. 10.1002/macp.201200332. [DOI] [Google Scholar]
- Beuhler A. A “Game-Changing” C18 Diacid for the Plastics Industry?. Plast. Eng. 2014, 70, 32–35. 10.1002/j.1941-9635.2014.tb01175.x. [DOI] [Google Scholar]
- O’Leary Havelka K.; Gerhardt G. E.. From Biorefinery to Performance Technology: Transforming Renewable Olefinic Building Blocks into Lubricants and Other High-Value Products. In Biobased Monomers, Polymers, and Materials ;Smith P. B., Gross R. A., Eds.; American Chemical Society: Washington, DC, 2012; pp 201–222. [Google Scholar]
- Quinzler D.; Mecking S. Linear Semicrystalline Polyesters from Fatty Acids by Complete Feedstock Molecule Utilization. Angew. Chem., Int. Ed. 2010, 49, 4306–4308. 10.1002/anie.201001510. [DOI] [PubMed] [Google Scholar]
- Jiménez-Rodriguez C.; Eastham G. R.; Cole-Hamilton D. J. Dicarboxylic Acid Esters from the Carbonylation of Unsaturated Esters under Mild Conditions. Inorg. Chem. Commun. 2005, 8, 878–881. 10.1016/j.inoche.2005.06.005. [DOI] [Google Scholar]
- Roesle P.; Caporaso L.; Schnitte M.; Goldbach V.; Cavallo L.; Mecking S. A Comprehensive Mechanistic Picture of the Isomerizing Alkoxycarbonylation of Plant Oils. J. Am. Chem. Soc. 2014, 136, 16871–16881. 10.1021/ja508447d. [DOI] [PubMed] [Google Scholar]
- Vondran J.; Furst M. R. L.; Eastham G. R.; Seidensticker T.; Cole-Hamilton D. J. Magic of Alpha: The Chemistry of a Remarkable Bidentate Phosphine, 1,2-Bis(di-tert-butylphosphinomethyl)benzene. Chem. Rev. 2021, 121, 6610–6653. 10.1021/acs.chemrev.0c01254. [DOI] [PubMed] [Google Scholar]
- Herrmann N.; Köhnke K.; Seidensticker T. Selective Product Crystallization for Concurrent Product Separation and Catalyst Recycling in the Isomerizing Methoxycarbonylation of Methyl Oleate. ACS Sustain. Chem. Eng. 2020, 8, 10633–10638. 10.1021/acssuschemeng.0c03432. [DOI] [Google Scholar]
- Christl J. T.; Roesle P.; Stempfle F.; Müller G.; Caporaso L.; Cavallo L.; Mecking S. Promotion of Selective Pathways in Isomerizing Functionalization of Plant Oils by Rigid Framework Substituents. ChemSusChem 2014, 7, 3491–3495. 10.1002/cssc.201402441. [DOI] [PubMed] [Google Scholar]
- Goldbach V.; Roesle P.; Mecking S. Catalytic Isomerizing ω-Functionalization of Fatty Acids. ACS Catal. 2015, 5, 5951–5972. 10.1021/acscatal.5b01508. [DOI] [Google Scholar]
- Pingen D.; Klinkenberg N.; Mecking S. Single-Step Catalytic Upgrading of Microalgae Biomass. ACS Sustain. Chem. Eng. 2018, 6, 11219–11221. 10.1021/acssuschemeng.8b02939. [DOI] [Google Scholar]
- Rist M.; Greiner A. Synthesis, Characterization, and the Potential for Closed Loop Recycling of Plant Oil-Based PA X.19 Polyamides. ACS Sustain. Chem. Eng. 2022, 10, 16793–16802. 10.1021/acssuschemeng.2c05176. [DOI] [Google Scholar]
- Witt T.; Häußler M.; Kulpa S.; Mecking S. Chain Multiplication of Fatty Acids to Precise Telechelic Polyethylene. Angew. Chem. 2017, 129, 7697–7702. 10.1002/ange.201702796. [DOI] [PubMed] [Google Scholar]
- Stempfle F.; Quinzler D.; Heckler I.; Mecking S. Long-Chain Linear C19 and C23 Monomers and Polycondensates from Unsaturated Fatty Acid Esters. Macromolecules 2011, 44, 4159–4166. 10.1021/ma200627e. [DOI] [Google Scholar]
- Saudan L. A.; Saudan C. M.; Debieux C.; Wyss P. Dihydrogen Reduction of Carboxylic Esters to Alcohols under the Catalysis of Homogeneous Ruthenium Complexes: High Efficiency and Unprecedented Chemoselectivity. Angew. Chem., Int. Ed. 2007, 119, 7617–7620. 10.1002/ange.200701015. [DOI] [PubMed] [Google Scholar]
- Picataggio S.; Rohrer T.; Deanda K.; Lanning D.; Reynolds R.; Mielenz J.; Eirich L. D. Metabolic Engineering of Candida Tropicalis for the Production of Long-Chain Dicarboxylic Acids. Nat. Biotechnol. 1992, 10, 894–898. 10.1038/nbt0892-894. [DOI] [PubMed] [Google Scholar]
- Huf S.; Krügener S.; Hirth T.; Rupp S.; Zibek S. Biotechnological Synthesis of Long-Chain Dicarboxylic Acids as Building Blocks for Polymers. Eur. J. Lipid Sci. Technol. 2011, 113, 548–561. 10.1002/ejlt.201000112. [DOI] [Google Scholar]
- Waltz E. Cathay Industrial Biotech. Nat. Biotechnol. 2012, 30, 480. 10.1038/nbt0612-480. [DOI] [PubMed] [Google Scholar]
- Lu W.; Ness J. E.; Xie W.; Zhang X.; Minshull J.; Gross R. A. Biosynthesis of Monomers for Plastics from Renewable Oils. J. Am. Chem. Soc. 2010, 132, 15451–15455. 10.1021/ja107707v. [DOI] [PubMed] [Google Scholar]
- Liu C.; Liu F.; Cai J.; Xie W.; Long T. E.; Turner S. R.; Lyons A.; Gross R. A. Polymers from Fatty Acids: Poly(ω-Hydroxyl Tetradecanoic Acid) Synthesis and Physico-Mechanical Studies. Biomacromolecules 2011, 12, 3291–3298. 10.1021/bm2007554. [DOI] [PubMed] [Google Scholar]
- Witt T.; Häußler M.; Mecking S. No Strain, No Gain? Enzymatic Ring-Opening Polymerization of Strainless Aliphatic Macrolactones. Macromol. Rapid Commun. 2017, 38, 5780–5785. 10.1002/marc.201600638. [DOI] [PubMed] [Google Scholar]
- Myers D.; Witt T.; Cyriac A.; Bown M.; Mecking S.; Williams C. K. Ring Opening Polymerization of Macrolactones: High Conversions and Activities Using an Yttrium Catalyst. Polym. Chem. 2017, 8, 5780–5785. 10.1039/C7PY00985B. [DOI] [Google Scholar]
- van der Meulen I.; Gubbels E.; Huijser S.; Sablong R.; Koning C. E.; Heise A.; Duchateau R. Catalytic Ring-Opening Polymerization of Renewable Macrolactones to High Molecular Weight Polyethylene-like Polymers. Macromolecules 2011, 44, 4301–4305. 10.1021/ma200685u. [DOI] [Google Scholar]
- Panten J.; Surburg H.; Hölscher B. New Oxa-Bridged Macrocycles. Chem. Biodivers. 2008, 5, 1011–1022. 10.1002/cbdv.200890081. [DOI] [PubMed] [Google Scholar]
- Pepels M. P. F.; Govaert L. E.; Duchateau R. Influence of the Main-Chain Configuration on the Mechanical Properties of Linear Aliphatic Polyesters. Macromolecules 2015, 48, 5845–5854. 10.1021/acs.macromol.5b01089. [DOI] [Google Scholar]
- Boden E. P.; Keck G. E. Proton-Transfer Steps in Steglich Esterification: a Very Practical New Method for Macrolactonization. J. Org. Chem. 1985, 50, 2394–2395. 10.1021/jo00213a044. [DOI] [Google Scholar]
- Witt T.; Mecking S. Large-Ring Lactones from Plant Oils. Green Chem. 2013, 15, 2361. 10.1039/c3gc40905h. [DOI] [Google Scholar]
- Zhou L.; Qin P.; Wu L.; Li B.-G.; Dubois P. Potentially Biodegradable “Short-Long” Type Diol-Diacid Polyesters with Superior Crystallizability, Tensile Modulus, and Water Vapor Barrier. ACS Sustain. Chem. Eng. 2021, 9, 17362–17370. 10.1021/acssuschemeng.1c06752. [DOI] [Google Scholar]
- Zhou L.; Wu L.; Qin P.; Li B.-G. Synthesis and Properties of Long Chain Polyesters from Biobased 1,5-Pentanediol and Aliphatic α,ω-Diacids with 10–16 Carbon Atoms. Polym. Degrad. Stab. 2021, 187, 109546. 10.1016/j.polymdegradstab.2021.109546. [DOI] [Google Scholar]
- Eck M.; Schwab S. T.; Nelson T. F.; Wurst K.; Iberl S.; Schleheck D.; Link C.; Battagliarin G.; Mecking S. Biodegradable High-Density Polyethylene-like Material. Angew. Chem., Int. Ed. 2023, 135, e202213438 10.1002/ange.202213438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häußler M.; Eck M.; Rothauer D.; Mecking S. Closed-Loop Recycling of Polyethylene-Like Materials. Nature 2021, 590, 423–427. 10.1038/s41586-020-03149-9. [DOI] [PubMed] [Google Scholar]
- Stempfle F.; Ritter B. S.; Mülhaupt R.; Mecking S. Long-Chain Aliphatic Polyesters from Plant Oil for injection molding, film extrusion and electrospinning. Green Chem. 2014, 16, 2008. 10.1039/c4gc00114a. [DOI] [Google Scholar]
- Menges M. G.; Penelle J.; Le Fevere de Ten Hove C.; Jonas A. M.; Schmidt-Rohr K. Characterization of Long-Chain Aliphatic Polyesters: Crystalline and Supramolecular Structure of PE22,4 Elucidated by X-ray Scattering and Nuclear Magnetic Resonance. Macromolecules 2007, 40, 8714–8725. 10.1021/ma071590p. [DOI] [Google Scholar]
- Pepels M. P. F.; Hansen M. R.; Goossens H.; Duchateau R. From Polyethylene to Polyester: Influence of Ester Groups on the Physical Properties. Macromolecules 2013, 46, 7668–7677. 10.1021/ma401403x. [DOI] [Google Scholar]
- Stempfle F.; Ortmann P.; Mecking S. Which Polyesters can Mimic Polyethylene?. Macromol. Rapid Commun. 2013, 34, 47–50. 10.1002/marc.201200611. [DOI] [PubMed] [Google Scholar]
- Häußler M.Polyethylene-like Building Blocks from Plant Oils for Recyclable Polymers, Nanocrystals and Ion-Conductive Materials, Dissertation; Universität Konstanz, 2020. [Google Scholar]
- Nelson T. F.; Rothauer D.; Sander M.; Mecking S. Degradable and Recyclable Polyesters from Multiple Chain Length Bio- and Waste-Sourceable Monomers. Angew. Chem., Int. Ed. Engl. 2023, 62, e202310729 10.1002/anie.202310729. [DOI] [PubMed] [Google Scholar]
- Ortmann P.; Mecking S. Long-Spaced Aliphatic Polyesters. Macromolecules 2013, 46, 7213–7218. 10.1021/ma401305u. [DOI] [Google Scholar]
- Ortmann P.; Heckler I.; Mecking S. Physical Properties and Hydrolytic Degradability of Polyethylene-like Polyacetals and Polycarbonates. Green Chem. 2014, 16, 1816. 10.1039/c3gc42592d. [DOI] [Google Scholar]
- Pepels M. P. F.; Koeken R. A. C.; van der Linden S. J. J.; Heise A.; Duchateau R. Mimicking (Linear) Low-Density Polyethylenes Using Modified Polymacrolactones. Macromolecules 2015, 48, 4779–4792. 10.1021/acs.macromol.5b00820. [DOI] [Google Scholar]
- Gigli M.; Genovese L.; Lotti N.; Munari A.; Dalla Rosa M.; Siracusa V. Gas Barrier and Thermal Behavior of Long Chain Aliphatic Polyesters after Stressed Treatments. Polym.-Plast. Technol. Eng. 2017, 56, 71–82. 10.1080/03602559.2016.1211686. [DOI] [Google Scholar]
- Saumer A.; Mecking S. Recyclable and Degradable Ionic-Substituted Long-Chain Polyesters. ACS Sustainable Chem. Eng. 2023, 11, 12414–12422. 10.1021/acssuschemeng.3c03141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill J. H. Formation of Spherulites in Polyamide Melts: Part III. Even-Even Polyamides. J. Polym. Sci. A-2 Polym. Phys. 1966, 4, 243–265. 10.1002/pol.1966.160040207. [DOI] [Google Scholar]
- Bennett C.; Kaya E.; Sikes A. M.; Jarrett W. L.; Mathias L. J. Synthesis and Characterization of Nylon 18 18 and Nylon 18 Adamantane. J. Polym. Sci. A-2 Polym. Chem. 2009, 47, 4409–4419. 10.1002/pola.23494. [DOI] [Google Scholar]
- Ortmann P.; Lemke T. A.; Mecking S. Long-Spaced Polyamides: Elucidating the Gap between Polyethylene Crystallinity and Hydrogen Bonding. Macromolecules 2015, 48, 1463–1472. 10.1021/acs.macromol.5b00060. [DOI] [Google Scholar]
- Kolb N.; Winkler M.; Syldatk C.; Meier M. A. Long-Chain Polyesters and Polyamides from Biochemically Derived Fatty Acids. Eur. Polym. J. 2014, 51, 159–166. 10.1016/j.eurpolymj.2013.11.007. [DOI] [Google Scholar]
- Haider T.; Shyshov O.; Suraeva O.; Lieberwirth I.; von Delius M.; Wurm F. R. Long-Chain Polyorthoesters as Degradable Polyethylene Mimics. Macromolecules 2019, 52, 2411–2420. 10.1021/acs.macromol.9b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraeva O.; Champanhac C.; Mailänder V.; Wurm F. R.; Weiss H.; Berger R.; Mezger M.; Landfester K.; Lieberwirth I. Vitamin C Loaded Polyethylene: Synthesis and Properties of Precise Polyethylene with Vitamin C Defects via Acyclic Diene Metathesis Polycondensation. Macromolecules 2020, 53, 2932–2941. 10.1021/acs.macromol.0c00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch H.; Schiebel E.; Sickinger A.; Mecking S. Ultralong-Chain-Spaced Crystalline Poly(H-phosphonate)s and Poly(phenylphosphonate)s. Macromolecules 2017, 50, 7901–7910. 10.1021/acs.macromol.7b01368. [DOI] [Google Scholar]
- Busch H.; Majumder S.; Reiter G.; Mecking S. Semicrystalline Long-Chain Polyphosphoesters from Polyesterification. Macromolecules 2017, 50, 2706–2713. 10.1021/acs.macromol.7b00243. [DOI] [Google Scholar]
- Tee H. T.; Koynov K.; Reichel T.; Wurm F. R. Noncovalent Hydrogen Bonds Tune the Mechanical Properties of Phosphoester Polyethylene Mimics. ACS Omega 2019, 4, 9324–9332. 10.1021/acsomega.9b01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider T. P.; Suraeva O.; Lieberwirth I.; Paneth P.; Wurm F. R. RNA-Inspired Intramolecular Transesterification Accelerates the Hydrolysis of Polyethylene-Like Polyphosphoesters. Chem. Sci. 2021, 12, 16054–16064. 10.1039/D1SC05509G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider T.; Suraeva O.; O’Duill M. L.; Mars J.; Mezger M.; Lieberwirth I.; Wurm F. R. Controlling the Crystal Structure of Precisely Spaced Polyethylene-Like Polyphosphoesters. Polym. Chem. 2020, 11, 3404–3415. 10.1039/D0PY00272K. [DOI] [Google Scholar]
- Tee H. T.; Lieberwirth I.; Wurm F. R. Aliphatic Long-Chain Polypyrophosphates as Biodegradable Polyethylene Mimics. Macromolecules 2019, 52, 1166–1172. 10.1021/acs.macromol.8b02474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemba A. G.; Flores J. A.; Miller S. A. Acetal Metathesis Polymerization (AMP): A Method for Synthesizing Biorenewable Polyacetals. Green Chem. 2013, 15, 325. 10.1039/c2gc36588j. [DOI] [Google Scholar]
- Chikkali S.; Stempfle F.; Mecking S. Long-Chain Polyacetals from Plant Oils. Macromol. Rapid Commun. 2012, 33, 1126–1129. 10.1002/marc.201200226. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Yue X.; Sun Y.; Zhang C.; Zhang X. Chemically Recyclable Polyethylene-like Sulfur-Containing Plastics from Sustainable Feedstocks. Angew. Chem., Int. Ed. 2023, 62, e202219251 10.1002/anie.202219251. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Qin K.-X.; Gan M.-X.; Xu J.; Li Z.-L.; Li Z.-C. Recyclable Biomass-Derived Polyethylene-Like Materials as Functional Coatings for Commercial Fabrics: Toward Upcycling of Waste Textiles. ACS Sustainable Chem. Eng. 2022, 10, 17187–17197. 10.1021/acssuschemeng.2c05080. [DOI] [Google Scholar]
- Benyathiar P.; Kumar P.; Carpenter G.; Brace J.; Mishra D. K. Polyethylene Terephthalate (PET) Bottle-to-Bottle Recycling for the Beverage Industry: A Review. Polymers 2022, 14, 2366. 10.3390/polym14122366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gutierrez P.; et al. Environmental and Economic Assessment of Plastic Waste Recycling: A Comparison of Mechanical, Physical, Chemical Recycling and Energy Recovery of Plastic Waste , Technical Report EUR 31423 EN; Publications Office of the European Union, 2023. 10.2760/0472 [DOI]
- Uekert T.; DesVeaux J. S.; Singh A.; Nicholson S. R.; Lamers P.; Ghosh T.; McGeehan J. E.; Carpenter A. C.; Beckham G. T. Life Cycle Assessment of Enzymatic Poly(Ethylene Terephthalate) Recycling. Green Chem. 2022, 24, 6531–6543. 10.1039/D2GC02162E. [DOI] [Google Scholar]
- Marathe M. N.; Dabholkar D. A.; Jain M. K.. Process for the Recovery of Dimethyl Terephthalate from Polyethylene Terephthalate: GB2041916A, 1980.
- Johnson A.; Johnson J. A. Deconstructable and Chemically Recyclable High-Density Polyethylene (HDPE)-Like Materials Based on Si-O Bonds. Angew. Chem., Int. Ed. Engl. 2023, 62, e202315085 10.1002/anie.202315085. [DOI] [PubMed] [Google Scholar]
- Mihut C.; Captain D. K.; Gadala-Maria F.; Amiridis M. D. Review: Recycling of Nylon from Carpet Waste. Polym. Eng. Sci. 2001, 41, 1457–1470. 10.1002/pen.10845. [DOI] [Google Scholar]
- Abel B. A.; Snyder R. L.; Coates G. W. Chemically Recyclable Thermoplastics from Reversible-Deactivation Polymerization of Cyclic Acetals. Science 2021, 373, 783–789. 10.1126/science.abh0626. [DOI] [PubMed] [Google Scholar]
- Hester H. G.; Abel B. A.; Coates G. W. Ultra-High-Molecular-Weight Poly(Dioxolane): Enhancing the Mechanical Performance of a Chemically Recyclable Polymer. J. Am. Chem. Soc. 2023, 145, 8800–8804. 10.1021/jacs.3c01901. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Zhang Z.; Shi C.; Scoti M.; Barange D. K.; Gowda R. R.; Chen E. Y.-X. Chemically Circular, Mechanically Tough, and Melt-Processable Polyhydroxyalkanoates. Science 2023, 380, 64–69. 10.1126/science.adg4520. [DOI] [PubMed] [Google Scholar]
- Schyns Z. O. G.; Shaver M. P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2021, 42, e2000415 10.1002/marc.202000415. [DOI] [PubMed] [Google Scholar]
- Soares C. T. d. M.; Ek M.; Östmark E.; Gällstedt M.; Karlsson S. Recycling of Multi-Material Multilayer Plastic Packaging: Current Trends and Future Scenarios. Resour. Conserv. Recycl. 2022, 176, 105905. 10.1016/j.resconrec.2021.105905. [DOI] [Google Scholar]
- Ait-Touchente Z.; Khellaf M.; Raffin G.; Lebaz N.; Elaissari A. Recent Advances in Polyvinyl Chloride (PVC) Recycling. Polym. Adv. Technol. 2024, 35, e6228 10.1002/pat.6228. [DOI] [Google Scholar]
- Hill M.; Barham P.; Keller A. Phase Segregation in Blends of Linear with Branched Polyethylene: The Effect of Varying the Molecular Weight of the Linear Polymer. Polymer 1992, 33, 2530–2541. 10.1016/0032-3861(92)91134-N. [DOI] [Google Scholar]
- Graziano A.; Jaffer S.; Sain M. Review on Modification Strategies of Polyethylene/Polypropylene Immiscible Thermoplastic Polymer Blends for Enhancing their Mechanical Behavior. J. Elastomers Plast. 2019, 51, 291–336. 10.1177/0095244318783806. [DOI] [Google Scholar]
- Eagan J. M.; Xu J.; Di Girolamo R.; Thurber C. M.; Macosko C. W.; LaPointe A. M.; Bates F. S.; Coates G. W. Combining Polyethylene and Polypropylene: Enhanced Performance with PE/iPP Multiblock polymers. Science 2017, 355, 814–816. 10.1126/science.aah5744. [DOI] [PubMed] [Google Scholar]
- Pepels M. P.; Kleijnen R. G.; Goossens J. G.; Spoelstra A. B.; Tandler R.; Martens H.; Soliman M.; Duchateau R. Compatibility and Epitaxial Crystallization between Poly(Ethylene) and Poly(Ethylene)-Like polyesters. Polymer 2016, 88, 63–70. 10.1016/j.polymer.2016.01.035. [DOI] [Google Scholar]
- Wang G.-X.; Huang D.; Ji J.-H.; Völker C.; Wurm F. R. Seawater-Degradable Polymers-Fighting the Marine Plastic Pollution. Adv. Sci. 2021, 8, 2001121. 10.1002/advs.202001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller R.-J. Biological Degradation of Synthetic Polyesters—Enzymes as Potential Catalysts for Polyester Recycling. Process Biochem. 2006, 41, 2124–2128. 10.1016/j.procbio.2006.05.018. [DOI] [Google Scholar]
- van der Meulen I.; de Geus M.; Antheunis H.; Deumens R.; Joosten E. A. J.; Koning C. E.; Heise A. Polymers from Functional Macrolactones as Potential Biomaterials: Enzymatic Ring Opening Polymerization, Biodegradation, and Biocompatibility. Biomacromolecules 2008, 9, 3404–3410. 10.1021/bm800898c. [DOI] [PubMed] [Google Scholar]
- Marten E.; Müller R.-J.; Deckwer W.-D. Studies on the Enzymatic Hydrolysis of Polyesters I. Low molecular Mass Model Esters and Aliphatic Polyesters. Polym. Degrad. Stab. 2003, 80, 485–501. 10.1016/S0141-3910(03)00032-6. [DOI] [Google Scholar]
- Zumstein M. T.; Rechsteiner D.; Roduner N.; Perz V.; Ribitsch D.; Guebitz G. M.; Kohler H.-P. E.; McNeill K.; Sander M. Enzymatic Hydrolysis of Polyester Thin Films at the Nanoscale: Effects of Polyester Structure and Enzyme Active-Site Accessibility. Environ. Sci. Technol. 2017, 51, 7476–7485. 10.1021/acs.est.7b01330. [DOI] [PubMed] [Google Scholar]
- Steinbach T.; Wurm F. R. Poly(phosphoester)s: A New Platform for Degradable Polymers. Angew. Chem., Int. Ed. 2015, 54, 6098–6108. 10.1002/anie.201500147. [DOI] [PubMed] [Google Scholar]
- Eck M.; Bernabeu L.; Mecking S. Polyethylene-Like Blends Amenable to Abiotic Hydrolytic Degradation. ACS Sustain. Chem. Eng. 2023, 11, 4523–4530. 10.1021/acssuschemeng.2c07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrish R. G. W.; Bamford C. H. Photo-Decomposition of Aldehydes and Ketones. Nature 1937, 140, 195–196. 10.1038/140195b0. [DOI] [Google Scholar]
- Hartley G. H.; Guillet J. E. Photochemistry of Ketone Polymers. I. Studies of Ethylene-Carbon Monoxide Copolymers. Macromolecules 1968, 1, 165–170. 10.1021/ma60002a012. [DOI] [Google Scholar]
- Geyer R.; Jambeck J. R.; Law K. L. Production, Use, and Fate of All Plastics Ever Made. Sci. Adv. 2017, 3, e1700782 10.1126/sciadv.1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Kim S.; Wahl L.; Khare R.; Hale L.; Hu J.; Camaioni D. M.; Gutiérrez O. Y.; Liu Y.; Lercher J. A. Low-Temperature Upcycling of Polyolefins into Liquid Alkanes via Tandem Cracking-Alkylation. Science 2023, 379, 807–811. 10.1126/science.ade7485. [DOI] [PubMed] [Google Scholar]
- Celik G.; Kennedy R. M.; Hackler R. A.; Ferrandon M.; Tennakoon A.; Patnaik S.; LaPointe A. M.; Ammal S. C.; Heyden A.; Perras F. A.; et al. Upcycling Single-Use Polyethylene into High-Quality Liquid Products. ACS Cent. Sci. 2019, 5, 1795–1803. 10.1021/acscentsci.9b00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Zeng M.; Yappert R. D.; Sun J.; Lee Y.-H.; LaPointe A. M.; Peters B.; Abu-Omar M. M.; Scott S. L. Polyethylene Upcycling to Long-Chain Alkylaromatics by Tandem Hydrogenolysis/Aromatization. Science 2020, 370, 437–441. 10.1126/science.abc5441. [DOI] [PubMed] [Google Scholar]
- Westlie A. H.; Chen E. Y.-X.; Holland C. M.; Stahl S. S.; Doyle M.; Trenor S. R.; Knauer K. M. Polyolefin Innovations toward Circularity and Sustainable Alternatives. Macromol. Rapid Commun. 2022, 43, e2200492 10.1002/marc.202200492. [DOI] [PubMed] [Google Scholar]
- Martín A. J.; Mondelli C.; Jaydev S. D.; Pérez-Ramírez J. Catalytic Processing of Plastic Waste on the Rise. Chem. 2021, 7, 1487–1533. 10.1016/j.chempr.2020.12.006. [DOI] [Google Scholar]
- Tennakoon A.; Wu X.; Paterson A. L.; Patnaik S.; Pei Y.; LaPointe A. M.; Ammal S. C.; Hackler R. A.; Heyden A.; Slowing I. I.; et al. Catalytic Upcycling of High-Density Polyethylene via a Processive Mechanism. Nat. Catal. 2020, 3, 893–901. 10.1038/s41929-020-00519-4. [DOI] [Google Scholar]
- Liu S.; Kots P. A.; Vance B. C.; Danielson A.; Vlachos D. G. Plastic Waste to Fuels by Hydrocracking at Mild Conditions. Sci. Adv. 2021, 7, eabf8283 10.1126/sciadv.abf8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X.; Qin C.; Friedberger T.; Guan Z.; Huang Z. Efficient and Selective Degradation of Polyethylenes into Liquid Fuels and Waxes under Mild Conditions. Sci. Adv. 2016, 2, e1501591 10.1126/sciadv.1501591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlanés N.; Kavitake S. G.; Rosenfeld D. C.; Basset J.-M. Alkane Cross-Metathesis Reaction between Light and Heavy Linear Alkanes, on a Silica Supported Well-Defined Single-Site Catalyst. ACS Catal. 2019, 9, 1274–1282. 10.1021/acscatal.8b02472. [DOI] [Google Scholar]
- Patil V. B.; Saliu K. O.; Jenkins R. M.; Carnahan E. M.; Kramer E. J.; Fredrickson G. H.; Bazan G. C. Efficient Synthesis of α,ω-Divinyl-Functionalized Polyolefins. Macromol. Chem. Phys. 2014, 215, 1140–1145. 10.1002/macp.201400139. [DOI] [Google Scholar]
- Zeng M.; Lee Y.-H.; Strong G.; LaPointe A. M.; Kocen A. L.; Qu Z.; Coates G. W.; Scott S. L.; Abu-Omar M. M. Chemical Upcycling of Polyethylene to Value-Added α,ω-Divinyl-Functionalized Oligomers. ACS Sustain. Chem. Eng. 2021, 9, 13926–13936. 10.1021/acssuschemeng.1c05272. [DOI] [Google Scholar]
- Fraser C.; Hillmyer M. A.; Gutierrez E.; Grubbs R. H. Degradable Cyclooctadiene/Acetal Copolymers: Versatile Precursors to 1,4-Hydroxytelechelic Polybutadiene and Hydroxytelechelic Polyethylene. Macromolecules 1995, 28, 7256–7261. 10.1021/ma00125a031. [DOI] [Google Scholar]
- Marmo J. C.; Wagener K. B. Metathetical Degradation and Depolymerization of Unsaturated Polymers. Rubber Chem. Technol. 1997, 70, 519–529. 10.5254/1.3538439. [DOI] [Google Scholar]
- Mathers R. T.; Coates G. W. Cross Metathesis Functionalization of Polyolefins. Chem. Commun. 2004, 422–423. 10.1039/b313954a. [DOI] [PubMed] [Google Scholar]
- Sinclair F.; Alkattan M.; Prunet J.; Shaver M. P. Olefin Cross Metathesis and Ring-Closing Metathesis in Polymer Chemistry. Polym. Chem. 2017, 8, 3385–3398. 10.1039/C7PY00340D. [DOI] [Google Scholar]
- Arroyave A.; Cui S.; Lopez J. C.; Kocen A. L.; LaPointe A. M.; Delferro M.; Coates G. W. Catalytic Chemical Recycling of Post-Consumer Polyethylene. J. Am. Chem. Soc. 2022, 144, 23280–23285. 10.1021/jacs.2c11949. [DOI] [PubMed] [Google Scholar]
- Ray A.; Kissin Y. V.; Zhu K.; Goldman A. S.; Cherian A. E.; Coates G. W. Catalytic Post-Modification of Alkene Polymers. J. Mol. Catal. A Chem. 2006, 256, 200–207. 10.1016/j.molcata.2006.04.017. [DOI] [Google Scholar]
- Choi J.; MacArthur A. H. R.; Brookhart M.; Goldman A. S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. 10.1021/cr1003503. [DOI] [PubMed] [Google Scholar]
- Wang N. M.; Strong G.; DaSilva V.; Gao L.; Huacuja R.; Konstantinov I. A.; Rosen M. S.; Nett A. J.; Ewart S.; Geyer R.; et al. Chemical Recycling of Polyethylene by Tandem Catalytic Conversion to Propylene. J. Am. Chem. Soc. 2022, 144, 18526–18531. 10.1021/jacs.2c07781. [DOI] [PubMed] [Google Scholar]
- Conk R. J.; Hanna S.; Shi J. X.; Yang J.; Ciccia N. R.; Qi L.; Bloomer B. J.; Heuvel S.; Wills T.; Su J.; et al. Catalytic Deconstruction of Waste Polyethylene with Ethylene to Form Propylene. Science 2022, 377, 1561–1566. 10.1126/science.add1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttker-Schnetmann I.; White P.; Brookhart M. Iridium Bis(Phosphinite) p-XPCP Pincer Complexes: Highly Active Catalysts for the Transfer Dehydrogenation of Alkanes. J. Am. Chem. Soc. 2004, 126, 1804–1811. 10.1021/ja0385235. [DOI] [PubMed] [Google Scholar]
- Allen K. E.; Heinekey D. M.; Goldman A. S.; Goldberg K. I. Regeneration of an Iridium(III) Complex Active for Alkane Dehydrogenation Using Molecular Oxygen. Organometallics 2014, 33, 1337–1340. 10.1021/om401241e. [DOI] [Google Scholar]
- Allen K. E.; Heinekey D. M.; Goldman A. S.; Goldberg K. I. Alkane Dehydrogenation by C-H Activation at Iridium(III). Organometallics 2013, 32, 1579–1582. 10.1021/om301267c. [DOI] [Google Scholar]
- Goldberg K. I.; Goldman A. S. Large-Scale Selective Functionalization of Alkanes. Acc. Chem. Res. 2017, 50, 620–626. 10.1021/acs.accounts.6b00621. [DOI] [PubMed] [Google Scholar]
- Guironnet D.; Peters B. Tandem Catalysts for Polyethylene Upcycling: A Simple Kinetic Model. J. Phys. Chem. 2020, 124, 3935–3942. 10.1021/acs.jpca.0c01363. [DOI] [PubMed] [Google Scholar]
- Ballard D.; Dawkins J. V. Preparation and Chain Extension of C100dicarboxylic acid. Eur. Polym. J. 1973, 9, 211–217. 10.1016/0014-3057(73)90128-6. [DOI] [Google Scholar]
- Guzik M. W.; Nitkiewicz T.; Wojnarowska M.; Sołtysik M.; Kenny S. T.; Babu R. P.; Best M.; O’Connor K. E. Robust Process for High Yield Conversion of Non-Degradable Polyethylene to a Biodegradable Plastic Using a Chemo-Biotechnological Approach. Waste Manage. 2021, 135, 60–69. 10.1016/j.wasman.2021.08.030. [DOI] [PubMed] [Google Scholar]
- Sullivan K. P.; Werner A. Z.; Ramirez K. J.; Ellis L. D.; Bussard J. R.; Black B. A.; Brandner D. G.; Bratti F.; Buss B. L.; Dong X.; et al. Mixed Plastics Waste Valorization through Tandem Chemical Oxidation and Biological Funneling. Science 2022, 378, 207–211. 10.1126/science.abo4626. [DOI] [PubMed] [Google Scholar]
- Alter H. Metal-Catalyzed Oxidation of Polyethylene. Ind. Eng. Chem. 1960, 52, 121–124. 10.1021/ie50602a026. [DOI] [Google Scholar]
- Palmer R. P.; Cobbold A. J. The Texture of Melt Crystallised Polythene as Revealed by Selective Oxidation. Makromol. Chem. 1964, 74, 174–189. 10.1002/macp.1964.020740114. [DOI] [Google Scholar]
- Keller A.; Udagawa Y. Preparation of Long-Chain Paraffins from Nitric Acid Degradation Products of Polyethylene; Some Consequences for Polymer Crystal Structure Studies. J. Polym. Sci. A-2 Polym. Phys. 1970, 8, 19–34. 10.1002/pol.1970.160080102. [DOI] [Google Scholar]
- Melby L. R. Nitric Acid Oxidation of High-Density Polyethylene. Organic Chemical Aspects. Macromolecules 1978, 11, 50–56. 10.1021/ma60061a010. [DOI] [Google Scholar]
- Yao J. Y.; Wang Y. W.; Muppaneni T.; Shrestha R.; Le Roy J.; Figuly G. D.. Products from the Decomposition of Plastic Waste: US11192999B2, 2019.
- Pinsuwan K.; Opaprakasit P.; Petchsuk A.; Dubas L.; Opaprakasit M. Chemical Recycling of High-Density Polyethylene (HDPE) Wastes by Oxidative Degradation to Dicarboxylic Acids and their Use as Value-Added Curing Agents for Acrylate-Based Materials. Polym. Degrad. Stab. 2023, 210, 110306. 10.1016/j.polymdegradstab.2023.110306. [DOI] [Google Scholar]
- Bäckström E.; Odelius K.; Hakkarainen M. Trash to Treasure: Microwave-Assisted Conversion of Polyethylene to Functional Chemicals. Ind. Eng. Chem. Res. 2017, 56, 14814–14821. 10.1021/acs.iecr.7b04091. [DOI] [Google Scholar]
- Pifer A.; Sen A. Chemical Recycling of Plastics to Useful Organic Compounds by Oxidative Degradation. Angew. Chem., Int. Ed. 1998, 37, 3306–3308. 10.1002/(SICI)1521-3773(19981217)37:23<3306::AID-ANIE3306>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Chow C.-F.; Wong W.-L.; Ho K. Y.-F.; Chan C.-S.; Gong C.-B. Combined Chemical Activation and Fenton Degradation to Convert Waste Polyethylene into High-Value Fine Chemicals. Chem.-Eur. J. 2016, 22, 9513–9518. 10.1002/chem.201600856. [DOI] [PubMed] [Google Scholar]
- Tomás R. A. F.; Bordado J. C. M.; Gomes J. F. P. p-Xylene Oxidation to Terephthalic Acid: A Literature Review Oriented toward Process Optimization and Development. Chem. Rev. 2013, 113, 7421–7469. 10.1021/cr300298j. [DOI] [PubMed] [Google Scholar]
- Ishii Y.; Sakaguchi S. Recent Progress in Aerobic Oxidation of Hydrocarbons by N-Hydroxyimides. Catal. Today 2006, 117, 105–113. 10.1016/j.cattod.2006.05.006. [DOI] [Google Scholar]
- Rabot C.; Chen Y.; Bijlani S.; Chiang Y.-M.; Oakley C. E.; Oakley B. R.; Williams T. J.; Wang C. C. C. Conversion of Polyethylenes into Fungal Secondary Metabolites. Angew. Chem., Int. Ed. 2023, 62, e202214609 10.1002/anie.202214609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häußler M.; Odenwald L.. Utilization of Polyethylene-Containing Mixtures to Form Long-Chain Alkyl Dicarboxylic Acids by Way of Oxidative Degradation: WO 2023/118135 A1, 2022.
- Zawadiak J.; Orlińska B.; Marek A. A. Catalytic Oxidation of Polyethylene with Oxygen in Aqueous Dispersion. J. Appl. Polym. Sci. 2013, 127, 976–981. 10.1002/app.37515. [DOI] [Google Scholar]
- Wang K.; Jia R.; Cheng P.; Shi L.; Wang X.; Huang L. Highly Selective Catalytic Oxi-upcycling of Polyethylene to Aliphatic Dicarboxylic Acid under a Mild Hydrogen-Free Process. Angew. Chem., Int. Ed. 2023, 62, e202301340 10.1002/anie.202301340. [DOI] [PubMed] [Google Scholar]
- Sanluis-Verdes A.; Colomer-Vidal P.; Rodriguez-Ventura F.; Bello-Villarino M.; Spinola-Amilibia M.; Ruiz-Lopez E.; Illanes-Vicioso R.; Castroviejo P.; Aiese Cigliano R.; Montoya M.; et al. Wax Worm Saliva and the Enzymes therein are the Key to Polyethylene Degradation by Galleria Mellonella. Nat. Commun. 2022, 13, 5568. 10.1038/s41467-022-33127-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampolli J.; Mangiagalli M.; Vezzini D.; Lasagni M.; Ami D.; Natalello A.; Arrigoni F.; Bertini L.; Lotti M.; Di Gennaro P. Oxidative Degradation of Polyethylene by two Novel Laccase-Like Multicopper Oxidases from Rhodococcus Opacus R7. Environ. Technol. Innov. 2023, 32, 103273. 10.1016/j.eti.2023.103273. [DOI] [Google Scholar]
- Knauer K. M.; Le Roy J.; Higginson C. J.; Pratt R. C.; Pilsk D. S.. Thermoplastic Polyurethane Compositions Comprising Nitro-Substituted Polyester Diols: US11028217B1, 2020.
- Xu Z.; Munyaneza N. E.; Zhang Q.; Sun M.; Posada C.; Venturo P.; Rorrer N. A.; Miscall J.; Sumpter B. G.; Liu G. Chemical upcycling of polyethylene, polypropylene, and mixtures to high-value surfactants. Science 2023, 381, 666–671. 10.1126/science.adh0993. [DOI] [PubMed] [Google Scholar]
- Guzik M. W.; Kenny S. T.; Duane G. F.; Casey E.; Woods T.; Babu R. P.; Nikodinovic-Runic J.; Murray M.; O’Connor K. E. Conversion of Post Consumer Polyethylene to the Biodegradable Polymer Polyhydroxyalkanoate. Appl. Microbiol. Biotechnol. 2014, 98, 4223–4232. 10.1007/s00253-013-5489-2. [DOI] [PubMed] [Google Scholar]
- Radecka I.; Irorere V.; Jiang G.; Hill D.; Williams C.; Adamus G.; Kwiecień M.; Marek A. A.; Zawadiak J.; Johnston B. Oxidized Polyethylene Wax as a Potential Carbon Source for PHA Production. Materials 2016, 9, 367. 10.3390/ma9050367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albonetti S.; Cavani F.; Trifirò F. Key Aspects of Catalyst Design for the Selective Oxidation of Paraffins. Catalysis Reviews 1996, 38, 413–438. 10.1080/01614949608006463. [DOI] [Google Scholar]
- Gregory G. J.; Wang C.; Sadula S.; Koval S.; Lobo R. F.; Vlachos D. G.; Papoutsakis E. T. Polyethylene Valorization by Combined Chemical Catalysis with Bioconversion by Plastic-Enriched Microbial Consortia. ACS Sustain. Chem. Eng. 2023, 11, 3494–3505. 10.1021/acssuschemeng.2c07461. [DOI] [Google Scholar]
- Roy P. K.; Hakkarainen M.; Varma I. K.; Albertsson A.-C. Degradable Polyethylene: Fantasy or Reality. Environ. Sci. Technol. 2011, 45, 4217–4227. 10.1021/es104042f. [DOI] [PubMed] [Google Scholar]
- Walsh A. N.; Reddy C. M.; Niles S. F.; McKenna A. M.; Hansel C. M.; Ward C. P. Plastic Formulation is an Emerging Control of Its Photochemical Fate in the Ocean. Environ. Sci. Technol. 2021, 55, 12383–12392. 10.1021/acs.est.1c02272. [DOI] [PubMed] [Google Scholar]
- European Commission, Directorate General for Environment . Turning the Tide on Single-Use Plastics ;Publications Office, 2019. 10.2779/294711 [DOI] [Google Scholar]
- Zumstein M. T.; Narayan R.; Kohler H.-P. E.; McNeill K.; Sander M. Dos and Do Nots When Assessing the Biodegradation of Plastics. Environ. Sci. Technol. 2019, 53, 9967–9969. 10.1021/acs.est.9b04513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.