

Abstract
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders characterized by a wide phenotypic range including severe motor and cognitive impairments, microcephaly, distinctive facial features, and other features according to the type. Several classes of PCH1 have been linked to mutations in the evolutionarily conserved RNA exosome complex that consists of 9 subunits (EXOSC1 to EXOSC9) and facilitates the degradation and processing of cytoplasmic and nuclear RNA from the 3’ end. Only a single individual with an EXOSC1 mutation was reported with clinical features of pontocerebellar hypoplasia type 1 (PCH1F). Here, we report a 3-month-old female with pontocerebellar hypoplasia and additional clinical features not previously reported to be associated with PCH1, including dilated cardiomyopathy. On assessment, failure to thrive, microcephaly, distinctive facial features, and bluish sclera, were noted. Whole-exome sequencing was performed and revealed a novel homozygous missense variant c.547C>T (p.Arg183Trp) in the EXOSC1 gene. Functional studies in a budding yeast model that expresses the human EXOSC1 variant Arg183Trp show a slow-growth phenotype, whereas the previously identified PCH1F allele EXOSC1-Ser35Leu is lethal, indicating impaired exosome function for both of these variants. The protein levels of both EXOSC1 variants are reduced compared to wild type when expressed in budding yeast. Herein, we ascertain the second case of pontocerebellar hypoplasia associated with a EXOSC1 variant that causes defects in RNA exosome function and provide a model organism system to distinguish between benign and pathogenic variants in EXOSC1.
Keywords: Pontocerebellar hypoplasia type 1, EXOSC1, dilated cardiomyopathy, exosomopathy
Introduction
Pontocerebellar hypoplasia (PCH) represents a heterogeneous group of rare neurodegenerative disorders characterized by severe hypoplasia or atrophy of the cerebellum and pons with variable involvement of supratentorial structures (Rudnik-Schöneborn et al., 2014). PCH follows an autosomal recessive hereditary model with 17 different types classified to date, resulting from biallelic pathogenic variants in at least 27 different genes according to Online Mendelian Inheritance in Man (OMIM.org) (as of Sep 2, 2022). Most affected individuals present with motor and cognitive impairments, microcephaly, epilepsy, and other features according to the type (Van Dijk et al., 2018). Diagnosis is based on genetic testing, neuroimaging studies, and neurological symptoms (Appelhof et al., 2019).
The RNA exosome is a ribonuclease complex that has a fundamental role in monitoring, processing, and degradation of different types of RNA in the nucleus and cytoplasm (Chlebowski et al., 2013; Januszyk & Lima, 2014). The RNA exosome comprises a core multiprotein complex that includes a six-subunit barrel-shaped central channel (EXOSC4–9) and a three-subunit cap (EXOSC 1–3). This core associates with 3’ to 5’ exoribonucleases DIS3/DIS3L, and EXOSC10. Multiple genes encoding RNA exosome subunits have been associated with PCH such as EXOSC3 (PCH1B), EXOSC8 (PCH1C), and EXOSC9 (PCH1D) (Wan et al. 2012; Boczonadi et al. 2014; Burns et al. 2017). In addition to PCH, mutations in EXOSC2 were linked to a distinct disease Short stature, Hearing loss, Retinitis pigmentosa and distinctive Facies (SHRF) (Di Donato et al. 2016), and mutations in EXOSC5 were linked to a disease Cerebellar Ataxia, Brain Abnormalities, and Cardiac conduction defects (CABAC) that appears to share some features with PCH (Slavotinek et al. 2020; Calame et al. 2021).
The EXOSC1 gene encodes one of the three cap subunits of core RNA exosome (Boczonadi et al., 2014; Burns et al., 2018; Somashekar et al., 2021; Wan et al., 2012). In a recent study, a bi-allelic missense variant, p.Ser35Leu in EXOSC1 was demonstrated as a novel candidate gene for PCH1F (Somashekar et al., 2021). Here, we present an additional individual with a novel missense variant in the EXOSC1 gene presented with PCH and additional clinical features. Our report expands the known phenotypic and genotypic spectrum associated with EXOSC1 gene. We further show that when heterologously expressed, both missense variants destabilize the EXOSC1 protein and cause loss of function.
Clinical report
Individual IV-1 is the first child of a healthy fourth-degree consanguineous Palestinian couple (Figure 1A), born at 38 weeks of gestational age by normal vaginal delivery, weighing 2140 g at birth. The APGAR score was 8/9 at 1 and 5 min, respectively. The mother was 20-year-old during her pregnancy, with uneventful pregnancy course, no relevant family history, and reassuring detailed fetal US at 18 weeks of gestation. The baby was assessed for the first time at age of 38 days of life due to failure to thrive despite adequate caloric intake. At evaluation, the weight was 3060 gm (−3 SD), the length was 51 cm (−3 SD), and the head circumference was 34 cm (−4 SD). Distinctive facial features were appreciated including triangular face, frontal bossing, deep set eyes, bluish sclera, telecanthus, bulbous nasal tip, depressed nasal root, long philtrum, and microretrognathia (Figure 1B). Neurological examination revealed generalized hypotonia. Otherwise, physical examination was unremarkable. Her basic biochemical lab tests including ammonia, lactate, plasma amino acid and urine organic acids were all normal.
Figure 1: Family pedigree, clinical and radiological findings in the proband.
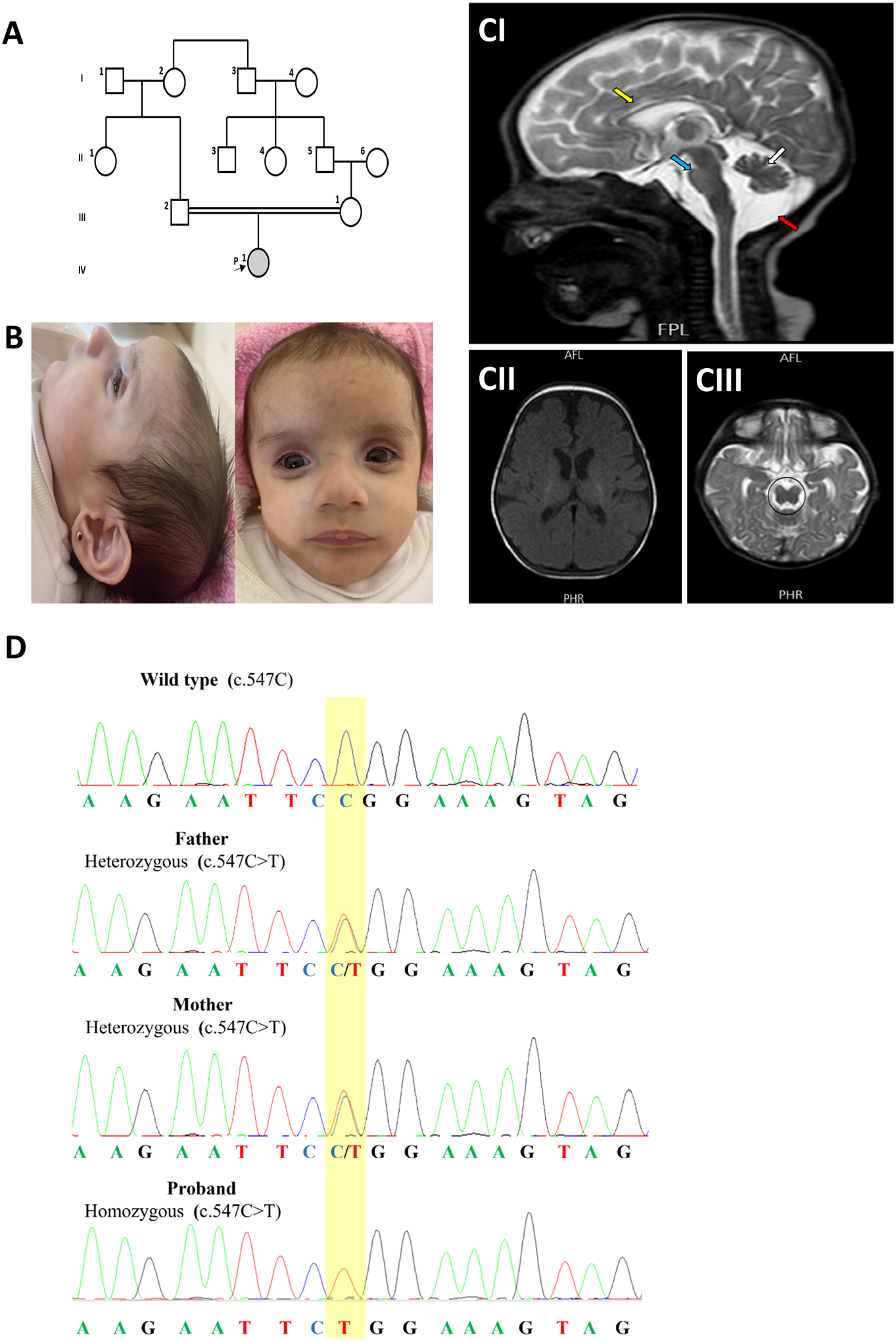
(A) pedigree of the family. (B) clinical photographs of the proband. (C) MRI of the brain at three months shows sagittal T2-weighted image showing cerebellar hypoplasia (white arrow), hypoplasia of the pons (blue arrow), thin corpus callosum (yellow arrow), enlarged posterior fossa CSF space (red arrow) (CI), “figure of 8” appearance of midbrain (CII), and Axial T1-weighted showing widening of extra axial CSF spaces (CIII). D. Sanger sequencing results of the EXOSC1 gene from the proband and parents.
Magnetic resonance imaging (MRI) of the brain showed pontocerebellar hypoplasia, thin corpus callosum, widening of extra-axial cerebrospinal fluid (CSF) spaces, and increased CSF intensity in the posterior fossa (Figure 1C). Myelination was grossly appropriate for the corresponding age without any calcifications with a pituitary gland of normal size and position. Echocardiography revealed the presence of dilated cardiomyopathy with a moderately reduced left ventricular ejection fraction (LVEF) of approximately 43% with normal coronaries. The ophthalmic examination disclosed the presence of mild atrophic fundi with mottling of pigment epithelium and pale disc. EEG, abdominal ultrasound, and skeletal survey were normal. Unfortunately, the baby died suddenly during her sleep at the age of 6 months.
Genetic analysis and functional validation
Singleton whole exome sequencing was performed and revealed the presence of a homozygous variant c.547C>T p.Arg183Trp in the EXOSC1 gene (NM_016046.5). This variant is present at low frequency in population databases (rs145472895, gnomAD 0.0000319). Algorithms developed to predict the effect of missense changes on protein structure and function agree on the potential damaging impact of this missense change (SIFT: “Deleterious”; PolyPhen-2: “probably_damaging”; CADD score: “32”). Sanger sequencing showed that both parents were heterozygous carriers for this variant (Figure 1D).
Structural analysis using a Cryo-EM Structure of the human nuclear exosome (6D6R (WEICK et al. 2018)) indicates the novel EXOSC1variant (p.Arg183Trp) is positioned in the carboxy terminal domain and the previously identified PCH linked variant p.Ser35Leu is present in the amino-terminal domain of the EXOSC1 protein (Figure 2A). To elucidate the effect of the novel and previously reported EXOSC1 variant, we performed functional studies using a budding yeast model. Deletion of the orthologous yeast gene (csl4Δ) is lethal but can be rescued by expressing the human EXOSC1 protein. This suggests that human EXOSC1 can form a functional RNA exosome complex with the other yeast subunits. (Allmang et al., 1999). Utilizing this complementation strategy, we expressed wild-type EXOSC1 and both variants and tested for growth defects. The strain that expressed the novel EXOSC1 variant Arg183Trp showed a slow-growth phenotype and the previously reported EXOSC1 variant p.Ser35Leu was lethal. In contrast, the strain expressing wild-type EXOSC1 grew well (Figure 2B), confirming previous results (Allmang et al., 1999). We assessed the protein level of both EXOSC1 variants by western blot analysis and observed a large reduction compared to wild type when expressed in budding yeast, indicating compromised protein stability of both variants (Figure 2C) and confirming the SIFT prediction. These data indicate that the PCH linked EXOSC1 variants lead to impairment of the essential function of the RNA exosome. Given our findings, the p.Arg183Trp variant was classified as likely pathogenic based on The American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) variant classification criteria (PS3, PM2, PP3).
Figure 2: Functional studies of EXOSC1 variants reveals impaired RNA exosome function.
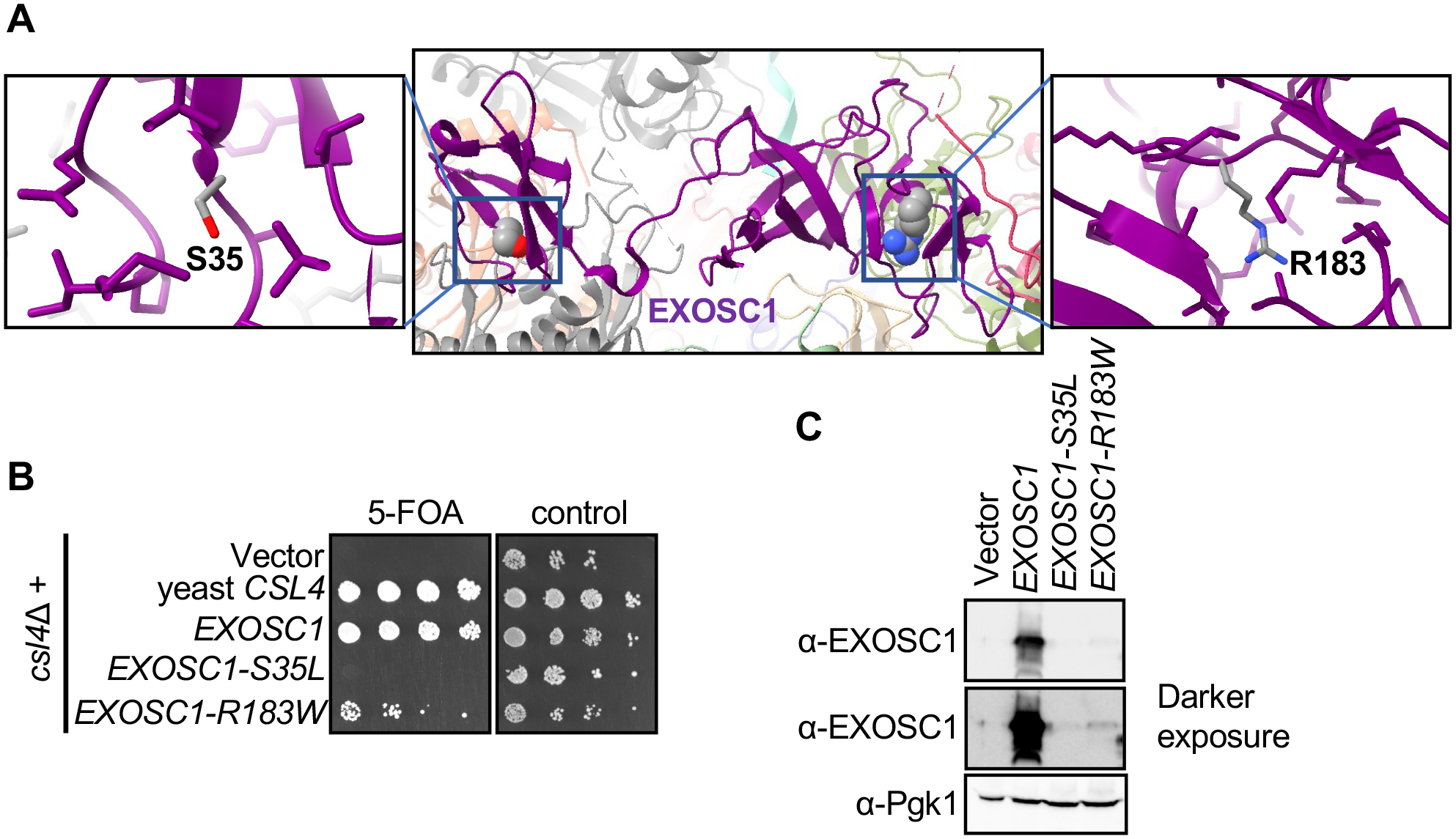
(A) Center: Structure of EXOSC1 in the context of the whole RNA exosome (6D6R) with EXOSC1 highlighted in purple. The panels on the left and right provide detailed views of S35 and R183 residues, with the side chains with 5 angstroms shown. Each of these side chains are from EXOSC1, indicating that the PCH-related residues are internal to EXOSC1. Structural analysis was performed UCSF ChimeraX version: 1.2.5. (B) serial dilution growth assay of yeast expressing wild-type human EXOSC1 (row 3) or variants (rows 4 and 5). Growth on the 5FOA-containing media (left) reflects the functionality of EXOSC1 (C) wild-type EXOSC1 expressed in yeast is readily detected by a polyclonal anti-EXOSC 1 antibody (lane 2) but the signal for the variant proteins (lanes 3 and 4) is strongly reduced. Pgk1 was used as a loading control.
Discussion
Pontocerebellar hypoplasia (PCH) is a rare, genetically heterogeneous, neurodegenerative disorder. Traditionally, PCH was classified in two major types based on the neuropathological findings, with muscular atrophy and hypotonia in PCH type 1, and dyskinesia and spasticity in PCH type 2 (Barth, 1993). Currently, there are 17 different types of PCH, and six subtypes (A, B, C, D, E, F) for each PCH type 1 and 2 based on the clinical, radiological, and genetic findings (Dabaj et al., 2022). PCH1 is characterized by anterior horn degeneration in the spinal cord with muscle weakness and hypotonia (Namavar et al., 2011a), and specific MRI findings of cerebellar hypoplasia with variable involvement of cerebellar hemispheres, cerebrum, and pons (Rudnik-Schöneborn et al., 2003). A wide range of clinical manifestations may affect individuals with PCH1 including developmental delay, psychomotor retardation, joint contractures, primary hypoventilation, feeding difficulties, visual and hearing impairment, seizures, and progressive microcephaly (Namavar et al., 2011b; Spyridakis et al., 2022). To date, single amino acid variants in four different subunits of the RNA exosome (EXOSC1, EXOSC3, EXOSC8 and EXOSC9) have been reported to be associated with PCH1 (Van Dijk et al., 2017, 2021), although previously only a single individual with an EXOSC1 variant was known. Single amino acid changes in EXOSC5 have been described in CABAC and single amino acids changes in EXOSC2 have been described in SHRF. PCH, CABAC and SHRF share that each include cerebellar atrophy which can be mild or borderline (in SHRF) or more severe. Mutations in two genes with no established connection to the RNA exosome (VRK1 and SLC25A46) have also been associated with PCH1. Clarification on the molecular causes of these disease will require a better understanding of the range of causal mutations and how they affect cellular function.
The first case of PCH1 due to EXOSC1 variant was reported in an eight-months-old male of a consanguineous couple (Somashekar et al., 2021). Here, we add the second case of PCH associated with the EXOSC1 gene to the literature. Our case had the typical clinical and radiological features of PCH type 1. Our proband presented with developmental delay, distinctive fascial features, and hypotonia. Additionally, she had dilated cardiomyopathy, which was not previously reported in individuals affected with PCH1. Whole exome sequencing (WES) was retrospectively reviewed for known genetic causes associated with cardiomyopathy but none could be identified, suggesting that the cardiac defect could be related to the EXOSC1 variant. Finally, there was no history of seizures or joint contractures in both cases.
Brain MRI showed hypoplasia of the cerebellum, thinning of the corpus callosum, increased CSF intensity in the posterior fossa, widening of the extra-axial CSF spaces, and pontine hypoplasia. Myelination was appropriate for age. The reported case by (Somashekar et al., 2021) showed similar radiological findings in addition to mild delay in myelination, with relative sparing of the pons. Ophthalmic evaluation in both cases revealed blue sclera. (Burns et al., 2018) also reported blue sclera in an individual with EXOSC9 variant.
We assessed the functional consequences of both the previously described EXOSC1 p.Ser35Leu variant and the novel EXOSC1 p.Arg183Trp variant. It was previously reported that expressing human EXOSC1 can compensate for the complete absence of the yeast ortholog, Csl4 (Allmang et al., 1999). Importantly, expressing either the previously described EXOSC1 p.Ser35Leu variant or the novel variant we discovered, EXOSC1 p.Arg183Trp, could not compensate for csl4 deletion, proving strong support that these variants disrupt the protein and are pathogenic. Both variants caused a strong reduction of the EXOSC1protein level when expressed in yeast. This confirms data from patient-derived cultured fibroblasts for the EXOSC1 p.Ser35Leu variant. Unfortunately, no patient-derived material is available for similar analysis of EXOSC1 p.Arg183Trp. We conclude that the EXOSC1 p.Ser35Leu and EXOSC1 p.Arg183Trp variants interfere with overall folding of the EXOSC1 subunit, which then likely leads to its degradation. EXOSC1 consists of two domains that are functionally redundant to each other, at least in yeast (Schaeffer et el., 2009). The EXOSC1 p.Ser35Leu and p.Arg183Trp mutations are in the N- and C-terminal domain, respectively but because they cause the degradation of the protein they would cause loss of function of both redundant domains.
In conclusion, our study reports a novel homozygous variant in the EXOSC1 gene in a Palestinian infant with clinical and neuroradiologic features that are consistent with type 1 pontocerebellar hypoplasia. To our knowledge, this is the second case of EXOSC1 variant-related PCH.
Materials and Methods
Whole exome sequencing:
Whole exome analysis Exonic sequences were enriched in the DNA sample of the patient using SureSelect Human All Exon ~51 Mb Kit V5 (Agilent Technologies, Santa Clara, California, United States). Sequences were determined by NOVASEQ 6000 (Illumina, San Diego, California, USA) as 100-bp paired-end runs. Data analysis including read alignment and variant calling was performed by DNAnexus software (Palo Alto, California, USA) using the default parameters with the human genome assembly hg19 (GRCh37) as reference.
Saccharomyces cerevisiae strains and plasmids:
The csl4 deletion strain yav1047 and EXOSC1 expression plasmid (pav314) have previously been described. (Schaeffer et al., 2009). The EXOSC1 S35L and R183W mutations were introduced into plasmid pav314 of by site directed mutagenesis using a QuikChange Lightning kit and the manufacturer’s protocol (Agilent).
Yeast growth assay:
The growth assay was performed as described in Schaeffer et al., 2009. In short, the wild-type and mutant EXOSC1 plasmids or an empty vector control were introduced into yeast strain yav1047 that contains a deletion of the endogenous CSL4 gene but expresses CSL4 from a plasmid that also contains a URA3 counter-selectable marker. Transformants were selected on SC-LEU-URA media. These transformants were then grown to O.D. ~0.8, serially diluted, and spotted on 5FOA-containing or control media. 5FOA selects for cells that have lost the CSL4, URA3 plasmid and thus growth on 5FOA media occurs only if the EXOSC1 plasmid supports viability.
Immunoblotting
For analysis of the EXOSC1 wild-type and mutant protein level, the transformants described above were used in immunoblotting. The EXOSC1 protein and Pgk1 (loading control) was detected with polyclonal anti-EXOSC1 antibody raised against the full-length protein (12585–1-AP Proteintech) and anti-Pgk1 antibody 22C5D8 (Invitrogen), respectively.
Acknowledgements
We are thankful to the family for their cooperation. This work was funded by NIH grant R35GM141710 awarded to AvH as well as a Dr. John J. Kopchick Research Award from the MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences to KSA.
Footnotes
Conflict of interest statement
Declaration of competing interest Authors declare no competing financial interests in relation to the work described in this paper.
Editorial policies and ethical considerations
Informed consent was obtained from the parents of our proband.
References
- Appelhof B, Barth PG, & Baas F (2019). Classification of Pontocerebellar Hypoplasia: Where does it End? August, 52–61. [Google Scholar]
- Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, Mitchell P. The yeast exosome and human PM-Scl are related complexes of 3’ --> 5’ exonucleases. Genes Dev. 1999. Aug 15;13(16):2148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG (1993). Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain & Development, 15(6), 411–422. 10.1016/0387-7604(93)90080-R [DOI] [PubMed] [Google Scholar]
- Boczonadi V, Müller JS, Pyle A, Munkley J, Dor T, Quartararo J, Ferrero I, Karcagi V, Giunta M, Polvikoski T, Birchall D, Princzinger A, Cinnamon Y, Lützkendorf S, Piko H, Reza M, Florez L, Santibanez-Koref M, Griffin H, … Horvath R (2014). EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nature Communications, 5. 10.1038/NCOMMS5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DT, Donkervoort S, Müller JS, Knierim E, Bharucha-Goebel D, Faqeih EA, Bell SK, AlFaifi AY, Monies D, Millan F, Retterer K, Dyack S, MacKay S, Morales-Gonzalez S, Giunta M, Munro B, Hudson G, Scavina M, Baker L, … Bönnemann CG (2018). Variants in EXOSC9 Disrupt the RNA Exosome and Result in Cerebellar Atrophy with Spinal Motor Neuronopathy. American Journal of Human Genetics, 102(5), 858. 10.1016/J.AJHG.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calame DG, Herman I, Fatih JM, Du H, Akay G et al. , 2021. Risk of sudden cardiac death in EXOSC5-related disease. Am J Med Genet A 185: 2532–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski A, Lubas M, Jensen TH, & Dziembowski A (2013). RNA decay machines: the exosome. Biochimica et Biophysica Acta, 1829(6–7), 552–560. 10.1016/J.BBAGRM.2013.01.006 [DOI] [PubMed] [Google Scholar]
- Dabaj I, Hassani A, Burglen L, Qebibo L, Guerrot A-M, Marret S, Tebani A, & Bekri S (2022). Pontocerebellar Hypoplasia Type 1D: A Case Report and Comprehensive Literature Review. Journal of Clinical Medicine, 11(15), 4335. 10.3390/JCM11154335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato N, Neuhann T, Kahlert AK, Klink B, Hackmann K et al. , 2016. Mutations in EXOSC2 are associated with a novel syndrome characterised by retinitis pigmentosa, progressive hearing loss, premature ageing, short stature, mild intellectual disability and distinctive gestalt. J Med Genet 53: 419–425. [DOI] [PubMed] [Google Scholar]
- Januszyk K, & Lima CD (2014). The eukaryotic RNA exosome. Current Opinion in Structural Biology, 0(1), 132. 10.1016/J.SBI.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namavar Y, Barth PG, Poll-The B, & Baas F (2011a). Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet Journal of Rare Diseases, 6(1). 10.1186/1750-1172-6-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namavar Y, Barth PG, Poll-The B, & Baas F (2011b). Classification, diagnosis and potential mechanisms in Pontocerebellar Hypoplasia. Orphanet Journal of Rare Diseases, 6(1), 1–14. 10.1186/1750-1172-6-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Barth PG, & Zerres K (2014). Pontocerebellar hypoplasia. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics, 166(2), 173–183. 10.1002/ajmg.c.31403 [DOI] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Sztriha L, Aithala GR, Houge G, Lægreid LM, Seeger J, Huppke M, Wirth B, & Zerres K (2003). Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. American Journal of Medical Genetics. Part A, 117A(1), 10–17. 10.1002/AJMG.A.10863 [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Misceo D, Htun S, Mathisen L, Frengen E et al. , 2020. Biallelic variants in the RNA exosome gene EXOSC5 are associated with developmental delays, short stature, cerebellar hypoplasia and motor weakness. Hum Mol Genet in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somashekar PH, Kaur P, Stephen J, Guleria VS, Kadavigere R, Girisha KM, Bielas S, Upadhyai P, & Shukla A (2021). Bi-allelic missense variant, p.Ser35Leu in EXOSC1 is associated with pontocerebellar hypoplasia. Clinical Genetics, 99(4), 594–600. 10.1111/CGE.13928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer D, Tsanova B, Barbas A, Reis FP, Dastidar EG, Sanchez-Rotunno M, Arraiano CM, van Hoof A. The exosome contains domains with specific endoribonuclease, exoribonuclease and cytoplasmic mRNA decay activities. Nat Struct Mol Biol. 2009. Jan;16(1):56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyridakis AC, Cao Y, & Litra F (2022). A Rare Case of Pontocerebellar Hypoplasia Type 1B With Literature Review. Cureus, 14(7). 10.7759/CUREUS.27098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk T, Baas F, Barth PG, & Poll-The BT (2018). What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet Journal of Rare Diseases, 13(1). 10.1186/S13023-018-0826-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk T, Barth P, Baas F, Reneman L, & Poll-The BT (2021). Postnatal Brain Growth Patterns in Pontocerebellar Hypoplasia. Neuropediatrics, 52(3), 163–169. 10.1055/S-0040-1716900 [DOI] [PubMed] [Google Scholar]
- Van Dijk T, Rudnik-Schöneborn S, Senderek J, Hajmousa G, Mei H, Dusl M, Aronica E, Barth P, & Baas F (2017). Pontocerebellar hypoplasia with spinal muscular atrophy (PCH1): identification of SLC25A46 mutations in the original Dutch PCH1 family. Brain : A Journal of Neurology, 140(8), e46. 10.1093/BRAIN/AWX147 [DOI] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, Seeman P, Von Moers A, Graul-Neumann L, Kornberg AJ, Castro-Gago M, Sobrido MJ, Sanefuji M, Shieh PB, Salamon N, … Jen JC (2012). Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nature Genetics, 44(6), 704–708. 10.1038/NG.2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick EM, Puno MR, Januszyk K, Zinder JC, DiMattia MA et al. , 2018. Helicase-Dependent RNA Decay Illuminated by a Cryo-EM Structure of a Human Nuclear RNA Exosome-MTR4 Complex. Cell 173: 1663–1677 e1621. [DOI] [PMC free article] [PubMed] [Google Scholar]