

Abstract
INTRODUCTION
With emergence of disease‐modifying therapies, efficient diagnostic pathways are critically needed to identify treatment candidates, evaluate disease severity, and support prognosis. A combination of plasma biomarkers and brief digital cognitive assessments could provide a scalable alternative to current diagnostic work‐up.
METHODS
We examined the accuracy of plasma biomarkers and a 10‐minute supervised tablet‐based cognitive assessment (Tablet‐based Cognitive Assessment Tool Brain Health Assessment [TabCAT‐BHA]) in predicting amyloid β positive (Aβ+) status on positron emission tomography (PET), concurrent disease severity, and functional decline in 309 older adults with subjective cognitive impairment (n = 49), mild cognitive impairment (n = 159), and dementia (n = 101).
RESULTS
Combination of plasma pTau181, Aβ42/40, neurofilament light (NfL), and TabCAT‐BHA was optimal for predicting Aβ‐PET positivity (AUC = 0.962). Whereas NfL and TabCAT‐BHA optimally predicted concurrent disease severity, combining these with pTau181 and glial fibrillary acidic protein was most accurate in predicting functional decline.
DISCUSSION
Combinations of plasma and digital cognitive markers show promise for scalable diagnosis and prognosis of ADRD.
Highlights
The need for cost‐efficient diagnostic and prognostic markers of AD is urgent.
Plasma and digital cognitive markers provide complementary diagnostic contributions.
Combination of these markers holds promise for scalable diagnosis and prognosis.
Future validation in community cohorts is needed to inform clinical implementation.
Keywords: Alzheimer's disease, blood‐based biomarkers, diagnosis, digital cognitive assessment, disease monitoring
1. BACKGROUND
The prevalence of Alzheimer's disease and related dementias (ADRD) continues to rise and is estimated to affect 153 million individuals worldwide by 2050. 1 Despite these alarming projections, the rates of missed, inaccurate, or delayed diagnoses of dementia remain high, 2 , 3 particularly in underserved populations, including racially and ethnically diverse individuals residing in high‐income countries and those residing in low‐ and middle‐income countries. 4 Access to timely and accurate diagnostic services remains limited, 5 , 6 and the need for globally scalable, accessible, and cost‐effective diagnostic markers of ADRD is urgent. Implementation of such markers is also critical for establishing the infrastructure for case identification and treatment management as disease‐modifying therapies become available.
Plasma biomarkers offer greater accessibility and cost effectiveness compared to the current gold standard clinical testing using cerebrospinal fluid (CSF) or positron emission tomography (PET). Among these, plasma amyloid β (Aβ), phosphorylated tau (pTau), neurofilament light (NfL) chain, and glial fibrillary acidic protein (GFAP) represent the most promising markers to date and are closest to clinical implementation. 7 , 8 , 9 Growing evidence supports the role of the ratio of plasma Aβ1‐42 to Aβ1‐40 (Aβ42/40) as a marker of cerebral Aβ pathology, 7 , 8 , 9 , 10 , 11 plasma pTau at threonine‐181 (pTau181) or at threonine‐217 (pTau217) as a marker AD‐related neuropathology, 7 , 8 , 9 , 12 , 13 , 14 , 15 , 16 , 17 , 18 plasma NfL as an etiologically non‐specific marker of neuroaxonal injury, 7 , 8 , 9 , 19 , 20 and plasma GFAP as a marker of reactive astrogliosis. 7 , 8 , 9 , 20 , 21 Given clinical and etiological heterogeneity of ADRD, a combination of different biomarkers is likely to offer greater accuracy and precision compared to any single marker, with each predictor explaining unique variance of an underlying disease process. Several studies support this premise showing that various combinations of plasma Aβ42/40, pTau, NfL, and GFAP showed highest accuracy in predicting Aβ positivity based on CSF 22 , 23 and PET 24 and differentiating clinical AD dementia from other neurodegenerative syndromes. 25 , 26 Combination of markers also exhibited highest test‐retest reliability over time, which is a critical consideration for clinical implementation. 22
Expert recommendations also highlight the importance of examining high‐performing combinations of plasma and other clinical markers, particularly cognitive assessments, to maximize their diagnostic and prognostic potential. 8 , 9 While scarce but promising evidence to date supports complementary contributions of paper‐and‐pencil neuropsychological and plasma markers to predicting conversion to AD dementia, 27 , 28 cognitive measures that could be used in combination with plasma biomarkers in novel clinical algorithms must have characteristics that support, not hinder, widescale implementation. In particular, use of most traditional assessments (e.g., the Mini‐Mental State Examination) in routine healthcare workflows has been limited by their poor sensitivity to milder stages of impairment, 29 limited validity in diverse populations, 30 and costly training, time, and administration demands. 31 , 32 To address these limitations, substantial progress has been made in development of sensitive and reliable digital cognitive assessments highlighting their potential as scalable and efficient clinical markers not requiring access to highly trained specialists, lengthy clinical evaluations, or complex scoring and interpretation algorithms. 31 , 32 Examining the combination of scalable digital cognitive and plasma markers, therefore, may provide critical insights for the development of novel efficient clinical algorithms.
This study evaluated the accuracy of a combination of plasma biomarkers (Aβ42/40, pTau181, NfL, and GFAP) and a 10‐minute examiner‐administered tablet‐based cognitive battery (Tablet‐based Cognitive Assessment Tool Brain Health Assessment, TabCAT‐BHA) in predicting Aβ‐PET positivity, disease severity, and functional decline. TabCAT‐BHA is a brief, multidomain battery that showed excellent sensitivity to mild cognitive impairment (MCI) and typical and atypical dementias, 33 , 34 , 35 high reliability in tracking change over time, 34 and associations with Aβ‐ and tau‐PET burden. 36 In accordance with the U.S. National Alzheimer's Project Act 37 to improve detection of cognitive impairment following a concern, the sample included older adult participants with subjective or objective cognitive impairment. Participants were racially and ethnically diverse individuals across diagnostic groups to support generalizability and applicability of findings in populations that have been historically excluded from ADRD biomarker research. 38
2. METHODS
2.1. Participants
The study was approved by the University of California San Francisco (UCSF) Committee on Human Research. All participants provided written informed consent. Participants were adults aged 50 or older enrolled in longitudinal observational studies at the UCSF Memory and Aging Center, who were clinically characterized as having subjective cognitive impairment (SCI) or met criteria for MCI or dementia. All participants underwent a comprehensive diagnostic evaluation consisting of neurological examination, multidomain neuropsychological testing, clinical interview with an informant including Clinical Dementia Rating Scale (CDR), 39 and structural neuroimaging. Clinical diagnoses were made in multidisciplinary consensus conferences based on published criteria as previously described. 12 , 33 , 34
The SCI group (n = 49) included older adults who endorsed worsening of their memory or other thinking abilities on clinical interview, but performed within normal limits on neurological evaluation, neuropsychological testing, and CDR. Among 159 participants with MCI, 72 (45%) were characterized with an AD spectrum clinical syndrome, including 58 with a multidomain amnestic syndrome, 40 10 with logopenic variant of primary progressive aphasia (lvPPA), 41 and 4 with posterior cortical atrophy (PCA). 42 The remaining participants with MCI were clinically diagnosed with a non‐AD spectrum syndromes, including non‐amnestic MCI (n = 56), corticobasal syndrome (CBS, n = 4), 43 non‐fluent variant of primary progressive aphasia (nfvPPA, n = 16), 41 progressive supranuclear palsy (PSP, n = 4), 44 semantic variant of primary progressive aphasia (svPPA, n = 5), 41 and traumatic encephalopathy syndrome (TES, n = 2). 45 Among 101 participants with dementia, 48 (48%) were clinically diagnosed with an AD spectrum syndrome, including 41 with AD amnestic dementia, 46 5 with lvPPA, 41 and 2 with PCA. 42 The remaining participants were diagnosed with behavioral variant frontotemporal dementia (n = 31), 47 CBS (n = 3), 43 dementia with Lewy bodies (n = 4), 48 nfvPPA (n = 2), 41 PSP (n = 6), 44 svPPA (n = 6), 41 and TES (n = 1). 45
All participants completed a blood draw and TabCAT‐BHA at baseline independent of standard diagnostic procedures. The average time difference between a blood draw and TabCAT‐BHA completion was 2.3 ± 7.7 days. We only included participants whose plasma biomarker measurements were successful. Additional exclusion criteria were presence of severe psychiatric illness, known non‐neurodegenerative neurological condition affecting cognition, or significant substance use disorder and/or systemic illness. Longitudinal disease severity data were available in a subsample of 32 SCI, 107 MCI, 46 dementia participants who completed the CDR at baseline and annual follow‐up visits (n = 185 with 2 visits; n = 88 with 3 visits; n = 38 with 4 or more visits). Average time between visits was 1.3 ± 0.6 years.
2.2. Measures and procedures
2.2.1. TabCAT‐BHA
TabCAT‐BHA is a 10‐min cognitive battery programmed in the TabCAT software platform (UCSF, San Francisco, CA). It is comprised of 2 required subtests: Favorites (associative memory) and Match (executive functioning and processing speed); and 2 optional subtests: Line Orientation (visuospatial) and Animal Fluency (language). 33 , 34 , 35 Detailed task descriptions are available at memory.ucsf.edu/tabcat and were described previously. 33 , 34 All participants completed the TabCAT‐BHA on a 9.7‐inch iPad with a trained examiner in a private examination room. Performance on the TabCAT‐BHA battery was included in all analyses in the form of a previously validated cognitive composite score, 34 which is based on the demographically adjusted (age, sex, education, testing language) Favorites and Match scores.
RESEARCH IN CONTEXT
Systematic review: We reviewed the literature on plasma biomarkers for diagnosis and prognosis of Alzheimer's disease and related dementias using traditional sources (e.g., PubMed). Supplementing plasma biomarkers with cognitive and other clinical tools offers greater accuracy, and there are ongoing efforts to examine high performing combinations of different markers to maximize diagnostic precision.
Interpretation: Our findings show that a combination of plasma and digital cognitive markers is highly accurate at predicting amyloid β‐positron emission tomography (Aβ‐PET) positivity, concurrent disease severity, and longitudinal functional decline.
Future directions: Replication of the results in more representative and socioeconomically diverse communities is needed to support future clinical implementation of these markers.
2.2.2. Plasma biomarker measurements
Blood samples were obtained by venipuncture in ethylenediaminetetraacetic acid (EDTA) tubes for plasma as previously described. 12 After centrifugation at 2000 g for 10 min at 4°C, plasma samples were aliquoted in polypropylene tubes and stored at −80°C, with an average needle‐to‐freezer time <2 h. Before analysis, samples underwent only one freeze‐thaw cycle. Biomarker concentrations were measured in duplicate using commercially available pTau181 V2 (pTau181; lot #503008) and Neurology 4‐PLEX E (Aβ40, Aβ42, NfL, and GFAP; lot #503105) Quanterix kits (Billerica, MA) on the Simoa HD‐X platform at UCSF. For each kit 100 microliters of plasma were diluted 1:4 by the instrument. The instrument operator was blinded to clinical variables. For pTau181, all samples were measured above the kit lower limit of quantification (LLOQ) of 0.085 pg mL−1 with the mean coefficient of variation (CV) of 7.3%. For Aβ40 and Aβ42, all samples were measured above the LLOQ of 1.02 pg mL−1 and 0.378 pg mL−1 (respectively) with the average CVs of 3.3% and 3.4% (respectively). For NfL and GFAP, all samples were measured above the LLOQ of 0.400 pg mL−1 and 2.89 pg mL−1 (respectively) with the mean CVs of 5.3% and 6.3% (respectively).
2.2.3. Aβ‐PET acquisition and processing
Aβ‐PET was performed independent of diagnostic procedures in a subset of 105 participants, including 6 with SCI, 64 with MCI (36 with AD spectrum, 28 with a non‐AD spectrum), and 35 with dementia (19 with AD spectrum, 16 with a non‐AD spectrum). Aβ‐PET acquired with 11C‐Pittsburgh Compound B (PIB, n = 82), 18F‐Florbetapir (n = 22), or 18F‐Florbetaben (n = 1) radiotracers. The average time between plasma sample collection and PET imaging was 136 ± 256 days. PET scans were acquired in list mode on PET‐CT scanners (GE Discovery VCT, n = 22; Siemens Biograph 6 Truepoint, n = 83) using standard acquisition protocols: 50‐70 min acquisition for Florbetapir and PIB and 90‐110 min post injection for Florbetaben. PET data were attenuation corrected using low dose CT and reconstructed as four five‐minute frames using an ordered subset expectation maximization algorithm. PET frames were realigned to the first one and averaged; this mean PET image was used to perform a visual read by a trained clinician following FDA‐approved guidelines for Florbetaben and Florbetapir, or internal reading guidelines for PIB (see 49 , 50 for validation of the PIB‐PET visual read approach in PET‐to‐autopsy studies).
2.2.4. Apolipoprotein E genotyping
Apolipoprotein E (APOE) genotyping was performed in 302 (98%) participants (48 SCI, 155 MCI, 99 dementia) and was based on DNA analysis from peripheral blood samples as described previously 51 APOE 𝜀4 status was coded as “1” for homozygotes and heterozygotes of 𝜀4 and as “0” otherwise. There were eight participants with a 𝜀2/𝜀4 genotype in the whole sample, whose APOE data were not used in the analyses.
2.3. Statistical analyses
Baseline group differences were determined using analyses of variance (ANOVA) for continuous variables and Fisher's exact tests for categorical variables. Pairwise comparisons were performed using the Bonferroni method. Values of raw concentrations of plasma pTau181 (skewness = 1.2, kurtosis = 1.4), NfL (skewness = 5.0, kurtosis = 36.2), and GFAP (skewness = 2.0, kurtosis = 5.6) were not normally distributed and were natural log‐transformed for baseline ANOVA and linear regression analyses. The distribution of Aβ42/40 ratio was within expectations for a normal distribution (skewness = 0.01, kurtosis = 0.34). Transformed values approximated normal distributions for each biomarker, including log‐transformed pTau181 (skewness = 0.0, kurtosis = −0.5), log‐transformed NfL (skewness = 0.6, kurtosis = 1.3), and log‐transformed GFAP (skewness = 0.2, kurtosis = −0.1).
We performed logistic regression models with receiver operating characteristic (ROC) curves to determine the accuracy of plasma biomarkers and TabCAT‐BHA in differentiating Aβ‐PET status at baseline. Individual models were run for each of the markers and the full model was comprised of all markers included simultaneously. Model selection was performed using the information‐theoretic approach based on the Akaike information criterion (AIC). 27 , 52 The optimal model was defined as having the lowest AIC and was considered to maximize the optimal trade‐off between model fit and sparsity. 52 Differences between areas under the ROC curves (AUC) were examined using DeLong tests. All models were performed with and without covariates for age (years), sex (coded as “1” for female and “0” for male), education (years), APOE 𝜀4 status (coded as “1” for presence of one or two 𝜀4 alleles and “0” for absence of an 𝜀4 allele), and time difference between PET acquisition and plasma collection (days).
Multiple linear regression models were used to examine baseline associations of plasma and TabCAT‐BHA markers with disease severity measured by the CDR Sum of Boxes score (CDR‐SB). All variables were included simultaneously, and unstandardized coefficients were reported. Linear mixed effect models with random intercepts and slopes were used to examine the associations between baseline values of plasma and TabCAT‐BHA markers with longitudinal changes on the CDR‐SB (number of visits: mean = 2.62, SD = 0.85). All models controlled for age, sex, education, and APOE 𝜀4 status; longitudinal analyses additionally controlled for time since baseline visit (years) and baseline clinical diagnosis. To account for phenotypic heterogeneity, we additionally repeated both baseline and longitudinal models in AD spectrum and FTLD spectrum clinical groups separately using CDR plus the National Alzheimer's Coordinating Center FTLD rating (CDR+NACC/FTLD) Sum of Boxes as an outcome. 53
All analyses were performed in R (v 4.2.3, R Project for Statistical Computing) with two‐tailed significance level set at P < 0.05. Linear models were checked for overdispersion, influential values, and multicollinearity. We report P values without adjusting for multiple comparisons as this methodology focuses on avoiding one or more results with P < 0.05 in the case where all differences are truly zero, which represents an unlikely hypothesis in our analyses. Therefore, we use scientific judgment rather than formal methods of adjustment to indicate where caution is warranted despite findings with P < 0.05.
3. RESULTS
3.1. Baseline differences
The study cohort was comprised of 309 older adults, including 49 participants with SCI, 159 with MCI, and 101 with dementia. Baseline demographic and clinical characteristics, plasma biomarker levels, and TabCAT‐BHA performance are reported in Table 1. Among 260 participants with MCI and dementia, 120 (46%) were diagnosed with an AD spectrum clinical syndrome, 77 (30%) with an FTLD spectrum syndrome, and 63 (24%) with other clinical syndromes (detailed description is provided in Methods). The SCI, MCI, and dementia groups did not significantly differ with regard to age, sex, APOE ε4 status, and AD spectrum clinical phenotype (Table 1). Participants in the SCI group had more years of education compared to the MCI group and included fewer individuals who identified as non‐Hispanic White compared to the MCI and dementia groups (Table 1).
TABLE 1.
Baseline demographic and clinical characteristics and biomarker values.
| SCI (n = 49) | MCI (n = 159) | Dementia (n = 101) | P‐Value | |
|---|---|---|---|---|
| Age (years), M (SD) | 68.9 (9.3) | 68.2 (9.3) | 67.4 (9.9) | 0.623 |
| Sex (female), n (%) | 26 (53%) | 82 (52%) | 43 (43%) | 0.311 |
| Education (years), M (SD) | 16.4 (3.6) | 14.8 (4.0) | 15.2 (3.0) | 0.033 |
| Race/ethnicity | 0.001 | |||
| Asian, n (%) | 12 (24%) | 21 (13%) | 8 (8%) | |
| Non‐Hispanic Black, n (%) | 2 (4%) | 6 (4%) | 3 (3%) | |
| Hispanic, n (%) | 13 (27%) | 25 (16%) | 12 (12%) | |
| Non‐Hispanic White, n (%) | 22 (45%) | 107 (67%) | 78 (77%) | |
| APOE e4 allele, n/total n (%) | 13/46 (28%) | 49/152 (32%) | 34/96 (35%) | 0.697 |
| AD spectrum syndrome, n (%) | – | 72 (45%) | 48 (48%) | 0.799 |
| No. of longitudinal visits, M (SD) | 2.8 (0.9) | 2.6 (0.8) | 2.3 (0.5) | <0.001 |
| Years since baseline (longitudinal visits only), M (SD) | 2.7 (1.6) | 1.9 (1.1) | 1.4 (0.7) | <0.001 |
| CDR‐SB, M (SD) | 0.26 (0.53) | 2.04 (1.42) | 5.89 (2.29) | <0.001 |
| Plasma Aβ42/40 (pg ml−1), M (SD) | 0.07 (0.01) | 0.07 (0.01) | 0.06 (0.01) | 0.029 |
| Plasma pTau181 (pg ml−1), M (SD) | 1.57 (0.69) | 2.03 (1.05) | 2.47 (1.41) | <0.001 * |
| Plasma NfL (pg ml−1), M (SD) | 23.59 (12.97) | 29.67 (24.77) | 42.36 (39.15) | <0.001 * |
| Plasma GFAP (pg ml−1), M (SD) | 126.52 (56.04) | 163.93 (106.54) | 206.59 (125.68) | <0.001 * |
| TabCAT‐BHA Cognitive Composite, M (SD) | −0.71 (1.19) | −2.30 (1.74) | −3.99 (1.95) | <0.001 |
Note: Group differences were examined using analyses of variance for continuous outcomes and Fisher's exact tests for categorical outcomes.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CDR‐SB, Clinical Dementia Rating Scale Sum of Boxes; M, mean; MCI, mild cognitive impairment; NfL, neurofilament light; SCI, subjective cognitive impairment; SD, standard deviation; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
P‐values are based on natural log‐transformed plasma pTau181, NfL, and GFAP values.
As shown in Table 1, post hoc pairwise comparisons revealed that plasma Aβ42/40 concentrations were higher in participants with dementia compared to those with MCI (P = 0.025) but not SCI (P = 0.222). Concentrations of plasma pTau181 were significantly different across groups with highest levels in participants with dementia (P vs. MCI = 0.034; P vs. SCI < 0.001) followed by those with MCI (P vs. SCI = 0.043). Plasma NfL was elevated in dementia when compared to MCI (P < 0.001) and SCI (P < 0.001); differences between MCI and SCI groups were not significant (P = 0.242). Similarly, plasma GFAP was higher in dementia participants compared to MCI (P = 0.002) and SCI (P < 0.001) with no significant differences between MCI and SCI groups (P = 0.094). Performance on the TabCAT‐BHA was significantly different across diagnostic groups with worst performance in participants with dementia (P vs. MCI < 0.001; P vs. SCI < 0.001) followed by MCI (P vs. SCI < 0.001).
3.2. Associations with Aβ‐PET at baseline
In a subset of 105 participants who completed Aβ‐PET (6 SCI, 64 MCI, 35 dementia), 58 (55%) were classified as Aβ positive based on expert visual read, 49 , 50 including 4/6 participants with SCI, 34/64 with MCI, and 20/35 with dementia. In individual models without covariates, plasma pTau181 had the highest AUC to differentiate Aβ+ vs. Aβ‐ in the total sample (n = 105; AUC = 0.895, 95% confidence interval [CI]: 0.832‐0.959) and SCI and MCI groups only (n = 70; AUC = 0.888, 95% CI: 0.808‐0.967). Inclusion of covariates for age, female sex, education, APOE 𝜀4 status, and time difference between PET acquisition and plasma collection improved discrimination for each of the markers (Table 2) with plasma pTau181 again exhibiting the highest AUC in the total sample (n = 101; AUC = 0.933, 95% CI: 0.885‐0.980) and in SCI and MCI groups only (n = 67; AUC = 0.939, 95% CI: 0.883‐0.995). Findings on the performance of individual plasma and TabCAT‐BHA markers in discriminating Aβ‐PET status without and with covariates are presented in Table 2.
TABLE 2.
Accuracy of individual plasma biomarkers and TabCAT‐BHA in discriminating Aβ‐PET status.
| AUC (95% CI) | SN | SP | PPV | NPV | |
|---|---|---|---|---|---|
| Total sample without covariates (47 Aβ‐PET negative vs. 58 Aβ‐PET positive) | |||||
| Aβ42/40 | 0.750 (0.655–0.845) | 0.690 | 0.766 | 0.784 | 0.667 |
| pTau181 | 0.895 (0.832–0.959) | 0.879 | 0.809 | 0.850 | 0.844 |
| NfL | 0.493 (0.367–0.619) | 0.931 | 0.319 | 0.628 | 0.789 |
| GFAP | 0.772 (0.678–0.866) | 0.862 | 0.596 | 0.725 | 0.778 |
| TabCAT‐BHA | 0.700 (0.602–0.799) | 0.345 | 1.000 | 1.000 | 0.553 |
| Total sample with covariates a (45 Aβ‐PET negative vs. 56 Aβ‐PET positive) | |||||
| Aβ42/40 | 0.886 (0.824–0.948) | 0.679 | 0.933 | 0.927 | 0.700 |
| pTau181 | 0.933 (0.885–0.980) | 0.804 | 0.933 | 0.938 | 0.792 |
| NfL | 0.864 (0.793–0.935) | 0.786 | 0.822 | 0.846 | 0.755 |
| GFAP | 0.895 (0.835–0.956) | 0.875 | 0.778 | 0.831 | 0.833 |
| TabCAT‐BHA | 0.918 (0.866–0.971) | 0.893 | 0.844 | 0.877 | 0.864 |
| SCI and MCI only without covariates (32 Aβ‐PET negative vs. 38 Aβ‐PET positive) | |||||
| Aβ42/40 | 0.790 (0.683–0.898) | 0.711 | 0.813 | 0.818 | 0.703 |
| pTau181 | 0.888 (0.808–0.967) | 0.921 | 0.750 | 0.814 | 0.889 |
| NfL | 0.527 (0.373–0.681) | 0.921 | 0.344 | 0.625 | 0.786 |
| GFAP | 0.772 (0.659–0.886) | 0.789 | 0.656 | 0.732 | 0.724 |
| TabCAT‐BHA | 0.642 (0.513–0.772) | 0.474 | 0.781 | 0.720 | 0.556 |
| SCI and MCI only with covariates a (31 Aβ‐PET negative vs. 36 Aβ‐PET positive) | |||||
| Aβ42/40 | 0.927 (0.867–0.986) | 0.806 | 0.935 | 0.935 | 0.806 |
| pTau181 | 0.939 (0.883–0.995) | 0.806 | 0.968 | 0.967 | 0.811 |
| NfL | 0.883 (0.798–0.967) | 0.889 | 0.774 | 0.821 | 0.857 |
| GFAP | 0.913 (0.843–0.983) | 0.889 | 0.839 | 0.865 | 0.867 |
| TabCAT‐BHA | 0.883 (0.805–0.960) | 0.667 | 0.935 | 0.923 | 0.707 |
Abbreviations: Aβ‐PET, amyloid β positron emission tomography; APOE, apolipoprotein E; AUC, area under the ROC curve; GFAP, glial fibrillary acidic protein; MCI, mild cognitive impairment; NfL, neurofilament light; NPV, negative predictive value; PPV, positive predictive value; SCI, subjective cognitive impairment; SN, sensitivity; SP, specificity; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
Covariates include age (years), sex (female), education (years), presence of APOE 𝜀4 allele (binary), and time difference between PET acquisition and plasma collection (days).
In a combined model, in which all markers were included simultaneously with demographic and clinical covariates, plasma Aβ42/40, pTau181, NfL, and TabCAT‐BHA cognitive composite were the only significant predictors of Aβ‐PET positivity in the total sample with an AUC of 0.971 (95% CI: 0.944‐0.997, Table 3). The optimal model offering the best trade‐off between model fit and sparsity based on the AIC included plasma Aβ42/40, pTau181, NfL, TabCAT‐BHA, and age (Table 3). Discrimination accuracy of the optimal model (AUC = 0.964, 95% CI: 0.936‐0.993) was significantly better compared to the reduced models including plasma pTau181, TabCAT‐BHA, and age (AUC = 0.919, 95% CI: 0.868‐0.970; P vs. optimal model = 0.023) and plasma pTau181 and age only (AUC = 0.897, 95%CI: 0.833‐0.960; P vs. optimal model = 0.011) using Delong tests. Performance of combined models is presented in Figure 1.
TABLE 3.
Results of logistic regression analyses including baseline values of scalable plasma and TabCAT‐BHA markers as predictors, demographic and clinical covariates, and Aβ‐PET status as an outcome (n = 105; 47 Aβ‐PET negative, 58 Aβ‐PET positive).
| β | SE | P‐Value | |
|---|---|---|---|
| Full model (AIC = 61) | |||
| Plasma Aβ42/40 | −94.009 | 47.496 | 0.048 |
| Plasma pTau181 | 2.202 | 0.714 | 0.002 |
| Plasma NfL | −0.081 | 0.034 | 0.017 |
| Plasma GFAP | 0.001 | 0.006 | 0.886 |
| TabCAT‐BHA | −0.827 | 0.368 | 0.025 |
| Age (years) | 0.132 | 0.068 | 0.050 |
| Sex (female) | −0.290 | 1.033 | 0.779 |
| Education (years) | 0.117 | 0.239 | 0.625 |
| APOE ε4 allele | 2.138 | 1.285 | 0.096 |
| PET/Plasma time difference (days) | 0.005 | 0.003 | 0.103 |
| Optimal model (AIC = 63) | |||
| Plasma Aβ42/40 | −94.597 | 34.328 | 0.006 |
| Plasma pTau181 | 2.435 | 0.559 | <0.001 |
| Plasma NfL | −0.088 | 0.031 | 0.005 |
| TabCAT‐BHA | −0.619 | 0.258 | 0.016 |
| Age (years) | 0.079 | 0.046 | 0.085 |
| pTau181, TabCAT‐BHA, age (AIC = 84) | |||
| Plasma pTau181 | 1.977 | 0.432 | <0.001 |
| TabCAT‐BHA | −0.525 | 0.195 | 0.007 |
| Age (years) | 0.054 | 0.037 | 0.139 |
| pTau181, age (AIC = 92) | |||
| Plasma pTau181 | 2.091 | 0.407 | <0.001 |
| Age (years) | 0.019 | 0.031 | 0.527 |
Abbreviations: Aβ‐PET, amyloid β positron emission tomography; AIC, Akaike information criterion; APOE, apolipoprotein E; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; PET, positron emission tomography; SE, standard error; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
FIGURE 1.
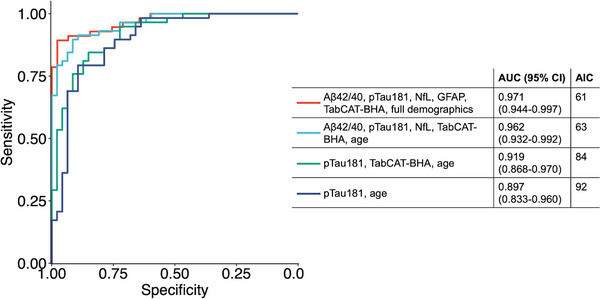
Comparative performance of models combining plasma biomarkers, TabCAT‐BHA, and demographic and clinical covariates in discriminating Aβ‐PET status. ROC curves are based on logistic regression models in 105 participants (47 Aβ‐PET negative, 58 Aβ‐PET positive). Full model (red) included all markers and demographic variables (age, sex, education, presence of APOE 𝜀4 allele, and time difference between PET acquisition and plasma collection) as predictors. Optimal model (teal) offered the best fit with lowest number of predictors. Aβ‐PET, amyloid β positron emission tomography; AIC, Akaike information criterion; APOE, apolipoprotein E; AUC, area under the ROC curve; CI, confidence interval; ROC, receiver operating characteristic; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
3.3. Associations among markers and disease severity at baseline
Plasma biomarkers were significantly correlated with each other with magnitudes of associations ranging from weak (Aβ42/40 with NfL, ρ = −0.169, P = 0.003) to strong (NfL with GFAP, ρ = −0.575, P < 0.001). TabCAT‐BHA cognitive composite showed moderate correlations with plasma GFAP (ρ = −0.307, P < 0.001) and weaker correlations with plasma Aβ42/40 (ρ = 0.196, P < 0.001), pTau181 (ρ = −0.271, P < 0.001), and NfL (ρ = −0.211, P < 0.001). Bivariate correlations among all scalable markers by diagnostic group are reported in Supplementary Table 1.
We performed individual multiple linear regression models to examine baseline associations between each of the scalable markers with Clinical Dementia Rating (CDR) Sum of Boxes. All markers were individually associated with disease severity when covarying for age, female sex, education, and presence of at least one APOE ε4 allele, including Aβ42/40 (β = −32.231, P = 0.005), pTau181 (β = 0.956, P = 0.002), NfL (β = 1.306, P < 0.001), GFAP (β = 1.479, P < 0.001), and TabCAT‐BHA (β = −0.719, P < 0.001). The results are illustrated in Figure 2.
FIGURE 2.
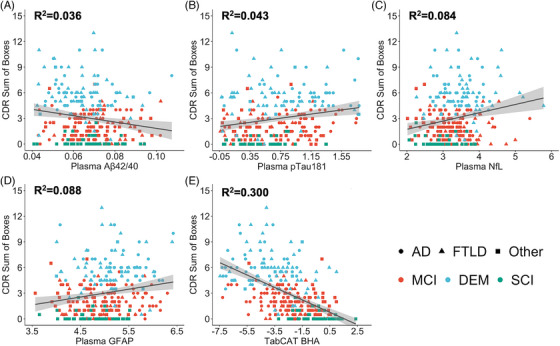
Baseline associations between plasma Aβ42/40 (A), pTau181 (B), NfL (C), GFAP (D), and TabCAT‐BHA cognitive composite (E) with the Clinical Dementia Rating Scale (CDR) Sum of Boxes score. Coefficients of determination are based on multiple linear models covarying for age, female sex, education, and presence of an APOE ε4 allele. Raw values of plasma pTau181, NfL, and GFAP concentrations were natural log‐transformed. Shapes represent clinical syndrome groups (circles = Alzheimer's disease spectrum, triangles = frontotemporal lobar degeneration spectrum, squares = other neurodegenerative syndromes) and colors represent diagnosis groups (red = mild cognitive impairment, teal = dementia, green = subjective cognitive impairment). Aβ, amyloid β; APOE, apolipoprotein E; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
In a linear model including all scalable markers as predictors simultaneously, only log‐transformed plasma NfL (β = 0.625, P = 0.034), TabCAT‐BHA (β = −0.688, P < 0.001), and female sex (β = −0.676, P = 0.013) were significantly associated with disease severity at baseline (detailed output is reported in Supplementary Table 2). A reduced model including log‐transformed plasma NfL, TabCAT‐BHA, and female sex as the only predictors did not result in a significantly worse model fit using likelihood ratio testing (χ2 = 2.377, P = 0.795).
3.4. Associations with changes in disease severity
In separate linear mixed effect models for each of the scalable markers, elevated baseline plasma pTau181 (β = 0.510, P < 0.001) and NfL (β = 0.021, P < 0.001) were associated with greater longitudinal increases on the CDR Sum of Boxes. Lower baseline TabCAT‐BHA cognitive composite values were associated with greater increases in disease severity (β = −0.389, P < 0.001). In contrast, neither baseline values of plasma Aβ42/40 (β = −4.490, P = 0.575) nor plasma GFAP (β = 0.0002, P = 0.829) were significantly associated with changes in CDR Sum of Boxes over time. Detailed outputs of each of the models are reported in Supplementary Tables 3‐7. ,
In a combined mixed model, in which baseline values of all scalable markers were included as predictors simultaneously along with demographic and clinical covariates, higher baseline plasma pTau181 (β = 0.357, P < 0.001) and NfL (β = 0.018, P < 0.001) and lower baseline GFAP (β = −0.002, P = 0.029) and TabCAT‐BHA (β = −0.320, P < 0.001) were independently associated with longitudinal increases in disease severity (Figure 3). Full model output of the combined model is presented in Table 4. Based on the analysis of marginal coefficients of determination for mixed models, 54 total variance explained by the combined effects of plasma pTau181, NfL, GFAP, and TabCAT‐BHA was 29% after accounting for fixed effects of age, sex, education, APOE ε4 status, and baseline diagnosis.
FIGURE 3.
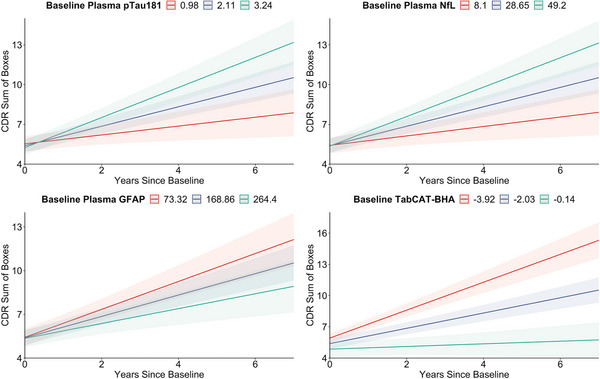
Modeled associations of baseline values of plasma pTau181, NfL, GFAP, and TabCAT‐BHA with longitudinal changes on the Clinical Dementia Rating (CDR) Sum of Boxes score. Regression lines are based on the results of linear mixed effect models with random intercepts and slopes in which all markers were simultaneously included as predictors covarying for age, sex, education, presence of APOE ε4 allele, baseline diagnostic group, and years since baseline. Colors represent values one standard deviation below the mean predictor value (red lines), the mean predictor value (blue lines), and one standard deviation above the mean predictor value (green lines). APOE, apolipoprotein E; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment.
TABLE 4.
Results of linear mixed model analyses including baseline values of all scalable plasma and TabCAT‐BHA markers as predictors, demographic and clinical covariates, and Clinical Dementia Rating Scale (CDR) Sum of Boxes as an outcome (n = 185; 32 SCI, 107 MCI, 46 dementia).
| β | SE | df | P | |
|---|---|---|---|---|
| Baseline plasma Aβ42/40 x Years since baseline | 2.060 | 5.937 | 99 | 0.729 |
| Baseline plasma pTau181 x Years since baseline | 0.357 | 0.082 | 103 | <0.001 |
| Baseline plasma NfL x Years since baseline | 0.018 | 0.004 | 90 | <0.001 |
| Baseline plasma GFAP x Years since baseline | −0.002 | 0.001 | 125 | 0.029 |
| Baseline TabCAT‐BHA x Years since baseline | −0.320 | 0.047 | 120 | <0.001 |
| Baseline plasma Aβ42/40 | −0.122 | 8.433 | 159 | 0.149 |
| Baseline plasma pTau181 | −0.128 | 0.117 | 162 | 0.274 |
| Baseline plasma NfL | −0.001 | 0.006 | 157 | 0.876 |
| Baseline plasma GFAP | −0.001 | 0.001 | 165 | 0.658 |
| Baseline TabCAT‐BHA | −0.283 | 0.081 | 166 | <0.001 |
| Age (years; centered) | 0.012 | 0.014 | 163 | 0.397 |
| Sex (female) | 0.163 | 0.228 | 164 | 0.476 |
| Education (years) | 0.025 | 0.037 | 165 | 0.500 |
| APOE ε4 allele | 0.209 | 0.251 | 166 | 0.408 |
| Baseline dementia diagnosis (reference) | – | – | – | – |
| Baseline MCI diagnosis | −3.487 | 0.302 | 165 | <0.001 |
| Baseline SCI diagnosis | −4.832 | 0.431 | 164 | <0.001 |
| Years since baseline | −0.940 | 0.477 | 96 | 0.052 |
Abbreviations: Aβ, amyloid β; APOE, apolipoprotein E; df, degrees of freedom; GFAP, glial fibrillary acidic protein; MCI, mild cognitive impairment; NfL, neurofilament light; SCI, subjective cognitive impairment; SE, standard error; TabCAT‐BHA, Tablet‐based Cognitive Assessment Tool Brain Health Assessment. Degrees of freedom are estimated using Satterthwaite's method.
3.5. Additional analyses
To account for phenotypic heterogeneity in our sample, we performed separate models examining cross‐sectional and longitudinal associations between a combination of scalable markers with disease severity in AD spectrum and FTLD spectrum subgroups using the CDR+NACC/FTLD 53 Sum of Boxes score as an outcome. In 120 participants with AD spectrum clinical syndromes (72 MCI, 48 dementia), TabCAT‐BHA (β = −1.346, P < 0.001), and female sex (β = −2.112, P = 0.027) were the only markers associated with CDR+NACC/FTLD Sum of Boxes score cross‐sectionally. In 77 individuals with FTLD spectrum diagnoses (29 MCI, 48 dementia), only TabCAT‐BHA (β = −1.341, P = 0.002) was significantly associated with the baseline CDR+NACC/FTLD, with plasma Aβ42/40 ratio approaching significance (β = −116.093, P = 0.062). The detailed results of cross‐sectional multiple regression models in AD and FTLD subgroups are presented in Supplementary Table 8.
Baseline plasma pTau181 (β = 0.536, P < 0.001), NfL (β = 0.015, P = 0.007), and TabCAT‐BHA (β = −0.299, P < 0.001) were significantly associated with longitudinal changes in CDR+NACC/FTLD in linear mixed models with all scalable markers and demographic and clinical variables included as predictors simultaneously in a subsample of participants with AD syndromes (n = 78; 51 MCI, 27 dementia). In the FTLD subsample (n = 33; 15 MCI, 18 dementia), only baseline TabCAT‐BHA (β = −0.572, P = 0.007) was significantly associated with longitudinal increases in the CDR+NACC/FTLD, with baseline plasma NfL (β = 0.039, P = 0.075) approaching significance. Detailed results from these models are reported in Supplementary Table 9.
4. DISCUSSION
This study examined the performance of a combination of scalable plasma biomarkers and brief digital cognitive battery TabCAT‐BHA in relation to ADRD diagnostic outcomes. Specifically, we investigated how well a combination of these markers was able to detect in vivo Aβ pathology on PET, reflect disease staging at baseline, and predict future changes in disease severity. When each biomarker and TabCAT‐BHA were examined individually, pTau181 showed the best accuracy in detecting Aβ‐PET positivity particularly when combined with demographic and clinical information, including age, sex, education, and APOE ε4 status (AUC = 0.933, 95% CI: 0.885‐0.980, Table 2). While these findings are largely consistent with prior reports on the performance of plasma pTau181 in clinical cohorts, 12 , 13 , 14 we found that combining TabCAT‐BHA with demographics and clinical data enhanced accuracy at discriminating Aβ status (AUC = 0.918, 95% CI: 0.866‐0.971, Table 2). When all markers were analyzed simultaneously, a combination of age with plasma Aβ42/40, pTau181, NfL, and TabCAT‐BHA had the highest accuracy against Aβ‐PET with an AUC of 0.962 (95% CI: 0.932‐0.992) within an optimal model selection paradigm (Figure 1). These results provide further support for superior performance of combinations compared to any single one of the included plasma biomarkers in relation to gold standard anchors 22 , 23 , 24 , 25 , 26 and constitute strong evidence for inclusion of scalable cognitive markers into future algorithms.
The added value of efficient cognitive assessment was most evident across findings related to disease severity outcomes. Although each of the scalable markers was associated with CDR Sum of Boxes score at baseline, TabCAT‐BHA cognitive composite showed the strongest magnitude of the association (Figure 2). When all markers were included simultaneously, only TabCAT‐BHA and plasma NfL were cross‐sectionally associated with disease severity (Supplementary Table 2), which is unsurprising given prior evidence of high sensitivity of these two markers to tracking disease severity. 33 , 55 , 56 While our findings are generally consistent with previously reported baseline associations between pTau181 and CDR Sum of Boxes, 12 these relationships were no longer significant when pTau181 was included with TabCAT‐BHA and plasma NfL in our sample (Supplementary Table 2). These results have important implications for clinical implementation algorithms and suggest that scalable plasma and digital cognitive markers play complementary roles for supporting ADRD diagnosis.
In our sample, baseline plasma pTau181, NfL, GFAP, and TabCAT‐BHA were significant predictors of future functional decline and increases in disease severity across SCI, MCI, and dementia groups (Table 4, Figure 3). Despite limited sample sizes, these results were mostly replicated in participants with AD spectrum clinical syndromes, aside from loss of significance for plasma GFAP (Supplementary Table 9). In a smaller subsample with FTLD clinical syndromes, only baseline TabCAT‐BHA reached significance while plasma NfL was trending (Supplementary Table 9). These results complement prior studies showing that the combination of plasma pTau and cognitive tests predicted conversion from SCI and MCI to AD dementia 27 , 28 and extend current knowledge by examining a broader combination of markers in a highly clinically heterogeneous cohort. The relationships between baseline plasma pTau181, NfL, and TabCAT‐BHA with longitudinal changes in CDR were in expected directions with higher baseline pTau181 and NfL and lower baseline TabCAT‐BHA predicting faster rates of decline (Table 4, Figure 3). We further found that the relationship between baseline plasma GFAP and CDR change was inverse when other scalable markers were included simultaneously (Table 4). This finding is likely related to the temporal dynamics of individual biomarkers along the disease continuum with recent evidence implying that plasma GFAP levels exhibited highest elevations in preclinical and early symptomatic stages of AD, 57 which may explain its negative associations in our total sample enriched for FTLD syndromes (Supplementary Table 9). Taken together, our results suggest that supplementing a plasma biomarker panel of pTau181, NfL, and GFAP with TabCAT‐BHA is accurate in predicting longitudinal changes in disease severity across AD and FTLD syndromes, making this combination promising for disease monitoring.
Key strengths of this study include its focus on a novel combination of highly scalable, cost‐effective, and accessible digital cognitive tools and plasma biomarkers, clinical heterogeneity of the study sample with regard to disease severity and neurodegenerative etiology, and greater representation of racially and ethnically diverse individuals compared to most prior studies. 8 At the same time, this study has a number of important limitations. First, not all participants in our sample completed Aβ‐PET, and the prevalence of Aβ‐PET positivity (55%) was higher compared to prior population‐based estimates in older adults without dementia (22%). 58 Along with the fact that the Aβ‐PET sample included only a few participants with SCI, these specifics of the study sample may limit generalizability of our findings to community settings. Our sample was also overrepresented for individuals with AD and FTLD, whereas other diseases such as Lewy body disease, which are relatively more common in general population, were not sufficiently represented. Second, we cannot exclude the possibility of our results being assay‐ and species‐specific since we did not have access to other high performing assays (e.g., mass spectrometry‐based Aβ 10 ) or other pTau species (e.g., pTau217 15 ). These limitations are important to consider given reported differences in performance between different Aβ assays 10 , 11 and plasma pTau species, 14 , 15 and more studies addressing the question of which combination of scalable plasma and clinical markers offers the best diagnostic and prognostic accuracy are needed. Also, the sample of this study was comprised of individuals with a low medical comorbidity burden, and given prior evidence suggesting differential performance of plasma biomarkers in individuals with certain comorbid conditions such as renal disease, stroke, or heart disease, 59 application of our findings to these clinical groups may be limited. Finally, while this study included a racially and ethnically diverse sample, replication of the results in more representative and socioeconomically diverse communities is needed to support the clinical validity of these markers across demographic groups.
In summary, our findings support complementary roles of scalable plasma and digital cognitive markers for detecting Aβ‐PET positivity and predicting disease severity and longitudinal functional decline in diverse older adults with subjective and objective cognitive impairment. These low‐cost, accessible technologies have the potential to complement or replace current diagnostic practices and can be used to inform future research on identification of optimal clinical implementation algorithms for early detection of ADRD.
CONFLICT OF INTEREST STATEMENT
None of the authors declare any competing financial interests relevant to the present study. J.C.R. is a site investigator for clinical trials sponsored by Eli Lilly and Eisai and receives funding from AlzOut and consulting fees from Roon Health, Inc. C.W. has received honoraria from the American Academy of Neurology and consulting fees from LCN Consulting. J.H.K. has received consulting fees from Biogen. BLM has received royalties from Cambridge University Press, Elsevier, Inc., Guilford Publications, Inc., Johns Hopkins Press, Oxford University Press, and Taylor & Francis Group; consulting fees from the Massachusetts General Hospital Alzheimer's Disease Research Center (ADRC) Scientific Advisory Board (SAB), Stanford University ADRC SAB, University of Washington ADRC SAB, Genworth SAB; and honoraria from the Global Summit on Neurodegenerative Diseases, Korean Dementia Society, Massachusetts General Hospital, National MS Society, Ochsner Neuroscience Institute, Providence Saint Joseph Medical Center, Taipei Medical University, University of California Irvine, University of California Los Angeles, and University of Texas. A.L.B. has served as a paid consultant to AGTC, Alector, Alzprotect, Amylyx, Arkuda, Arrowhead, Arvinas, Aviado, Boehringer Ingelheim, Denali, Eli Lilly, GSK, Humana, Life Edit, Merck, Modalis, Oligomerix, Oscotec, Roche, Transposon and Wave; has served as a site investigator for clinical trials sponsored by Biogen, Eisai, and Regeneron; and has received research support from the Rainwater Charitable Foundation, Bluefield Project to Cure FTD, GHR Foundation, Association for Frontotemporal Degeneration, Gates Ventures, Alzheimer's Drug Discovery Foundation, UCSF Parkinson's Spectrum Disorders Center and the University of California Cures AD Program. G.D.R. has received research funding from the Rainwater Charitable Foundation, Avid Radiopharmaceuticals, GE Healthcare, Genentech, Life Molecular Imaging; has served as a consultant for Alector, Eli Lilly, Genentech, GE Healthcare, Roche, Johnson & Johnson, and Merck; and is an Associate Editor for JAMA Neurology. K.L.P. has received research funding from Quest Diagnostics. ET, RLJ, LVV, CY, BC, ALL, AMR, CAG, SJE, BLT, SL, and PDA report no disclosures. Author disclosures are available in the supporting information.
CONSENT STATEMENT
The study was approved by the University of California San Francisco (UCSF) Committee on Human Research. All participants provided written informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors are grateful for the invaluable contributions of the study participants and families and research teams at the University of California, San Francisco Alzheimer's Disease Research Center. This work was supported by the National Institute on Aging [P30AG062422, R35AG072362], the National Institute of Neurological Diseases and Stroke [UG3NS105557], and the Alzheimer's Association [AARF‐21‐851552]. The funding sources had no role in any of the aspects of study design or performance, including collection, analysis, and interpretation of data, preparation and writing of the manuscript, and decision to submit this work for publication.
Tsoy E, La Joie R, VandeVrede L, et al. Scalable plasma and digital cognitive markers for diagnosis and prognosis of Alzheimer's disease and related dementias. Alzheimer's Dement. 2024;20:2089–2101. 10.1002/alz.13686
REFERENCES
- 1. GBD 2019 Dementia Forecasting Collaborators . Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7:e105‐e125. doi:10.1016/S2468‐2667(21)00249‐8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bradford A, Kunik ME, Schulz P, Williams SP, Singh H. Missed and delayed diagnosis of dementia in primary care: prevalence and contributing factors. Alzheimer Dis Assoc Disord. 2009;23:306‐314. doi: 10.1097/WAD.0b013e3181a6bebc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27:954‐963. doi: 10.1038/s41591-021-01382-x [DOI] [PubMed] [Google Scholar]
- 4. Gauthier S, Rosa‐Neto P, Morais JA, Webster C. World Alzheimer report 2021: journey through the diagnosis of dementia. Alzheimer's Disease International; 2021. [Google Scholar]
- 5. Murchison CF, Kennedy RE, McConathy JE, Roberson ED. Racial differences in Alzheimer's disease specialist encounters are associated with usage of molecular imaging and dementia medications: an enterprise‐wide analysis using i2b2. J Alzheimers Dis. 2021;79:543‐557. doi: 10.3233/JAD-200796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsoy E, Kiekhofer RE, Guterman EL, et al. Assessment of racial/ethnic disparities in timeliness and comprehensiveness of dementia diagnosis in California. JAMA Neurol. 2021;78:657‐665. doi: 10.1001/jamaneurol.2021.0399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood‐based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022;21:66‐77. doi:10.1016/S1474‐4422(21)00361‐6 [DOI] [PubMed] [Google Scholar]
- 8. Hansson O, Edelmayer RM, Boxer AL, et al. The Alzheimer's Association appropriate use recommendations for blood biomarkers in Alzheimer's disease. Alzheimers Dement. 2022;18:2669‐2686. doi: 10.1002/alz.12756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Angioni D, Delrieu J, Hansson O, et al. Blood biomarkers from research use to clinical practice: what must be done? A report from the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2022;9:569‐579. doi: 10.14283/jpad.2022.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐β 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78:1375‐1382. doi: 10.1001/jamaneurol.2021.3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Y, Schindler SE, Bollinger JG, et al. Validation of plasma amyloid‐β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology. 2022;98:e688‐e699. doi: 10.1212/WNL.0000000000013211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. doi: 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26:379‐386. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 14. Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20:739‐752. doi:10.1016/S1474‐4422(21)00214‐3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janelidze S, Bali D, Ashton NJ, et al. Head‐to‐head comparison of 10 plasma phospho‐tau assays in prodromal Alzheimer's disease. Brain. 2023;146:1592‐1601. doi: 10.1093/brain/awac333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ashton NJ, Janelidze S, Mattsson‐Carlgren N, et al. Differential roles of Aβ42/40, p‐tau231 and p‐tau217 for Alzheimer's trial selection and disease monitoring. Nat Med. 2022;28:2555‐2562. doi: 10.1038/s41591-022-02074-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janelidze S, Berron D, Smith R, et al. Associations of plasma phospho‐tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021;78:149‐156. doi: 10.1001/jamaneurol.2020.4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324:772‐781. doi: 10.1001/jama.2020.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashton NJ, Janelidze S, Al Khleifat A, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021;12:3400. doi: 10.1038/s41467-021-23620-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johansson C, Thordardottir S, Laffita‐Mesa J, et al. Plasma biomarker profiles in autosomal dominant Alzheimer's disease. Brain. 2023;146:1132‐1140. doi: 10.1093/brain/awac399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144:3505‐3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cullen NC, Janelidze S, Mattsson‐Carlgren N, et al. Test‐retest variability of plasma biomarkers in Alzheimer's disease and its effects on clinical prediction models. Alzheimers Dement. 2022. doi: 10.1002/alz.12706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palmqvist S, Stomrud E, Cullen N, et al. An accurate fully automated panel of plasma biomarkers for Alzheimer's disease. Alzheimers Dement. 2023;19:1204‐1215. doi: 10.1002/alz.12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benedet AL, Brum WS, Hansson O, et al. The accuracy and robustness of plasma biomarker models for amyloid PET positivity. Alzheimers Res Ther. 2022;14:26. doi:10.1186/s13195‐021‐00942‐0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarto J, Ruiz‐García R, Guillén N, et al. Diagnostic performance and clinical applicability of blood‐based biomarkers in a prospective memory clinic cohort. Neurology. 2023;100:e860‐e873. doi: 10.1212/WNL.0000000000201597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thijssen EH, Verberk IMW, Kindermans J, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alzheimers Dement (Amst). 2022;14:e12285. doi: 10.1002/dad2.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer's disease dementia using plasma phospho‐tau combined with other accessible measures. Nat Med. 2021;27:1034‐1042. doi: 10.1038/s41591-021-01348-z [DOI] [PubMed] [Google Scholar]
- 28. Pichet Binette A, Palmqvist S, Bali D, et al. Combining plasma phospho‐tau and accessible measures to evaluate progression to Alzheimer's dementia in mild cognitive impairment patients. Alzheimers Res Ther. 2022;14:46. doi: 10.1186/s13195-022-00990-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petrazzuoli F, Vestberg S, Midlöv P, Thulesius H, Stomrud E, Palmqvist S. Brief cognitive tests used in primary care cannot accurately differentiate mild cognitive impairment from subjective cognitive decline. J Alzheimers Dis. 2020;75:1191‐1201. doi: 10.3233/JAD-191191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsoy E, Sideman AB, Piña Escudero SD, et al. Global perspectives on brief cognitive assessments for dementia diagnosis. J Alzheimers Dis. 2021;82:1001‐1013. doi: 10.3233/JAD-201403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mattke S, Batie D, Chodosh J, et al. Expanding the use of brief cognitive assessments to detect suspected early‐stage cognitive impairment in primary care. Alzheimers Dement. 2023;19(9):4252‐4259. doi: 10.1002/alz.13051 [DOI] [PubMed] [Google Scholar]
- 32. Sabbagh MN, Boada M, Borson S, et al. Early detection of mild cognitive impairment (MCI) in primary care. J Prev Alzheimers Dis. 2020;7:165‐170. doi: 10.14283/jpad.2020.21 [DOI] [PubMed] [Google Scholar]
- 33. Possin KL, Moskowitz T, Erlhoff SJ, et al. The Brain Health Assessment for detecting and diagnosing neurocognitive disorders. J Am Geriatr Soc. 2018;66:150‐156. doi: 10.1111/jgs.15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsoy E, Erlhoff SJ, Goode CA, et al. BHA‐CS: a novel cognitive composite for Alzheimer's disease and related disorders. Alzheimers Dement (Amst). 2020;12(1):e12042. doi: 10.1002/dad2.12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodríguez‐Salgado AM, Llibre‐Guerra JJ, Tsoy E, et al. A brief digital cognitive assessment for detection of cognitive impairment in Cuban older adults. J Alzheimers Dis. 2021;79:85‐94. doi: 10.3233/JAD-200985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsoy E, Strom A, Iaccarino L, et al. Detecting Alzheimer's disease biomarkers with a brief tablet‐based cognitive battery: sensitivity to Aβ and tau PET. Alzheimers Res Ther. 2021;13:36. doi: 10.1186/s13195-021-00776-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. National Alzheimer's Project Act, S 3036, 111th Cong; 2011.
- 38. Barnes LL. Biomarkers for Alzheimer dementia in diverse racial and ethnic minorities – a public health priority. JAMA Neurol. 2019;76:251‐253. doi: 10.1001/jamaneurol.2018.3444 [DOI] [PubMed] [Google Scholar]
- 39. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 40. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging – Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006‐1014. doi: 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13:870‐884. doi: 10.1016/j.jalz.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496‐503. doi: 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32:853‐864. doi: 10.1002/mds.26987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katz DI, Bernick C, Dodick DW, et al. National Institute of Neurological Disorders and Stroke consensus diagnostic criteria for traumatic encephalopathy syndrome. Neurology. 2021;96:848‐863. doi: 10.1212/WNL.0000000000011850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging – Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456‐2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863‐1872. doi: 10.1212/01.wnl.0000187889.17253.b1 [DOI] [PubMed] [Google Scholar]
- 49. La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB‐PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement. 2019;15:205‐216. doi: 10.1016/j.jalz.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lesman‐Segev OH, La Joie R, Iaccarino L, et al. Diagnostic accuracy of amyloid versus 18F‐fluorodeoxyglucose positron emission tomography in autopsy‐confirmed dementia. Ann Neurol. 2021;89:389‐401. doi: 10.1002/ana.25968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Staffaroni AM, Brown JA, Casaletto KB, et al. The longitudinal trajectory of default mode network connectivity in healthy older adults varies as a function of age and is associated with changes in episodic memory and processing speed. J Neurosci. 2018;38:2809‐2817. doi: 10.1523/JNEUROSCI.3067-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information‐theoretic approach, 2nded. Springer‐Verlag; 2002. [Google Scholar]
- 53. Miyagawa T, Brushaber D, Syrjanen J, et al. Utility of the global CDR® plus NACC FTLD rating and development of scoring rules: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16:106‐117. doi: 10.1002/alz.12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakagawa S, Schielzeth H. A general and simple method for obtaining R2 from generalized linear mixed‐effects models. Methods Ecol Evol. 2013;4:133‐142. doi: 10.1111/j.2041-210x.2012.00261.x [DOI] [Google Scholar]
- 55. Rojas JC, Wang P, Staffaroni AM, et al. Plasma neurofilament light for prediction of disease progression in familial frontotemporal lobar degeneration. Neurology. 2021;96:e2296‐e2312. doi: 10.1212/WNL.0000000000011848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Illán‐Gala I, Lleo A, Karydas A, et al. Plasma tau and neurofilament light in frontotemporal lobar degeneration and Alzheimer disease. Neurology. 2021;96:e671‐e683. doi: 10.1212/WNL.0000000000011226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guo Y, Shen XN, Wang HF, et al. The dynamics of plasma biomarkers across the Alzheimer's continuum. Alzheimers Res Ther. 2023;15:31. doi: 10.1186/s13195-023-01174-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roberts RO, Aakre JA, Kremers WK, et al. Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population‐based setting. JAMA Neurol. 2018;75:970‐979. doi: 10.1001/jamaneurol.2018.0629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mielke MM, Dage JL, Frank RD, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat Med. 2022;28:1398‐1405. doi: 10.1038/s41591-022-01822-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information