

Abstract
Introduction
Hereditary transthyretin-mediated amyloidosis (ATTRv, v for variant) is a progressive disease caused by mutations in the TTR gene, leading to sensory-motor, axonal and length-dependent neuropathy. However, some patients may show variable electrophysiological pattern. The aim of this study was to evaluate the electrophysiological features of TTR amyloid neuropathy at the time of the first nerve conduction study (NCS) to assess whether there were distinguishing features useful for early diagnosis.
Methods
We retrospectively revised the first electrophysiological findings of ATTRv patients, and we categorized the neuropathy based on nerve conduction slowing, type of involved fibres and distribution pattern of PNS involvement. Cluster analysis was performed to evaluate the prevalence of neuropathy features between the early and late stage of disease, based on disease duration and disability burden assessed by NIS.
Results
We recruited 33 patients (27 males) with mean age 63.9 ± 10.8 years, mean disease duration 2.8 ± 2.4 years and mean NIS 47.6 ± 41.8. Overall, the frequency analysis showed that the most common features of ATTRv neuropathy included the categories of axonal, sensory-motor and neuronopathic-like pattern. This electrophysiological pattern of PNS involvement was constant in patients in late stage of disease, whereas ATTRv patients in early stage of disease displayed variable electrophysiological pattern of PNS involvement.
Discussion
Our findings demonstrated that ATTRv neuropathy may present at first NCS in a variable way, and it changes over the course of disease. Such heterogeneity makes the suspicion of ATTRv even more challenging at the time of first electrophysiological examination.
Keywords: Amyloidosis, TTR-related neuropathy, Heterogeneous neuropathy, Early-stage disease
Introduction
Hereditary transthyretin amyloidosis (ATTRv, v for variant) is a multisystemic disorder caused by mutation in the transthyretin (TTR) gene with a progressive involvement of peripheral nervous system (PNS). The mutated TTR tetramer dissociates in monomers that misfold and aggregate in amyloid fibrils accumulating in PNS [1]. In detail, amyloid fibril deposit starts in the dorsal root ganglia (DRG) and autonomic ganglia, causing an early axonal loss of sensory nerve fibres through a dying-back degeneration. Subsequently, TTR deposition may involve nerve roots and peripheral nerves leading to a progressive sensory-motor axonal neuropathy [2].
Accordingly, the neuropathy in ATTRv was labelled as a progressive, length-dependent, sensory or sensory-motor axonal neuropathy. However, ATTRv diagnosis is challenging with high rate of misdiagnosis, inappropriate treatment and diagnostic delay [3]. In the era of different disease modifying treatments, a proper and early diagnosis is essential to limit the disability burden and assure the halting of disease progression [4].
The aim of this study was to evaluate the electrophysiological features of neuropathy in 33 ATTRv patients at the time of the first neurophysiological evaluation and to assess whether there were distinguishing features useful for early diagnosis.
Methods
This is a monocentric and retrospective study that re-evaluated the first neurophysiological evaluation of ATTRv patients. We included patients carrying a pathogenic mutation in TTR gene that underwent nerve conduction study (NCS) at our third level center from 2011 to 2023. Patients with incomplete clinical and electrophysiological data were excluded from the analysis.
Electrophysiological data was re-evaluated from internal database and was collected to establish the features of neuropathy based on nerve conduction slowing, type of involved fibres and distribution pattern of PNS involvement.
In detail, in the “nerve conduction slowing” feature, we included three categories: (1) demyelinating neuropathy: motor (MNCV) and/or sensory nerve conduction velocities (SNCV) met the EAN/PNS criteria of demyelination [5], (2) axonal neuropathy: reduction of compound action potential (CMAP)/sensory action potential (SAP) amplitude and MNCV/SNCV normal or slightly slowed but not fulfilling demyelinating criteria [5] and (3) intermediate neuropathy: normal CMAP/SAP amplitude and MNCV/SNCV reduced but not fulfilling demyelinating criteria [5].
In the “type of involved fibres” feature, we included three categories: (1) sensory predominant neuropathy: SAPs absent or reduced ≥ 30% of lower limit normal value (LLN) with normal o slightly reduced CMAPs, (2) motor predominant neuropathy: CMAPs absent or reduced ≥ 30% of LLN with normal o slightly reduced SAPs and (3) sensory-motor neuropathy: SAPs and CMAPs similarly impaired.
In the “distribution pattern” feature, we included four categories: (1) length-dependent neuropathy: lower limb nerves more impaired than upper limb nerves, (2) upper limb predominant neuropathy: upper limb nerves more impaired than lower limb nerves, (3) neuronopathic-like: upper and lower limb nerves equally impaired and (4) multi-neuropathy: side-to-side nerve CMAP/SAP asymmetry (≥ 50%) or inhomogeneous involvement of two contiguous nerves (not explained by entrapment neuropathy).
Lastly, gender, mutation, age at first nerve conduction study (NCS), disease duration, familial amyloid polyneuropathy (FAP) stage and Neuropathy Impairment Score (NIS) were recorded in a minimal clinical dataset.
Statistical analysis
Descriptive analyses, based on the mean ± standard deviation (SD) for numerical variables and percentage for categorical data, were used to define the prevalence of the most frequent features of ATTRv neuropathy.
In order to evaluate whether the neuropathy features may change over time, we used K-mean cluster analysis [6] to divide our cohort in two clusters—the cluster 1 (early stage) and cluster 2 (late stage). We set up the criteria to recognize early vs late-onset cluster in our cohort based on disease duration and disability burden assessed by NIS. We consequently analyzed the prevalence of neuropathy features of each cluster.
Analyses were performed using the statistical software IBM SPSS Statistic version 25.
Results
We included 33 ATTRv patients (M/F = 27/6) belonging to 25 families. Fourteen patients carried V30M variant, 15 F64L, 3 E54L and 1 V122I. Twenty of them were in FAP stage 1, 8 in FAP stage 2 and 5 in FAP stage 3. Patients presented at first NCS a mean age of 63.9 ± 10.8 years, a mean disease duration of 2.8 ± 2.4 years and a disability burden assessed by NIS of 47.6 ± 41.8 points (Table 1).
Table 1.
Clinical and electrophysiological findings
| Age at first NCS (years) | 63.9 ± 10.8 (31–76) |
| Disease duration (years) | 2.8 ± 2.4 (0–10) |
| Mutation | |
|
V30M F64L E54L V122I |
14 (42.4%) 15 (45.6%) 3 (9%) 1 (3%) |
| FAP stage | |
|
FAP 1 FAP 2 FAP 3 |
20 (60.6%) 8 (24.2%) 5 (15.2%) |
| NIS | 47.6 ± 41.8 (0–147) |
| Nerve conduction slowing | |
|
Axonal Demyelinating Intermediate |
29 (87.8%) 2 (6.1%) 2 (6.1%) |
| Type of involved fibres | |
|
Sensory Motor Sensory-motor |
6 (18.1%) 2 (6.1%) 25 (75.8%) |
| Distribution pattern | |
|
Length-dependent Upper limb predominant Neuronopathic-like Multiplex neuropathy |
10 (30.3%) 3 (9.1%) 16 (48.5%) 4 (12.1%) |
NCS nerve conduction study, FAP familial amyloid polyneuropathy, NIS Neuropathy Impairment Score
The analysis of NCS features revealed that the most frequent “nerve conduction slowing” category was axonal (87.8% vs demyelinating 6.1%; intermediate 6.1%), the most frequent “type of involved fibres” category was sensory-motor (75.8% vs sensory 18.1%; motor 6.1%) and the most frequent “distribution pattern” category was neuronopathic-like (48.5% vs length-dependent 30.3%; upper limb 9.1%; multi-neuropathy 12.1%) (Table 1).
The K-mean analysis showed the presence in our cohort of the two distinct clusters (p < 0.001): in the early-stage patients had a disease duration of 1.8 ± 1.5 years and NIS of 23.2 ± 16.8 points, while the late-stage cluster showed a disease duration of 5.2 ± 2.5 years and disability burden of 103.8 ± 22.6 points (Fig. 1). The frequency analysis showed that neuropathy features were different between the two clusters. In the late-stage cluster, the neuropathy was exclusively axonal, sensory-motor and with neuronopathic-like pattern, whereas in the early stage, neuropathy features were heterogenous, including intermediate NCV, sensory and/or motor predominant nerve fibre involvement and variable distribution pattern (length-dependent, upper limb predominant, multi-neuropathy) (Table 2).
Fig. 1.
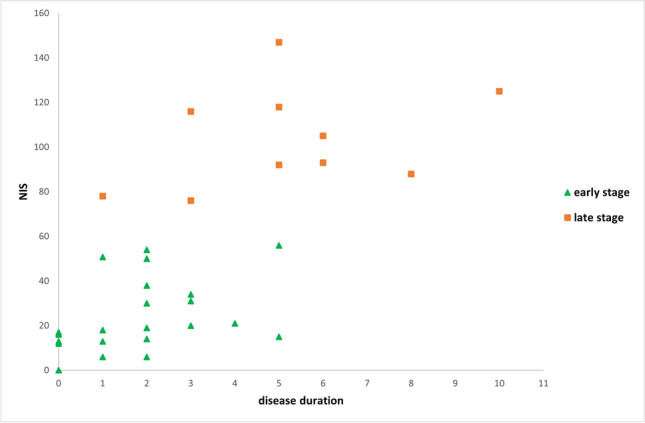
Early and late stage clusters. Figure showed early and late stage clusters divided according to disease duration and disability burden (assessed through the NIS). NIS, Neuropathy Impairment Score
Table 2.
Prevalence of electrophysiological findings between the early and late stage cluster
| Cluster 1 (early stage) | Cluster 2 (late stage) | |
|---|---|---|
| Number of patients | 23 | 10 |
| Age at first NCS | 61.9 ± 12.3 (31–76) | 68.6 ± 3.5 (64–75) |
| Disease duration | 1.8 ± 1.5 (0–5) | 5.2 ± 2.5 (1–10) |
| NIS | 23.2 ± 16.8 (0–56) | 103.8 ± 22.6 (76–147) |
| Nerve conduction slowing | ||
|
Axonal Demyelinating Intermediate |
20 (86.9%) 1 (4.4%) 2 (8.7%) |
9 (90%) 1 (10%) 0 (0%) |
| Type of involved fibres | ||
|
Sensory Motor Sensory-motor |
6 (26.1%) 2 (8.7%) 15 (65.2%) |
0 (0%) 0 (0%) 10 (100%) |
| Distribution pattern | ||
|
Length-dependent Upper limb Neuronopathic Multiplex neuropathy |
10 (43.5%) 3 (13.1%) 6 (26.1%) 4 (17.3%) |
0 (0%) 0 (0%) 10 (100%) 0 (0%) |
NCS nerve conduction study, NIS Neuropathy Impairment Score
Discussion
ATTRv neuropathy is a rare, multisystemic and fatal disease. In the last decade, several disease modifying treatments have become available, and they are more effective in slowing or halting disease progression, the earlier they are administrated [7–9]. Therefore, early diagnosis is crucial for limiting disability burden.
However, the diagnosis of ATTRv neuropathy is still delayed and patients may experience misdiagnosis and sometimes inappropriate therapy as well [10]. The aim of this study was to define the heterogeneity of ATTRv neuropathy at first NCS.
Overall, the frequency analysis showed that the most common features of ATTRv neuropathy included the categories of axonal, sensory-motor and neuronopathic-like pattern.
The neuropathy was classifiable as primary axonal in most patients, and noteworthy, CMAP/SAP amplitude was constantly reduced in patients displaying demyelinating features as well [11], suggesting that a severe axonal loss in patient with a diagnosis of CIDP with a progressive course and a poor response to immunomodulatory therapy should raise the suspicion for ATTRv neuropathy. Indeed, demyelination may occur in ATTRv patients as amyloid deposit may cause myelin sheet detachment and nodal enlargement [12]. Interestingly, 6% of our patients displayed slightly reduced NCV not fulfilling EAN/PNS criteria [5] and normal CMAP amplitude (intermediate category). In such patients, we might speculate that TTR oligomers interact with ion channels and, hence, alter the homeostasis of the nerve membrane and impair NCV before nerve damage has occurred [13, 14].
The most frequent “type of involved fibres” category was sensory-motor neuropathy, while sensory or motor predominant neuropathy appeared rarer in our cohort. If sensory predominant neuropathy can be explained by early involvement of DRG, for predominant motor neuropathy, the pathomechanism is more difficult to figure out. Anyway, TTR-related predominant motor neuropathy should not be overlooked as it can be also misdiagnosed as lower motor neuron disease [10, 15].
Lastly, 50% of patients had a neuropathy with neuronopathic-like pattern. Such findings fit with the notion that nerve damage starts proximally (DRG and nerve roots) and, accordingly, the involvement of upper limb nerves can be more precocious than in length-dependent neuropathy [16]. In keeping with this, skin biopsy demonstrated an early and highly frequent of peripheral nerve involvement with a non-length dependent fashion (ganglionopathy) in ATTRv patients and carriers [17, 18]. Moreover, the rapidly progressive feature of ATTRv neuropathy means that even a neuropathy starting as length-dependent may soon involve the upper limbs, thus, resulting in a neuronopathic-like pattern.
Interestingly, we also observed upper limb predominant neuropathy in 10% and multi-neuropathy in 12% of our cohort of patients. Upper limb onset (ULO) of ATTRv may be quite common in non-endemic areas [19], and the pathomechanism underling the nerve damage seems to be different from other form of ATTRv neuropathy. Pathological data from radial nerve biopsy showed that ULO patients had a significant loss of large myelinated fibres with conspicuous amyloid deposits in endoneurium as well as in small epineurial vessels walls, leading to vessel obstruction and eventually to chronic ischaemia due to amyloid angiopathy [19].
On the other hand, multi-neuropathic involvement of PNS has been only anecdotally reported and such involvement could reflect a random heterogeneous deposition of amyloid along the nerve [20].
The cluster analysis showed that ATTRv patients in late stage of disease (longer disease duration and higher disability) had a constant electrophysiological pattern of PNS involvement consistent with an axonal, sensory-motor and neuronopathic-like neuropathy. Conversely, ATTRv patients in early stage of disease (shorter disease duration and lower disability) displayed variable electrophysiological pattern of PNS involvement for “nerve conduction slowing”, “type of involved fibres” and “distribution pattern”.
In conclusion, although the limits of the study (retrospective design and relatively small sample), our findings demonstrated why ATTRv diagnosis is so challenging since the neuropathy may present at first NCS in variable way and it changes over the course of disease. If in early stage of the disease, neuropathy can be extremely heterogeneous, it can rapidly evolve in a severe polyneuropathy characterized by a sensory-motor axonal involvement with a neuronopathic-like pattern.
Generally, in clinical practice, patients with progressive axonal and sensory-motor neuropathy are addressed to genetic analysis if at least one red flag is detected (e.g., carpal tunnel syndrome, lumbar stenosis, cardiac, ocular, renal involvement, and autonomic symptoms). However, clinicians should not overlook the hypothesis of ATTRv neuropathy when facing with different electrophysiological features. Clinicians should perform the TTR genetic analysis in patients with neuronopathic-like polyneuropathy, while in patients with other type of polyneuropathy, they have to look for ATTRv multisystemic involvement (red flags) in order to suspect ATTRv disease and thus perform an early diagnosis.
Lastly, such heterogeneity at the time of presentation should be carefully considered when investigating presymptomatic subjects, as if an extensive NCS is not performed (bilaterally upper and lower limb nerves), PNS involvement may be missed, resulting in a delay of treatment.
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Declarations
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki. Ethical review and approval were waived for this study due to the retrospective nature of the study, and all the procedures being performed were part of the routine care.
Conflict of interest
S.T. received personal fees for scientific events from Alnylam Pharmaceuticals, Amicus Therapeutics and Takeda and travel grants to attend scientific meetings from Akcea Therapeutics; D.S. received a travel grant to attend scientific meetings from SOBI; F.M. received personal fees for scientific events from Alfa-Sigma, Alnylam Pharmaceuticals and Akcea Therapeutics and has received a travel grant to attend scientific meetings from CSL Behring.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Manganelli F, Fabrizi GM, Luigetti M, Mandich P, Mazzeo A, Pareyson D. Hereditary transthyretin amyloidosis overview. Neurol Sci. 2022;43(Suppl 2):595–604. doi: 10.1007/s10072-020-04889-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tozza S, Severi D, Spina E, Iovino A, Aruta F, Ruggiero L, Dubbioso R, Iodice R, Nolano M, Manganelli F. The neuropathy in hereditary transthyretin amyloidosis: a narrative review. J Peripher Nerv Syst. 2021;26(2):155–159. doi: 10.1111/jns.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gertz M, Adams D, Ando Y, Beirão JM, Bokhari S, Coelho T, Comenzo RL, Damy T, Dorbala S, Drachman BM, Fontana M, Gillmore JD, Grogan M, Hawkins PN, Lousada I, Kristen AV, Ruberg FL, Suhr OB, Maurer MS, Nativi-Nicolau J, Quarta CC, Rapezzi C, Witteles R, Merlini G. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020;21(1):198. doi: 10.1186/s12875-020-01252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll A, Dyck PJ, de Carvalho M, Kennerson M, Reilly MM, Kiernan MC, Vucic S. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2022;93(6):668–678. doi: 10.1136/jnnp-2021-327909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Vankrunkelsven P, Allen JA, Attarian S, Blomkwist-Markens PH, Cornblath DR, Eftimov F, Goedee HS, Harbo T, Kuwabara S, Lewis RA, Lunn MP, Nobile-Orazio E, Querol L, Rajabally YA, Sommer C, Topaloglu HA (2021) European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force-second revision. J Peripher Nerv Syst 26(3):242–268. 10.1111/jns.12455. Erratum in: J Peripher Nerv Syst. 2022;27(1):94. Erratum in: Eur J Neurol. 2022;29(4):1288
- 6.Selim SZ, Ismail MA. K-means-type algorithms: a generalized convergence theorem and characterization of local optimality. IEEE Trans Pattern Anal Mach Intell. 1984;6(1):81–87. doi: 10.1109/tpami.1984.4767478. [DOI] [PubMed] [Google Scholar]
- 7.Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, Conceiçao I, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Grogan DR. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–2814. doi: 10.1007/s00415-013-7051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Stefano V, Fava A, Gentile L, Guaraldi P, Leonardi L, Poli L, Tagliapietra M, Vastola M, Fanara S, Ferrero B, Giorgi M, Perfetto F, Russo M, Russo D. Italian real-life experience of patients with hereditary transthyretin amyloidosis treated with patisiran. Pharmgenomics Pers Med. 2022;12(15):499–514. doi: 10.2147/PGPM.S359851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luigetti M, Antonini G, Di Paolantonio A, Gentile L, Grandis M, Leonardi L, Lozza A, Manganelli F, Mazzeo A, Mussinelli R, My F, Obici L, Maria Pennisi E, Romozzi M, Russo M, Sabatelli M, Salvalaggio A, Tagliapietra M, Tozza S. Real-life experience with inotersen in hereditary transthyretin amyloidosis with late-onset phenotype: data from an early-access program in Italy. Eur J Neurol. 2022;29(7):2148–2155. doi: 10.1111/ene.15325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortese A, Vegezzi E, Lozza A, Alfonsi E, Montini A, Moglia A, Merlini G, Obici L. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017;88(5):457–458. doi: 10.1136/jnnp-2016-315262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohashi N, Kodaira M, Morita H, Sekijima Y. Electrophysiological demyelinating features in hereditary ATTR amyloidosis. Amyloid. 2019;26(1):15–23. doi: 10.1080/13506129.2018.1564903. [DOI] [PubMed] [Google Scholar]
- 12.Luigetti M, Romozzi M, Bisogni G, Cardellini D, Cavallaro T, Di Paolantonio A, Fabrizi GM, Fenu S, Gentile L, Grandis M, Marucci G, Massucco S, Mazzeo A, Pareyson D, Romano A, Russo M, Schenone A, Tagliapietra M, Tozza S, Vita G, Sabatelli M. ATTRv pathology: nerve biopsy results from Italian referral centers. Brain Sci. 2020;10(11):780. doi: 10.3390/brainsci10110780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou X, Parkington HC, Coleman HA, Mechler A, Martin LL, Aguilar MI, Small DH. Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. J Neurochem. 2007;100(2):446–457. doi: 10.1111/j.1471-4159.2006.04210.x. [DOI] [PubMed] [Google Scholar]
- 14.Gasperini RJ, Hou X, Parkington H, Coleman H, Klaver DW, Vincent AJ, Foa LC, Small DH (2011) TRPM8 and Nav1.8 sodium channels are required for transthyretin-induced calcium influx in growth cones of small-diameter TrkA-positive sensory neurons. Mol Neurodegener 6(1):19. 10.1186/1750-1326-6-19 [DOI] [PMC free article] [PubMed]
- 15.Salvi F, Pastorelli F, Plasmati R, Bartolomei I, Dall'Osso D, Rapezzi C. Genotypic and phenotypic correlation in an Italian population of hereditary amyloidosis TTR-related (HA-TTR): clinical and neurophysiological aids to diagnosis and some reflections on misdiagnosis. Amyloid. 2012;19(Suppl 1):58–60. doi: 10.3109/13506129.2012.682187. [DOI] [PubMed] [Google Scholar]
- 16.Tozza S, Severi D, Spina E, Di Paolantonio A, Iovino A, Guglielmino V, Aruta F, Nolano M, Sabatelli M, Santoro L, Luigetti M, Manganelli F. A compound score to screen patients with hereditary transthyretin amyloidosis. J Neurol. 2022;269(8):4281–4287. doi: 10.1007/s00415-022-11056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonardi L, Galosi E, Vanoli F, Fasolino A, Di Pietro G, Luigetti M, Sabatelli M, Fionda L, Garibaldi M, Alfieri G, Lauletta A, Morino S, Salvetti M, Truini A, Antonini G. Skin biopsy and quantitative sensory assessment in an Italian cohort of ATTRv patients with polyneuropathy and asymptomatic carriers: possible evidence of early non-length dependent denervation. Neurol Sci. 2022;43(2):1359–1364. doi: 10.1007/s10072-021-05434-5. [DOI] [PubMed] [Google Scholar]
- 18.Leonardi L, Costanzo R, Forcina F, Morino S, Antonini G, Salvetti M, Luigetti M, Romano A, Primiano G, Guglielmino V, Fionda L, Garibaldi M, Lauletta A, Rossini E, Tufano L, Ceccanti M, Esposito N, Falco P, di Pietro G, Truini A, Galosi E. Quantitative sensory testing and skin biopsy findings in late-onset ATTRv presymptomatic carriers: relationships with predicted time of disease onset (PADO) J Peripher Nerv Syst. 2023;28(3):390–397. doi: 10.1111/jns.12586. [DOI] [PubMed] [Google Scholar]
- 19.Théaudin M, Lozeron P, Algalarrondo V, Lacroix C, Cauquil C, Labeyrie C, Slama MS, Adam C, Guiochon-Mantel A, Adams D; French FAP Network (CORNAMYL) Study Group (2019) Upper limb onset of hereditary transthyretin amyloidosis is common in non-endemic areas. Eur J Neurol 26(3):497–e36. 10.1111/ene.13845 [DOI] [PubMed]
- 20.Guy CD, Jones CK. Abdominal fat pad aspiration biopsy for tissue confirmation of systemic amyloidosis: specificity, positive predictive value, and diagnostic pitfalls. Diagn Cytopathol. 2001;24(3):181–185. doi: 10.1002/1097-0339(200103)24:3<181::aid-dc1037>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]