

Abstract
Osteoporosis results from overactivation of osteoclasts. There are currently few drug options for treatment of this disease. Since the successful development of allosteric inhibitors, phosphatases have become attractive therapeutic targets. Protein phosphatase 1, regulatory subunit 15 A (PPP1R15A), is a stress-responsive protein, which promotes the UPR (unfolded protein response) and restores protein homeostasis. In this study we investigated the role of PPP1R15A in osteoporosis and osteoclastogenesis. Ovariectomy (OVX)-induced osteoporosis mouse model was established, osteoporosis was evaluated in the left femurs using micro-CT. RANKL-stimulated osteoclastogenesis was used as in vitro models. We showed that PPP1R15A expression was markedly increased in BMMs derived from OVX mice and during RANKL-induced osteoclastogenesis in vitro. Knockdown of PPP1R15A or application of Sephin1 (a PPP1R15A allosteric inhibitor in a phase II clinical trial) significantly inhibited osteoclastogenesis in vitro. Sephin1 (0.78, 3.125 and 12.5 μM) dose-dependently mitigated the changes in NF-κB, MAPK, and c-FOS and the subsequent nuclear factor of activated T cells 1 (NFATc1) translocation in RANKL-stimulated BMMs. Both Sephin1 and PPP1R15A knockdown increased the phosphorylated form of eukaryotic initiation factor 2α (eIF2α); knockdown of eIF2α reduced the inhibitory effects of Sephin1 on NFATc1-luc transcription and osteoclast formation. Furthermore, Sephin1 or PPP1R15A knockdown suppressed osteoclastogenesis in CD14+ monocytes from osteoporosis patients. In OVX mice, injection of Sephin1 (4, 8 mg/kg, i.p.) every two days for 6 weeks significantly inhibited bone loss, and restored bone destruction and decreased TRAP-positive cells. This study has identified PPP1R15A as a novel target for osteoclast differentiation, and genetic inhibition or allosteric inhibitors of PPP1R15A, such as Sephin1, can be used to treat osteoporosis.
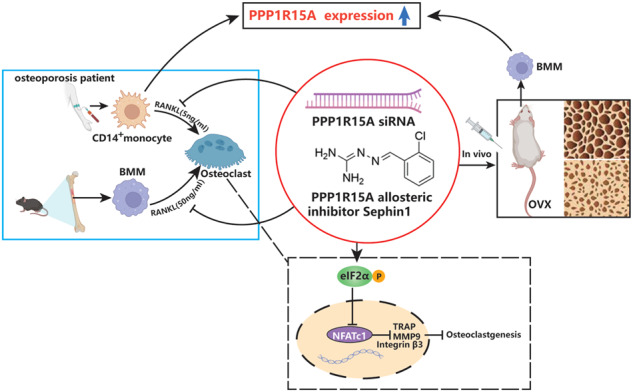
This study revealed that PPP1R15A expression was increased in osteoporosis in both human and mice. Inhibition of PPP1R15A by specific knockdown or an allosteric inhibitor Sephin1 mitigated murine osteoclast formation in vitro and attenuated ovariectomy-induced osteoporosis in vivo. PPP1R15A inhibition also suppressed pathogenic osteoclastogenesis in CD14+ monocytes from osteoporosis patients. These results identify PPP1R15A as a novel regulator of osteoclastogenesis and a valuable therapeutic target for osteoporosis.
Keywords: osteoporosis, PPP1R15A, Sephin1, osteoclastogenesis, RANKL, eIF2α
Introduction
Osteoporosis is a critical public health problem that reduces quality of life, and there are more than 275 million people with osteoporosis worldwide [1]. An imbalance between bone formation and bone resorption causes osteoporosis; thus, targeting osteoclastogenesis is considered an effective therapeutic strategy for osteoporosis [2]. Currently, denosumab and bisphosphonates are widely used to inhibit osteoclasts in the clinic [3]. However, these drugs inhibit both pathological and physiological osteoclasts and their function, which can result in severe side effects, such as osteonecrosis of the jaw (ONJ) and atypical femoral fractures [4–6]. According to a multi-institutional retrospective cohort study, denosumab and bisphosphonate-treated osteoporosis patients had incidence densities of 1.47 and 2.49 ONJ events, respectively (per 1000 person-years) [7]. Patients with ONJ suffer from disability and a serious reduction in quality of life. Therefore, alternative strategies to address excessive osteoclast activity and subsequent bone destruction are urgently needed.
Osteoclasts (OCs) are differentiated from bone marrow-derived macrophages (BMMs) in response to stimulation with nuclear factor-κB ligand receptor activator (RANKL) and macrophage colony-stimulating factor (M-CSF) [8]. When RANK-expressing BMMs are activated by RANKL, it triggers a cascade of intracellular signaling pathways, such as NF-κB and MAPK [9], resulting in the activation of the transcription factors c-FOS and NFATc1 [10, 11]. The expression of a series of osteoclast marker genes, such as matrix metalloprotease 9 (MMP-9), DC-specific transmembrane protein, cathepsin K, and tartrate-resident alkaline phosphatase (ACP5), is upregulated, ultimately inducing the formation of mature osteoclasts [8].
Phosphatase is an enzyme that dephosphorylates proteins as a substrate. Recent studies have shown that functional abnormalities of some protein phosphatases are closely related to tumors, diabetes, and autoimmune diseases [12, 13]. In contrast to kinases, which have been used for a long time as drug targets with over 3000 FDA-approved and tested agents, phosphatases were previously thought to be impossible as a drug target to develop safe inhibitors [14]. The recently developed allosteric inhibitors of phosphatase exhibit the ability to bypass highly conserved protein phosphatase catalytic sites, which enhances the selectivity of small molecules for specific phosphatases while reducing the molecule polarity and charge, and they show increased solubility, permeability, and bioavailability and overcome the challenges associated with the “undruggable” nature of phosphatases [14]. The development of allosteric phosphatase inhibitors has become a hot field in pharmaceutical enterprises [14]. Several phosphatase inhibitors are now in clinical trials, including the SHP2 inhibitors TNO155 [15] and RMC-4630 [14] and the PPP1R15A inhibitor Sephin1 [16]. Sephin1 is effective in ameliorating amyotrophic lateral sclerosis (ALS), Charcot–Marie–Tooth 1B [16], multiple sclerosis [17], and prion disease [18]. Sephin1 is a newly synthesized selective allosteric inhibitor of stress-induced PPP1R15A, and it shows no effect on the constitutive PPP1R15B as a protein phosphatase 1 [13].
Endoplasmic reticulum (ER) stress occurs when misfolded proteins accumulate, activating the unfolded protein response (UPR) aimed at restoring protein homeostasis [19]. The resolution of the UPR is directed by PPP1R15A, and the UPR is involved in the pathogenesis of diseases such as neurological disorders and metabolic disorders [20, 21]. PPP1R15A also serves as a therapeutic target for diseases, and several PPP1R15A inhibitors are now in clinical trials and used as potential treatments in neuron motor- and proteostasis-related diseases [16, 22]. To date, the roles of PPP1R15A in osteoclastogenesis and osteoporosis have not been defined. Thus, we attempted to determine the effects of PPP1R15A on the pathogenesis and treatment of osteolytic diseases.
Here, we revealed that PPP1R15A expression was increased in osteoporosis in both humans and mice. Inhibition of PP1R15A by specific knockdown or an allosteric inhibitor mitigated murine osteoclast formation in vitro and attenuated osteoporosis induced by ovariectomy in mice. Targeted inhibition of PPP1R15A also suppressed osteoclastogenesis in CD14+ monocytes from osteoporosis patients. These results identify PPP1R15A as a novel regulator of osteoclastogenesis and a valuable therapeutic target for osteoporosis.
Materials and methods
Reagents and antibodies
Specific mPPP1R15A and hPPP1R15A siRNAs were obtained from Genepharma (Shanghai, China). The PPP1R15A antibody was purchased from Proteintech (Wuhan, China). Sephin1 (purity ≥99%) was supplied by APExBIO (Houston, TX, USA). M-CSF and RANKL were supplied by R&D (Minneapolis, MN, USA). Fetal bovine serum and alpha-MEM were supplied by Gibco (Rockville, MD, USA). GoTaq® qPCR Master Mix and luciferase reagents were acquired from Promega (Madison, WI, USA). Lipofectamine RNAiMAX and LipofectamineTM 2000 were obtained from Invitrogen (Carlsbad, CA, USA). Oestradiol (E2), MTT, TRAP kit, BSA, and DAPI were supplied by Sigma (St Louis, MO, USA). Primary antibodies against eIF2α, p-eIF2α, NFATc1, p65, c-FOS, Integrin β3, MMP-9, phosphorylated (p-)IκBα, and phospho-MAPKs were supplied by Cell Signaling Technology (Boston, MA, USA)).
Osteoclastic TRAP staining
The peripheral blood of osteoporosis patients was isolated for CD14+ monocytes by a Human Monocyte Isolation Kit (STEMCELL Technologies, Canada). Sorted CD14+ monocytes were maintained with M-CSF (30 ng/mL) for 1 day and then cultured with Sephin1 (0.78, 3.125, 12.5 µM) or E2 (0.1 µM) under RANKL (5 ng/mL) stimulation for 6 days until osteoclasts formed for the TRAP assay. This study was approved by the Ethics Committees of Nanfang Hospital of Southern Medical University (NFEC-202101-K14-01), the patients with osteoporosis participating in the study were informed in advance and signed informed consent forms.
BMMs were prepared as osteoclast precursors as described previously [23]. Cells were flushed from the tibia and femur using complete α-MEM and incubated overnight. Nonadherent cells were lysed with red blood cell lysis buffer to remove red blood cells and cultured with M-CSF (30 ng/mL). After 2 days, 8 × 103 BMMs were seeded overnight and treated with Sephin1 or E2 in 96-well plates for 5 days under RANKL (50 ng/mL) stimulation. The differentiated OCs were stained for the TRAP assay.
Bone resorption assay
BMMs or CD14+ monocytes from osteoporosis patients were seeded into plates coated with hydroxyapatite separately with Sephin1 (0.78, 3.125, 12.5 µM) or E2 (0.1 µM) for seven days. Finally, the plates were washed to eliminate the cells using a 10% bleach solution. The resorption areas of hydroxyapatite were quantified.
Cytotoxicity assay
The cytotoxic effects of Sephin1 on BMMs and calvarial osteoblasts were detected by CCK-8 assays. BMMs and calvarial osteoblasts were cultured in 96-well plates and treated with 0.78, 3.125, or 12.5 µM Sephin1 for 4 or 5 days, and the absorbance at 490 nm was determined.
Luciferase reporter assays of NF-κB and NFATc1
RAW264.7 cells transfected with NFATc1-Luc and NF-κB-Luc were cultured as described previously [24]. For analysis of the effects of Sephin1 on the transcriptional activation of NFATc1 and NF-κB, RAW264.7 cells (1 × 104 cells) were pretreated with 0.78, 3.125, or 12.5 µM Sephin1, and RANKL (50 ng/mL) was then added for 24 h to determine luciferase activity.
Immunofluorescence staining of PPP1R15A, NFATc1, NF-κB p65, and the F-actin ring
Paraformaldehyde (4%) was used to fix the osteoclasts induced from BMMs at 4 °C for 20 min, followed by permeabilization with 0.1% Triton X-100 for 5 min and blocking with 10% goat serum for 1 h. The cells were immunostained using rhodamine phalloidin to observe the F-actin ring. Furthermore, PPP1R15A, NFATc1, and p65 protein expression was determined by primary antibody and secondary antibody conjugated with Alexa Fluor 488. DAPI solution was applied for the staining of cell nuclei. The cells were observed by fluorescence microscopy.
Quantitative real-time PCR
The RNeasy Mini Kit (Promega, Madison, WI, USA) was used to extract total RNA from CD14+ monocytes of osteoporosis patients or BMMs, and cDNA was prepared from 1 μg of total RNA using reverse transcription (TaKaRa Biotechnology, Japan). Quantitative real-time PCR was conducted using SYBR Premix ExTaqTM II (Promega, Madison, WI, USA) as previously described [23]. Values were normalized to internal control Gapdh expression. The primer sequences are listed in Table 1.
Table 1.
Design of the primer’s sequences (5′→3′) for RT-PCR.
| Genes | Forward primer | Reverse primer |
|---|---|---|
| mPpp1r15a | GACCCCTCCAACTCTCCTTC | CTTCCTCAGCCTCAGCATTC |
| hPPP1R15A | AGCCACGGAGGATAAAAGA | CTGAACGATACTCCCAGGACC |
| Acp5 | TGTGGCCATCTTTATGCT | GTCATTTCTTTGGGGCTT |
| Mmp-9 | CGTGTCTGGAGATTCGACTTGA | TTGGAAACTCACACGCCAGA |
| Integrin β3 | TGACTCGGACTGGACTGGCTA | CACTCAGGCTCTTCCACCACA |
| eIF2α-siRNA | UCGAGCAGAUAUUGAAGUA | CAUGAUUCUUCUUAGUGAA |
| mPPP1R15A-siRNA | CAGGAGCAGAUCAGAUAGATT | UCUAUCUGAUCUGCUCCUGTT |
| hPPP1R15A-siRNA | GAGGAUAAGGAAGAUGAUUTT | AAUCAUCUUCCUUAUCCUCTT |
Western blot analysis
In six-well plates, BMMs (1 × 106 cells) were seeded overnight. Sephin1 (0.78, 3.125, and 12.5 µM) was added, and then, the cells were stimulated with RANKL for 5 h or 72 h for Western blot analysis. Total cellular proteins were prepared as previously described [25]. After blocking for 1 h with 5% skim milk, different membranes were incubated with primary antibodies (1:1000) at 4 °C for more than 12 h with gentle shaking. Afterwards, TBST was used to wash the membranes for half an hour, and the membranes were incubated with secondary antibodies (1:3000). Proteins were tested by an ECL system (Fdbio Science, Hangzhou, China), which was visualized by a multifunctional imaging analysis system (Protein Simple, USA).
Knockdown of PPP1R15A or eIF2α by siRNA
NFAT-luc RAW264.7 cells, BMMs or CD14+ monocytes from osteoporosis patients were transfected with siRNA (GenePharma Co., Shanghai, China) in Opti-MEM serum-free medium with Lipofectamine RNAiMAX (Invitrogen, CA, USA). The efficiency of the knockdown of PPP1R15A or eIF2α was evaluated by qRT-PCR or Western blots after 48 h of transfection. The selected target sequences are listed in Table 1.
Ovariectomy (OVX)-induced osteoporosis mouse model and micro-CT scanning
The experiments were performed using C57BL/6 mice as previously described [25]. Seven days after the OVX operation, the mice were injected intraperitoneally with physiological saline solution (sham and OVX groups), Sephin1 (low-dose and high-dose groups) or E2 every 2 days for 6 weeks. Three-dimensional reconstructions of left femurs from each group were obtained from high-resolution microcomputed tomography (micro-CT) (SkyScan, Belgium) using the following settings: 500 μA, 50 kV, and 0.7 rotation step. Trabecular bone was analyzed in a region of interest 0.5 mm above and 1 mm in height above the growth plate. The parameters of trabecular analysis included bone mineral density (BMD), trabecular space (Tb.Sp), relative bone volume (BV/TV), and trabecular number (Tb.N). The right femurs were decalcified in EDTA decalcification solution (Servicebio, Wuhan, China) for 4 weeks and then embedded in paraffin blocks. Bone sections were sectioned at a thickness of 5 µm for H&E and TRAP staining.
Animal study approval
Female C57BL/6 mice for BMMs and OVX models were provided by the Guangdong Medical Laboratory Animal Center. The mice were caged under a 12 h light/dark cycle. All animal studies were conducted in accordance with the protocols approved by the Institutional Animal Care and Use Committee of Southern Medical University (Registration number: L2019205, registered on 8 July 2019).
Statistical analysis
Experimental data are presented as the mean ± SD. GraphPad Prism 8.0 was used to analyze data by one-way analysis of variance (ANOVA) or t test. Statistical significance was defined as P < 0.05.
Results
PPP1R15A is increased in osteoporotic mice and RANKL-induced osteoclastogenesis
To study the role of PPP1R15A in the pathogenesis of osteolysis diseases, we assessed PPP1R15A expression in preosteoclastic BMMs in vivo and in vitro. First, we found that Ppp1r15a and Acp5 gene expression was upregulated in BMMs derived from ovariectomized mice in vivo in comparison with those derived from healthy mice (Fig. 1a). Later, PPP1R15A protein expression was observed to be dramatically upregulated in BMMs during RANKL-induced osteoclastogenesis in mice in vitro, as shown by Western blotting (Fig. 1b) and fluorescence microscopy assays (Fig. 1c).
Fig. 1. PPP1R15A is increased in osteoclastogenesis, and inhibition of PPP1R15A suppresses RANKL-induced osteoclast formation in mice.

a The mRNA expression of Acp5 and Ppp1r15a in BMMs derived from ovariectomized mice was determined by qRT-PCR (n = 6). Data are expressed as the mean ± SD. **P < 0.01. b The protein level of PPP1R15A in BMMs stimulated with RANKL was determined by Western blotting (n = 3). Data are expressed as the mean ± SD. *P < 0.05. c Immunofluorescence analysis of PPP1R15A expression after RANKL stimulation in BMMs (n = 3). Data are expressed as the mean ± SD. *P < 0.05. d The efficiency of Ppp1r15a knockdown by siRNA was measured by qRT-PCR (n = 3). Data are expressed as the mean ± SD. ***P < 0.001. e Quantitative analysis and representative pictures of osteoclast differentiation in the TRAP assay after mPPP1R15A knockdown in RANKL-stimulated BMMs (n = 3). Data are expressed as the mean ± SD. *P < 0.05. f The viability of BMMs incubated with Sephin1 for 96 h was determined by CCK-8 assays (n = 3). g Images of TRAP staining of RANKL-induced osteoclasts in BMMs treated with Sephin1 and the quantitative analysis of TRAP-positive cells (nuclei >3) (scale bar=200 μm) (n = 3). h Representative pictures and analysis of RANKL-induced osteoclasts treated with 12.5 µM Sephin1 at different times. Data are expressed as the mean ± SD. *P < 0.05, ***P < 0.001 vs. the RANKL group.
PPP1R15A inhibition by specific siRNA or the allosteric inhibitor Sephin1 suppresses murine osteoclastogenesis in vitro
As PPP1R15A increased during osteoclastogenesis in mice, we assessed the role of PPP1R15A in osteoclastogenesis by knocking down Ppp1r15a with specific siRNA (Fig. 1d). mPPP1R15A siRNA obviously suppressed RANKL-induced osteoclastogenesis, as shown by the TRAP assay (Fig. 1e). Next, we investigated whether the PPP1R15A allosteric inhibitor Sephin1 suppressed osteoclastogenesis. BMMs were treated with different concentrations of Sephin1 and then stimulated with RANKL. The results of TRAP staining indicated that 0.78, 3.125, and 12.5 µM Sephin1 significantly inhibited the formation of osteoclasts (Fig. 1g) without cytotoxicity (Fig. 1f). E2 was the positive control drug used in this experiment and had an inhibitory effect on osteoclast formation in mice (Fig. 1g). Furthermore, we investigated the effects of adding Sephin1 at different times on the formation of osteoclasts. The number of osteoclasts was significantly decreased after Sephin1 (12.5 µM) administration at different stages of osteoclast differentiation, and the effect of Sephin1 was particularly pronounced when added at the early stage of osteoclastogenesis (Fig. 1h). In addition, Sephin1 prompted osteoblast differentiation in mice in vitro (Supplementary Fig. S1), and this function as well as its inhibition of osteoclasts can be beneficial for increasing bone mass. Overall, inhibition of PPP1R15A suppresses RANKL-induced osteoclast differentiation in mice in vitro.
Allosteric inhibition of PPP1R15A mitigates the bone resorption function of osteoclasts and disturbs F-actin ring formation
To determine the function of osteoclasts in response to PPP1R15A inhibition, we examined the resorptive activity of osteoclasts by using a hydroxyapatite resorption assay. Administration of the allosteric PPP1R15A inhibitor Sephin1 led to the suppression of bone resorption pits after RANKL stimulation (Fig. 2a). F-actin ring formation is a crucial component of bone resorption during the maturation of functional osteoclasts [26]. The nuclear number and F-actin ring area of osteoclasts were decreased with 12.5 µM Sephin1 treatment (Fig. 2b). In addition, osteoclast-specific genes, such as Acp5, Mmp-9, and Integrin β3, were downregulated by Sephin1 at concentrations of 0.78–12.5 μM (Fig. 2c–e). Moreover, the PPP1R15A allosteric inhibitor Sephin1 suppressed the elevation of MMP-9 and Integrin β3 protein levels, all of which are critical for the generation and function of osteoclasts (Fig. 2f).
Fig. 2. Allosteric inhibition of PPP1R15A reduces RANKL-induced F-actin ring formation and osteoclastic bone resorption.

a Representative pictures of bone resorption pits with or without Sephin1 treatment and the quantitative analysis of the resorbed hydroxyapatite area in each group. b Representative micrographs of F-actin rings (red) in BMMs stimulated with RANKL and the quantitative analysis of nuclear number and average osteoclast area per osteoclast. c–e qRT-PCR analysis of the mRNA expression of osteoclast-specific genes. f Protein levels of MMP-9 and Integrin β3 by Western blotting. Scale bar = 100 μm. Data are expressed as the mean ± SD, n = 3; ###P < 0.001 vs. the normal group, *P < 0.05, **P < 0.01, ***P < 0.001, ns (not significant) vs. the RANKL group.
Allosteric inhibition of PPP1R15A blocks RANKL-induced NF-κB and MAPK signaling pathways
The NF-κB pathway is considered a major signaling pathway activated early during osteoclastogenesis [27]. We observed that the PPP1R15A allosteric inhibitor Sephin1 inhibited NF-κB activity induced by RANKL through luciferase reporter gene assays (Fig. 3a). The phosphorylation of IκBα was suppressed by various concentrations of Sephin1 (Fig. 3b). Next, a significant reduction in the nuclear translocation of p65 with 12.5 μM Sephin1 was observed by fluorescence microscopy (Fig. 3c). Collectively, these results showed that Sephin1 inhibited NF-κB activation by blocking p-IκBα as well as the nuclear translocation of p65 during osteoclastogenesis induced by RANKL. To determine the impact of Sephin1 on the MAPK pathway, we investigated the expression of p-JNK, p-P38, and p-ERK. As demonstrated in Fig. 3d, the phosphorylation of JNK and p38 but not ERK was inhibited significantly by Sephin1 treatment in BMMs (Fig. 3e-g). c-FOS is commonly thought to be activated through the MAPK pathway [28]. The expression of c-FOS was significantly suppressed after treatment with the PPP1R15A allosteric inhibitor Sephin1 during osteoclastogenesis (Fig. 3h).
Fig. 3. Allosteric inhibition of PPP1R15A blocks NF-κB and MAPK signaling pathways induced by RANKL.

a Luciferase activity of RAW264.7 cells with the NF-κB reporter gene under RANKL stimulation for 6 h with or without Sephin1. b Western blot analysis of p-IκBα after RANKL stimulation for 30 min. c Immunofluorescence analysis of NF-κB p65 nuclear translocation. d Representative immunoblots of MAPK proteins after Sephin1 treatment and (e–g) the analysis of p-P38, p-JNK, and p-ERK expression. h Representative immunoblots and analysis of c-FOS protein. Scale bar = 200 μm. Data are expressed as the mean ± SD, n = 3; #P < 0.05, ###P < 0.001 vs. the normal group, *P < 0.05, **P < 0.01, ***P < 0.001, ns (not significant) vs. the RANKL group.
Allosteric inhibition of PPP1R15A suppresses NFATc1 activation during osteoclastogenesis
NFATc1 is considered a critical transcriptional regulator in the formation of osteoclasts [11]. Here, we examined the impact of the PPP1R15A allosteric inhibitor Sephin1 on NFATc1 activation. Sephin1 inhibited the transcriptional activity and protein expression of NFATc1 in a dose-dependent manner (Fig. 4a, b) and reduced NFATc1 nuclear translocation induced by RANKL at a dose of 12.5 μM (Fig. 4c). Consequently, these findings indicate that the PPP1R15A allosteric inhibitor Sephin1 exhibits a suppressive effect on NFATc1 activation in RANKL-induced osteoclastogenesis in vitro.
Fig. 4. Allosteric inhibition of PPP1R15A suppresses NFATc1 activity induced by RANKL.

a Luciferase activity of the NFATc1 reporter gene in RANKL-activated NFATc1-luc-RAW264.7 cells. b Representative immunoblots and analysis of NFATc1 protein in RANKL-stimulated BMMs. c Representative pictures of NFATc1 translocation to the nucleus after RANKL stimulation in BMMs were observed using a fluorescence microscope. Scale bar = 200 μm. Data are expressed as the mean ± SD (n = 3); ##P < 0.01, ###P < 0.001 vs. the normal group, **P < 0.01, ***P < 0.001, ns (not significant) vs. the RANKL group.
The effects of PPP1R15A inhibition on osteoclastogenesis are driven by eIF2α phosphorylation
PPP1R15A is a phosphatase that dephosphorylates p-eIF2α [29]. The PPP1R15A allosteric inhibitor Sephin1 was reported to prolong eIF2α phosphorylation in stressed cells [30]. We next assessed the effect of PPP1R15A on eIF2α phosphorylation in preosteoclastic BMMs exposed to RANKL. PPP1R15A knockdown by mPPP1R15A-siRNA promoted the phosphorylation form of eIF2α during osteoclastogenesis (Fig. 5a). In addition, Sephin1 treatment increased the phosphorylation of eIF2α, but it did not affect the expression of PPP1R15A, as it is an allosteric inhibitor of PPP1R15A (Fig. 5b, c). To explore whether p-eIF2α is a key molecule downstream of PPP1R15A during osteoclastogenesis, we silenced eIF2α with specific siRNA in BMMs during osteoclastogenesis. Silencing of eIF2α reduced not only the protein level of eIF2α but also the level of phosphorylated eIF2α (Fig. 5d). The suppressive effects of Sephin1 (12.5 μM) on RANKL-induced osteoclastogenesis were reduced by eIF2α silencing (Fig. 5e–g). Furthermore, the inhibition of RANKL-induced NFATc1 luciferase activity by Sephin1 was abrogated after decreasing the level of phosphorylated eIF2α in preosteoclastic RAW264.7 cells by eIF2α-siRNA (Fig. 5h, i). These findings indicate that the suppression of osteoclastogenesis by inhibiting PPP1R15A is partly dependent on eIF2α phosphorylation.
Fig. 5. The suppressive effect of PPP1R15A inhibitors on osteoclastogenesis is reduced after eIF2α interference.

a The knockdown efficiency of mPPP1R15A-siRNA and the expression of phosphorylated eIF2α were detected by Western blotting in BMMs (n = 3). b, c Phosphorylated PPP1R15A and eIF2α protein levels were assessed by Western blots after Sephin1 treatment under RANKL (50 ng/mL) stimulation for 5 h in BMMs (n = 3). d Phosphorylated eIF2α was assessed by Western blotting in BMMs transfected with eIF2α-siRNA. e TRAP staining of BMMs transfected with eIF2α siRNA for 12 h and stimulated with RANKL with or without Sephin1 (12.5 μM) for 5 days. f TRAP-positive polynuclear cells (nuclei>3) and (g) the inhibition rate of osteoclastogenesis by Sephin1 in the NC-siRNA and eIF2α-siRNA treatment groups are shown. h The knockdown efficiency of eIF2α-siRNA was detected by Western blots in NFATc1-luc-RAW264.7 cells. i Luciferase activity of the NFATc1 reporter gene in NFATc1-luc-RAW264.7 cells transfected with eIF2α-siRNA for 12 h and then treated with RANKL and Sephin1 (12.5 μM) for 24 h (n = 3). Data are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant.
Allosteric inhibition of PPP1R15A ameliorates bone loss induced by OVX in vivo
PPP1R15A inhibition can suppress osteoclast formation and bone resorption in vitro. We next observed the effects of the PPP1R15A allosteric inhibitor Sephin1 in an OVX model in vivo. There were no notable adverse effects or fatalities found during Sephin1 treatment (Supplementary Fig. S2). Sephin1 treatment inhibited bone loss in OVX mice, as shown by micro-CT (Fig. 6a). Quantitative morphometric analysis indicated that the bone parameters Tb.Sp, BV/TV, Tb.N, and BMD were significantly improved in the 4 and 8 mg/kg Sephin1- and E2-treated mice (Fig. 6b). Sephin1 (4 and 8 mg/kg) exhibited similar protective effects to 1 mg/kg E2 on bone loss in OVX mice. Consistently, quantitative analysis of H&E and TRAP staining shows that Sephin1 administration restored bone destruction (Fig. 6c) and simultaneously decreased TRAP-positive cells in OVX mice (Fig. 6d). These results suggest a promising therapeutic application of the PPP1R15A allosteric inhibitor Sephin1 in osteolytic bone diseases in vivo.
Fig. 6. A PPP1R15A allosteric inhibitor alleviates ovariectomy-induced osteoporosis in vivo.

a Three-dimensional micro-CT reconstruction of longitudinal (a, lower) and transverse (a, upper) sections of the femurs in each group. b Quantification of the trabecular microstructural indices, including BMD, BV/TV, Tb.N, and Tb.Sp. c, d Typical pictures of H&E and TRAP staining of decalcified bone. BV/TV and osteoclast number/bone surface (N. Oc/BS) were quantitatively analysed by BIOQUANT OSTEO software. Scale bar = 500 μm. Sephin1-L,low dose of Sephin1(4 mg/kg) ; Sephin1-H,high dose of Sephin1(8 mg/kg). Data are expressed as the mean ± SD (n = 5); ##P < 0.01, ###P < 0.001 vs. the sham group, *P < 0.05, **P < 0.01 ***P < 0.001 vs. the OVX group.
PPP1R15A is a positive regulator of human osteoclastogenesis
To explore the role of PPP1R15A expression in osteolysis patients, we subjected isolated preosteoclastic CD14+ monocytes from osteoporosis patients to qRT-PCR. Compared with healthy individuals, osteoporosis patients showed increased PPP1R15A expression (Fig. 7a). Strikingly, reduction of PPP1R15A expression by specific hPPP1R15A-siRNA resulted in diminished osteoclast differentiation in CD14+ monocytes from osteoporosis patients (Fig. 7b, c). Next, 0.78, 3.125, and 12.5 µM of the allosteric PPP1R15A inhibitor Sephin1 significantly inhibited both osteoclast differentiation (Fig. 7d) and bone resorption function in CD14+ monocytes from osteoporosis patients in vitro (Fig. 7e). These results demonstrate that targeted inhibition of PPP1R15A suppresses osteoclastogenesis and bone resorption in osteoporosis patients as well as in mice.
Fig. 7. Targeted inhibition of PPP1R15A represses osteoclast differentiation and bone resorption in monocytes from osteoporosis patients.

a The mRNA expression of PPP1R15A in CD14+ monocytes from osteoporosis patients and healthy control was determined by qRT-PCR (n = 5). Data are expressed as the mean ± SD. *P < 0.05. b The knockdown efficiency of hPPP1R15A-siRNA in monocytes. c TRAP staining of osteoclasts in CD14+ monocytes from osteoporosis patients transfected with hPPP1R15A-siRNA (n = 3). Data are expressed as the mean ± SD. *P < 0.05. d TRAP staining of osteoclasts (nuclei > 3) and (e) the resorbed hydroxyapatite area in CD14+ monocytes from osteoporosis patients treated with Sephin1 (n = 5). Data are expressed as the mean ± SD, ***P < 0.001 vs. the model group.
Discussion
Osteoporosis is a metabolic skeletal disease characterized by bone microstructure degeneration and bone mass loss, which directly increases the risk of fracture and decreases life expectancy [31]. Hyperactivation of osteoclasts is known as the hallmark of both postmenopausal and senile osteoporosis [5, 32]. Inhibiting the overactivity of osteoclasts has been a direct strategy for osteoporosis patients in recent years [33, 34]. Bisphosphonates and denosumab are used extensively as osteoclast inhibitors in the treatment of osteoporosis and have been shown to be effective in reducing vertebral and nonvertebral fractures. However, these types of drugs both inhibit physiological osteoclast formation, have withdrawal effects such as atypical femoral fractures (AFF), and induce rare but unacceptable disabling side effects such as ONJ [7, 35–37]. Thus, identification of new targets and drugs has therapeutic value for mitigating pathological osteoclast formation. Here, we reveal that the phosphatase PPP1R15A is a new target for osteoporosis treatment. Inhibition of PPP1R15A by specific siRNA or an allosteric inhibitor suppresses osteoclast formation and osteoporosis.
PPP1R15A is an attractive allosteric drug target, and it results in hyperosmotic stress via its well-known role in promoting the UPR (unfolded protein response) [16]. PPP1R15A prompts cell death only in stressed cells which induced by ionizing radiation (IR) [38]. Genetic studies have shown that aberrant UPR signaling is associated with defective skeletal development [39, 40]. Abnormal phosphatases in different environments can lead to the pathogenesis of diseases [41]. For the first time, we observed that PPP1R15A was increased in RANKL-induced osteoclastogenesis and elevated in BMMs from OVX mice. Subsequently, we observed that mPPP1R15A-siRNA and the allosteric PPP1R15A inhibitor Sephin1 suppressed osteoclastogenesis and bone resorption in osteoclastic precursor cells from mice in vitro and attenuated OVX-induced bone loss in vivo without toxicity. These results indicate that PPP1R15A participates in osteoclastogenesis and shows potential to serve as a new target for osteoporosis.
NFATc1 is essential in the late phase of osteoclast differentiation [42]. Once NFATc1 is activated, it translocates into the nucleus and initiates osteoclast-related genes, such as ACP5, MMP-9 and ITGB3 [43]. We observed that Sephin1 repressed the expression and nuclear translocation of NFATc1 and the subsequent osteoclast-related genes. NF-κB activation is induced in the early phase of osteoclast differentiation [27], and downstream MAPK and c-FOS activation are also closely related to osteoclastogenesis [44]. We also found that inhibition of PPP1R15A by the allosteric inhibitor Sephin1 suppressed RANKL-induced NF-κB and the downstream expression of MAPK/c-Fos. These results demonstrated that an allosteric PPP1R15A inhibitor can suppress the early signal NF-κB and then the late signal NFATc1 to inhibit osteoclastogenesis.
An increased level of p-eIF2α has been reported to expedite the recovery process of bone injuries and promote the growth of new bone tissue [45]. We next found that the allosteric PPP1R15A inhibitor Sephin1 did not inhibit the protein expression of PPP1R15A, but it increased the levels of p-eIF2α as well as that of PPP1R15A-siRNA in RANKL-activated preosteoclastic cells. Previous studies have shown that eIF2α phosphorylation triggers an adaptive UPR program, which has the potential to inhibit NF-κB activation [46]. Thus, a PPP1R15A inhibitor might inhibit NF-κB activation during osteoclastogenesis via an increase in eIF2α phosphorylation. To further determine whether the effects of the PPP1R15A inhibitor on osteoclastogenesis depend on eIF2α, we reduced the phosphorylation of eIF2α by eIF2α-siRNA and observed that this treatment attenuated the inhibitory effects of the PPP1R15A inhibitor Sephin1 on osteoclast formation and NFATc1-luc transcription after silencing eIF2α. Above all, our findings suggest that the targeted inhibition of PPP1R15A to suppress osteoclastogenesis occurs partly via its action on eIF2α.
The mechanism of the PPP1R15A allosteric inhibitor Sephin1 was first observed in endoplasmic reticulum (ER)-stressed neurons to elevate eIF2α after binding to PPP1R15A [16]. Recently, Sephin1 was also reported to protect neurons against excitotoxicity independent of eIF2α dephosphorylation [47], and this molecule was identified as a modulator of acid-sensing ion channel 3 that relieves pain [48]. However, regardless of Sephin1’s precise mode of action, its presence clearly results in prolonged and elevated levels of phosphorylated eIF2α in stressed cells, and this phenomenon minimizes any concerns about deleterious effects on unstressed cells since they only affect cells under cytotoxic stress [16, 49]. In support of this hypothesis, cells with mutated PPP1R15A and treated with a PPP1R15A inhibitor exhibit regular levels of p-eIF2α and translational activity without stress [17, 50]. As an inhibitor of protein phosphatase 1 (PP1), salubrinal shows an inhibitory effect on osteoclastogenesis and osteoblastogenesis [45], and it also disrupts the nonstress-related phosphatase CReP-PP1c complex, which may result in cell death [51]. As a newly synthesized selective allosteric inhibitor of PPP1R15A, unlike salubrinal, Sephin1 shows no effect on constitutive PPP1R15B, which prolongs the benefit of an adaptive phospho-signaling pathway and protects cells from lethal protein misfolding stress [16]. This drug also has no effects on the α2-adrenergic receptor in vitro or in vivo; thus, it avoids α2-adrenergic receptor-related side effects [16]. In our study, Sephin1 also increased osteoblast activity, which can facilitate an increase in bone mass. As described above, PPP1R15A-mediated p-eIF2α has minimal effects on nonstressed healthy cells; thus, our data showed that Sephin1 increases p-eIF2α by acting on PPP1R15A to inhibit osteoclastogenesis with the predicted minimal impact on healthy cells.
ER stress is associated with the pathological development of osteoporosis [52]. During osteoclastogenesis, many active proteins are synthesized and secreted, potentially triggering ER stress and UPR, promoting osteoclast differentiation and bone resorption function [53]. In addition, the expression of ER stress marker genes can be detected during osteoclast differentiation, indicating that osteoclastogenesis is accompanied by ER stress [54]. Thus, in a translational study, we sorted CD14+ monocytes from osteoporosis patients to establish pathological osteoclast formation to test the therapeutic effect of a PPP1R15A inhibitor. Intriguingly, PPP1R15A inhibition by hPPP1R15A-siRNA or 0.78–12.5 μM Sephin1 strikingly suppressed osteoclast formation and bone absorption in CD14+ monocytes sorted from osteoporosis patients. Sephin1, renamed IFB-088, has recently been licenced to InFlectis BioScience and is currently in phase II clinical trials for treating genetic primary motor sensory neuropathy (Charcot-Marie-Tooth disease) [14] and for treating patients with bulbar-onset amyotrophic lateral sclerosis in combination with riluzole (NCT03276832). Thus, PPP1R15A inhibitors have the potential to be developed as drugs to treat osteoporosis clinically. However, the in-depth molecular effects of PPP1R15A inhibitors on osteoclasts are still worthy of exploration in the future.
In conclusion, our study reveals that the elevated level of the phosphatase PPP1R15A is a valuable target for osteolytic diseases such as osteoporosis. Inhibition of PPP1R15A suppresses osteoclast differentiation by increasing the phosphorylated form of eIF2α and inhibiting the subsequent activation of NFATc1, attenuates bone loss in OVX mice in vivo, and restrains osteoclastogenesis in osteoporosis patients. Therefore, Sephin1 and its derivatives specifically targeting allosteric PPP1R15A may be used as novel candidates to treat osteoclast-overactivated bone loss diseases such as osteoporosis. Moreover, more osteoclast inhibitors can be designed by targeting PPP1R15A to treat bone loss diseases in the future.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82173825, 81773740, and 81972064) and the Science and Technology Plan Project of Guangzhou City (201804010027).
Author contributions
This study was designed by ZBD, YC, XJL, and conducted by ZBD, YC, YRZ, YYW, QY, JHZ, WDD, ZYC, and LHL. The manuscript was written by ZBD and YC, and revised by HJ and XJL.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Zong-bao Ding, Yan Chen, Yu-rong Zheng
Contributor Information
Hui Jiang, Email: jianghui@smu.edu.cn.
Xiao-juan Li, Email: lixiaoj@smu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01209-0.
References
- 1.Ukon Y, Makino T, Kodama J, Tsukazaki H, Tateiwa D, Yoshikawa H, et al. Molecular-based treatment strategies for osteoporosis: a literature review. Int J Mol Sci. 2019;20:2557. doi: 10.3390/ijms20102557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noh JY, Yang Y, Jung H. Molecular mechanisms and emerging therapeutics for osteoporosis. Int J Mol Sci. 2020;21:7623. doi: 10.3390/ijms21207623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dömötör ZR, Vörhendi N, Hanák L, Hegyi P, Kiss S, Csiki E, et al. Oral treatment with bisphosphonates of osteoporosis does not increase the risk of severe gastrointestinal side effects: a meta-analysis of randomized controlled trials. Front Endocrinol. 2020;11:573976. doi: 10.3389/fendo.2020.573976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin VA, Jiang X, Kagan R. Estrogen therapy for osteoporosis in the modern era. Osteoporos Int. 2018;29:1049–55. doi: 10.1007/s00198-018-4414-z. [DOI] [PubMed] [Google Scholar]
- 5.Khosla S, Hofbauer LC. Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol. 2017;5:898–907. doi: 10.1016/S2213-8587(17)30188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamy O, Stoll D, Aubry-Rozier B, Rodriguez EG. Stopping Denosumab. Curr Osteoporos Rep. 2019;17:8–15. doi: 10.1007/s11914-019-00502-4. [DOI] [PubMed] [Google Scholar]
- 7.Liu FC, Luk KC, Chen YC. Risk comparison of osteonecrosis of the jaw in osteoporotic patients treated with bisphosphonates vs. denosumab: a multi-institutional retrospective cohort study in Taiwan. Osteoporos Int. 2023;34:1729–37. doi: 10.1007/s00198-023-06818-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ono T, Nakashima T. Recent advances in osteoclast biology. Histochem Cell Biol. 2018;149:325–41. doi: 10.1007/s00418-018-1636-2. [DOI] [PubMed] [Google Scholar]
- 9.Xiao L, Zhong M, Huang Y, Zhu J, Tang W, Li D, et al. Puerarin alleviates osteoporosis in the ovariectomy-induced mice by suppressing osteoclastogenesis via inhibition of TRAF6/ROS-dependent MAPK/NF-κB signaling pathways. Aging. 2020;12:21706–29. doi: 10.18632/aging.103976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Wang C, Wang G, Sun Y, Deng Z, Chen L, et al. Loureirin B suppresses RANKL-induced osteoclastogenesis and ovariectomized osteoporosis via attenuating NFATc1 and ROS activities. Theranostics. 2019;9:4648–62. doi: 10.7150/thno.35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231:241–56. doi: 10.1111/j.1600-065X.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- 12.Vainonen JP, Momeny M, Westermarck J. Druggable cancer phosphatases. Sci Transl Med. 2021;13:eabe2967. doi: 10.1126/scitranslmed.abe2967. [DOI] [PubMed] [Google Scholar]
- 13.Krzyzosiak A, Sigurdardottir A, Luh L, Carrara M, Das I, Schneider K, et al. Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell. 2018;174:1216–28. doi: 10.1016/j.cell.2018.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullard A. Phosphatases start shedding their stigma of undruggability. Nat Rev Drug Discov. 2018;17:847–9. doi: 10.1038/nrd.2018.201. [DOI] [PubMed] [Google Scholar]
- 15.LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J Med Chem. 2020;63:13578–94. doi: 10.1021/acs.jmedchem.0c01170. [DOI] [PubMed] [Google Scholar]
- 16.Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348:239–42. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Podojil JR, Kunjamma RB, Jones J, Weiner M, Lin W, et al. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain. 2019;142:344–61. doi: 10.1093/brain/awy322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thapa S, Abdelaziz DH, Abdulrahman BA, Schatzl HM. Sephin1 reduces prion infection in prion-infected cells and animal model. Mol Neurobiol. 2020;57:2206–19. doi: 10.1007/s12035-020-01880-y. [DOI] [PubMed] [Google Scholar]
- 19.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–99. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 20.Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, et al. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in Perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/S1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 22.Sun X, Aimé P, Dai D, Ramalingam N, Crary JF, Burke RE, et al. Guanabenz promotes neuronal survival via enhancement of ATF4 and parkin expression in models of Parkinson disease. Exp Neurol. 2018;303:95–107. doi: 10.1016/j.expneurol.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou B, Zheng J, Deng W, Tan Y, Jie L, Qu Y, et al. Kirenol inhibits RANKL-induced osteoclastogenesis and prevents ovariectomized-induced osteoporosis via suppressing the Ca2+-NFATc1 and Cav-1 signaling pathways. Phytomedicine. 2021;80:153377. doi: 10.1016/j.phymed.2020.153377. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Steer JH, Joyce DA, Yip KH, Zheng MH, Xu J. 12-O-tetradecanoylphorbol-13-acetate (TPA) inhibits osteoclastogenesis by suppressing RANKL-induced NF-kappaB activation. J Bone Min Res. 2003;18:2159–68. doi: 10.1359/jbmr.2003.18.12.2159. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Liu Q, Yang M, Wang T, Yao J, Cheng J, et al. Dihydroartemisinin, an anti-Malaria drug, suppresses estrogen deficiency-induced osteoporosis, osteoclast formation, and RANKL-induced signaling pathways. J Bone Min Res. 2016;31:964–74. doi: 10.1002/jbmr.2771. [DOI] [PubMed] [Google Scholar]
- 26.Zalli D, Neff L, Nagano K, Shin NY, Witke W, Gori F, et al. The actin-binding protein cofilin and its interaction with cortactin are required for podosome patterning in osteoclasts and bone resorption in vivo and in vitro. J Bone Min Res. 2016;31:1701–12. doi: 10.1002/jbmr.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abu-Amer Y. NF-κB signaling and bone resorption. Osteoporos Int. 2013;24:2377–86. doi: 10.1007/s00198-013-2313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardo VG, Boland R, de Boland AR. 1alpha,25(OH)2-Vitamin D3 stimulates intestinal cell p38 MAPK activity and increases c-Fos expression. Int J Biochem Cell Biol. 2006;38:1181–90. doi: 10.1016/j.biocel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 29.Carrara M, Sigurdardottir A, Bertolotti A. Decoding the selectivity of eIF2α holophosphatases and PPP1R15A inhibitors. Nat Struct Mol Biol. 2017;24:708–16. doi: 10.1038/nsmb.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fusade-Boyer M, Dupré G, Bessière P, Khiar S, Quentin-Froignant C, Beck C, et al. Evaluation of the antiviral activity of Sephin1 treatment and its consequences on eIF2α phosphorylation in response to viral infections. Front Immunol. 2019;10:134. doi: 10.3389/fimmu.2019.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276–87. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25:2359–81. doi: 10.1007/s00198-014-2794-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blangy A, Bompard G, Guerit D, Marie P, Maurin J, Morel A, et al. The osteoclast cytoskeleton—current understanding and therapeutic perspectives for osteoporosis. J Cell Sci. 2020;133:jcs244798. doi: 10.1242/jcs.244798. [DOI] [PubMed] [Google Scholar]
- 34.Cheng C, Wentworth K, Shoback DM. New frontiers in osteoporosis therapy. Annu Rev Med. 2020;71:277–88. doi: 10.1146/annurev-med-052218-020620. [DOI] [PubMed] [Google Scholar]
- 35.Querrer R, Ferrare N, Melo N, Stefani CM, Dos Reis PED, Mesquita CRM, et al. Differences between bisphosphonate-related and denosumab-related osteonecrosis of the jaws: a systematic review. Support Care Cancer: Off J Multinatl Assoc Support Care Cancer. 2021;29:2811–20. doi: 10.1007/s00520-020-05855-6. [DOI] [PubMed] [Google Scholar]
- 36.Madeira M, Rocha AC, Moreira CA, Aguiar ÁMM, Maeda SS, Cardoso AS, et al. Prevention and treatment of oral adverse effects of antiresorptive medications for osteoporosis - a position paper of the Brazilian society of endocrinology and metabolism (SBEM), Brazilian society of stomatology and oral pathology (Sobep), and Brazilian association for bone evaluation and osteometabolism (Abrasso) Arch Endocrinol Metab. 2021;64:664–72. doi: 10.20945/2359-3997000000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari S, Lewiecki EM, Butler PW, Kendler DL, Napoli N, Huang S, et al. Favorable skeletal benefit/risk of long-term denosumab therapy: a virtual-twin analysis of fractures prevented relative to skeletal safety events observed. Bone. 2020;134:115287. doi: 10.1016/j.bone.2020.115287. [DOI] [PubMed] [Google Scholar]
- 38.Hollander MC, Sheikh MS, Yu K, Zhan Q, Iglesias M, Woodworth C, et al. Activation of Gadd34 by diverse apoptotic signals and suppression of its growth inhibitory effects by apoptotic inhibitors. Int J cancer. 2001;96:22–31. doi: 10.1002/1097-0215(20010220)96:1<22::AID-IJC3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Zheng C, Wang C, Jie Q, Yang L. Analyze Mouse knockout models of UPR pathway elements. Methods Mol Biol. 2022;2378:205–19. doi: 10.1007/978-1-0716-1732-8_13. [DOI] [PubMed] [Google Scholar]
- 40.Briggs MD, Dennis EP, Dietmar HF, Pirog KA. New developments in chondrocyte ER stress and related diseases. F1000Res. 2020;9:F1000. doi: 10.12688/f1000research.22275.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov. 2022;21:115–40. doi: 10.1038/s41573-021-00320-3. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Q, Wang X, Liu Y, He A, Jia R. NFATc1: functions in osteoclasts. Int J Biochem Cell Biol. 2010;42:576–79. doi: 10.1016/j.biocel.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 43.Sitara D, Aliprantis AO. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol Rev. 2010;233:286–300. doi: 10.1111/j.0105-2896.2009.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kittaka M, Mayahara K, Mukai T, Yoshimoto T, Yoshitaka T, Gorski JP, et al. Cherubism mice also deficient in c-Fos exhibit inflammatory bone destruction executed by macrophages that express MMP14 despite the absence of TRAP+ osteoclasts. J Bone Min Res. 2018;33:167–81. doi: 10.1002/jbmr.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Li X, Liu D, Hamamura K, Wan Q, Na S, et al. eIF2α signaling regulates autophagy of osteoblasts and the development of osteoclasts in OVX mice. Cell Death Dis. 2019;10:921. doi: 10.1038/s41419-019-2159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitamura M. Biphasic, bidirectional regulation of NF-kappaB by endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11:2353–64. doi: 10.1089/ars.2008.2391. [DOI] [PubMed] [Google Scholar]
- 47.Ruiz A, Zuazo J, Ortiz-Sanz C, Luchena C, Matute C, Alberdi E. Sephin1 protects neurons against excitotoxicity independently of the integrated stress response. Int J Mol Sci. 2020;21:6088. doi: 10.3390/ijms21176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callejo G, Pattison LA, Greenhalgh JC, Chakrabarti S, Andreopoulou E, Hockley JRF, et al. In silico screening of GMQ-like compounds reveals guanabenz and Sephin1 as new allosteric modulators of acid-sensing ion channel 3. Biochem Pharmacol. 2020;174:113834. doi: 10.1016/j.bcp.2020.113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun. 2015;6:6532. doi: 10.1038/ncomms7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 51.Teng Y, Gao M, Wang J, Kong Q, Hua H, Luo T, et al. Inhibition of eIF2α dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014;5:e1060. doi: 10.1038/cddis.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong M, Wu Z, Chen Z, Ren Q, Zhou J. Advances in the interaction between endoplasmic reticulum stress and osteoporosis. Biomed Pharmacother. 2023;165:115134. doi: 10.1016/j.biopha.2023.115134. [DOI] [PubMed] [Google Scholar]
- 53.Huang W, Gong Y, Yan L. ER stress, the unfolded protein response and osteoclastogenesis: a review. Biomolecules. 2023;13:1050. doi: 10.3390/biom13071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo J, Ren R, Sun K, Yao X, Lin J, Wang G, et al. PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. Cell Death Dis. 2020;11:847. doi: 10.1038/s41419-020-03046-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.