

Abstract
Two phase-III clinical trials with anti-amyloid peptide antibodies have met their primary goal, i.e. slowing of Alzheimer’s disease (AD) progression. However, antibody therapy may not be the optimal therapeutic modality for AD prevention, as we will discuss in the context of the earlier small molecules described as “γ-secretase modulators” (GSM). We review here the structure, function, and pathobiology of γ-secretases, with a focus on how mutations in presenilin genes result in early-onset AD. Significant progress has been made in generating compounds that act in a manner opposite to pathogenic presenilin mutations: they stabilize the proteinase-substrate complex, thereby increasing the processivity of substrate cleavage and altering the size spectrum of Aβ peptides produced. We propose the term “γ-secretase allosteric stabilizers” (GSAS) to distinguish these compounds from the rather heterogenous class of GSM. The GSAS represent, in theory, a precision medicine approach to the prevention of amyloid deposition, as they specifically target a discrete aspect in a complex cell biological signalling mechanism that initiates the pathological processes leading to Alzheimer’s disease.
Keywords: Presenilin, Gamma-secretase-modulator, γ-Secretase Allosteric Stabilizer, Alzheimer, Cancer
Subject terms: Cancer, Neuroscience, Pharmacology & Drug Discovery
This review explains how allosteric stabilizers of γ-secretase may prevent Alzheimer’s disease by specifically exerting opposite effects as pathogenic presenilin mutations.
Introduction
Alzheimer’s disease (AD) arguably represents one of the biggest unmet medical needs of our time. It is a complex disorder that evolves from a clinically silent biochemical phase during which amyloid plaques accumulate, to a cellular phase driven by neuroinflammation and Tau aggregation, ultimately causing synaptic dysfunction and cell death (De Strooper and Karran, 2016). The clinical phase of the disease starts decades after the initial biochemical phase and is characterized by progressive cognitive deterioration leading to dementia and death. Therapeutic strategies are likely to produce greater clinical benefit if they are administered as early as possible in the disease process, as recent successful trials with anti-amyloid antibodies (Sims et al, 2023; van Dyck et al, 2023) have demonstrated. Ideally, therapeutic intervention would prevent the first pathognomonic sign of AD—amyloid aggregation and the early cellular response to it—thus preventing irreversible damage to the brain.
The discovery that presenilins (PSEN) are responsible for the γ-secretase-mediated cleavage of the membrane carboxyterminal fragments (Fig. 1) of the amyloid precursor protein (APP) to the generation of the amyloid peptide (Aβ) provided a breakthrough in AD research (De Strooper et al, 1998). This finding unified the two major causes of inherited familial AD (FAD), i.e. mutations in the APP or the PSEN genes, in one molecular process: Aβ generation. It seemed also to provide a clear drug target for the field. At about the same time, it was demonstrated that presenilins/γ-secretases release the intracellular domain of Notch, which then travels to the nucleus and regulates Notch signaling (De Strooper et al, 1999; Struhl and Greenwald, 1999; Levitan and Greenwald, 1995). It transpired that Notch signaling was only one of the many signaling functions of the γ-secretases, contributing to the concept of regulated intramembrane proteolysis (Brown et al, 2000).
Figure 1. Processing of amyloid precursor protein to long and short amyloid peptides (Aβ).

Amyloid precursor protein (APP) is a type I transmembrane protein, indicated at the top. The transmembrane domain (inserted in the membrane, blue) and the position of the β- and γ-secretase cleavage sites are indicated schematically. Beta-secretase is a type I transmembrane aspartyl-protease. We use in the text another name for β-secretase i.e., BACE1 (β-site amyloid precursor protein cleaving enzyme), because this name is preferred in the clinical trial literature. The β-secretase yields a soluble APPsβ that is secreted in the medium, and a membrane-bound APP carboxyterminal fragment that is 99 amino acid-long (APP-CTF99) and remains membrane bound. The primary amino acid sequence of APP-CTF99 is displayed and the precise positions of the β-secretase and the consecutive γ-secretase cleavage sites are indicated. The combined actions of β- and γ-secretases generate Aβ peptides of different lengths, as indicated by the black lines above and below the primary amino acid sequence. The γ-secretase complex (Fig. 2), which is also an aspartyl-protease, cleaves APP-CTF99 first close to the intracellular site of the cell membrane to generate Aβ49 and AICD50-99 (49-production line), or Aβ48 and AICD49-99 (48-production line). The AICD is released in the cytoplasm, while the Aβ48 and Aβ49 remain associated with the γ-secretase complex. The Aβ peptides are further trimmed by consecutive cleavages by γ-secretase removing tri- or tetrapeptides at each step (Takami et al, 2009). Alzheimer’s disease-causing mutations in the presenilin subunit of the γ-secretase complex destabilize the interaction with the Aβ peptides increasing the chance of premature release of incompletely digested Aβ species. This results in relative shifts of long versus short Aβ peptides, as explained in the text. Under physiological conditions, the most abundant peptide generated in this process is Aβ40, and clinical mutations increase the release of Aβ42 and/or Aβ43. However, currently, only Aβ37, Aβ38, Aβ40, Aβ42, and Aβ43 can be efficiently measured by ELISA, and the existence of other longer peptides in vivo remains to be proven. Evidence as discussed in the main text indicates, however, that the ratio of short (Aβ37 + Aβ38+Aβ40) over long (Aβ42 + Aβ43) correlates linearly (R2 = 0.78, p < 0.0001) with the age of onset of familial Alzheimer’s disease in presenilin mutation carriers (Petit et al, 2022a).
While these early breakthroughs created enthusiasm for the development of γ-secretase inhibitors (GSI), a deep understanding of the fundamental biology of this intriguing class of proteases was lacking. Ultimately this contributed to their failure in the clinic, largely due to unexpected adverse drug effects on the skin, the vascular and immune system, and cognition (Doody et al, 2013; Coric et al, 2015). A fundamental problem was the lack of selectivity of γ-secretase inhibitors that target all four γ-secretase isoforms. This blanket inhibition affected the proteolytic processing of hundreds of substrates in parallel (Hou et al, 2023), making it impossible to find an acceptable efficacy/side effect therapeutic window. Unfortunately, little was learned from the failed trials (De Strooper, 2014), and γ-secretases were largely deprioritized as drug targets for the industry in the context of AD. Interestingly, the capacity of GSIs to block Notch signaling (De Strooper et al, 1999) remained all this time of interest to companies in the context of cancer and chronic inflammation (McCaw et al, 2021; Christopoulos et al, 2021). Nirogacestat (Ogsiveo) recently became the first GSI to reach regulatory approval for rare desmoid tumors that are non-cancerous growths of the connective tissue (Gounder et al, 2023) (see Box 1).
Recent success with anti-amyloid antibodies (Sims et al, 2023; van Dyck et al, 2023) in AD have considerably changed the perspective on the potential of Aβ lowering as a possible therapy for this devastating disorder (Karran et al, 2011; Karran and De Strooper, 2022). However, various aspects of anti-amyloid immunotherapy may not make them the optimal therapeutic choice for primary prevention clinical studies. A possible approach to primary prevention might be the suppression of Aβ production so that the key Aβ plaque-seeding peptides are not produced in sufficient concentrations to initiate plaque formation. While inhibition of γ-secretase or of BACE1, the other protease that is needed to release Aβ from APP (Fig. 1), remain formal possibilities, both approaches suffer from the same dilemma: can a therapeutic window be established sparing the normal functions of both enzymes? To administer agents that already carry known adverse events to normal individuals lacks clinical equipoise. Gamma-secretase allosteric stabilizers (GSAS), in contrast to γ-secretase inhibitors (GSI), theoretically allow normal physiological processing of the many substrates of the γ-secretases, while enhancing the processive cleavage of the Aβ peptide, favouring the generation of shorter forms as we will discuss below. The GSASs avoid the mechanism-based side effects that precluded the clinical development of GSIs and revert the production of all long Aβ peptides to shorter forms, in contrast to the earlier γ-secretase modulators (GSM) that were directed to Aβ42 lowering (Weggen et al, 2001; Luo and Li, 2022).
In the first part of this review, we will discuss our current understanding of γ-secretases and the effect of clinical mutations on their activity, resolving ongoing controversy in the field. We will stress how novel insights from cryo-EM structures (Yang et al, 2021) and a better understanding of function (Petit et al, 2022a; Szaruga et al, 2017) and cell biology (Sannerud et al, 2016) of the γ-secretases have allowed insights into the mode of action of GSASs. In the second part of the review, we will use these insights to explain the mode of action of GSASs and to discuss the roadblocks to further clinical development of GSAS-based precision medicine approaches for the prevention of AD.
Box 1 Complex-specific γ-secretase inhibitors (GSI) for the treatment of cancer and other diseases.
One of the major aims of the therapeutic development of GSIs in AD was to separate Notch from APP processing. The putative Notch sparing inhibitor avagacestat (Coric et al, 2015) made it to a phase-II clinical trial but was halted because of Notch and other side effects. Recent cryo-EM structures show that semagacestat and avagacestat bind close to the critical mixed β-sheath that stabilizes APP and Notch in the catalytic cleft of γ-secretase (Fig. 3) (Zhou et al, 2019; Yang et al, 2019). While it seems unlikely that GSIs will be reconsidered in the future for the treatment of AD because of these side effects, (selective) γ-secretase inhibition might be useful in other therapeutic areas (Jurisch-Yaksi et al, 2013), for instance, various cancers (Habets et al, 2019; Ranganathan et al, 2011) and hearing loss (Tona et al, 2014) among others, especially when Notch signaling is involved (Christopoulos et al, 2021; McCaw et al, 2021). Actually, nirogacestat (Ogsiveo) is the first GSI that reached regulatory approval for rare desmoid tumors (Gounder et al, 2023). While developing such Notch inhibitors, it is important to take into account that at least four different γ-secretase complexes exist (see main text: The discovery of γ-secretases) resulting from the four possible combinations of the constituting subunits PSEN1 or 2, APH1A or B, NCT, and PSENEN. These complexes have tissue-specific roles in Notch signaling as demonstrated by experiments with MRK560 (Churcher et al, 2006; Best et al, 2006), a GSI that suppresses T-cell Acute lymphoblastic leukemia (T-ALL) (Habets et al, 2019) without the classical Notch-related toxicity seen with broad spectrum GSIs in gut, skin or thymus. The compound MRK560 is selective for PSEN1 above PSEN2 γ-secretase, and PSEN2-γ-secretase is able to maintain the Notch physiological function in the gut and skin. The compound MRK560 was not further developed as a clinical candidate because of CYP2C9 inhibition liability (Zhao et al, 2015). Recent structural information explains the specificity of MRK560 at the molecular level (Guo et al, 2022; Serneels et al, 2023). It is thus possible to exploit the relatively small differences between the different complexes to make selective and more safe medication. In a recent comparison of 12 GSIs (Serneels et al, 2023), a few showed slight preference for APH1B over APH1A. New cryo-EM structures should explore the structural differences between APH1A or APH1B-containing complexes, which should help to generate specific drugs against APH1B-containing complexes; these are involved in amyloid plaque generation in an AD mouse model (Serneels et al, 2009).
The discovery of γ-secretases
A pivotal study linked inherited forms of AD to missense mutations in S182, a gene of unknown function at that time (Sherrington et al, 1995). Soon thereafter, mutations in a second, homologous gene were identified (Rogaev et al, 1995). The genes were called PSEN1 (abbreviation for Presenilin 1) and PSEN2 to indicate their relationship with “presenile” early-onset AD (Rogaev et al, 1995). The function of PSENs was completely unknown, but similarities with other genes encoding multi-transmembrane domain proteins led to speculations about their potential roles in vesicle trafficking, ion channel activity, and Ca2+ signaling, among other possibilities (Sherrington et al, 1995). Some of these ideas are still prevalent in the field, although the first evidence for their function from work in Caenorhabditis elegans (Levitan and Greenwald, 1995) showed that the presenilin orthologue SEL-12 is essential for Notch (Lin) signaling. The authors noted that the remarkable conservation of SEL-12 and S182 does not provide any immediate indication of the function of S182 in the Alzheimer’s disease process. Nevertheless, speculations about the role of Notch signaling in AD still remain. A more direct clue to their role in AD was the finding that presenilin missense mutations, when expressed in cell cultures, increased the generation of long, more aggregation-prone Aβ42(43) peptides (Scheuner et al, 1996; Duff et al, 1996), but the underlying mechanism remained elusive. Finally, a simple knock-out experiment of PSEN1 provided clarity showing that PS1 (Presenilin 1) is involved in γ-secretase-mediated proteolytic cleavage of the C-terminal transmembrane fragments of APP after their generation by α- and β-secretase(s) (De Strooper et al, 1998). Further work (Wolfe et al, 1999; Struhl and Greenwald, 1999; De Strooper et al, 1999) confirmed this conclusion and demonstrated “a more direct role for PSEN1 as a regulatory or catalytic component of the protease(s) that cleave(s) Notch-1 and APP” (De Strooper et al, 1999). The unequivocal proof that presenilin was indeed the catalytic protease cleaving Notch and APP was delivered when the aspartyl-protease transition state inhibitor L-685,458 was cross-linked to PSEN1 (Li et al, 2000).
Presenilins only become active proteases when integrated into the γ-secretase complexes (Takasugi et al, 2003; Edbauer et al, 2003) (Fig. 2). These tetrameric complexes contain three other proteins, i.e., nicastrin (NCT or NCSTN), presenilin enhancer 2 (Pen-2 or PSENSEN), and anterior pharynx homolog 1 (APH-1) (reviewed in (De Strooper, 2003)). The assembly occurs stepwise with dimer formation in the endoplasmic reticulum and full assembly in the Golgi where the NCT subunit becomes fully glycosylated (Wouters et al, 2021). The first substrate of γ-secretase is the PSEN subunit itself, which becomes activated by auto-proteolytic cleavage (Thinakaran et al, 1996). As there are two PSEN genes and two APH1 genes, four different γ-secretase combinations exist (De Strooper, 2003). Additional complexity comes from alternative splicing, posttranslational modification, and transient association with other proteins and lipids, some of which regulate γ-secretase activity (Wong et al, 2020). The different complexes have rather divergent biological functions and therapeutic applications (see Box 1), as illustrated by the array of phenotypes caused by different genetic knockouts of the subunits. For instance, APH1A knock-out causes lethal Notch phenotypes during embryogenesis, while APH1B causes a mild behavioral phenotype in adulthood. PSEN1 and PSEN2 are present in different subcellular compartments. Complexes of PSEN1 are recycled between the cell surface and endosomes, while PSEN2 complexes are directed to late endosomes and lysosomes via binding to Adapter protein complex 1 (Sannerud et al, 2016). A more complete understanding of the heterogeneity in the structure, function, tissue distribution, and cellular compartmentalization of γ-secretases was not available during the preclinical and clinical development of the first GSIs (Doody et al, 2013; Coric et al, 2015).
Figure 2. Gamma-secretase complex embedded in the cell membrane.
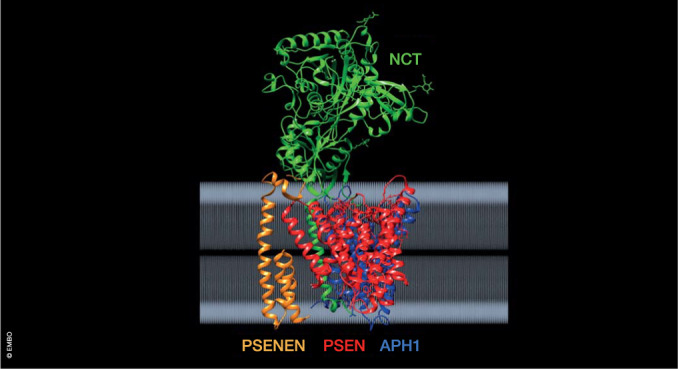
The four subunits are indicated in green (Nicastrin - NCT), red (Presenilin - PSEN), blue (Anterior pharynx defective - APH1), and orange (Presenilin enhancer - PSENEN). The structural coordinates can be found at 10.2210/pdb7D8X/pdb (Yang et al, 2021). Analysis of the data was done using the chimera package (v1. 17.3) (Pettersen et al, 2004).
Missense mutations in presenilins cause Alzheimer’s disease
Presenilins PSEN1 and PSEN2 provide the catalytic cores to the different γ-secretases (De Strooper et al, 1998; Struhl and Greenwald, 1999; De Strooper et al, 1999; Wolfe et al, 1999; Li et al, 2000). Hundreds of different AD-causing mutations have been identified in the PSENs, but—remarkably enough—not in the other subunits of the complex (Rogaev et al, 1995; Sherrington et al, 1995). In fact, haploinsufficiency of PSEN and other subunits of the γ-secretase complex (Fig. 2) cause a skin disorder called hidradenitis suppurativa (Wang et al, 2010), a chronic skin disease characterized by painful, acne-like lesions and abscesses (Vellaichamy et al, 2021). There is no increased incidence of AD associated with this skin disease (Theut Riis et al, 2017; Garg and Strunk, 2017). In contrast to AD-causing mutations that affect only PSEN, the hidradenitis suppurativa—causing mutations occur mostly in NCTSN or PSENEN, the other subunits of the γ-secretase complex, and such mutations have never been found in AD patients. Deficient NOTCH signaling in hair follicles might explain the skin phenotypes, but this is far from settled (Zhang and Sisodia, 2015; Shi et al, 2023). It is also unclear why only skin is affected: deficient NOTCH signaling should affect many organs. The proteolytic function of γ-secretase certainly plays a role in the disease, as 9 out of 17 patients treated with the γ-secretase inhibitor niragacestat for desmoid tumor/aggressive fibromatosis developed hidradenitis suppurativa-like lesions (O’Sullivan Coyne et al, 2018). The lesions disappeared after halting the treatment. In summary, while these classical loss-of-function mutations cause hidradenitis suppurativa, the FAD mutations, in contrast, alter γ-secretase function without profoundly affecting its role in multiple signaling processes.
Heterozygous carriers of PSEN1 mutations present with dominantly inherited early-onset AD (mean age of onset: 43.6 ± 7.2 years), and they display all cardinal features of sporadic AD with the full spectrum of Tau pathology, neuronal loss, and dementia. Some patients present additional atypical clinical phenotypes, such as myoclonus, seizures, pyramidal and extrapyramidal signs, and atypical neuropathology, such as cotton wool amyloid plaques (Ryan et al, 2016; Bergmans and De Strooper, 2010), but not hidradenitis suppurativa. Alzheimer’s disease caused by PSEN2 mutations is extremely rare, and the age of onset is rather variable (45–88 years) (Sherrington et al, 1996). It is possible that less APP is processed via the PSEN2-γ-secretase complex, and that the production of pathological Aβ is quantitatively less important than in the PSEN1 mutations.
While both PSEN1 and PSEN2 mutations are very rare causes of AD (Campion et al, 1999), their study has provided crucial insights into what constitutes pathological amyloid peptide (Aβ) generation (Szaruga et al, 2017; Veugelen et al, 2016; Kretner et al, 2016; Duff et al, 1996; Scheuner et al, 1996; Wagner et al, 2012), which has direct relevance to the understanding of sporadic AD (SAD), as we will discuss below. However, in our view, the genetic information was not, at the time, interpreted correctly by many in the field. The early-onset forms of AD caused by PSEN mutations were often referred to as being “aggressive” forms of the disease, and there was a belief that they led to overproduction of the Aβ42 peptide. In fact, nevertheless, there is only limited data currently supporting that FAD progresses more rapidly than SAD (Ryman et al, 2014), and in most cases, FAD PSEN mutations reduce, in some cases very significantly, the overall production of Aβ (Veugelen et al, 2016; Szaruga et al, 2015; Chávez-Gutiérrez et al, 2012; Shen and Kelleher, 2007; Xia et al, 2015).
Therefore, the consensus view is that the mutations cause a partial loss of PSEN function (Baumeister et al, 1997; De Strooper, 2007; Wolfe, 2007; Sun et al, 2017; Szaruga et al, 2015; Shen and Kelleher, 2007; Baumeister et al, 1997) and affect APP processing. The question of how this loss of function leads to AD remains, however, a contentious issue. The underlying conundrum is whether Aβ peptide and amyloid plaques are sufficient to trigger AD (“amyloid first”) (Selkoe and Hardy, 2016; Karran et al, 2011), or whether PSEN dysfunction itself is the cause of neuronal dyshomeostasis and neurodegeneration (“presenilin first”) (Shen and Kelleher, 2007). The latter hypothesis aligns with a school of thought that has criticized the amyloid hypothesis for AD and the idea that Aβ is the trigger of the disease. The “presenilin first” hypothesis is, however, largely based on experiments in conditional knock-out forebrain neurons or interneurons with total loss of PSEN1. This leads indeed to progressive neurodegeneration (Zhang et al, 2009; Xia et al, 2015; Watanabe et al, 2014), but in none of these conditions are amyloid plaques or neuronal tangles observed. Moreover, conditional knockouts of the other subunits of γ-secretase, i.e., Ncstn (Tabuchi et al, 2009) and Aph1a and b (Acx et al, 2017), mutations of which have not been associated with FAD, cause similar neurodegeneration. Membrane-bound fragments generated from App, Aplp1, Nrg1, Dcc, and other γ-secretase substrates (Acx et al, 2017) accumulate >10-fold in the targeted neurons. It is no surprise that this leads to severe disturbances of neuronal membrane functions, including synaptic transmission. Clearly, the “presenilin first” hypothesis fails to provide a consistent explanation for the neuropathology that characterizes FAD patients.
Familial Alzheimer’s disease mutations in presenilins reveal the importance of long Aβ-seeds
Importantly, the loss-of-function mutations causing FAD are not null mutations and they always target the enzymatically active PSEN subunit. Gamma-secretases cleave their substrates in two steps (Quintero-Monzon et al, 2011). The first endoproteolytic cleavage occurs close to the cytoplasmic side of the transmembrane domain and is referred to as the ε-cleavage. This is the cleavage that releases the intracellular domains of APP, Notch, and other substrates and enables intracellular signaling (Jurisch-Yaksi et al, 2013) (Fig. 1). The remaining Aβ48/49 is further trimmed by consecutive γ-cleavages that progressively shorten the membrane-bound part of Aβ until it is released into the extracellular space (Takami et al, 2009). Every cleavage step requires the progressive unwinding of the transmembrane helix, the reengagement of the catalytic site, and the formation of a new enzyme-substrate complex. Recent structural studies show how APP is anchored in the complex via an induced mixed β-sheet structure formed between the carboxyterminal Aβ region of APP and two additional peptide-strands of PSEN. This positions the cleavage site of the APP substrate into the catalytic site and makes the first γ-cleavage of APP possible. A partial unwinding of the resulting Aβ48/49 is needed to expose the new cleavage site to the catalytic site of the protease (Zhou et al, 2019; Bhattarai et al, 2022). The carboxyterminal amino acids of Aβ48/49 are likely to form a new short β-strand to anchor itself in a new mixed β-sheet structure allowing the next cleavage to occur (Yang et al, 2019) (Fig. 1). Computer modeling shows that minimally three amino acids are required to stabilize this β-sheet, explaining why tripeptides are generated in the consecutive cleavage steps (Chen et al, 2023). It is assumed that the consecutive enzyme-substrate complexes become thermodynamically less and less stable as the Aβ sequence shortens (Szaruga et al, 2017) to Aβ45/Aβ46, Aβ42/Aβ43, and finally, to Aβ40, Aβ37, and Aβ38, which can all be detected in the extracellular medium (Fig. 1) (Funamoto et al, 2004; Takami et al, 2009; Sato et al, 2003; Quintero-Monzon et al, 2011; Bhattarai et al, 2022).
This model predicts that small alterations destabilizing the consecutive enzyme-substrate complexes will lead to premature release of long Aβ peptides: this has been demonstrated experimentally by increasing the incubation temperature of an in vitro γ-secretase assay and measuring the shift in the Aβ peptide length profile (Szaruga et al, 2017). Clinical mutations in PSEN1 also destabilize the enzyme-substrate interactions, as shown by their increased temperature sensitivity (Szaruga et al, 2017). Thus, these mutations cause loss of γ-secretase function, which primarily affects the carboxypeptidase-like processing, and leads to the production of aggregation-prone long Aβ peptide. It is likely that the clinical PSEN mutations also affect the carboxypeptidase-mediated cleavages of transmembrane domains of other substrates (Weber et al, 2022), but only the Aβ peptide apparently has the property to form β-sheet fibrillar structures, explaining why PSEN mutations cause AD.
It should be stressed that most human carriers of PSEN mutations are heterozygous. Therefore, normal PSEN1 and PSEN2 alleles provide normal Notch and other signaling to the tissues, but also normal levels of Aβ peptides. The small amounts of longer Aβ peptide produced by the mutant PSEN allele, as discussed above, provide “seeds” that subsequently induce the aggregation of the Aβ generated from the normal γ-secretases into amyloid plaques (Veugelen et al, 2016). The more unstable the mutant complex, the less it contributes to the total amount of cellular Aβ produced, but the greater the proportion and concentration of the longer seeding peptides.
Total short-versus-long Aβ peptide predicts disease onset in familial Alzheimer’s disease patients
A recent elegant study analyzing 25 different FAD mutations in PSEN1 provides strong support for the “amyloid first” hypothesis. A linear correlation (R2 = 0.78, p < 0.0001) was established between age at onset of AD in the different carriers and the relative ratio of short-versus-long Aβ ((Aβ37 + Aβ38 + Aβ40)/(Aβ43 + Aβ42) (Petit et al, 2022a)). The data strongly corroborate the hypothesis that destabilizing effects of clinical mutations on the enzyme-substrate complex lead to the release of long (seeding) Aβ peptide (Szaruga et al, 2017), as illustrated by the increases in Aβ43 in several PSEN mutants. Importantly, an independent study confirmed a similar relationship between age at onset and long-versus-short Aβ in 162 PSEN1 FAD-causing variants (Schultz et al, 2023). It should be noted that robust assays for Aβ>43 (longer than 43 amino acids), which are likely generated by the most severe mutations, are currently lacking. A method to measure these longer Aβ peptides remains a priority for the field.
In conclusion, the study of gain and loss of function of the γ-secretases have yielded a plethora of insights into the normal and pathological functions of these intriguing enzymes in embryogenesis and in adulthood. These enzymes are essential for the homeostasis of tissues with the intestine, skin, immune system, and brain being particularly sensitive to alterations in their function. Clinical trials have demonstrated that a therapeutic window for γ-secretase inhibition is absent in AD. The evidence that PSEN mutations cause the generation of longer Aβ-peptides, however, suggests that an inverse mechanism, shifting the production to smaller peptides by stabilizing the APP-γ-secretase substrate-enzyme interaction could have a profound protective effect on the pathogenesis of AD. These studies also suggest that the focus of the field on Aβ42 production alone has been an oversimplification (see Box 2), as other long Aβ peptides are also produced and could, even when their total production is very low, have a catalytic effect on Aβ aggregation and amyloid plaque formation (Saito et al, 2011).
Box 2 Criticism of the “amyloid first” hypothesis.
The most serious criticism of the “amyloid first” hypothesis came from a comprehensive surview of the activities of 138 different PSEN1 mutations tested in two cell-free reconstitution assays (Sun et al, 2017). The authors found that eight of the mutations (V96F, Y154N, K155_insFI, V261F, G394V, C410Y, L435F, and DeltaT440) abrogated endoproteolytic maturation of the complex and eliminated Aβ40 and Aβ42 production. Furthermore, a large majority of 104 out of 138 variants were producing lower levels of Aβ40 and Aβ42 peptides. As there was no significant correlation between the Aβ42/Aβ40 ratio produced by the different reconstituted γ-secretases and mean age at the onset of disease in patients carrying those mutations (Sun et al, 2017), the authors concluded that PSEN mutations do not cause AD via the production of long Aβ and that other mechanisms must play a role. However, when one extreme outlier (A260V) was removed from the data set a highly significant correlation between age of onset and Aβ ratio was restored (Tang and Kepp, 2018). In addition, as only two Aβ species were measured, Aβ43, which is increased in PSEN1 mutations L166R, L235P, G266S, R278T, L282R, and A431E (Petit et al, 2022a), was not accounted for in these experiments. Finally, detergents or liposomes, used for cell-free assays of γ-secretase, have a destabilizing effect on the enzyme-substrate interaction needed to process Aβ peptides. This could explain why the 8 most extreme variants (V96F, Y154N, K155_insFI, V261F, G394V, C410Y, L435F, and DeltaT440) did not show enzymatic activity in these assays. In support of this explanation: two of these mutants, when tested in cell culture, i.e., C410Y and L435F, produce small amounts of Aβ43 peptide and are thus active (Veugelen et al, 2016; Kretner et al, 2016). It is important to note that such very severe mutations like R278I (Saito et al, 2011), L435F (Veugelen et al, 2016; Kretner et al, 2016), and C410Y (Veugelen et al, 2016) affect not only the γ-cleavages but also decrease the ε-cleavage. In homozygote knock-in mice, this results in deficient Notch signaling and lethality during embryogenesis (Saito et al, 2011; Xia et al, 2015). However, they are not catalytically inert as they release small amounts of long Aβ in cell culture (Veugelen et al, 2016) and animals (Saito et al, 2011).
Elucidation of the structure of γ-secretases facilitates new drug development
The elucidation of the atomic structures of γ-secretase (Lu et al, 2014) and γ-secretase bound to APP or Notch substrates (Zhou et al, 2019; Yang et al, 2019) have provided a molecular basis to understand the mechanism of these enzymes. The transmembrane domain of tetrameric γ-secretase consists of 20 transmembrane domain helices. This domain is organized in a horseshoe conformation (Bai et al, 2015b). The catalytic aspartates (Wolfe et al, 1999) D257 and D385 reside on the transmembrane domain (TM)6 and TM7, respectively, of PSEN, and they are located at the concave site of the horseshoe. The structure is topped by the large ectodomain of NCSTN that overlays the whole hydrophobic domain of γ-secretase on the extracellular surface (Fig. 1). To obtain structures from the in principle unstable enzyme-substrate complexes, Shi and colleagues (Yang et al, 2019; Zhou et al, 2019) introduced cross-linking cysteine mutations in both substrate and protease, and created a catalytically inactive enzyme by substituting aspartate D385 with Alanine. This, of course, blocks the autocatalytic activation of PSEN1. To mimic the effects of the autocatalytic PSEN activation, the PSEN1-NTF and -CTF were expressed separately.
The resulting cryo-EM structures (Zhou et al, 2019; Yang et al, 2019) are of remarkable quality, with resolutions of 2.8 and 2.7 Å, respectively. The novel structures reveal that several transmembrane domains of PSEN1 become reorganized and that unstructured parts of the enzyme become ordered when binding to its substrates (Yang et al, 2019; Zhou et al, 2019). A very interesting feature is the induction of a β-sheet structure between PSEN1 and its substrates that stabilizes the substrates in the catalytic cleft. This β-sheet consists of a β1 and a β2 strand at the end of transmembrane domains 6 and in the loop 2 domain of the PSEN protein, and an induced β3-strand in the Notch or APP substrates. Numerous other local rearrangements occur, for instance, the two catalytic aspartate residues, which are remote in the substrate-free structure become aligned and placed 6–7 Å away from the scissile peptide bond in Notch (Yang et al, 2019). Both APP-C83 and Notch-C100 substrates are accommodated in the same cut-through channel in the transmembrane part of γ-secretase (Zhou et al, 2019). Based on the steric constraints from the structure, it appears that substrates can only reach the catalytic channel via “lateral gating” between TM2 and TM6 of PSEN and threading their small extracellular fragment through the extended loop 1 between TM1 and TM2 (Yang et al, 2019).
In addition, the substrates undergo major conformational changes. A small N-terminal loop of Notch interacts with residues at the extracellular surface of PSEN and NCSTN. This loop domain is followed by a TM helix, which is on its C-terminal part unwound over one helical turn. This is followed by the already mentioned β3-strand. The unwinding of the helix exposes the scissile bonds of the substrate to the catalytic aspartates. The PAL motif in PSEN transmembrane domain 9 (Tomita et al, 2001), previously shown to be essential for catalytic activity (Wang et al, 2004), interacts with this unwounded part of the helix. Upon cleavage and release of the intracellular domain, the carboxyterminal end of the Aβ peptide is proposed to unwind over one helix and to reform again a new carboxyterminal β strand, initiating a new cycle of cleavage (Yang et al, 2019). This unwinding model, albeit not yet proven, could provide a mechanism for the consecutive cleavages of Aβ, which has tantalized the field for twenty years (Sato et al, 2003; Takami et al, 2009). It is logical to assume that the autocatalytic “presenilinase” cleavage (Thinakaran et al, 1996) of loop 2 also involves the induction of a third β-strand, provided by the PSEN-loop itself during assembly of the complex (Yang et al, 2019).
The different Aβ species are generated from APP by the progressive cutting of its transmembrane domain, which implies that it unwinds while in the γ-secretase catalytic cleft. The three helix-disrupting glycines in its transmembrane domain facilitate this process. Moreover, 11 out of the 18 amino acids in this domain are β-branched amino acids (e.g., Val, Ile) that might also contribute to the unwinding propensity of this domain (Yang et al, 2019). The consecutive cleavages result in a spectrum of Aβ peptides ranging from long (initially Aβ49, Aβ48, and then Aβ46, Aβ45, Aβ43, Aβ42) to short Aβ (Aβ40, Aβ38, and Aβ37) (Takami et al, 2009). At every step of this progressive shortening of the Aβ substrate a new enzyme-substrate complex is formed that is less stable, reflected in the stochastic release of the different peptides from the complex. Destabilization of the complex by clinical mutations (Szaruga et al, 2017) as explained, but also by alterations in the lipid composition in the membrane microenvironment of the enzyme (Winkler et al, 2012), or by external compounds (inverse modulators (Kukar et al, 2005)), increase the relative amount of long Aβ over short Aβ. Longer Aβ peptides are more prone to aggregate and to form oligomers and amyloid plaques, and they are therefore postulated here as the initial triggers of AD (Veugelen et al, 2016). The goal of γ-secretase allosteric modulation is to invert this process by stabilizing the complex so that shorter Aβ peptides are produced. As such, this should leave the initial ε-cleavage intact and, as a consequence, the signaling function of γ-secretase.
Targeting γ-secretases for Alzheimer’s disease
The amyloid hypothesis (Selkoe and Hardy, 2016) is linear, quantitative, and neurocentric (De Strooper and Karran, 2016), leading to the assumption that simple lowering of Aβ (or Aβ42) is necessary and sufficient to reverse the cognitive deficits in patients. However, the amyloid hypothesis does not consider the complex, decade-long cellular disease process—involving glia and vascular cells- that underlies AD (De Strooper and Karran, 2016), and therefore the possibility that amyloid is necessary, but not sufficient to cause neurodegeneration. The cellular hypothesis proposes instead that the reaction of the brain cells to amyloid, determined by genetics and environment, leads to neurodegeneration and dementia. This hypothesis suggests many targets for disease modification in the cellular phase of the disease, downstream of the amyloid plaques. The cellular hypothesis, however, also implies that once neurodegeneration has been initiated, amyloid therapies will have only a limited effect on the clinical outcome. The recent clinical data from the phase-III trials with lecanemab (van Dyck et al, 2023) and donanemab (Sims et al, 2023) clearly show a separation of the placebo and treated arms indicating a genuine disease-modifying effect, but it is very unclear from the available data (Sims et al, 2023; van Dyck et al, 2023) whether the divergence is sustained or whether the curves become parallel, which is predicted by the cellular hypothesis. The cellular hypothesis also strongly suggests that primary prevention of oligomer and amyloid plaque generation before the occurrence of brain inflammation and damage should have a major impact on the incidence of AD.
An AD prevention therapy could be envisaged with a modest, and perhaps intermittent, dosing of an amyloid-clearing antibody, such that amyloid plaques or oligomers are removed early on, or, even better, prevented from developing. The three available drugs (aducanumab, lecanemab, and likely soon, donanemab) all cause, with varying incidence, brain vascular extravasation, microhaemorrhages, and rarely severe brain hemorrhages (Solopova et al, 2023; Castellani et al, 2023). This might be mediated by a vascular inflammatory response to amyloid angiopathy aggravated by anti-Aβ antibody. If this holds true, it would seem unlikely that a similar response would be seen in people who have low or no amyloid in their brains. Perhaps a more concerning aspect is the anti-amyloid antibody-mediated increase in brain atrophy, as evidenced by MRI (Alves et al, 2023). The time course of this atrophy is discordant with the time course of amyloid removal, which is evident in the study with bapineuzumab, that barely removed any amyloid from the brain (Salloway et al, 2014). The atrophy appears to relate to the presence of a plaque-binding antibody in the brain. In fact, it is not clear whether this atrophy is even a measure of neurodegeneration in this particular context. Nevertheless, this might present an unquantifiable risk in a primary amyloid prevention study with anti-amyloid antibodies and will require post-treatment monitoring.
Current agents are administered intravenously (although subcutaneous administration is being investigated). This somewhat burdensome feature is not optimal for subjects entering a primary prevention study for which multiple-year studies will be required, and does not compare favorably with the potential of a once-a-day tablet. Using these antibodies for primary prevention of AD seems, therefore, to be logistically very challenging.
The insights gained by studying the FAD mutations in PSEN are pivotal in this regard, as they show that the age of disease onset is determined by the relative ratio of long Aβ≥42 to short Aβ≤40. Allosteric modulators of γ-secretase shift Aβ peptide production from long to short peptides, and are, therefore, fundamentally acting inversely to the causal mutations in FAD. As the mutations bring the age of onset forward, such treatment should postpone (indefinitely) the onset of disease.
The evolution of γ-secretase modulators to γ-secretase allosteric stabilizers
Gamma-secretase modulation was first demonstrated with non-steroidal anti-inflammatory drugs (NSAID), such as ibuprofen, indomethacine, and sulindac sulfide (Luo and Li, 2022; Weggen et al, 2001). At very high concentrations (25–300 µm) and independently from cyclooxygenase (COX) inhibition, these drugs lower significantly Aβ42, increase Aβ38, and do not affect Aβ40 in cell cultures and mice (Weggen et al, 2001). Tarenflurbil (R-flurbiprofen) was tested in a large phase-III trial, without success however (Green et al, 2009), likely because brain concentrations of the agent were insufficient to mediate a pharmacological effect (Karran and Hardy, 2014). In general, the first GSM generation lacked potency, and, besides some academic work (Saretz et al, 2021), this chemical space (Santiago et al, 2021; Mekala et al, 2020) has largely been abandoned.
A second generation of GSMs was developed based on the carboxyl-acid moiety of the NSAID scaffold. These compounds, similar to the first-generation GSMs, selectively affect Aβ42/Aβ38, while sparing Aβ40. Examples of the most potent compounds in these series are EVP-0015962 (Rogers et al, 2012), GSM2 (Kretner et al, 2011), and BIIB042 (Scannevin et al, 2016), all with EC50 values for Aβ42 lowering in the double-digit nM range. However, their drug-like properties were suboptimal and there are no reports of their effects in human studies (Mekala et al, 2020; Santiago et al, 2021).
These first and second generations of GSMs were screened empirically using cell-based assays and focusing on their Aβ42-lowering properties and their capacity to maintain the ε-cleavage of γ-secretases that is critical for intracellular signaling pathways. However, the mechanism of action and the precise target of these drugs remained unclear (Kounnas et al, 2010; Kukar et al, 2008; Kretner et al, 2011; Wanngren et al, 2012). The term “GSM” is used for any compound that decreases the Aβ42/Aβ40 ratio (Golde et al, 2013). This nomenclature, while useful at the outset to distinguish them from GSIs, fails to encompass our new understanding of γ-secretase enzymatic function.
The identification of the third generation of GSMs provided a major step forward and the first examples of GSASs. The prototype, introduced in 2006 by Eisai (Yu et al, 2014), targets an allosteric site in the PSEN subunit (Cai et al, 2017; Yang et al, 2021). The GSASs stabilize the enzyme-substrate complex to increase Aβ peptide processivity (Fig. 1), lowering the ratio of long Aβ≥42 to short Aβ≤40 peptides (Szaruga et al, 2017; Chávez-Gutiérrez et al, 2012; Petit et al, 2022b). In essence, they act in the inverse manner of FAD mutations, as discussed earlier. The properties of GSASs are summarized in Table 1. We propose to restrict the use of the name γ-secretase allosteric stabilizer (GSAS) strictly to this class of novel molecules (Table 1 and Table 2). Several molecules of this class are confirmed to bind to the same allosteric site on PSEN (Takeo et al, 2014; Ebke et al, 2011; Yang et al, 2021; Petit et al, 2022b).
Table 1.
Characteristics of a gamma-secretase allosteric modulator (GAM).
| Pharmacological properties | |||
| Does not affect the epsilon cleavage of substrates | |||
| Binds an allosteric site in gamma-secretase | |||
| No effect on total Abeta | |||
| Lowering effect on Abeta42 and 43 | |||
| No or moderate effects on Abeta 40 | |||
| Increasing effect on Abeta37/38 | |||
| No accumulation of APP-CTF | |||
| Maintenance of Notch and other signaling pathways | |||
| Drug properties | |||
| Property | Good | Acceptable | Poor |
| TPSA | 40–90 | 90–120 | <20 |
| 20–40 | >120 | ||
| H-Bond | <1 | 1–2 | >2 |
| Donor | |||
| H-Bond | <4 | 4–8 | >8 |
| Acceptor | |||
| MW | <360 | 360–500 | >500 |
| ClogP | <3 | 3–5 | >5 |
| ClogD | <2 | 2–4 | >4 |
| pKa | <8 | 8–10 | >10 |
| Kp,uu (brain) | >0.3 | ||
The essential pharmacological properties are summarized and defined as whether a compound can be considered an allosteric modulator of γ-secretase. All criteria should be checked and fulfilled. The drug properties are important for any drug targeted to the central nervous system.
TPSA, topical polar surface area in Å2; H-Bond, hydrogen-bond; MW, molecular weight; ClogP, calculated logarithm of partition coefficient between n-octanol and water; ClogD, calculated logarithm of distribution coefficient between n-octanol and water at pH7.4; pKa, -logarithm acid dissociation constant; Kp,uu, ratio of unbound drug in brain versus plasma.
Table 2.
Examples of ʏ-secretase allosteric modulators.
| GAM or GSM | Chemical structure | Clinical trials | IC50* | Effects on Abeta | Notes and ref |
|---|---|---|---|---|---|
| E2012 | aryl-imidazole | phase-I stop | ↓ Aβ40, Aβ42 ↑ Aβ37, Aβ38 | lenticular opacities and cholesterol metabolism problem in rat (EISAI)1 | |
| E2212 | phenylimidazole | phase-I | 9.0 nm | ↓ Aβ40, Aβ42 | EISAI2 |
| NGP555, (compound 4) | thiazole | phase Ia + Ib | 9 nm | lowers Aβ42 and plaques in rodents | Docked in ʏ-secretase complex (NeuroGenetic Pharmaceuticals)3 |
| PF-06648671 | pyridopyrazine-1,6-dione | Three phase-I 120 persons |
↓ Aβ40, Aβ42, ↑Aβ37,Aβ38 →total Aβ (human csf) |
Discontinued (Pfizer) 4 | |
| BMS-932481 (compound 12) | pyrimidine based | phase-I acute and chronic dosing/ | 6 nm |
↓ Aβ40, Aβ42, ↑Aβ37, Aβ38 →total Aβ (human csf) |
liver problems, discontinued (BMS)5 |
| RO7101556 | triazolo-azepine | no clinical data reported | 10–20 mg/kg (mouse) | ↓ Aβ40, Aβ42, ↑ Aβ38 | Hoffmann-Laroche6 |
| RG6289 | n.a. |
Phase I 180 persons |
n.a. | ↓ Aβ42 | Hoffmann-Laroche9 |
| FRM-024 (compound 41) | phenyl-oxadiazine | no clinical data reported | 9 nm |
↓ Aβ42 ↑Aβ37 |
Forum Pharmaceuticals7 |
| UCSD-776,890 (compound 1) | heterocyclic, imidazole. pyridazine | no clinical data reported | 5 nM | ↓ Aβ40, Aβ42, ↑Aβ37, Aβ38 | University of California and Massachusetts General Hospital8 |
The GSASs that have been tested in patients are indicated. The information was derived from the following references: 1(Nakano-Ito et al, 2014); 2(Yu et al, 2014); 3(Kounnas et al, 2017); 4(Ahn et al, 2020); 5(Soares et al, 2016); 6(Ratni et al, 2020); 7(Bursavich et al, 2021); 8(Rynearson et al, 2021) and 9https://www.alzforum.org/news/conference-coverage/second-generation-g-secretase-modulator-heads-phase-2.
The prototypes E2012 and E2212 have been tested in human phase-I trials demonstrating target engagement (Table 1). Trialing of E2012 was halted because of non-mechanism-based toxicity (cataracts in rats). Although E2122 was initially proposed as a safer alternative (Yu et al, 2014), undisclosed reasons have also halted the clinical development of E2122. A large variety of similar heterocyclic phenylimidazole compounds have been generated, but clinical progress was limited mostly because of non-mechanism-based liver toxicity (Mekala et al, 2020). Nevertheless, as indicated in Table 2, these are potent compounds (low single-digit nanomolar range in cell culture experiments), which decrease plaque formation and improve behavior in preclinical models of AD (for example (Kounnas et al, 2010; Rynearson et al, 2021)), while some show little preclinical toxicity as well. The compound NGP555 (Kounnas et al, 2019) demonstrated moderate pharmacodynamic effects in phase-I studies with a significant increase in Aβ38 but only a tendency to lower Aβ42. The compound BMS-932,481 (Soares et al, 2016) had the desired effects on Aβ: decreased Aβ42 and Aβ40, and increased Aβ37 and Aβ38 without effecting total Aβ in humans. However, liver toxicity limited the dose, and further clinical development has stopped. From the published work, compound PF-06648671 (Ahn et al, 2020) appears promising. Three phase-I clinical trials involving in total of 120 patients exposed to single and multiple-ascending doses have been reported (Ahn et al, 2020). Overall, an acceptable safety profile and a reduction in Aβ42 and Aβ40, with concomitant increases in Aβ37 and Aβ38 in the cerebrospinal fluid (CSF), without any effects on overall Aβ production were observed. This compound, at face value, has an appropriate GSAS profile (Table 1). In 2018, the company that had generated this compound announced that it was terminating its internal research efforts in AD (https://www.reuters.com/article/us-pfizer-alzheimers-idUSKBN1EW0TNf), and the fate of this compound series is unclear. Recently, phase-I data on RG6289, a novel GSAS, were presented (CTAD 2023), demonstrating robust reductions in CSF Aβ42 and Aβ40 levels with elevations in Aβ38 and Aβ37 (Table 2).
In conclusion, the available chemical, preclinical, and patient data suggest that it is possible to generate effective GSASs that have the desired effects in the human brain with likely a reasonable safety profile, although currently data on chronic dosing are missing. For many of these compounds, information on the criteria outlined in Table 1 remains only partially available, most crucially regarding the maintenance of the signaling function of the ʏ-secretases (Hou et al, 2023).
The mechanism of action of γ-secretase allosteric stabilizers
In addition to having good drug-like properties, a critical feature of a GSAS is to leave the initial ε-cleavage by γ-secretases intact (Lessard et al, 2015, 2020; Page et al, 2008; Golde et al, 2013). This maintains the signaling function of γ-secretases and avoids the unwanted accumulation of unprocessed APP-CTF fragments (Im et al, 2023) in the membrane (Table 1). The GSASs should not alter the total Aβ generated, a potential advantage given the possible physiological roles of Aβ in synaptic function (Abramov et al, 2009; Fogel et al, 2014). As GSASs affect the processivity of the γ-secretase, strong lowering of Aβ42 and Aβ43, moderate or no lowering of Aβ40, and increases in Aβ38 and Aβ37 should be demonstrated.
The imidazole-based allosteric GSMIII increases Aβ cleavage processivity, but at high concentrations (3 µM) it increases also the initial ε-cleavage. While this—as the authors noted (Petit et al, 2022b)—might be an additional advantage considering the potential pathogenicity of APP-CTF (Im et al, 2023), it is unclear whether and how this would increase other signaling pathways in which γ-secretase is involved. It is likely better to optimize GSASs along the criteria set out in Table 1. As already mentioned above, GSMIII and probably other GSMs can cause shifting of the production line of Aβ peptides. The GSMIII favors Aβ42 and Aβ38 as end products (Petit et al, 2022b). The acidic GSMI also increases mainly Aβ42 to Aβ38 turnover. Its binding mode (and allosteric pocket) is different from the imidazole GSMIII binding pocket (Petit et al, 2022b). Clearly, a variety of agents produce a range of pharmacological profiles of γ-secretase function.
The discovery of GSMs and GSASs was challenging from the compound screening and medicinal chemistry perspectives. The target does not lend itself to a facile biochemical screen, such as the one that was used successfully with BACE1 inhibitors. The measurement of compound activity was also complex: a range of Aβ proteoforms has to be measured to ensure the correct profile of activity is achieved. However, while challenging, by the time this research area was being deprioritized in many pharmaceutical companies, sufficient progress had been made to give confidence that clinically developable compounds might be found. The availability of highly resolved γ-secretase structures (Fig. 3) (Yang et al, 2021) opens the path to computer modeling, in silico screening, and rational drug design approaches (Ioppolo et al, 2021). The binding site of the prototype imidazole-based GSAS E2012 (Yang et al, 2021; Cai et al, 2017) is at the interface between PSEN1 and NCSTN on the extracellular side of the complex and partially covers the substrate-binding tunnel (Fig. 3). This overlaps with the binding mode of the amino-terminal portion of the APP substrate (Petit et al, 2022b; Yang et al, 2021). The compound NGP555 was computationally docked into the same site (Kounnas et al, 2017) based on an earlier, less well-resolved structure. Further exciting work (Petit et al, 2022b) using site-directed mutagenesis and in silico modeling demonstrated that another imidazole-based GSAS also potentially binds to the same site. Based on further functional work, the authors proposed a dual mode of action for this GSMIII: allosteric facilitation of the transition state and stabilization of the E-S complex by increased hydrophobic interactions of the shorter Aβ substrates with the complex (Petit et al, 2022b). The narrow structural and functional link between the substrate-binding site and the allosteric modulation site (Yang et al, 2021; Petit et al, 2022b) suggests that substrates and GSASs influence each other’s binding, stressing the importance of studying the molecular dynamics of the complex.
Figure 3. Binding sites of allosteric and orthosteric γ-secretase modulators and inhibitors.

The ribbon structure of the γ-secretase complex is displayed and turned to show the binding sites of the allosteric modulator E2012 (red) and the inhibitors L625,458 (red), Semagacestat (yellow), and Avagacestat (magenta). The three inhibitors bind closely to each other at the catalytic site of the protease, while the modulator occupies the allosteric site in the complex. The structural coordinates can be found at 10.2210/pdbxxxx/pdb using PDB6LR4, 7D8X, and 6LGQ as pdb coordinates (Yang et al, 2021). Analysis of the data was done using the chimera package (v1. 17.3) (Pettersen et al, 2004).
Challenges to γ-secretase allosteric stabilizers as a precision medicine approach to Alzheimer’s disease prevention
Demonstrating AD prevention, defined in this context as the prevention of amyloid deposition, is a time-consuming and expensive clinical endeavor, no matter the therapeutic modality employed. Normally, one would expect to demonstrate clinical efficacy in AD before moving to secondary (treating amyloid-positive subjects) and primary prevention. For GSASs, however, it seems unlikely that they would show efficacy in those patients where significant levels of amyloid are already deposited. From the verubecestat phase-III studies, it is known that profound suppression of all Aβ species did not result in a robust reduction in plaques over 18 months (Egan et al, 2019). Most GSMs tested in preclinical studies in transgenic mice harboring FAD mutations that cause amyloid deposition have demonstrated prevention of amyloid deposition, i.e., tested compounds were administered prior to plaque deposition (Rogers et al, 2012; Van Broeck et al, 2011; Imbimbo et al, 2009; Scannevin et al, 2016; Brendel et al, 2015) rather than in mice with pre-existing amyloid pathology (Murakami et al, 2016) (Egan et al, 2019). Therefore, clinical development would have to focus on primary prevention: how could this be envisioned?
For the purposes of this discussion, let us assume a GSAS has demonstrated the appropriate effect on Aβ peptide production in preclinical cell and human APP transgenic mouse models; further, that there is a sufficient efficacy:safety margin based on toxicological findings.
Firstly, there is the critical importance of sustaining clinical equipoise. For whom would this approach be appropriate, and who would be motivated to participate in clinical studies? Clearly, PSEN mutation carriers would likely be enthusiastic participants, in theory. But with the advent of anti-amyloid antibodies (lecanemab and donanemab), such patients have alternative options. Indeed, lecanemab is currently undergoing clinical testing under the aegis of the DIAN-TU consortium (ClinicalTrials.gov Identifier: NCT05269394) in mutation carriers with separate symptomatic and non-symptomatic cohorts and in combination with an anti-tau antibody. Another concern is that some PSEN mutations confer resistance to GSMs and possibly GSASs, likely because they affect the binding pocket (Lessard et al, 2015; Page et al, 2008).
Another appropriate cohort may be amyloid-negative, ApoE4 carriers who are at higher risk of developing AD, but for whom disease symptomatology is not an inevitable consequence. The burden of receiving biweekly or monthly intravenous infusions (or, in the future, subcutaneous injections) of an anti-amyloid antibody for several years in a preventative study might lead to a significant drop-out rate, as compared, say, to a daily tablet. In a primary prevention study, however, it seems likely that a low antibody dose, perhaps administered on a much longer time interval, would be sufficient to keep amyloid levels very low, thereby diminishing the participant burden. For example, this is the approach being taken in the AHEAD 3-45 studies with lecanemab (Rafii et al, 2023). Besides some of the reservations discussed above regarding potential issues when using the existing antibodies in prevention trials, being able to take a daily tablet would likely be significantly preferred to a regular subcutaneous injection over a duration of many years.
A third potential path to the clinic is a secondary intervention trial where patients, after treatment with one of the amyloid plaque-clearing antibodies and after becoming amyloid PET-negative, are transferred to a trial with GSASs to prevent re-accumulation of the amyloid plaques. The Donanemab phase-III trial shows a path forward to such a trial, as the antibody treatment was stopped once the patients became amyloid PET scan-negative (Sims et al, 2023). While BACE1 inhibitors could be considered for such a combination therapy as well, there are serious mechanism-based side effects that might limit the dose used for chronic BACE1 inhibition, as discussed above. Furthermore, GSASs have the distinct advantages of not changing the level of the Aβ species in contrast to BACE1 inhibition, to avoid the mechanistic side effects associated with pure inhibition of proteases, and finally, to do precisely what is crucial: lowering all long Aβ species.
A GSAS clinical study would first proceed via a traditional single ascending dose (SAD) and multiple-ascending dose (MAD) approach to assess safety, pharmacokinetics, and pharmacodynamic effects in small numbers of healthy controls. Cerebrospinal fluid sampling would be taken to establish that GSAS therapy alters the ratio of Aβ proteoforms appropriately. Cells transfected with PSEN mutants secrete Aβ proteoforms and the short/long Aβ ratios changes from the wild type correlate well with the age of disease onset in patients harboring each mutation (Petit et al, 2022a). From this analysis, it was demonstrated that a 25% change in the Aβ (Aβ37 + Aβ38 + Aβ40)/Aβ42 + Aβ43) ratio in favor of the longer proteoforms compared to the wild type correlated with a 13-year lowering in the average age of disease onset. It is not unreasonable, therefore, to expect that a similar change in Aβ proteoform ratio in favor of the shorter forms would result in a 13-year delay in disease onset of sporadic AD—a 20-year delay would effectively prevent AD. This 25% change could be set as the minimum pharmacodynamic effect evidenced in a MAD study that would provide confidence to embark on a phase-II dose-ranging study. However, if no adverse side effects were experienced, the phase-I MAD study could increase the drug exposure levels until the pharmacodynamic effect of increasing the ratio of shorter/longer Aβ proteoforms reached an asymptote.
A phase-II study should seek to demonstrate the safety of the therapeutic approach, evidence of efficacy or target engagement, and provide sufficient data to be able to either accept the null hypothesis or power a phase-III study in terms of therapeutic dose, group size, clinical trial duration, and primary outcome measure. A critical aspect of a GSAS phase-II study is the participant brain amyloid status at baseline. The GSAS therapeutic hypothesis posits that the initial amyloid seeding events will be prevented or significantly delayed. What is not known is whether GSAS therapy will, by suppressing the synthesis of the most amyloidogenic species, prevent further deposition in a brain with a low level of brain amyloid or even facilitate plaque resolution over time. In this context, “amyloid negativity” needs to be carefully defined (see Box 3).
By the time a phase-III study could be run, the regulatory environment may have changed such that prevention of amyloid accrual would be a surrogate endpoint. If cognitive and functional endpoints are required by regulatory bodies, an expensive trial will be needed. From longitudinal data on amyloid accrual (Sperling et al, 2023; LaPoint et al, 2022; Villemagne et al, 2013; van der Kall et al, 2021), an amyloid primary prevention study would have to be of many years’ duration with large group sizes even using the sensitive Preclinical Alzheimer Cognitive Composite (PACC) 5 assessment scale. An issue yet to be resolved is whether the magnitude of deflection of cognitive decline in very early disease will be deemed as being clinically meaningful by regulators and healthcare providers more generally (Insel et al, 2019).
Nevertheless, we should take lessons from other areas of medicine development. Large-scale clinical trials have been cornerstones for trialing in cardiovascular medicine and have delivered the evidence base for treatments currently saving many years of life throughout the population. A large-scale cardiovascular study enrolls typically 5000–20,000 patients and might take 7–10 years to complete (Packer and Pitt, 2018). Of note, approval for the first statin for human use in 1987 was based on studies showing that it lowers plasma LDL and is well-tolerated. There was no evidence that it could prevent heart attacks. This evidence was delivered only in 1994 with a second-generation statin in a large follow-up study (Goldstein and Brown, 2015).
The precision medicine approach that GSASs represent is theoretically very attractive, while the clinical trial execution would be very challenging. A therapeutic that will be administered to “at risk” individuals has to be very safe, especially as the risk, in this case, amyloid deposition, might lead to clinical symptoms only many years later. Longer-term clinical trials have not taken place with GSASs, and thus their adverse event profile and efficacy remain unknown. It is mandatory that further preclinical and clinical research is performed to explore this highly interesting avenue toward preventive therapy (see Box 4).
Box 3 Assessing amyloid negativity in a γ-secretase allosteric stabilizer (GSAS) trial.
Various cut-offs for amyloid negativity have been assigned for different amyloid PET ligands using various methodologies (La Joie et al, 2019; Landau et al, 2014; Jack et al, 2017a,b). However, brains below that cut-off will be a mixture of genuinely amyloid-negative brains and brains with low levels of amyloid. The Centiloid project has provided guidance on how to convert the amyloid plaques signals obtained with different amyloid PET ligands into a common, 100-point scale (Klunk et al, 2015). Two studies that examined the issue (Salvadó et al, 2019; Farrell et al, 2021) determined that an appropriate Centiloid (CL) cut-off below which brains could be confidently assigned as being without amyloid deposits was 12 CL or <15–18 CL, respectively. Trial participant inclusion could be initiated by genotyping cognitively normal subjects between the ages of 60–67 years and selecting ApoE4 positives (Burnham et al, 2020; Bilgel et al, 2016; Jansen et al, 2022). Currently, a range of plasma biomarkers are being explored that correlate well with brain amyloid status (Jack et al, 2023). Recently, also an elevation in plasma pTau231 has shown utility in identifying amyloid-positive subjects, and plasma pTau231 levels with Z-scores = 2 correlated with an amyloid level of 26 CLs (Milà-Alomà et al, 2022). Thus, including participants with pTau231 Z- scores >1 < 2 could be employed as a secondary filter. The amyloid PET ligand with the best sensitivity and signal-to-noise characteristics is 18F-NAV4694 (Krishnadas et al, 2022; Rowe et al, 2016), which can detect very early amyloid deposition. Trial participants would have their amyloid PET status confirmed using 18F-NAV4694, with patients with >30 Centiloids being excluded. Based on the ApoE4-positive control subjects with baseline amyloid levels <12 CL, a group size of 143 subjects would be required to demonstrate a 75% reduction of amyloid accrual in a study of 2 years’ duration1.
Box 4 Questions and future directions.
Gamma-secretase research peaked in 2012 with 765 publications, but has drastically slowed down over the last years. It seems crucial that this work gets again the attention needed as understanding the role of these complexes in Alzheimer’s disease and Cancer could provide therapeutics in two major areas of medical need. Important areas of research are:
The (patho-)biological function of the small peptides generated by γ-secretases from its many substrates. Accurate measurement methods are available only for Aβ, with the exception of Aβ≥43 forms. Sensitive assays of the whole spectrum of Aβ peptides and an understanding of their physiological roles are needed. Additionally, more work on the hundreds of γ-secretase substrates and their metabolites are required (Hou et al, 2023).
A major question is how γ-secretase processivity is affected by endogenous or exogenous stimuli. The membrane localization of γ-secretase determines its activity (Thathiah et al, 2013). In neurons, electrophysiological activity can modulate long versus short Aβ ratio (Dolev et al, 2013). The innate immunity protein IFITM3 regulates its activity (Hur et al, 2020). The potential relevance of all these effects remains, however, largely unknown.
While an understanding of the heterogeneity of the γ-secretases exists (Box 1), there is a lack of insight into the biological relevance of different γ-secretase complexes in health and disease.
Patients with familial Alzheimer’s disease have the classical signs of Alzheimer’s disease, but many present with additional phenotypes. Understanding whether other substrates are affected by the presenilin mutations will help us to understand better the normal functions of γ-secretases and will help to develop better medication for these patients.
Dynamic studies of the γ-secretase complexes and how allosteric modulation affects the enzymatic activity of those enzymes are needed. The cryo-EM structures have provided deep insight into the previously elusive intramembrane proteolysis of APP and Notch. Nevertheless, these structures provide only snapshots of a complex multi-step process. Intermediary structures must exist, for instance, when the substrate docks to the complex. Cryo-EM approaches might elucidate such intermediary structures (Bai et al, 2015a), but kinetic modeling combined with informative site-directed mutagenesis will be important to fully unravel the function of these remarkable structures (Bhattarai et al, 2022; Chen et al, 2023; Petit et al, 2022b).
Alzheimer’s disease prevention would have a profound effect on global societal well-being. Therapeutic agents are now becoming available that have the potential to realize this ambition. In the history of medicine, it is rare that the first available agents are the best. With this in mind, it is important that the field continues to treat existing agents as a benchmark from which to improve, rather than as being a final answer.
Conclusion
In many ways, the history of γ-secretases seems a repetition of other histories in medicine. By substituting long Aβ for LDL, we could just repeat what Noble laureates Brown and Goldstein wrote 8 years ago reviewing a century of cholesterol and coronaries: from plaques to genes to statins (Goldstein and Brown, 2015):
Few, if any, chronic diseases have been subjected to such intensive scrutiny, and rarely has the cause and the approach to prevention been documented so convincingly. It does not seem an exaggeration to state that targeted application of an LDL-lowering regimen may eventually curtail one of the major killers of the last century. The key questions for the 21st century are who to target and when. Ideally, LDL-lowering therapy should be initiated before atherosclerotic plaques develop or at least before they develop their most threatening features.
We know that AD presents an increasingly important healthcare problem, and the advent of the first disease-modifying drugs represents a towering breakthrough: the combined effort of thousands of basic and clinical scientists in academia and industry, and most importantly, the willingness of patients to enter clinical studies. However, the major growth in AD patients worldwide is going to be in low and middle-income countries (Martin Prince et al, 2015). The healthcare infrastructure of these nations is not yet sufficiently mature for large-scale deployment of antibody therapeutics. In summary, it seems sensible to have more, rather than fewer, therapeutic options available.
Acknowledgements
BDS is supported by the European Research Council (ERC) (grant agreement no. ERC-834682 CELLPHASE_AD), a Methusalem grant from KU Leuven and the Flemish Government, by the UKRI and the Medical Research Council (MR/Y014847/1), the Fonds voor Wetenschappelijk Onderzoek (Flanders); The Queen Elisabeth Medical Foundation for Neurosciences (Belgium), the Belgian Alzheimer Research Foundation (SAO-FRA); the Alzheimer’s Association USA and Opening the Future (OfT) of KU Leuven. BDS holds the Bax-Vanluffelen Chair for Alzheimer’s Disease.
Author contributions
Bart De Strooper: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Writing—original draft; Project administration; Writing—review and editing. Eric Karran: Funding acquisition; Investigation; Writing—original draft; Writing—review and editing.
Disclosure and competing interests statement
EK is an employee and stockholder of AbbVie. BDS is or has been a consultant for Eli Lilly, Biogen, Janssen Pharmaceutica, Eisai, AbbVie, and other companies. BDS is also a scientific founder of Augustine Therapeutics and a scientific founder and stockholder of Muna Therapeutics. BDS is a member of the Advisory Editorial Board of The EMBO Journal. This has no bearing on the editorial consideration of this article for publication.
Footnotes
The estimated sample size required for a primary prevention trial in Alzheimer’s Disease (AD) was determined by selecting healthy control, APOE4-positive carrier subjects with baseline centiloid values below 12 from the ADNI database. These subjects had baseline centiloid values of 0.77 ± 6.76 (mean ± std) and exhibited a 2.85 ± 6.45 (mean ± std) increase in centiloid over a 2-year period. A power analysis of this data suggests that a primary prevention trial to detect a 75% slowing of amyloid accrual would require 143 subjects per group for a 2-year trial using a two-group t-test with 80% power and a 0.05 two-sided significance level, calculated using the nQuery 8.1.2.0 software.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-Β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Acx H, Serneels L, Radaelli E, Muyldermans S, Vincke C, Pepermans E, Müller U, Chávez‐Gutiérrez L, De Strooper B. Inactivation of γ‐secretases leads to accumulation of substrates and non‐Alzheimer neurodegeneration. EMBO Mol Med. 2017;9:1088–1099. doi: 10.15252/emmm.201707561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, et al. Pharmacokinetic and pharmacodynamic effects of a γ-secretase modulator, PF-06648671, on CSF amyloid-β peptides in randomized phase I studies. Clin Pharmacol Ther. 2020;107:211–220. doi: 10.1002/cpt.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves F, Kalinowski P, Ayton S. Accelerated brain volume loss caused by anti-β-amyloid drugs: a systematic review and meta-analysis. Neurology. 2023;100:e2114–e2124. doi: 10.1212/WNL.0000000000207156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Rajendra E, Yang G, Shi Y, Scheres SHW. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife. 2015;4:e11182. doi: 10.7554/eLife.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW, Shi Y. An atomic structure of human γ-secretase. Nature. 2015;525:212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister R, Leimer U, Zweckbronner I, Jakubek C, Grünberg J, Haass C. Human presenilin-1, but not familial Alzheimer’s disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct. 1997;1:149–159. doi: 10.1046/j.1365-4624.1997.00012.x. [DOI] [PubMed] [Google Scholar]
- Bergmans BA, De Strooper B. γ-secretases: from cell biology to therapeutic strategies. Lancet Neurol. 2010;9:215–226. doi: 10.1016/S1474-4422(09)70332-1. [DOI] [PubMed] [Google Scholar]
- Best JD, Jay MT, Otu F, Churcher I, Reilly M, Morentin-Gutierrez P, Pattison C, Harrison T, Shearman MS, Atack JR (2006) In vivo characterization of Aβ(40) changes in brain and cerebrospinal fluid using the novel γ-secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1- trifluoromethanesulfonamide (MRK-560) in the rat. J Pharmacol Exp Ther 317:786–790 [DOI] [PubMed]
- Bhattarai A, Devkota S, Do HN, Wang J, Bhattarai S, Wolfe MS, Miao Y. Mechanism of tripeptide trimming of amyloid β-peptide 49 by γ-secretase. J Am Chem Soc. 2022;144:6215–6226. doi: 10.1021/jacs.1c10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgel M, An Y, Zhou Y, Wong DF, Prince JL, Ferrucci L, Resnick SM. Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimers Dement. 2016;12:373–379. doi: 10.1016/j.jalz.2015.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Jaworska A, Herms J, Trambauer J, Rötzer C, Gildehaus F-J, Carlsen J, Cumming P, Bylund J, Luebbers T, et al. Amyloid-PET predicts inhibition of de novo plaque formation upon chronic γ-secretase modulator treatment. Mol Psychiatry. 2015;20:1179–1187. doi: 10.1038/mp.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Fandos N, Fowler C, Pérez-Grijalba V, Dore V, Doecke JD, Shishegar R, Cox T, Fripp J, Rowe C, et al. Longitudinal evaluation of the natural history of amyloid-β in plasma and brain. Brain Commun. 2020;2:fcaa041. doi: 10.1093/braincomms/fcaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursavich MG, Harrison BA, Acharya R, Costa DE, Freeman EA, Hrdlicka LA, Jin H, Kapadnis S, Moffit JS, Murphy D, et al. Discovery of the oxadiazine FRM-024: a potent CNS-penetrant gamma secretase modulator. J Med Chem. 2021;64:14426–14447. doi: 10.1021/acs.jmedchem.1c00904. [DOI] [PubMed] [Google Scholar]
- Cai T, Yonaga M, Tomita T. Activation of γ-secretase trimming activity by topological changes of transmembrane domain 1 of presenilin 1. J Neurosci. 2017;37:12272–12280. doi: 10.1523/JNEUROSCI.1628-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F et al (1999) Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65:664–670 [DOI] [PMC free article] [PubMed]
- Castellani RJ, Shanes ED, McCord M, Reish NJ, Flanagan ME, Mesulam M-M, Jamshidi P. Neuropathology of anti-amyloid-β immunotherapy: a case report. J Alzheimers Dis. 2023;93:803–813. doi: 10.3233/JAD-221305. [DOI] [PubMed] [Google Scholar]
- Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-Y, Feilen LP, Chávez-Gutiérrez L, Steiner H, Zacharias M. Enzyme-substrate hybrid β-sheet controls geometry and water access to the γ-secretase active site. Commun Biol. 2023;6:670. doi: 10.1038/s42003-023-05039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos PF, Gjølberg TT, Krüger S, Haraldsen G, Andersen JT, Sundlisæter E. Targeting the Notch signaling pathway in chronic inflammatory diseases. Front Immunol. 2021;12:668207. doi: 10.3389/fimmu.2021.668207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churcher I, Beher D, Best JD, Castro JL, Clarke EE, Gentry A, Harrison T, Hitzel L, Kay E, Kerrad S, et al. 4-Substituted cyclohexyl sulfones as potent, orally active γ-secretase inhibitors. Bioorg Med Chem Lett. 2006;16:280–284. doi: 10.1016/j.bmcl.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Coric V, Salloway S, Van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, et al. Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 2015;72:1324–1333. doi: 10.1001/jamaneurol.2015.0607. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell. 2014;159:721–726. doi: 10.1016/j.cell.2014.10.016. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Rayk WJ, et al. A presenilin-1-dependent g-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Dolev I, Fogel H, Milshtein H, Berdichevsky Y, Lipstein N, Brose N, Gazit N, Slutsky I. Spike bursts increase amyloid-β 40/42 ratio by inducing a presenilin-1 conformational change. Nat Neurosci. 2013;16:587–595. doi: 10.1038/nn.3376. [DOI] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Ebke A, Luebbers T, Fukumori A, Shirotani K, Haass C, Baumann K, Steiner H. Novel γ-secretase enzyme modulators directly target presenilin protein. J Biol Chem. 2011;286:37181–37186. doi: 10.1074/jbc.C111.276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Eng J Med. 2019;380:1408–1420. doi: 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell ME, Jiang S, Schultz AP, Properzi MJ, Price JC, Becker JA, Jacobs HIL, Hanseeuw BJ, Rentz DM, Villemagne VL, et al. Defining the lowest threshold for amyloid-PET to predict future cognitive decline and amyloid accumulation. Neurology. 2021;96:e619–e631. doi: 10.1212/WNL.0000000000011214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O’Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, et al. APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep. 2014;7:1560–1576. doi: 10.1016/j.celrep.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y. Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry. 2004;43:13532–13540. doi: 10.1021/bi049399k. [DOI] [PubMed] [Google Scholar]
- Garg A, Strunk A. Risk of Alzheimer’s disease is not increased among patients with hidradenitis suppurativa: a retrospective population-based cohort analysis. J Am Acad Dermatol. 2017;77:176–177. doi: 10.1016/j.jaad.2017.02.055. [DOI] [PubMed] [Google Scholar]
- Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828:2898–2907. doi: 10.1016/j.bbamem.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounder M, Ratan R, Alcindor T, Schöffski P, van der Graaf WT, Wilky BA, Riedel RF, Lim A, Smith LM, Moody S, et al. Nirogacestat, a γ-secretase inhibitor for desmoid tumors. N Engl J Med. 2023;388:898–912. doi: 10.1056/NEJMoa2210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, Tarenflurbil Phase 3 Study Group Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang Y, Zhou J, Jin C, Wang J, Jia B, Jing D, Yan C, Lei J, Zhou R, et al. Molecular basis for isoform-selective inhibition of presenilin-1 by MRK-560. Nat Commun. 2022;13:6299. doi: 10.1038/s41467-022-33817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets RA, De Bock CE, Serneels L, Lodewijckx I, Verbeke D, Nittner D, Narlawar R, Demeyer S, Dooley J, Liston A, et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci Transl Med. 2019;11:eaau6246. doi: 10.1126/scitranslmed.aau6246. [DOI] [PubMed] [Google Scholar]
- Hou P, Zielonka M, Serneels L, Martinez-Muriana A, Fattorelli N, Wolfs L, Poovathingal S, T’Syen D, Balusu S, Theys T, et al. The γ-secretase substrate proteome and its role in cell signaling regulation. Mol Cell. 2023;83:4106–4122.e10. doi: 10.1016/j.molcel.2023.10.029. [DOI] [PubMed] [Google Scholar]
- Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T, Nie P, Zhai Y, et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586:735–740. doi: 10.1038/s41586-020-2681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im E, Jiang Y, Stavrides PH, Darji S, Erdjument-Bromage H, Neubert TA, Choi JY, Wegiel J, Lee J-H, Nixon RA. Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr682-phosphorylated APP βCTF. Sci Adv. 2023;9:eadg1925. doi: 10.1126/sciadv.adg1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Hutter-Paier B, Villetti G, Facchinetti F, Cenacchi V, Volta R, Lanzillotta A, Pizzi M, Windisch M. CHF5074, a novel gamma-secretase modulator, attenuates brain beta-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2009;156:982–993. doi: 10.1111/j.1476-5381.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PS, Weiner M, Mackin RS, Mormino E, Lim YY, Stomrud E, Palmqvist S, Masters CL, Maruff PT, Hansson O, et al. Determining clinically meaningful decline in preclinical Alzheimer disease. Neurology. 2019;93:e322–e333. doi: 10.1212/WNL.0000000000007831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioppolo A, Eccles M, Groth D, Verdile G, Agostino M. Evaluation of virtual screening strategies for the identification of γ-secretase inhibitors and modulators. Molecules. 2021;27:176. doi: 10.3390/molecules27010176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Algeciras-Schimnich A, Figdore DJ, Schwarz CG, Lowe VJ, Ramanan VK, Vemuri P, Mielke MM, Knopman DS, et al. Predicting amyloid PET and tau PET stages with plasma biomarkers. Brain. 2023;146:2029–2044. doi: 10.1093/brain/awad042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol. 2017;16:435–444. doi: 10.1016/S1474-4422(17)30077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13:205–216. doi: 10.1016/j.jalz.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Janssen O, Tijms BM, Vos SJB, Ossenkoppele R, Visser PJ, Amyloid Biomarker Study Group, Aarsland D, Alcolea D, Altomare D, et al. Prevalence estimates of amyloid abnormality across the Alzheimer disease clinical spectrum. JAMA Neurol. 2022;79:228–243. doi: 10.1001/jamaneurol.2021.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurisch-Yaksi N, Sannerud R, Annaert W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim Biophys Acta. 2013;1828:2815–2827. doi: 10.1016/j.bbamem.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014;76:185–205. doi: 10.1002/ana.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, Strooper B, De The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022;21:306–318. doi: 10.1038/s41573-022-00391-w. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1-15):e1–e4. doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, Ahn K, Nguyen P, Comer D, Mao L, et al. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Durakoglugil MS, Herz J, Comer WT. NGP 555, a γ-secretase modulator, shows a beneficial shift in the ratio of amyloid biomarkers in human cerebrospinal fluid at safe doses. Alzheimer’s Dement. 2019;5:458–467. doi: 10.1016/j.trci.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Lane-Donovan C, Nowakowski DW, Herz J, Comer WT. NGP 555, a γ-secretase modulator, lowers the amyloid biomarker, Aβ42 in cerebrospinal fluid while preventing Alzheimer’s disease cognitive decline in rodents. Alzheimer’s Dement. 2017;3:65–73. doi: 10.1016/j.trci.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretner B, Fukumori A, Gutsmiedl A, Page RM, Luebbers T, Galley G, Baumann K, Haass C, Steiner H. Attenuated Abeta42 responses to low potency gamma-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J Biol Chem. 2011;286:15240–15251. doi: 10.1074/jbc.M110.213587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretner B, Trambauer J, Fukumori A, Mielke J, Kuhn P, Kremmer E, Giese A, Lichtenthaler SF, Haass C, Arzberger T, et al. Generation and deposition of Aβ43 by the virtually inactive presenilin‐1 L435F mutant contradicts the presenilin loss‐of‐function hypothesis of Alzheimer’s disease. EMBO Mol Med. 2016;8:458–465. doi: 10.15252/emmm.201505952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnadas N, Doré V, Laws SM, Porter T, Lamb F, Bozinovski S, Villemagne VL, Rowe CC. Exploring discordant low amyloid beta and high neocortical tau positron emission tomography cases. Alzheimers Dement. 2022;14:e12326. doi: 10.1002/dad2.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, et al. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, et al. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, Doré V, Grinberg LT, Huang E, Hwang J-H, et al. Multisite study of the relationships between antemortem [11 C]PIB-PET Centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement. 2019;15:205–216. doi: 10.1016/j.jalz.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Thomas BA, Thurfjell L, Schmidt M, Margolin R, Mintun M, Pontecorvo M, Baker SL, Jagust WJ, Alzheimer’s Disease Neuroimaging Initiative Amyloid PET imaging in Alzheimer’s disease: a comparison of three radiotracers. Eur J Nucl Med Mol Imaging. 2014;41:1398–1407. doi: 10.1007/s00259-014-2753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPoint MR, Baker SL, Landau SM, Harrison, TM, Jagust WJ (2022) Rates of β-amyloid deposition indicate widespread simultaneous accumulation throughout the brain. Neurobiol Aging 115:1–11 [DOI] [PMC free article] [PubMed]
- Lessard CB, Cottrell BA, Maruyama H, Suresh S, Golde TE, Koo EH. γ-secretase modulators and APH1 isoforms modulate γ-secretase cleavage but not position of ε-cleavage of the amyloid precursor protein (APP) PLoS ONE. 2015;10:e0144758. doi: 10.1371/journal.pone.0144758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard CB, Rodriguez E, Ladd TB, Minter LM, Osborne BA, Miele L, Golde TE, Ran Y. γ-Secretase modulators exhibit selectivity for modulation of APP cleavage but inverse γ-secretase modulators do not. Alzheimers Res Ther. 2020;12:61. doi: 10.1186/s13195-020-00622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D, Greenwald I. Facilitation of lin-12-mediated signaling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SHW, et al. Three-dimensional structure of human γ-secretase. Nature. 2014;512:166–170. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JE, Li Y-M. Turning the tide on Alzheimer’s disease: modulation of γ-secretase. Cell Biosci. 2022;12:2. doi: 10.1186/s13578-021-00738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Prince A, Wimo A, Guerchet M, Gemma-Claire Ali M, Wu Y-T, Prina M, Yee Chan K, Xia Z, World Alzheimer Report (2015) The Global Impact of Dementia An AnalysIs of Prevalence, Incidence, Cost and Trends. Alzheimer’s Disease International (ADI), London
- McCaw TR, Inga E, Chen H, Jaskula-Sztul R, Dudeja V, Bibb JA, Ren B, Rose JB. Gamma secretase inhibitors in cancer: a current perspective on clinical performance. Oncologist. 2021;26:e608–e621. doi: 10.1002/onco.13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekala S, Nelson G, Li Y-M. Recent developments of small molecule γ-secretase modulators for Alzheimer’s disease. RSC Med Chem. 2020;11:1003–1022. doi: 10.1039/d0md00196a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milà-Alomà M, Ashton NJ, Shekari M, Salvadó G, Ortiz-Romero P, Montoliu-Gaya L, Benedet AL, Karikari TK, Lantero-Rodriguez J, Vanmechelen E, et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med. 2022;28:1797–1801. doi: 10.1038/s41591-022-01925-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Watanabe T, Koike T, Kamata M, Igari T, Kondo S. Pharmacological properties of a novel and potent γ-secretase modulator as a therapeutic option for the treatment of Alzheimer’s disease. Brain Res. 2016;1633:73–86. doi: 10.1016/j.brainres.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Nakano-Ito K, Fujikawa Y, Hihara T, Shinjo H, Kotani S, Suganuma A, Aoki T, Tsukidate K. E2012-induced cataract and its predictive biomarkers. Toxicol Sci. 2014;137:249–258. doi: 10.1093/toxsci/kft224. [DOI] [PubMed] [Google Scholar]
- O’Sullivan Coyne G, Woodring TS, Lee CCR, Chen AP, Kong HH. Hidradenitis suppurativa-like lesions associated with pharmacologic inhibition of gamma-secretase. J Invest Dermatol. 2018;138:979–981. doi: 10.1016/j.jid.2017.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M, Pitt B. Future large-scale clinical trials in cardiovascular medicine: challenges and uncertainties. Eur J Heart Fail. 2018;20:1645–1648. doi: 10.1002/ejhf.1360. [DOI] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Pérez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, et al. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- Petit D, Fernández SG, Zoltowska KM, Enzlein T, Ryan NS, O’Connor A, Szaruga M, Hill E, Vandenberghe R, Fox NC, et al. Aβ profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol Psychiatry. 2022;27:2821–2832. doi: 10.1038/s41380-022-01518-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit D, Hitzenberger M, Koch M, Lismont S, Zoltowska KM, Enzlein T, Hopf C, Zacharias M, Chávez‐Gutiérrez L. Enzyme–substrate interface targeting by imidazole‐based γ‐secretase modulators activates γ‐secretase and stabilizes its interaction with APP. EMBO J. 2022;41:e111084. doi: 10.15252/embj.2022111084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry. 2011;50:9023–9035. doi: 10.1021/bi2007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, Dhadda S, Sethuraman G, Kramer LD, Swanson CJ, et al. The AHEAD 3-45 study: design of a prevention trial for Alzheimer’s disease. Alzheimers Dement. 2023;19:1227–1233. doi: 10.1002/alz.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- Ratni H, Alker A, Bartels B, Bissantz C, Chen W, Gerlach I, Limberg A, Lu M, Neidhart W, Pichereau S, et al. Discovery of RO7185876, a highly potent γ-secretase modulator (GSM) as a potential treatment for Alzheimer’s disease. ACS Med Chem Lett. 2020;11:1257–1268. doi: 10.1021/acsmedchemlett.0c00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376:775–778 [DOI] [PubMed]
- Rogers K, Felsenstein KM, Hrdlicka L, Tu Z, Albayya F, Lee W, Hopp S, Miller M-J, Spaulding D, Yang Z, et al. Modulation of γ-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol Neurodegener. 2012;7:61. doi: 10.1186/1750-1326-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Jones G, Doré V, Pejoska S, Margison L, Mulligan RS, Chan JG, Young K, Villemagne VL. Standardized expression of 18F-NAV4694 and 11C-PiB β-amyloid PET results with the centiloid scale. J Nucl Med. 2016;57:1233–1237. doi: 10.2967/jnumed.115.171595. [DOI] [PubMed] [Google Scholar]
- Ryan NS, Nicholas JM, Weston PSJ, Liang Y, Lashley T, Guerreiro R, Adamson G, Kenny J, Beck J, Chavez-Gutierrez L, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 2016;15:1326–1335. doi: 10.1016/S1474-4422(16)30193-4. [DOI] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JBS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rynearson KD, Ponnusamy M, Prikhodko O, Xie Y, Zhang C, Nguyen P, Hug B, Sawa M, Becker A, Spencer B, et al. Preclinical validation of a potent γ-secretase modulator for Alzheimer’s disease prevention. J Exp Med. 2021;218:e20202560. doi: 10.1084/jem.20202560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, et al. Potent amyloidogenicity and pathogenicity of Aβ 243. Nat Neurosci. 2011;14:1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Eng J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadó G, Molinuevo JL, Brugulat-Serrat A, Falcon C, Grau-Rivera O, Suárez-Calvet M, Pavia J, Niñerola-Baizán A, Perissinotti A, Lomeña F, et al. Centiloid cut-off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther. 2019;11:27. doi: 10.1186/s13195-019-0478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud R, Esselens C, Ejsmont P, Mattera R, Rochin L, Tharkeshwar AK, De Baets G, De Wever V, Habets R, Baert V, et al. Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell. 2016;166:193–208. doi: 10.1016/j.cell.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago Á, Guzmán-Ocampo DC, Aguayo-Ortiz R, Dominguez L. Characterizing the chemical space of γ-secretase inhibitors and modulators. ACS Chem Neurosci. 2021;12:2765–2775. doi: 10.1021/acschemneuro.1c00313. [DOI] [PubMed] [Google Scholar]
- Saretz S, Basset G, Useini L, Laube M, Pietzsch J, Drača D, Maksimović-Ivanić D, Trambauer J, Steiner H, Hey-Hawkins E. Modulation of γ-secretase activity by a carborane-based flurbiprofen analogue. Molecules. 2021;26:2843. doi: 10.3390/molecules26102843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, Maruyama H, Koo EH, Haass C, Takio K, et al. Potential link between amyloid β-protein 42 and C-terminal fragment γ 49-99 of β-amyloid precursor protein. J Biol Chem. 2003;278:24294–20301. doi: 10.1074/jbc.M211161200. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Chollate S, Brennan MS, Snodgrass-Belt PA, Peng H, Xu L, Jung M-Y, Bussiere T, Arastu MF, Talreja T, et al. BIIB042, a novel γ-secretase modulator, reduces amyloidogenic Aβ isoforms in primates and rodents and plaque pathology in a mouse model of Alzheimer’s disease. Neuropharmacology. 2016;103:57–68. doi: 10.1016/j.neuropharm.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schultz S, Liu L, Schultz AP, Fitzpatrick CD, Levin R (2023) Functional variations in gamma-secretase activity are critical determinants of the clinical, biomarker, and cognitive progression of autosomal domainant Alzheimer’s disease. Preprint at bioRxiv 10.1101/2023.07.04.547688
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Houtvin TVan, Esselmann H, Paul S, Schäfer MK, Berezovska O, et al. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science. 2009;324:639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serneels L, Narlawar R, Perez-Benito L, Municoy M, Guallar V, T’Syen D, Dewilde M, Bischoff F, Fraiponts E, Tresadern G, et al. Selective inhibitors of the PSEN1-gamma-secretase complex. J Biol Chem. 2023;299:104794. doi: 10.1016/j.jbc.2023.104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, Levesque G, Rogaev EI, Lin C, Liang Y, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5:985–988. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shi TW, Cao W, Zhao QZ, Yu HX, Zhang SS, Hao YB (2023) Effects of NCSTN Mutation on Hair Follicle Components in Mice. Dermatology 239:60–71 [DOI] [PubMed]
- Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, Wessels AM, Shcherbinin S, Wang H, Monkul Nery ES, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330:512–527. doi: 10.1001/jama.2023.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares HD, Gasior M, Toyn JH, Wang JS, Hong Q, Berisha F, Furlong MT, Raybon J, Lentz KA, Sweeney F, et al. The γ-secretase modulator, BMS-932481, modulates Aβ peptides in the plasma and cerebrospinal fluid of healthy volunteerss. J Pharmacol Exp Ther. 2016;358:138–150. doi: 10.1124/jpet.116.232256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solopova E, Romero-Fernandez W, Harmsen H, Ventura-Antunes L, Wang E, Shostak A, Maldonado J, Donahue MJ, Schultz D, Coyne TM, et al. Fatal iatrogenic cerebral β-amyloid-related arteritis in a woman treated with lecanemab for Alzheimer’s disease. Nat Commun. 2023;14:8220. doi: 10.1038/s41467-023-43933-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Donohue MC, Raman R, Rafii MS, Johnson K, Masters CL, van Dyck CH, Iwatsubo T, Marshall GA, Yaari R, et al. Trial of solanezumab in preclinical Alzheimer’s disease. N Engl J Med. 2023;389:1096–1110. doi: 10.1056/NEJMoa2305032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci USA. 2017;114:E476–E485. doi: 10.1073/pnas.1618657114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, et al. Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell. 2017;170:443–456.e14. doi: 10.1016/j.cell.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S, et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J Exp Med. 2015;212:2003–2013. doi: 10.1084/jem.20150892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J Neurosci. 2009;29:7290–7301. doi: 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo K, Tanimura S, Shinoda T, Osawa S, Zahariev IK, Takegami N, Ishizuka-Katsura Y, Shinya N, Takagi-Niidome S, Tominaga A, et al. Allosteric regulation of γ-secretase activity by a phenylimidazole-type γ-secretase modulator. Proc Natl Acad Sci USA. 2014;111:10544–10549. doi: 10.1073/pnas.1402171111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N, Kepp KP. Aβ42/Aβ40 ratios of presenilin 1 mutations correlate with clinical onset of Alzheimer’s disease. J Alzheimers Dis. 2018;66:939–945. doi: 10.3233/JAD-180829. [DOI] [PubMed] [Google Scholar]
- Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, De Kloe G, Munck S, De Strooper B. β-Arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat Med. 2013;19:44–49. doi: 10.1038/nm.3023. [DOI] [PubMed] [Google Scholar]
- Theut Riis P, Egeberg A, Gislason GH, Jemec GB. Patients with hidradenitis suppurativa have no increased risk of Alzheimer disease. Br J Dermatol. 2017;177:273–275. doi: 10.1111/bjd.15064. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Tomita T, Watabiki T, Takikawa R, Morohashi Y, Takasugi N, Kopan R, De Strooper B, Iwatsubo T. The first proline of PALP motif at the C terminus of presenilins is obligatory for stabilization, complex formation, and gamma-secretase activities of presenilins. J Biol Chem. 2001;276:33273–33281. doi: 10.1074/jbc.M011152200. [DOI] [PubMed] [Google Scholar]
- Tona Y, Hamaguchi K, Ishikawa M, Miyoshi T, Yamamoto N, Yamahara K, Ito J, Nakagawa T. Therapeutic potential of a gamma-secretase inhibitor for hearing restoration in a guinea pig model with noise-induced hearing loss. BMC Neurosci. 2014;15:66. doi: 10.1186/1471-2202-15-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Broeck B, Chen J-M, Tréton G, Desmidt M, Hopf C, Ramsden N, Karran E, Mercken M, Rowley A. Chronic treatment with a novel γ-secretase modulator, JNJ-40418677, inhibits amyloid plaque formation in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2011;163:375–389. doi: 10.1111/j.1476-5381.2011.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kall LM, Truong T, Burnham SC, Doré V, Mulligan RS, Bozinovski S, Lamb F, Bourgeat P, Fripp J, Schultz S, Lim YY, Laws SM, Ames D, Fowler C, Rainey-Smith SR, Martins RN, Salvado O, Robertson J, Maruff P, Masters CL, Villemagne VL, Rowe CC (2021) Association of β-Amyloid Level, Clinical Progression, and Longitudinal Cognitive Change in Normal Older Individuals. Neurology 96:e662–e670 [DOI] [PMC free article] [PubMed]
- van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9–21. doi: 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- Vellaichamy G, Dimitrion P, Zhou L, Ozog D, Lim HW, Liao W, Hamzavi IH, Mi QS. Insights from γ-secretase: functional genetics of hidradenitis suppurativa. J Invest Dermatol. 2021;141:1888–1896. doi: 10.1016/j.jid.2021.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelen S, Saito T, Saido TC, Chávez-Gutiérrez L, De Strooper B. Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron. 2016;90:410–416. doi: 10.1016/j.neuron.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL (2013) Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12:357–367 [DOI] [PubMed]
- Wagner SL, Tanzi RE, Mobley WC, Galasko D. Potential use of γ-secretase modulators in the treatment of Alzheimer disease. Arch Neurol. 2012;69:1255–1258. doi: 10.1001/archneurol.2012.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, et al. Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330:1065. doi: 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- Wang J, Brunkan AL, Hecimovic S, Walker E, Goate A. Conserved ‘PAL’ sequence in presenilins is essential for gamma-secretase activity, but not required for formation or stabilization of gamma-secretase complexes. Neurobiol Dis. 2004;15:654–666. doi: 10.1016/j.nbd.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Wanngren J, Ottervald J, Parpal S, Portelius E, Strömberg K, Borgegård T, Klintenberg R, Juréus A, Blomqvist J, Blennow K, et al. Second generation γ-secretase modulators exhibit different modulation of Notch β and Aβ production. J Biol Chem. 2012;287:32640–32650. doi: 10.1074/jbc.M112.376541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Iqbal M, Zheng J, Wines-Samuelson M, Shen J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J Neurosci. 2014;34:15912–15922. doi: 10.1523/JNEUROSCI.3261-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber TA, Lundkvist J, Wanngren J, Kvartsberg H, Jin S, Larssen P, Wu D, Oliveira DV, Minta K, Brinkmalm G, et al. γ-Secretase modulators show selectivity for γ-secretase-mediated amyloid precursor protein intramembrane processing. J Cell Mol Med. 2022;26:880–892. doi: 10.1111/jcmm.17146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Winkler E, Kamp F, Scheuring J, Ebke A, Fukumori A, Steiner H. Generation of Alzheimer disease-associated amyloid β 42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J Biol Chem. 2012;287:21326–21334. doi: 10.1074/jbc.M112.356659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:136–140. doi: 10.1038/sj.embor.7400896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wong E, Frost GR, Li YM (2020) γ-Secretase modulatory proteins: the guiding hand behind the running scissors. Front Aging Neurosci 12:614690 [DOI] [PMC free article] [PubMed]
- Wouters R, Michiels C, Sannerud R, Kleizen B, Dillen K, Vermeire W, Ayala AE, Demedts D, Schekman R, Annaert W. Assembly of γ-secretase occurs through stable dimers after exit from the endoplasmic reticulum. J Cell Biol. 2021;220:e201911104. doi: 10.1083/jcb.201911104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial alzheimer’s disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhou R, Guo X, Yan C, Lei J, Shi Y. Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell. 2021;184:521–533.e14. doi: 10.1016/j.cell.2020.11.049. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, Shi Y. Structural basis of Notch recognition by human γ-secretase. Nature. 2019;565:192–197. doi: 10.1038/s41586-018-0813-8. [DOI] [PubMed] [Google Scholar]
- Yu Y, Logovinsky V, Schuck E, Kaplow J, Chang MK, Miyagawa T, Wong N, Ferry J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects. J Clin Pharmacol. 2014;54:528–536. doi: 10.1002/jcph.249. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Sisodia SS. Acne inversa caused by missense mutations in NCSTN is not fully compatible with impairments in Notch signaling. J Invest Dermatol. 2015;135:618–620. doi: 10.1038/jid.2014.399. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Pissarnitski DA, Josien HB, Wu WL, Xu R, Li H, Clader JW, Burnett DA, Terracina G, Hyde L, et al. Discovery of a novel, potent spirocyclic series of γ-secretase inhibitors. J Med Chem. 2015;58:8806–8817. doi: 10.1021/acs.jmedchem.5b00774. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y. Recognition of the amyloid precursor protein by human g-secretase. Science. 2019;363:eaaw0930. doi: 10.1126/science.aaw0930. [DOI] [PubMed] [Google Scholar]