

Abstract
Cognitive dysfunction can occur both in normal aging and age-related neurological disorders, such as mild cognitive impairment and Alzheimer's disease (AD). These disorders have few treatment options due to side effects and limited efficacy. New approaches to slow cognitive decline are urgently needed. Dietary interventions (nutraceuticals) have received considerable attention because they exhibit strong neuroprotective properties and may help prevent or minimize AD symptoms. Biological aging is driven by a series of interrelated mechanisms, including oxidative stress, neuroinflammation, neuronal apoptosis, and autophagy, which function through various signaling pathways. Recent clinical and preclinical studies have shown that dietary small molecules derived from natural sources, including flavonoids, carotenoids, and polyphenolic acids, can modulate oxidative damage, cognitive impairments, mitochondrial dysfunction, neuroinflammation, neuronal apoptosis, autophagy dysregulation, and gut microbiota dysbiosis. This paper reviews research on different dietary small molecules and their bioactive constituents in the treatment of AD. Additionally, the chemical structure, effective dose, and specific molecular mechanisms of action are comprehensively explored. This paper also discusses the advantages of using nanotechnology-based drug delivery, which significantly enhances oral bioavailability, safety, and therapeutic effect, and lowers the risk of adverse effects. These agents have considerable potential as novel and safe therapeutic agents that can prevent and combat age-related AD.
Keywords: Drug discovery, Dietary small molecules, Neuroprotection, Cognitive function, Neuroinflammation, Alzheimer's disease
Graphical abstract
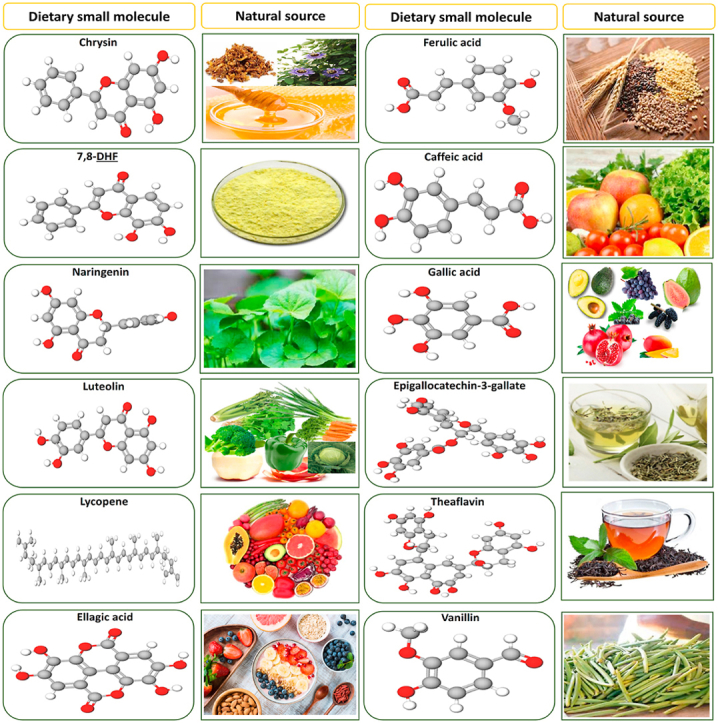
Dietary small molecules sources and their respective structures.
Highlights
-
•
Different molecular mechanisms of dietary small molecules against AD are discussed.
-
•
Dietary small molecules bioactive constituents having neuroprotective efficacy are discussed.
-
•
Nano-formulated dietary small molecules cross the BBB and improving drug delivery are discussed.
-
•
Nano-formulated dietary small molecules enhance the drug efficacy, bioavailability, and safety in AD are discussed.
-
•
Drug development, advantages, future prospects of dietary small molecules in the treatment of AD are discussed.
1. Introduction
Alzheimer's disease (AD) is a multifactorial neurodegenerative disease (NDD). It is the most common cause of dementia and one of the most serious problems facing the world's older adult population [1]. The World Health Organization (WHO) estimates that approximately 50 million people have AD or AD-related dementia, potentially accounting for 60–70% of all dementia cases [2]. This number is expected to reach 82 million by 2030 and 152 million by 2050. In the US, approximately 6.2 million people have AD, while in South Korea more than 1 million people are affected [3,4]. AD has a major impact on quality of life in affected individuals and imposes a significant economic burden on their families and society [5]. As a result, increasing attention is being paid to the pathogenesis and treatment of AD.
Formation of extracellular insoluble amyloid-beta (Aβ) plaques and intracellular aggregation of hyperphosphorylated tau protein in neurofibrillary tangles in the brain are typical pathological features of AD [6]. Tau proteins are a group of structural proteins that function predominantly in microtubules; when hyperphosphorylated, they become toxic and insoluble, forming aggregates that contribute to the pathophysiology of AD [7]. Tau hyperphosphorylation can lead to abnormal folding, fragmentation, aggregation, and/or the development of deposits known as neurofibrillary tangles. Extracellular Aβ plaques and intracellular tangles in brain hippocampal and cortical areas are hallmarks of AD [8]. These changes lead to AD symptoms, such as progressive memory loss, difficulty remembering new information, cognitive decline, behavioral and emotional changes, loss of motor coordination, confusion, speech problems, and mood swings [9,10]. Although the underlying mechanisms leading to AD development have not been clearly defined, many risk factors have been identified, including age, genetic predisposition, environmental factors such as exposure to metals, lifestyle, malnutrition, traumatic brain injury, immune system dysfunction, infectious agents, vascular disease, and psychological impairment [11,12]. Recent research has demonstrated that sustained oxidative stress, altered lipid metabolism, cholinergic dysfunction, protein homeostasis, mitochondrial energy depletion, synaptic dysfunction, and subsequent neuronal loss also have prominent roles in AD progression [13,14]. Moreover, previous studies have demonstrated that elevated pro-inflammatory mediators like cytokines, chemokines, and interleukins (ILs) make major contributions to neuroinflammation-induced AD progression [15,16].
Cholinergic neurons synthesize acetylcholine (ACh), which plays a crucial role in learning and memory [17]. AD has been characterized by the loss of cholinergic neurons in both the cortical and hippocampal regions [18,19]. Serotonin (5-HT) is an important neuromodulator involved in various physiological functions including cognition, emotion, sexual function, and sleep [20,21]. In addition, 5-HT reductions in the hippocampus and frontal cortex are responsible for short- and long-term memory impairment [21]. Along with 5-HT, the gamma-aminobutyric acid (GABA [A]) receptor is altered in AD. The greater the expression of the GABA (A) receptor, the more neuropathological changes are apparent in people with AD [22]. Glutamatergic neurotransmission is another important mechanism involved in learning and memory. In AD, Aβ induces oxidative stress, which interferes with glutamatergic neurotransmission, ultimately leading to its failure and attendant impairments of cognition, learning, and memory in patients [23].
The limited number of effective treatment options for AD has encouraged researchers to explore both potent pharmacological agents and a broad range of other biological activities as ways to prevent NDDs. Consequently, dietary small molecules from natural sources were investigated and shown to have potential therapeutic benefits in various NDDs, including AD. These molecules (e.g., flavonoids, phenolic acids, and carotenoids) have been widely reported as anti-AD agents both in vivo and in vitro, with only minor side effects. In this paper, we summarize recent advances in dietary small molecules and their bioactive compounds as potential alternative therapies for the prevention and treatment of AD. We highlight the chemical structure, mechanism, and effective dose of each phytochemical. In addition, current challenges and prospects are discussed. The systematic literature search was conducted using Google Scholar, SciFinder, Science Direct, PubMed, Web of Science, EBSCO, Scopus, JSTOR and other web sources. The scientific literature preferably on natural dietary small molecules and their bioactive constituents in context to their neuroprotective properties and their mechanism of action were selected. Literature with scientific rigor published up to 2023 was included.
2. Molecular pathogenesis of AD
The molecular pathogenesis of AD remains under active investigation. The sporadic and familial forms of the disease, which are histopathologically indistinguishable, are characterized by neurodegeneration in the hippocampus and cerebral cortex that is associated with numerous aggregated Aβ plaques and tau hyperphosphorylation (Fig. 1). Moreover, neuroinflammation mediated by activated microglia has increasingly received attention as a key pathogenic mechanism in AD [24].
Fig. 1.

The current knowledge of the factors contributing to AD pathogenesis. Ach: acetylcholine; ROS: reactive oxygen species; RNS: reactive nitrogen species.
Oxidative stress plays an important role in the initiation and progression of aging and age-associated AD. It is caused by redox imbalance, antioxidant dysregulation, and the production of reactive oxygen species (ROS) [25]. A large body of research suggests that oxidative stress changes vital cellular elements within the brain, such as proteins, lipids, and DNA [26]. These changes lead to deleterious alterations in brain neurotransmitter levels, modulation of intracellular signaling pathways, and ultimately attacks on neurons and glial cells, leading to increased neuronal apoptosis [27,28]. Moreover, the extensive oxidative damage observed in mild cognitive impairment (MCI) suggests that oxidative stress may be an early event in the progression from normal aging to AD pathology [29]. Several preclinical studies have demonstrated that oxidative stress plays a key role in the initiation and progression of cognitive deficits and contributes to AD pathogenesis [30,31]. Other studies have demonstrated that increased ROS production induces synaptic loss and promotes the accumulation and aggregation of Aβ plaques in AD brains [32,33]. In addition, postmortem studies have revealed that age-dependent increases in oxidative damage and mitochondrial dysfunction may contribute to increased oxidative stress in AD [[34], [35], [36]]. Moreover, increased ROS levels are strongly associated with tau aggregates, tau phosphorylation, and neuronal cell death [37].
Senile plaques are mainly composed of Aβ peptides, which are produced by amyloidogenic proteolytic processing of amyloid precursor protein (APP), a transmembrane protein with a large luminal domain and a short cytoplasmic domain [38]. Aβ is generated from APP by consecutive cleavage by β‐secretase and γ‐secretase, while non-amyloidogenic processing is initiated by α-secretase cleavage of APP within the Aβ domain, followed by γ‐secretase cleavage. Aberrant APP processing may result in an imbalance between the aggregation and clearance of Aβ peptides, leading to the formation of toxic oligomers, fibrils, and senile plaques associated with neurotoxicity. However, previous studies have indicated that cognitive dysfunction in AD is linked to the accumulation of extracellular soluble oligomeric Aβ species rather than amyloid plaque deposition in the brain [[38], [39], [40]]. It has been reported that oligomeric Aβ can disrupt synaptic plasticity, induce synaptic degeneration, and reduce long-term potentiation (LTP), all of which contribute to AD progression [41]. Neurofibrillary tangles (NFTs) are fibrillar aggregates composed of paired helical filaments (PHFs) of abnormally hyperphosphorylated tau protein. Tau is a highly soluble, microtubule-associated phosphoprotein that regulates microtubule stabilization and polymerization and plays an important physiological role in microtubule dynamics and axonal transport [7,42]. In AD, aberrantly phosphorylated tau redistributes from the axon to the soma, inducing microtubule network breakdown and axonal transport deficits, resulting in neuronal atrophy and cell death. Moreover, when detached from microtubules, hyperphosphorylated tau is highly likely to self-aggregate into neurotoxic PHFs [43]. There is a strong relationship between the tau aggregation associated with NFT formation and neuronal loss in the brain, and this contributes to disease progression in AD [44].
Among the neurotoxic factors involved in AD progression, neuroinflammation has a crucial role. Microglia and astrocytes are the main components of the immune system within the central nervous system (CNS), and they play a vital role in the development and progression of neuroinflammation. While microglial cells typically protect and maintain homeostasis in the brain, their effects can be beneficial or detrimental to neurons in the CNS. Normally, microglia clear abnormal Aβ protein aggregates by phagocytosis and degradation [45,46]. It has been reported that the loss of phagocytic efficiency by activated microglia is a key contributing factor to Aβ accumulation [47]. In the presence of neuroinflammation caused by aging, AD, and other neuropathological conditions, chronic abnormal activation of microglia disrupts the CNS microenvironment, leading to the release of various pathogenic proinflammatory mediators including IL-1β, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) [[48], [49], [50]]. Activated microglia also induce IL-6 and tumor necrosis factor-alpha (TNF-α), two important proinflammatory cytokines that are directly involved in AD pathogenesis [51]. In patients with AD, changes have been observed in IL-6 and TNF-α levels in the brain and serum, which are associated with increasing Aβ accumulation and APP expression. These in turn lead to AD pathology with clinical symptoms presenting later in life, suggesting a direct relationship between the expression of inflammatory markers and cognitive ability [52,53]. Evidence suggests that Aβ accumulation involves several intracellular signaling pathways, including phosphorylation cascades leading to mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB activation. Moreover, previous studies have suggested that NF-κB activation is mediated by microglial activation and that it significantly increases the expression of the inhibitor of NF-κB p-IκBα/IκBα and the NF-κB family protein p-p65/p65 [54,55]. Additionally, activation of NF-κB and MAPK signaling helps trigger tau protein aggregation and tau hyperphosphorylation, which are responsible for AD development [56].
It is well known that synaptic alteration and dysfunction are among the earliest events in the initiation and progression of AD and are closely related to spatial memory impairment, neuronal loss, and cognitive decline [57]. Histological studies of brain tissue from patients with AD as well as AD mouse models have demonstrated that synaptic alterations include changes in dendrite and spine structure, loss of dendritic spines, aberrant sprouting and curvature of dendritic processes, dystrophic dendrites, compromised synaptic transmission, and reduced synaptic plasticity [58,59]. Increased levels of soluble Aβ and tau hyperphosphorylation in the brain are strongly correlated with synaptic degeneration and loss of dendritic spines in humans and animal models of AD [[60], [61], [62], [63]]. Evidence suggests that neuronal loss and synaptic degeneration have the most robust correlation with cognitive decline in AD [64]. Accordingly, preventing synaptic degeneration and restoring synaptic function may be a novel, early therapeutic approach to slow AD progression and preserve memory function.
3. Challenges for AD therapy and alternative treatment strategies
Currently, two types of drugs are approved by the United States Food and Drug Administration (FDA) for treating AD symptoms: acetylcholinesterase (AChE) inhibitors including galantamine (RAZADYN, REMINYL), donepezil (ARICEPT, ARICEPT ODT), rivastigmine (EXELON), and the N-methyl-d-aspartate (NMDA) receptor antagonist memantine (NAMENDA) [65,66]. Recent clinical trials in AD have shown some efficacy for antibody therapies in removing neuritic plaques. For example, a phase II study by Eisai and Biogen showed that the humanized IgG1 antibody lecanemab (BAN2401) significantly reduced Aβ plaques, as did donanemab (LY3002813), another humanized IgG1 antibody developed by Eli Lilly. Some evidence of cognitive stabilization was seen in patients treated with these drugs during the trials, but there was also an increased incidence of amyloid-related imaging abnormalities-cerebral edema (ARIA-E). A similar antibody developed by Biogen (aducanumab [BIIB037]) was submitted to the FDA but was rejected for its weak efficacy.
Synthetic drugs for AD have side effects and do not halt disease progression. The discovery of medications that slow the progression of the disease has advanced in recent years. There are now particular therapeutics that can prevent the production of Aβ, discontinue the activation mechanism of brain inflammation, improve cognitive and learning abilities, and be tolerated over an extended period. The process of creating new, efficient medications is challenging, time-consuming, and has a relatively high failure rate. Drug repurposing has made it possible to find effective treatments for a variety of ailments. The fact that the candidate compound's safety has been established is a key benefit of this strategy because it eliminates the need for additional preclinical safety testing, chemical optimization, or toxicology studies, which significantly cuts down on the time and expense needed to advance potential treatment into clinical studies. A reasonable safety database developed from prior registrational procedures, post-marketing experience, and safety surveillance is likely to exist for medications that have been put on the market. It is urgently necessary to find novel treatments and targets for creating more potent medications to slow AD progression and recover cognitive function [67].
The need for more effective AD therapies has led to the extensive study of natural dietary products and their isolated compounds. In general, natural dietary phytochemicals can bind with numerous proteins in the body and modify transporters, enzymes, hormones, and DNA, as well as chelate heavy metals and scavenge free radicals; therefore, many of these agents have strong antioxidant, anti-inflammatory, and neuroprotective properties [68,69]. Dietary interventions can be a greatly accepted, inexpensive, and efficacious option to preserve against age-related cognitive decline and neurodegeneration, leading to important personal and societal benefits (67). Synchronously, considering the transformation at the colonic level and absorption, the good bioavailability of some dietary small molecules owing to the ability to pass the blood–brain barrier (BBB), to a various extent, facilitated the interest in these molecules as fresh neuroprotective agents against NDDs (68,69). Recent studies have shown that regular use of nutraceuticals, including dietary supplements and herbal medicines, has the potential to enhance cognitive ability in humans and treat many diseases [70,71]. Possible mechanisms for these effects include inhibitory effects on neuroinflammation-induced neuronal apoptosis, oxidative stress, key enzymes involved in Aβ plaque production, tau phosphorylation, and other pathological products [72]. Furthermore, dietary small molecules thus exert their neuroprotective effects by maintaining neuronal quality and number in key brain areas, thus preventing the onset or progression of diseases responsible for decreased cognitive function. In the following sections, we will discuss the effects of dietary small molecules and their main constituents on spatial learning and memory, tau phosphorylation, the Aβ plaque pathway, brain oxidative damage, neuroinflammation, neurotoxicity, and AD treatment.
4. Dietary small molecules and neuroprotection – a overview of potential molecular mechanisms
4.1. Neuroprotective mechanisms of chrysin in AD
Chrysin is a promising, naturally occurring polyphenolic flavone with a 15-carbon backbone; it is well-studied and known to be beneficial to human health. Chrysin is abundant in medicinal herbs, honey, fruits, flowers, dietary supplements, and many plant species. Structurally, chrysin has a lack of oxygenation in B and C rings, which is associated with its pharmacological activity and includes anti-inflammatory, antioxidant, anticancer, antiallergic, cardioprotective, hepatoprotective, and neuroprotective effects [73,74]. Chrysin's maximum plasma concentration and oral bioavailability have been estimated at 12–64 nM and 0.003–0.02%, respectively [75,76]. Opposing effects have not been reported with the usual daily oral dosage of 400–500 mg [77,78]. The recommended daily dose of chrysin is reported to be 0.5–3 g [79,80]. In a rat model, toxicological experiments showed that chrysin (1000 mg/kg) induced major changes in blood chemistry (albumin, bilirubin, alanine transaminase, aspartate aminotransferase, creatinine, and gamma-glutamyl transferase) and hematology parameters (red blood cells, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, total leucocyte count, lymphocytes, and neutrophils). In addition, chrysin (1000 mg/kg) administration significantly increased oxido-nitrosative stress associated with the kidney and liver. The no observed adverse effect level (NOAEL) for chrysin was 500 mg/kg and the lowest observed adverse effect level was 1000 mg/kg following administration to both sexes [81].
Recent in vitro and in vivo studies have shown that chrysin has antioxidant and neuroprotective activity against AlCl3-induced neurotoxicity and related pathological changes in a Swiss mouse AD model. In vitro, chrysin (5 μM) reduced early oxidative stress, significantly decreased proinflammatory mediator levels (iNOS, IL-1β, and TNFα), and attenuated AlCl3-induced late necrotic cell death in neuronal SH-SY5Y cells. Oral administration of chrysin (10, 30, and 100 mg/kg body weight [bw]) for 90 days in mice with AlCl3-induced AD led to reduced cognitive impairment, decreased neuronal degeneration, and normalized AChE and butyrylcholinesterase (BChE) activity (Fig. 2). In addition, there was a reduction in oxidative damage that was shown by a decrease in lipid peroxidation and enhanced activity of antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH) in the cerebral cortex and hippocampus [82]. In a d-galactose-induced rat model of aging, chrysin treatment (20 mg/kg bw) for 8 weeks significantly raised the activity levels of enzymatic and non-enzymatic antioxidants such as CAT, SOD, glutathione peroxidase (GPx), glutathione reductase (GR), glutathione-S-transferase (GST), GSH, glucose-6-phosphate dehydrogenase, and vitamins C and E; malondialdehyde (MDA) levels and protein carbonylation were diminished [83]. Similarly, oral administration of chrysin (10 or 30 mg/kg bw) for 28 days can attenuate spatial learning and recognition memory impairments in rats with d-galactose-induced aging. Chrysin also induces cell proliferation, supports cell survival, and regulates the depletion of hippocampal neurogenesis in these rats [84]. It has also been reported that oral administration of chrysin (125 or 250 mg/kg bw) for 8 weeks can regulate d-galactose-induced changes in behavior, brain biochemistry, neuroinflammation, and histopathology in aging mice. It was also demonstrated that chrysin elevated the percentage of spontaneous alternation, and increased locomotor activity and the discrimination ratio in novel object recognition (NOR) memory in d-galactose-treated mice. In addition, brain levels of adenosine monophosphate-activated protein kinase (AMPK), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), liver kinase B1 (LKB1), neurotrophin-3 (NT-3), NAD (P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), and serotonin (5-HT) were increased and the proinflammatory mediators TNF-α, NF-κB, and glial fibrillary acidic protein (GFAP) were reduced. Chrysin also alleviated neuronal degeneration in the white matter and cerebral cortex [85].
Fig. 2.

Illustration of dietary small molecules inhibiting/reducing the AChE activity in cholinergic pathway involved in AD pathogenesis. Acetylcholine after being synthesized is released from the vesicles and supports memory formation. AChE causes the breakdown of acetylcholine. So, dietary small molecules like chrysin, 7.8-DHE, naringenin, luteolin, lycopene, CA, GA, EGCG, TFs, and vanillin are blocking AChE thus preventing acetylcholinesterase breakdown, Aβ aggregation, neuroinflammation, and supporting memory formation. Ach: acetylcholine; Aβ: amyloid beta; AchE: acetylcholinesterase; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-6: interleukin-6; NF-κB: nuclear factor kappa B.
A recent study has shown that chrysin treatment (25, 50, and 100 mg/kg bw) for 7 days inhibits sevoflurane-induced hippocampal oxidative stress and spatial navigation impairments in the elevated plus maze, NOR, and Morris water maze (MWM) tests in aged rats, suggesting that chrysin has potential as a memory-enhancing agent for AD [86]. Aged mice were investigated to identify the effects of chrysin on cognitive function. Oral administration of chrysin (1 or 10 mg/kg bw) for 60 days decreased ROS levels, restored SOD, CAT, and GPx activity, and increased levels of Na+, K+-ATPase activity, and brain-derived neurotrophic factor (BDNF) expression in the hippocampus and cortex of aged mice [87]. Similarly, in C57BL/6 AD mice with methimazole-induced AD, treatment with chrysin (10 and 20 mg/kg bw) for 28 days improved spatial learning and cognitive function in the MWM and increased levels of the neurotrophins BDNF and nerve growth factor (NGF) in the hippocampus and prefrontal cortex. Additionally, molecular docking analysis demonstrated that chrysin effectively binds to the active site of tropomyosin receptor kinase (Trk) A and TrkB, and the p75 neurotrophin receptor [88]. Further experiments have demonstrated that chrysin treatment (10 and 20 mg/kg bw) for 28 days ameliorated long-term spatial learning and memory deficits, reduced glutamate levels, and decreased Na+, K+-ATPase activity in both the hippocampus and prefrontal cortex in C57BL/6 mice with euthyroid and methimazole-induced AD (Fig. 3) [89].
Fig. 3.

Molecular pathways are involved in the neuroprotective, antioxidant, and promoting neuronal cell survival of dietary small molecules. The PI3K/Akt pathway destabilizes Nrf2-Keap1 complex, which under basal conditions leads to the ubiquitination and degradation of Nrf2 by proteasomal system, and thus, promotes the nuclear translocation of Nrf2. This, in turn, activates the Nrf2-ARE antioxidant system and results in the expressions of multiple genes that encode antioxidant enzymes responsible for redox metabolism and GSH synthesis and metabolism. Activated Akt also regulates cell survival by maintaining a balance between pro-apoptotic and anti-apoptotic proteins. Furthermore, MAPK signaling pathways regulate neuronal survival and the transcriptions of CREB-dependent genes that encode BDNF, and other proteins required for synaptic plasticity and neurogenesis. Dietary small molecules may promote cell survival by activating TrkB signaling by functioning as BDNF mimetics or by promoting Akt phosphorylation or inhibiting Nrf2-Keap1 complex, and thus, activating the antioxidant defense system. TrkB signaling and the Nrf2-ARE antioxidant system are complementary to each other, and simultaneous activation of these pathways has been shown to confer neuroprotection against oxidative stress and to attenuate memory and cognition impairments in patients with AD or NDDs brain injury. BDNF: brain derived neurotrophic factor; CREB: cAMP response element-binding protein; TrkB: tropomyosin receptor kinase; Nrf2: nuclear factor erythroid 2-related factor 2; HO-1: heme oxygenase 1; (PI3K)/AKT: phosphoinositol 3-kinase; SOD: superoxide dismutase; GST: glutathione-S-transferase; MDA: malondialdehyde; ERK1/2: extracellular signal-regulated kinase 1/2; CaMK: Ca2+/calmodulin-dependent protein kinase; PKC: protein kinase-C.
4.2. Neuroprotective mechanisms of 7,8-dihydroxyflavone in AD
The flavone 7,8-dihydroxyflavone (7,8-DHF) is a natural flavonoid known to have neuroprotective activity. It is found in Tridax procumbens, Godmania aesculifolia, and various species of Primula. Preparations from these plants, such as decoctions, powdered leaves, teas, leaf juice, and oils, have been used as drugs or food supplements and are reported to have antifungal, antioxidant, anti-hepatotoxic, antimicrobial, immunomodulatory, analgesic, anti-inflammatory, and antidiabetic properties [90,91]. However, research into the relationships between structure and activity showed that a catechol group gives 7,8-DHF suboptimal oral bioavailability and brain exposure, suggesting that 7,8-DHF can cross the BBB and is orally bioactive [92,93]. Furthermore, a toxicity study found that following 7,8-DHF (3 mg/kg) treatment for 3 weeks and (2.4 μg) for 20 weeks, male and female C57BL/6 mice had normal blood cells counts and there were no adverse pathological effects in drug-treated liver, muscle, lung, kidney, heart, intestine, testis, spleen, hippocampus, or cortex [92].
Recent in vivo and in vitro pharmacological studies have shown that 7,8-DHF might be useful in treating AD. A study has shown the neuroprotective effect of 7,8-DHF (5 μM) in ΔK280 TauRD-DsRed-induced neurotoxicity using SH-SY5Y cells as an in vitro model of AD. Treatment with 7,8-DHF reduced tau aggregation, oxidative stress, and caspase-1 activity, and also improved neurite outgrowth, restored the reduced heat shock protein beta-1 (HSPB1) and nuclear factor erythroid 2-related factor 2 (Nrf2) expression, and activated TrkB-mediated extracellular signal-regulated kinase (ERK) signaling to upregulate cAMP response element-binding protein (CREB) and its downstream antiapoptotic B-cell lymphoma 2 (Bcl2) protein expression in SH-SY5Y cells expressing ΔK280 TauRD-DsRed [94]. This neuroprotective effect of 7,8-DHF has also been detected in 6-OHDA-induced PC-12 cells. Interestingly, 7,8-DHF (1–100 μM) pretreatment protected PC12 cells against neuronal cell death by enhancing SOD activity and direct free radical scavenging, and inhibiting oxidative injury, mitochondrial dysfunction, and apoptosis [95]. In an intracerebroventricular (ICV)-streptozotocin (STZ)-induced AD mouse model, treatment with 7,8-DHF (5, 10, and 20 mg/kg bw) for 3 weeks attenuated cognitive deficits in MWM and NOR, increased activity of the antioxidant enzymes GSH, CAT, SOD, GPx, and decreased MDA, protein carbonyl (PCO), and nitrite levels. In addition, AChE activity was inhibited, NADPH and mitochondrial complex I, II, III, and IV were reduced, and p-tau protein expression was significantly lowered along with neuronal cell death in the cortex and hippocampus [96]. Chen et al. [97] observed that oral administration of 7,8-DHF (1 mg/kg bw) for 4 weeks improved spatial learning and cognitive function by blocking cholinergic signaling and reducing oxidative stress, synaptic degeneration, and Aβ deposition in the hippocampus of rats with scopolamine-induced AD. Gao et al. [98] investigated the effects of 7,8-DHF in the Tg2576 AD mouse model and found that chronic administration of 7,8-DHF (5 mg/kg bw) for 4 weeks rescued spine density in CA1 pyramidal neurons and synaptoneurosomal fractions, and improved cognitive function. In addition, chronic 7,8-DHF treatment increased the levels of reduced proteins GluA1 and GluA2, two subunits of the glutamate AMPA receptor, and activation of TrkB (Y816) and its downstream phosphoinositol 3-kinase (PI3K)/AKT, Ras/ERK, and PLCγ (phospholipase C-γ)/Ca2+/calmodulin-dependent protein kinase-II (CaMKII) signaling pathways (Fig. 3).
Zhang et al. [99] found that 7,8-DHF treatment (1 mg/kg bw) activated TrkB signaling, restored synapse number and synaptic plasticity, and prevented Aβ deposition, the loss of hippocampal synapses, and memory deficits in 5XFAD transgenic mice. Similarly, 7,8-DHF treatment (5 mg/kg bw) for 10 days rescued memory deficits, restored deficient TrkB signaling without affecting endogenous BDNF levels, blocked beta-secretase 1 (BACE1) elevations, and lowered levels of the β-secretase-cleaved C-terminal fragment of amyloid precursor protein (C99), Aβ40, and Aβ42 in 5XFAD mice [100]. Another study reported that 7,8-DHF treatment (5 mg/kg bw) decreased cortical Aβ plaque deposition and protected cortical neurons against reduced dendritic arbor complexity, hippocampal increases in choline-containing compounds, and glutamate loss, but had no significant impact on dendritic spine density or hippocampal neurogenesis in 5XFAD mice (Fig. 4) [101]. Furthermore, in a rat model of age-related memory loss, 7,8-DHF treatment (5 mg/kg bw) for 4 weeks prevented declines in the performance of fear conditioning tasks and cognition; significantly enhanced the activation of phosphorylated TrkB at the Y515 and Y816 sites; increased spine density and number in several brain regions that process fear memory, including the amygdala, hippocampus, and prefrontal cortex; and facilitated basolateral amygdalar synaptic plasticity [102]. Treatment with 7,8-DHF (5 mg/kg bw) for 28 days attenuated alcohol and high-fat diet (HFD)-induced memory loss, restored GSH levels, reduced nitrite and MDA levels and AChE activity, downregulated caspase-3, IL-1β, iNOS, and NF-κB, and upregulated Nrf2, HO-1, and BDNF mRNA levels in the rat hippocampus (Fig. 3) [103]. However, more recent studies concluded that 7,8-DHF administration significantly and selectively increased thin-spine density in the CA1 region, increased the number of newly formed oligodendrocytes in the corpus callosum, and significantly improved spatial learning and memory in MyRF−/− and CaM/Tet-DTA mice [104,105].
Fig. 4.

Represents the potential role of dietary small molecules in promoting autophagy and inhibiting Aβ plaques and NFT induced neurotoxicity and inflammatory responses in AD. Aβ protein is generated through a series of proteolytic digestions of the single-pass transmembrane, APP. First β secretase (BACE) cleaves APP, releasing the soluble Appβ fragment (sAPPβ) into the extracellular space and leaves the C terminal fragment (CTFβ) attached to the membrane. Subsequently, CTFβ is cleaved by an intramembrane-cleaving aspartyl protease complex, γ secretase to generate APP intracellular domain (AICD) and Aβ isoforms (Aβ40 or Aβ42). The major product is a 40 amino acid long Aβ40 while the 42 amino acid residues Aβ42 oligomerize faster and form Aβ plaques in Alzheimer's brains. Mechanism of aberrant hyper-phosphorylation of tau protein that leads to microtubule depolymerization. Further, aggregation of oligomerized hyper-phosphorylated tau forms NFTs which accounts for neuronal death. Autophagy is suggested to involve in the clearance of Aβ and hyper-phosphorylated tau aggregates, restraining generation of Aβ plaques and NFTs, respectively. Autophagy inducers dietary small molecules can potentially enhance autophagic degradation of aggregated Aβ and tau proteins and inhibit neuroinflammatory responses. ROS: reactive oxygen species; Aβ: amyloid beta; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-6: interleukin-6; iNOS: inducible nitric oxide synthase; NF-κB: nuclear factor kappa B; BACE-1: beta-secretase 1; APP: amyloid precursor protein; NFTs: neurofibrillary tangles.
4.3. Neuroprotective mechanisms of naringenin in AD
Naringenin is a flavanone that is highly lipophilic due to its chemical structure. It is found in oranges, grapefruits, tangerines, raw lemon peel, and cherries, as well as in vegetables, especially tomatoes [106]. Interestingly, grapefruits and oranges contain large amounts of naringenin, ranging from 14.17 to 53 mg/100g in grapefruits and from 1.47 to 11.15 mg/100 g in oranges [106]. Naringenin has strong antioxidant, immunomodulatory, antidepressant, anti-inflammatory, cardioprotective, neuroprotective, and antiaging properties [107]. It is also readily distributed in the stomach, small intestine, liver, brain, heart, kidney, and spleen through the intestinal epithelium via passive diffusion into enterocytes [108]. According to previous in vitro and animal studies, naringenin has high BBB permeability [109,110].
An in vitro study in PC12 cells revealed that naringenin performs an important function in reducing apoptosis and neurotoxicity in Aβ-induced AD. The suppression of caspase-3, activation of PI3K/AKT, and regulation of glycogen synthase kinase-3 beta (GSK3β) signaling pathways are among the intracellular processes responsible for naringenin's anti-apoptotic and neuroprotective actions [111]. It also abrogated the Aβ-induced increase in ROS and decreased Aβ toxicity in a concentration-dependent manner in PC12 cells [112]. A study using Neuro2a cells and primary mouse neurons revealed that naringenin treatment can induce autophagy-promoting proteins such as ULK1, beclin1, ATG5, and ATG7 and that naringenin restored transcript levels of AMPK after siRNA-AMPK knockdown, reduced Aβ levels to a nontoxic concentration, maintained the mitochondrial membrane potential, and resisted ROS production in Aβ1-40-induced neuronal cells (Fig. 4) [113]. In lipopolysaccharide (LPS)-stimulated BV-2 microglial and N2a cells, naringenin pretreatment inhibited NO release, the expression of iNOS and COX-2, as well as the expression of pro-inflammatory cytokines induced by the activation of suppressor of cytokine signaling-3 (SOCS-3) through the AMPKα and protein kinase C-delta (PKCδ) signaling pathways (Fig. 5) [114].
Fig. 5.

Molecular pathways are involved in the anti-inflammatory effect of dietary small molecules. LPS is a potent activator of the NF-кB, MAPK, and IRF3 signaling pathways. Phosphorylation of IкB by IKK results in the release of cytoplasmic NF-кB and subsequently its translocation into the nucleus. In the nucleus, NF-кB activates the expression of the pro-inflammatory gene. NF-кB can also be activated by MAPK and IRF3 signaling pathways. Dietary small molecules mainly inhibit the secretion and expression of related inflammatory factors through multiple molecular pathways. LPS: lipopolysaccharide; TLR4: toll-like receptor 4; ERK1/2: extracellular signal-regulated kinase 1/2; JNK: c-Jun N-terminal kinase; p38: NF-κB: nuclear factor kappa B; MAPK: mitogen-activated protein kinase.
A study in mice with LPS-induced AD demonstrated that naringenin treatment (10 and 20 mg/kg bw) had significant protective effects on microglial activation and improved motor coordination, as well as influencing anti-neuroinflammatory responses in the hippocampus and cerebral cortex [114]. In mice with Aβ-induced AD, naringenin treatment (100 mg/kg bw) for 1 day improved spatial learning and memory function, lowering hippocampal MDA and NO levels and successfully restoring SOD levels and neuronal loss [115]. In ICR mice with scopolamine-induced amnesia, naringenin treatment (1, 3, and 4.5 mg/kg bw) for 3 weeks significantly ameliorated spatial learning and memory deficits as measured in both the passive avoidance and Y-maze tests, and inhibited AChE activity [116]. Similarly, in rats with ICV-STZ-induced AD, 2 weeks of naringenin treatment (50 mg/kg bw) significantly decreased non-enzymatic 4-hydroxynonenal (4-HNE), MDA, thiobarbituric acid reactive substances (TBARS), H2O2, and PCO levels, and increased GSH levels and GPx, GR, GST, SOD, CAT, and Na(+)/K(+)-ATPase activity; loss of choline acetyltransferase (ChAT)-positive neurons in the hippocampus was attenuated and there was significant improvement in spatial learning and memory (Fig. 2, Fig. 3) [117]. Another study found that ICV-STZ-induced rats receiving naringenin supplements (25, 50 mg, 100 mg/kg bw) for 3 weeks had better learning and memory performance. This was accompanied by increased mRNA expression of insulin and insulin receptors, and reduced tau hyperphosphorylation and Aβ upregulation in both the hippocampus and cerebral cortex through downregulation of GSK3-β activity. In addition, naringenin enhanced insulin signaling in the brain and the peroxisome proliferator-activated receptor gamma (PPAR-λ) in these rats [118].
However, in a study of iron-dextran-induced neurotoxicity in AD rats, administration of naringenin (50 mg/kg bw) for 28 days significantly improved antioxidant enzyme activity (SOD, CAT, and GPx) and attenuated oxidative damage by decreasing TBARS and PCO levels, and DNA apoptosis, as well as reducing AChE activity and reducing the density of degenerating and necrotic cells in the cerebral cortex [119]. In rats, oral administration of naringenin (50 mg/kg bw) for 2 weeks successfully improved spatial learning and recognition memory and significantly ameliorated MDA, AChE, and 5-hydroxyindoleacetic acid levels by modulating GSH, SOD, CAT, and GPx levels in the hippocampus and cortex while simultaneously increasing ACh, 5-HT, and dopamine levels as well as maintaining normal neuronal morphology in AlCl3-induced AD rats [120]. Oral administration of naringenin (25, 50, and 100 mg/kg bw) for 7 days can prevent LPS-induced spatial memory deficits, oxidative damage, and improve levels of antioxidant enzymes such as SOD, CAT, and GSH in the hippocampus. In addition, naringenin treatment at the same doses significantly increased Nrf2 and decreased NF-κB, TNF-α, toll-like receptor 4 (TLR4), GFAP, iNOS, and COX2 in the hippocampus in LPS-induced rats [121]. Furthermore, naringenin (200 mg/kg bw) for 12 weeks can prevent memory impairment and neuroinflammation; these effects were associated with the inhibition of Aβ production, oxidative stress, and tau hyperphosphorylation in the brain in mice with HFD-induced AD [122]. Another study reported that naringenin (25, 50, or 100 mg/kg bw) for 3 weeks significantly improved learning capacity and memory retention, effectively ameliorated cognitive dysfunction, reduced isoflurane-induced apoptosis, and modulated the PI3/Akt/PTEN and NF-κB signaling pathways in isoflurane-induced rats [123].
4.4. Neuroprotective mechanisms of luteolin in AD
Luteolin is a flavonoid biophenol compound found in vegetables and fruits, including black and white pepper, broccoli, thyme, olive oil, pomegranate, cocoa, cucumber, buckwheat sprouts, and celery [124]. Evidence suggests that it has anticancer, cardioprotective, antioxidant, neuroprotective, and anti-inflammatory properties [124]. Luteolin has been commercially developed as a dietary supplement and can be found in food and cosmetic products because of its safety profile, with nontoxic side effects reported in mice at 2500 mg/kg and rats at 5000 mg/kg; this is equivalent to 219.8–793.7 mg/kg in humans [125]. In animals, glycosides of luteolin (10−70 mg/kg bw) can modulate systemic and brain insulin resistance, decrease Aβ deposition by activating the gut-liver-brain axis, and prevent AD development (Fig. 6) [126]. A luteolin intake of about 2–125 mg per day is generally recommended as healthy [127].
Fig. 6.

Regulative role of dietary small molecules in gut–brain axis. Dietary small molecules (luteolin, EGCG, and TFs) modulate gut microbiota diversity and subsequently involved in gut–brain axis; Dietary small molecules are related with the neurotrophic and neurotransmitter system and ultimately contributes to the regulation between gut homeostasis and brain function. SCFA: short-chain fatty acids; EGCG: epigallocatechin-3-gallate; TFs: theaflavins.
In Aβ25-35-induced PC-12 cells, luteolin treatment significantly prevented the Aβ25-35-induced decrease in cell viability and inhibited apoptosis while significantly upregulating Bcl-2 expression and downregulating Bcl-2-associated X protein (Bax) and caspase-3. In addition, luteolin treatment significantly upregulated the expression of estrogen receptor beta (Erβ) and p-ERK1/2 in Aβ25-35-induced PC-12 cells [128]. Zheng et al. [129] found that luteolin pretreatment suppressed the activation of the BACE1 promoter and inhibited NF-κB signaling by both directly and indirectly disrupting p65 complex formation, and inhibiting APP, C99 protein, and Aβ1-42 aggregation in lipofectamine 2000-transfected HEK293 and SH-SY5Y cells. Luteolin has also been used to investigate cellular toxicity, barrier function, cytokine production, and inflammation-related intracellular signaling pathways in Aβ1-40-induced human brain microvascular endothelial cells (hBMECs) and human astrocytes (hAs). The results demonstrated that luteolin increased cell viability and protected barrier function by preserving transendothelial electrical resistance and relieving aggravated permeability in the human BBB. Luteolin also reduced the production of inflammatory mediators and cytokines, including COX-2, TNF-α, IL-1β, IL-6, and IL-8 in hBMECs and hAs. In addition, luteolin treatment inhibited p-p38, p-ERK, and p-c-Jun N-terminal kinase (p-JNK) activation, downregulated p-IκB kinase (p-IKK), relieved inhibitory IκBα degradation, and blocked NF-κB p65 nuclear translocation in Aβ1-40-induced hBMECs and hAs (Fig. 5) [130]. Similarly, in APP-overexpressed SH-SY5Y cells, luteolin treatment increased cell viability, reduced intracellular ROS generation, enhanced SOD activity, and reversed mitochondrial membrane potential dissipation. Furthermore, luteolin effectively inhibited caspase-mediated apoptosis, downregulated AβPP expression, and lowered Aβ₁₋₄₂ secretion in APP-overexpressed SH-SY5Y cells [131]. In another study, luteolin decreased zinc-induced tau hyperphosphorylation at the Ser262/356 epitope by regulating phosphorylation/dephosphorylation in SH-SY5Y cells [132]. In LPS-induced rat C6 glioma cells, pretreatment with luteolin (1 and 10 μM) inhibited cell proliferation, reduced the release of TNF-α, IL-1β, and IL-6, and decreased the endoplasmic reticulum (ER) stress marker GRP78 (Fig. 5) [133].
In a mouse model of AD, luteolin administration (20 and 40 mg/kg bw) for 3 weeks improved spatial memory learning and cognitive function, and ameliorated Aβ-plaque formation; this was accompanied by inhibition of both GFAP expression and neuroinflammation (TNF-α, IL-1β, IL-6, NO, COX-2, iNOS, p-p38, and p-NF-κB) as well as decreased expression of GRP78 and inositol-requiring enzyme type 1α (IRE1α) in brain tissue [133]. In an APP23 AD mouse model, treatment with luteolin (100 mg/kg bw) for 29 weeks ameliorated depression-like behavior and improved spatial learning, suppressed IL1-β production by reducing endoplasmic reticulum (ER) stress, and reduced brain levels of activation factor 4 (ATF4), Iba-1, and CD68 [134]. Similarly, in rats with STZ-induced AD, luteolin treatment (10 and 20 mg/kg bw) for 1 week significantly ameliorated spatial learning and memory impairment and increased the thickness of the hippocampal CA1 pyramidal layer [135]. Another study revealed that luteolin treatment (50, 100, and 200 mg/kg bw) of Aβ-induced AD rats for 17 days ameliorated Aβ-induced learning and memory impairment; the ChAT, SOD and GPx activity was increased, there was a reversal of the increase in AChE and MDA activity, as well as increase the Bcl2/Bax ratio in the hippocampus [136].
A very recent study demonstrated that oral administration of luteolin (20 or 40 mg/kg bw) for 8 weeks significantly ameliorated memory and cognitive impairment and provided neuroprotection by inhibiting Aβ generation, restoring mitochondrial damage, reducing neuronal apoptosis and promoting PPARγ expression and function in 3 × Tg-AD mice [137]. In an Aβ-induced AD mouse model, luteolin (80 mg/kg bw) for 2 weeks promoted Aβ clearance by reducing levels of p-JNK, p-p38, GFAP, and Iba-1, and decreased the inflammatory markers p-NF-kB p65, TNF-α, IL-1β and COX-2 in the cortex and hippocampus. Luteolin exerted its anti-AD effects in Aβ1-42-injected mice by enhancing synaptic proteins PSD-95 and SNAP-25 expression through reductions in BACE-1 and Aβ1-42 expression and regulation of proapoptotic and antiapoptotic protein expression via decreased Bax and caspase-3 and increased Bcl2 in the cortex and hippocampus (Fig. 4, Fig. 7) [138]. In a transgenic Drosophila AD model, luteolin treatment (5, 10, 15, and 20 μM)—in a concentration-dependent manner—reduced Aβ aggregation, LPO, protein carbonyl content (PCC), GST, AChE, caspase-9, and caspase-3 activity and increased SOD, CAT, and GSH expression while increasing the life span and climbing ability of the flies [139]. However, more recent studies found that administration of luteolin combined with palmitoylethanolamide (5 mg/kg bw) attenuated the activity of Aβ-induced proinflammatory mediators (iNOS, COX-2, IL-1β, IL-6, IL-10, and TNF-α), inhibited astrogliosis and microgliosis activation, and upregulated mRNA expression for neurotrophic markers such as BDNF and GDNF, as well as promoting neuronal survival [140,141].
Fig. 7.

Molecular pathways are involved in the anti-apoptotic effect of dietary small molecules. Apoptosis is conducted through intrinsic and extrinsic pathways in the pathogenesis of AD. Inflammatory cytokines, such as TNFα and IL-1β, can trigger the neuronal apoptosis through membrane receptor or extrinsic pathway (i.e., TNFα/caspase-8/caspase-3). The intrinsic pathway of neuronal apoptosis is activated by intracellular insults including tau aggregates, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, free radicals, etc. Mitochondrial cytochrome c (Cyt c) is released to initiate apoptosis signaling through the Cyto c/caspase-9/caspase-3 cascade. Dietary small molecules (7,8-DHE, luteolin, lycopene, FA, EGCG, and vanillin) can take an anti-apoptotic action by mediating different signaling targets. ROS: reactive oxygen species; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; Bax: Bcl-2-associated X protein; Bcl2: B-cell lymphoma 2; ER: endoplasmic reticulum.
4.5. Neuroprotective mechanisms of lycopene in AD
Lycopene is a lipophilic aliphatic hydrocarbon carotenoid that can be obtained from many fruits and vegetables, including tomatoes, watermelons, pink guavas, papayas, pink grapefruits, and apricots. Tomato is a major source of lycopene and is also a considerable source of vitamins C, K, A, carbohydrates, and fiber, as well as potassium, iron, sulfur, and phosphorus [142]. Previous studies have shown that lycopene is an antioxidant that is cardioprotective, neuroprotective, anticancer, anti-inflammatory, and antihypertensive and that it improves memory [143]. Investigators recommend that a daily lycopene intake of about 5–10 mg is healthy [144]. No known adverse events were observed following rodent consumption of up to 3 g/kg per day of either dietary or formulated lycopene [145,146].
Recent pharmacological studies have shown that lycopene might be useful in treating AD. Fang et al. recently reported that lycopene may have neuroprotective activity in Aβ-overexpressed M146L cells. Their results showed that lycopene treatment alleviated oxidative stress and apoptosis, activated the PI3K/Akt/Nrf2 signaling pathway, upregulated antioxidant and antiapoptotic proteins, downregulated proapoptotic proteins, and inhibited APP and BACE activity in M146L cells [147]. Another in vitro study reported that lycopene improved cell viability, reduced intracellular and mitochondrial ROS levels, and inhibited apoptosis and NF-κB-target gene expression in Aβ-treated SH-SY5Y cells [148]. Similarly, Lim et al. reported that lycopene treatment increased mitochondrial respiration, mitochondrial membrane potential, and glycolytic function by reducing intracellular and mitochondrial ROS levels, DNA fragmentation, caspase-3 activation, cytochrome c release, and inhibited cell death and NF-κB activation in RCAN1-overexpressing SH-SY5Y cells [149]. Lycopene protected cells from LPS-stimulated neurotoxicity and attenuated neuroinflammation by inhibiting the Iba-1, COX-2, p-p38, p-Erk, p-JNK, and p-Akt and p-IκB-α and p-NF-κB signaling pathways, and significantly elevated expression of Nrf2 in the nucleus and cytosol, HO-1, and NQO-1, and reduced intracellular ROS production in BV-2 microglial cells (Fig. 5) [150]. Huang et al. [151] reported that lycopene treatment promoted the secretion of neuroprotective NGF, BDNF, vascular endothelial growth factor (VEGF), and increased Bcl2 expression by inhibiting the expression of Bax, cleaved caspase-3, and cytochrome c. It has also been reported that lycopene increased expression of the synaptic proteins PSD-95 and synaptophysin (SYP) and activated the PI3K/Akt pathway in a tert-butyl hydroperoxide (t-BHP)-induced AD primary culture model. Qu et al. [152] observed that lycopene treatment efficiently attenuated Aβ-induced neurotoxicity in a primary culture AD model as evidenced by improved cell viability, inhibited ROS generation and mitochondrial membrane potential depolarization, restored levels of proapoptotic Bax and antiapoptotic Bcl-2, and inhibited caspase-3 activation (Fig. 7). Another study showed that lycopene attenuated Aβ-induced oxidative stress by decreasing intracellular and mitochondrial ROS production and mitochondria-derived superoxide production, opening mitochondrial permeability transition pores leading to cytochrome c release and ameliorating mitochondrial morphological dysfunction. Lycopene also restored ATP levels, improved mitochondrial complex activities, prevented mitochondrial DNA damage, and increased levels of mitochondrial transcription factor A within mitochondria in an Aβ-induced primary culture AD model [153].
In rats with Aβ1-42-induced AD, lycopene treatment (2.5 and 5 mg/kg bw) improved memory retention in MWM and markedly attenuated oxidative stress as indicated by significant decreases in MDA, LPO, and NO activity, reduced mitochondrial damage, inhibition of caspase-3 activity, IL-6, and TNF-α, and restoration of BDNF levels [154]. Similarly, treatment with lycopene (1, 2, and 4 mg/kg bw) reduced mitochondrial dysfunction, inhibited caspase-3 activity associated with neuronal cell death, and improved spatial memory learning and memory by reducing levels of the proinflammatory cytokines TNF-α, TGF-β, and IL-1β as well as NF-κB in the hippocampus and cerebral cortex in Aβ1-42-induced AD rats [155]. Recently, Xu et al. observed that lycopene treatment (4 mg/kg bw) improved learning and memory function, induced liver X receptor (LXR) expression and activated LXR-PI3K-AKT signaling, and inhibited Aβ-plaque formation and neuroinflammation in APP/PS1 mice [156]. Other research found that lycopene administration (4 mg/kg bw) effectively restored cognitive and memory impairment by diminishing Aβ deposition, reducing inflammatory responses by blocking inflammatory cytokine production and NF-κB p65 and TLR4 protein expression in the hippocampus of Aβ-induced AD rats (Fig. 4) [157]. In mice with LPS-induced AD, 5 weeks of lycopene treatment (30 mg/kg) lowered APP levels, prevented accumulation of Aβ, and suppressed neuronal BACE1 and elevated expression of the α-secretase ADAM10. Furthermore, lycopene downregulated Iba-1 expression, reduced inflammatory mediator levels, and inhibited oxidative stress in the hippocampus (Fig. 4) [150].
Furthermore, in a fructose-induced mouse model of AD investigating lycopene treatment (4 mg/kg) for 10 weeks, spatial learning and memory were improved, oxidative stress was reduced, and AChE activity was reduced in both the hippocampus and cerebral cortex. In addition, lycopene also significantly reduced insulin resistance and the expression of insulin-like growth factor 1 receptor, PI3K, and p-AKT proteins, significantly reduced plasma insulin and Homeostatic Model Assessment for Insulin Resistance; it also reduced TNF-α, IL-1β and NF-κB and elevated PPARγ protein expression in both the hippocampus and cerebral cortex [158]. Xu et al. [159] showed that the administration of lycopene (5 mg/kg), human amniotic epithelial cell transplantation, and a combination of the two significantly improved cognitive function and spatial learning, decreased TNF-α and IL-1β levels, downregulated TLR4 and NF-κB p65 mRNA and protein expression, and increased levels of the anti-inflammatory markers IL-10 and TGF-β1 in the cerebrospinal fluid and hippocampal tissue in a rat model with Aβ1-42-induced AD at the choroid plexus.
4.6. Neuroprotective mechanisms of ferulic acid in AD
Ferulic acid (FA) is a natural phenolic compound found in oranges, wheat, peanuts, apples, rice, coffee, barley, vanilla beans, and many other foods [160]. FA has been confirmed to have anti-inflammatory, antiapoptotic, antidiabetic, anticancer, antioxidant, and neuroprotective properties [161]. In the United States, Japan, and most European countries, FA has been approved as a food additive and is also added to beverages and cosmetics [162]. Li et al. [163] reported that FA (300 μg/mL) had no toxic effects on red blood cells, white blood cells, or platelets. Recommendations for a healthy daily intake of FA are lacking, but consumption from food sources can be estimated at approximately 200–1000 mg per day.
FA has been suggested as a possible neuroprotective strategy against AD due to its antioxidant and anti-inflammatory properties. Several studies have investigated FA using in vitro and in vivo AD models. Huang et al. [164] reported that FA treatment was neuroprotective by significantly reducing the production of TNF-α and IL-1β and upregulating CREB and p-CREB as well as phosphodiesterase 4 (PDE4) activity in LPS-stimulated PC12 cells (Fig. 3). Additionally, FA pretreatment of SH-SY5Y cells inhibited the transition of Aβ42 from monomers to oligomers by preventing Aβ42 from forming fibrils [165]. Similarly, another study in SH-SY5Y cells examined the effect of FA on Aβ1–40-induced hallmarks of AD such as Aβ aggregation, cell death, and ROS formation, and reported significant decreases in Aβ aggregation and intracellular and mitochondrial ROS formation, as well as membrane damage repair [166]. A comparable study revealed that the anti-inflammatory effect of FA in LPS-stimulated BV-2 microglial cells was mediated via significant suppression of IL-1β, prostaglandin E2 (PGE2), and NO release, a reduction of COX-2 and iNOS expression, and the subsequent inhibition of TLR4 activity in a concentration-dependent manner [167]. The most recent study demonstrated that FA pretreatment inhibited TNF-α, IL-1β, and IL-6 production and significantly decreased the expression of autophagy-related protein p62 by increasing beclin1, p-ULK1, p-AMPK, LC3, transcription factor EB, and lysosomal-associated membrane protein 2A in LPS-stimulated BV-2 microglial cells (Fig. 5) [168].
Kim et al. [169] highlighted that FA administration (20 or 50 mg/kg bw) for 28 days improved trimethyltin-induced cognitive deficits and increased ChAT activity. Most recently, FA improved behavioral and cognitive function and effectively attenuated Aβ-induced upregulation of intracellular Ca2+, inhibited PP1, and activated dopamine and cAMP regulated phosphoprotein 32 kDa (DARPP-32) by increasing synapsin1 and synaptic protein PSD-95 and improving LTP, as well as preventing the loss of GluN2B phosphorylation and decreasing Aβ accumulation in APP/PS1 mice [170]. Wang et al. [171] reported that FA (20 mg/kg bw) for 30 days reduced Aβ plaque deposition and increased the diameter and density of hippocampal capillaries, thus facilitating the supply of oxygen and nutrients to the brain and the removal of metabolic waste; this led to improved spatial memory in the APP/PS1 AD mouse model. Mori et al. [172] also used transgenic APP/PS1 mice and found that FA (30 mg/kg bw) for 6 months decreased oxidative stress associated with Aβ production and inhibited synaptotoxicity, amyloidogenic APP processing, microgliosis, astrocytosis, and neuroinflammation, leading to improvements in cognitive tasks. Yan et al. [173] showed that oral administration of FA (5.3 and 16 mg/kg bw) for 6 months significantly reduced neuroinflammation, which was associated with reduced Aβ₁₋₄0 and Aβ₁₋₄₂ deposition and IL-1β levels in the hippocampus and frontal cortex as well as enhanced cognitive performance. Furthermore, Jin et al. [174] observed that treatment with FA (50, 100, and 250 mg/kg bw) for 3 weeks led to a strong IL-1β reduction, inhibition of GFAP, decreased phosphorylation of both ERK1/2 and p-p38 and inhibition of the apoptotic cascade, particularly caspase-3 and FasL protein expression in the hippocampus of rats induced with Aβ1–40. A further study concluded that FA (1–100 μM) inhibited AD-like symptoms induced by Aβ neurotoxicity through clearance of Aβ aggregation, reduction in lipid levels, and activation of autophagy pathways in a C. elegans AD model [175].
4.7. Neuroprotective mechanisms of ellagic acid in AD
Ellagic acid (EA) is a natural polyphenol compound found in vegetables and fruits such as persimmons, black raspberries, wolfberries, strawberries, pomegranates, cranberries, walnuts, pecans, plums, and peaches, as well as in other plant foods [176]. It has shown anticancer, antioxidant, antidiabetic, antimutagenic, anti-inflammatory, antiaging, and neuroprotective properties [177]. A previous study observed that the no observable effect level was estimated at 3011 mg/kg bw per day for male rats and less than 778 mg/kg bw per day for females. Meanwhile, the lowest observed adverse effect level was estimated at 3254 mg/kg bw per day for male and female rats [178]. At present, standard dosing recommendations for EA are lacking, suggesting that the correct dose depends on several factors, including the age and health of the consumer.
Recent in vitro results showed that EA treatment improves cell viability and reduces cytotoxicity by reducing Aβ fibril formation and oligomer loss in Aβ42-induced SH-SY5Y cells [179]. Shen et al. [180] observed that EA treatment of H2O2-induced PC12 cells reduced cell damage and enhanced cell viability through two key mechanisms: inhibiting ROS production and reducing calcium ion influx. In another in vitro experiment using SH-SY5Y cells induced by d-galactose, EA exhibited antioxidant and anti-inflammatory effects by inhibiting ROS production and reducing MDA levels and neuroinflammation by targeting β-galactosidase, advanced glycation end products, and TNF-α activity, and by increasing GSH and improving cell viability [181]. A recent study demonstrated that the anti-inflammatory effect of EA in LPS-induced BV-2 microglial cells was associated with the downregulation of inflammatory mediators such as TNF-α, NO, and p-ERK1/2 (Fig. 5) [182].
Interestingly, Zhong et al. [183] reported that EA treatment (50 mg/kg bw) for 2 months ameliorated spatial learning and memory impairment, exhibited antiapoptotic effects by inhibiting caspase-3 activation and neuronal apoptosis, significantly inhibited Aβ deposition and hyperphosphorylation of tau, and decreased GSK3-β activity in the hippocampus of APP/PS1 mice. Jha et al. [184] previously investigated the anti-AD effect of a 5-week EA treatment (50 mg/kg bw) in STZ-induced AD rats. Their results showed that EA significantly diminished oxidative stress, AChE levels, GFAP, and C-reactive protein expression, decreased the Aβ plaque level, improved synaptic connectivity, and improved motor function and learning. Similarly, oral EA administration 50 mg/kg bw in AlCl3-induced AD rats significantly improved episodic memory and decreased APP and TBARS levels, increased CAT, SOD, and GSH activity, and reduced NFTs and neuritic plaques as well as downregulated caspase-3 and APP expression in entorhinal cortex sections (Fig. 3, Fig. 4) [185]. Oral EA administration (10, 50, and 100 mg/kg bw) for 1 week provided neuroprotection, including remarkable decreases in levels of AChE, BChE, and oxidative stress markers, while increasing learning and memory and modulating the NF-κB/Nrf2/TLR4 signaling pathway in Aβ-induced AD rats [186]. Assaran et al. subsequently reported that administration of EA (25, 50, and 100 mg/kg bw) for 3 weeks prevented learning and memory deficits and significantly reduced MDA activity but significantly increased SOD and CAT activity in the hippocampus and cortex in scopolamine-induced AD rats [187].
4.8. Neuroprotective mechanisms of caffeic acid in AD
Caffeic acid (CA) is a phenolic compound widely distributed in vegetables and medicinal plants such as cabbage, turmeric, mushrooms, cauliflower, radishes, kale, and apples, and in plant-derived liquids and beverages like olive oil, wine, coffee, and tea [188,189]. It is reported to have antiviral, anticancer, anti-inflammatory, immunomodulatory, antioxidant, and neuroprotective properties [190,191]. Drinking a single cup of coffee provides as much as 70–350 mg of CA [192], and the daily intake of CA has been estimated at 500–1000 mg in humans through consumption of fruits, vegetables, and CA-containing beverages [161]. A reproductive and toxicity study in mice revealed that the NOAEL was 150 mg/kg bw per day [193].
Recent pharmacological animal and in vitro studies have shown that CA has beneficial effects on the brain, including protection from neuronal damage induced by a variety of neurotoxic agents, and may be useful in treating AD. Furthermore, pretreatment of Aβ1–42-induced PC12 cells with CA significantly reversed Aβ-induced neurotoxicity by increasing cell viability, efficiently scavenging free radicals, attenuating increases in intracellular calcium and tau phosphorylation, and significantly decreasing the phosphorylation of GSK-3β [194]. Another in vitro study reported that CA pretreatment improved cell viability and attenuated acrolein-induced neurotoxicity, ROS accumulation, GSH depletion, and mitochondrial membrane potential dysfunction; CA also modulated MAPKs and Akt/GSK3β signaling pathways, and subsequently restored the changes to BACE-1 and ADAM-10 induced by acrolein [195].
A recent study in HFD-induced AD rats showed that CA treatment (30 mg/kg bw) for 30 weeks enhanced SOD and CAT activity, increased the expression of p-GSK3β and synaptophysin proteins, and reduced phosphorylated-tau (p-tau [Ser 396], p-tau [Ser 404], and p-tau [Thr 181]) expression in the hippocampus and cortex; it also attenuated APP and BACE1 expression, which led to the lowering of Aβ1-42 and drebrin accumulation in the hippocampus and cortex (Fig. 4) [196]. Using Aβ-induced AD rats, Wang et al. [197] found that oral administration of CA (100 mg/kg bw) for 2 weeks significantly rescued learning deficits and increased cognitive function by reducing oxidative stress, increasing NO and AChE activity, and suppressing inflammation. This was accompanied by the inhibition of NF-κB-p65, p53, and p-p38 MAPK as well as decreased expression of caspase-3 in the hippocampus. Deshmukh et al. [198] found that CA (10, 20, and 40 mg/kg bw) for 3 weeks attenuated STZ-induced behavioral changes and cognitive function. Their results showed that CA treatment abrogated STZ-induced increases in AChE, NO, and PCO levels, reduced MDA, and modulated oxidative stress indicators in the hippocampus and cortex in AD rats.
Khan et al. [199] demonstrated that CA (50 mg/kg bw) for 2 weeks exerted neuroprotective effects against Aβ1–42-induced AD in mice via improvements in spatial learning, memory, and cognitive abilities, markedly reduced levels of ROS and TBARS, and increased expression of Nrf2 and HO-1 in the hippocampus. CA also has anti-inflammatory properties and has been shown to downregulate Iba-1 and GFAP over-expression, and reduce the proinflammatory cytokines IL-1β and TNF-α in the hippocampus of AD mice. It also activated NF-κB and increased expression of the synaptosomal-associated proteins SNAP-25 and SNAP-23, SYN, and postsynaptic density protein 95, and significantly increased p-PI3K/p-AKT and BDNF proteins in these mice (Fig. 3, Fig. 4). In an ApoE−/− AD mouse model, treatment with CA (20 mg/kg bw) for 2 months alleviated spatial learning impairment and cognitive decline, reduced Aβ accumulation, upregulated mRNA levels for ATP-binding cassette (ABC) transporters ABCA1 and ABCG1, and elevated serum HDL cholesterol concentration, as well as reducing levels of serum TNF-α, monocyte chemoattractant protein-1, and IL-6 and decreasing local inflammation in the hippocampus [200]. A recent C. elegans AD model showed that CA treatment significantly alleviated Aβ-induced toxicity, increased life span, decreased body paralysis, and improved reproductive defects. In addition, the treatment increased resistance to heat and oxidative stress and reduced polyglutamine (polyQ40) aggregate formation in strains N2 and CL4176. These protective effects were exerted via activation of transcription factor DAF-16 and its downstream targets SOD-3 and GST-4, which subsequently upregulated mRNA expression for heat shock proteins HSP-1 and HSP-16.2 in CL4176 [201].
4.9. Neuroprotective mechanisms of gallic acid in AD
Gallic acid (GA) is a phenolic small molecule found naturally in numerous medicinal plants and a wide range of fruits, such as blueberries, strawberries, blackberries, mangoes, grapes, and pomegranates. It is also found in walnuts, wine, tea, cashew nuts, hazelnuts, plums, and other foods [202]. The therapeutic potential of GA is due to its antioxidative, anti-proliferative, chemoprotective, antimicrobial, antidiabetic, anti-inflammatory, antidepressant, and neuroprotective properties [202,203].
Recent in vitro and in vivo studies have drawn attention to the neuroprotective effects of GA. In an in vitro AD model, pretreatment with GA suppressed Aβ-induced neuronal cell death, efficiently suppressed the expression of proinflammatory cytokines (iNOS, COX-2, and IL-1β), and greatly reduced the levels of nuclear p65 and p65 acetylation in BV-2 microglial cells and Neuro-2A cells (Fig. 5) [204].
Moreover, treatment with GA (10 and 20 mg/kg) for 28 days ameliorated spatial working memory and cognitive dysfunction, as well as significantly inhibiting the expression of IL-1β, TNF-α, iNOS, and COX-2 in the hippocampus and cortex of Aβ-induced AD mice. GA was also shown to restore Aβ-induced NF-κB acetylation, substantially inhibit nuclear NF-κB activation, and dramatically suppress Aβ-mediated neuronal cell death in AD mice [204]. Yu et al. [205] studied the anti-AD effects of GA in APP/PS1 mouse models and found that oral administration of GA (30 mg/kg) improved spatial learning and reference memory in both 4-month-old and 9-month-old APP/PS1 mice. Furthermore, GA treatment significantly elevated hippocampal LTP, increased expression of synaptic protein PSD-95, and decreased Iba-1 and GFAP protein expression in 9-month-old APP/PS1 mice. Further, fluorescent microscopy showed that GA significantly reduced Aβ1–42 aggregation and Aβ1–42-induced intracellular calcium influx in both the hippocampus and cortex (Fig. 4). Another study reported that GA supplementation (20 mg/kg) for 6 months in APP/PS1 mice mitigated cerebral amyloidosis, including brain parenchymal and cerebrovascular Aβ deposits, and decreased cerebral Aβ proteins; reduced amyloidogenic and elevated nonamyloidogenic APP processing simultaneously elevated α- and reduced β-secretase activity, inhibited neuroinflammation, and stabilized brain oxidative stress [206].
Similarly, in an AlCl3-induced AD rat model, treatment with GA (100 mg/kg) for 2 months decreased spatial memory impairment and learning deficits, significantly increased antioxidant enzymes (CAT, GSH, and SOD), serum electrolytes (except K+) and neurotransmitter levels (except norepinephrine) with a corresponding decrease in stress markers (MDA, H2O2, and NO) in the hippocampus. In addition, histological studies showed that GA administration protected NFTs and amyloid plaques in the external granular layer in these rats (Fig. 3) [207]. GA treatment (30 mg/kg) for 26 days enhanced spatial memory performance and significantly prevented cognitive deficits associated with increased enzymatic SOD, CAT, and GPx activity and decreased TBARS levels in the hippocampus and cerebral cortex of ICV-STZ-induced AD rats [208]. In a sodium arsenite-induced memory model, co-administration of GA (50 and 1000 mg/kg) for 4 weeks produced antioxidant effects by normalizing oxidative stress indicators (SOD, CAT, and GPx), reducing AChE and MDA activity, and improving spatial learning and memory [209]. Finally, a small study showed that administration of (50, 100, and 200 mg/kg) for 10 days reduced neural damage and brain amyloid neuropathology and improved cognitive function via free radical scavenging and inhibition of Aβ oligomerization in the hippocampus of rats with Aβ-induced AD [210].
4.10. Neuroprotective mechanisms of epigallocatechin-3-gallate in AD
Epigallocatechin-3-gallate (EGCG) is a green tea polyphenol that exerts metal-chelating, anti-inflammatory, free-radical-scavenging, antioxidant, anticancer, anti-apoptotic, and neuroprotective activity by modulating neuroprotective proteins, transcription factors, and other important neurotrophic growth factors [211,212]. Notably, in vitro and animal studies suggest that EGCG is highly absorbed, has moderate oral bioavailability, and can easily cross the BBB [213,214]. Food supplements containing green tea catechins provide a daily EGCG dose in the range of 5–1000 mg/day for adults [161]. Furthermore, a toxicity study showed that the NOAEL was 500 mg/kg bw per day following a 290-day green tea (EGCG) intervention in rats and mice [215,216], while a few 4-week oral intervention studies reported a NOAEL of 2500 mg/kg bw per day [217].
There is evidence that EGCG can decrease AChE activity and inhibit the Aβ aggregation and tau phosphorylation associated with AD. An in vitro study showed that EGCG pretreatment effectively ameliorated Aβ-induced cytotoxicity, suppressed increases in iNOS expression and subsequent production of NO and peroxynitrite, enhanced antioxidant enzyme levels, and altered the expression of proapoptotic signals in BV-2 microglial cells [218]. A similar study reported that EGCG administration significantly inhibited IL-6, IL-8, VEGF, and PGE2 production, decreased COX-2 expression, and inhibited NF-κB activation, with subsequent inhibition of phosphorylation of p38, JNK, and ERK in IL-1β+Aβ(25-35)-induced U373MG cells [219]. Furthermore, in microglial BV-2 cells stimulated with LPS, pretreatment with EGCG significantly inhibited the production of the proinflammatory mediator NO, decreased the release of proinflammatory cytokine IL-6 while increasing TNF-α, downregulated MIF, CCL-2, and CSF2 and upregulated IL-3, IL-11, and TNFS10, significantly downregulated mTOR, NF-κB2, STAT1, Akt3, CCL5, and SMAD3 while significantly upregulating the expression of mRNA of Ins2, Pld2, A20/TNFAIP3, and GAB1 [220]. Similarly, EGCG is associated with protection against Aβ and LPS-induced neurotoxicity in SH-SY5Y and BV-2 microglial cells through the inhibition of proinflammatory cytokines (IL-1β and IL-18), the reduction in Iba-1 over-expression, and the suppression of the co-localization of caspase-1 and Iba-1 activation as shown by immunofluorescence staining. In addition, EGCG treatment reversed the increased expression of NLRP3 and significantly reduced the expression of TLR4, p-IKK/IKK, and p-NF-κB/NF-κB in LPS + Aβ-induced BV-2 microglial cells [221].
Zhong et al. [221] observed that the administration of EGCG (2 mg/kg bw) for 28 days reduced microglial inflammation and neurotoxicity by suppressing the activation of the canonical NLRP3 and noncanonical caspase-11-dependent inflammasome via the TLR4/NF-κB pathway in LPS + Aβ-induced rat primary microglia and in the hippocampus of APP/PS1 mice. Furthermore, EGCG supplementation (50 mg/kg bw) for 2 months had anti-AD activity, with significant attenuation of cognitive deficits, improvement in dendritic integrity and expression of synaptic proteins, decreased pro-inflammatory cytokine IL-1β and increased anti-inflammatory cytokines IL-4, IL-10, and IL-13, and subsequent alleviation of microglial activation in the brains of APP/PS1 mice. In addition, EGCG treatment markedly reduced Aβ plaque formation and tau phosphorylation in the hippocampus of APP/PS1 mice [222]. Similarly, Nan et al. [223] reported that EGCG (100, 250, and 625 mg/kg bw) for 9 weeks increased ACh levels by inhibiting AChE activity, decreasing tau hyperphosphorylation and BACE1 expression, and suppressing Aβ1-42 expression in the hippocampus. In addition, EGCG treatment decreased the escape latency period while increasing the time at the target quadrant among AD rats. Interestingly, Lee et al. [224] reported that EGCG treatment (1.5 and 3.0 mg/kg bw) for 4 weeks prevented LPS-induced memory impairment and apoptotic neuronal cell death, suppressed Aβ accumulation and the expression of APP, BACE1, and its product C99, while reducing the proinflammatory cytokines IL-1β, TNF-α, IL-16, iNOS, COX-2, and activating astrocytes as well as reducing amyloidogenesis.
Similarly, Ettcheto et al. [225] observed that EGCG administration (40 mg/kg bw) for 3 months improved peripheral parameters such as insulin sensitivity and liver insulin pathway signaling, as well as central memory deficits. It also markedly increased synaptic markers and CREB phosphorylation rates, because of a decrease in unfolded protein response activation through a reduction in ATF4 levels and subsequent downregulation of protein tyrosine phosphatase 1B. In addition, EGCG significantly reduced brain Aβ production and plaque burden by increasing ADAM10 levels in C57BL/6 WT and APP/PS1 mice; neuroinflammation was subsequently reduced as suggested by the decrease in astrocyte reactivity and TLR4 levels. In mice with estrogen deficiency and HFD-induced AD, EGCG administration (45 mg/kg bw) for 2 months improved object recognition and spatial memory, restored the diversity and structure of the gut microbiome in HFOVX mice, with increased Prevotella and inhibition of Bifidobacteriales but no effect on the decreases in Alloprevotella and Lactobacillaceae or on the increase in Enterorhabdus (Fig. 6) [226]. Soto-Mercado et al. [227] subsequently showed that EGCG treatment significantly inhibited the aggregation of (i)sAPPβf, blocked p-tau, increased ΔΨm, decreased oxidation of DJ-1 at residue Cys106-SH, and inhibited the activation of transcription factor c-JUN and P53, PUMA, and caspase-3 in mutant cholinergic-like neurons (ChLNs) compared to wild type. In addition, it reversed the Ca2+ influx associated with dysregulation as a response to ACh stimuli in PSEN1 E280A ChLNs, inhibited NF-κB activation, and reduced the secretion of pro-inflammatory IL-6 in wild-type astrocyte-like cells when exposed to mutant ChLNs culture supernatant.
4.11. Neuroprotective mechanisms of theaflavins in AD
Theaflavins (TFs) are major polyphenols found in black tea and include theaflavin-3, 3′-digallate, theaflavin-3-gallate, and theaflavin-3′-gallate [228]. TFs have anticancer, anti-obesity, antiviral, antioxidant, antidiabetic, anti-inflammatory, and neuroprotective properties [229]. A pharmacokinetic study in humans demonstrated that consumption of 700 mg of TFs once a day (corresponding to about 30 cups of black tea) resulted in maximum blood plasma concentration and improved oral bioavailability [230].
Cao et al. [231] reported that theaflavin-3,3′-digallate treatment (15, 30, and 60 mg/kg bw) improved learning and memory in the NOR and Y-maze tests, increased the brain/body mass ratio, decreased glutamate concentration and AChE activity and increased ACh concentration in the brain, and enhanced ChAT, glutaminase, and glutamine synthetase activity. It has also been reported that theaflavin-3,3′-digallate treatment of d-galactose-induced AD mice increased activity of the antioxidant enzymes GPx and SOD, while the concentration of MDA was reduced; the antioxidant genes Nrf2, Prx2, GSH-px1, and SOD1 were subsequently upregulated in the brain. Another study reported that administration of TFs (10 and 50 mg/kg bw) for 3 days to C57BL/6J mice with LPS-induced AD significantly increased spontaneous alternations in the Y-maze test and reduced immobility in the tail suspension test, indicating improved spatial memory and cognitive function. The results also showed suppressed production of inflammatory cytokines and the prevention of dendritic atrophy and spine loss in the brain [232]. A very recent study demonstrated that TFs effectively upregulate brain neurotrophic factors, improving behavioral function by maintaining gut homeostasis, boosting the immune response and antioxidant activity, restructuring the gut microbiome, strengthening tight junction proteins, and altering core microbiota metabolites, i.e., short-chain fatty acids (SCFAs) and essential amino acids (Fig. 6) [233].
4.12. Neuroprotective mechanisms of vanillin in AD
Vanillin is an important phenolic flavoring agent naturally found in plant species including vanilla tahitensis, vanilla planifolia, and vanilla pompona. It is widely used in the production of foods, pharmaceuticals, beverages, cosmetics, and perfumes [234]. Vanillin has anti-mutagenic, antioxidant, antidepressant, and anti-inflammatory properties [234], and has neuroprotective effects in several neuropathological conditions, including AD [235]. For a person weighing 70 kg, the recommended daily dose of vanillin is 10 mg/kg, which has been approved by the Food and Agriculture Organization of the United Nations/WHO and the European Union [161]. Moreover, toxicology studies in rodents receiving intraperitoneal and oral vanillin confirmed that it is safe at a high concentration of 300 mg/kg and produced no adverse effects on blood cells or the liver or kidneys [236].
An in vitro study examined the neuroprotective effects of ethyl vanillin on Aβ1-42-induced oxidative injury in PC12 cells. The results revealed that vanillin treatment increased cell viability, markedly alleviated intracellular lipid peroxidation levels, and ROS production, induced the antioxidative enzymes SOD, CAT, and GPx, increased Bcl2 expression, and reduced Bax and caspase-3 expression in Aβ1-42-induced PC12 cells [237]. NO release and protein and mRNA expression of the pro-inflammatory cytokines IL-1β, TNF-α, IL-6, COX-2, and iNOS were significantly reduced by vanillin in LPS-activated BV-2 microglial cells in a dose-dependent manner. In addition, vanillin treatment effectively inhibited the phosphorylation of MAPK and NF-κB signaling [238].
Anand et al. [239] demonstrated that vanillin administration (10 and 20 mg/kg bw) for 10 days significantly improved memory and behavioral functions, inhibited AChE activity and TBARS levels, enhanced activity of the antioxidant enzymes GSH and CAT, and significantly decreased the levels of IL-6 and TNF-α in the cortical and hippocampal regions of the brain in scopolamine-induced AD mice. Vanillin was also shown to prevent amyloidogenesis by inhibiting AChE activity, BACE1, and caspase-3, enhancing degradation of Aβ and elevating BDNF levels in the hippocampus and cortex of AlCi3-and d-galactose-induced AD mice [240]. Moreover, two animal studies have shown that vanillin (40 mg/kg bw) administered for 28 days markedly attenuated cognitive deficits, increased ID-1 protein expression, and effectively restored the number of Ki-76, DCX, ID-1-immunoreactive cells as well as increasing the number of NeuN-immunoreactive neurons in the hippocampus of mice with scopolamine-induced AD [241,242]. Table 1 summarizes the latest preclinical research findings on the neuroprotective efficacy of dietary small molecules against AD.
Table 1.
Summarizes the latest preclinical research findings on the neuroprotective efficacy of dietary small molecules against AD.
| Cell line/animal | Neurotoxin/duration | Drug concentration/duration | Behavioral parameters | Behavioral changes | Mechanism of action | Reference |
|---|---|---|---|---|---|---|
| Chrysin – in vitro studies | ||||||
| SH-SY5Y cells | AlCl3 (50 μM for 24 h) | 50 μM for 24 h | – | – | Oxidative damage ↓ iNOS, IL-1β, and TNFα ↓ Cell death ↓ |
[82] |
| Chrysin – in vivo studies | ||||||
| Swiss mice | AlCl3 (100 mg/kg bw for 90 days) | 10, 30, and 100 mg/kg bw for 90 days | Open-field test, chimney test, and step-down avoidance test | Amelioration of behavioral impairments | SOD, CAT, and GSH ↑ AChE, BChE, and lipid peroxidation ↓ Neuronal degeneration ↓ |
[82] |
| Albino Wistar rats | d-galactose (50 mg/kg bw for 8 weeks) | 20 mg/kg bw for 8 weeks | – | – | SOD, CAT, GPx, GST, GR, G6PD, GSH, vitamins C and E ↑ MDA and protein carbonylation ↓ |
[83] |
| Sprague Dawley rats | d-galactose (50 mg/kg bw for 8 weeks) | 10 and 30 mg/kg bw for 8 weeks | NOL and NOR | Improvement in spatial and recognition memory abilities | Cell proliferation, cell survival, and hippocampal neurogenesis ↑ | [84] |
| C57BL/6 mice | d-galactose (200 mg/kg bw for 8 weeks) | 125 and 250 mg/kg bw for 8 weeks | NOR and Y-maze | Elevation in the percentages of spontaneous alternation, locomotor activity, and spatial learning and memory abilities | AMPK, PGC1α, LKB1, NT-3, NQO1, HO-1, and 5-HT ↑ TNF-α, NF-κB, and GFAP ↓ Neuronal degeneration ↓ |
[85] |
| Albino Wistar rats | Sevoflurane anesthesia for 2 h | 25, 50, and 100 mg/kg bw for 8 weeks | NOL, NOR, MWM, and elevated plus maze | Amelioration of anxiety and explorative behavior and improvements in spatial learning and memory function | Protein and mRNA expression of Nrf2, HO-1, and NQO1 ↑ | [86] |
| Swiss Albino mice | – | 1 or 10 mg/kg bw for 60 days | Open-field test and MWM | Improvements in spatial learning, cognitive function, and motor function | ROS level ↓ SOD, CAT, GPx, and Na+, K+-ATPase activity ↑ BDNF expression ↑ |
[87] |
| C57BL/6 mice | Methimazole (0.1% for 31 days) | 10 or 20 mg/kg bw for 28 days | Open-field test and MWM | Prevention of cognitive dysfunction but no effects on locomotor activity | BDNF and NGF expression ↑ | [88] |
| 7,8-DHF – in vitro studies | ||||||
| SH-SY5Y cells | ΔK280 TauRD-DsRed for 7 days | 5 μM for 7 days | – | – | Tau aggregation, oxidative stress, and caspase-1 activity ↓ Neurite outgrowth, HSPB1, Nrf2, CREB, and Bcl2 ↑ |
[94] |
| PC12 cells | 6-OHDA (100 μM for 24 h) | 1–100 μM for 24 h | – | – | Cell viability, MMP, and SOD ↑ Oxidative injury, MDA, caspase-3, and cell death ↓ |
[95] |
| 7,8-DHF – in vivo studies | ||||||
| Wistar rats | ICV-STZ (3 mg/kg for 3 weeks) | 5, 10, and 20 mg/kg bw for 21 days | NOR and MWM | Recovery of spatial learning and memory function | GSH, SOD, CAT, and GPx ↑ NADPH activity and mitochondrial complexes I, II, III, and IV ↑ MDA, PCO, AChE, p-tau, and neuronal cell death ↓ |
[96] |
| Sprague-Dawley rats | Scopolamine (1 mg/kg for 2 weeks) | 1 mg/kg bw for 4 weeks | MWM | Attenuation of learning and memory deficits | GSH and SOD activities ↑ p-TrkB, p-Akt, and p-Erk ↑ MDA, Aβ40, and Aβ42 levels ↓ |
[97] |
| Tg2576 (Tg) mice and C57BL/6 N mice | – | 5 mg/kg bw for 4 weeks | MWM, elevated plus maze, and open-field test | Improvement in learning and memory abilities | GluA1, GluA2, and AMPA receptor ↑ TrkB, PI3K/Akt, Ras/ERK, and PLCc/CaMKII signaling ↑ |
[98] |
| 5XFAD mice and C57BL/6J mice | – | 5 mg/kg bw for 8 weeks | MWM | Improvement in spatial learning and memory function | TrkB signaling, synapse number, and synaptic plasticity ↑ Aβ deposition and synapse loss ↓ |
[99] |
| 5XFAD mice | – | 5 mg/kg bw for 10 days | Y-maze | Improvement in spatial working memory function | p-TrkB signaling and BDNF ↑ BACE1, C99, and flAPP expression ↓ |
[100] |
| 5XFAD mice | – | 5 mg/kg bw 3 days a week for 8 weeks | – | – | Cortical neurons ↑ Aβ plaque deposition ↓ |
[101] |
| Sprague-Dawley rats | – | 5 mg/kg bw for 28 days | Fear conditioning test | Improvement in memory function | p-TrkB signaling and BDNF ↑ p-CaMKII, p-Erk1/2, p-CREB, and p-GluR1 ↑ Dendritic spines, synapse formation, and synaptic plasticity ↑ |
[102] |
| Wistar rats | Alcohol/HFD (3–15% alcohol in 0.2% saccharin solution) for 12 weeks | 5 mg/kg bw for 28 days | MWM | Attenuation of impaired spatial learning and memory retention | GSH content, Nrf2, and HO-1↑ BDNF expression ↑ MDA, nitrite level, and AChE ↓ Caspase-3, iNOS, IL-1β, and NF-κB expression ↓ |
[103] |
| Naringenin – in vitro studies | ||||||
| PC12 cells | Aβ25-35 (20 μM) for 24 h | 0.4 μM for 24 h | – | – | Cell viability ↑ p-Akt and p-GSK3-β ↑ Cell apoptosis and caspase-3 ↓ |
[111] |
| PC12 cells | H2O2 (100 μM) for 24 h | 25–100 μM for 24 h | – | – | Cell viability ↑ ROS and LDH levels ↓ |
[112] |
| Neuro2a cells | Aβ25-35 (10 μM) for 24 h | 50 μM for 24 h | – | – | Cell viability ↑ p-AMPK, p-ULK1, and LC3II ↑ LDH, SQSTM1, Aβ accumulation ↓ |
[113] |
| BV-2 and N2a cells | LPS (100 ng/mL) for 24 h | 5, 10, and 20 μM for 24 h | – | – | Cell viability ↑ SOCS-3 and PKCδ activation ↑ NO, iNOS, and COX-2 ↓ |
[114] |
| Naringenin – in vivo studies | ||||||
| Swiss mice | LPS (20 mg/kg bw) for 1 day | 10 or 20 mg/kg bw for 3 days | Rotarod | Amelioration of motor impairment | Microglial activation ↓ | [114] |
| Wistar rats | Aβ (4 μL) for 1 day | 100 mg/kg bw for 1 day | Y-maze, radial arm maze, and passive avoidance test | Improvement in spatial learning and memory function | SOD levels ↑ MDA, NO, and neuronal loss ↓ |
[115] |
| ICR mice | Scopolamine (1 mg/kg) for 30 min | 100 mg/kg bw for 1 day | Y-maze and passive avoidance test | Amelioration of spatial learning and memory deficits | AChE levels ↓ | [116] |
| Swiss Albino mice | ICV-STZ (3 mg/kg for 7 days) | 50 mg/kg bw for 14 days | MWM and passive avoidance test | Prevention of spatial learning and cognitive function deficits | 4-HNE, MDA, TBARS, H2O2, and protein carbonyl ↓ GSH, GPx, GR, GST, SOD, and CAT ↑ Na(+)/K(+)-ATPase activity and ChAT-positive neuron ↑ |
[117] |
| Sprague-Dawley rats | ICV-STZ (3 mg/kg bw for 21 days) | 25, 50, and 100 mg/kg bw for 21 days | MWM | Improvement in learning and memory abilities | INS and INSR ↑ p-Tua and p-GSK3-β ↓ | [118] |
| Iron (Fe-dextran) (50 mg/kg) 5 days a week for 4 weeks | 50 mg/kg bw for 28 days | – | – | SOD, CAT, and GPx TBARS, protein carbonyl, and DNA apoptosis ↓ AChE and neuron density loss ↓ |
[119] | |
| Albino Wistar rats | AlCl3 + D-gal (150 mg/kg + 300 mg/kg bw) | 50 mg/kg bw for 14 days | MWM and NOR | Improvement in spatial learning and recognition memory | GSH, SOD, CAT, and GPx ↑ ACh, 5-HT, and DA ↑ Protected neurons ↑ MDA, AChE, and 5-HIAA ↓ |
[120] |
| Albino Wistar rats | LPS (167 μg/kg bw) for 7 days | 25, 50, and 100 mg/kg bw for 7 days | Y-maze, novel object discrimination, passive avoidance test | Amelioration of memory dysfunction | SOD, CAT, GSH, and Nrf2 ↑ NF-κB, TNF-α, TLR4, GFAP, iNOS, and COX2 ↓ |
[121] |
| SAMP8 mice | HFD for 12 weeks | 200 mg/kg bw for 12 weeks | Barnes maze and MWM test | Dramatic improvement in cognitive performance | GSH, SOD, and IL-10 ↑ MDA, NO, TNF-α, and IL-1β ↓ Aβ, APP, BACE1, GFAP ↓ Aβ40, and Aβ42 ↓ p-tau and p-GSK ↓ |
[122] |
| Sprague-Dawley rats | Isoflurane (0.75% for 1 week) | 25, 50 or 100 mg/kg bw for 3 weeks | Y-maze, elevated plus maze, open-field test, and fear conditioning test | Improvement in learning capacity and memory retention and amelioration of cognitive dysfunction | Bcl-xL, Bcl-2, and PI3K/Akt ↑ NF-κB, TNF-α, IL-6, and IL-1β ↓ Caspase-3, Bad, and Bax ↓ |
[123] |
| Luteolin – in vitro studies | ||||||
| PC-12 cells | Aβ25-35 (20 μM/L for 24 h) | 10, 1, 10−1, 10−2, 10−3, 10−4, 10−5, and 10−6 μM/L | – | – | Cell viability and Bcl2 ↑ Bax, caspase-3, p-ERK1/2 and cell death ↓ |
[128] |
| HEK293 and SH-SY5Y cells | Lipofectamine 2000 | 5, 15, and 30 μM for 24 h | – | – | BACE1 and p-NF-κB ↓ APP, C99, and Aβ 1-42 ↓ |
[129] |
| hBMEC and hA cells | fAβ1-42 (20 μM for 72 h) | 3, 10, and 30 μM for 72 h | – | – | Cell viability ↑ ROS levels ↓ TNF-α, IL-6, IL-8, and IL-1β ↓ p-p38, p-Erk1/2, and p-JNK ↓ p-IκB-α and p-NF-κB ↓ |
[130] |
| SH-SY5Y cells | Aβ₁₋₄₂ | Neo and APPsw transfected cells | – | – | Cell viability ↑ ROS levels ↓ SOD and MMP↑ Caspase 3 and caspase-9 ↓ |
[131] |
| SH-SY5Y cells | Zinc (0, 100, 200, 300, or 400 μM) for 1 h | 5–100 μM for 1 h | – | – | CaMKII and p-CaMKII ↑ p-tau at Ser262/356 ↓ | [132] |
| Luteolin – in vivo studies | ||||||
| 3 × Tg-AD and WT, C57BL/6J mice | – | 20 and 40 mg/kg bw for 3 weeks | MWM | Improvement in spatial learning and amelioration of memory deficits | GRP78, IRE1α, and p-p38 ↓ TNF-α, IL-1β, IL-6, and NO ↓ COX-2, iNOS, and p-NF-κB ↓ Aβ-plaque formation ↓ |
[133] |
| APP23 mice | Human APP751 | 100 mg/kg bw for 29 weeks | Y-maze, fear conditioning test, tail suspension test, and forced swimming test | Amelioration of depressive-like behaviors and improvement in spatial learning and memory function | eIF2α, p-eIF2α, and PS1C ↓ IL-1β, Iba-1, and ATF4 ↓ |
[134] |
| Sprague-Dawley rats | STZ (3 mg/kg on the fourth and sixth days) | 10 and 20 mg/kg bw for 1 week | MWM | Amelioration of spatial learning and memory impairment | CA1 pyramidal layer ↑ | [135] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (2 μl for 1 day) | 50, 100, and 200 mg/kg bw for 17 days | MWM and passive avoidance test | Improvement in spatial learning and spatial working memory | SOD level and GSH content ↑ ChAT and Ach content ↑ Bax and Bcl2 ratio ↑ MDA and AChE activity ↓ |
[136] |
| 3 × Tg-AD mice and WT mice | – | 20 or 40 mg/kg bw for 8 weeks | NOR, open-field test, and step-down avoidance test | Recovery of cognitive deficits and amelioration of anxiety, locomotor, and exploration abilities | IDE and UCP2 ↑ SOD level and GSH content ↑ PGC-α, Nrf1, Nrf2, and TFAM ↑ PPARγ, Bcl2, and Mfn2 ↑ APP, BACE1, and Aβ₁₋₄₂ ↓ ROS and MDA ↓ Drp1 and Fis1 ↓ Bax, caspase-3, caspase-9, and cyt-C ↓ |
[137] |
| C57BL/6 N mice | Aβ₁₋₄₂ (5 μl for one day) | 80 mg/kg bw for 2 weeks | – | – | Bcl2, PSD95, and SNAP25 ↑ BACE1 and Aβ₁₋₄₂ ↓ p-p38, p-JNK, Iba-1, and GFAP ↓ TNF-α, IL-1β, and p-NF-κB ↓ Bax, caspase-3, and COX-2 ↓ |
[138] |
| Lycopene – in vitro studies | ||||||
| M146L cells | Aβ-overexpression | 10 μM for 24 h | – | – | Gclc and Gclm expression ↑ Nrf2, p-Akt, p-GSK-3β, and Bcl2 ↑ ROS and MDA levels ↓ APP, BACE1, and caspase-3 ↓ |
[147] |
| SH-SY5Y cells | Aβ₁₋₄₂ (20 μl for 24 h) | 0.2 or 0.5 μM for 24 h | – | – | Cell viability ↑ Bcl2 and MMP ↑ ROS, Bax, p53, and caspase-3 ↓ p-IκBα and p-NF-κB ↓ |
[148] |
| SH-SY5Y cells | RCAN1-overexpressing | 1–2 μM for 24 h | – | – | Cell viability ↑ RCAN1 and MMP ↑ ROS, caspase-3, and cyt-C ↓ p-IκBα and p-NF-κB ↓ |
[149] |
| BV-2 cells | LPS (1 μg/mL 24 h) | 12.5, 25, and 50 μM for 24 h | – | – | Nrf2, HO-1, and NQO-1 ↑ ROS levels ↓ Iba-1 and COX-2 ↓ p-p38, p-Erk, p-JNK, and p-Akt ↓ p-IκB-α and p-NF-κB ↓ |
[150] |
| Primary culture | t-BHP (2 μM for 24 h) | 0.1–16 μM for 24 h | – | – | Cell viability ↑ NGF, VEGF, and BDNF ↑ Bcl2 and MMP ↑ SYP and PSD95 ↑ p-PI3K and p-Akt ↑ ROS, Bax, caspase-3, and cyt-C ↓ |
[151] |
| Primary culture | Aβ25–35 (25 μM for 24 h) | 0.1–10 μM for 24 h | – | – | Cell viability ↑ Bcl2 and MMP ↑ ROS, Bax, and caspase-3 ↓ |
[152] |
| Primary culture | Aβ25–35 (10 μM for 24 h) | 0.1–5 μM for 24 h | – | – | Mitochondrial complex I, II, III, and IV activity and ATP content ↑ Mito Tfam levels and Mito-cyt-C ↑ mtDNA copy numbers ↑ mtDNA transcript levels ↑ 8-OHdG and cytosolic-C ↓ Intracellular ROS level ↓ Mitochondrial ROS level ↓ |
[153] |
| Lycopene-in vivo studies | ||||||
| Sprague-Dawley rats | Aβ₁₋₄₂ (3 μl for one day) | 2.5 or 5 mg/kg bw for 3 weeks | MWM | Improvement in spatial learning performance and alteration in locomotor activity | Mitochondrial complex I, II, III, and IV activity and BDNF ↑ CAT, SOD, and GSH ↑ NO, MDA, and LPO levels ↓ AChE, TNF-α, and IL-6 ↓ Caspase-3 activity ↓ |
[154] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (10 μl for one day) | 1, 2, and 4 mg/kg bw for 3 weeks | MWM, elevated plus maze, and actophotometer test | Attenuation of poor latency performance and improvement in learning and memory performance | Mitochondrial complex I, II, III, and IV activity ↑ CAT, SOD, and GSH ↑ NO, MDA, and LPO levels ↓ AChE, TNF-α, TGF-β1, and IL-6 ↓ Caspase-3 activity ↓ |
[155] |
| APP/PS1 mice and WT C57/BL6J mice | – | 4 mg/kg bw | MWM | Amelioration of spatial learning and memory impairments | CD105, VEGF, and NeuN ↑ p-Akt, LXRα, and LXRβ ↑ Aβ aggregation, GFAP, and iNOS ↓ |
[156] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (3 μl for 1 day) | 5 mg/kg bw for 3 weeks | MWM | Improvement in spatial learning and cognitive function | TNF-α, IL-1β, and IL-6 ↓ TLR4 and p-NF-κB ↓ APP and PS1 ↓ |
[157] |
| C57BL/6J mice | LPS (0.25 mg/kg bw for 9 days) | 30 mg/kg bw for 5 weeks | MWM and Y-maze test | Prevention of losses in spatial learning and memory | IL10 and MMP3 ↑ GSH, CAT, and SOD ↑ APP and BACE1 ↓ iNOS, COX-2, IL-6, and MMP9 ↓ Aβ aggregation ↓ Iba-1 activation ↓ |
[150] |
| Albino Wistar rats | Fructose (10% for 16 weeks) | 4 mg/kg bw for 10 weeks | MWM | Recovery of impaired learning and memory | IR, IGF-1R, PI3K, and p-Akt ↑ SOD, CAT, GPx, and GSH ↑ PPARγ expression ↑ ROS, AChE, LPO, and PCC ↓ TNF-α, IL-1β, and p-NF-κB ↓ |
[158] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (10 μl for 14 days) | 5 mg/kg bw for 5 weeks | MWM | Mitigation of spatial memory and learning dysfunction | IL-10 and TGF-β1 levels ↑ Aβ accumulation ↓ TNF-α and IL-1β levels ↓ TLR4 and p-NF-κB ↓ |
[159] |
| Ferulic acid – in vitro studies | ||||||
| PC12 cells | LPS (1 μg/mL for 24 h) | 2.5, 5, 10, 20 and 40 μM for 24 h | – | – | CREB, p-CREB, and PDE4 ↑ TNF-α, IL-1β, and IL-6 ↓ |
[164] |
| SH-SY5Y cells | Aβ₁₋₄₂ (20 μl for 72 h) | 10–50 μM for 72 h | – | – | Aβ42 aggregation ↓ | [165] |
| SH-SY5Y cells | Aβ₁₋₄₂ (5 μl for 24 h) | 1–50 μM for 24 h | – | – | Cell viability ↑ Aβ42 aggregation ↓ Intracellular ROS formation ↓ Mitochondrial ROS formation ↓ Repair of membrane damage ↓ |
[166] |
| BV-2 cells | LPS (100 ng/mL for 24 h) | 2.5, 7.5, and 22.5 μM for 24 h | – | – | Cell viability ↑ PGE2, IL-1β, and NO ↓ iNOS, COX-2, and TLR4 ↓ |
[167] |
| BV-2 cells | LPS (1 μg/mL for 12 h) | 55 μM for 24 h | – | – |
p-AMPK, p-ULK1, and Beclin1 ↑ LC3, LAMP2A, and TFEB ↑ IL-6, IL-1β, TNF-α, and p-65 ↓ |
[168] |
| Ferulic acid – in vivo studies | ||||||
| ICR mice | Trimethyltin chloride (2.5 mg/kg bw for one day) | 20 or 50 mg/kg bw for 4 weeks | Y-maze and passive avoidance test | Attenuation of memory impairment and behavioral disabilities | ChAT activity ↑ | [169] |
| Male APP/PSI mice | – | 50 mg/kg bw for 4 months | MWM, Open-field test, NOR, and fear conditioning test | Recovery of impaired learning and memory | PSD95 and Syn1 ↑ APP and APPβ ↓ Neuronal loss ↓ BACE-1 and PS1 ↓ |
[170] |
| APP/PS1 transgenic mice and C57 mice | – | 20 mg/kg bw for 30 days | MWM | Reduction in spatial memory deficits | Cerebral blood flow ↑ Aβ plaque deposition ↓ APP, BACE1, and p-NF-κB ↓ |
[171] |
| PSAPP transgenic mice and WT mice | – | 30 mg/kg bw for 90 days | Y-maze, NOR, and radial arm water maze test | Recovery of episodic memory, spatial working memory, and spatial reference learning and memory deficits | Synaptophysin ↑ Oxidative stress ↓ Aβ deposits ↓ C99 and BACE1 ↓ IL-1β, TNF-α, Iba-1 and GFAP ↓ |
[172] |
| APP/PS1 mice | – | 5.3 or 16 mg/kg bw for 6 months | Y-maze test and NOR | Enhancement of behavioral performance and memory function | Aβ1–42 deposits ↓ Aβ1–40 deposits ↓ IL-1β levels ↓ |
[173] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (10 μl for 7 days) | 50, 100, and 250 mg/kg bw for 3 weeks | – | – | PKB and p-PKB ↑ IL-1β and GFAP activation ↓ p-p38 and p-ERK1/2 expression ↓ Caspase-3 and FasL expression ↓ |
[174] |
| Transgenic C. elegans | Aβ₁₋₄₂ over-expression | 1, 10, and 100 μM for 24–96 h (different assays at different time points) | – | – | HLH-30 activation ↑ Aβ monomers, oligomers, and deposits ↓ PolyQ40 aggregation ↓ |
[175] |
| Ellagic acid – in vitro studies | ||||||
| SH-SY5Y cells | Aβ₁₋₄₂ (10 μl for 12 h) | 100–300 μM for 24 h | – | – | Cell viability ↑ Morphology of Aβ42 aggregates ↓ Aβ oligomer levels ↓ |
[179] |
| PC-12 cells | H2O2 (40 μM) and Aβ25-35 (0.01 μM) for 24 h | 0.5–5.0 μg/ml for 24 h | – | – | Cell viability ↑ Intracellular ROS ↓ calcium ion influx ↓ |
[180] |
| SH-SY5Y cells | d-galactose (200 mM for 24 h) | 0.01–10 μM for 24 h | – | – | Cell viability ↑ GSH content ↑ MDA and ROS formation ↓ β-GAL, AGEs, and TNF-α level ↓ |
[181] |
| BV-2 cells | LPS (1 μg/mL for 24 h) | 25, 50, and 100 μM for 24 h | – | – | p-p38 and p-ERK1/2 expression ↓ TNF-α and NO activity ↓ |
[182] |
| Ellagic acid – in vivo studies | ||||||
| APP/PS1 double-transgenic and WT-C57BL/6 mice | – | 50 mg/kg bw for 2 months | MWM | Improvement in learning and memory performance | Aβ₁₋₄₂ deposits ↓ BACE1, APP and pThr668-APP ↓ pSer199-tau and pSer396-tau ↓ pTyr216-GSK3β activation ↓ Percentage of apoptotic cells ↓ Caspase-3 expression ↓ |
[183] |
| Sprague-Dawley rats | STZ (3 mg/kg for 1 day) | 25 and 50 mg/kg bw for 5 weeks | Y-maze and radial arm maze test | Improvement in memory score and cognitive performance | CAT and GSH content ↑ Synaptophysin ↑ MDA, CRP, and AChE activity ↓ GFAP and Aβ-plaques ↓ |
[184] |
| Sprague-Dawley rats | AlCl3 (50 mg/kg for 4 weeks) | 50 mg/kg bw for 4 weeks | NOR | Improvement in episodic memory | SOD and GSH content ↑ TBARS activity ↓ Neuronal loss ↓ NFTs and NPs ↓ APP and caspase-3 expression ↓ |
[185] |
| Sprague-Dawley rats | Aβ₁₋₄₂ (10 μg/kg for one day) | 10, 50, and 100 mg/kg bw for 7 days | Y-maze, radial arm maze test, NOR, and passive avoidance test | Prevention of learning and memory deficits | CAT, GSH, and Nrf2 ↑ MDA, NO, AChE, and BChE ↓ TLR4 and NF-κB ↓ CA1 neuronal loss ↓ |
[186] |
| Sprague-Dawley rats | Scopolamine (2 mg/kg for 1 day) | 25, 50, and 100 mg/kg bw for 3 weeks | MWM and passive avoidance test | Attenuation of learning and memory deficits | CAT and SOD ↑ MDA content ↓ |
[187] |
| Caffeic acid – in vitro studies | ||||||
| PC12 cells | Aβ1–42 (10 μl for 24 h) | 10 or 20 μg/mL for 24 h | – | – | Cell viability ↑ Calcium levels ↓ AT8 and phosphorylation of tau ↓ Phosphorylation of GSK-3β ↓ |
[194] |
| HT22 cells | Acrolein (25 μl for 24 h) | 5–50 μM for 24 h | – | – | Cell viability ↑ GSH levels and MMP ↑ ADAM-10, p-Akt, and LR-11 ↑ p-Erk and p-GSK-3β ↑ Intracellular ROS production ↓ p-p38 and p-JNK1 ↓ RAGE and BACE1 ↓ |
[195] |
| Caffeic acid – in vivo studies | ||||||
| Sprague-Dawley rats | HFD for 30 weeks | 200 mg/kg bw for 12 weeks | MWM | Amelioration of learning and memory impairments | SOD and CAT levels ↑ p-GSK-3β and synaptophysin ↑ GSSG activity ↓ APP and BACE1 ↓ Aβ₁₋₄₂, PSD95, and drebrin ↓ p-tau (Ser 396), p-tau (Ser 404), and p-tau (Thr 181) ↓ |
[196] |
| Sprague-Dawley rats | Aβ1–40 (5 μl for 1 day) | 100 mg/kg bw for 2 weeks | MWM | Prevention of learning deficits and increased cognitive function | GSH and CAT levels ↑ AChE activity and NO level ↓ IL-6 and TNF-α activities ↓ p53, p-p38, and p-NF-κB-p65 ↓ Caspase 3 protein expression ↓ |
[197] |
| Sprague-Dawley rats | ICV-STZ (3 mg/kg for 1 and 3 days) | 10, 20, and 40 mg/kg bw for 3 weeks | MWM, object recognition test, and spontaneous locomotor activity | Improvement in discriminating ability and alleviation of spatial learning and memory deficits | GSH levels ↑ AChE activity and NO level ↓ MDA content and protein carbonyl ↓ |
[198] |
| C57BL/6 mice | Aβ1–40 (5 μl for one day) | 50 mg/kg bw for 2 weeks | MWM and Y-maze | Improvement in spatial working memory and cognitive performance | Nrf2 and HO-1 expression ↑ p-PI3K and BDNF expression ↑ SYN, SNAP-23, and SNAP-23 ↑ PSD-95 expression ↑ BACE1 and Aβ₁₋₄₂ deposits ↓ GFAP and Iba-1 expression ↓ IL-1β, TNF-α, and p-NF-κB-p65 ↓ |
[199] |
| ApoE−/− mice and WT-C57BL/6J | – | 20 mg/kg bw for 8 weeks | MWM and Y-maze | Amelioration of cognitive decline and spatial memory impairment | ABCA1 and ABCG1 ↑ Aβ accumulation ↓ TNF-α, IL-6, and MCP-1 ↓ |
[200] |
| Gallic acid – in vitro studies | ||||||
| Neuro-2A cells and BV-2 cells | Aβ1–40 (5 μl for 24 h) | 5, 25, and 50 μM for 24 h | – | – | Cell viability ↑ IL-1β and TNF-α expression ↓ COX-2 and iNOS expression ↓ p-NF-κB-p65 expression ↓ |
[204] |
| Gallic acid – in vivo studies | ||||||
| ICR mice | Aβ1–40 (5 μl for 7 days) | 10 or 30 mg/kg bw for 4 weeks | Y-maze and passive avoidance test | Restoration of memory deficits and cognitive function | IL-1β and TNF-α expression ↓ COX-2 and iNOS expression ↓ p-NF-κB-p65 expression ↓ |
[204] |
| APP/PS1 and WT-C57BL/6 N mice | – | 30 mg/kg bw for 4 weeks | MWM, NOR, and Y-maze | Mitigation of spatial learning and reference memory deficits | PSD-95 expression ↑ GFAP and Iba-1 expression ↓ Aβ accumulation ↓ |
[205] |
| APP/PS1 mice | – | 20 mg/kg bw for 6 months | Y-maze, radial arm maze test, and NOR | Attenuation of spatial learning and working memory impairment and reversal of episodic memory impairment | APP, C99, and BACE1 ↓ GFAP and Iba-1 expression ↓ Aβ deposition |
[206] |
| Sprague-Dawley rats | AlCl3 (200 mg/kg bw for 60 days) | 100 or 200 mg/kg bw for 8 weeks | Y-maze and MWM | Improvement in learning and memory status | SOD, CAT, and GSH content ↑ Na+, Ca2+, and Zn2+ ↑ Dopamine and serotonin ↑ Norepinephrine ↓ K+ and CU2+ ↓ H2O2, MDA, and NO levels ↓ NFTs and NPs ↓ |
[207] |
| Sprague-Dawley rats | ICV-STZ (3 mg/kg bw for 1 and 3 days) | 100 or 200 mg/kg bw for 26 days | MWM and passive avoidance test | Mitigation of spatial learning and memory impairment | SOD, CAT, and GPx content ↑ MDA levels ↓ |
[208] |
| Sprague-Dawley rats | Sodium arsenite (2.5 mg/kg for 28 days) | 50 or 100 mg/kg bw for 4 weeks | MWM, elevated plus maze, forced swim test, and light-dark activity box test | Attenuation of anxiety-like behavior and improvement in learning and memory performance | SOD, CAT, and GPx content ↑ AChE activity and MDA levels ↓ |
[209] |
| Sprague-Dawley rats | Aβ1–40 (1 μg/1 μL for 1 day) | 50, 100, and 200 mg/kg bw for 10 days | – | – | Aβ accumulation ↓ | [210] |
| Epigallocatechin-3-gallate – in vitro studies | ||||||
| BV2-cells | Aβ25₋35 (25 μM for 24 h) | 2, 5, and 10 μM for 24 h | – | – | Cell viability ↑ Bcl2 and GSH levels ↑ MDA and ROS production ↓ iNOS and NO production ↓ Bax expression ↓ |
[218] |
| U373MG cells | Aβ25–35 (30 μM for 24 h) and IL-1β (10 ng/mL for 24 h) | 0.2–200 μM for 24 h | – | – | Cell viability ↑ MKP-1 expression ↑ IL-6, IL-8, and VEGF ↓ PGE2, COX-2, and p-NF-κB-p65 ↓ p-p38 and p-JNK ↓ |
[219] |
| BV2-cells | LPS (1 μg/mL for 1 h) | 0–350 μM for 24 h | – | – | Cell viability ↑ IL-6, TNF-α, and NO production ↓ MIF, CCL2, and TNFS10 ↓ STAT1, TRAF2, and TRAF3 ↓ TRAF5 and NFKΒ2 ↓ AKT3, mTOR, and NF-κB2 |
[220] |
| SH-SY5Y cell and BV-2 cells | Aβ1–40 (10 μM for 6 h) and LPS (1 μg/mL for 1 h) | 10 μM for 24 h | – | – | Cell viability ↑ IL-1β, IL-18, and Iba-1 expression ↓ Aβ accumulation ↓ Caspase-1 and caspase-1 p20 ↓ Caspase-11 p26 and NLRP3 ↓ p-NF-κB-p65, p-I κB-α, and TLR4 ↓ |
[221] |
| Epigallocatechin-3-gallate – in vivo studies | ||||||
| APP/PS1 mice and WT mice | – | 2 mg/kg bw for 28 days | – | – | IL-1β, IL-18, and Iba-1 expression ↓ Aβ accumulation ↓ Caspase-1 and Caspase-1 p20 ↓ Caspase-11 p26 and NLRP3 ↓ p-NF-κB-p65, p-I κB-α, and TLR4 ↓ |
[221] |
| APP/PS1 mice and WT mice | – | 50 mg/kg bw for 8 weeks | MWM and NOR | Alleviation of memory and cognitive deficits | Synapsin-1 and synaptophysin ↑ Synaptotagmin and PSD93 ↑ PSD95, MAP-2, and GluR1 ↑ IL-4, IL-10, and IL-13 ↑ Aβ accumulation ↓ Tau phosphorylation ↓ Iba-1, IL-6, TNF-α, and IL-1β ↓ |
[222] |
| Sprague-Dawley rats | Aβ1–40 (1 nM/μL for 1 day) | 100, 250, and 625 mg/kg bw for 8 weeks | MWM | Improvement in learning and memory function | Ach, GPx and SOD content ↑ Tau aggregation ↓ Tau phosphorylation and BACE1 ↓ Aβ1–42 expression ↓ MDA and AChE ↓ |
[223] |
| ICR mice | LPS (0.250 mg/kg bw for 7 days) | 15 or 3 mg/kg bw for 4 weeks | MWM | Amelioration of memory impairment and cognitive dysfunction | Aβ accumulation ↓ APP, C99, and BACE1 ↓ iNOS and COX-2 expression ↓ IL-1β, TNF-α, and IL-16 ↓ GFAP and caspase-3 expression ↓ |
[224] |
| APP/PS1 mice and WT mice | HFD for 12 weeks | 40 mg/kg bw for 12 weeks | MWM and NOR | Improvement in spatial learning and memory function | ADAM10, p-CDK5, and pAKT ↑ GSK3β and pGSK3β, and p-CREB ↑ APP and BACE1 ↓ GFAP, ATF4, and CHOP ↓ p-JNK, p-ERK, and TRL4 ↓ IRE1/p-IRE1 and ATF6 ↓ |
[225] |
| C57BL/6 mice | HFD for 8 weeks | 5, 15, and 45 mg/kg bw for 8 weeks | Barnes maze and NOR | Improvement in cognitive function and amelioration of cognitive decline | Prevotella ↑ Gut function and diversity ↑ Bifidobacteriales ↓ |
[226] |
| Theaflavins – in vivo studies | ||||||
| ICR mice | d-galactose (120 mg/kg bw for 56 days) | 15, 30, and 60 mg/kg bw for 8 weeks | Y-maze and NOR | Alleviation of memory and cognitive deficits |
Ach and ChAT activity ↑ SOD, GSH, GS, and GLS ↑ Nrf2, Prx2, GSH-px1, and SOD 1 ↑ MDA, GLU, and AChE activity ↓ |
[231] |
| C57BL/6J mice | LPS (10 μg for 1 h) | 10 or 50 mg/kg bw for 3 days | Y-maze and tail suspension test | Attenuation of concomitant cognitive impairment and Alzheimer-like behavior |
Dendritic spine density ↑ MIP-1α- and TNF-α ↓ |
[232] |
| Vanillin – in vitro studies | ||||||
| PC12 cells | Aβ1–40 (20 μM for 12 h) | 0, 10, 30, and 100 μM for 24 h | – | – | Cell viability ↑ SOD, CAT, and GSH ↑ MMP and Bcl2 ↑ LDH leakage ↓ MDA and ROS production ↓ Caspase-3, Bax, and apoptosis ↓ |
[237] |
| BV2-cells | LPS (200 ng/mL for 24 h) | 0–400 μM for 24 h | – | – | Cell viability ↑ iNOS and COX-2 expression ↓ IL-1β, TNF-α, and IL-16 ↓ p-p38, p-JNK, and p-ERK ↓ p-NF-κB-p65, and p-I κB-α ↓ |
[238] |
| Vanillin-in vivo studies | ||||||
| Swiss Albino mice | Scopolamine (2 mg/kg bw for 10 days) | 10 or 20 mg/kg bw for 10 days | Passive avoidance, elevated plus maze, and MWM | Reversal of memory and behavioral deficits |
GSH and CAT ↑ AChE and TBARS ↓ IL-6 and TNF-α ↓ |
[239] |
| Swiss Albino mice | AlCl3 (5 mg/kg for 90 days) and d-galactose (60 mg/kg bw for 90 days) | 40 mg/kg bw for 3 months | NOR, elevated plus maze, and MWM | Mitigation of memory and behavioral deficits |
GSH and CAT ↑ BDNF and neuron density ↑ AChE and TBARS ↓ BACE1 and caspase-3 ↓ Aβ accumulation ↓ |
[240] |
| ICR mice | Scopolamine (1 mg/kg bw for 28 days) | 40 mg/kg bw for 4 weeks | Passive avoidance and MWM | Attenuation of spatial learning impairment and cognitive dysfunction | ID-1 protein expression ↑ ID-1-immunoreactive cells ↑ NeuN-immunoreactive neurons ↑ DCX-immunoreactive cells ↑ Ki-76-immunoreactive cells ↑ |
[241,242] |
5. Neuroprotective mechanisms of dietary small molecules analogs in AD
Recent in vitro and in silico studies reported the neuroprotective effects of the chrysin derivative 4-oxo-2-phenyl-4H-chromene-5,7-diyl bis(piperidine-1-carboxylate). The in vitro study demonstrated that the derivative had good anti-cholinesterase and antioxidant activity, appropriate BBB penetration, some inhibitory activity against Aβ aggregation, as well as high selective inhibition of butyrylcholinesterase (BuChE) and AChE activity. In addition, in silico results showed that 4-oxo-2-phenyl-4H-chromene-5,7-diyl bis(piperidine-1-carboxylate) had high hydrophobicity and was predicted to have good BBB penetration and oral bioavailability [243]. Analogs of 7,8-DHF, including 7-O-β-d-glucosyl-8-hydroxyflavone, 7-hydroxy-8-O-β-d-glucosyl flavone, and 7,8-di-O-β-d-glucosylflavone, showed anti-AD effects through interactions with amino acids of the human APP active site and suppressing the BACE1 enzyme in an in silico study [244]. Another in vitro study examined the naringenin derivatives compounds 3c and 5a in H2O2-induced HT22 cells. Interestingly, 3c and 5a could cross the BBB, considerably inhibited self-induced Aβ1-42 aggregation, activated the ubiquitin-proteasome system degradation pathway, and cleared the aggregated tau and APP proteins in HT22 cells. The same study also revealed that compound 3c showed good anti-neuroinflammatory effects by inhibiting the release of NO and suppressing TNF-α production in LPS-induced BV-2 microglial cells [245]. Table 2 summarizes the latest preclinical research findings on the neuroprotective efficacy of dietary small molecules bioactive compounds against AD.
Table 2.
Summarizes the latest preclinical research findings on the effect of dietary small molecules bioactive compounds against AD.
| Dietary small molecules | Bioactive constituents | BBB crossing ability | Type of study | Observed effects | Reference |
|---|---|---|---|---|---|
| Chrysin | 4-oxo-2-phenyl-4H-chromene-5,7-diyl bis(piperidine-1-carboxylate) | Able | In vitro and in silico | Oral bioavailability ↑ Antioxidant activity ↑ AChE and BuChE ↓ Aβ aggregation ↓ |
[243] |
| 7,8-DHF | 7-O-β-d-glucosyl-8-hydroxyflavone, 7-hydroxy-8-O-β-d-glucosyl flavone, and 7,8-di-O-β-d-glucosylflavone | Not able | In vitro and in silico | APP and BACE inhibition ↓ | [244] |
| Naringenin | Compound 3c and 5a | Able | In vitro and in silico | Cell viability ↑ Antioxidant activity ↑ AChE and BuChE ↓ Aβ aggregation ↓ Tau phosphorylation ↓ NO and TNF-α ↓ |
[245] |
| Naringenin | Compounds 5f and 7k | Able | In vitro, in vivo, and in silico | Cell viability ↑ Antioxidant activity ↑ AChE and BuChE ↓ Aβ aggregation ↓ NO and TNF-α ↓ Cognitive function ↑ |
[246] |
| Naringenin | 5-hydroxy-2-(4-hydroxyphenyl)-4-oxochroman-7-yl piperidine-1-carboxylate | Able | In vitro and in silico | Oral bioavailability ↑ Antioxidant activity ↑ AChE and BuChE ↓ Aβ aggregation ↓ |
[247] |
| FA | Compounds 4g and 7f | Able | In vitro, in vivo, and in silico | Cell viability ↑ Antioxidant activity ↑ AChE and BuChE ↓ Aβ aggregation ↓ Cognitive function ↑ |
[248,249] |
| FA | TM-10 | Able | In vitro, in vivo, and in silico | Cell viability ↑ Antioxidant activity ↑ Autophagy activation ↑ AChE and BuChE ↓ MAO-A and MAO-B ↓ Acute toxicity ↓ Aβ aggregation ↓ Cognitive function ↑ |
[250] |
| FA | Compounds 6j and 6k | Able | In vitro, in vivo, and in silico | Cell viability ↑ Antioxidant activity ↑ MDA, AChE, and BuChE ↓ BACE-1 activity ↓ Acute toxicity ↓ Cognitive function ↑ Oral bioavailability ↑ |
[251] |
| FA | Compounds FA-G1 and FA-DG1 | Not able | In vitro and in vivo | Cell viability ↑ Aβ aggregation ↓ NO production ↓ iNOS and p-NF-κB-p65 ↓ Neuronal cell death ↓ |
[252,253] |
| CA | CAPE | Not able | In vitro and in vivo | NF200 and NeuN ↑ BDNF, nestin, and NeuN ↑ Neuromuscular activity ↑ Cognitive function ↑ PSD95, MAP-2, and GFAP ↓ |
[254] |
| CA | CAPE | Not able | In vitro and in vivo | Memory function ↑ Neuronal density ↑ Nrf2 and HO-1 ↑ GSK3β and p-GSK3β Oxidative stress ↓ GFAP and Iba-1 expression Caspase-9 expression ↓ |
[255] |
| CA | FA-97 | Not able | In vitro and in vivo | Cell viability ↑ GSH and SOD activity ↑ Nrf2, HO-1, and NQO-1 ↑ Neuronal density ↑ Bcl2 expression ↑ LDH leakage ↓ MDA and ROS production ↓ Bax, caspase-9, and cyt-C ↓ Memory function ↑ |
[256] |
| Vanillin | Compounds 9e, 9h, 9 l, 9t and 9z | Not able | In vitro and in silico | Cell viability ↑ AChE, BChE, and DPPH ↓ Aβ aggregation ↓ Neuronal cell death ↓ |
[257] |
Yang et al. investigated the neuroprotective and anti-inflammatory properties of naringenin derivatives in H2O2-induced PC12 and LPS-induced BV-2 microglial cells. They found that two bioactive compounds, 5f and 7k (naringenin-O-carbamate derivatives), were primarily responsible for naringenin's neuroprotective and anti-inflammatory activity by inhibiting NO release and suppressing TNF-α production in LPS-induced BV-2 microglial cells. Moreover, 5f and 7k could cross the BBB, significantly inhibited self-induced Aβ1-42 aggregation and huAChE-Aβ1–40 aggregation as well as selective metal chelators, and remarkably inhibited Cu2+-induced Aβ1–42 aggregation. In addition, the in vivo study reported that compound 5f significantly improved spatial learning and memory in scopolamine-induced AD mice [246]. Naringenin analog 5-hydroxy-2-(4-hydroxyphenyl)-4-oxochroman-7-yl piperidine-1-carboxylate, showed anti-AD effects by suppressing BuChE and AChE activity and scavenging OH and displayed better inhibitory potency on self-induced Aβ1-42 aggregation. In addition, a pharmacokinetics study demonstrated that 5-hydroxy-2-(4-hydroxyphenyl)-4-oxochroman-7-yl piperidine-1-carboxylate can cross the BBB and has improved oral bioavailability [247].
In previous studies, the FA derivative compounds 4g and 7f had neuroprotective effects on H2O2-induced and Aβ-induced PC12 cell damage and could cross the BBB. It has also been demonstrated that they have considerable ability to inhibit hAChE and hBuChE, good Aβ aggregation inhibition, and good antioxidant activity. Furthermore, compound 7f significantly reversed scopolamine-induced memory deficits in AD mice [248,249]. The FA analog TM-10 displayed neuroprotective and cognitive effects in Aβ1-42-induced SH-SY5Y cells, AlCl3-induced zebrafish, and scopolamine-induced AD mice. In vitro results showed that TM-10 treatment had good penetration of the BBB, inhibited BuChE activity, and remarkably inhibited monoamine oxidases A and B, as well as inhibiting self-induced Aβ aggregation; TM-10 subsequently exhibited potent antioxidant activity and had autophagic activator activity in Aβ1-42-induced SH-SY5Y cells. In addition, TM-10 exhibited a favorable dyskinesia recovery rate and response efficiency in an AlCl3-induced zebrafish AD model. A further in vivo study demonstrated that TM-10 had low acute toxicity, and the step-down passive avoidance test indicated that this compound improved spatial learning and memory function in scopolamine-induced AD mice [250].
Recent in vitro and in vivo findings suggested that two bioactive FA analogs—methoxy-4-(2-(5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)vinyl)phenol (6j) and 2-methoxy-4-(2-(5-(4-(trifluoromethoxy)phenyl)-1,3,4-oxadiazol-2-yl)vinyl)phenol (6k)—have potent neuroprotective and memory-enhancing effects in Aβ-induced SH-SY5Y cells and in a Wister rat model. An in vitro study showed that compounds 6j and 6k had excellent BBB permeability and that they inhibited AChE, BChE, and BACE-1 activity and self-induced Aβ aggregation in SH-SY5Y cells. Moreover, the in vivo study showed that compounds 6j and 6k induced brain AChE inhibition, enhanced antioxidant activity, and improved learning and memory behavior in the Aβ-induced AD rat model [251]. Recent in vitro and in vivo studies have shown that the water-soluble FA analogs 1-feruloyl glycerol (FA-G1) and 1-feruloyl diglycerol (FA-DG1) inhibit NO production, suppress iNOS expression, and inhibit the NF-κB pathway in a concentration-dependent manner. In addition, treatment with these analogs inhibited Aβ-induced neuronal cell death and Aβ aggregation, destabilized pre-aggregated Aβ to a similar extent, and subsequently protected hippocampal neurons [252,253].
Konar et al. reported that CA phenethyl ester (CAPE) had potent neuroprotective effects in a Drosophila AD model and a scopolamine-induced model of amnesia. Their results showed that CAPE treatment increased levels of the neurotrophin BDNF, the neural progenitor marker nestin, and the differentiation marker NeuN in the cerebral cortex and hippocampus [254]. Similarly, intraperitoneal administration of CAPE prevented oxidative stress and was accompanied by the induction of Nrf2 and HO-1 via the modulation of GSK3β in the hippocampus. Additionally, CAPE treatment in Aβ-induced AD mice decreased Aβ-induced neuronal apoptosis and neuroinflammation, and improved spatial learning and memory, protecting the mice against cognitive deficits [255]. Another study reported that CAPE-4-O-glucoside (FA-97) attenuated oxidative stress by inhibiting ROS, MDA, and PCOation levels, as well as inducing cellular GSH and SOD and apoptosis in H2O2-induced SH-SY5Y and PC12 cells. Mechanistically, FA-97 promotes the nuclear translocation and transcriptional activity of Nrf2 that is associated with upregulated expression of HO-1 and NQO-1. In addition, FA-97 prevents scopolamine-induced learning and memory impairments by reducing oxidative stress and neuronal apoptosis, protecting against cholinergic system dysfunction, and increasing the expression of HO-1, NQO-1, and nuclear Nrf2 in the hippocampus and cortex of AD mice [256]. Yelamanda Rao et al. [257] observed that the five acrylamide analogs 9e, 9h, 9 l, 9t, and 9z from vanillin protected against H2O2-induced SK-N-SH cell death. The results showed that these analogs had excellent neuroprotective effects, inhibited AChE and BChE activity, and had good radical-scavenging activity. In addition, acrylamide analogs were strong modulators of Aβ aggregation; they irretrievably disrupted Aβ42 fibril morphology, decreased the aggregates, and reduced Aβ-induced toxicity in neurons.
6. Potential of dietary small molecule-based nanotherapy in AD
Nanocarriers can increase a drug's bioavailability and lipophilicity, which can improve its therapeutic effect. Drug stability has been another target for nanocarriers; they were designed to improve encapsulation efficiency and allow for more precise control over drug release, which is essential for treating a variety of diseases [258,259]. Therefore, nanotechnology has the potential to improve the therapeutic efficacy of medicines by producing the desired pharmacological response in humans. Several nanodelivery systems with dietary small molecule payloads have demonstrated effectiveness in modulating oxidative stress, Aβ deposition, and hyperphosphorylation of tau, chronic neuroinflammation, and various signaling pathways that mediate aging-associated NDDs, particularly in AD (Fig. 8).
Fig. 8.

Illustrates the potential uses of dietary small molecules nanoformulations in AD. Dietary small molecules lower BBB permeability, poor solubility, and low bioavailability, they have failed to reduce disease progression and treat AD. The development of dietary small molecules-loaded nanocarrier systems can reduce these barriers while improving neuroprotective benefits. BBB: blood brain barrier.
In aged female mice with Aβ1–42-induced AD, treatment with chrysin-loaded lipid-core nanocapsules (LNCs) (1 and 5 mg/kg) for 2 weeks provided gradual release and increased bioavailability as well as greater accumulation in brain tissues compared with free chrysin. Furthermore, LNC administration improved spatial learning and memory, decreased non-protein thiols levels (NPSH) and reactive species (RS), and increased GR, GPx, GST, and CAT activity in the prefrontal cortex and hippocampus. Treatment also upregulated BDNF expression, reduced proinflammatory cytokines such as TNF-α, IL-1β and increased expression of the anti-inflammatory cytokine IL-10 in the prefrontal cortex and hippocampus of the mice [260]. Similarly, chrysin-loaded chitosan nanoparticles (Chr-Chi NPs) were evaluated for neuroprotection activity in an Aβ-induced zebrafish AD model. The results demonstrated that Chr-Chi NP treatment effectively improved memory, cognition, and synaptic connections and reduced Aβ aggregates, thus decreasing neuronal cell death and the generation of ROS [261]. Another study reported that the administration of chrysin in an oil-in-water nanoemulsion (chrysin-NE) was easy to digest, was significantly absorbed by human intestinal cells (Caco-2), and enhanced the bioavailability of chrysin [262]. Recently, chrysin-loaded solid lipid nanoparticles (CN-SLNPs) showed enhanced oral bioavailability and increased the encapsulating capacity of chrysin in SLNs in Aβ-induced AD rats at low concentrations. In addition, CN-SLNP treatment significantly increased all the antioxidant and non-antioxidant enzymes while significantly reducing TBARS and AChE activity in the hippocampus of Aβ-induced AD rats. Furthermore, histopathological evidence and behavioral analysis showed that CN-SLNPs restored hippocampal neuronal loss and prevented memory impairments in Aβ-induced AD rats [263]. Table 3 summarizes the latest preclinical research findings on the neuroprotective effect of nano-formulated dietary small molecules against AD.
Table 3.
Summarizes the latest preclinical research findings on the effect of nano-formulated dietary small molecules against AD.
| Dietary small molecules | Limitation | Nanocarrier | Nanocarrier size (nm) | Neurotoxin/duration | Drug concentration/duration | Mechanism of action | Reference |
|---|---|---|---|---|---|---|---|
| Chrysin | Poor oral bioavailability | Lipid-core nanocapsule | 368–419 | Aβ1–40 (3 μL for 1 day) | 1 or 5 mg/kg bw for 14 days | Oral bioavailability ↑ Memory function ↑ NPSH, GSH, SOD, and CAT ↑ BDNF and IL-10 ↑ IL-1β and TNF-α ↓ |
[260] |
| Chrysin | Chitosan | Aβ1–40 (3 μL for 1 day) | [261] | ||||
| Chrysin | Low bioaccessibility | Nanoemulsion | 161 | – | – | Bioaccessibility ↑ Cell viability ↑ Antioxidant activity ↑ AChE and BChE activity ↓ |
[262] |
| Chrysin | Poor pharmacokinetic profile | Solid lipid nanoparticles | 240.0 ± 4.79 | Aβ1–40 (3 μg for 1 day) | 50 mg/kg bw for 21 days | Cognitive performance ↑ GSH, SOD, CAT, and GPx ↑ Vitamin-C ↑ TBARS and AChE ↓ Neuronal cell loss ↓ GFAP-positive astrocyte ↓ |
[263] |
| Naringenin | Low aqueous solubility and low bioavailability | Nanoemulsion | 113.83 ± 3.35 | Aβ1–40 (2 μM for 84 h) | 0.125, 0.250, 0.5 μM for 24 h | Solubility ↑ Cell viability ↑ ROS production ↓ APP and BACE1 ↓ Aβ level, tau, and pT231 ↓ |
[264] |
| Luteolin | Low solubility and low permeability | Chitosan | 412.8 ± 3.28 | ICV-STZ (3 mg/kg for 1 day) | 50 mg/kg bw for 21 days | Cognitive performance ↑ Spatial learning and memory ↑ GSH, Nrf2, and p-CREB ↑ Neuronal survival ↑ MDA, iNOS, and COX-2 ↓ MMP9, TNF-α, and NF-κB-p65 ↓ Aβ level and Tau ↓ Neuronal damage ↓ Aβ-plaques formation ↓ GFAP-positive astrocyte ↓ |
[265] |
| Luteolin | Low solubility and low permeability | Nanobilosomes | 153.2 ± 0.98 | ICV-STZ (3 mg/kg for 1 day) | 50 mg/kg bw for 21 days | Cognitive performance ↑ Spatial learning and memory ↑ Neuronal survival ↑ MMP9 and TNF-α ↓ Aβ level and Tau ↓ Neuronal damage ↓ Aβ-plaques formation ↓ GFAP-positive astrocyte ↓ Iba-1-positive astrocyte ↓ |
[266] |
| Lycopene | Poor oral bioavailability | Microemulsion | <100 | Aβ1–40 (10 μM for 1 day) | 4 mg/kg bw for 21 days | Cognitive performance ↑ Spatial learning and memory ↑ Reelin, nestin, and Pax6 ↑ BrdU+, Dcx+, and BrdU+ ↑ Neun+, BrdU+, and Dcx+ ↑ p-GSK-3β and p-β-Catenin ↑ |
[267] |
| Lycopene | Microemulsion | Aβ1–40 (10 μM for 1 day) | 4 mg/kg bw for 21 days | Memory function ↑ GSH, SOD, CAT, and GPx ↑ MDA levels ↓ |
[268] | ||
| FA | Poor aqueous solubility | Chitosan | 185 | ICV-STZ (3 mg/kg for 1 day) | 80 mg/kg bw for 28 days | Solubility ↑ Oral bioavailability ↑ Memory function ↑ GSH and SOD content ↑ MDA, NO, and AChE activity ↓ |
[269] |
| EA | Low absorption and low permeability | Poly (lactic-co-glycolic acid) | 277 | AlCl3 (50 mg/kg for 28 days) | 10 mg/kg bw for 14 days | Solubility ↑ Oral bioavailability ↑ Memory function ↑ Neuronal survival ↑ GSH and SOD content ↑ TBARS content ↓ Neuronal damage ↓ NFT and Aβ-plaque formation ↓ |
[270] |
| CA | Poor oral bioavailability and drug instability | Liposome | 140 | Aβ1–40 (10 μM for 1 h) | 0.8 μM for 2 h | Oral bioavailability ↑ Stability ↑ Aβ aggregation ↓ Aβ fibril formation ↓ |
[271] |
| EGCG | Instability and low bioavailability | Poly (lactic-co-glycolic acid) | 124.8 ± 5.2 | Transgenic model | 40 mg/kg bw for 90 days | Physicochemical stability ↑ Synaptic neurons ↑ Memory function ↑ Aβ-plaque and GFAP ↓ |
[272] |
| EGCG | Poor bioavailability and inefficient delivery | Nanolipids | <100 | – | 3.125–30 μM for 18 h and 100 mg/kg bw for 8 h | Oral bioavailability ↑ Stability ↑ α-secretase ↑ |
[273] |
Md et al. observed that naringenin-loaded nanoemulsion significantly alleviated the direct neurotoxic effects of Aβ on SH-SY5Y cells; this was associated with a downregulation of APP and BACE expression, indicating reduced amyloidogenesis, and decreased the levels of phosphorylated tau in Aβ-exposed SH-SY5Y cells [264].
Recent studies found that luteolin-loaded Chi and bilosome NPs provided obvious improvement in the acquisition of short- and long-term spatial memory, increased neuronal survival rate with a reduction in the number of Aβ-plaques, improved antioxidant enzyme status, and reduced pro-inflammatory mediator levels. In addition, luteolin-loaded Chi NPs suppressed both Aβ aggregation and hyperphosphorylated tau protein in the hippocampus and cerebral cortex in ICV-STZ-induced AD mice [265,266]. Interestingly, Ning et al. [267] demonstrated that in Aβ-induced AD rats, lycopene-loaded microemulsion (LME-NPs) improved spatial learning and memory, significantly increased reelin, nestin, and Pax6 gene expression, which regulate neurogenesis, as well as increased BrdU+, Dcx+, BrdU+/Neun+, and BrdU+/Dcx+ cells in the dentate gyrus of the hippocampus and subventricular regions, thus promoting neurogenesis. Treatment also reduced the number of Iba1+ and Iba1+/BrdU+ cells, thus reducing the neuroinflammatory response. In addition, LME-NPs upregulated the Wnt/β-catenin pathway by upregulating Wnt3a, β-catenin, Disheveled (Dvl), and p-GSK3β and downregulating p-β-catenin and GSK3β. More recently, LME-NP supplementation dramatically decreased MDA production and restored antioxidant capacity, alleviated neuronal loss, attenuated astrocytosis and microgliosis, normalized apoptotic parameters, inhibited amyloidogenic processing, and activated the non-amyloidogenic pathway, together with upregulating synaptic protein expression and restoring synaptic plasticity [268].
Another recent study demonstrated that FA-coated Chi-SLNPs successfully improved anti-AD efficacy, provided superior nasal mucoadhesion and permeation, and extended drug release. It has also been reported that FA-coated Chi-SLNPs improved cognitive ability in ICV-STZ-induced AD rats by reducing escape latency, significantly improving body weight gain and various other biochemical parameters; histopathological changes were also observed in the hippocampus and cerebral cortex [269]. Moreover, poly (lactic-co-glycolic acid)-EA-NPs significantly enhanced antioxidant activity and reduced brain neurotoxicity in rats with AlCl3-induced AD by reducing oxidative stress, reducing the AD-vacuolation of neurons, chromatolysis, NFTs, and senile plaques in the brain and restoring Nissl granules as well as modulating behavioral performance [270]. Transferrin-targeted CA-loaded liposome-NPs showed suitable encapsulation efficiency and physical stability for at least 2 months. They prevented Aβ aggregation, fibril formation, and disaggregation of mature fibrils [271]. Furthermore, dual-drug-loaded PEGylated PLGA NPs (EGCG/AA NPs) induced tight junction disruption and opened the BBB in vitro and ex vivo. In addition, EGCG/AA NP treatment produced a marked increase in synapses as judged by synaptophysin expression, reduced neuroinflammation, Aβ plaque formation, and cortical levels of soluble and insoluble Aβ(1-42) peptide; this subsequently enhanced spatial learning and memory in APP/PS1 AD mice [272]. Finally, Smith et al. found that nanolipid EGCG NPs improved oral bioavailability and enhanced neuronal α-secretase in SweAPP N2a cells [273].
7. Combination therapeutic effects of dietary small molecules in AD
Accumulating evidence has suggested that multidrug therapeutic targets associated with multifactorial molecular mechanisms could prove to be effective in treating AD. A recent study indicated that flavones such as apigenin, 7,8-DHF, quercetin, and wogonin were investigated for Tau aggregation inhibitory activity and neuroprotection in SH-SY5Y cells. Among the four tested flavones, 7,8-DHF, apigenin, and quercetin reduced oxidative stress, Tau aggregation, and caspase-1 activity by improving the expression of HSPB1 and Nrf2 and activated TRKB-mediated ERK signaling as well as upregulate CREB and its downstream antiapoptotic Bcl2 expression in SH-SY5Y cells expressing ΔK280 TauRD-DsRed folding reporter [94]. Tao et al. [274] found that luteolin (100 mg/kg bw) combined with exercise training significantly improved learning and memory, reversed the increase in Aβ, and inhibited the activation of astrocytes and microglia (as shown by GFAP and Iba-1 markers, respectively) in a mouse AD model induced by Aβ1-42 oligomers. This model also showed decreased levels of autophagy markers (p-ULK1 and p62) as well as increases in autophagy-related proteins LC3I and LC3II in the hippocampus and cortex following luteolin treatment. Notably, luteolin combined with exercise therapy led to more intact hippocampal and cortical areas, with a greater improvement than that seen with monotherapy. However, luteolin and l-theanine can act together in the anti-Alzheimer's effects by improving memory function, norepinephrine metabolisms, hippocampal insulin signaling, and reducing neuroinflammation in Aβ-induced AD rats [275]. In vitro and in vivo research findings reported that combined treatment with palmitoylethanolamide and luteolin (co-ultra PEALut) promoted neuronal survival, upregulated the protein and mRNA BDNF and GDNF expression, significantly reduced iNOS and GFAP expression, and reduced the neuronal apoptosis as well as neuroinflammation [140,276].
Interestingly, combination treatment with lycopene and vitamin E significantly improved spatial and passive memory, decreased MDA levels and increased GPx activity in the serum, and decreased tau phosphorylation at Thr231/Ser235, Ser262, and Ser396 in the brains of P301L transgenic mice [277]. Ohashi et al. reported that FA combination with curcumin treatment was confirmed to effectively inhibit Aβ aggregation, decrease mitochondrial and extracellular ROS levels by increasing the cell viability, and improve the mitochondrial function in Aβ exposure SH-SY5Y cells. Combination treatment produced an overall higher neuroprotective effect than treatment with FA or curcumin alone [166]. In another study, it was found that a combination treatment of FA, atractylenolide III, and paeoniflorin can exert neuroprotective and anti-inflammatory effects on LPS-induced autophagy and neuroinflammation in BV-2 microglial cells by significantly increasing the expression of autophagy-related proteins p-AMPK, Beclin1, p-ULK1, LC3, and TFEB and decreased the expression of p62 and significantly reduced the production of inflammatory factors such as IL-6, IL-1β, and TNF-α [168]. A similar study has also been reported that FA, curcumin, and phosphatidylserine exposed to APPswe/PS1dE9 transgenic mice and evaluated neuroprotective and cognitive function. The results showed that FA, curcumin, and phosphatidylserine treatment exhibited a significant improvement in memory function, BDNF expression, and acetylcholine level and strongly inhibited aggregation of Aβ-plague and phosphorylated tau formation and reduced the proinflammatory cytokine IL-1β [278].
Interestingly, nutraceutical bioactive products such as EGCG, curcumin, α-lipoic acid, and docosahexaenoic acid appeared to have the most potent anti-inflammatory and neuroprotective effects by suppressing the Aβ-induced BACE-1 upregulation, attenuated ROS production and beta-sheet structure formation; and strongly reduction of Aβ-plague aggregation and inhibition of microglial activation [279,280]. In hAbKI mice, combined treatment with EGCG and urolithin A ameliorated behavioral deficits by upregulating the levels of protein and mRNA expression of synaptic function, mitochondrial fusion, autophagy, and mitophagy function, increased dendritic spine length and downregulating the mitochondrial fission. In addition, EGCG combination with urolithin A strongly inhibited Aβ-plague aggregation, reduced mitochondrial fragmentation, and increased mitochondrial length and mitophagosomal formations [281]. A similar study reported that both EGCG and voluntary exercise were able to attenuate nest building and Barnes maze performance deficits and lowered soluble Aβ1-42 levels in the cortex and hippocampus [282]. Moreover, PEGylated-loaded EGCG/ascorbic acid nanoparticles resulted in a significantly enhanced spatial learning and memory and a marked increase in synaptophysin (SYP) expression, synapse function, and reduction of Aβ-plaque burden and cortical levels of soluble and insoluble Aβ(1-42) peptide as well as neuroinflammation in APP/PS1 AD mice [272]. Ali et al. [283] conducted a study in AlCl3-induced AD rats to evaluate the neuroprotective and inhibitory effects of Aβ-plague aggregation. The results showed that combination administration of EGCG, Coenzyme COQ10, Vitamin E and Selenium significant decrease in Aβ-plague aggregation and AChE activity, suppressed TNF-α, IL-1β expression, and increased the BDNF protein expression as well as antioxidant enzymes activities (MDA, SOD, TAC).
8. Clinical aspects and dietary small molecules
A 12-month, open-label, randomized controlled trial in 40 patients with MCI showed that physical exercise combined with lipophilic micronutrients including lutein and lycopene reduced cognitive impairment, suggesting a potential benefit from multifactorial lifestyle changes. Therefore, combinations of nutritional and physical interventions may have greater effects on cognition in individuals at risk for AD [284]. A randomized placebo-controlled trial in patients with MCI showed that citrus peel extract (0.1, 3, and 400 mg) containing naringenin and auraptene administered over 36 weeks reduced neuronal damage (BDNF, IGF-1, and NF-light), oxidative stress (NO, TBARS, and SOD), and cytokines (pro-inflammatory: CXCL8/IL-8, IL-1β, IL-6, IL-17, TNF-α, IP10, MIP-1α, CCL2/MCP-1, CCL5/RANTES, IL-18; anti-inflammatory: IL-10, IL1-Ra, soluble TNF receptor I, soluble TNF receptor II) [285]. In a study of the effects of EA treatment (50 mg/kg for 12 weeks) in patients with mild age-related cognitive decline, EA improved blood lipid metabolism, with declines in total cholesterol, triglycerides, and low-density lipoprotein, as well as increased high-density lipoprotein and plasma BDNF levels, and enhanced cognitive function [286]. A 12-month randomized controlled study conducted between September 2016 and August 2017 included 200 older adults with MCI and reported that a dietary supplement containing FA 200 mg/kg and A. archangelica extract 40 mg/kg showed clinical effectiveness for cognitive functioning [287].
9. Advantages of dietary small molecules and their bioactive constituents
Since ancient times, natural products (e.g., fruits, vegetables, and other medicinal plants) have been used to prevent and treat human diseases such as NDDs. Most modern medicines are derived from herbs, with many based on traditional knowledge and practices [288]. As reported by the WHO, approximately 80% of individuals in developing countries are currently using natural plant-based medicine as well as food products to meet their primary healthcare needs [289]. The science of natural small molecule drug discovery is currently among the most interesting areas of research. Over the last 20 years, research in this field has led to the filing of more than 200,000 patents and the completion of several dozen clinical trials [290]. Almost 25% of the major pharmaceutical compounds (and bioactive derivatives) available today are acquired from natural resources [291]. This is mainly because natural agents have unique advantages, such as reduced toxicity and minimal side effects, considerable chemical diversity, and low cost, as well as favorable chemical and pharmacological properties, including preventive effects on disease. While the value of dietary small molecules and their bioactive derivatives as replacements for pharmaceuticals has been questioned, studies have shown that they are at least as effective as commercially available pharmaceuticals or possibly more effective [292]. In addition, bioactive dietary components do not require extensive toxicity testing and rigorous human trials, which can significantly diminish their time to market [293].
Plant-based foods containing the most bioactive dietary components (cereals, vegetables, fruits, and other medicinal plant sources) have been investigated to assess their activity in the body, and functional foods have emerged as prospective preventive and therapeutic options [294]. Bioactive dietary small molecules have a range of metabolic activities that may be useful for treating illness. These chemicals are secondary metabolites in medicinal plants; they have antimicrobial, neuroprotective, antioxidative, and anti-inflammatory activity and can perform a variety of biological activities, including regulating the immune system; reducing platelet aggregation; regulating hormone, glucose, and lipid metabolism; controlling enzyme detoxification and digestion, and delaying aging [294,295]. However, the biological processes through which plant-based bioactive dietary small molecules promote health remain a mystery. However, these molecules operate in distinct or comparable locations. Bioactive dietary elements have been shown to lower the risk of developing NDDs and infectious diseases [296,297]. Individuals with risk factors for early death can benefit from regular intake of foods rich in polyphenols, flavonoids, and carotenoids since these bioactive dietary small molecules improve memory and cognitive function, decrease neuroinflammation and oxidative stress, and contribute to clearing aggregates of Aβ plaques and tangles by promoting neuronal growth and survival, and reducing AD-related manifestations [298,299]. Importantly, bioactive dietary small molecules can interact with gut microbes and host and/or modify the microbial metabolites that are available to the host. SCFAs promote digestive wellness, metabolic management, immune regulation, glucose and lipid composition, and microbiome antioxidant and neuroprotective activity [297,300]. In recent years, dietary small molecules have been promoted as dietary supplements for hypertension and obesity and as anti-aging preparations [301]. Interestingly, the future use of dietary small molecules and their bioactive compounds as anti-Alzheimer's as anti-obesity agents is the use of combination therapies. By combining dietary small molecules, herbal supplement, and other drug combinations that target different aspects of neuroprotective signaling pathways, it may be possible to achieve synergistic effects and improve overall therapeutic efficacy. This could lead to favors for targeted drug development that modulate early-stage mild cognitive impairment associated with AD, offering more precise and effective treatment options.
Natural dietary products can also be used in the food and cosmetics industries. Several dairy products with dietary supplements have already been commercialized [302]. The antioxidant potential of dietary small molecules is also of great interest in food packaging and storage because they can prevent food spoilage, with antioxidants used in a controlled atmosphere to limit oxygen availability [303]. Green tea (EGCG and TFs) is becoming a very popular beverage worldwide because of its health benefits, attributed to its high polyphenol content, especially the monomeric catechins. Enzymes have been used in tea processing mainly to improve the quality of tea beverages. Generally, cell-wall-digesting enzymes improved the extraction efficiency including polyphenols and thereby the antioxidant capacity of the extract. It can be seen that green tea consumption may protect cognitive function by reducing AD pathology and improving anti-oxidative stress capacity. Green tea extract can also be used in lipid-bearing foods to delay lipid oxidation and to enhance the shelf-life of various food products [211,212,228]. Dietary small molecules have become a modern food trend to add nutritional fortification to foods to increase the content of essential micro-nutrients and improve their nutritional quality. Cakes contain high carbohydrates and fats, while other nutrients, including proteins, minerals, and vitamins are fewer. Adding available, cheap, and nutritious edible plants to the cakes can enhance their health and nutritional safety. Many studies have shown that cake made with dietary small molecules powder is rich in protein, vitamins, and dietary fiber. It has high nutritional value and good taste [304]. Recently, dairy products-derived snacks mixed with other plant materials have received extensive attention. For example, sweet and salty biscuits rich in calcium and protein were prepared by mixing dietary supplements. Fermented snacks made from cassava flour, soybean flour, and horseradish leaf powder rich in Vitamin A provided a new strategy for improving the diet of people with nutritional deficiencies. Snacks are one of the fastest-growing segments of the food industry. However, providing nutrition through the inclusion of fruits and vegetables in puffed snacks is a relatively new concept. Importantly, dietary supplements can be easily blended with oats to create extruded puffed snacks with a significantly improved nutrient profile compared to that of commercially available snacks [301]. However, more attention should be paid to other phenolic compounds that are not regulated by current legislation and that have the potential to be used as food additives, such as phenolic acids and other classes of dietary products. Dietary small molecules could also be useful in cosmetics and could be used to prevent skin aging and cutaneous disorders since ROS are key drivers of aging [305]. In addition, their ability to prevent lipid peroxidation allows them to extend shelf-life, as they do for foods [306]. Dietary small molecules could also be used in cosmetic formulations such as body lotion, shampoo, hair restorer, hair color, and soaps [307]. Since dietary small molecules are safe for consumption, their use as dietary supplements in natural, green, and healthy foods may soon become a trend in the food and drug development industry.
10. Limitation and future directions
Indeed, dietary small molecules and their bioactive constituents are an important natural compound that has multiple health benefits. However, the use of dietary small molecules as targeted AD therapy has some limitations. The advances, challenges, and future perspectives that remain to be considered include the following. (1) Molecules like polyphenols are unstable when pH, temperature, humidity, or light level changes and they cause toxicity and adverse events at specific concentrations. Therefore, future research must focus on improving the extraction yield and stability of these molecules, minimizing side effects, and increasing health-promoting activity. In addition, since natural dietary small molecules are usually safe, recommendations on daily dosing and treatment duration must be provided for safe and effective use. (2) The pharmacokinetic and pharmacodynamic features of dietary small molecules can compromise their therapeutic efficacy. These include poor water solubility, low bioavailability, rapid metabolism, and susceptibility to physical and chemical degradation. Therefore, further pharmacokinetic and pharmacological studies are needed to improve these features. (3) Based on the rich functionality of dietary small molecules, separation of functional components, discovery of new structural compounds, structural identification, active function research, and structure-activity relationship studies should be investigated. (4) The BBB is a biological membrane that restricts the entry of compounds, thus hindering treatments from reaching the brain. Characteristics of the BBB should be thoroughly investigated to facilitate the discovery of structural compounds from dietary small molecules that can successfully cross the barrier and target the disease. (5) Nanotechnology is a novel and promising solution for improving oral bioavailability and providing other health-promoting and neuroprotective effects. However, it is difficult to determine the range of useful doses, treatment durations, and drug-delivery methods. Extensive research is needed to investigate these areas (6) Dietary small molecules (e.g., luteolin, EGCG, and TFs). can modulate the gut microbiota and exert a neuroprotective effect through the gut–brain axis. However, studies are limited to the neuroprotective molecular signaling pathways involved. More work is needed to elucidate how dietary small molecules influence the gut microbiota and induce anti-AD effects. (7) Finally, as most in vivo studies are currently based on rat or mouse models, well-designed human intervention studies are urgently required to verify the safe and effective dosage of dietary small molecules and their bioactive constituents.
Apart from a few limitation, the main concern is high lipophilicity. Lipophilicity is an important physicochemical property of a compound that has parabolic relationship with in vivo brain penetration. High lipophilic natural phytochemicals often experience low BBB access owing to increased non-specific plasma protein bindings and vulnerability to P450 metabolism that led to rapid clearance of compounds. Because of this high lipophilic property, a low concentration of dietary small molecule (naringenin) is observed in the brain, limiting its neuroprotective effect at a low dose. Recent study has addressed this issue and resolved it on their own. For example, nanoemulsion-loaded with naringenin showed higher efficacy over free drug without affecting naringenin neuroprotective function restoration effect in an AD model (264). However, dietary small molecules are a potential bioenhancer, it could be administered with other drugs or herbs to potentiate their therapeutic efficacy in NDDs, like with resveratrol, curcumin, quercetin, and Vitamin E.
11. Conclusion
AD is associated with cognitive impairment in a substantial proportion of people, and age-related cognitive decline is associated with alterations in the brain, particularly in the hippocampus and cerebral cortex. These include moderate but chronic neuroinflammation, increased oxidative stress, mitochondrial dysfunction, diminished autophagy, waning neurogenesis, and neuronal synaptic loss. Aβ plaque deposition contributes to the accumulation of neurofibrillary tangles and the progression of neuroinflammation induced by activated microglia; these processes lead to synaptic loss and neurodegeneration. Extensive in vitro and animal studies have provided strong evidence that dietary small molecules and their main bioactive ingredients can improve AD pathology via multi-target signaling, making them suitable for multi-target drug design. Dietary small molecules and their bioactive ingredients can inhibit AChE activity, reducing Aβ deposition and inhibiting tau hyperphosphorylation to rescue synaptic dysfunction. In addition, they enhance mitochondrial activity, activate autophagy, and modulate the gut microbiome, as well as having anti-apoptosis, anti-oxidation, and anti-inflammatory activity. Importantly, evidence shows that dietary small molecules and their main bioactive ingredients play an essential role in improving spatial learning and memory in animal models of AD.
Ethical approval and consent to participate
Not applicable.
CRediT authorship contribution statement
Rengasamy Balakrishnan: Writing – review & editing, Writing – original draft, Validation, Investigation, Conceptualization. Khoshnur Jannat: Resources, Formal analysis. Dong-Kug Choi: Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00208350).
Data availability
Data will be made available on request.
References
- 1.Hickman R.A., Faustin A., Wisniewski T. Alzheimer disease and its growing epidemic. Neurol. Clin. 2016;34:941–953. doi: 10.1016/j.ncl.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization . 2019. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines. [PubMed] [Google Scholar]
- 3.Baek M.S., Kim H.-K., Han K., Kwon H.-S., Na H.K., Lyoo C.H., Cho H. Annual trends in the incidence and prevalence of Alzheimer's disease in South Korea: a nationwide cohort study. Front. Neurol. 2022;13 doi: 10.3389/fneur.2022.883549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paramanick D., Singh V.D., Singh V.K. Neuroprotective effect of phytoconstituents via nanotechnology for treatment of Alzheimer diseases. J. Contr. Release. 2022;351:638–655. doi: 10.1016/j.jconrel.2022.09.058. [DOI] [PubMed] [Google Scholar]
- 5.Aranda M.P., Kremer I.N., Hinton L., Zissimopoulos J., Whitmer R.A., Hummel C.H., Trejo L., Fabius C. Impact of dementia: health disparities, population trends, care interventions, and economic costs. J. Am. Geriatr. Soc. 2021;69:1774–1783. doi: 10.1111/jgs.17345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harper L.C. 2020. Alzheimer's Association Facts and Figures. [Google Scholar]
- 7.Gong C.-X., Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rawat P., Sehar U., Bisht J., Selman A., Culberson J., Reddy P.H. Phosphorylated tau in Alzheimer's disease and other Tauopathies. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232112841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerejeira J., Lagarto L., Mukaetova-Ladinska E.B. Behavioral and psychological symptoms of dementia. Front. Neurol. 2012;3 doi: 10.3389/fneur.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bessey L.J., Walaszek A. Management of behavioral and psychological symptoms of dementia. Curr. Psychiatr. Rep. 2019;21:66. doi: 10.1007/s11920-019-1049-5. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong R.A. Risk factors for Alzheimer's disease. Folia Neuropathol. 2019;57:87–105. doi: 10.5114/fn.2019.85929. [DOI] [PubMed] [Google Scholar]
- 12.Ballatore C., Lee V.M.-Y., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 13.Sotiropoulos I., Sousa N. Tau as the converging protein between chronic stress and Alzheimer's disease synaptic pathology. Neurodegener. Dis. 2016;16:22–25. doi: 10.1159/000440844. [DOI] [PubMed] [Google Scholar]
- 14.Balakrishnan R., Azam S., Kim I.-S., Choi D.-K. Neuroprotective effects of black pepper and its bioactive compounds in age-related neurological disorders. Aging Dis. 2023;14:750. doi: 10.14336/AD.2022.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azizi G., Khannazer N., Mirshafiey A. The potential role of chemokines in Alzheimer's disease pathogenesis. Am. J. Alzheimer's Dis. Other Dementias. 2014;29:415–425. doi: 10.1177/1533317513518651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azizi G., Mirshafiey A. The potential role of proinflammatory and antiinflammatory cytokines in Alzheimer disease pathogenesis. Immunopharmacol. Immunotoxicol. 2012;34:881–895. doi: 10.3109/08923973.2012.705292. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z.-R., Huang J.-B., Yang S.-L., Hong F.-F. Role of cholinergic signaling in Alzheimer's disease. Molecules. 2022;27:1816. doi: 10.3390/molecules27061816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arendt T., Brückner M.K., Morawski M., Jäger C., Gertz H.-J. Early neurone loss in Alzheimer's disease: cortical or subcortical? Acta Neuropathol Commun. 2015;3:10. doi: 10.1186/s40478-015-0187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arora R.B., Kumar K., Deshmukh R.R. FK506 attenuates intracerebroventricular streptozotocin-induced neurotoxicity in rats. Behav. Pharmacol. 2013;24:580–589. doi: 10.1097/FBP.0b013e32836546db. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez J.J., Noristani H.N., Verkhratsky A. The serotonergic system in ageing and Alzheimer's disease. Prog. Neurobiol. 2012;99:15–41. doi: 10.1016/j.pneurobio.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Cifariello A., Pompili A., Gasbarri A. 5-HT7 receptors in the modulation of cognitive processes. Behav. Brain Res. 2008;195:171–179. doi: 10.1016/j.bbr.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Mizukami K., Ikonomovic M.D., Grayson D.R., Sheffield R., Armstrong D.M. Immunohistochemical study of GABAA receptor α1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 1998;799:148–155. doi: 10.1016/S0006-8993(98)00437-5. [DOI] [PubMed] [Google Scholar]
- 23.Butterfield D.A., Pocernich C.B. The glutamatergic system and Alzheimer???s disease. CNS Drugs. 2003;17:641–652. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- 24.Karran E., De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J. Neurochem. 2016;139:237–252. doi: 10.1111/jnc.13632. [DOI] [PubMed] [Google Scholar]
- 25.Huang W.-J., Zhang X., Chen W.-W. Role of oxidative stress in Alzheimer's disease. Biomed Rep. 2016;4:519–522. doi: 10.3892/br.2016.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misrani A., Tabassum S., Yang L. Mitochondrial dysfunction and oxidative stress in Alzheimer's disease. Front. Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.617588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uttara B., Singh A., Zamboni P., Mahajan R. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilgun-Sherki Y., Melamed E., Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–975. doi: 10.1016/S0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- 29.Huang X., Zhao X., Li B., Cai Y., Zhang S., Yu F., Wan Q. Biomarkers for evaluating the effects of exercise interventions in patients with MCI or dementia: a systematic review and meta-analysis. Exp. Gerontol. 2021;151 doi: 10.1016/j.exger.2021.111424. [DOI] [PubMed] [Google Scholar]
- 30.Thingore C., Kshirsagar V., Juvekar A. Amelioration of oxidative stress and neuroinflammation in lipopolysaccharide-induced memory impairment using Rosmarinic acid in mice. Metab. Brain Dis. 2021;36:299–313. doi: 10.1007/s11011-020-00629-9. [DOI] [PubMed] [Google Scholar]
- 31.Mecocci P., Boccardi V., Cecchetti R., Bastiani P., Scamosci M., Ruggiero C., Baroni M. A long journey into aging, brain aging, and Alzheimer's disease following the oxidative stress tracks. J. Alzheim. Dis. 2018;62:1319–1335. doi: 10.3233/JAD-170732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karisetty B.C., Bhatnagar A., Armour E.M., Beaver M., Zhang H., Elefant F. Amyloid-β peptide impact on synaptic function and neuroepigenetic gene control reveal new therapeutic strategies for alzheimer's disease. Front. Mol. Neurosci. 2020;13 doi: 10.3389/fnmol.2020.577622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amin F.U., Shah S.A., Kim M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017;7 doi: 10.1038/srep40753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi J., Levey A.I., Weintraub S.T., Rees H.D., Gearing M., Chin L.-S., Li L. Oxidative Modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and alzheimer's diseases. J. Biol. Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 35.Wang X., Su B., Zheng L., Perry G., Smith M.A., Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J. Neurochem. 2009;109:153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu X., Smith M.A., Perry G., Aliev G. Mitochondrial failures in Alzheimer's disease. Am. J. Alzheimer's Dis. Other Dementias. 2004;19:345–352. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashok A., Andrabi S.S., Mansoor S., Kuang Y., Kwon B.K., Labhasetwar V. Antioxidant therapy in oxidative stress-induced neurodegenerative diseases: role of nanoparticle-based drug delivery systems in clinical translation. Antioxidants. 2022;11:408. doi: 10.3390/antiox11020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Thompson R., Zhang H., Xu H. APP processing in Alzheimer's disease. Mol. Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Dyck C.H. Anti-Amyloid-β monoclonal antibodies for Alzheimer's disease: pitfalls and promise. Biol. Psychiatr. 2018;83:311–319. doi: 10.1016/j.biopsych.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy M.P., LeVine H. Alzheimer's disease and the amyloid-β peptide. J. Alzheim. Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu S., Okamoto S., Lipton S.A., Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kametani F., Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer's disease. Front. Neurosci. 2018;12 doi: 10.3389/fnins.2018.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mocanu M.-M., Nissen A., Eckermann K., Khlistunova I., Biernat J., Drexler D., Petrova O., Schönig K., Bujard H., Mandelkow E., Zhou L., Rune G., Mandelkow E.-M. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous tau in inducible mouse models of tauopathy. J. Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duan Y., Dong S., Gu F., Hu Y., Y, Zhao Z. Advances in the pathogenesis of Alzheimer's disease: focusing on tau-mediated neurodegeneration. Transl. Neurodegener. 2012;1:1–7. doi: 10.1186/2047-9158-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wendimu M.Y., Hooks S.B. Microglia phenotypes in aging and neurodegenerative diseases. Cells. 2022;11:2091. doi: 10.3390/cells11132091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ries M., Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 2016;8 doi: 10.3389/fnagi.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thériault P., ElAli A., Rivest S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimer's Res. Ther. 2015;7:41. doi: 10.1186/s13195-015-0125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai Y., Liu J., Wang B., Sun M., Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer's disease and related therapeutic targets. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.856376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwon H.S., Koh S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 2020;9:42. doi: 10.1186/s40035-020-00221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachiller S., Jiménez-Ferrer I., Paulus A., Yang Y., Swanberg M., Deierborg T., Boza-Serrano A. Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 2018;12 doi: 10.3389/fncel.2018.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erta M., Quintana A., Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012;8:1254–1266. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carta M.G., Serra P., Ghiani A., Manca E., Hardoy M.C., Del Giacco G.S., Diaz G., Carpiniello B., Manconi P.E. Chemokines and pro-inflammatory cytokines in down's syndrome: an early marker for Alzheimer-type dementia? Psychother. Psychosom. 2002;71:233–236. doi: 10.1159/000063649. [DOI] [PubMed] [Google Scholar]
- 53.Wilcock D.M., Griffin W.S.T. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation. 2013;10:864. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Longpre F., Garneau P., Christen Y., Ramassamy C. Protection by EGb 761 against β-amyloid-induced neurotoxicity: involvement of NF-κB, SIRT1, and MAPKs pathways and inhibition of amyloid fibril formation. Free Radic. Biol. Med. 2006;41:1781–1794. doi: 10.1016/j.freeradbiomed.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Cai X., Chen Y., Xie X., Yao D., Ding C., Chen M. Astaxanthin prevents against lipopolysaccharide-induced acute lung injury and sepsis via inhibiting activation of MAPK/NF-κB. Am J Transl Res. 2019;11:1884–1894. [PMC free article] [PubMed] [Google Scholar]
- 56.Urrutia P.J., Hirsch E.C., González‐Billault C., Núñez M.T. Hepcidin attenuates amyloid beta‐induced inflammatory and pro‐oxidant responses in astrocytes and microglia. J. Neurochem. 2017;142:140–152. doi: 10.1111/jnc.14005. [DOI] [PubMed] [Google Scholar]
- 57.Ribarič S. Detecting early cognitive decline in Alzheimer's disease with brain synaptic structural and functional evaluation. Biomedicines. 2023;11:355. doi: 10.3390/biomedicines11020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urbanc B., Cruz L., Le R., Sanders J., Ashe K.H., Duff K., Stanley H.E., Irizarry M.C., Hyman B.T. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai J., Grutzendler J., Duff K., Gan W.-B. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 60.Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palop J.J., Chin J., Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 62.Lue L.-F., Kuo Y.-M., Roher A.E., Brachova L., Shen Y., Sue L., Beach T., Kurth J.H., Rydel R.E., Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/S0002-9440(10)65184-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J., Dickson D.W., Trojanowski J.Q., Lee V.M.-Y. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 64.Subramanian J., Savage J.C., Tremblay M.-È. Synaptic loss in Alzheimer's disease: mechanistic insights provided by two-photon in vivo imaging of transgenic mouse models. Front. Cell. Neurosci. 2020;14 doi: 10.3389/fncel.2020.592607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yaari R., Tariot P.N., Schneider L.S. Psychiatry. Wiley; 2008. Cognitive enhancers and treatments for Alzheimer's disease; pp. 2294–2317. [DOI] [Google Scholar]
- 66.Lleo A. Current therapeutic options for Alzheimers disease. Curr. Genom. 2007;8:550–558. doi: 10.2174/138920207783769549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kennedy D.O., Wightman E.L. Herbal extracts and phytochemicals: plant secondary metabolites and the enhancement of human brain function. Adv. Nutr. 2011;2:32–50. doi: 10.3945/an.110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bjørklund G., Shanaida M., Lysiuk R., Butnariu M., Peana M., Sarac I., Strus O., Smetanina K., Chirumbolo S. Natural compounds and products from an anti-aging perspective. Molecules. 2022;27:7084. doi: 10.3390/molecules27207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh Makhaik M., Shakya A.K., Kale R. Antioxidants - Benefits, Sources, Mechanisms of Action. IntechOpen; 2021. Dietary phytochemicals: as a natural source of antioxidants. [DOI] [Google Scholar]
- 70.Socci V., Tempesta D., Desideri G., De Gennaro L., Ferrara M. Enhancing human cognition with cocoa flavonoids. Front. Nutr. 2017;4 doi: 10.3389/fnut.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bakoyiannis I., Daskalopoulou A., Pergialiotis V., Perrea D. Phytochemicals and cognitive health: are flavonoids doing the trick? Biomed. Pharmacother. 2019;109:1488–1497. doi: 10.1016/j.biopha.2018.10.086. [DOI] [PubMed] [Google Scholar]
- 72.Williams R.J., Spencer J.P.E. Flavonoids, cognition, and dementia: actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012;52:35–45. doi: 10.1016/j.freeradbiomed.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 73.Talebi M., Talebi M., Farkhondeh T., Kopustinskiene D.M., Simal-Gandara J., Bernatoniene J., Samarghandian S. An updated review on the versatile role of chrysin in neurological diseases: chemistry, pharmacology, and drug delivery approaches. Biomed. Pharmacother. 2021;141 doi: 10.1016/j.biopha.2021.111906. [DOI] [PubMed] [Google Scholar]
- 74.Gao S., Siddiqui N., Etim I., Du T., Zhang Y., Liang D. Developing nutritional component chrysin as a therapeutic agent: bioavailability and pharmacokinetics consideration, and ADME mechanisms. Biomed. Pharmacother. 2021;142 doi: 10.1016/j.biopha.2021.112080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walle T., Otake Y., Brubaker J.A., Walle U.K., Halushka P.V. Disposition and metabolism of the flavonoid chrysin in normal volunteers. Br. J. Clin. Pharmacol. 2001;51:143–146. doi: 10.1111/j.1365-2125.2001.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kao Y.C., Zhou C., Sherman M., Laughton C.A., Chen S. Molecular basis of the inhibition of human aromatase (estrogen synthetase) by flavone and isoflavone phytoestrogens: a site-directed mutagenesis study. Environ. Health Perspect. 1998;106:85–92. doi: 10.1289/ehp.9810685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tobin P.J., Beale P., Noney L., Liddell S., Rivory L.P., Clarke S. A pilot study on the safety of combining chrysin, a non-absorbable inducer of UGT1A1, and irinotecan (CPT-11) to treat metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2006;57:309–316. doi: 10.1007/s00280-005-0053-0. [DOI] [PubMed] [Google Scholar]
- 78.Gambelunghe C., Rossi R., Sommavilla M., Ferranti C., Rossi R., Ciculi C., Gizzi S., Micheletti A., Rufini S. Effects of chrysin on urinary testosterone levels in human males. J. Med. Food. 2003;6:387–390. doi: 10.1089/109662003772519967. [DOI] [PubMed] [Google Scholar]
- 79.Samarghandian S., Farkhondeh T., Azimi-Nezhad M. Protective effects of chrysin against drugs and toxic agents. Dose Response. 2017;15 doi: 10.1177/1559325817711782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naz S., Imran M., Rauf A., Orhan I.E., Shariati M.A., Iahtisham-Ul-Haq, IqraYasmin, Shahbaz M., Qaisrani T.B., Shah Z.A., Plygun S., Heydari M. Chrysin: pharmacological and therapeutic properties. Life Sci. 2019;235 doi: 10.1016/j.lfs.2019.116797. [DOI] [PubMed] [Google Scholar]
- 81.Yao W., Cheng J., Kandhare A.D., Mukherjee-Kandhare A.A., Bodhankar S.L., Lu G. Toxicological evaluation of a flavonoid, chrysin: morphological, behavioral, biochemical and histopathological assessments in rats. Drug Chem. Toxicol. 2021;44:601–612. doi: 10.1080/01480545.2019.1687510. [DOI] [PubMed] [Google Scholar]
- 82.Campos H.M., da Costa M., da Silva Moreira L.K., da Silva Neri H.F., Branco da Silva C.R., Pruccoli L., dos Santos F.C.A., Costa E.A., Tarozzi A., Ghedini P.C. Protective effects of chrysin against the neurotoxicity induced by aluminium: in vitro and in vivo studies. Toxicology. 2022;465 doi: 10.1016/j.tox.2021.153033. [DOI] [PubMed] [Google Scholar]
- 83.Anand K.V., Mohamed Jaabir M.S., Thomas P.A., Geraldine P. Protective role of chrysin against oxidative stress in <scp>d</scp> ‐galactose‐induced aging in an experimental rat model. Geriatr. Gerontol. Int. 2012;12:741–750. doi: 10.1111/j.1447-0594.2012.00843.x. [DOI] [PubMed] [Google Scholar]
- 84.Prajit R., Sritawan N., Suwannakot K., Naewla S., Aranarochana A., Sirichoat A., Pannangrong W., Wigmore P., Welbat J.U. Chrysin protects against memory and hippocampal neurogenesis depletion in D-galactose-induced aging in rats. Nutrients. 2020;12:1100. doi: 10.3390/nu12041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salama A., Elgohary R. Influence of chrysin on D‐galactose induced‐aging in mice: up regulation of AMP kinase/liver kinase B1/peroxisome proliferator‐activated receptor‐γ coactivator 1‐α signaling pathway. Fundam. Clin. Pharmacol. 2023;37:947–959. doi: 10.1111/fcp.12895. [DOI] [PubMed] [Google Scholar]
- 86.Chen C., Zeng J., Luo B., Li S. Chrysin attenuates oxidative stress to alleviate sevoflurane-induced cognitive impairments in aged rats. Psychiatry Investig. 2023;20:430–438. doi: 10.30773/pi.2022.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Souza L.C., Antunes M.S., Filho C.B., Del Fabbro L., de Gomes M.G., Goes A.T.R., Donato F., Prigol M., Boeira S.P., Jesse C.R. Flavonoid Chrysin prevents age-related cognitive decline via attenuation of oxidative stress and modulation of BDNF levels in aged mouse brain. Pharmacol. Biochem. Behav. 2015;134:22–30. doi: 10.1016/j.pbb.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 88.Bortolotto V.C., Araujo S.M., Pinheiro F.C., Poetini M.R., Meichtry L.B., Fronza M.G., Boeira S.P., Savegnago L., Prigol M. Chrysin restores memory deficit in hypothyroidism mice: behavioral, neurochemical and computational approaches involving the neurotrophinergic system. J. Psychiatr. Res. 2021;144:225–233. doi: 10.1016/j.jpsychires.2021.10.018. [DOI] [PubMed] [Google Scholar]
- 89.Bortolotto V.C., Araujo S.M., Pinheiro F.C., Poetini M.R., de Paula M.T., Meichtry L.B., de Almeida F.P., Musachio E.A.S., Guerra G.P., Prigol M. Modulation of glutamate levels and Na+,K+-ATPase activity contributes to the chrysin memory recovery in hypothyroidism mice. Physiol. Behav. 2020;222 doi: 10.1016/j.physbeh.2020.112892. [DOI] [PubMed] [Google Scholar]
- 90.Emili M., Guidi S., Uguagliati B., Giacomini A., Bartesaghi R., Stagni F. Treatment with the flavonoid 7,8-Dihydroxyflavone: a promising strategy for a constellation of body and brain disorders. Crit. Rev. Food Sci. Nutr. 2022;62:13–50. doi: 10.1080/10408398.2020.1810625. [DOI] [PubMed] [Google Scholar]
- 91.Paul R., Nath J., Paul S., Mazumder M.K., Phukan B.C., Roy R., Bhattacharya P., Borah A. Suggesting 7,8-dihydroxyflavone as a promising nutraceutical against CNS disorders. Neurochem. Int. 2021;148 doi: 10.1016/j.neuint.2021.105068. [DOI] [PubMed] [Google Scholar]
- 92.Liu X., Chan C.-B., Jang S.-W., Pradoldej S., Huang J., He K., Phun L.H., France S., Xiao G., Jia Y., Luo H.R., Ye K. A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 2010;53:8274–8286. doi: 10.1021/jm101206p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murota K., Nakamura Y., Uehara M. Flavonoid metabolism: the interaction of metabolites and gut microbiota. Biosci. Biotechnol. Biochem. 2018;82:600–610. doi: 10.1080/09168451.2018.1444467. [DOI] [PubMed] [Google Scholar]
- 94.Chiang N.-N., Lin T.-H., Teng Y.-S., Sun Y.-C., Chang K.-H., Lin C.-Y., Hsieh-Li H.M., Su M.-T., Chen C.-M., Lee-Chen G.-J. Flavones 7,8-DHF, quercetin, and apigenin against tau toxicity via activation of TRKB signaling in ΔK280 TauRD-DsRed SH-SY5Y cells. Front. Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.758895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han X., Zhu S., Wang B., Chen L., Li R., Yao W., Qu Z. Antioxidant action of 7,8-dihydroxyflavone protects PC12 cells against 6-hydroxydopamine-induced cytotoxicity. Neurochem. Int. 2014;64:18–23. doi: 10.1016/j.neuint.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 96.Akhtar A., Dhaliwal J., Sah S.P. 7,8-Dihydroxyflavone improves cognitive functions in ICV-STZ rat model of sporadic Alzheimer's disease by reversing oxidative stress, mitochondrial dysfunction, and insulin resistance. Psychopharmacology (Berl) 2021;238:1991–2009. doi: 10.1007/s00213-021-05826-7. [DOI] [PubMed] [Google Scholar]
- 97.Chen C., Li X.-H., Zhang S., Tu Y., Wang Y.-M., Sun H.-T. 7,8-Dihydroxyflavone ameliorates scopolamine-induced Alzheimer-like pathologic dysfunction. Rejuvenation Res. 2014;17:249–254. doi: 10.1089/rej.2013.1519. [DOI] [PubMed] [Google Scholar]
- 98.Gao L., Tian M., Zhao H., Xu Q., Huang Y., Si Q., Tian Q., Wu Q., Hu X., Sun L., McClintock S.M., Zeng Y. TrkB activation by 7, 8‐dihydroxyflavone increases synapse AMPA subunits and ameliorates spatial memory deficits in a mouse model of Alzheimer's disease. J. Neurochem. 2016;136:620–636. doi: 10.1111/jnc.13432. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Z., Liu X., Schroeder J.P., Chan C.-B., Song M., Yu S.P., Weinshenker D., Ye K. 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2014;39:638–650. doi: 10.1038/npp.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Devi L., Ohno M. 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2012;37:434–444. doi: 10.1038/npp.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aytan N., Choi J.-K., Carreras I., Crabtree L., Nguyen B., Lehar M., Blusztajn J.K., Jenkins B.G., Dedeoglu A. Protective effects of 7,8-dihydroxyflavone on neuropathological and neurochemical changes in a mouse model of Alzheimer's disease. Eur. J. Pharmacol. 2018;828:9–17. doi: 10.1016/j.ejphar.2018.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zeng Y., Liu Y., Wu M., Liu J., Hu Q. Activation of TrkB by 7,8-dihydroxyflavone prevents fear memory defects and facilitates amygdalar synaptic plasticity in aging. J. Alzheim. Dis. 2012;31:765–778. doi: 10.3233/JAD-2012-120886. [DOI] [PubMed] [Google Scholar]
- 103.Pandey S.N., Kwatra M., Dwivedi D.K., Choubey P., Lahkar M., Jangra A. 7,8-Dihydroxyflavone alleviated the high-fat diet and alcohol-induced memory impairment: behavioral, biochemical and molecular evidence. Psychopharmacology (Berl) 2020;237:1827–1840. doi: 10.1007/s00213-020-05502-2. [DOI] [PubMed] [Google Scholar]
- 104.Castello N.A., Nguyen M.H., Tran J.D., Cheng D., Green K.N., LaFerla F.M. 7,8-Dihydroxyflavone, a small molecule TrkB agonist, improves spatial memory and increases thin spine density in a mouse model of Alzheimer disease-like neuronal loss. PLoS One. 2014;9 doi: 10.1371/journal.pone.0091453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rawlings-Mortimer F., Johansen-Berg H. Rescue of long-term spatial memory by 7,8-dihydroxyflavone in mice with reduced oligodendrogenesis. ENeuro. 2023;10 doi: 10.1523/ENEURO.0498-22.2023. ENEURO.0498-22.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Calderaro A., Patanè G.T., Tellone E., Barreca D., Ficarra S., Misiti F., Laganà G. The neuroprotective potentiality of flavonoids on Alzheimer's disease. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232314835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meng-zhen S., Ju L., Lan-chun Z., Cai-feng D., Shu-da Y., Hao-fei Y., Wei-yan H. Potential therapeutic use of plant flavonoids in AD and PD. Heliyon. 2022;8 doi: 10.1016/j.heliyon.2022.e11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hernández-Aquino E., Muriel P. Beneficial effects of naringenin in liver diseases: molecular mechanisms. World J. Gastroenterol. 2018;24:1679–1707. doi: 10.3748/wjg.v24.i16.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tsai T.-H. Determination of naringin in rat blood, brain, liver, and bile using microdialysis and its interaction with cyclosporin A, a P-glycoprotein modulator. J. Agric. Food Chem. 2002;50:6669–6674. doi: 10.1021/jf020603p. [DOI] [PubMed] [Google Scholar]
- 110.Youdim K.A., Dobbie M.S., Kuhnle G., Proteggente A.R., Abbott N.J., Rice‐Evans C. Interaction between flavonoids and the blood–brain barrier: in vitro studies. J. Neurochem. 2003;85:180–192. doi: 10.1046/j.1471-4159.2003.01652.x. [DOI] [PubMed] [Google Scholar]
- 111.Zhang N., Hu Z., Zhang Z., Liu G., Wang Y., Ren Y., Wu X., Geng F. Protective role of naringenin against Aβ25-35-caused damage via ER and PI3K/Akt-Mediated pathways. Cell. Mol. Neurobiol. 2018;38:549–557. doi: 10.1007/s10571-017-0519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Heo H.J., Kim M.-J., Lee J.-M., Choi S.J., Cho H.-Y., Hong B., Kim H.-K., Kim E., Shin D.-H. Naringenin from Citrus junos has an inhibitory effect on acetylcholinesterase and a mitigating effect on amnesia. Dement. Geriatr. Cogn. Disord. 2004;17:151–157. doi: 10.1159/000076349. [DOI] [PubMed] [Google Scholar]
- 113.Ahsan A.U., Sharma V.L., Wani A., Chopra M. Naringenin upregulates AMPK-mediated autophagy to rescue neuronal cells from β-amyloid (1–42) evoked neurotoxicity. Mol. Neurobiol. 2020;57:3589–3602. doi: 10.1007/s12035-020-01969-4. [DOI] [PubMed] [Google Scholar]
- 114.Wu L.-H., Lin C., Lin H.-Y., Liu Y.-S., Wu C.Y.-J., Tsai C.-F., Chang P.-C., Yeh W.-L., Lu D.-Y. Naringenin suppresses neuroinflammatory responses through inducing suppressor of cytokine signaling 3 expression. Mol. Neurobiol. 2016;53:1080–1091. doi: 10.1007/s12035-014-9042-9. [DOI] [PubMed] [Google Scholar]
- 115.Ghofrani S., Joghataei M.-T., Mohseni S., Baluchnejadmojarad T., Bagheri M., Khamse S., Roghani M. Naringenin improves learning and memory in an Alzheimer's disease rat model: insights into the underlying mechanisms. Eur. J. Pharmacol. 2015;764:195–201. doi: 10.1016/j.ejphar.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 116.Heo H.J., Kim D.-O., Shin S.C., Kim M.J., Kim B.G., Shin D.-H. Effect of antioxidant flavanone, naringenin, from Citrus junos on neuroprotection. J. Agric. Food Chem. 2004;52:1520–1525. doi: 10.1021/jf035079g. [DOI] [PubMed] [Google Scholar]
- 117.Khan M.B., Khan Mohd.M., Khan A., Ahmed Md.E., Ishrat T., Tabassum R., Vaibhav K., Ahmad A., Islam F. Naringenin ameliorates Alzheimer's disease (AD)-type neurodegeneration with cognitive impairment (AD-TNDCI) caused by the intracerebroventricular-streptozotocin in rat model. Neurochem. Int. 2012;61:1081–1093. doi: 10.1016/j.neuint.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 118.Yang W., Ma J., Liu Z., Lu Y., Hu B., Yu H. Effect of naringenin on brain insulin signaling and cognitive functions in ICV-STZ induced dementia model of rats. Neurol. Sci. 2014;35:741–751. doi: 10.1007/s10072-013-1594-3. [DOI] [PubMed] [Google Scholar]
- 119.Chtourou Y., Fetoui H., Gdoura R. Protective effects of naringenin on iron-overload-induced cerebral cortex neurotoxicity correlated with oxidative stress. Biol. Trace Elem. Res. 2014;158:376–383. doi: 10.1007/s12011-014-9948-0. [DOI] [PubMed] [Google Scholar]
- 120.Haider S., Liaquat L., Ahmad S., Batool Z., Siddiqui R.A., Tabassum S., Shahzad S., Rafiq S., Naz N. Naringenin protects AlCl3/D-galactose induced neurotoxicity in rat model of AD via attenuation of acetylcholinesterase levels and inhibition of oxidative stress. PLoS One. 2020;15 doi: 10.1371/journal.pone.0227631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khajevand-Khazaei M.-R., Ziaee P., Motevalizadeh S.-A., Rohani M., Afshin-Majd S., Baluchnejadmojarad T., Roghani M. Naringenin ameliorates learning and memory impairment following systemic lipopolysaccharide challenge in the rat. Eur. J. Pharmacol. 2018;826:114–122. doi: 10.1016/j.ejphar.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 122.Zhou T., Liu L., Wang Q., Gao Y. Naringenin alleviates cognition deficits in high‐fat diet‐fed SAMP8 mice. J. Food Biochem. 2020;44 doi: 10.1111/jfbc.13375. [DOI] [PubMed] [Google Scholar]
- 123.Hua F.-Z., Ying J., Zhang J., Wang X.-F., Hu Y.-H., Liang Y.-P., Liu Q., Xu G.-H. Naringenin pre-treatment inhibits neuroapoptosis and ameliorates cognitive impairment in rats exposed to isoflurane anesthesia by regulating the PI3/Akt/PTEN signalling pathway and suppressing NF-κB-mediated inflammation. Int. J. Mol. Med. 2016;38:1271–1280. doi: 10.3892/ijmm.2016.2715. [DOI] [PubMed] [Google Scholar]
- 124.Nabavi S.F., Braidy N., Gortzi O., Sobarzo-Sanchez E., Daglia M., Skalicka-Woźniak K., Nabavi S.M. Luteolin as an anti-inflammatory and neuroprotective agent: a brief review. Brain Res. Bull. 2015;119:1–11. doi: 10.1016/j.brainresbull.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 125.Wang Z., Zeng M., Wang Z., Qin F., Chen J., He Z. Dietary luteolin: a narrative review focusing on its pharmacokinetic properties and effects on glycolipid metabolism. J. Agric. Food Chem. 2021;69:1441–1454. doi: 10.1021/acs.jafc.0c08085. [DOI] [PubMed] [Google Scholar]
- 126.Daily J.W., Kang S., Park S. Protection against Alzheimer's disease by luteolin: role of brain glucose regulation, anti‐inflammatory activity, and the gut microbiota‐liver‐brain axis. Biofactors. 2021;47:218–231. doi: 10.1002/biof.1703. [DOI] [PubMed] [Google Scholar]
- 127.Nishitani Y., Yamamoto K., Yoshida M., Azuma T., Kanazawa K., Hashimoto T., Mizuno M. Intestinal anti‐inflammatory activity of luteolin: role of the aglycone in NF‐κB inactivation in macrophages co‐cultured with intestinal epithelial cells. Biofactors. 2013;39:522–533. doi: 10.1002/biof.1091. [DOI] [PubMed] [Google Scholar]
- 128.Wang H.-R., Pei S.-Y., Fan D.-X., Liu Y.-H., Pan X.-F., Song F.-X., Deng S.-H., Qiu H.-B., Zhang N. Luteolin protects pheochromocytoma (PC-12) cells against Aβ25-35-induced cell apoptosis through the ER/ERK/MAPK signalling pathway. Evid. base Compl. Alternative Med. 2020;2020:1–8. doi: 10.1155/2020/2861978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zheng N., Yuan P., Li C., Wu J., Huang J. Luteolin reduces BACE1 expression through NF-κB and estrogen receptor mediated pathways in HEK293 and SH-SY5Y cells. J. Alzheim. Dis. 2015;45:659–671. doi: 10.3233/JAD-142517. [DOI] [PubMed] [Google Scholar]
- 130.Zhang J.-X., Xing J.-G., Wang L.-L., Jiang H.-L., Guo S.-L., Liu R. Luteolin inhibits fibrillary β-amyloid1–40-induced inflammation in a human blood-brain barrier model by suppressing the p38 MAPK-mediated NF-κB signaling pathways. Molecules. 2017;22:334. doi: 10.3390/molecules22030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu R., Meng F., Zhang L., Liu A., Qin H., Lan X., Li L., Du G. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules. 2011;16:2084–2096. doi: 10.3390/molecules16032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou F., Chen S., Xiong J., Li Y., Qu L. Luteolin reduces zinc-induced tau phosphorylation at Ser262/356 in an ROS-dependent manner in SH-SY5Y cells. Biol. Trace Elem. Res. 2012;149:273–279. doi: 10.1007/s12011-012-9411-z. [DOI] [PubMed] [Google Scholar]
- 133.Kou J., Shi J., He Y., Hao J., Zhang H., Luo D., Song J., Yan Y., Xie X., Du G., Pang X. Luteolin alleviates cognitive impairment in Alzheimer's disease mouse model via inhibiting endoplasmic reticulum stress-dependent neuroinflammation. Acta Pharmacol. Sin. 2022;43:840–849. doi: 10.1038/s41401-021-00702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tana T. Nakagawa. Luteolin ameliorates depression-like behaviors by suppressing ER stress in a mouse model of Alzheimer's disease. Biochem. Biophys. Res. Commun. 2022;588:168–174. doi: 10.1016/j.bbrc.2021.12.074. [DOI] [PubMed] [Google Scholar]
- 135.Wang H., Wang H., Cheng H., Che Z. Ameliorating effect of luteolin on memory impairment in an Alzheimer's disease model. Mol. Med. Rep. 2016;13:4215–4220. doi: 10.3892/mmr.2016.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yu T.-X., Zhang P., Guan Y., Wang M., Zhen M.-Q. Protective effects of luteolin against cognitive impairment induced by infusion of Aβ peptide in rats. Int. J. Clin. Exp. Pathol. 2015;8:6740–6747. [PMC free article] [PubMed] [Google Scholar]
- 137.He Z., Li X., Wang Z., Cao Y., Han S., Li N., Cai J., Cheng S., Liu Q. Protective effects of luteolin against amyloid beta-induced oxidative stress and mitochondrial impairments through peroxisome proliferator-activated receptor γ-dependent mechanism in Alzheimer's disease. Redox Biol. 2023;66 doi: 10.1016/j.redox.2023.102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ahmad S., Jo M.H., Ikram M., Khan A., Kim M.O. Deciphering the potential neuroprotective effects of luteolin against Aβ1–42-induced Alzheimer's disease. Int. J. Mol. Sci. 2021;22:9583. doi: 10.3390/ijms22179583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ali F., Rahul, Jyoti S., Naz F., Ashafaq Mo., Shahid M., Siddique Y.H. Therapeutic potential of luteolin in transgenic Drosophila model of Alzheimer's disease. Neurosci. Lett. 2019;692:90–99. doi: 10.1016/j.neulet.2018.10.053. [DOI] [PubMed] [Google Scholar]
- 140.Facchinetti R., Valenza M., Bronzuoli M.R., Menegoni G., Ratano P., Steardo L., Campolongo P., Scuderi C. Looking for a treatment for the early stage of Alzheimer's disease: preclinical evidence with Co-ultramicronized palmitoylethanolamide and luteolin. Int. J. Mol. Sci. 2020;21:3802. doi: 10.3390/ijms21113802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Facchinetti R., Valenza M., Gomiero C., Mancini G.F., Steardo L., Campolongo P., Scuderi C. Co-ultramicronized palmitoylethanolamide/luteolin restores oligodendrocyte homeostasis via peroxisome proliferator-activated receptor-α in an in vitro model of Alzheimer's disease. Biomedicines. 2022;10:1236. doi: 10.3390/biomedicines10061236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Khan U.M., Sevindik M., Zarrabi A., Nami M., Ozdemir B., Kaplan D.N., Selamoglu Z., Hasan M., Kumar M., Alshehri M.M., Sharifi-Rad J. Lycopene: food sources, biological activities, and human health benefits. Oxid. Med. Cell. Longev. 2021;2021:1–10. doi: 10.1155/2021/2713511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen D., Huang C., Chen Z. A review for the pharmacological effect of lycopene in central nervous system disorders. Biomed. Pharmacother. 2019;111:791–801. doi: 10.1016/j.biopha.2018.12.151. [DOI] [PubMed] [Google Scholar]
- 144.Rao A.V., Shen H. Effect of low dose lycopene intake on lycopene bioavailability and oxidative stress. Nutr. Res. 2002;22:1125–1131. doi: 10.1016/S0271-5317(02)00430-X. [DOI] [Google Scholar]
- 145.Gustin D.M., Rodvold K.A., Sosman J.A., Diwadkar-Navsariwala V., Stacewicz-Sapuntzakis M., Viana M., Crowell J.A., Murray J., Tiller P., Bowen P.E. Single-dose pharmacokinetic study of lycopene delivered in a well-defined food-based lycopene delivery system (Tomato Paste-oil Mixture) in healthy adult male subjects, cancer epidemiology. Biomarkers & Prevention. 2004;13:850–860. doi: 10.1158/1055-9965.850.13.5. [DOI] [PubMed] [Google Scholar]
- 146.Trumbo P.R. Are there adverse effects of lycopene exposure? J. Nutr. 2005;135:2060S–2061S. doi: 10.1093/jn/135.8.2060S. [DOI] [PubMed] [Google Scholar]
- 147.Fang Y., Ou S., Wu T., Zhou L., Tang H., Jiang M., Xu J., Guo K. Lycopene alleviates oxidative stress via the PI3K/Akt/Nrf2pathway in a cell model of Alzheimer's disease. PeerJ. 2020;8 doi: 10.7717/peerj.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hwang S., Lim J., Kim H. Inhibitory effect of lycopene on amyloid-β-induced apoptosis in neuronal cells. Nutrients. 2017;9:883. doi: 10.3390/nu9080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lim S., Hwang S., Yu J.H., Lim J.W., Kim H. Lycopene inhibits regulator of calcineurin 1‐mediated apoptosis by reducing oxidative stress and down‐regulating Nucling in neuronal cells. Mol. Nutr. Food Res. 2017;61 doi: 10.1002/mnfr.201600530. [DOI] [PubMed] [Google Scholar]
- 150.Wang J., Li L., Wang Z., Cui Y., Tan X., Yuan T., Liu Q., Liu Z., Liu X. Supplementation of lycopene attenuates lipopolysaccharide-induced amyloidogenesis and cognitive impairments via mediating neuroinflammation and oxidative stress. J. Nutr. Biochem. 2018;56:16–25. doi: 10.1016/j.jnutbio.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 151.Huang C., Gan D., Fan C., Wen C., Li A., Li Q., Zhao J., Wang Z., Zhu L., Lu D. The secretion from neural stem cells pretreated with lycopene protects against tert -butyl hydroperoxide-induced neuron oxidative damage. Oxid. Med. Cell. Longev. 2018;2018:1–14. doi: 10.1155/2018/5490218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Qu M., Li L., Chen C., Li M., Pei L., Chu F., Yang J., Yu Z., Wang D., Zhou Z. Protective effects of lycopene against amyloid β-induced neurotoxicity in cultured rat cortical neurons. Neurosci. Lett. 2011;505:286–290. doi: 10.1016/j.neulet.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 153.Qu M., Jiang Z., Liao Y., Song Z., Nan X. Lycopene prevents amyloid [Beta]-Induced mitochondrial oxidative stress and dysfunctions in cultured rat cortical neurons. Neurochem. Res. 2016;41:1354–1364. doi: 10.1007/s11064-016-1837-9. [DOI] [PubMed] [Google Scholar]
- 154.Prakash A., Kumar A. Implicating the role of lycopene in restoration of mitochondrial enzymes and BDNF levels in β-amyloid induced Alzheimer׳s disease. Eur. J. Pharmacol. 2014;741:104–111. doi: 10.1016/j.ejphar.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 155.Sachdeva A.K., Chopra K. Lycopene abrogates Aβ(1–42)-mediated neuroinflammatory cascade in an experimental model of Alzheimer's disease. J. Nutr. Biochem. 2015;26:736–744. doi: 10.1016/j.jnutbio.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 156.Xu X., Teng Y., Zou J., Ye Y., Song H., Wang Z. Effects of lycopene on vascular remodeling through the LXR–PI3K–AKT signaling pathway in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2020;526:699–705. doi: 10.1016/j.bbrc.2020.02.063. [DOI] [PubMed] [Google Scholar]
- 157.Liu C.-B., Wang R., Yi Y.-F., Gao Z., Chen Y.-Z. Lycopene mitigates β-amyloid induced inflammatory response and inhibits NF-κB signaling at the choroid plexus in early stages of Alzheimer's disease rats. J. Nutr. Biochem. 2018;53:66–71. doi: 10.1016/j.jnutbio.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 158.Yin Q., Ma Y., Hong Y., Hou X., Chen J., Shen C., Sun M., Shang Y., Dong S., Zeng Z., Pei J.-J., Liu X. Lycopene attenuates insulin signaling deficits, oxidative stress, neuroinflammation, and cognitive impairment in fructose-drinking insulin resistant rats. Neuropharmacology. 2014;86:389–396. doi: 10.1016/j.neuropharm.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 159.Xu Z., Liu C., Wang R., Gao X., Hao C., Liu C. A combination of lycopene and human amniotic epithelial cells can ameliorate cognitive deficits and suppress neuroinflammatory signaling by choroid plexus in Alzheimer's disease rat. J. Nutr. Biochem. 2021;88 doi: 10.1016/j.jnutbio.2020.108558. [DOI] [PubMed] [Google Scholar]
- 160.Zhao Z., Moghadasian M.H. Chemistry, natural sources, dietary intake and pharmacokinetic properties of ferulic acid: a review. Food Chem. 2008;109:691–702. doi: 10.1016/j.foodchem.2008.02.039. [DOI] [PubMed] [Google Scholar]
- 161.Balakrishnan R., Azam S., Cho D.-Y., Su-Kim I., Choi D.-K. Natural phytochemicals as novel therapeutic strategies to prevent and treat Parkinson's disease: current knowledge and future perspectives. Oxid. Med. Cell. Longev. 2021;2021:1–32. doi: 10.1155/2021/6680935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ou S., Kwok K. Ferulic acid: pharmaceutical functions, preparation and applications in foods. J. Sci. Food Agric. 2004;84:1261–1269. doi: 10.1002/jsfa.1873. [DOI] [Google Scholar]
- 163.Li D., Rui Y., Guo S., Luan F., Liu R., Zeng N. Ferulic acid: a review of its pharmacology, pharmacokinetics and derivatives. Life Sci. 2021;284 doi: 10.1016/j.lfs.2021.119921. [DOI] [PubMed] [Google Scholar]
- 164.Huang H., Hong Q., Tan H., Xiao C., Gao Y. Ferulic acid prevents LPS-induced up-regulation of PDE4B and stimulates the cAMP/CREB signaling pathway in PC12 cells. Acta Pharmacol. Sin. 2016;37:1543–1554. doi: 10.1038/aps.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cui L., Zhang Y., Cao H., Wang Y., Teng T., Ma G., Li Y., Li K., Zhang Y. Ferulic acid inhibits the transition of amyloid-β42 monomers to oligomers but accelerates the transition from oligomers to fibrils. J. Alzheim. Dis. 2013;37:19–28. doi: 10.3233/JAD-130164. [DOI] [PubMed] [Google Scholar]
- 166.Ohashi H., Tsuji M., Oguchi T., Momma Y., Nohara T., Ito N., Yamamoto K., Nagata M., Kimura A.M., Kiuchi Y., Ono K. Combined treatment with curcumin and ferulic acid suppressed the Aβ-induced neurotoxicity more than curcumin and ferulic acid alone. Int. J. Mol. Sci. 2022;23:9685. doi: 10.3390/ijms23179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Huang F., Deng H.M., Zhu M.M., Xiao F., Yang L., Zhang Z.J. Inhibitory effect of ferulic acid on in? ammatory response in microglia induced by lipopolysaccharides. Zool. Res. 2011;32:311–316. doi: 10.3724/SP.J.1141.2011.03311. [DOI] [PubMed] [Google Scholar]
- 168.Zhou X., Chen X., Cheng X., Lin L., Quan S., Li S., Zhan R., Wu Q., Liu S. Paeoniflorin, ferulic acid, and atractylenolide III improved LPS-induced neuroinflammation of BV2 microglia cells by enhancing autophagy. J. Pharmacol. Sci. 2023;152:151–161. doi: 10.1016/j.jphs.2023.04.007. [DOI] [PubMed] [Google Scholar]
- 169.Kim M.J., Choi S.J., Lim S.-T., Kim H.K., Heo H.J., Kim E.-K., Jun W.J., Cho H.Y., Kim Y.J., Shin D.-H. Ferulic acid supplementation prevents trimethyltin-induced cognitive deficits in mice. Biosci. Biotechnol. Biochem. 2007;71:1063–1068. doi: 10.1271/bbb.60564. [DOI] [PubMed] [Google Scholar]
- 170.Mahaman Y.A.R., Huang F., Salissou M.T.M., Yacouba M.B.M., Wang J.-Z., Liu R., Zhang B., Li H.-L., Zhu F., Wang X. Ferulic acid improves synaptic plasticity and cognitive impairments by alleviating the PP2B/DARPP-32/PP1 axis-mediated STEP increase and Aβ burden in Alzheimer's disease. Neurotherapeutics. 2023;20:1081–1108. doi: 10.1007/s13311-023-01356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang N.-Y., Li J.-N., Liu W.-L., Huang Q., Li W.-X., Tan Y.-H., Liu F., Song Z.-H., Wang M.-Y., Xie N., Mao R.-R., Gan P., Ding Y.-Q., Zhang Z., Shan B.-C., Chen L.-D., Zhou Q.-X., Xu L. Ferulic acid ameliorates Alzheimer's disease-like pathology and repairs cognitive decline by preventing capillary hypofunction in APP/PS1 mice. Neurotherapeutics. 2021;18:1064–1080. doi: 10.1007/s13311-021-01024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mori T., Koyama N., Tan J., Segawa T., Maeda M., Town T. Combination therapy with octyl gallate and ferulic acid improves cognition and neurodegeneration in a transgenic mouse model of Alzheimer's disease. J. Biol. Chem. 2017;292:11310–11325. doi: 10.1074/jbc.M116.762658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yan J.-J., Jung J.-S., Kim T.-K., Hasan Md.A., Hong C.-W., Nam J.-S., Song D.-K. Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull. 2013;36:140–143. doi: 10.1248/bpb.b12-00798. [DOI] [PubMed] [Google Scholar]
- 174.Jin Y., Yan E., Fan Y., Zong Z., Qi Z., Li Z. Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus1. Acta Pharmacol. Sin. 2005;26:943–951. doi: 10.1111/j.1745-7254.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- 175.Wang N., Zhou Y., Zhao L., Wang C., Ma W., Ge G., Wang Y., Ullah I., Muhammad F., Alwayli D., Zhi D., Li H. Ferulic acid delayed amyloid β-induced pathological symptoms by autophagy pathway via a fasting-like effect in Caenorhabditis elegans. Food Chem. Toxicol. 2020;146 doi: 10.1016/j.fct.2020.111808. [DOI] [PubMed] [Google Scholar]
- 176.Priyadarsini K.I., Khopde S.M., Kumar S.S., Mohan H. Free radical studies of ellagic acid, a natural phenolic antioxidant. J. Agric. Food Chem. 2002;50:2200–2206. doi: 10.1021/jf011275g. [DOI] [PubMed] [Google Scholar]
- 177.Sharifi-Rad J., Quispe C., Castillo C.M.S., Caroca R., Lazo-Vélez M.A., Antonyak H., Polishchuk A., Lysiuk R., Oliinyk P., De Masi L., Bontempo P., Martorell M., Daştan S.D., Rigano D., Wink M., Cho W.C. Ellagic acid: a review on its natural sources, chemical stability, and therapeutic potential. Oxid. Med. Cell. Longev. 2022;2022:1–24. doi: 10.1155/2022/3848084. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 178.Tasaki M., Umemura T., Maeda M., Ishii Y., Okamura T., Inoue T., Kuroiwa Y., Hirose M., Nishikawa A. Safety assessment of ellagic acid, a food additive, in a subchronic toxicity study using F344 rats. Food Chem. Toxicol. 2008;46:1119–1124. doi: 10.1016/j.fct.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 179.Feng Y., Yang S., Du X., Zhang X., Sun X., Zhao M., Sun G., Liu R. Ellagic acid promotes Aβ42 fibrillization and inhibits Aβ42-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2009;390:1250–1254. doi: 10.1016/j.bbrc.2009.10.130. [DOI] [PubMed] [Google Scholar]
- 180.Shen Y.-C., Juan C.-W.J., Juan C.-W.J., Lin C.-S., Lin C.-S., Chen C.-C., Chen C.-C., Chang C.-L., Chang C.-L. Neuroprotective EFfect of Terminalia Chebula extracts and ellagic acid in PC12 cells. Afr. J. Tradit., Complementary Altern. Med. 2017;14:22–30. doi: 10.21010/ajtcam.v14i4.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rahimi V.B., Askari V.R., Mousavi S.H. Ellagic acid reveals promising anti-aging effects against d-galactose-induced aging on human neuroblastoma cell line, SH-SY5Y: a mechanistic study. Biomed. Pharmacother. 2018;108:1712–1724. doi: 10.1016/j.biopha.2018.10.024. [DOI] [PubMed] [Google Scholar]
- 182.Liu Y.-L., Huang H.-J., Sheu S.-Y., Liu Y.-C., Lee I.-J., Chiang S.-C., Lin A.M.-Y. Oral ellagic acid attenuated LPS-induced neuroinflammation in rat brain: MEK1 interaction and M2 microglial polarization. Exp. Biol. Med. 2023;248:656–664. doi: 10.1177/15353702231182230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhong L., Liu H., Zhang W., Liu X., Jiang B., Fei H., Sun Z. Ellagic acid ameliorates learning and memory impairment in APP/PS1 transgenic mice via inhibition of β-amyloid production and tau hyperphosphorylation. Exp. Ther. Med. 2018 doi: 10.3892/etm.2018.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jha A.B., Panchal S.S., Shah A. Ellagic acid: insights into its neuroprotective and cognitive enhancement effects in sporadic Alzheimer's disease. Pharmacol. Biochem. Behav. 2018;175:33–46. doi: 10.1016/j.pbb.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 185.Ramadan W.S., Alkarim S. Ellagic acid modulates the amyloid precursor protein gene via superoxide dismutase regulation in the entorhinal cortex in an experimental Alzheimer's model. Cells. 2021;10:3511. doi: 10.3390/cells10123511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kiasalari Z., Heydarifard R., Khalili M., Afshin-Majd S., Baluchnejadmojarad T., Zahedi E., Sanaierad A., Roghani M. Ellagic acid ameliorates learning and memory deficits in a rat model of Alzheimer's disease: an exploration of underlying mechanisms. Psychopharmacology (Berl) 2017;234:1841–1852. doi: 10.1007/s00213-017-4589-6. [DOI] [PubMed] [Google Scholar]
- 187.Assaran A.H., Akbarian M., Amirahmadi S., Salmani H., Shirzad S., Hosseini M., Beheshti F., Rajabian A. Ellagic acid prevents oxidative stress and memory deficits in a rat model of scopolamine-induced Alzheimer's disease. Cent. Nerv. Syst. Agents Med. Chem. 2022;22:214–227. doi: 10.2174/1871524923666221027100949. [DOI] [PubMed] [Google Scholar]
- 188.Marques V., Farah A. Chlorogenic acids and related compounds in medicinal plants and infusions. Food Chem. 2009;113:1370–1376. doi: 10.1016/J.FOODCHEM.2008.08.086. [DOI] [Google Scholar]
- 189.Wang X., Stavchansky S., Kerwin S.M., Bowman P.D. Structure–activity relationships in the cytoprotective effect of caffeic acid phenethyl ester (CAPE) and fluorinated derivatives: effects on heme oxygenase-1 induction and antioxidant activities. Eur. J. Pharmacol. 2010;635:16–22. doi: 10.1016/J.EJPHAR.2010.02.034. [DOI] [PubMed] [Google Scholar]
- 190.Kim J., Lee K.W. 2015. Coffee and its Active Compounds Are Neuroprotective, Coffee in Health and Disease Prevention; pp. 423–427. [DOI] [Google Scholar]
- 191.Chu Y.F., editor. vol. 59. John Wiley & Sons; 2012. https://www.wiley.com/en-us/Coffee%3A+Emerging+Health+Effects+and+Disease+Prevention-p-9781119949893 (Coffee: Emerging Health Effects and Disease Prevention). [Google Scholar]
- 192.Manach C., Scalbert A., Morand C., Rémésy C., Jiménez L. Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr. 2004;79:727–747. doi: 10.1093/AJCN/79.5.727. [DOI] [PubMed] [Google Scholar]
- 193.Liu Y., Qiu S., Wang L., Zhang N., Shi Y., Zhou H., Liu X., Shao L., Liu X., Chen J., Hou M. Reproductive and developmental toxicity study of caffeic acid in mice. Food Chem. Toxicol. 2019;123:106–112. doi: 10.1016/J.FCT.2018.10.040. [DOI] [PubMed] [Google Scholar]
- 194.Sul D., Kim H.S., Lee D., Joo S.S., Hwang K.W., Park S.Y. Protective effect of caffeic acid against beta-amyloid-induced neurotoxicity by the inhibition of calcium influx and tau phosphorylation. Life Sci. 2009;84:257–262. doi: 10.1016/J.LFS.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 195.Huang Y., Jin M., Pi R., Zhang J., Chen M., Ouyang Y., Liu A., Chao X., Liu P., Liu J., Ramassamy C., Qin J. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci. Lett. 2013;535:146–151. doi: 10.1016/J.NEULET.2012.12.051. [DOI] [PubMed] [Google Scholar]
- 196.Chang W., Huang D., Lo Y.M., Tee Q., Kuo P., Wu J.S., Huang W., Shen S. Protective effect of caffeic acid against Alzheimer's disease pathogenesis via modulating cerebral insulin signaling, β-amyloid accumulation, and synaptic plasticity in hyperinsulinemic rats. J. Agric. Food Chem. 2019;67:7684–7693. doi: 10.1021/acs.jafc.9b02078. [DOI] [PubMed] [Google Scholar]
- 197.Wang Y., Wang Y., Li J., Hua L., Han B., Zhang Y., Yang X., Zeng Z., Bai H., Yin H., Lou J. Effects of caffeic acid on learning deficits in a model of Alzheimer's disease. Int. J. Mol. Med. 2016;38:869–875. doi: 10.3892/ijmm.2016.2683. [DOI] [PubMed] [Google Scholar]
- 198.Deshmukh R., Kaundal M., Bansal V. Samardeep, Caffeic acid attenuates oxidative stress, learning and memory deficit in intra-cerebroventricular streptozotocin induced experimental dementia in rats. Biomed. Pharmacother. 2016;81:56–62. doi: 10.1016/J.BIOPHA.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 199.Khan A., Park J.S., Kang M.H., Lee H.J., Ali J., Tahir M., Choe K., Kim M.O. Caffeic acid, a polyphenolic micronutrient rescues mice brains against Aβ-induced neurodegeneration and memory impairment. Antioxidants. 2023;12:1284. doi: 10.3390/antiox12061284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sun R., Wu T., Xing S., Wei S., Bielicki J.K., Pan X., Zhou M., Chen J. Caffeic acid protects against atherosclerotic lesions and cognitive decline in ApoE−/− mice. J. Pharmacol. Sci. 2023;151:110–118. doi: 10.1016/j.jphs.2022.12.006. [DOI] [PubMed] [Google Scholar]
- 201.Li H., Yu X., Li C., Ma L., Zhao Z., Guan S., Wang L. Caffeic acid protects against Aβ toxicity and prolongs lifespan in Caenorhabditis elegans models. Food Funct. 2021;12:1219–1231. doi: 10.1039/D0FO02784G. [DOI] [PubMed] [Google Scholar]
- 202.Bhuia Md.S., Rahaman Md.M., Islam T., Bappi M.H., Sikder Md.I., Hossain K.N., Akter F., Al Shamsh Prottay A., Rokonuzzman Md., Gürer E.S., Calina D., Islam M.T., Sharifi-Rad J. Neurobiological effects of gallic acid: current perspectives. Chin. Med. 2023;18:27. doi: 10.1186/s13020-023-00735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cirillo G., Kraemer K., Fuessel S., Puoci F., Curcio M., Spizzirri U.G., Altimari I., Iemma F. Biological activity of a gallic Acid−Gelatin conjugate. Biomacromolecules. 2010;11:3309–3315. doi: 10.1021/bm100760x. [DOI] [PubMed] [Google Scholar]
- 204.Kim M., Seong A., Yoo J., Jin C., Lee Y., Kim Y.J., Lee J., Jun W.J., Yoon H. Gallic acid, a histone acetyltransferase inhibitor, suppresses β‐amyloid neurotoxicity by inhibiting microglial‐mediated neuroinflammation. Mol. Nutr. Food Res. 2011;55:1798–1808. doi: 10.1002/mnfr.201100262. [DOI] [PubMed] [Google Scholar]
- 205.Yu M., Chen X., Liu J., Ma Q., Zhuo Z., Chen H., Zhou L., Yang S., Zheng L., Ning C., Xu J., Gao T., Hou S.-T. Gallic acid disruption of Aβ1–42 aggregation rescues cognitive decline of APP/PS1 double transgenic mouse. Neurobiol. Dis. 2019;124:67–80. doi: 10.1016/j.nbd.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 206.Mori T., Koyama N., Yokoo T., Segawa T., Maeda M., Sawmiller D., Tan J., Town T. Gallic acid is a dual α/β-secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer mice. J. Biol. Chem. 2020;295:16251–16266. doi: 10.1074/jbc.RA119.012330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ogunlade B., Adelakun S.A., Agie J.A. Nutritional supplementation of gallic acid ameliorates Alzheimer-type hippocampal neurodegeneration and cognitive impairment induced by aluminum chloride exposure in adult Wistar rats. Drug Chem. Toxicol. 2022;45:651–662. doi: 10.1080/01480545.2020.1754849. [DOI] [PubMed] [Google Scholar]
- 208.Mansouri M.T., Naghizadeh B., Ghorbanzadeh B., Farbood Y., Sarkaki A., Bavarsad K. Gallic acid prevents memory deficits and oxidative stress induced by intracerebroventricular injection of streptozotocin in rats. Pharmacol. Biochem. Behav. 2013;111:90–96. doi: 10.1016/j.pbb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 209.Samad N., Jabeen S., Imran I., Zulfiqar I., Bilal K. Protective effect of gallic acid against arsenic-induced anxiety−/depression- like behaviors and memory impairment in male rats. Metab. Brain Dis. 2019;34:1091–1102. doi: 10.1007/s11011-019-00432-1. [DOI] [PubMed] [Google Scholar]
- 210.Hajipour S., Sarkaki A., Farbood Y., Eidi A., Mortazavi P., Valizadeh Z. Effect of gallic acid on dementia type of Alzheimer disease in rats: electrophysiological and histological studies. Basic and Clinical Neuroscience Journal. 2016;7 doi: 10.15412/J.BCN.03070203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mandel S.A., Avramovich-Tirosh Y., Reznichenko L., Zheng H., Weinreb O., Amit T., Youdim M.B.H. Multifunctional activities of green tea catechins in neuroprotection. Neurosignals. 2005;14:46–60. doi: 10.1159/000085385. [DOI] [PubMed] [Google Scholar]
- 212.Singh R., Akhtar N., Haqqi T.M. Green tea polyphenol epigallocatechi3-gallate: inflammation and arthritis. Life Sci. 2010;86:907–918. doi: 10.1016/J.LFS.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Unno K., Pervin M., Nakagawa A., Iguchi K., Hara A., Takagaki A., Nanjo F., Minami A., Nakamura Y. Blood–brain barrier permeability of green tea catechin metabolites and their neuritogenic activity in human neuroblastoma SH‐SY5Y cells. Mol. Nutr. Food Res. 2017;61 doi: 10.1002/mnfr.201700294. [DOI] [PubMed] [Google Scholar]
- 214.Lin L.-C., Wang M.-N., Tseng T.-Y., Sung, Tsai T.-H. Pharmacokinetics of (−)-Epigallocatechin-3-gallate in conscious and freely moving rats and its brain regional distribution. J. Agric. Food Chem. 2007;55:1517–1524. doi: 10.1021/jf062816a. [DOI] [PubMed] [Google Scholar]
- 215.Chan P.C., Ramot Y., Malarkey D.E., Blackshear P., Kissling G.E., Travlos G., Nyska A. Fourteen-week toxicity study of green tea extract in rats and mice. Toxicol. Pathol. 2010;38:1070–1084. doi: 10.1177/0192623310382437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Isbrucker R.A., Edwards J.A., Wolz E., Davidovich A., Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 2: dermal, acute and short-term toxicity studies. Food Chem. Toxicol. 2006;44:636–650. doi: 10.1016/j.fct.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 217.Hsu Y.-W., Tsai C.-F., Chen W.-K., Huang C.-F., Yen C.-C. A subacute toxicity evaluation of green tea (Camellia sinensis) extract in mice. Food Chem. Toxicol. 2011;49:2624–2630. doi: 10.1016/j.fct.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 218.Kim C.-Y., Lee C., Park G.H., Jang J.-H. Neuroprotective effect of epigallocatechin-3-gallate against β-amyloid-induced oxidative and nitrosative cell death via augmentation of antioxidant defense capacity. Arch Pharm. Res. (Seoul) 2009;32:869–881. doi: 10.1007/s12272-009-1609-z. [DOI] [PubMed] [Google Scholar]
- 219.Kim S.-J., Jeong H.-J., Lee K.-M., Myung N.-Y., An N.-H., Mo Yang W., Kyu Park S., Lee H.-J., Hong S.-H., Kim H.-M., Um J.-Y. Epigallocatechin-3-gallate suppresses NF-κB activation and phosphorylation of p38 MAPK and JNK in human astrocytoma U373MG cells. J. Nutr. Biochem. 2007;18:587–596. doi: 10.1016/j.jnutbio.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 220.Payne A., Taka E., Adinew G.M., Soliman K.F.A. Molecular mechanisms of the anti-inflammatory effects of epigallocatechin 3-gallate (EGCG) in LPS-activated BV-2 microglia cells. Brain Sci. 2023;13:632. doi: 10.3390/brainsci13040632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhong X., Liu M., Yao W., Du K., He M., Jin X., Jiao L., Ma G., Wei B., Wei M. Epigallocatechin‐3‐Gallate attenuates microglial inflammation and neurotoxicity by suppressing the activation of canonical and noncanonical inflammasome via TLR4/NF‐κB pathway. Mol. Nutr. Food Res. 2019;63 doi: 10.1002/mnfr.201801230. [DOI] [PubMed] [Google Scholar]
- 222.Bao J., Liu W., Zhou H., Gui Y., Yang Y., Wu M., Xiao Y., Shang J., Long G., Shu X. Epigallocatechin-3-gallate alleviates cognitive deficits in APP/PS1 mice. Curr Med Sci. 2020;40:18–27. doi: 10.1007/s11596-020-2142-z. [DOI] [PubMed] [Google Scholar]
- 223.Nan S., Wang P., Zhang Y., Fan J. Epigallocatechin-3-Gallate provides protection against Alzheimer's disease-induced learning and memory impairments in rats. Drug Des. Dev. Ther. 2021;15:2013–2024. doi: 10.2147/DDDT.S289473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee Y.J., Choi D.Y., Yun Y.P., Han S.B., Oh K.W., Hong J.T. Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloidogenesis via its anti-neuroinflammatory properties. J. Nutr. Biochem. 2013;24:298–310. doi: 10.1016/J.JNUTBIO.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 225.Ettcheto M., Cano A., Manzine P.R., Busquets O., Verdaguer E., Castro-Torres R.D., García M.L., Beas-Zarate C., Olloquequi J., Auladell C., Folch J., Camins A. Epigallocatechin-3-Gallate (EGCG) improves cognitive deficits aggravated by an obesogenic diet through modulation of unfolded protein response in APPswe/PS1dE9 mice. Mol. Neurobiol. 2020;57:1814–1827. doi: 10.1007/s12035-019-01849-6. [DOI] [PubMed] [Google Scholar]
- 226.Qu Y., Wu Y., Cheng W., Wang D., Zeng L., Wang Y., Li T., Zhang L., Yang J., Sun L., Ai J. Amelioration of cognitive impairment using epigallocatechin-3-gallate in ovariectomized mice fed a high-fat diet involves remodeling with Prevotella and Bifidobacteriales. Front. Pharmacol. 2023;13 doi: 10.3389/fphar.2022.1079313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Soto-Mercado V., Mendivil-Perez M., Velez-Pardo C., Jimenez-Del-Rio M. (−)-Epigallocatechin-3-Gallate diminishes intra-and extracellular amyloid-induced cytotoxic effects on cholinergic-like neurons from familial Alzheimer's disease PSEN1 E280A. Biomolecules. 2021;11:1845. doi: 10.3390/biom11121845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Schuh C., Schieberle P. Characterization of the key aroma compounds in the beverage prepared from Darjeeling black tea: quantitative differences between tea leaves and infusion. J. Agric. Food Chem. 2006;54:916–924. doi: 10.1021/jf052495n. [DOI] [PubMed] [Google Scholar]
- 229.Takemoto M., Takemoto H. Synthesis of theaflavins and their functions. Molecules. 2018;23:918. doi: 10.3390/molecules23040918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mulder T.P.J., Van Platerink C.J., Wijnand Schuyl P.J., Van Amelsvoort J.M.M. Analysis of theaflavins in biological fluids using liquid chromatography–electrospray mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001;760:271–279. doi: 10.1016/S0378-4347(01)00285-7. [DOI] [PubMed] [Google Scholar]
- 231.Cao Y., Zhang Y., Jia Z., Jia H., Sun Y., Yuan H., Bian Y., Xu B., Fu J., Qin F. Theaflavin-3,3′-digallate ameliorates learning and memory impairments in mice with premature brain aging induced by D-galactose. Physiol. Behav. 2023;261 doi: 10.1016/j.physbeh.2023.114077. [DOI] [PubMed] [Google Scholar]
- 232.Ano Y., Ohya R., Kita M., Taniguchi Y., Kondo K. Theaflavins improve memory impairment and depression-like behavior by regulating microglial activation. Molecules. 2019;24:467. doi: 10.3390/molecules24030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Li M., Zhang C., Xiao X., Zhu M., Quan W., Liu X., Zhang S., Liu Z. Theaflavins in black tea mitigate aging-associated cognitive dysfunction via the microbiota–gut–brain Axis. J. Agric. Food Chem. 2023;71:2356–2369. doi: 10.1021/acs.jafc.2c06679. [DOI] [PubMed] [Google Scholar]
- 234.Walton N.J., Mayer M.J., Narbad A., Vanillin Phytochemistry. 2003;63:505–515. doi: 10.1016/S0031-9422(03)00149-3. [DOI] [PubMed] [Google Scholar]
- 235.Iannuzzi C., Liccardo M., Sirangelo I. Overview of the role of vanillin in neurodegenerative diseases and neuropathophysiological conditions. Int. J. Mol. Sci. 2023;24:1817. doi: 10.3390/ijms24031817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ho K., Yazan L.S., Ismail N., Ismail M. Toxicology study of vanillin on rats via oral and intra-peritoneal administration. Food Chem. Toxicol. 2011;49:25–30. doi: 10.1016/j.fct.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 237.Zhong L., Tong Y., Chuan J., Bai L., Shi J., Zhu Y. Protective effect of ethyl vanillin against Aβ-induced neurotoxicity in PC12 cells via the reduction of oxidative stress and apoptosis. Exp. Ther. Med. 2019 doi: 10.3892/etm.2019.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kim M.E., Na J.Y., Park Y.-D., Lee J.S. Anti-neuroinflammatory effects of vanillin through the regulation of inflammatory factors and NF-κB signaling in LPS-stimulated microglia. Appl. Biochem. Biotechnol. 2019;187:884–893. doi: 10.1007/s12010-018-2857-5. [DOI] [PubMed] [Google Scholar]
- 239.Anand A., Khurana N., Ali N., AlAsmari A.F., Alharbi M., Waseem M., Sharma N. Ameliorative effect of vanillin on scopolamine-induced dementia-like cognitive impairment in a mouse model. Front. Neurosci. 2022;16 doi: 10.3389/fnins.2022.1005972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Anand A., Khurana N., Kaur S., Ali N., AlAsmari A.F., Waseem M., Iqbal M., Alzahrani F.M., Sharma N. The multifactorial role of vanillin in amelioration of aluminium chloride and D-galactose induced Alzheimer's disease in mice. Eur. J. Pharmacol. 2023;954 doi: 10.1016/j.ejphar.2023.175832. [DOI] [PubMed] [Google Scholar]
- 241.Lee J., Kim I.H., Cho J.H., Lee T., Park J.H., Ahn J.H., Shin B.N., Yan B.C., Kim J., Jeon Y.H., Lee Y.J., Won M., Kang I.J. Vanillin improves scopolamine-induced memory impairment through restoration of ID1 expression in the mouse hippocampus. Mol. Med. Rep. 2018 doi: 10.3892/mmr.2018.8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kim Y.H., Park J.H. Vanillin and 4-hydroxybenzyl alcohol attenuate cognitive impairment and the reduction of cell proliferation and neuroblast differentiation in the dentate gyrus in a mouse model of scopolamine-induced amnesia. Anat Cell Biol. 2017;50:143. doi: 10.5115/acb.2017.50.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Liu C., Kou X., Wang X., Wu J., Yang A., Shen R. Novel chrysin derivatives as hidden multifunctional agents for anti-Alzheimer’s disease: design, synthesis and in vitro evaluation. Eur. J. Pharmaceut. Sci. 2021;166 doi: 10.1016/j.ejps.2021.105976. [DOI] [PubMed] [Google Scholar]
- 244.Pandey R.P., Parajuli P., Pokhrel A.R., Sohng J.K. Biosynthesis of novel 7,8‐dihydroxyflavone glycoside derivatives and in silico study of their effects on BACE1 inhibition. Biotechnol. Appl. Biochem. 2018;65:128–137. doi: 10.1002/bab.1570. [DOI] [PubMed] [Google Scholar]
- 245.Mi J., He Y., Yang J., Zhou Y., Zhu G., Wu A., Liu W., Sang Z. Development of naringenin-O-carbamate derivatives as multi-target-directed liagnds for the treatment of Alzheimer's disease. Bioorg. Med. Chem. Lett. 2022;60 doi: 10.1016/j.bmcl.2022.128574. [DOI] [PubMed] [Google Scholar]
- 246.Yang J., Zhou Y., Ban Y., Mi J., He Y., Li X., Liu Z., Wang K., Zhu G., Liu W., Tan Z., Sang Z. Development of naringenin- O -alkylamine derivatives as multifunctional agents for the treatment of Alzheimer's disease. J. Enzym. Inhib. Med. Chem. 2022;37:792–816. doi: 10.1080/14756366.2022.2041627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wu J., Kou X., Ju H., Zhang H., Yang A., Shen R. Design, synthesis and biological evaluation of naringenin carbamate derivatives as potential multifunctional agents for the treatment of Alzheimer's disease. Bioorg. Med. Chem. Lett. 2021;49 doi: 10.1016/J.BMCL.2021.128316. [DOI] [PubMed] [Google Scholar]
- 248.Lan J.-S., Zeng R.-F., Jiang X.-Y., Hou J., Liu Y., Hu Z.-H., Li H.-X., Li Y., Xie S.-S., Ding Y., Zhang T. Design, synthesis and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. Bioorg. Chem. 2020;94 doi: 10.1016/j.bioorg.2019.103413. [DOI] [PubMed] [Google Scholar]
- 249.Sang Z., Pan W., Wang K., Ma Q., Yu L., Yang Y., Bai P., Leng C., Xu Q., Li X., Tan Z., Liu W. Design, synthesis and evaluation of novel ferulic acid- O -alkylamine derivatives as potential multifunctional agents for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 2017;130:379–392. doi: 10.1016/j.ejmech.2017.02.039. [DOI] [PubMed] [Google Scholar]
- 250.Sang Z., Wang K., Han X., Cao M., Tan Z., Liu W. Design, synthesis, and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. ACS Chem. Neurosci. 2019;10:1008–1024. doi: 10.1021/acschemneuro.8b00530. [DOI] [PubMed] [Google Scholar]
- 251.Tripathi A., Choubey P.K., Sharma P., Seth A., Saraf P., Shrivastava S.K. Design, synthesis, and biological evaluation of ferulic acid based 1,3,4-oxadiazole hybrids as multifunctional therapeutics for the treatment of Alzheimer's disease. Bioorg. Chem. 2020;95 doi: 10.1016/j.bioorg.2019.103506. [DOI] [PubMed] [Google Scholar]
- 252.Kikugawa M., Tsutsuki H., Ida T., Nakajima H., Ihara H., Sakamoto T. Water-soluble ferulic acid derivatives improve amyloid-β-induced neuronal cell death and dysmnesia through inhibition of amyloid-β aggregation. Biosci. Biotechnol. Biochem. 2016;80:547–553. doi: 10.1080/09168451.2015.1107463. [DOI] [PubMed] [Google Scholar]
- 253.Kikugawa M., Ida T., Ihara H., Sakamoto T. Ferulic acid and its water-soluble derivatives inhibit nitric oxide production and inducible nitric oxide synthase expression in rat primary astrocytes. Biosci. Biotechnol. Biochem. 2017;81:1607–1611. doi: 10.1080/09168451.2017.1336925. [DOI] [PubMed] [Google Scholar]
- 254.Konar A., Kalra R.S., Chaudhary A., Nayak A., Guruprasad K.P., Satyamoorthy K., Ishida Y., Terao K., Kaul S.C., Wadhwa R. Identification of caffeic acid phenethyl ester (CAPE) as a potent neurodifferentiating natural compound that improves cognitive and physiological functions in animal models of neurodegenerative diseases. Front. Aging Neurosci. 2020;12 doi: 10.3389/fnagi.2020.561925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Morroni F., Sita G., Graziosi A., Turrini E., Fimognari C., Tarozzi A., Hrelia P. Neuroprotective effect of caffeic acid phenethyl ester in A mouse model of Alzheimer's disease involves Nrf2/HO-1 pathway. Aging Dis. 2018;9:605. doi: 10.14336/AD.2017.0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wan T., Wang Z., Luo Y., Zhang Y., He W., Mei Y., Xue J., Li M., Pan H., Li W., Wang Q., Huang Y. FA-97, a new synthetic caffeic acid phenethyl ester derivative, protects against oxidative stress-mediated neuronal cell apoptosis and scopolamine-induced cognitive impairment by activating Nrf2/HO-1 signaling. Oxid. Med. Cell. Longev. 2019;2019:1–21. doi: 10.1155/2019/8239642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Yelamanda Rao K., Jeelan Basha S., Monika K., Naidu Gajula N., Sivakumar I., Kumar S., Vadde R., Aramati B.M.R., Subramanyam R., Damu A.G. Development of quinazolinone and vanillin acrylamide hybrids as multi-target directed ligands against Alzheimer's disease and mechanistic insights into their binding with acetylcholinesterase. J. Biomol. Struct. Dyn. 2023;41:11148–11165. doi: 10.1080/07391102.2023.2203255. [DOI] [PubMed] [Google Scholar]
- 258.Quispe C., Herrera-Bravo J., Javed Z., Khan K., Raza S., Gulsunoglu-Konuskan Z., Daştan S.D., Sytar O., Martorell M., Sharifi-Rad J., Calina D. Therapeutic applications of curcumin in diabetes: a review and perspective. BioMed Res. Int. 2022;2022:1–14. doi: 10.1155/2022/1375892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Quispe C., Herrera-Bravo J., Khan K., Javed Z., Semwal P., Painuli S., Kamiloglu S., Martorell M., Calina D., Sharifi-Rad J. Therapeutic applications of curcumin nanomedicine formulations in cystic fibrosis. Prog Biomater. 2022;11:321–329. doi: 10.1007/s40204-022-00198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Giacomeli R., de Gomes M.G., Reolon J.B., Haas S.E., Colomé L.M., Jesse C.R. Chrysin loaded lipid-core nanocapsules ameliorates neurobehavioral alterations induced by β-amyloid1-42 in aged female mice. Behav. Brain Res. 2020;390 doi: 10.1016/j.bbr.2020.112696. [DOI] [PubMed] [Google Scholar]
- 261.Saleem S., Banerjee R., Rajesh Kannan R. Chrysin-loaded chitosan nanoparticle-mediated neuroprotection in Aβ 1–42 -induced neurodegenerative conditions in zebrafish. ACS Chem. Neurosci. 2022;13:2017–2034. doi: 10.1021/acschemneuro.2c00240. [DOI] [PubMed] [Google Scholar]
- 262.Ting P., Srinuanchai W., Suttisansanee U., Tuntipopipat S., Charoenkiatkul S., Praengam K., Chantong B., Temviriyanukul P., Nuchuchua O. Development of chrysin loaded oil-in-water nanoemulsion for improving bioaccessibility. Foods. 2021;10:1912. doi: 10.3390/foods10081912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Vedagiri A., Thangarajan S. Mitigating effect of chrysin loaded solid lipid nanoparticles against Amyloid β25–35 induced oxidative stress in rat hippocampal region: an efficient formulation approach for Alzheimer's disease. Neuropeptides. 2016;58:111–125. doi: 10.1016/j.npep.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 264.Md S., Gan S.Y., Haw Y.H., Ho C.L., Wong S., Choudhury H. In vitro neuroprotective effects of naringenin nanoemulsion against β-amyloid toxicity through the regulation of amyloidogenesis and tau phosphorylation. Int. J. Biol. Macromol. 2018;118:1211–1219. doi: 10.1016/j.ijbiomac.2018.06.190. [DOI] [PubMed] [Google Scholar]
- 265.Abbas H., El Sayed N.S., Youssef N.A.H.A., Gaafar P.M.E., Mousa M.R., Fayez A.M., Elsheikh M.A. Novel luteolin-loaded chitosan decorated nanoparticles for brain-targeting delivery in a sporadic Alzheimer's disease mouse model: focus on antioxidant, anti-inflammatory, and amyloidogenic pathways. Pharmaceutics. 2022;14:1003. doi: 10.3390/pharmaceutics14051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Elsheikh M.A., El-Feky Y.A., Al-Sawahli M.M., Ali M.E., Fayez A.M., Abbas H. A brain-targeted approach to ameliorate memory disorders in a sporadic Alzheimer's disease mouse model via intranasal luteolin-loaded nanobilosomes. Pharmaceutics. 2022;14:576. doi: 10.3390/pharmaceutics14030576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ning W., Lv R., Xu N., Hou X., Shen C., Guo Y., Fan Z., Cao N., Liu X.-P. Lycopene-loaded microemulsion regulates neurogenesis in rats with Aβ-induced Alzheimer's disease rats based on the Wnt/β-catenin pathway. Neural Plast. 2021;2021:1–15. doi: 10.1155/2021/5519330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Guo Y., Fan Z., Zhao S., Yu W., Hou X., Nie S., Xu S., Zhao C., Han J., Liu X. Brain-targeted lycopene-loaded microemulsion modulates neuroinflammation, oxidative stress, apoptosis and synaptic plasticity in β-amyloid-induced Alzheimer's disease mice. Neurol. Res. 2023;45:753–764. doi: 10.1080/01616412.2023.2203615. [DOI] [PubMed] [Google Scholar]
- 269.Saini S., Sharma T., Jain A., Kaur H., Katare O.P., Singh B. Systematically designed chitosan-coated solid lipid nanoparticles of ferulic acid for effective management of Alzheimer's disease: a preclinical evidence. Colloids Surf. B Biointerfaces. 2021;205 doi: 10.1016/j.colsurfb.2021.111838. [DOI] [PubMed] [Google Scholar]
- 270.Harakeh S., Qari M.H., Ramadan W.S., Al Jaouni S.K., Almuhayawi M.S., Al Amri T., Ashraf G.Md., Bharali D.J., Mousa S.A. A novel nanoformulation of ellagic acid is promising in restoring oxidative homeostasis in rat brains with Alzheimer's disease. Curr. Drug Metabol. 2021;22:299–307. doi: 10.2174/1389200221666201216170851. [DOI] [PubMed] [Google Scholar]
- 271.Andrade S., Pereira M.C., Loureiro J.A. Caffeic acid loaded into engineered lipid nanoparticles for Alzheimer's disease therapy. Colloids Surf. B Biointerfaces. 2023;225 doi: 10.1016/j.colsurfb.2023.113270. [DOI] [PubMed] [Google Scholar]
- 272.Cano A., Ettcheto M., Chang J.-H., Barroso E., Espina M., Kühne B.A., Barenys M., Auladell C., Folch J., Souto E.B., Camins A., Turowski P., García M.L. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer's disease mice model. J. Contr. Release. 2019;301:62–75. doi: 10.1016/j.jconrel.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Smith A., Giunta B., Bickford P.C., Fountain M., Tan J., Shytle R.D. Nanolipidic particles improve the bioavailability and α-secretase inducing ability of epigallocatechin-3-gallate (EGCG) for the treatment of Alzheimer's disease. Int. J. Pharm. 2010;389:207–212. doi: 10.1016/j.ijpharm.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Tao X., Zhang R., Wang L., Li X., Gong W. Luteolin and exercise combination therapy ameliorates amyloid-β1-42 oligomers-induced cognitive impairment in AD mice by mediating neuroinflammation and autophagy. J. Alzheim. Dis. 2023;92:195–208. doi: 10.3233/JAD-220904. [DOI] [PubMed] [Google Scholar]
- 275.Park S., Kim D.S., Kang S., Kim H.J. The combination of luteolin and l-theanine improved Alzheimer disease–like symptoms by potentiating hippocampal insulin signaling and decreasing neuroinflammation and norepinephrine degradation in amyloid-β–infused rats. Nutr. Res. 2018;60:116–131. doi: 10.1016/j.nutres.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 276.Paterniti I., Cordaro M., Campolo M., Siracusa R., Cornelius C., Navarra M., Cuzzocrea S., Esposito E. Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer's disease models: the control of neuroinflammation. CNS Neurol. Disord.: Drug Targets. 2014;13:1530–1541. doi: 10.2174/1871527313666140806124322. [DOI] [PubMed] [Google Scholar]
- 277.Yu L., Wang W., Pang W., Xiao Z., Jiang Y., Hong Y. Dietary lycopene supplementation improves cognitive performances in tau transgenic mice expressing P301L mutation via inhibiting oxidative stress and tau hyperphosphorylation. J. Alzheim. Dis. 2017;57:475–482. doi: 10.3233/JAD-161216. [DOI] [PubMed] [Google Scholar]
- 278.Okuda M., Fujita Y., Sugimoto H. The additive effects of low dose intake of ferulic acid, phosphatidylserine and curcumin, not alone, improve cognitive function in APPswe/PS1dE9 transgenic mice. Biol. Pharm. Bull. 2019;42:1694–1706. doi: 10.1248/bpb.b19-00332. [DOI] [PubMed] [Google Scholar]
- 279.Sharman M.J., Gyengesi E., Liang H., Chatterjee P., Karl T., Li Q.-X., Wenk M.R., Halliwell B., Martins R.N., Münch G. Assessment of diets containing curcumin, epigallocatechin-3-gallate, docosahexaenoic acid and α-lipoic acid on amyloid load and inflammation in a male transgenic mouse model of Alzheimer's disease: are combinations more effective? Neurobiol. Dis. 2019;124:505–519. doi: 10.1016/j.nbd.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 280.Shimmyo Y., Kihara T., Akaike A., Niidome T., Sugimoto H. Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregulation. Neuroreport. 2008;19:1329–1333. doi: 10.1097/WNR.0b013e32830b8ae1. [DOI] [PubMed] [Google Scholar]
- 281.Kshirsagar S., Alvir R.V., Pradeepkiran J.A., Hindle A., Vijayan M., Ramasubramaniam B., Kumar S., Reddy A.P., Reddy P.H. A combination therapy of urolithin A+EGCG has stronger protective effects than single drug urolithin A in a humanized amyloid beta knockin mice for late-onset Alzheimer's disease. Cells. 2022;11:2660. doi: 10.3390/cells11172660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Walker J.M., Klakotskaia D., Ajit D., Weisman G.A., Wood W.G., Sun G.Y., Serfozo P., Simonyi A., Schachtman T.R. Beneficial effects of dietary EGCG and voluntary exercise on behavior in an Alzheimer's disease mouse model. J. Alzheim. Dis. 2015;44:561–572. doi: 10.3233/JAD-140981. [DOI] [PubMed] [Google Scholar]
- 283.Ali A.A., Khalil M.G., Abd El-latif D.M., Okda T., Abdelaziz A.I., Abu-Elfotuh karema, Kamal M.M., Wahid A. The influence of vinpocetine alone or in combination with Epigallocatechin-3-gallate, Coenzyme COQ10, Vitamin E and Selenium as a potential neuroprotective combination against aluminium-induced Alzheimer's disease in Wistar Albino Rats. Arch. Gerontol. Geriatr. 2022;98 doi: 10.1016/j.archger.2021.104557. [DOI] [PubMed] [Google Scholar]
- 284.Weigert H., Stuckenschneider T., Pickert L., Rossi A., Meyer A.M., Nelles G., Schulz R.-J., Stahl W., Schneider S., Polidori M.C. Influence of a 12-month structured exercise program on the micronutrient-cognitive fitness-physical association profiles in mild cognitive impairment. J Alzheimers Dis Rep. 2022;6:711–722. doi: 10.3233/ADR-220039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Galluzzi S., Zanardini R., Ferrari C., Gipponi S., Passeggia I., Rampini M., Sgrò G., Genovese S., Fiorito S., Palumbo L., Pievani M., Frisoni G.B., Epifano F. Cognitive and biological effects of citrus phytochemicals in subjective cognitive decline: a 36-week, randomized, placebo-controlled trial. Nutr. J. 2022;21:64. doi: 10.1186/s12937-022-00817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Liu Y., Yu S., Wang F., Yu H., Li X., Dong W., Lin R., Liu Q. Chronic administration of ellagic acid improved the cognition in middle-aged overweight men. Appl. Physiol. Nutr. Metabol. 2018;43:266–273. doi: 10.1139/apnm-2017-0583. [DOI] [PubMed] [Google Scholar]
- 287.Kudoh C., Hori T., Yasaki S., Ubagai R., Tabira T. Effects of ferulic acid and angelica archangelica extract (Feru-guard ®) on mild cognitive impairment: a multicenter, randomized, double-blind, placebo-controlled prospective trial. J Alzheimers Dis Rep. 2020;4:393–398. doi: 10.3233/ADR-200211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Swamy M.K., Sinniah U.R. Patchouli (Pogostemon cablin Benth.): botany, agrotechnology and biotechnological aspects. Ind. Crops Prod. 2016;87:161–176. doi: 10.1016/J.INDCROP.2016.04.032. [DOI] [Google Scholar]
- 289.Robinson M.M., Zhang X. World Health Organization; Geneva: 2011. The World Medicines Situation 2011, Traditional Medicines: Global Situation, Issues and Challenges; pp. 1–2. [Google Scholar]
- 290.Liu Y., Sun Z., Da Z., Sun R. World traditional medicine patent database and its applications. World Patent Inf. 2014;36:40–46. doi: 10.1016/J.WPI.2013.12.001. [DOI] [Google Scholar]
- 291.Mohanty S.K., Swamy M.K., Sinniah U.R., Anuradha M. Leptadenia reticulata (Retz.) Wight & Arn. (Jivanti): Botanical, Agronomical, phytochemical, pharmacological, and biotechnological aspects. Molecules. 2017;22:1019. doi: 10.3390/molecules22061019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Atanasov A.G., Zotchev S.B., Dirsch V.M., Supuran C.T. Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov. 2021;20:200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Lila M.A., Raskin I. Health-related interactions of phytochemicals. J. Food Sci. 2005;70:R20–R27. doi: 10.1111/j.1365-2621.2005.tb09054.x. [DOI] [Google Scholar]
- 294.Echegaray N., Gullón B., Pateiro M., Amarowicz R., Misihairabgwi J.M., Lorenzo J.M. Date fruit and its by-products as promising source of bioactive components: a review. Food Rev. Int. 2023;39:1411–1432. doi: 10.1080/87559129.2021.1934003. [DOI] [Google Scholar]
- 295.Poudel P., Petropoulos S.A., Di Gioia F. Natural Secondary Metabolites. Springer International Publishing; Cham: 2023. Plant tocopherols and phytosterols and their bioactive properties; pp. 285–319. [DOI] [Google Scholar]
- 296.Jiménez-Moreno N., Esparza I., Bimbela F., Gandía L.M., Ancín-Azpilicueta C. Valorization of selected fruit and vegetable wastes as bioactive compounds: opportunities and challenges. Crit. Rev. Environ. Sci. Technol. 2020;50:2061–2108. doi: 10.1080/10643389.2019.1694819. [DOI] [Google Scholar]
- 297.Tariq A., Sahar A., Usman M., Sameen A., Azhar M., Tahir R., Younas R., Issa Khan M. Extraction of dietary fiber and polyphenols from mango peel and its therapeutic potential to improve gut health. Food Biosci. 2023;53 doi: 10.1016/j.fbio.2023.102669. [DOI] [Google Scholar]
- 298.Khan F., Joshi A., Devkota H.P., Subramaniyan V., Kumarasamy V., Arora J. Dietary glucosinolates derived isothiocyanates: chemical properties, metabolism and their potential in prevention of Alzheimer's disease. Front. Pharmacol. 2023;14 doi: 10.3389/fphar.2023.1214881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Grodzicki W., Dziendzikowska K. The role of selected bioactive compounds in the prevention of Alzheimer's disease. Antioxidants. 2020;9:229. doi: 10.3390/antiox9030229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Di Pede G., Mena P., Bresciani L., Almutairi T.M., Del Rio D., Clifford M.N., Crozier A. Human colonic catabolism of dietary flavan-3-ol bioactives. Mol. Aspect. Med. 2023;89 doi: 10.1016/j.mam.2022.101107. [DOI] [PubMed] [Google Scholar]
- 301.de Araújo F.F., de Paulo Farias D., Neri-Numa I.A., Pastore G.M. Polyphenols and their applications: an approach in food chemistry and innovation potential. Food Chem. 2021;338 doi: 10.1016/J.FOODCHEM.2020.127535. [DOI] [PubMed] [Google Scholar]
- 302.Korhonen H., Pihlanto A. Bioactive peptides: production and functionality. Int. Dairy J. 2006;16:945–960. doi: 10.1016/J.IDAIRYJ.2005.10.012. [DOI] [Google Scholar]
- 303.Gómez-Estaca J., López-de-Dicastillo C., Hernández-Muñoz P., Catalá R., Gavara R. Advances in antioxidant active food packaging. Trends Food Sci. Technol. 2014;35:42–51. doi: 10.1016/J.TIFS.2013.10.008. [DOI] [Google Scholar]
- 304.Arfaoui L. Dietary plant polyphenols: effects of food processing on their content and bioavailability. Molecules. 2021;26:2959. doi: 10.3390/molecules26102959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kammeyer A., Luiten R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015;21:16–29. doi: 10.1016/j.arr.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 306.Tian F., Decker E.A., Goddard J.M. Controlling lipid oxidation of food by active packaging technologies. Food Funct. 2013;4:669. doi: 10.1039/c3fo30360h. [DOI] [PubMed] [Google Scholar]
- 307.Chuarienthong P., Lourith N., Leelapornpisid P. Clinical efficacy comparison of anti‐wrinkle cosmetics containing herbal flavonoids. Int. J. Cosmet. Sci. 2010;32:99–106. doi: 10.1111/j.1468-2494.2010.00522.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.