

Abstract
Continually repeating outbreaks of pathogenic viruses necessitate the construction of effective antiviral strategies. Therefore, the development of new specific antiviral drugs in a well-established and efficient manner is crucial. Taking into account the strong ability of viruses to change, therapies with diversified molecular targets must be sought. In addition to the widely explored viral enzyme inhibitor approach, inhibition of protein–protein interactions is a very valuable strategy. In this Perspective, protein–protein interaction inhibitors targeting HIV, SARS-CoV-2, HCV, Ebola, Dengue, and Chikungunya viruses are reviewed and discussed. Antibodies, peptides/peptidomimetics, and small molecules constitute three classes of compounds that have been explored, and each of them has some advantages and disadvantages for drug development.
Significance
Protein–protein interaction inhibitors are a highly important and emerging group of antiviral agents.
Numerous successful examples of compounds targeting HIV, SARS-CoV-2, HCV, Ebola, Dengue, and Chikungunya viruses are discussed.
Improving methodologies of protein–protein interaction inhibitor design and development make this group of molecules more attractive in comparison to widely explored enzyme inhibitors.
Introduction
Viruses have coexisted with humans forever, and they cause epidemics of various geographical ranges and intensities in a regular fashion.1 Recently, these outbreaks have included severe acute respiratory syndrome (SARS) (2002), influenza A/H1N1 (2009), Ebola (2013), and SARS-CoV-2 (2019) viruses. Due to the high propensity of viruses to mutate, new viral pathogens can be expected to continuously threaten humans.2 Therefore, the development of antiviral strategies is and will remain highly important in the future.3,4 Taking into consideration the molecular mode of action, enzyme inhibitors are studied most intensively due to the importance of enzymes in the viral replication process as well as the well-established experimental and computational methodologies of enzyme inhibitor discovery.5,6 However, fast changes in viruses necessitate the application of various strategies; thus, other options, including protein–protein interaction inhibitors, are also of high importance.
Protein–protein interactions (PPIs) regulate a large number of processes in biological systems and thus are often targets for the treatment of various diseases.7,8 Although major efforts in this field are focused on the development of PPI inhibitors with anticancer activity, other applications of this strategy are also of high importance. The discovery of antiviral PPIs has already been significant, over a 20-year-long history, with the first drug (Enfuvirtide) approved by the FDA in 2003. The use of PPI inhibitors in the treatment of various viral infections has already been shown.
The most explored strategy is related to the construction of virus entry inhibitors and, thus, the interaction of human cell surface proteins with virus capsid proteins. It has already been shown to be effective and marketed for anti-HIV treatment, while drug candidates for HCV, SARS-CoV-2, and other viruses have been found. The HIV fusion process includes interactions of gp41 and gp120 of HIV and CD4, CCR5, and CXCR4 of humans. In the case of SARS-CoV-2, the interaction of the viral spike protein with human ACE2 is crucial for entry. HCV interacts with several surface human proteins, including SR-BI, CD81, CLDN1, and OCLN. Notably, other types of protein–protein interactions that can be targeted by antivirals; e.g., inhibitors of HIV protease or reverse transcriptase dimerization or oligomerization of integrase, were investigated.
Three major approaches for the development of antiviral protein–protein interaction inhibitors can be distinguished, namely, low-molecular-weight compounds,9,10 peptide-based inhibitors,11,12 and antibodies. Small molecules are usually challenging to develop because of the discrepancy between the large surface of the interaction between proteins and their size. In particular, small molecules can bind to one hot spot, unless other hot spots are nearby. However, there are some successful stories in this area, and two drugs, maraviroc, which binds to CCR5, and fostemsavir, which interacts with gp120, have been marketed. On the other hand, medium-sized compounds, including peptides and peptidomimetics, have been effectively explored and provide highly active drug candidates. Their size allows for reaching several spatially separated hot spots. In recent years, antiviral peptides have gained increasing interest as highly specific and effective potential therapeutics of natural or computational origin with diverse activities and minimal toxicity. Enfuvirtide, a 36-mer peptide, is the first marketed drug of this type. Finally, antibodies, in spite of their disadvantages, can also be used to develop inhibitors of protein–protein interactions, for example, ibalizumab, an anti-HIV drug binding to CD4.
In summary, various strategies for the construction of protein–protein interaction inhibitors of antiviral activity will be discussed in this perspective article. This concerns both the analysis of possible molecular targets and the possibility of the application of compounds derived from various classes.
HIV Inhibitors
Acquired immunodeficiency syndrome (AIDS) develops from the infection and subsequent depletion of T lymphocytes caused by the human immunodeficiency viruses (HIV-1 and HIV-2).13,14 With more than 38 million people living with HIV, this global epidemic continues to present a significant global health problem, and the development of effective new treatment strategies and medicines remains a critically important challenge.15 The greater infectivity of the HIV-1 type, which is more readily transmitted, makes it predominant and responsible for the AIDS pandemic.16
The process of HIV-1 entering and infecting a target cell starts with the interaction between an HIV-1 envelope glycoprotein (HIV-1 Env), gp120, and a cellular receptor protein, CD4. Mutual attachment leads to conformational changes in gp120 that prompt its binding to a coreceptor on the cell surface (C–C chemokine receptor type 5, CCR5 or C-X-C chemokine receptor type 4, CXCR4). This binding triggers a conformational rearrangement of another HIV-1 envelope glycoprotein, gp41, causing an interaction with the host cell membrane and resulting in the fusion of the viral envelope with the cellular membrane.17,18 Consequently, HIV-1 invasion can be divided into four stages: (1) viral attachment to the host cell; (2) gp120-CD4 binding; (3) gp120-coreceptor binding; and (4) fusion between the virus and cell membrane. Therefore, gp120, CD4, coreceptors (CCR5 or CXCR4), and gp41 are all essential elements in the entry process and represent attractive drug targets.19 Antiviral agents that may disrupt any step of the presented process can prevent viral entry and are categorized as HIV-1 virus entry inhibitors. It is important to point out that entry inhibitors are typically used in combination with other antiretroviral drugs to form highly active antiretroviral therapy (HAART) regimens, predominantly for “heavily treatment experienced” (HTE) individuals with limited treatment options, usually due to extensive drug resistance. According to the steps of the viral entry process, entry inhibitors can be broadly classified into four categories: (1) preattachment inhibitors, blocking the first phase of viral attachment; (2) postattachment inhibitors, binding to CD4 receptors and inducing conformational changes in the CD4–gp120 complex; (3) CCR5 antagonists, binding to the coreceptor CCR5 and preventing gp120–coreceptor attachment; and (4) fusion inhibitors, interacting with gp41, preventing viral and host cell membranes from coming into close proximity and disabling membrane fusion. Four HIV-1 entry-inhibitor-based medicines have been approved by the U.S. Food and Drug Administration (FDA) (Table 1).
Table 1. FDA-Approved HIV-1 Entry Inhibitors.
| generic name | structure type | drug class |
|---|---|---|
| Ibalizumab | antibody | postattachment inhibitor |
| Enfuvirtide | peptide | fusion inhibitor |
| Fostemsavir | small molecule | preattachment inhibitor |
| Maraviroc | small molecule | CCR5 antagonist |
Antibody-Based Inhibitors
Antibody-based therapies possess several advantages over small molecule-based treatment in various aspects, such as a high degree of specificity for the target and safety.20 Treatment with a monoclonal antibody (mAb) brings several advantages for HIV therapy, including a unique mechanism of action, capacity to restore CD4 T-cell counts, prolonged time to resistance development, and a low potential for toxicities.
Ibalizumab (TNX-355) is a humanized IgG4 mAb that targets the CD4 receptor and blocks HIV-1 infection.21 A phase 3 trial demonstrated that ibalizumab is a safe and well-tolerated drug and can be used as a monotherapy, especially for multidrug-resistant HIV patients with limited treatment options.22 Researchers are currently developing a new formulation of ibalizumab for intramuscular injection use. Studies suggest that ibalizumab does not inhibit the process of binding of gp120 to CD4, which occurs in the domain 1 region. It appears to exert its antiviral effect by postbinding conformational effects that prevent CD4-bound gp120 from interacting with CCR5 or CXCR4 without interfering with major histocompatibility complex class II (MHC-II)-mediated immune function.23 This is consistent with the fact that ibalizumab is well tolerated when introduced to patients and has demonstrated promising anti-HIV-1 activity without causing immunosuppression. The crystal structure of the ibalizumab Fab fragment in complex with the two N-terminal domains of human CD4 was reported and confirmed binding of ibalizumab on the opposite side of both gp120 and MHC class II binding sites (Figure 1).24
Figure 1.

Crystal structure of the ibalizumab-CD4 complex (PDB id: 3O2D) (A) and a fragment of CD4 with selected interacting residues of the antibody (B). CD4 is shown as a cyan ribbon or as a solvent-accessible surface colored by interpolated charge (blue–positive, gray–neutral, red–negative). The antibody is shown as a green or orange ribbon for the light and heavy chains, respectively. Interacting residues are shown in stick representation, with carbon atoms colored the same as the parent chain. Intermolecular interactions are shown as dashed lines: green–hydrogen bonds, orange–charge-assisted hydrogen bonds, pink–hydrophobic interactions, and white–hydrogen bond donor/π interactions.
Broad neutralizing antibodies (bnAbs) are capable of neutralizing multiple HIV-1 viral strains by targeting conserved epitopes of viruses and have significantly contributed to HIV vaccine development.25 However, none of the FDA-approved therapeutic antibodies are used for HIV treatment. One of the bnAbs, VRC01, effectively suppressed plasma viremia below detectable concentrations in a phase I trial, but the emergence of VRC01-resistant HIV after exposure is the main concern.26 The crystal structure of VRC01 in complex with an HIV-1 gp120 core showed that the heavy chain of VRC01 interacts with gp120 in a manner similar to CD4 (Figure 2).27 The results of a phase IIa trial using the HIV Env-specific antibody 3BNC117 showed that the antibody exerts strong selective pressure on HIV-1 emerging from latent reservoirs.28 In comparison to VRC01 and other less potent antibodies, 3BNC117 exhibited higher efficiency, which could be justified by its increased potency and/or a longer half-life.29,30 Research on anti-HIV therapeutic antibodies is continuing, and many of them have successfully advanced to human clinical trials.31
Figure 2.

Crystal structure of the VRC01 antibody-gp120 complex (PDB id: 5CD5) (A) and a fragment of gp120 with selected interacting residues of the antibody (B). gp120 is shown as a cyan ribbon or as a solvent-accessible surface colored by interpolated charge (blue–positive, gray–neutral, red–negative). The antibody is shown as violet or dark blue ribbons for the light and heavy chains, respectively. Interacting residues are shown in stick representation with carbon atoms colored the same as the parent chain. Intermolecular interactions are shown as dashed lines in the same color as in Figure 1.
Various antibodies targeting CCR5, such as HGS004, PRO140, and cenicriviroc, have shown promising antiviral effects and are now being evaluated in clinical trials.32−34 An anti-PDL1 antibody, BMS-936559, has been subjected to a phase I trial on HIV patients, showing that the immunologic check point inhibitor could enhance HIV-specific immunity in a subset of participants included in the trial.35
Peptide-Based Inhibitors
Peptide-based inhibitors, compared to small molecules, have the potential to be more efficient and specific due to larger surface areas, better target surface recognition, and potentially lower toxicity.36 Nevertheless, there are serious disadvantages limiting the application of peptides, such as proteolytic instability, conformational flexibility, and poor cellular penetration.37
Several synthetic peptides derived from the NHR (N-terminal heptad repeat regions) and CHR (C-terminal heptad repeat regions) of gp41 can proficiently inhibit HIV-1 infection by competitively binding to the exposed NHR or CHR in the gp41 prehairpin state, therefore blocking six-helix bundle (6-HB) formation in a dominant-negative manner.38,39 Several pairs of protease-resistant N- and C-peptides from gp41, including N36 and C34, which were later found to form the stable fusogenic core, were identified.40 The peptide C34 is more potent in inhibiting HIV-1 fusion than both SJ-217641 and T-20.42 It was proposed that C-peptide (e.g., C34) may be involved in blocking the formation of the fusion-active core of gp41 and inhibiting the fusion between the viral and target cell membranes.43 Enfuvirtide (T20/Fuzeon), a native CHR-derived peptide with 36 amino acids, was approved for clinical use in 2003 as a first member of a new class of anti-HIV drugs - HIV fusion inhibitors.44 However, it was determined that this peptide drug easily induces resistance in both clinical settings and laboratory studies.45 As a response to the drug resistance problem, many efforts have been undertaken to design and develop new anti-HIV agents including fusion inhibitors with improved stability and potency. The problem of fibrillation as a common issue in the development of therapeutic peptides has been addressed recently, and cucurbit[7]urils (CB[7]), a group of water-soluble macrocycles, was reported. These compounds are capable of modulating fibrillation behavior of the HIV fusion inhibitor enfuvirtide by specifically binding to the C-terminal Phe residue.46 The second-generation peptide, the fusion inhibitor T1249, exhibited enhanced antiviral potency, but its clinical development was discontinued owing to the drug formulation problem.47 Sifuvirtide (SFT) is a third-generation peptide-based HIV-1 fusion inhibitor approved for phase III clinical trials in China.48 It is an electrostatically constrained α-helical peptide fusion inhibitor showing potent anti-HIV activity, good safety, and pharmacokinetic profiles. The crystal structure of SFT in complex with its target NHR sequence was solved by using the peptide N36 as a surrogate (Figure 3).49 This reveals that SFT adopts a fully helical conformation stabilized by multiple engineered salt bridges that involve, in addition to the residues at the i and i + 4 positions, other charged residues contributing to stabilization.
Figure 3.
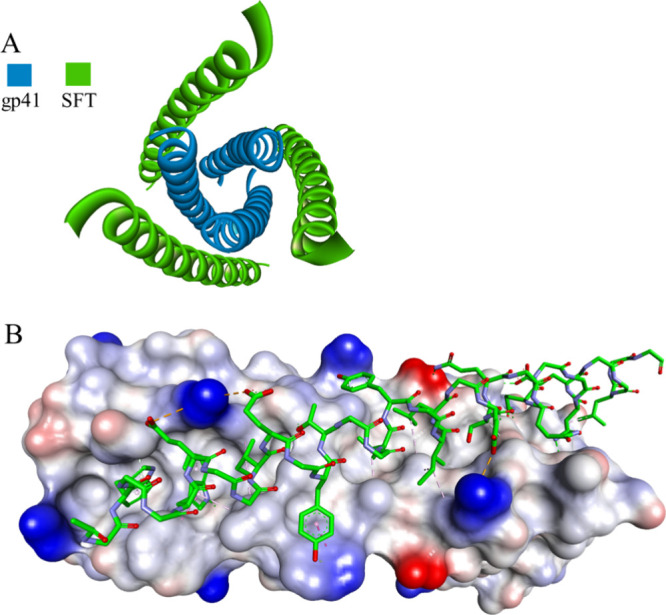
Crystal structure of the SFT-gp41 complex: top (A) and side view (B) (PDB id: 3VIE). gp41 is shown as a blue ribbon or as a solvent-accessible surface colored by interpolated charge (blue–positive, gray–neutral, red–negative). The SFT peptide is shown as a green ribbon or in stick representation colored according to atom types, and noninteracting side chains are hidden for clarity. Intermolecular interactions are shown as dashed lines in the same color as in Figure 1.
The crystal structure of another CHR-based peptide, CP32, with high efficiency against both enfuvirtide- and C34-resistant HIV-1 strains,50 showed that the residues Met115 and Thr116 preceding the pocket-binding domain (PBD) of the peptide form a hook-like structure, named the M-T hook (Figure 4).51
Figure 4.
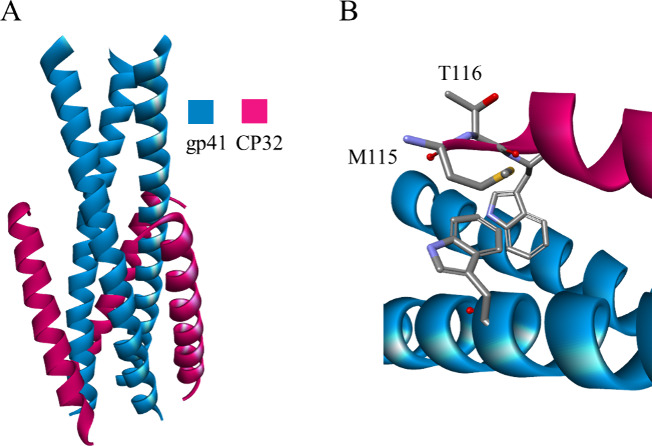
Crystal structure of the CP32 peptide-gp41 complex (A) and enlargement of the hook region (B) (PDB id: 3VTP). Gp41 and CP32 are shown as blue and red ribbons, respectively. The hook region is shown in stick representation colored according to atom types.
The structure of this molecule was modified with 11 of 32 residues mutated, giving peptide CP32M, which exhibited significant anti-HIV activity against enfuvirtide-resistant viruses.52 Positively or negatively charged residues were introduced to promote the formation of ion pairs (salt bridges) or to increase the hydrophilicity of the peptide. The findings related to the structure of CP32 also led to the introduction of the M-T hook into the Sifuvirtide structure, and the resulting peptide (MT-SFT) showed enhanced binding affinity and anti-HIV activity compared to Sifuvirtide, as well as an increased genetic barrier to the development of drug resistance.53 This strategy was also adopted in designing other short-peptide fusion inhibitors, such as a 24-residue peptide named MT-SC22EK by adding two hook residues to the N-terminus of the poorly active short-peptide SC22EK, which did show high activity against diverse HIV-1 variants.54 The crystal structures of these two peptides led to the development of a highly potent short-peptide inhibitor named HP23,55 whose structure was improved by the substitution of the methionine residue in the M-T hook structure with leucine to avoid potential oxidation problems, thus resulting in the inhibitor HP23L.56 Additionally, different lipids were introduced into the C-terminus of this inhibitor through a PEG linker, and one of these derivatives, termed LP-11, demonstrated strong anti-HIV activity. The M-T hook strategy has also been employed to design a fusion inhibitor, a 23-mer helical peptide named 2P23 that was effective against both HIV-1 and HIV-2 isolates.57 A lipopeptide derivative, LP-19, with improved binding stability and antiviral activity, was subsequently obtained by adding a fatty acid group to the C-terminus of 2P23.58 Moreover, it was shown that LP-19 exhibits a broad spectrum anti-HIV activity, and high drug resistance barrier compared to C34.59
Sulfonyl-γ-AApeptides that were designed to mimic MT-SC22EK, and showed comparable activity to this peptide, exhibited exceptional resistance to proteolysis, favorable PAMPA (parallel artificial membrane permeability assay) permeability, as well as promising oral bioavailability.60 Further investigations indicated that these sulfonyl-γ-AApeptides act by mimicking the CHR of gp41 and tightly bind NHR, resulting in inhibition of the 6-HB structure formation.
The crystal structures of HP23L and LP-11 bound to a target-mimic NHR peptide with the wild-type sequence or resistant mutations were determined (Figure 5A).61 The interhelical hydrogen bonds and salt bridges between HP23L and gp41 critically determined the binding of the inhibitors, and rich hydrophobic interactions played fundamental roles in stabilizing the whole structure (Figure 5B). Leu-115 and Thr-116 adopted a hooklike (designated L-T hook) structure, with Leu-115 forming numerous hydrophobic interactions with multiple pocket-forming residues, strengthening the stability of the L-T hook structure and the binding of inhibitors. Additionally, hydrogen bonds in the L-T hook region increased the binding stability of the hook and pocket region (Figure 5C).
Figure 5.

Crystal structure of the HP23L-gp41 complex (PDB id 5YB3) (A). Intermolecular interactions stabilizing the complex (B) and the structure of the L-T hook (C). gp41 is shown as a blue ribbon or as a solvent-accessible surface colored by interpolated charge (blue–positive, gray–neutral, red–negative). The HP23L peptide is shown as an orange ribbon or in stick representation colored according to atom types, and noninteracting side chains are hidden for clarity. Intermolecular interactions are shown as dashed lines in the same color as in Figure 1.
A multitarget-directed ligand strategy was used to design entry inhibitors that combine the pharmacological activities of a peptide fusion inhibitor disrupting HIV-1 gp41 glycoprotein hexameric coiled-coil assembly and a small-molecule CCR5 antagonist.62 Among these, dual-target 23-residue peptides SP12T and SP12L showed significantly increased inhibitory activities against HIV-1 replication in comparison to enfuvirtide. The same research group designed and engineered a chimeric peptide-based bifunctional HIV-1 entry inhibitor, AP3P4E, by incorporating the TAK-220 structure (Figure 6) to an artificially designed gp41-binding peptide, resulting in improved antiviral capabilities compared to parent components, as well as HIV-1 fusion inhibitor, enfuvirtide.
Figure 6.

Structures of small molecule HIV entry inhibitors.
Small-Molecule Inhibitors
Small-molecule HIV-1 entry inhibitors targeting early events in the virus life cycle represent an important class of valuable drugs. Their application has certain advantages in terms of lower cost, longer half-life in vivo, and oral availability, making them more convenient to use than, for example, peptide-based therapeutics. The first in action, preattachment inhibitors, are responsible for gp120–CD4 interaction inhibition, thus preventing the first stage of viral attachment to the target cell. One of the most effective entry inhibitor classes targeting the HIV-1 Env gp120 subunit was developed by Bristol-Myers Squibb and includes a group of compounds sharing a piperazine core motif (Figure 6).63
From this class of inhibitors, temsavir (BMS-626529) showed potency against many of the major subtypes of HIV-1, but due to its low solubility, it showed poor bioavailability. Fostemsavir (BMS-663068), a phosphooxymethyl prodrug of temsavir designed to address dissolution- and solubility-limited absorption issues, binds to gp120, disabling the conformational rearrangements normally activated by CD4 binding and ultimately resulting in fusion of the virus to the target cell.64 Seventeen clinical studies on the pharmacodynamics, pharmacokinetics, drug–drug interactions, safety, and bioavailability of fostemsavir and temsavir have been completed, with five ongoing clinical studies, including one phase IV clinical trial.65 Fostemsavir has performed well in clinical trials and was approved for use by the FDA in 2020. However, studies indicated suboptimal solubility after cleavage of the prodrug and scope concerns for specific subtypes of HIV-1. For this reason, fostemsavir is only suggested for treatment-experienced patients with limited therapeutic options and developed resistance to multiple existing antiretroviral drugs. The X-ray structure of the cocrystal complex of gp120 with temsavir revealed an induced binding pocket under the β20–21 loop (Figure 7), distinct from the docking studies that place this type of compound in the Phe43 cavity.66
Figure 7.

Mode of binding of temsavir to gp120: the temsavir-gp120 complex (A) and closeup of the binding cavity of gp120 with interactions between temsavir and the target (PDB id 5U7O). gp120 is shown as a cyan ribbon with β20-β12 and CD4-binding loops colored red and yellow, respectively. Interacting residues (B) are shown in stick representation colored according to atom type and carbon atom color that matches the parent chain color. Temsavir is shown as space-filling balls. Intermolecular interactions are shown as dashed lines in the same color as in Figure 1.
CD4 mimetics (CD4m) are a class of compounds designed to competitively bind to HIV gp120 protein, effectively blocking its interaction with the CD4 receptor and preventing viral entry. Keeping in mind that the Phe43 cavity of the HIV-1 gp120 envelope glycoprotein plays a crucial role in the interaction with the CD4 receptor during HIV entry, there is continued interest in developing CD4 mimetics that specifically target the Phe43 pocket to inhibit viral entry. CD4m compounds were initially identified with the discovery of NBD-556 and NBD-557 (Figure 6), two small molecules composed of an aromatic ring, an oxalamide linker, and a piperidine moiety.67 These molecules inhibit HIV-1 entry into cells expressing CD4 and a coreceptor.68 The cocrystal structure of NBD-556 bound to the extended gp120 core demonstrated that the oxalamide moiety formed two hydrogen bonds with backbone carbonyls of residues on opposite sides of the Phe43 cavity.69 Further modification of the NBD-556 structure led to new CD4 mimics, such as YIR-821 (Figure 6), containing a cyclohexane group in a spiro attachment to a piperidine ring and a guanidino group on the piperidine nitrogen atom.70 These molecules were designed to establish hydrophobic and electrostatic interactions with Val430 and Asp368 located in the entrance of the Phe43 cavity of gp120. These interactions were confirmed by the molecular modeling of YIR-821 docked into gp120. This compound exhibited remarkable synergistic anti-HIV activity when coadministered with the neutralizing antibody KD-247, thus representing a promising lead, especially for use with neutralizing antibodies in combination therapy. To improve the low water solubility of the NBD-556 molecule that might correlate with relatively strong cytotoxicity, the hydrophobic aromatic ring was replaced with various pyridine-type moieties (Figure 6).71 Some of these derivatives showed high anti-HIV activity, indicating that a halogen substituent on the carbon closest to the nitrogen atom in the pyridine ring can conserve anti-HIV activity. In addition, the CD4ms that have a cyclohexyl group instead of a tetramethyl group on the piperidine ring also showed high anti-HIV activity and no significant cytotoxicity. Recently, TKB-002, a hybrid CD4m containing a polyethylene glycol unit attached through an uncleavable linker, was developed and showed high anti-HIV activity and low cytotoxicity (Figure 6).72 This hybrid compound showed a more effective pharmacokinetic profile when tested in a rhesus macaque than did the parent compound, YIR-821. In addition, novel compounds were designed and synthesized, in which the phenyl group of YIR-821 and TKB-002 was replaced by a halopyridyl group with the halogen atoms and the nitrogen atom on the pyridine ring at different positions.73 Additionally, compounds without cyclohexane groups on the piperidine ring were prepared to increase the water solubility and decrease the hydrophobicity and cytotoxicity. Two of these compounds, 22a and 23, maintained promising anti-HIV activity but below that of YIR-821 (Figure 6).
The development of NBD-556-based entry inhibitors has been compromised after the discovery of its properties as CD4 agonist and its ability to promote HIV-1 infectivity in CD4-CCR5+ cells.74 A successful structure-based modification of the oxalamide midregion was achieved, and a CD4-antagonist named NBD-11021 was obtained (Figure 6, IC50 against Env-pseudotyped HIV-1 as low as 270 nM).75
The X-ray structure of NBD-11021 prompted further modification of the molecules in which the piperazine ring was replaced with an amine, giving NBD-14010 (Figure 8). The X-ray structure of NBD-14010 implied the modification of the thiazole ring substituents to obtain stronger interactions with the target protein.76 This led to the development of the hypothesis of the CH2OH “positional switch” that resulted in more potent antivirals, such as NBD-14136 (IC50 = 0.27 μM), NBD-14168 (IC50 = 0.28 μM), and NBD-14189 (IC50 = 0.089 μM).77 It was confirmed by X-ray analysis that the CH2OH group at position 5 in the thiazole ring showed no interactions, while switching of the CH2OH to position 4 in the thiazole ring, provides H bonding with Met426 and Gly431 (NBD-14189).78
Figure 8.
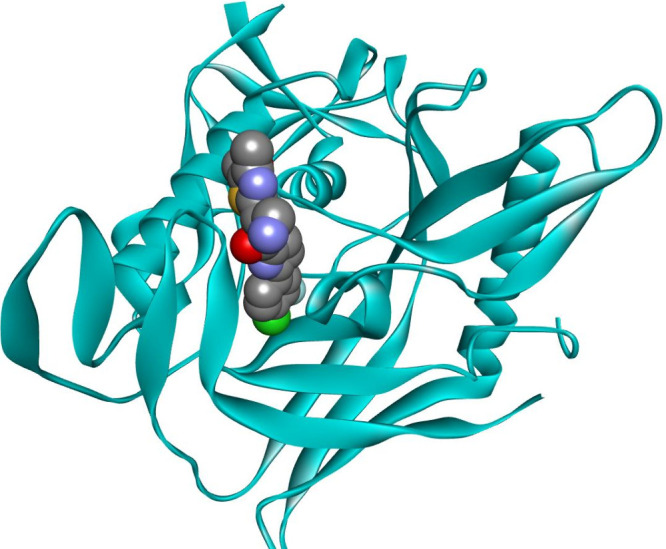
NBD-14010/CD120 complex, CD120 protein is shown as a cyan ribbon, and the inhibitor is shown as space-filling balls colored according to atom type.
To overcome the existing shortcomings of NBD-14189, such as high cytotoxicity and relatively poor aqueous solubility, the phenyl ring was replaced with a pyridine moiety.79 One of the new analogues, NBD-14270 (Figure 6), showed a noticeable enhancement in cytotoxicity, with 58-fold improvements in the selectivity index (SI) value as well as improvements in aqueous solubility compared to NBD-11021. A recent study that explored the replacement of the thiazole ring in NBD-14270 with two other positional thiazole isomers, revealed that the original scaffold provided the best HIV-1 inhibitors with a higher potency and better SI.80
The CD4m small molecule inhibitors BNM-III-170 and BNM-IV-147 were also developed to bind the Phe43 pocket but also to prevent infection of cells lacking CD4.81 Using single-particle cryo-EM, it was found that this inhibitor binds to the native-like BG505 Env trimer, resulting in its opening and structural rearrangements similar to those provoked by the CD4 host receptor.82
A carbamoyl derivative, named TAK-220 (Figure 6), showed good metabolic stability, potent binding affinity to CCR5 (IC50 = 3.5 nM), and excellent inhibition of membrane fusion (IC50 = 0.42 nM).83 For these reasons, it was further selected for Phase I clinical trials as a promising candidate for the treatment of HIV-1 infection.84
Maraviroc (Figure 6), a CCR5 antagonist, is a selective imidazopyridine molecule that received FDA approval for clinical use in 200785 and is currently being used to treat patients with resistance to multiple HIV drugs but has also been recently approved for first-line treatment regimens.86 This compound displayed good antiretroviral activity, without blocking potassium channels, and good absorption.87 It is also characterized by a good pharmacokinetic profile, relatively low protein binding, and high bioavailability.88 The structural details of the maraviroc-CCR5 complex have been determined through X-ray crystallography, providing insights into the mechanism of allosteric inhibition of chemokine signaling and viral entry (Figure 9).89 Vicriviroc is another CCR5 inhibitor that has entered phase III clinical trials.90 However, due to the inferior results of this compound compared to other drugs, clinical studies in treatment-naive patients have been stopped, but the development of Vicriviroc for treatment-experienced patients continues.
Figure 9.
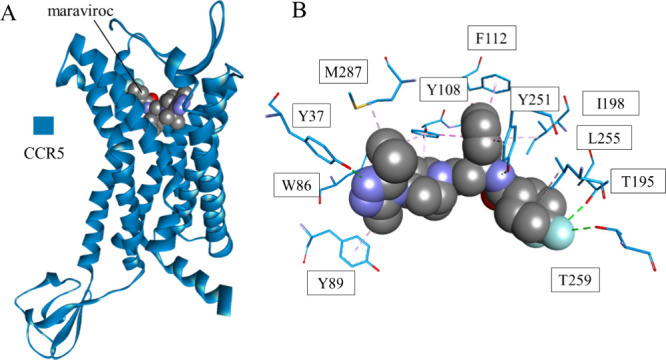
Crystal structure of maraviroc bound to CCR5 (PDB id: 4MBS) (A) and closeup of the interaction site of the drug (B). CCR5 is shown as a blue ribbon, and maraviroc is shown as space-filling balls colored according to the atom type. Interacting residues (B) are shown in stick representation colored according to the atom type, and the carbon atom color matches the parent chain color. Interactions are shown as dashed lines with the same color scheme as in Figure 1.
Recently, the structure of the CCR5-maraviroc complex (Figure 9) was used for the design and synthesis of novel tropane derivatives.91 Two of them, compounds 26 and 25 (Figure 6), showed activity comparable to maraviroc, without significant cytochrome P450 (CYP450) inhibition. Furthermore, docking analysis of these compounds showed a new binding mode with CCR5 in comparison to maraviroc-CCR5, which implies that this could be a strategy to overcome the problem of drug resistance derived from maraviroc.
Broadly neutralizing antibodies (bnAbs) of HIV-1, such as VRC01, possess exceptional potency against variant strains of HIV-1.92 An in silico study on natural product-derived compounds that mimic VRC01 has delivered molecules that could serve as templates for the design of next-generation HIV-1 inhibitors, giving novel insights into the binding mechanisms using molecular dynamics simulations.93
SARS-CoV INHIBITORS
Coronaviruses are a group of related enveloped, single-stranded, positively sensed RNA viruses. Although some coronaviruses can cause the common cold, with mild symptoms, others are highly pathogenic and have led to several outbreaks in recent years.94−96 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the pathogen responsible for COVID-19, which was declared a pandemic and a global emergency shortly after it began in late 2019. Therapeutics currently used for COVID-19 treatment include convalescent plasma, neutralizing antibodies, and repurposed drugs. Coronavirus entry inhibitors play an important role in the treatment of coronavirus diseases alone or in combination with other drugs. They are designed to block various processes of viral entry, including receptor binding, proteolytic activation of spike protein, or virus–cell membrane fusion. The SARS-CoV-2 entry pathways are well understood, in part due to the close similarity of SARS-CoV-2 to SARS-CoV, the causative agent for a global outbreak in 2002–2003.97 SARS-CoV-2 Spike (S protein), a homotrimeric protein found on the surface of the viral membrane, mediates the main entry steps, including receptor binding and membrane fusion.98 Each monomer consists of two noncovalently associated subunits, S1, which is responsible for binding the receptor, and S2, which mediates membrane fusion (Figure 10).
Figure 10.
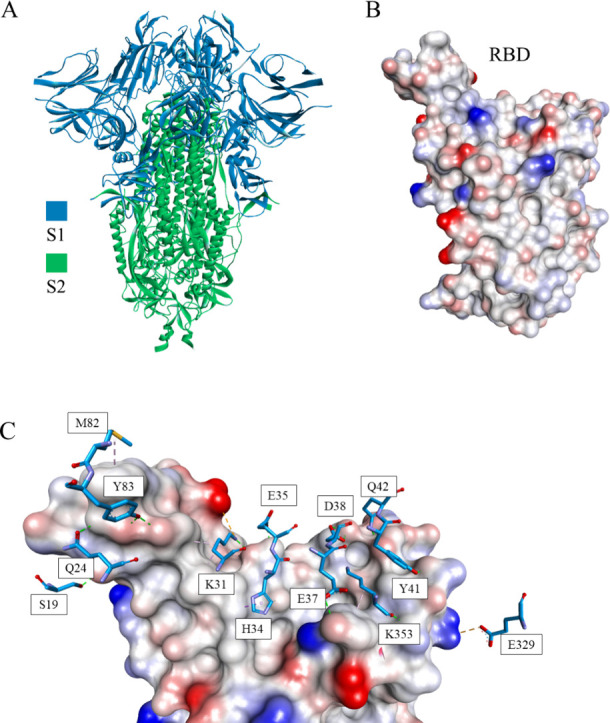
Crystal structure of the S protein (PDB id 6VXX) (A) and its receptor-binding domain (RBD) (B). S protein is shown in ribbon representation colored in blue and green for S1 and S2 chains, respectively. (C) The interactions between RBD and the S-protein have been shown with S1 represented as sticks (PDB ID: 6VW1).99 RBD is presented as a solvent-accessible surface colored by interpolated charge: red–negative, blue–positive, gray–neutral.
The S1 subunit consists of an N-terminal domain (NTD) and a receptor binding domain (RBD), which contains a subdomain called the receptor binding motif (RBM).100 The first step in viral entry, and an attractive therapeutic target, is the interaction of S1 RBD and angiotensin converting enzyme-2 (ACE-2) of the host cell.101 The next step is mediated by the S2 subunit and involves fusion with the cell membrane, an entry point that can also be selectively targeted. The S2 subunit is located at the C-terminus and is built from a fusion peptide (FP), two α-helical heptad repeats (HR1 and HR2), a loop region, a transmembrane (TM) domain, and a cytoplasmic tail (CT). While the S1–S2 boundary is cleaved by furin in the virus-producing cell, the S2′ site cleavage requires target cell proteases, and serine 2 transmembrane protease (TMPRSS2) and cathepsin L are the two main proteases involved in the activation of the S protein. After the formation of the S-ACE2 complex, four pairs of disulfide bonds stabilize the RBD structure, while the RBM forms a concave outer surface to accommodate the N-terminal helix of ACE2. Additionally, the complex is further stabilized through 13 hydrogen bonds, two salt bridges (between K417 of the RBD and D30 of ACE2), and several hydrophobic interactions (between F486 of the RBD and L79, M82, and Y83 of ACE2).102,103 Among these, salt bridge interactions between K417 of the SARS-CoV-2 S protein and D30 of ACE2 are not present in SARS-CoV. To date, none of the examined SARS-CoV-2 entry inhibitors have been approved by the FDA for treatment. However, to help strengthen public health protection against the spread of COVID-19 infection, the FDA has issued an emergency use authorization (EUA) for a few diverse agents with different mechanisms of action.104
Antibody-Based Inhibitors
The role of monoclonal antibodies (mAbs) in the inhibition of SARS-CoV-2 is to prevent viral attachment mostly by binding to a nonoverlapping epitope on the surface spike protein RBD with high affinity, thus preventing the virus from binding to the human ACE2 (hACE2) receptor.105 However, the application of mAbs has been limited due to the development of SARS-CoV-2 variants resistant to existing treatments.106 Although the majority of SARS-CoV-neutralizing human mAbs (hmAbs) specifically bind to the RBD of the S protein,107,108 there are hmAbs that do not use the same mechanism of inhibition as RBD-specific hmAbs and can bind to, for example, the S2 domain and neutralize the virus in the postbinding step of viral entry.109 This finding indicates that a mixture of antibodies recognizing distinct regions and targeting more than one step in viral entry is expected to be more efficient in neutralizing the virus and conquering the generation of escape mutants.
The high binding affinity of the antibody named P2B-2F6 to the RBD of SARS-CoV-2 (Kd = 5.14 nM) was found to be similar to that between ACE2 and the RBD (Kd = 4.70 nM), indicating ACE2 receptor engagement, which was also supported by the strong P2B-2F6 competition with ACE2.110 The crystal structure of P2B-2F6 in complex with the SARS-CoV-2 RBD indicates that P2B-2F6 attachment uses hydrophobic interactions around RBD residues Y449, L452, and F490 and hydrophilic interactions at the interface (Figure 11).
Figure 11.

Crystal structure of the P2B-2F6 antibody - RBD of the S protein complex (PDB id: 8DCC): the whole structure (A) and closeup showing intermolecular interactions (B). The antibody shown in the ribbon representation is colored orange and yellow for light and heavy chains, respectively. The RBD is presented as a solvent-accessible surface colored by interpolated charge: red–negative, blue–positive, gray–neutral (A) or green ribbon (B). Interacting residues are shown as sticks colored by atom type, and the colors of the carbon atoms match that of the ribbon of the same chain. Interactions are shown as dashed lines with the same color scheme shown in Figure 1.
Bamlanivimab (LY-CoV555 or LY3819253) is a potent neutralizing monoclonal antibody that targets the RBD of the SARS-CoV-2 S protein and has been shown to potently neutralize SARS-CoV-2.111 Structural analysis using X-ray crystallography and cryoelectron microscopy (cryo-EM) indicated that LY-CoV555 binds to an epitope overlapping the ACE2 binding site, and binding to the S protein RBD was observed in both up and down conformations (Figure 12). Bamlanivimab received EUA by the FDA at the end of 2020 for clinical use in nonhospitalized patients with mild to moderate conditions, but this decision was revoked after six months due to the emergence of SARS-CoV-2 variants resistant to bamlanivimab monotherapy.112 However, it is still used in combination with etesevimab, another monoclonal antibody.113
Figure 12.

Crystal structure of bamlanivimab-RBD of the S protein complex (PDB id: 7KMG): the whole structure (A) and closeup showing intermolecular interactions (B). The antibody shown in ribbon representation is colored in orange and yellow for light and heavy chains, respectively. The RBD is presented as a solvent-accessible surface colored by interpolated charge: red–negative, blue–positive, gray–neutral (A) or as a green ribbon (B). Interacting residues are shown as sticks colored by atom type, and the color of the carbon atoms matches that of the ribbon of the same chain. Interactions are shown as dashed lines with the same color scheme shown in Figure 1.
Bebtelovimab (LY-CoV1404) is a neutralizing monoclonal antibody targeting the S protein RBD of the SARS-CoV-2 virus and demonstrates broad neutralizing activity against all SARS-CoV-2 variants of concern (VOCs).114 Binding kinetic studies revealed that Bebtelovimab shows high affinity toward the S protein of D614G (Kd values between 790 pM and 4 nM). Both potent activity and structural analysis of binding of LY-CoV1404 to the RBD indicate that this antibody binds uniquely to an epitope with a low frequency of mutations. Bebtelovimab is equally effective at viral neutralization against all tested variants and several times more potent in viral neutralization assays than the antibody named VIR-7831, which also binds to an epitope of the SARS-CoV-2 S protein distinct from current VOCs and has clinical efficacy at a 500 mg dose.115
Peptide-Based Inhibitors
Although highly effective experimental and computational methodologies for the development of peptide-based protein binders are available, the discovery of S protein–human ACE2 interaction inhibitors was found to be nontrivial.116
Highly active small proteins (56- and 64-amino acid long sequences) incorporating three helices were designed de novo using computational methodology.117 Two approaches were applied: (a) incorporation of the helical fragment of ACE2 into the binder structure and (b) building new proteins from scratch. A high number of computationally designed molecules were initially screened to observe their interaction with fluorescently labeled RBD displayed on the surface of yeast cells. The second optimization step relied on the production of saturation mutagenesis libraries and checking of the enrichment after fluorescence-activated cell sorting (FACS). One of the obtained proteins, LCB3, exhibited high conformational stability (Tm > 95 °C) combined with RBD binding Kd values below 1 nM and live virus neutralization EC50 values below 50 pM. The cryo-EM structure of the LCB3-S protein complex revealed numerous intermolecular interactions mediated mainly by two or three helices of LCB3 (Figure 13). Notably, its N-terminal helix positioning is similar to that of helix 1 of ACE2.
Figure 13.
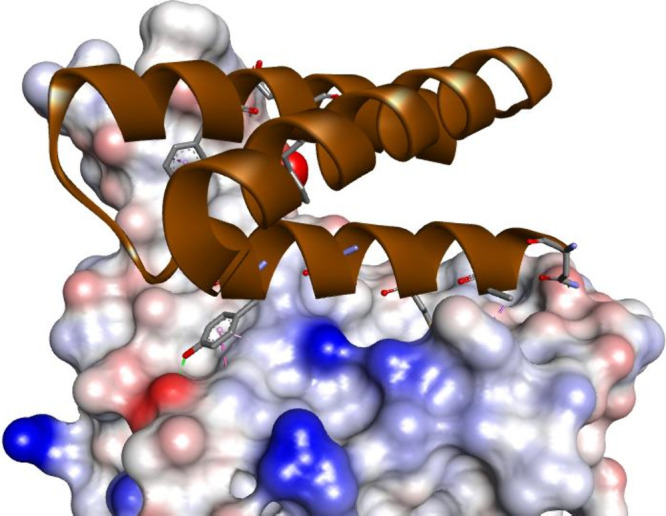
Cryo-EM structure of the small protein LCB3 bound to the RBD of the S protein (PDB id: 7JZM). LBC3 protein is shown as a brown ribbon with interacting residues represented as sticks colored according to the atom type. The RBD of the S protein is shown as a solvent-accessible surface that is colored according to the interpolated charge (red–negative, blue–positive, gray–neutral). Interactions are shown as dashed lines with the color scheme shown in Figure 1.
The discovery of shorter peptides that could inhibit ACE2–S protein interactions was difficult. Molecules that bind to RBD with Kd values in the nanomolar range up to Kd = 80 nM were identified by screening libraries containing 800 million synthetic peptides with 13 residue-long sequences using affinity selection-mass spectrometry.118 However, the developed peptide does not inhibit interaction of the RBD with ACE2.
Stapling of peptides—covalently joining residues close in a space in a specific conformation—can be efficiently applied to rigidify helical peptidomimetics that could bind RBD. Although initial attempts to staple ACE2 fragments were unsuccessful,119 further studies provided molecules with micromolar potency.120−122 Moreover, macrocyclic peptides were also discovered using epitope-directed selection in phage display.123
On the basis of the sequence of the helical fragment of the LCB3 protein, conformationally constrained peptides were developed.124 Incorporation of a cyclopentane-based β-amino acid residue using an ααβαααβ sequence pattern resulted in peptides retaining a helical conformation in solution. After four rounds of optimization, the peptide showed binding affinity to the RBD of the S protein, Kd = 650 nM. Moreover, the HTRF assay allowed us to confirm the inhibition of the S protein–human ACE2 interaction by engineered peptides with an IC50 equal to 1.3 μM. Modeling studies revealed the importance of two Trp residues that hook the peptide in the RBD, which were accompanied by several charge-assisted hydrogen bonds (Figure 14).
Figure 14.
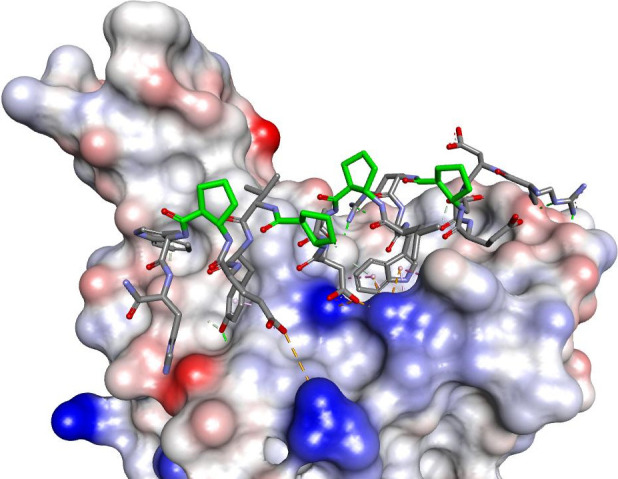
Modeled complex of peptide-RBD of the S protein. The inhibitory peptide is shown in stick representation colored according to atom type, and β-amino acid carbon atoms are colored in green. The RBD of the S protein is shown as a solvent-accessible surface that is colored according to interpolated charge (red–negative, blue–positive, gray–neutral). Interactions are shown as dashed lines with the same color scheme as shown in Figure 1.
Two peptides, HR1–1 and HR2–18, designed to inhibit the interaction between the HR1 and HR2 domains, thus blocking the formation of the six-helix bundle of the S protein, were identified as potential inhibitors, with EC50 values of 0.14 and 1.19 μM, respectively, as determined using the wild-type SARS-CoV assay.125 Additionally, two peptides derived from ACE2, containing charged amino acids (residues 22–44 and 22–57), possess SARS-CoV antiviral activity with IC50 values of 50 and 6 μM, respectively.126 However, another peptide comprising two ACE2 segments (residues 22–44 and 351–357) linked by glycine showed stronger activity in the inhibition of SARS-CoV infection, with an IC50 of approximately 0.1 μM.
The first pan-CoV fusion inhibitor named EK1 was developed on the basis of the HR2 sequence in the S protein of HCoV-OC439.127 This peptide was effective in inhibiting various HCoVs by forming a stable six-helix bundle with the HR1 domain, as confirmed by crystal structures. The EK1 peptide was further modified by the introduction of polyethylene glycol (PEG) and cholesterol, resulting in lipopeptide EK1C4 with significantly improved inhibitory activity against all HCoVs tested, including SARS-CoV-2.128,129 EK1C4 effectively inhibits SARS-CoV-2 replication with an EC50 value of 36.5 nM, which is 67-fold more potent than EK1 (EC50 = 2.47 μM) in the same assay. However, due to the existing side effects and risks involved with the use of PEGylated drugs,130,131 a highly stable dePEGylated lipopeptide, EKL1C, was developed with broad-spectrum anti-HCoV activity, a high genetic barrier for drug resistance, improved resistance to proteolytic enzymes, and higher thermostability.132 In addition, fusion inhibitor peptides targeting the spike S2 subunit were studied, revealing a long 36-mer peptide with potent inhibition of SARS-CoV-2 S-mediated cell–cell fusion at a 1 μM concentration.133 Another study showed that the combination of the fusion inhibitor EK1 and an antiviral lectin, griffithsin (GRFT),134 displays an excellent synergistic effect against SARS-CoV-2 pseudovirus infection, giving IC50 values of 49 nmol/L and 24 nmol/L, respectively.135 It is suggested that high inhibition potency is a result of GRFT and EK1 having different target sites, where GRFT inhibits SARS-CoV-2 infection by targeting the glycosylation sites in the S1 subunit, possibly the RBD, of the SARS-CoV-2 S protein.
The HR2 sequence-based lipopeptide fusion inhibitor IPB02 has high activity in inhibiting SARS-CoV-2, with an IC50 of 0.025 μM for the inhibition by IPB02 on SARS-CoV-2 S protein-mediated cell–cell fusion and IC50 values of 0.08 μM for the inhibition of SARS-CoV-2 pseudovirus infection. A structure–activity relationship study of IPB02 indicated the importance of the N- and C-terminal amino acids for binding and antiviral capacities.
An in silico study examined the potential of the HIV-1 fusion inhibitor Enfuvirtide for drug repurposing.136 The MD simulation study showed that interactions between Enfuvirtide and the most important residues of the HR2 domain of the SARS-CoV-2 S2 protein were remarkably stable, thus showing good potential to act as a fusion inhibitor of this virus.
Small-Molecule Inhibitors
Modern discovery and development of safe drugs are most often a laborious and time-consuming process, often accompanied by a discouraging failure rate at the stage of clinical trials.137 To avoid these setbacks and accelerate the discovery of suitable therapeutics, research is directed to the repurposing of well-established drugs. The fast spread of COVID-19 infection has prompted the emergency use authorization of the antiviral drug remdesivir, even with its modest benefits.138,139
Ivermectin (Figure 15), a macrocyclic lactone derived from Streptomyces avermitilis and an FDA-approved antiparasitic, was found to inhibit the replication of SARS-CoV-2 in vitro,140 as successfully confirmed by MD simulation and docking calculations, which indicated that the R403, R405, Y449, L455, G496 and Y505 residues from ACE2 are the residues that interact the most with ligands.141 However, no clinical evidence was found showing that it is effective in COVID-19 treatment.142 Furthermore, an anti-influenza drug, umifenovir, was found to efficiently inhibit SARS-CoV-2 infection with an EC50 of 4.11 μM.143 MD simulation-based analyses demonstrated a high affinity binding of umifenovir at the RBD/ACE2 interface (Figure 15), inducing structural rigidity in the viral glycoprotein, thus limiting the conformational rearrangements needed for membrane fusion and virus entry.144
Figure 15.

Structures of small molecules identified as S protein/ACE2 interaction inhibitors.
Numerous in silico and in vitro screening studies, considering both FDA-approved drugs and newly designed structures, were performed to identify highly bioactive molecules that could efficiently suppress COVID-19 infection.145,146 Natural products, such as alkaloids, could have good potential for treating different diseases with mild side effects. An investigation was conducted to evaluate the potential of alkaloids to interact with and alter the binding function of the SARS-CoV-2 S protein and block the receptor function of ACE2.147
In a computer-aided drug design approach, two potential inhibitors of the S protein RBD and ACE2 interaction, named MU-UNMC-1 and MU-UNMC-2 (Figure 15), were identified and validated.148 In SARS-CoV-2 infection assays, both compounds showed antiviral activity, with IC50 values of 0.67 and 1.72 mM, respectively. Moreover, MU-UNMC-2 has a synergistic effect when combined with remdesivir, implying the possibility of developing a combination therapy to treat COVID-19 patients.
Compounds (Figure 15) isolated from the stromata of Hypocrella bambusae were characterized as potential SARS-CoV-2 entry inhibitors, showing potent activity against live SARS-CoV-2 infection with EC50 values of 0.22 and 0.21 μM, respectively.149 Moreover, cell–cell fusion assays, surface plasmon resonance assays, and molecular docking studies revealed that these compounds could bind with the RBD of the SARS-CoV-2 S protein to prevent its interaction with the human ACE2 receptor.
Recently, several highly efficient small molecule pan-coronavirus inhibitors were discovered.150 Among them, the compound NBCoV63 (Figure 15) showed a high potency against SARS-CoV-2 (IC50 = 55 nM), SARS-CoV (IC50 = 59 nM), and MERS-CoV (IC50 = 75 nM) with excellent SI values and favorable in vitro ADME properties.
Hepatitis C Virus Inhibitors
The hepatitis C virus causes an infection that can have life-long consequences, becoming chronic and eventually leading to liver cirrhosis and hepatocellular carcinoma.151 It is an enveloped ssRNA virus with seven recognized genotypes and high genetic diversity, making it a difficult target for vaccine and drug development.151,152 The current treatment employs FDA-approved direct-acting antivirals (DAAs) and their pangenotypic combinations. Nonetheless, this solution has two significant drawbacks: the high price of the treatment and the emergence of resistance-associated variants.151−153
An alternative approach is focused on the inhibition of the entry of viruses into hepatocytes. It is a complicated, multistep mechanism involving many host factors, such as the tetraspanin CD81, scavenger receptor class B member 1 (SR-BI), claudin-1 (CLDN1), and occludin.154 It requires two viral envelope glycoproteins, E1 and E2, which form a heterodimer on the surface of the virus. Although only E2 interacts directly with the entry factors, E1 assists it at different stages of the process; hence, it may be crucial to consider these two together as a complex.155,156
The development of entry inhibitors is hampered by the lack of crystal structures of the complexes involved. The most well-studied host factor is CD81, a transmembrane protein from the tetraspanin family, which interacts directly with E2 glycoprotein. Unfortunately, CD81 is ubiquitously expressed in the human body and has multiple functions, raising doubts about the potential toxicity of its inhibitors. The problem is further complicated by the conformational flexibility of both CD81 and E2. Based on the changing position of the large external loop (Figure 16) on results from X-ray crystallography, CD81 is thought to adopt three conformations—open, closed, and intermediate.161 It is possible that E2 and CD81 undergo conformational changes upon binding.161,162 To date, there is no solved structure of the complex.163 Nonetheless, attempts to dock the interacting domains using molecular modeling methods have been made,164 and a structure of the complex with tamarin CD81 has been resolved (Figure 16C).159 It has been determined that the glycoprotein’s amino acid residues W420, Y527, W529, G530, and D535 are crucial for the CD81-E2 interaction (Figure 16B)158 and that the E2 502–520 aa segment is conserved and necessary for HCV infectivity.165 Additionally, molecular dynamics (MD) simulations predict that E2 binds to CD81 at site 1 (422–444 aa) at first, undergoes conformational changes, and then binds at site 2 (521–537 aa, Figure 16A).164
Figure 16.

(A) The crystal structure of the truncated glycoprotein E2 shown in green in ribbon representation with fragments containing the predicted binding sites in yellow and magenta, as well as a conserved region in cyan (PDB id: 6MEH).157 (B) Amino acid residues crucial for the E2-CD81 interaction, presented as sticks colored according to atom type.158 (C) The interaction between tamarin CD81 (pink) and E2 (green) with the interacting residues carbon atoms’ colors matching the ribbon color of the same chain (PDB id: 7MWX).159 (D) The full structure of human tetraspanin CD81 in the closed conformation, including the transmembrane helices with a cholesterol binding pocket.160 The large extracellular loop is highlighted in pink (PDB id: 5TCX).160
CD81 is not the only promising target for the treatment of HCV infection. SR-BI (also known as SRB1) is an HDL receptor participating in the bidirectional transport of cholesterol. Its involvement in cell entry can be 2-fold: direct interaction with the E2 glycoprotein and with lipoproteins associated with the virion.166,167
The tight junction proteins start playing a larger role in the late stages of entry. CLDN1 is a tight junction protein that mediates paracellular permeability and is highly expressed in the liver. It forms a coreceptor with CD81 after the formation of the HCV-CD81 complex.168 The disruption of the CD81-CLDN1 interaction has become one of the targets for drug development. While a full structure of CLDN1 has not been solved, claudins are composed of two extracellular loops and four transmembrane helices.169 Mutations of amino acid residues 32 and 48 of CLDN1 extracellular loop 1 have revealed their importance for HCV infectivity.170
Antibody-Based Inhibitors
The glycoprotein E2 is highly immunogenic; hence, triggering the natural body response has become the focal point of vaccine development. E2 has multiple regions with high sequence variability, facilitating virus escape from neutralization. Structural studies of the E2 glycoprotein point to a couple of neutralization epitopes with conserved residues, the identification of which has led to the development of broadly neutralizing antibodies, such as HC84.26.5D,171 AR3A,172,173 and AP33 (Table 2).153,174 While many of the developed antibodies are in late-stage development, there are still questions regarding their long-term usefulness. It is possible that further viral evolution and mutation of the conserved residues will lead to the emergence of new resistance-associated variants.171
Table 2. Antibodies Targeting HCV Entry, Their Epitopes, and the Reported IC50.
| name | target | epitope | IC50 |
|---|---|---|---|
| AR3A172,173 | E2 | L427, N428, C429, N430, D431, T439, F442, Y443, W529, E531 | 0.5 nM < 0.002 μM |
| HC84.26.5D171 | W437, F442, Y443, K446 | 0.0013–0.0082 μMa | |
| AP33153,174 | 412–423 aa (QLINTNGSWHIN) | 0.013–0.69 μMa | |
| K21175 | CD81 | LEL | 56 ± 11 ng/mL |
| JS81175 | LEL | 144 ± 43 ng/mL | |
| mAb151176 | SRBI | Not determined | 0.5 nM |
| 7A5177 | CLDN1 | M152 (second extracellular loop) | 0.23 μg/mL |
Depending on the HCV variant.
Host factors have also been targeted for vaccine development. Monoclonal antibodies against CD81,175 SR-BI,176 and CLDN1177 have been developed (Figure 17). This approach should prevent the appearance of resistance-associated variants. However, the initial function of the proteins could be impacted, so the binding site should be chosen carefully. Antibodies are also much better at preventing infections than at clearing an existing one, and their cost and instability may limit the availability of the treatment.
Figure 17.
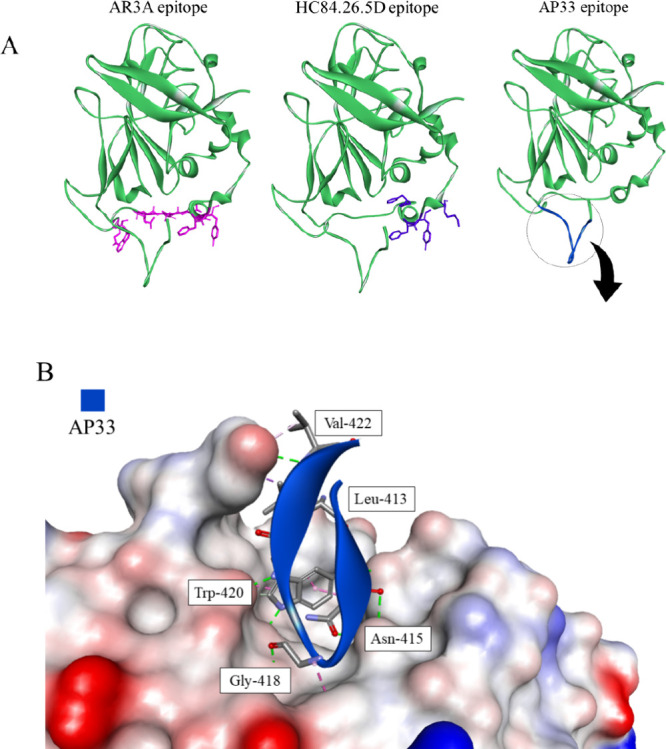
(A) The epitopes of the E2-targeting antibodies. The proteins are shown in cartoon representation with the epitope amino acid side chains displayed as sticks (PDB id: 6MEI).157 (B) The intermolecular interactions between AP33 and its epitope are shown in two ways: on the left, the epitope is represented as a blue ribbon with the interacting amino acid residues as sticks. On the right, the antibody is the solvent accessible surface colored according to interpolated charge (red–negative, blue–positive, gray–neutral), with the interacting residues of E2 displayed as blue sticks. Interactions are shown as dashed lines with the same color scheme shown in Figure 1 (PDB id: 4GAG).153
Peptide-Based Inhibitors
Much research has been performed in the pursuit of peptide-based HCV entry inhibitors. The rational design of inhibitors is often based on the structure of the binding partner. The peptide 710–725 is based on the C-terminal sequence of E2 and has been determined to inhibit the CD81-E2 interaction, showing that this region of the glycoprotein also participates in binding to CD81 (Table 3).178 This methodology was also applied to create the CLDN1 N-terminus-derived peptide CL58, which prevents the formation of the CD81-CLDN1 complex.179 Furthermore, screening of a library of peptides made out of 15-amino acid fragments of E1E2 yielded the peptide E2–42 (E2 residues 548–562), which can interact with both glycoproteins.180
Table 3. Peptide-Based Inhibitors of HCV Entry, Their Sequences, and the Available Inhibition Data.
| target | name | sequence | efficiency of inhibition |
|---|---|---|---|
| CD81 | 710–725178 | ASWAIKWEYVVLLFLL | ∼60% relative luciferase activity at 51 μMa |
| CD81-binding nonapeptide182 | SPQYWTGA motif | NA | |
| CL58179 | MANAGLQLLGFILAFLGW | IC50 = 2.1 ± 0.5 μM | |
| E2 | hCD81-like nonapeptide184 | ATWVCGPCT | E2 inhibits binding of C9 clone to anti-hCD81 JS-81 with inhibition ratio of 67.5% |
| SAHH-5181 | PSGSNXSNIISNXFRED | EC50 = 17–39 μMb | |
| C18183 | WPWHNHR | Relative level of HCV RNA ∼ 0.5 at 97 μMc | |
| E2–42180 | QGSWFGCTWMNSTGF | % of infectivity at C = 5 nM: 50–60% |
Luciferase activity in Huh7.5 cells compared to the activity in PBS buffer.
Value depending on the HCV subtype.
In Huh7.5 cells infected with HCVcc.
The properties of peptide inhibitors can be further improved by adding stabilizing modifications. SAHH-5, a peptide based on the large extracellular loop of CD81, was stapled through a covalent bond between two amino acid side chains, ensuring proper folding and high stability against proteolysis.181
Another way to find active anti-HCV compounds is through phage display selection. Curiously, CD81-binding nonapeptides selected from a random peptide library did not share any sequence similarities with E2. The structural motif present in all of the CD81-binding peptides found in the study is speculated to be a mimotope of an E2 epitope.182 Similarly, a set of short peptides was found to bind to E2.183
Small Molecule-Based Inhibitors
The recent trend is to aim for naturally sourced inhibitors characterized by their affordability and availability even in disadvantaged communities. Such molecules have also been found to inhibit the protein–protein interactions occurring during HCV entry. Trachelogenin (Figure 18), a natural lignan compound obtained from Caulis trachelospermi, disrupts the E2-CD81 interaction by binding to the large extracellular loop of CD81.185 According to MD simulations, it interacts with residues T166, N184, K187, and E188.185
Figure 18.

Small molecule inhibitors of HCV entry and available inhibition data.
Saikoponin B2 (Figure 18) is a terpenoid that can be extracted from the roots of Bupleurum kaoi. It targets the E2 glycoprotein, effectively preventing its binding to CD81. Furthermore, its effect may be pangenetic.186 The plant alkaloid berberine (Figure 18) has a similar mode of action and blocks the entry of HCV into cells. Its binding site may be close to site 1, which is involved in the formation of the complex with CD81.187
ITX5061 (Figure 18), an SR-BI antagonist, has reached phase I clinical trials. The arylketoamide hampers the activity of SR-BI but also stops the binding of a soluble, truncated form of E2 to the receptor. Whether SR-BI binding with E2 directly occurs during the entry process is disputed.188
Ebola Virus
The 2014–2016 outbreak brought much attention to the Ebola virus. The enveloped filovirus causes a severe hemorrhagic fever in humans. Currently, there are three FDA-approved vaccines against it; nonetheless, studies of entry inhibition have also progressed. The virus has one glycoprotein (GP) on its surface, which is cleaved into two separate parts postattachment.189,190 The first part, GP-1, is involved in receptor interactions and has a Niemann-Pick C1 receptor-binding domain (Figure 19A,B). GP-2 is crucial for the membrane fusion step. The antibodies isolated from vaccinated individuals, such as the 1T0227 antibody, have been proven to bind at the receptor binding domain of GP-1 (Figure 19C,D).190 Furthermore, the structure of the resolved NPC1-GP complex has been used to develop a set of macrocyclic peptide analogs of loop 2 of the host factor. The highest inhibitory activity was achieved with Pep3.3 (IC50 = 5.1 μM, Ac-cyclo(CEYFFWYC)-NH2).191 Two small molecule inhibitors of the interaction have been found through library screening; the compounds, benzylpiperazine adamantane diamide (compound 3.0, IC50 = 1.6 μM) and its derivative, compound 3.47 (IC50 = 130 nM), target NPC1 directly, but the exact mode of binding is unknown (Figure 20).192 Many studies have focused on inhibitors targeting a loop at the interface between GP1/GP2, blocking the fusion of the virus and the cell. Nonetheless, the conformational changes and the structural destabilization of GP can disrupt the interaction with NPC1 as well.193 For example, a benzodiazepine derivative (compound 7, IC50 = 10 μM, Figure 20) targets a hydrophobic pocket at the interface of this interaction. Whether it affects the glycoprotein or only prevents its binding to NPC1 is yet unclear.194
Figure 19.

Glycoprotein GP in the complex with the Niemann-Pick C1 receptor (A) (PDB id: 5F1B)195 and closeup showing intermolecular interactions (B). The glycoprotein GP in the complex with the IT0227 antibody (C), with a closeup of the interactions shown in (D) (PDB id: 6S8D).196 The proteins are shown in ribbon representation colored by chain with magenta for GP-2, navy for GP-1, cyan for NPC1, red for the light chain of IT0227, and green for the heavy chain of IT01227. Interacting residues are shown as sticks colored by atom type, and the color of the carbon atoms matches that of the ribbon of the same chain. Interactions are shown as dashed lines with the same color scheme shown in Figure 1.
Figure 20.

Small molecule inhibitors of PPI related to the Ebola virus.
Dengue Virus
Dengue fever, despite its annual 390 million human infections,197 continues to be classified as a neglected tropical disease.198 It predominantly affects low-income and developing regions of the world.199 The severe form of the disease can be life-threatening and can lead to dengue hemorrhagic fever or dengue shock syndrome. The risk is elevated for individuals who have already been infected by a different strain of the virus possibly due to antibody-dependent enhancement of infection (ADE). The existence of four distinct serotypes with sequence similarity of 60–70% is the main challenge for vaccine and treatment development.199 Currently, two vaccines have been authorized, but none have been authorized by the FDA. The European Medicines Agency approved the Qdenga tetravalent attenuated vaccine in 2022.200 Another vaccine, Dengvaxia, has been available since 2016.201 The latter is recommended only for previously infected individuals and populations with high infection rates.
Dengue virus (DENV) is an enveloped flavivirus that encodes three structural proteins: capsid (C), premembrane (prM), and envelope (E). The spikes on the virion particles are composed of three prM:E heterodimers. The envelope glycoprotein has three domains. Domain III (DIII) contains immunogenic epitopes and receptor-binding motifs, while the hydrophobic membrane fusion loop is located in domain II (DII) (Figure 21A).202 The entry route of DENV seems to be serotype dependent. Multiple host receptors have been identified for their role in the process, including DC-SIGN, CLEC5A, mannose receptor, CD14, heat-shock proteins 70 and 90, ER chaperonin GRP78, and prohibitin.203
Figure 21.
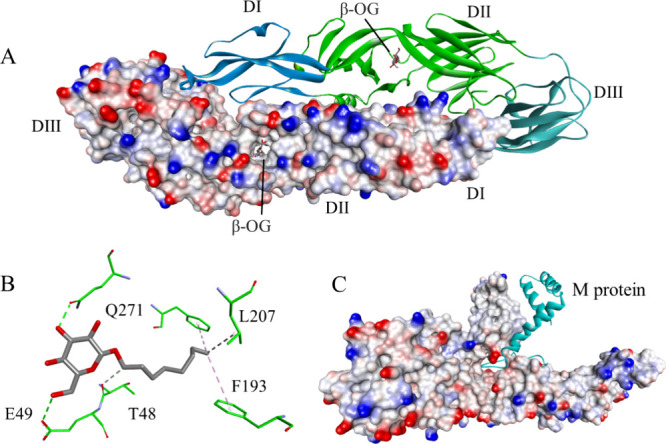
(A) The crystal structure of a DENV2 envelope glycoprotein homodimer (PDB id: 1OKE).202 One chain is shown in the ribbon representation with blue marking the DI, green – DII, and cyan – DIII. The other chain is shown as a solvent-accessible surface colored according to the interpolated charge (red–negative, blue–positive, gray–neutral). The binding sites of n-octyl-β-d-glucoside are indicated with an arrow (“β-OG”). (B) Interactions within the n-octyl-β-d-glucoside-binding pocket; the ligand and the amino acid residues are shown in stick representation, with colors corresponding to atom type. The protein’s carbon atom color corresponds to the location of the binding site (green - DII). Interactions are shown as dashed lines with the same color scheme shown in Figure 1. (C) The E protein–M protein complex. Both envelope protein chains are depicted in the solvent accessible surface representation, while the M protein is shown as a cyan ribbon (PDB id: 3J27).206
It is crucial to immunize the patient against all DENV strains to prevent the onset of severe disease. The murine broadly neutralizing antibody 4E11 (Figure 22) exhibits a binding affinity to the DIII region of four DENV serotypes (DENV1 to DENV4), although with comparatively weaker binding to DENV4.204 This antibody recognizes a conserved fragment within DIII, encompassing residues K310, L/I387, L389, and W391.204 It was further re-engineered to enhance its binding capabilities, resulting in the 4E5A antibody. Finally, further optimization of 4E5A yielded Ab513 with EC50 values below 200 ng/mL for all serotypes.205
Figure 22.
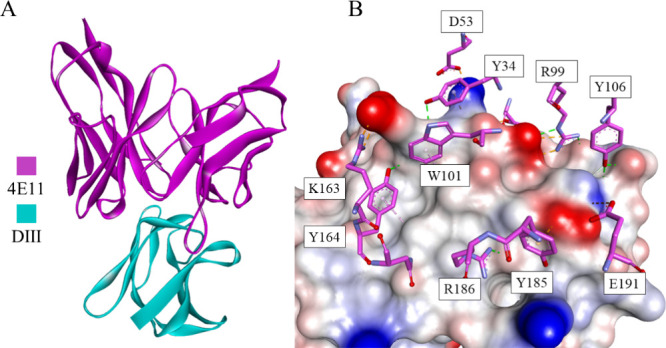
(A) The 4E11 bNAb-DIII complex (DENV4) is shown in the ribbon representation, where 4E11 is colored magenta, while DIII is colored cyan. (B) A closeup showing the intermolecular interactions. DIII is shown as a solvent-accessible surface colored according to interpolated charge (red–negative, blue–positive, gray–neutral). The interacting residues are displayed as sticks colored by atom type, and the colors of the carbon atoms match that of the ribbon of the same chain. Interactions are shown as dashed lines with the same color scheme shown in Figure 1 (PDB ID: 3UYP).207
Due to the uncertainty in the role of the suspected host factors as well as the serotype dependency of the entry mechanism, most work on DENV entry inhibition has been focused on the structural proteins of the virus.
During maturation in the trans-Golgi network, the prM protein is cleaved by a protease into two parts: the M protein and the pr peptide. This triggers a conformational change in E and primes the particle for membrane fusion.203,202 The crystal structure of the envelope protein revealed an n-octyl-β-d-glucoside (β-OG)-binding hydrophobic pocket that is essential for the structural transition (Figure 21B).202 Targeting this pocket has become one of the main strategies for entry inhibition. The EF dipeptide targets this site, resulting in an IC50 of 96.8 μM (DENV2),208 as does Compound 6 (Figure 23) with an EC50 of 0.068 μM (DENV2).209 Another site is targeted by 1OAN1, a de novo peptide binding to the 41–60 amino acid fragment of the envelope protein. In silico optimization of the sequence resulted in an IC50 of 7 μM (DENV2).210
Figure 23.

Inhibitors of PPI involved in dengue infection.
MLH40, a peptide created from the conserved ectodomain of the M protein, interferes with the M-E interaction, changing the viral interface and hence blocking entry at the attachment stage. The value of IC50 depended on the serotype, with results between 24 and 31 μM.211
Chikungunya Virus
Chikungunya virus (CHIKV) is an enveloped alphavirus originating from Africa. The disease is characterized by high fever and joint and skeletal muscle pain. While it is not deadly to otherwise healthy individuals, it can become chronic and significantly lower the quality of life.212 Currently, there is no vaccine or specific treatment for CHIKV; nonetheless, some potential vaccines are in clinical trials and on their way to approval, such as live-attenuated VLA1553.213,214
CHIKV enters host cells through pH-dependent receptor-mediated endocytosis. The viral glycoproteins E1 and E2 form heterodimeric spikes on their surfaces, with the former participating in membrane fusion and the latter interacting directly with host factors. The acidic pH of endosomes is speculated to cause a conformational change in the E1E2 complex (Figure 24), resulting in exposure of the hydrophobic fusion loop of E1, which acts as an anchor in the cell membrane.212
Figure 24.

(A) The E1E2 complex of CHIKV. The structure is represented as ribbons, with green indicating the E2 glycoprotein, blue indicating the E1 glycoprotein, and red indicating the fusion loop. The amino acid residues involved in the intramolecular interactions between E1 and E2 are shown as sticks. (B) The MXRA8 receptor (magenta ribbon) binds to the E1E2 complex. The transmembrane part of E1E2 is included (“TM”) (PDB id: 6JO8).223
E2 is composed of three domains, one of which—domain A—is responsible for receptor binding. The C9 (IC50= 51 ng/mL) and IM-CKV063 (7.4 ng/mL) neutralizing antibodies interact with the domain of one E2 glycoprotein and domains A and B of a neighboring E2 in the spike.215 It has also been reported that one residue, E2 W64, is especially important and may be an attractive target for drug and vaccine design.215 E2 has also been targeted by small molecule inhibitors, such as suramin (IC50 = 5.68 μM)212 and Arbidol (IC50 < 20.9 μM) (Figure 25).216 It has been found that the N5R H18Q mutation of E2 results in a suramin-resistant variant, while the G407R substitution causes Arbidol resistance.212,216 Furthermore, the N-terminal loop and domain A of E2 have been identified in silico as the most likely suramin binding site.212
Figure 25.

Small molecule inhibitors of PPI related to Chikungunya virus infection.
Phospholipase A2, a snake venom protein, is a strong CHIKV entry inhibitor with an EC50 of 44.6 nM; according to MD simulations, it may bind to E1, forming approximately 30 hydrophobic interactions.217 Three amphiphilic peptides, GA-Hecate (gallic acid-FALALKALKKALKKLKKALKKAL-CONH2) and its analogs, PSSct1910 and PSSct1905, have been reported to interfere with virus attachment and internalization into cells. Among these peptides, PSSct1910 showed the most promising results, with an EC50 of 1.1 μM in BHK-21 cells and 0.2 μM in Huh-7 cells.218
To date, several possible host receptors have been proposed, including the MXRA8 receptor (Figure 24B), prohibitin-1, TIM1, DC-SIGN, and the basigin CD147.219,220 The E2/prohibitin-1 interaction was inhibited by the small molecule drug flavagline FL3, which binds prohibitin-1 with an IC50 of 22.4 nM (Figure 25).221 Despite the recognition of MXRA8 as a host factor in CHIKV entry, its decrease in concentration or absence did not fully stop the infection, suggesting the existence of an MXRA8-independent entry pathway.222
Summary and Outlook
Usually, in the case of newly emerging viral diseases, enzyme inhibitors are considered as a first choice due to their high efficiency combined with well-developed methodologies of their discovery. However, due to the high potential of viruses to change, exploration of various molecular targets is necessary. In particular, inhibitors of protein–protein interactions can be very valuable in the treatment of viral infections. In the majority of cases, interactions between the virus and human proteins are targeted, which affects the entry of the virus into the host cell. To efficiently create PPI inhibitors in these cases, detailed knowledge of the virus entry process is needed, and several proteins of both interacting partners are often involved. Analogous methodologies are applied to develop protein–protein inhibitors against any studied viruses, although the progress of projects is constrained by the availability of structural data and a comprehensive understanding of the viral entry mechanism. Furthermore, the lack of funding for research on rarely spreading diseases and/or in less developed countries poses an additional challenge.
Although the methodologies of development of viral PPI inhibitors are based on the same principles as those in other cases, some specific points can be indicated. The major difficulty of treating viral infections is related to fast mutations of viruses that can lead to changes in the structure of molecular targets and, subsequently, weakening of drug-target interactions. This problem can be addressed on two different levels, namely, molecule design and therapy development. First, drugs can be designed to bind regions of viral proteins that are less prone to mutations or target human proteins that interact with viral ones. Importantly, such options are not available in the case of enzyme inhibitor design. Second, antiviral therapy can be constructed in a way that drugs targeting various proteins are used at the same time. In this case, PPI and enzyme inhibitors can work cooperatively.
Usually, developing specific antibodies toward selected PPIs is the quickest option for obtaining highly active molecules, due to the availability of effective experimental methods. However, the high costs of antibody-based therapies and other disadvantages of antibodies are problematic in this case. Most often, peptides and peptidomimetics are the second choice. Recently, experimental and computational techniques have been significantly improved, and the effective development of compounds from this class is possible. Their medium size allows them to create compounds with numerous interaction sites with the protein target; thus, they can show high potency. On the other hand, the usual problems encountered in the medical application of peptides, e.g., poor proteolytic stability, can be feasibly solved by a wide range of known modifications leading to peptidomimetics. Finally, small molecules can also be applied as PPI inhibitors; however, their development is usually slower and requires more effort than that of the two classes discussed above. This is related to the necessity of finding a strong binder for large protein surfaces, which subsequently leads to extended compounds that are synthetically demanding.
Numerous successful stories presented in this Perspective confirm the high potential of PPI inhibitors in treating viral diseases. Both marketed drugs and drug candidates at various stages of clinical trials are known and constitute therapies that can be effectively applied alternatively or in combination with other approaches, particularly enzyme inhibitors. Advancements in methodologies of discovery and better possibilities to avoid virus resistance further enhance potential antiviral PPI inhibitors. Therefore, we can expect an increasing number of drugs derived from this field.
Acknowledgments
The work was financially supported by the National Science Centre, Poland, Grant No. 2020/01/0/ST4/00064 (to Ł.B.).
Glossary
Abbreviations Used
- ACE2
angiotensyn converting enzyme 2
- AIDS
acquired immunodeficiency syndrome
- CCR5
C–C chemokine receptor type 5
- CHR
C-terminal heptad repeat regions
- CHIKV
Chikungunya virus
- CXCR4
C-X-C chemokine receptor type 4
- DAA
direct-acting antivirals
- DENV
Dengue virus
- FDA
Food and Drug Administration
- HIV
human immunodeficiency virus
- NHR
N-terminal heptad repeat regions
- NTD
N-terminal domain
- RBD
receptor binding domain
- SARS
severe acute respiratory syndrome
- PPI
protein–protein interaction
Biographies
Violeta Marković received her Ph.D. in Chemistry from the Faculty of Science, University of Kragujevac (Serbia) in 2012 (on the synthesis and evaluation of pyrazole and pyrazolone derivatives). Under the Basileus postdoctoral scholarship program at Sapienza, University of Rome (Italy), she studied the total synthesis of natural compounds and their corresponding synthetic analogs. During her second postdoctoral research study at Wrocław University of Technology (Poland) under the NAWA (Polish National Agency for Academic Exchange) scholarship program, she was involved in the synthesis and evaluation of miniproteins as organocatalysts and peptidomimetics as inhibitors of protein–protein interactions. Currently, she is an Assistant Professor at the Faculty of Science, University of Kragujevac, and her research interest is focused on the synthesis and structural characterization of potentially bioactive organic molecules.
Anna Szczepańska is a Ph.D. student at the Faculty of Chemistry, Wrocław University of Science and Technology (Poland). She does her PhD studies under the supervision of Prof. Ł. Berlicki. Her major scientific interests include the design and synthesis of functional peptides.
Łukasz Berlicki received his Ph.D. in Chemistry from Wrocław University of Technology (Poland) in 2004 (on the design and synthesis of glutamine synthetase inhibitors, under the supervision of Professor P. Kafarski). Under a Marie Curie Fellowship at the University of Regensburg (Germany), he studied the structural and biological aspects of peptides constrained with cyclopentane-containing amino acid residues. Presently, he works as a Professor at Wrocław University of Science and Technology as the head of the Department of Bioorganic Chemistry. His research focuses on the structure and functions of peptidomimetics, constrained peptides and miniproteins, in particular, targeting protein–protein interactions of medicinal relevance.
The authors declare no competing financial interest.
References
- Ye S.; Lu C.; Qiu Y.; Zheng H.; Ge X.; Wu A.; Xia Z.; Jiang T.; Zhu H.; Peng Y. An Atlas of Human Viruses Provides New Insights into Diversity and Tissue Tropism of Human Viruses. Bioinformatics 2022, 38 (11), 3087–3093. 10.1093/bioinformatics/btac275. [DOI] [PubMed] [Google Scholar]
- Ye S.; Lu C.; Qiu Y.; Zheng H.; Ge X.; Wu A.; Xia Z.; Jiang T.; Zhu H.; Peng Y. An Atlas of Human Viruses Provides New Insights into Diversity and Tissue Tropism of Human Viruses. Bioinformatics 2022, 38 (11), 3087–3093. 10.1093/bioinformatics/btac275. [DOI] [PubMed] [Google Scholar]
- Müller B.; Kräusslich H.-G. Antiviral Strategies. Handb Exp Pharmacol 2009, 189 (189), 1–24. 10.1007/978-3-540-79086-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Ding D.; Zhang X.; Sun L.; Kang D.; Huang B.; Liu X.; Zhan P. Newly Emerging Strategies in Antiviral Drug Discovery: Dedicated to Prof. Dr. Erik De Clercq on Occasion of His 80th Anniversary. Molecules 2022, 27 (3), 850. 10.3390/molecules27030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa D. R.; Immanuel A.; Srikanth S.; Kadhirvel S. Trends and Strategies to Combat Viral Infections: A Review on FDA Approved Antiviral Drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. 10.1016/j.ijbiomac.2021.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa D. R.; Immanuel A.; Srikanth S.; Kadhirvel S. Trends and Strategies to Combat Viral Infections: A Review on FDA Approved Antiviral Drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. 10.1016/j.ijbiomac.2021.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K.; Sheynkman G. M.; Zhang I.; Vidal M. Proteome-Scale Human Interactomics. Trends Biochem. Sci. 2017, 42 (5), 342–354. 10.1016/j.tibs.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M.; Cusick M. E.; Barabási A.-L. Interactome Networks and Human Disease. Cell 2011, 144 (6), 986–998. 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L.; Wang W.; Fang G. Targeting Protein-Protein Interaction by Small Molecules. Annu. Rev. Pharmacol Toxicol 2014, 54, 435–456. 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- Falchi F.; Caporuscio F.; Recanatini M. Structure-Based Design of Small-Molecule Protein-Protein Interaction Modulators: The Story so Far. Future Med. Chem. 2014, 6 (3), 343–357. 10.4155/fmc.13.204. [DOI] [PubMed] [Google Scholar]
- Berlicki Ł. Tailoring Peptides and Peptidomimetics for Targeting Protein-Protein Interactions. Peptide and Peptidomimetic Therapeutics: From Bench to Bedside 2022, 57. 10.1016/B978-0-12-820141-1.00009-1. [DOI] [Google Scholar]
- Ciesiołkiewicz A.; Lizandra Perez J.; Berlicki Ł. Miniproteins in Medicinal Chemistry. Bioorg. Med. Chem. Lett. 2022, 71, 128806. 10.1016/j.bmcl.2022.128806. [DOI] [PubMed] [Google Scholar]
- Barré-Sinoussi F.; Chermann J. C.; Rey F.; Nugeyre M. T.; Chamaret S.; Gruest J.; Dauguet C.; Axler-Blin C.; Vézinet-Brun F.; Rouzioux C.; Rozenbaum W.; Montagnier L. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220 (4599), 868–871. 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- Gallo R. C.; Salahuddin S. Z.; Popovic M.; Shearer G. M.; Kaplan M.; Haynes B. F.; Palker T. J.; Redfield R.; Oleske J.; Safai B.; White G.; Foster P.; Markham P. D. Frequent Detection and Isolation of Cytopathic Retroviruses (HTLV-III) from Patients with AIDS and at Risk for AIDS. Science 1984, 224 (4648), 500–503. 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- https://www.unaids.org/en.
- Nyamweya S.; Hegedus A.; Jaye A.; Rowland-Jones S.; Flanagan K. L.; Macallan D. C. Comparing HIV-1 and HIV-2 Infection: Lessons for Viral Immunopathogenesis. Rev. Med. Virol. 2013, 23, 221–240. 10.1002/rmv.1739. [DOI] [PubMed] [Google Scholar]
- Cooley L. A.; Lewin S. R. HIV-1 Cell Entry and Advances in Viral Entry Inhibitor Therapy 2003, 26, 121. 10.1016/S1386-6532(02)00111-7. [DOI] [PubMed] [Google Scholar]
- Nowak S. A.; Chou T. Mechanisms of Receptor/Coreceptor-Mediated Entry of Enveloped Viruses. Biophys. J. 2009, 96 (7), 2624–2636. 10.1016/j.bpj.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. New Anti-HIV Agents and Targets. Medicinal Research Reviews. 2003, 34, 531–565. 10.1002/chin.200303248. [DOI] [PubMed] [Google Scholar]
- Salazar G.; Zhang N.; Fu T. M.; An Z. Antibody Therapies for the Prevention and Treatment of Viral Infections. npj Vaccines 2017, 2, 19 10.1038/s41541-017-0019-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov A. Ibalizumab, a CD4-Specific MAb to Inhibit HIV-1 Infection. Curr. Opin Investig. Drugs 2007, 8 (8), 653–661. [PubMed] [Google Scholar]
- Iacob S. A.; Iacob D. G.. Ibalizumab Targeting CD4 Receptors, An Emerging Molecule in HIV Therapy. Front Microbiol 2017, 8 ( (NOV), ). 10.3389/fmicb.2017.02323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R.; Franco D.; Kao C.-Y.; Yu F.; Huang Y.; Ho D. D. Epitope Mapping of Ibalizumab, a Humanized Anti-CD4Monoclonal Antibody with Anti-HIV-1 Activity in Infected Patients. J. Virol 2010, 84 (14), 6935–6942. 10.1128/JVI.00453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M. M.; Seaman M. S.; Rits-Volloch S.; Hong X.; Kao C. Y.; Ho D. D.; Chen B. Crystal Structure of HIV-1 Primary Receptor CD4 in Complex with a Potent Antiviral Antibody. Structure 2010, 18 (12), 1632–1641. 10.1016/j.str.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton D. R.; Hangartner L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu. Rev. Immunol. 2016, 34, 635–659. 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar K. J.; Sneller M. C.; Harrison L. J.; Justement J. S.; Overton E. T.; Petrone M. E.; Salantes D. B.; Seamon C. A.; Scheinfeld B.; Kwan R. W.; Learn G. H.; Proschan M. A.; Kreider E. F.; Blazkova J.; Bardsley M.; Refsland E. W.; Messer M.; Clarridge K. E.; Tustin N. B.; Madden P. J.; Oden K.; O’Dell S. J.; Jarocki B.; Shiakolas A. R.; Tressler R. L.; Doria-Rose N. A.; Bailer R. T.; Ledgerwood J. E.; Capparelli E. V.; Lynch R. M.; Graham B. S.; Moir S.; Koup R. A.; Mascola J. R.; Hoxie J. A.; Fauci A. S.; Tebas P.; Chun T.-W. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. New England Journal of Medicine 2016, 375 (21), 2037–2050. 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Georgiev I.; Wu X.; Yang Z.-Y.; Dai K.; Finzi A.; Do Kwon Y.; Scheid J. F.; Shi W.; Xu L.; Yang Y.; Zhu J.; Nussenzweig M. C.; Sodroski J.; Shapiro L.; Nabel G. J.; Mascola J. R.; Kwong P. D. Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science (1979) 2010, 329 (5993), 811–817. 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid J. F.; Horwitz J. A.; Bar-On Y.; Kreider E. F.; Lu C. L.; Lorenzi J. C. C.; Feldmann A.; Braunschweig M.; Nogueira L.; Oliveira T.; Shimeliovich I.; Patel R.; Burke L.; Cohen Y. Z.; Hadrigan S.; Settler A.; Witmer-Pack M.; West A. P.; Juelg B.; Keler T.; Hawthorne T.; Zingman B.; Gulick R. M.; Pfeifer N.; Learn G. H.; Seaman M. S.; Bjorkman P. J.; Klein F.; Schlesinger S. J.; Walker B. D.; Hahn B. H.; Nussenzweig M. C.; Caskey M. HIV-1 Antibody 3BNC117 Suppresses Viral Rebound in Humans during Treatment Interruption. Nature 2016, 535 (7613), 556–560. 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch R. M.; Boritz E.; Coates E. E.; DeZure A.; Madden P.; Costner P.; Enama M. E.; Plummer S.; Holman L.; Hendel C. S.; Gordon I.; Casazza J.; Conan-Cibotti M.; Migueles S. A.; Tressler R.; Bailer R. T.; McDermott A.; Narpala S.; O’Dell S.; Wolf G.; Lifson J. D.; Freemire B. A.; Gorelick R. J.; Pandey J. P.; Mohan S.; Chomont N.; Fromentin R.; Chun T.-W.; Fauci A. S.; Schwartz R. M.; Koup R. A.; Douek D. C.; Hu Z.; Capparelli E.; Graham B. S.; Mascola J. R.; Ledgerwood J. E.. Virologic Effects of Broadly Neutralizing Antibody VRC01 Administration during Chronic HIV-1 Infection. Sci. Transl Med. 2015, 7 ( (319), ). 10.1126/scitranslmed.aad5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M.; Klein F.; Lorenzi J. C. C.; Seaman M. S.; West A. P.; Buckley N.; Kremer G.; Nogueira L.; Braunschweig M.; Scheid J. F.; Horwitz J. A.; Shimeliovich I.; Ben-Avraham S.; Witmer-Pack M.; Platten M.; Lehmann C.; Burke L. A.; Hawthorne T.; Gorelick R. J.; Walker B. D.; Keler T.; Gulick R. M.; Fätkenheuer G.; Schlesinger S. J.; Nussenzweig M. C. Viraemia Suppressed in HIV-1-Infected Humans by Broadly Neutralizing Antibody 3BNC117. Nature 2015, 522 (7557), 487–491. 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis D. M.; Koup R. A.; Ferrari G.. HIV Antibodies for Treatment of HIV Infection. Immunological Reviews.; Blackwell Publishing Ltd January 1, 2017; pp 313–323. 10.1111/imr.12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalezari J.; Yadavalli G. K.; Para M.; Richmond G.; DeJesus E.; Brown S. J.; Cai W.; Chen C.; Zhong J.; Novello L. A.; Lederman M. M.; Subramanian G. M. Safety, Pharmacokinetics, and Antiviral Activity of HGS004, a Novel Fully Human IgG4Monoclonal Antibody against CCR5, in HIV-1-Infected Patients. Journal of Infectious Diseases 2008, 197 (5), 721–727. 10.1086/527327. [DOI] [PubMed] [Google Scholar]
- Dhody K.; Pourhassan N.; Kazempour K.; Green D.; Badri S.; Mekonnen H.; Burger D.; Maddon P. J. PRO 140, a Monoclonal Antibody Targeting CCR5, as a Long-Acting, Single-Agent Maintenance Therapy for HIV-1 Infection. HIV Clin Trials 2018, 19 (3), 85–93. 10.1080/15284336.2018.1452842. [DOI] [PubMed] [Google Scholar]
- Klibanov O. M.; Williams S. H.; Iler C. A. Cenicriviroc, an Orally Active CCR5 Antagonist for the Potential Treatment of HIV Infection. Curr. Opin Investig. Drugs 2010, 11 (8), 940–950. [PubMed] [Google Scholar]
- Gay C. L.; Bosch R. J.; Ritz J.; Hataye J. M.; Aga E.; Tressler R. L.; Mason S. W.; Hwang C. K.; Grasela D. M.; Ray N.; Cyktor J. C.; Coffin J. M.; Acosta E. P.; Koup R. A.; Mellors J. W.; Eron J. J. Clinical Trial of the Anti-PD-L1 Antibody BMS-936559 in HIV-1 Infected Participants on Suppressive Antiretroviral Therapy. Journal of Infectious Diseases 2017, 215 (11), 1725–1733. 10.1093/infdis/jix191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno B. J.; Miller G. D.; Lim C. S. Basics and Recent Advances in Peptide and Protein Drug Delivery. Therapeutic Delivery 2013, 4, 1443–1467. 10.4155/tde.13.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalainen M.; Mönkäre J.; Riikonen J.; Pesonen U.; Vlasova M.; Salonen J.; Lehto V. P.; Järvinen K.; Herzig K. H. Novel Delivery Systems for Improving the Clinical Use of Peptides. Pharmacological Reviews 2015, 67, 541–546. 10.1124/pr.113.008367. [DOI] [PubMed] [Google Scholar]
- Baldwin C. E.; Sanders R. W.; Berkhout B.. Inhibiting HIV-1 Entry with Fusion Inhibitors; 2003; Vol. 10. www.berkhoutlab.com. [DOI] [PubMed]
- He Y.; Cheng J.; Li J.; Qi Z.; Lu H.; Dong M.; Jiang S.; Dai Q. Identification of a Critical Motif for the Human Immunodeficiency Virus Type 1 (HIV-1) Gp41 Core Structure: Implications for Designing Novel Anti-HIV Fusion Inhibitors. J. Virol 2008, 82 (13), 6349–6358. 10.1128/JVI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Kim P. S. A Trimeric Structural Subdomain of the Hiv-1 Transmembrane Glycoprotein. J. Biomol Struct Dyn 1997, 15 (3), 465–471. 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- Jiang S.B.; Lin K.; Strick N.; Neurath A.R. Inhibition of HIV-1 Infection by a Fusion Domain Binding Peptide from the HIV-1 Envelope Glycoprotein Gp41. Biochem. Biophys. Res. Commun. 1993, 195 (2), 533. 10.1006/bbrc.1993.2078. [DOI] [PubMed] [Google Scholar]
- Wild C.; Greenwell T.; Matthews T. A Synthetic Peptide from HIV-1 Gp41 Is a Potent Inhibitor of Virus-Mediated Cell—Cell Fusion. AIDS Res. Hum. Retroviruses 1993, 9 (11), 1051–1053. 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- Chan D. C.; Kim P. S. HIV Entry and Its Inhibition. Cell 1998, 93 (5), 681–684. 10.1016/S0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- Kilby J. M.; Hopkins S.; Venetta T. M.; DiMassimo B.; Cloud G. A.; Lee J. Y.; Alldredge L.; Hunter E.; Lambert D.; Bolognesi D.; Matthews T.; Johnson M. R.; Nowak M. A.; Shaw G. M.; Saag M. S. Potent Suppression of HIV-1 Replication in Humans by T-20, a Peptide Inhibitor of Gp41-Mediated Virus Entry. Nature America Inc. 1998, 4, 1302–1307. 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- Wei X.; Decker J. M.; Liu H.; Zhang Z.; Arani R. B.; Kilby J. M.; Saag M. S.; Wu X.; Shaw G. M.; Kappes J. C. Emergence of Resistant Human Immunodeficiency Virus Type 1 in Patients Receiving Fusion Inhibitor (T-20) Monotherapy. Antimicrob. Agents Chemother. 2002, 46 (6), 1896–1905. 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Morales M.; van der Walle C. F.; Derrick J. P. Modulation of the Fibrillation Kinetics and Morphology of a Therapeutic Peptide by Cucurbit[7]Uril. Mol. Pharmaceutics 2023, 20 (7), 3559–3569. 10.1021/acs.molpharmaceut.3c00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Carbonero L. Discontinuation of the Clinical Development of Fusion Inhibitor T-1249. AIDS Rev. 2004, 6 (1), 61. [PubMed] [Google Scholar]
- He Y.; Xiao Y.; Song H.; Liang Q.; Ju D.; Chen X.; Lu H.; Jing W.; Jiang S.; Zhang L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2008, 283 (17), 11126–11134. 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- Yao X.; Chong H.; Zhang C.; Waltersperger S.; Wang M.; Cui S.; He Y. Broad Antiviral Activity and Crystal Structure of HIV-1 Fusion Inhibitor Sifuvirtide. J. Biol. Chem. 2012, 287 (9), 6788–6796. 10.1074/jbc.M111.317883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Cheng J.; Li J.; Qi Z.; Lu H.; Dong M.; Jiang S.; Dai Q. Identification of a Critical Motif for the Human Immunodeficiency Virus Type 1 (HIV-1) Gp41 Core Structure: Implications for Designing Novel Anti-HIV Fusion Inhibitors. J. Virol 2008, 82 (13), 6349–6358. 10.1128/JVI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H.; Yao X.; Qiu Z.; Qin B.; Han R.; Waltersperger S.; Wang M.; Cui S.; He Y. Discovery of Critical Residues for Viral Entry and Inhibition through Structural Insight of HIV-1 Fusion Inhibitor CP621–652. J. Biol. Chem. 2012, 287 (24), 20281–20289. 10.1074/jbc.M112.354126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Cheng J.; Lu H.; Li J.; Hu J.; Qi Z.; Liu Z.; Jiang S.; Dai Q. Potent HIV Fusion Inhibitors against Enfuvirtide-Resistant HIV-1 Strains. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16332. 10.1073/pnas.080733510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H.; Yao X.; Qiu Z.; Sun J.; Qiao Y.; Zhang M.; Wang M.; Cui S.; He Y. The M-T Hook Structure Increases the Potency of HIV-1 Fusion Inhibitor Sifuvirtide and Overcomes Drug Resistance. J. Antimicrob. Chemother. 2014, 69 (10), 2759–2769. 10.1093/jac/dku183. [DOI] [PubMed] [Google Scholar]
- Chong H.; Yao X.; Qiu Z.; Sun J.; Zhang M.; Waltersperger S.; Wang M.; Liu S. L.; Cui S.; He Y. Short-Peptide Fusion Inhibitors with High Potency against Wild-Type and Enfuvirtide-Resistant HIV-1. FASEB J. 2013, 27 (3), 1203–1213. 10.1096/fj.12-222547. [DOI] [PubMed] [Google Scholar]
- Chong H.; Qiu Z.; Su Y.; Yang L.; He Y. Design of a Highly Potent HIV-1 Fusion Inhibitor Targeting the Gp41 Pocket. AIDS 2015, 29 (1), 13–21. 10.1097/QAD.0000000000000498. [DOI] [PubMed] [Google Scholar]
- Chong H.; Wu X.; Su Y.; He Y. Development of Potent and Long-Acting HIV-1 Fusion Inhibitors. AIDS 2016, 30 (8), 1187–1196. 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- Xiong S.; Borrego P.; Ding X.; Zhu Y.; Martins A.; Chong H.; Taveira N.; He Y.. A Helical Short-Peptide Fusion Inhibitor with Highly Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol 2017, 91 ( (1), ). 10.1128/JVI.01839-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H.; Xue J.; Xiong S.; Cong Z.; Ding X.; Zhu Y.; Liu Z.; Chen T.; Feng Y.; He L.; Guo Y.; Wei Q.; Zhou Y.; Qin C.; He Y.. A Lipopeptide HIV-1/2 Fusion Inhibitor with Highly Potent In Vitro, Ex Vivo, and In Vivo Antiviral Activity. J. Virol. 2017, 91( (11), ), 10.1128/JVI.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Wang C.; Zhang Y.; Chong H.; Hu X.; Li D.; Xing H.; He Y.; Shao Y.; Hong K.; Ma L.. Broad-Spectrum Anti-HIV Activity and High Drug Resistance Barrier of Lipopeptide HIV Fusion Inhibitor LP-19. Front Immunol 2023, 14. 10.3389/fimmu.2023.1199938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S.; Xu W.; Wang L.; Xu L.; Calcul L.; Teng P.; Lu L.; Jiang S.; Cai J. Rational Design of Sulfonyl-γ-AApeptides as Highly Potent HIV-1 Fusion Inhibitors with Broad-Spectrum Activity. J. Med. Chem. 2023, 66 (18), 13319–13331. 10.1021/acs.jmedchem.3c01412. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhu Y.; Hu H.; Zhang S.; Wang P.; Chong H.; He J.; Wang X.; He Y.. Structural Insights into the Mechanisms of Action of Short-Peptide HIV-1 Fusion Inhibitors Targeting the Gp41 Pocket. Front Cell Infect. Microbiol. 2018, 8 ( (FEB), ). 10.3389/fcimb.2018.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Wang H.; Wang X.; Sun L.; Wang Q.; Li Q.; Liang R.; Dou D.; Yu F.; Lu L.; Jiang S. Multitargeted Drug Design Strategy for Discovery of Short-Peptide-Based HIV-1 Entry Inhibitors with High Potency. Eur. J. Med. Chem. 2023, 252, 115294. 10.1016/j.ejmech.2023.115294. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhang Z.; Wallace O. B.; Deshpande M.; Fang H.; Yang Z.; Zadjura L. M.; Tweedie D. L.; Huang S.; Zhao F.; Ranadive S.; Robinson B. S.; Gong Y. F.; Ricarrdi K.; Spicer T. P.; Deminie C.; Rose R.; Wang H. G. H.; Blair W. S.; Shi P. Y.; Lin P. F.; Colonno R. J.; Meanwell N. A. Discovery of 4-Benzoyl-1-[(4-Methoxy-1H-Pyrrolo[2,3-b]Pyridin-3-Yl)Oxoacetyl]-2-(R) -Methylpiperazine (BMS-378806): A Novel HIV-1 Attachment Inhibitor That Interferes with CD4-Gp120 Interactions. J. Med. Chem. 2003, 46 (20), 4236–4239. 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A.; Krystal M. R.; Nowicka-Sans B.; Langley D. R.; Conlon D. A.; Eastgate M. D.; Grasela D. M.; Timmins P.; Wang T.; Kadow J. F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and Its Prodrug Fostemsavir. J. Med. Chem. 2018, 61 (1), 62–80. 10.1021/acs.jmedchem.7b01337. [DOI] [PubMed] [Google Scholar]
- https://www.clinicaltrials.gov/.
- Pancera M.; Lai Y. T.; Bylund T.; Druz A.; Narpala S.; O’Dell S.; Schön A.; Bailer R. T.; Chuang G. Y.; Geng H.; Louder M. K.; Rawi R.; Soumana D. I.; Finzi A.; Herschhorn A.; Madani N.; Sodroski J.; Freire E.; Langley D. R.; Mascola J. R.; McDermott A. B.; Kwong P. D. Crystal Structures of Trimeric HIV Envelope with Entry Inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13 (10), 1115–1122. 10.1038/nchembio.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ma L.; Jiang S.; Lu H.; Liu S.; He Y.; Strick N.; Neamati N.; Debnath A. K. Identification of N-Phenyl-N′-(2,2,6,6-Tetramethyl-Piperidin-4-Yl)-Oxalamides as a New Class of HIV-1 Entry Inhibitors That Prevent Gp120 Binding to CD4. Virology 2005, 339 (2), 213–225. 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Courter J. R.; Madani N.; Sodroski J.; Schön A.; Freire E.; Kwong P. D.; Hendrickson W. A.; Chaiken I. M.; Lalonde J. M.; Smith A. B. Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist. Acc. Chem. Res. 2014, 47 (4), 1228–1237. 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. Do; Finzi A.; Wu X.; Dogo-Isonagie C.; Lee L. K.; Moore L. R.; Schmidt S. D.; Stuckey J.; Yang Y.; Zhou T.; Zhu J.; Vicic D. A.; Debnath A. K.; Shapiro L.; Bewley C. A.; Mascola J. R.; Sodroski J. G.; Kwong P. D. Unliganded HIV-1 Gp120 Core Structures Assume the CD4-Bound Conformation with Regulation by Quaternary Interactions and Variable Loops. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (15), 5663–5668. 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi N.; Harada S.; Mizuguchi T.; Irahara Y.; Yamada Y.; Kotani M.; Nomura W.; Matsushita S.; Yoshimura K.; Tamamura H. Small-Molecule CD4Mimics Containing Mono-Cyclohexyl Moieties as HIV Entry Inhibitors. ChemMedChem. 2016, 11 (8), 940–946. 10.1002/cmdc.201500590. [DOI] [PubMed] [Google Scholar]
- Kobayakawa T.; Konno K.; Ohashi N.; Takahashi K.; Masuda A.; Yoshimura K.; Harada S.; Tamamura H. Soluble-Type Small-Molecule CD4Mimics as HIV Entry Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29 (5), 719–723. 10.1016/j.bmcl.2019.01.011. [DOI] [PubMed] [Google Scholar]
- Kobayakawa T.; Tsuji K.; Konno K.; Himeno A.; Masuda A.; Yang T.; Takahashi K.; Ishida Y.; Ohashi N.; Kuwata T.; Matsumoto K.; Yoshimura K.; Sakawaki H.; Miura T.; Harada S.; Matsushita S.; Tamamura H. Hybrids of Small-Molecule CD4Mimics with Polyethylene Glycol Units as HIV Entry Inhibitors. J. Med. Chem. 2021, 64 (3), 1481–1496. 10.1021/acs.jmedchem.0c01153. [DOI] [PubMed] [Google Scholar]
- Tsuji K.; Kobayakawa T.; Konno K.; Masuda A.; Takahashi K.; Ohashi N.; Yoshimura K.; Kuwata T.; Matsushita S.; Harada S.; Tamamura H. Exploratory Studies on Soluble Small Molecule CD4Mimics as HIV Entry Inhibitors. Bioorg. Med. Chem. 2022, 56, 116616. 10.1016/j.bmc.2022.116616. [DOI] [PubMed] [Google Scholar]
- Schön A.; Madani N.; Klein J. C.; Hubicki A.; Ng D.; Yang X.; Smith A. B.; Sodroski J.; Freire E. Thermodynamics of Binding of a Low-Molecular-Weight CD4Mimetic to HIV-1 Gp120. Biochemistry 2006, 45 (36), 10973–10980. 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Kwon Y. Do; Zhang H.; Scacalossi D.; Belov D. S.; Tikhonov A. A.; Andreev I. A.; Altieri A.; Kurkin A. V.; Kwong P. D.; Debnath A. K. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58 (17), 6909–6927. 10.1021/acs.jmedchem.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Kwon Y. Do; Belov D. S.; Ramesh R. R.; Kurkin A. V.; Altieri A.; Kwong P. D.; Debnath A. K. Synthesis, Antiviral Potency, in Vitro ADMET, and X-Ray Structure of Potent CD4Mimics as Entry Inhibitors That Target the Phe43 Cavity of HIV-1 Gp120. J. Med. Chem. 2017, 60 (7), 3124–3153. 10.1021/acs.jmedchem.7b00179. [DOI] [PubMed] [Google Scholar]
- Curreli F.; Belov D. S.; Kwon Y. Do; Ramesh R.; Furimsky A. M.; O’Loughlin K.; Byrge P. C.; Iyer L. V.; Mirsalis J. C.; Kurkin A. V.; Altieri A.; Debnath A. K. Structure-Based Lead Optimization to Improve Antiviral Potency and ADMET Properties of Phenyl-1H-Pyrrole-Carboxamide Entry Inhibitors Targeted to HIV-1 Gp120. Eur. J. Med. Chem. 2018, 154, 367–391. 10.1016/j.ejmech.2018.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Ahmed S.; Benedict Victor S. M.; Iusupov I. R.; Spiridonov E. A.; Belov D. S.; Altieri A.; Kurkin A. V.; Debnath A. K. Design, Synthesis, and Antiviral Activity of a Series of CD4-Mimetic Small-Molecule HIV-1 Entry Inhibitors. Bioorg. Med. Chem. 2021, 32, 116000. 10.1016/j.bmc.2021.116000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli F.; Ahmed S.; Benedict Victor S. M.; Iusupov I. R.; Belov D. S.; Markov P. O.; Kurkin A. V.; Altieri A.; Debnath A. K. Preclinical Optimization of Gp120 Entry Antagonists as Anti-HIV-1 Agents with Improved Cytotoxicity and ADME Properties through Rational Design, Synthesis, and Antiviral Evaluation. J. Med. Chem. 2020, 63 (4), 1724–1749. 10.1021/acs.jmedchem.9b02149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkin A. V.; Curreli F.; Iusupov I. R.; Spiridonov E. A.; Ahmed S.; Markov P. O.; Manasova E. V.; Altieri A.; Debnath A. K. Design, Synthesis, and Antiviral Activity of the Thiazole Positional Isomers of a Potent HIV-1 Entry Inhibitor NBD-14270. ChemMedChem. 2022, 17 (22), e202200344 10.1002/cmdc.202200344. [DOI] [PubMed] [Google Scholar]
- Melillo B.; Liang S.; Park J.; Schön A.; Courter J. R.; Lalonde J. M.; Wendler D. J.; Princiotto A. M.; Seaman M. S.; Freire E.; Sodroski J.; Madani N.; Hendrickson W. A.; Smith A. B. Small-Molecule CD4-Mimics: Structure-Based Optimization of HIV-1 Entry Inhibition. ACS Med. Chem. Lett. 2016, 7 (3), 330–334. 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette C. A.; Barnes C. O.; Kirk S. M.; Melillo B.; Smith A. B.; Bjorkman P. J.. Cryo-EM Structures of HIV-1 Trimer Bound to CD4-Mimetics BNM-III-170 and M48U1 Adopt a CD4-Bound Open Conformation. Nat. Commun. 2021, 12 ( (1), ). 10.1038/s41467-021-21816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura S.; Ichikawa T.; Nishikawa Y.; Kanzaki N.; Takashima K.; Niwa S.; Iizawa Y.; Baba M.; Sugihara Y. Discovery of a Piperidine-4-Carboxamide CCR5 Antagonist (TAK-220) with Highly Potent Anti-HIV-1 Activity. J. Med. Chem. 2006, 49 (9), 2784–2793. 10.1021/jm051034q. [DOI] [PubMed] [Google Scholar]
- Imamura S.; Ichikawa T.; Nishikawa Y.; Kanzaki N.; Takashima K.; Niwa S.; Iizawa Y.; Baba M.; Sugihara Y. Discovery of a Piperidine-4-Carboxamide CCR5 Antagonist (TAK-220) with Highly Potent Anti-HIV-1 Activity. J. Med. Chem. 2006, 49 (9), 2784–2793. 10.1021/jm051034q. [DOI] [PubMed] [Google Scholar]
- https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022128_selzentry_toc.cfm.
- Woollard S. M.; Kanmogne G. D.. Maraviroc: A Review of Its Use in HIV Infection and Beyond. Drug Design, Development and Therapy; Dove Medical Press Ltd. October 1, 2015; pp 5447–5468. 10.2147/DDDT.S90580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.; Armour D. The Discovery of the CCR5 Receptor Antagonist, UK-427,857, A New Agent for the Treatment of HIV Infection and AIDS. Progress in Medicinal Chemistry 2005, 43, 239–271. 10.1016/S0079-6468(05)43007-6. [DOI] [PubMed] [Google Scholar]
- Abel S.; Russell D.; Whitlock L. A.; Ridgway C. E.; Nedderman A. N. R.; Walker D. K. Assessment of the Absorption, Metabolism and Absolute Bioavailability of Maraviroc in Healthy Male Subjects. Br. J. Clin. Pharmacol. 2008, 65 (SUPPL. 1), 60–67. 10.1111/j.1365-2125.2008.03137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science (1979) 2013, 341 (6152), 1387–1390. 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürmann D.; Fätkenheuer G.; Reynes J.; Michelet C.; Raffi F.; Van Lier J.; Caceres M.; Keung A.; Sansone-Parsons A.; Dunkle L. M.; Hoffmann C. Antiviral Activity, Pharmacokinetics and Safety of Vicriviroc, an Oral CCR5 Antagonist, during 14-Day Monotherapy in HIV-Infected Adults. AIDS 2007, 21 (10), 1293–1299. 10.1097/QAD.0b013e3280f00f9f. [DOI] [PubMed] [Google Scholar]
- Xie X.; Zheng Y. G.; Chen H.; Li J.; Luo R. H.; Chen L.; Zheng C. B.; Zhang S.; Peng P.; Ma D.; Yang L. M.; Zheng Y. T.; Liu H.; Wang J. Structure-Based Design of Tropane Derivatives as a Novel Series of CCR5 Antagonists with Broad-Spectrum Anti-HIV-1 Activities and Improved Oral Bioavailability. J. Med. Chem. 2022, 65 (24), 16526–16540. 10.1021/acs.jmedchem.2c01383. [DOI] [PubMed] [Google Scholar]
- Wibmer C. K.; Moore P. L.; Morris L.. HIV Broadly Neutralizing Antibody Targets. Current Opinion in HIV and AIDS; Lippincott Williams and Wilkins, 2015; pp 135–143. 10.1097/COH.0000000000000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugwu-Korie N.; Quaye O.; Wright E.; Languon S.; Agyapong O.; Broni E.; Gupta Y.; Kempaiah P.; Kwofie S. K. Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry. Molecules 2023, 28 (2), 474. 10.3390/molecules28020474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C.; Günther S.; Preiser W.; van der Werf S.; Brodt H.-R.; Becker S.; Rabenau H.; Panning M.; Kolesnikova L.; Fouchier R. A. M.; Berger A.; Burguière A.-M.; Cinatl J.; Eickmann M.; Escriou N.; Grywna K.; Kramme S.; Manuguerra J.-C.; Müller S.; Rickerts V.; Stürmer M.; Vieth S.; Klenk H.-D.; Osterhaus A. D. M. E.; Schmitz H.; Doerr H. W. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. New England Journal of Medicine 2003, 348 (20), 1967–1976. 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Zaki A. M.; van Boheemen S.; Bestebroer T. M.; Osterhaus A. D. M. E.; Fouchier R. A. M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. New England Journal of Medicine 2012, 367 (19), 1814–1820. 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Lu R.; Zhao X.; Li J.; Niu P.; Yang B.; Wu H.; Wang W.; Song H.; Huang B.; Zhu N.; Bi Y.; Ma X.; Zhan F.; Wang L.; Hu T.; Zhou H.; Hu Z.; Zhou W.; Zhao L.; Chen J.; Meng Y.; Wang J.; Lin Y.; Yuan J.; Xie Z.; Ma J.; Liu W. J.; Wang D.; Xu W.; Holmes E. C.; Gao G. F.; Wu G.; Chen W.; Shi W.; Tan W. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395 (10224), 565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. D. The Chronology of the 2002–2003 SARS Mini Pandemic. Paediatr Respir Rev. 2004, 5 (4), 262–269. 10.1016/j.prrv.2004.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C.-L.; Abiona O.; Graham B. S.; Mclellan J. S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation 2019, 367, 1260. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581 (7807), 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Yang C.; Xu X.-f.; Xu W.; Liu S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacologica Sinica 2020, 41, 1141–1149. 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N. H.; Nitsche A.; Müller M. A.; Drosten C.; Pöhlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181 (2), 271–280. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181 (2), 281–292. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581 (7807), 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- https://www.fda.gov/drugs/emergency-preparedness-drugs/emergency-use-authorizations-drugs-and-non-vaccine-biological-products.
- Liu L.; Wang P.; Nair M. S.; Yu J.; Rapp M.; Wang Q.; Luo Y.; Chan J. F. W.; Sahi V.; Figueroa A.; Guo X. V.; Cerutti G.; Bimela J.; Gorman J.; Zhou T.; Chen Z.; Yuen K. Y.; Kwong P. D.; Sodroski J. G.; Yin M. T.; Sheng Z.; Huang Y.; Shapiro L.; Ho D. D. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584 (7821), 450–456. 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Harvey W. T.; Carabelli A. M.; Jackson B.; Gupta R. K.; Thomson E. C.; Harrison E. M.; Ludden C.; Reeve R.; Rambaut A.; Peacock S. J.; Robertson D. L. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nature Reviews Microbiology 2021, 19, 409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink E. N.; ter Meulen J.; Cox F.; Jongeneelen M. A. C.; Thijsse A.; Throsby M.; Marissen W. E.; Rood P. M. L.; Bakker A. B. H.; Gelderblom H. R.; Martina B. E.; Osterhaus A. D. M. E.; Preiser W.; Doerr H. W.; de Kruif J.; Goudsmit J. Molecular and Biological Characterization of Human Monoclonal Antibodies Binding to the Spike and Nucleocapsid Proteins of Severe Acute Respiratory Syndrome Coronavirus. J. Virol 2005, 79 (3), 1635–1644. 10.1128/JVI.79.3.1635-1644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenough T. C.; Babcock G. J.; Roberts A.; Hernandez H. J.; Thomas W. D. Jr.; Coccia J. A.; Graziano R. F.; Srinivasan M.; Lowy I.; Finberg R. W.; Subbarao K.; Vogel L.; Somasundaran M.; Luzuriaga K.; Sullivan J. L.; Ambrosino D. M. Development and Characterization of a Severe Acute Respiratory Syndrome-Associated Coronavirus-Neutralizing Human Monoclonal Antibody That Provides Effective Immunoprophylaxis in Mice. J. Infect Dis 2005, 191 (4), 507–514. 10.1086/427242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin M. M.; Babcook J.; Prabhakar B. S. Human Monoclonal Antibodies to SARS-Coronavirus Inhibit Infection by Different Mechanisms. Virology 2009, 394 (1), 39–46. 10.1016/j.virol.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B.; Zhang Q.; Ge J.; Wang R.; Sun J.; Ge X.; Yu J.; Shan S.; Zhou B.; Song S.; Tang X.; Yu J.; Lan J.; Yuan J.; Wang H.; Zhao J.; Zhang S.; Wang Y.; Shi X.; Liu L.; Zhao J.; Wang X.; Zhang Z.; Zhang L. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584 (7819), 115–119. 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- Jones B. E.; Brown-Augsburger P. L.; Corbett K. S.; Westendorf K.; Davies J.; Cujec T. P.; Wiethoff C. M.; Blackbourne J. L.; Heinz B. A.; Foster D.; Higgs R. E.; Balasubramaniam D.; Wang L.; Bidshahri R.; Kraft L.; Hwang Y.; Žentelis S.; Jepson K. R.; Goya R.; Smith M. A.; Collins D. W.; Hinshaw S. J.; Tycho S. A.; Pellacani D.; Xiang P.; Muthuraman K.; Sobhanifar S.; Piper M. H.; Triana F. J.; Hendle J.; Pustilnik A.; Adams A. C.; Berens S. J.; Baric R. S.; Martinez D. R.; Cross R. W.; Geisbert T. W.; Borisevich V.; Abiona O.; Belli H. M.; de Vries M.; Mohamed A.; Dittmann M.; Samanovic M.; Mulligan M. J.; Goldsmith J. A.; Hsieh C.-L.; Johnson N. V.; Wrapp D.; McLellan J. S.; Barnhart B. C.; Graham B. S.; Mascola J. R.; Hansen C. L.; Falconer E.. LY-CoV555, a Rapidly Isolated Potent Neutralizing Antibody, Provides Protection in a Non-Human Primate Model of SARS-CoV-2 Infection. bioRxiv, 2020, 10.1101/2020.09.30.318972 [DOI]
- Chen P.; Nirula A.; Heller B.; Gottlieb R. L.; Boscia J.; Morris J.; Huhn G.; Cardona J.; Mocherla B.; Stosor V.; Shawa I.; Adams A. C.; Van Naarden J.; Custer K. L.; Shen L.; Durante M.; Oakley G.; Schade A. E.; Sabo J.; Patel D. R.; Klekotka P.; Skovronsky D. M. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. New England Journal of Medicine 2021, 384 (3), 229–237. 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb R. L.; Nirula A.; Chen P.; Boscia J.; Heller B.; Morris J.; Huhn G.; Cardona J.; Mocherla B.; Stosor V.; Shawa I.; Kumar P.; Adams A. C.; Van Naarden J.; Custer K. L.; Durante M.; Oakley G.; Schade A. E.; Holzer T. R.; Ebert P. J.; Higgs R. E.; Kallewaard N. L.; Sabo J.; Patel D. R.; Klekotka P.; Shen L.; Skovronsky D. M. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 2021, 325 (7), 632–644. 10.1001/jama.2021.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf K.; Žentelis S.; Wang L.; Foster D.; Vaillancourt P.; Wiggin M.; Lovett E.; van der Lee R.; Hendle J.; Pustilnik A.; Sauder J. M.; Kraft L.; Hwang Y.; Siegel R. W.; Chen J.; Heinz B. A.; Higgs R. E.; Kallewaard N. L.; Jepson K.; Goya R.; Smith M. A.; Collins D. W.; Pellacani D.; Xiang P.; de Puyraimond V.; Ricicova M.; Devorkin L.; Pritchard C.; O’Neill A.; Dalal K.; Panwar P.; Dhupar H.; Garces F. A.; Cohen C. A.; Dye J. M.; Huie K. E.; Badger C. V.; Kobasa D.; Audet J.; Freitas J. J.; Hassanali S.; Hughes I.; Munoz L.; Palma H. C.; Ramamurthy B.; Cross R. W.; Geisbert T. W.; Menachery V.; Lokugamage K.; Borisevich V.; Lanz I.; Anderson L.; Sipahimalani P.; Corbett K. S.; Yang E. S.; Zhang Y.; Shi W.; Zhou T.; Choe M.; Misasi J.; Kwong P. D.; Sullivan N. J.; Graham B. S.; Fernandez T. L.; Hansen C. L.; Falconer E.; Mascola J. R.; Jones B. E.; Barnhart B. C. LY-CoV1404 (Bebtelovimab) Potently Neutralizes SARS-CoV-2 Variants. Cell Rep 2022, 39 (7), 110812. 10.1016/j.celrep.2022.110812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathcart A. L.; Havenar-Daughton C.; Lempp F. A.; Ma D.; Schmid M. A.; Agostini M. L.; Guarino B.; Di iulio J.; Rosen L. E.; Tucker H.; Dillen J.; Subramanian S.; Sloan B.; Bianchi S.; Pinto D.; Saliba C.; Culap K.; Wojcechowskyj J. A.; Noack J.; Zhou J.; Kaiser H.; Lee S.; Farhat N.; Chase A.; Montiel-Ruiz M.; Dellota E.; Park A.; Spreafico R.; Lauron E. J.; Czudnochowski N.; Cameroni E.; Ledoux S.; Werts A.; Colas C.; Soriaga L.; Telenti A.; Purcell L. A.; Snell G.; Virgin H. W.; Corti D.; Hebner C. M.. The Dual Function Monoclonal Antibodies VIR-7831 and VIR-7832 Demonstrate Potent in Vitro and 2 in Vivo Activity against SARS-CoV-2. bioRxiv, 2021, 10.1101/2021.03.09.434607. [DOI]
- Panchal D.; Kataria J.; Patel K.; Crowe K.; Pai V.; Azizogli A.; Kadian N.; Sanyal S.; Roy A.; Dodd-o J.; Acevedo-Jake A. M.; Kumar V. A. Peptide-Based Inhibitors for SARS-CoV-2 and SARS-CoV. Adv. Ther (Weinh) 2021, 4 (10), 2100104. 10.1002/adtp.202100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Goreshnik I.; Coventry B.; Case J. B.; Miller L.; Kozodoy L.; Chen R. E.; Carter L.; Walls A. C.; Park Y.-J.; Strauch E.-M.; Stewart L.; Diamond M. S.; Veesler D.; Baker D. De Novo Design of Picomolar SARS-CoV-2 miniprotein Inhibitors. Science (1979) 2020, 370 (6515), 426–431. 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomplun S.; Jbara M.; Quartararo A. J.; Zhang G.; Brown J. S.; Lee Y. C.; Ye X.; Hanna S.; Pentelute B. L. De Novo Discovery of High-Affinity Peptide Binders for the SARS-CoV-2 Spike Protein. ACS Cent Sci. 2021, 7 (1), 156–163. 10.1021/acscentsci.0c01309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D. C.; Morris C.; Mahindra A.; Blair C. M.; Tejeda G.; Herbert I.; Turnbull M. L.; Lieber G.; Willett B. J.; Logan N.; Smith B.; Tobin A. B.; Bhella D.; Baillie G.; Jamieson A. G. Stapled ACE2 Peptidomimetics Designed to Target the SARS-CoV-2 Spike Protein Do Not Prevent Virus Internalization. Pept Sci. (Hoboken) 2021, 113 (4), e24217 10.1002/pep2.24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T.; Namii K.; Seko K.; Kakehi S.; Moro G.; Mihara H. Pattern Enrichment Analysis for Phage Selection of Stapled Peptide Ligands. Chem. Sci. 2022, 13 (43), 12634. 10.1039/D2SC04058A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas M. N.; Hintzen J. C. J.; Löffler P. M. G.; Mecinović J. Targeting SARS-CoV-2 Spike Protein by Stapled HACE2 Peptides. Chem. Commun. (Camb) 2021, 57 (26), 3283–3286. 10.1039/D0CC08387A. [DOI] [PubMed] [Google Scholar]
- Calugi L.; Sautariello G.; Lenci E.; Mattei M. L.; Coppa C.; Cini N.; Contini A.; Trabocchi A. Identification of a Short ACE2-Derived Stapled Peptide Targeting the SARS-CoV-2 Spike Protein. Eur. J. Med. Chem. 2023, 249, 115118. 10.1016/j.ejmech.2023.115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton J. T.; Lalonde T. J.; Tharp J. M.; Kurra Y.; Alugubelli Y. R.; Roundy C. M.; Hamer G. L.; Xu S.; Liu W. R. Novel Regioselective Approach to Cyclize Phage-Displayed Peptides in Combination with Epitope-Directed Selection to Identify a Potent Neutralizing Macrocyclic Peptide for SARS-CoV-2. ACS Chem. Biol. 2022, 17 (10), 2911–2922. 10.1021/acschembio.2c00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marković V.; Shaik J. B.; Ożga K.; Ciesiołkiewicz A.; Lizandra Perez J.; Rudzińska-Szostak E.; Berlicki Ł. Peptide Foldamer-Based Inhibitors of the SARS-CoV-2 S Protein-Human ACE2 Interaction. J. Enzyme Inhib Med. Chem. 2023, 38 (1), 2244693. 10.1080/14756366.2023.2244693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan K.; Yi L.; Chen J.; Qu X.; Qing T.; Rao X.; Jiang P.; Hu J.; Xiong Z.; Nie Y.; Shi X.; Wang W.; Ling C.; Yin X.; Fan K.; Lai L.; Ding M.; Deng H. Suppression of SARS-CoV Entry by Peptides Corresponding to Heptad Regions on Spike Glycoprotein. Biochem. Biophys. Res. Commun. 2004, 319 (3), 746–752. 10.1016/j.bbrc.2004.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D. P.; Penn-Nicholson A.; Cho M. W. Identification of Critical Determinants on ACE2 for SARS-CoV Entry and Development of a Potent Entry Inhibitor. Virology 2006, 350 (1), 15–25. 10.1016/j.virol.2006.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Yan L.; Xu W.; Agrawal A. S.; Algaissi A.; Tseng C.-T. K.; Wang Q.; Du L.; Tan W.; Wilson I. A.; Jiang S.; Yang B.; Lu L.. A Pan-Coronavirus Fusion Inhibitor Targeting the HR1 Domain of Human Coronavirus Spike; 2019. www.who.int/emergencies/. [DOI] [PMC free article] [PubMed]
- Xia S.; Liu M.; Wang C.; Xu W.; Lan Q.; Feng S.; Qi F.; Bao L.; Du L.; Liu S.; Qin C.; Sun F.; Shi Z.; Zhu Y.; Jiang S.; Lu L. Inhibition of SARS-CoV-2 (Previously 2019-NCoV) Infection by a Highly Potent Pan-Coronavirus Fusion Inhibitor Targeting Its Spike Protein That Harbors a High Capacity to Mediate Membrane Fusion. Cell Res. 2020, 30 (4), 343–355. 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries R. D.; Schmitz K. S.; Bovier F. T.; Predella C.; Khao J.; Noack D.; Haagmans B. L.; Herfst S.; Stearns K. N.; Drew-Bear J.; Biswas S.; Rockx B.; McGill G.; Valerio Dorrello N.; Gellman S. H.; Alabi C. A.; de Swart R. L.; Moscona A.; Porotto M. Intranasal Fusion Inhibitory Lipopeptide Prevents Direct-Contact SARS-CoV-2 Transmission in Ferrets 2021, 371, 1379. 10.1126/science.abf4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay R. P.; El-Gewely R.; Armstrong J. K.; Garratty G.; Richette P. Antibodies against Polyethylene Glycol in Healthy Subjects and in Patients Treated with PEG-Conjugated Agents. Expert Opin Drug Deliv 2012, 9 (11), 1319–1323. 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- Povsic T. J.; Lawrence M. G.; Lincoff A. M.; Mehran R.; Rusconi C. P.; Zelenkofske S. L.; Huang Z.; Sailstad J.; Armstrong P. W.; Steg P. G.; Bode C.; Becker R. C.; Alexander J. H.; Adkinson N. F.; Levinson A. I. Pre-Existing Anti-PEG Antibodies Are Associated with Severe Immediate Allergic Reactions to Pegnivacogin, a PEGylated Aptamer. Journal of Allergy and Clinical Immunology 2016, 138 (6), 1712–1715. 10.1016/j.jaci.2016.04.058. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Xu W.; Liu Z.; Wang C.; Xia S.; Lan Q.; Cai Y.; Su S.; Pu J.; Xing L.; Xie Y.; Lu L.; Jiang S.; Wang Q. A Highly Potent and Stable Pan-Coronavirus Fusion Inhibitor as a Candidate Prophylactic and Therapeutic for COVID-19 and Other Coronavirus Diseases. Acta Pharm. Sin B 2022, 12 (4), 1652–1661. 10.1016/j.apsb.2021.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M.; Yamamoto M.; Tani H.; Kobayashi A.; Gohda J.; Kawaguchi Y.; Park B. K.; Kwon H. J.; Inoue J. I.; Alkattan A. Discovery of New Fusion Inhibitor Peptides against Sars-Cov-2 by Targeting the Spike S2 Subunit. Biomol Ther (Seoul) 2021, 29 (3), 282–289. 10.4062/biomolther.2020.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusvarghi S.; Bewley C. A. Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential. Viruses 2016, 8, 296. 10.3390/v8100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Xu W.; Gu C.; Cai X.; Qu D.; Lu L.; Xie Y.; Jiang S. Griffithsin with A Broad-Spectrum Antiviral Activity by Binding Glycans in Viral Glycoprotein Exhibits Strong Synergistic Effect in Combination with A Pan-Coronavirus Fusion Inhibitor Targeting SARS-CoV-2 Spike S2 Subunit. Virologica Sinica 2020, 35, 857–860. 10.1007/s12250-020-00305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi K.; Farasat A.; Rostamian M.; Johari B.; Madanchi H. Enfuvirtide, an HIV-1 Fusion Inhibitor Peptide, Can Act as a Potent SARS-CoV-2 Fusion Inhibitor: An in Silico Drug Repurposing Study. J. Biomol Struct Dyn 2022, 40 (12), 5566–5576. 10.1080/07391102.2021.1871958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowden H.; Munro J.. Trends in Clinical Success Rates and Therapeutic Focus. Nature reviews. Drug discovery. May 8, 2019; pp 495–496. 10.1038/d41573-019-00074-z. [DOI] [PubMed] [Google Scholar]
- Wang M.; Cao R.; Zhang L.; Yang X.; Liu J.; Xu M.; Shi Z.; Hu Z.; Zhong W.; Xiao G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Research 2020, 30, 269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E. S.; Dufort E. M.; Udo T.; Wilberschied L. A.; Kumar J.; Tesoriero J.; Weinberg P.; Kirkwood J.; Muse A.; Dehovitz J.; Blog D. S.; Hutton B.; Holtgrave D. R.; Zucker H. A. Association of Treatment with Hydroxychloroquine or Azithromycin with In-Hospital Mortality in Patients with COVID-19 in New York State. JAMA - Journal of the American Medical Association 2020, 323 (24), 2493–2502. 10.1001/jama.2020.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly L.; Druce J. D.; Catton M. G.; Jans D. A.; Wagstaff K. M.. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 in Vitro. Antiviral Res. 2020, 178, 104787. 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira O. V.; Rocha G. B.; Paluch A. S.; Costa L. T. Repurposing Approved Drugs as Inhibitors of SARS-CoV-2 S-Protein from Molecular Modeling and Virtual Screening. J. Biomol Struct Dyn 2021, 39 (11), 3924–3933. 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garegnani L. I.; Madrid E.; Meza N. Misleading Clinical Evidence and Systematic Reviews on Ivermectin for COVID-19. BMJ. Evid Based Med. 2022, 27 (3), 156–158. 10.1136/bmjebm-2021-111678. [DOI] [PubMed] [Google Scholar]
- Wang X.; Cao R.; Zhang H.; Liu J.; Xu M.; Hu H.; Li Y.; Zhao L.; Li W.; Sun X.; Yang X.; Shi Z.; Deng F.; Hu Z.; Zhong W.; Wang M. The Anti-Influenza Virus Drug, Arbidol Is an Efficient Inhibitor of SARS-CoV-2 in Vitro. Cell Discovery 2020, 6, 28 10.1038/s41421-020-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A. K.; Seal A.; Khan J. M.; Ahamed M.; Tripathi T. Unraveling the Mechanism of Arbidol Binding and Inhibition of SARS-CoV-2: Insights from Atomistic Simulations. Eur. J. Pharmacol. 2021, 894, 173836. 10.1016/j.ejphar.2020.173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Wang Y.; Chen C. Z.; Xu M.; Guo H.; Itkin M.; Zheng W.; Shen M. Identification of SARS-CoV-2 Viral Entry Inhibitors Using Machine Learning and Cell-Based Pseudotyped Particle Assay. Bioorg. Med. Chem. 2021, 38, 116119. 10.1016/j.bmc.2021.116119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Z.; Xu M.; Pradhan M.; Gorshkov K.; Petersen J. D.; Straus M. R.; Zhu W.; Shinn P.; Guo H.; Shen M.; Klumpp-Thomas C.; Michael S. G.; Zimmerberg J.; Zheng W.; Whittaker G. R. Identifying SARS-CoV-2 Entry Inhibitors through Drug Repurposing Screens of SARS-S and MERS-S Pseudotyped Particles. ACS Pharmacol Transl Sci. 2020, 3 (6), 1165–1175. 10.1021/acsptsci.0c00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi G. A.; Adegunloye A. P.; Ibrahim I. M.; Ogunyemi O. M.; Afolabi S. O.; Ogunro O. B. Prevention of SARS-CoV-2 Cell Entry: Insight from in Silico Interaction of Drug-like Alkaloids with Spike Glycoprotein, Human ACE2, and TMPRSS2. J. Biomol Struct Dyn 2022, 40 (5), 2121–2145. 10.1080/07391102.2020.1835726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya A.; Pandey K.; Thurman M.; Klug E.; Trivedi J.; Sharma K.; Lorson C. L.; Singh K.; Byrareddy S. N.. Discovery and Evaluation of Entry Inhibitors for SARS-CoV-2 and Its Emerging Variants. J. Virol 2021, 95 ( (24), ). 10.1128/JVI.01437-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. T.; Yang C.; Wu Y.; Lv J. J.; Feng X.; Tian X.; Zhou Z.; Pan X.; Liu S.; Tian L. W. Axial Chiral Binaphthoquinone and Perylenequinones from the Stromata of Hypocrella Bambusae Are SARS-CoV-2 Entry Inhibitors. J. Nat. Prod 2021, 84 (2), 436–443. 10.1021/acs.jnatprod.0c01136. [DOI] [PubMed] [Google Scholar]
- Curreli F.; Chau K.; Tran T.-T.; Nicolau I.; Ahmed S.; Das P.; Hillyer C. D.; Premenko-Lanier M.; Debnath A. K. Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors. Viruses 2023, 15 (4), 1001. 10.3390/v15041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X. J.; Zhu Y. Z.; Zhao P.; Qi Z. T. Entry Inhibitors: New Advances in HCV Treatment. Emerg. Microbes Infect. 2016, 5 (2016), e3 10.1038/emi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaan A. A.; Al-ahmed S. H.; Bazzi A. M.; Alfouzan W. A.; Alsuliman S. A.; Aldrazi F. A.; Haque S. Overview of Hepatitis C Infection, Molecular Biology, and New Treatment. J. Infect Public Health 2020, 13 (5), 773–783. 10.1016/j.jiph.2019.11.015. [DOI] [PubMed] [Google Scholar]
- Potter J. A.; Owsianka A. M.; Jeffery N.; Matthews D. J.; Keck Z.-Y.; Lau P.; Foung S. K. H.; Taylor G. L.; Patel A. H. Toward a Hepatitis C Virus Vaccine: The Structural Basis of Hepatitis C Virus Neutralization by AP33, a Broadly Neutralizing Antibody. J. Virol 2012, 86 (23), 12923–12932. 10.1128/JVI.02052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpitts C. C.; Baumert T. F. Hepatitis C Virus Cell Entry: A Target for Novel Antiviral Strategies to Address Limitations of Direct Acting Antivirals. Hepatol Int. 2016, 10 (5), 741–748. 10.1007/s12072-016-9724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y.; Zhong J.; Lavillette D.; Li Q.. Role of Hepatitis C Virus Envelope Glycoprotein E1 in Virus Entry and Assembly. Front. Immunol. 2018, 9 ( (June), ), 1–7. 10.3389/fimmu.2018.01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L.; Quinn E. R.; Flint M.; Hadlock K. G.; Foung S. K. H.; Levy S. Recognition of Native Hepatitis C Virus E1E2 Heterodimers by a Human Monoclonal Antibody. J. Virol 2003, 77 (2), 1604–1609. 10.1128/JVI.77.2.1604-1609.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak A. I.; Ruiz S.; Colbert M.; Luong T.; Crowe J. E. C. Jr.; Bailey J. R.; Bjorkman P. J. HCV Broadly Neutralizing Antibodies against HCV Use a CDRH3 Disulfide Motif to Recognize an E2 Glycoprotein Site That Can Be Targeted for Vaccine Design. Cell Host Microbe 2019, 24 (5), 703–716. 10.1016/j.chom.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owsianka A. M.; Timms J. M.; Tarr A. W.; Brown R. J. P.; Hickling T. P.; Szwejk A.; Bienkowska-Szewczyk K.; Thomson B. J.; Patel A. H.; Ball J. K. Identification of Conserved Residues in the E2 Envelope Glycoprotein of the Hepatitis C Virus That Are Critical for CD81 Binding. J. Virol 2006, 80 (17), 8695–8704. 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Hossain R. A.; Yost S. A.; Bu W.; Wang Y.; Dearborn A. D.; Grakoui A.; Cohen J. I.; Marcotrigiano J. Structural Insights into Hepatitis C Virus Receptor Binding and Entry. Nature 2021, 598 (7881), 521–525. 10.1038/s41586-021-03913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B.; Kelly B.; Mcmillan B. J.; Seegar T. C. M. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041. 10.1016/j.cell.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha E. S.; Sfriso P.; Rojas A. L.; Roversi P.; Hospital A.; Orozco M. Mechanism of Structural Tuning of the Hepatitis C Virus Human Cellular Receptor CD81 Large Article Mechanism of Structural Tuning of the Hepatitis C Virus Human Cellular Receptor CD81 Large Extracellular Loop. Structure/Folding Des. 2017, 25 (1), 53–65. 10.1016/j.str.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Sharma N. R.; Mateu G.; Dreux M.; Arash Grakoui F.-L. C.; Melikyan G. B. Hepatitis C Virus Is Primed by CD81 Protein for Low pH-Dependent Fusion. J. Biol. Chem. 2011, 286 (35), 30361–30376. 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearborn A. D.; Marcotrigiano J.. Hepatitis C Virus Structure: Defined by What It Is Not. Cold Spring Harb Perspect Med. 2020, 10 ( (1), ). 10.1101/cshperspect.a036822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. C.; Hsu H. J.; Yen J. H.; Lo S. Y.; Liou J. W. A Sequence in the Loop Domain of Hepatitis C Virus E2 Protein Identified in Silico as Crucial for the Selective Binding to Human CD81. PLoS One 2017, 12 (5), 1–22. 10.1371/journal.pone.0177383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie M.; Sarrazin S.; Montserret R.; Descamps V.; Baumert T. F.; Duverlie G.; Séron K.; Penin F.; Dubuisson J. Identification of Conserved Residues in Hepatitis C Virus Envelope Glycoprotein E2 That Modulate Virus Dependence on CD81 and SRB1 Entry Factors. J. Virol 2014, 88 (18), 10584–10597. 10.1128/JVI.01402-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syder A. J.; Lee H.; Zeisel M. B.; Grove J.; Soulier E.; MacDonald J.; Chow S.; Chang J.; Baumert T. F.; McKeating J. A.; McKelvy J.; Wong-Staal F. Small Molecule Scavenger Receptor BI Antagonists Are Potent HCV Entry Inhibitors. J. Hepatol 2011, 54 (1), 48–55. 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Bankwitz D.; Vieyres G.; Hueging K.; Bitzegeio J.; Doepke M.; Chhatwal P.; Haid S.; Catanese M. T.; Zeisel M. B.; Nicosia A.; Baumert T. F.; Kaderali L.; Pietschmann T. Role of Hypervariable Region 1 for the Interplay of Hepatitis C Virus with Entry Factors and Lipoproteins. J. Virol 2014, 88 (21), 12644–12655. 10.1128/JVI.01145-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H. J.; Davis C.; Mullins J. G. L.; Hu K.; Goodall M.; Farquhar M. J.; Mee C. J.; McCaffrey K.; Young S.; Drummer H.; Balfe P.; McKeating J. A. Claudin Association with CD81 Defines Hepatitis C Virus Entry. J. Biol. Chem. 2010, 285 (27), 21092–21102. 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzel D.; Fromm M. Claudins and Other Tight Junction Proteins. Compr Physiol 2012, 2 (3), 1819–1852. 10.1002/cphy.c110045. [DOI] [PubMed] [Google Scholar]
- Evans M. J.; Von Hahn T.; Tscherne D. M.; Syder A. J.; Panis M.; Wölk B.; Hatziioannou T.; McKeating J. A.; Bieniasz P. D.; Rice C. M. Claudin-1 Is a Hepatitis C Virus Co-Receptor Required for a Late Step in Entry. Nature 2007, 446 (7137), 801–805. 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- Keck Z.-Y.; Wang Y.; Lau P.; Lund G.; Rangarajan S.; Fauvelle C.; Liao G. C.; Holtsberg F. W.; Warfield K. L.; Aman M. J.; Pierce B. G.; Fuerst T. R.; Bailey J. R.; Baumert T. F.; Mariuzza R. A.; Kneteman N. M.; Foung S. K.H. Affinity Maturation of a Broadly Neutralizing Human Monoclonal Antibody That Prevents Acute HCV Infection. Hepatology 2016, 64 (6), 1922–1933. 10.1002/hep.28850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M.; Maruyama T.; Lewis J.; Giang E.; Tarr A. W.; Stamataki Z.; Gastaminza P.; Chisari F. V.; Jones I. M.; Fox R. I.; Ball J. K.; McKeating J. A.; Kneteman N. M.; Burton D. R. Broadly Neutralizing Antibodies Protect against Hepatitis C Virus Quasispecies Challenge. Nat. Med. 2008, 14 (1), 25–27. 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- Tzarum N.; Giang E.; Kong L.; He L.; Prentoe J.; Augestad E.; Hua Y.; Castillo S.; Lauer G. M.; Bukh J.; Zhu J.; Wilson I. A.; Law M. Genetic and Structural Insights into Broad Neutralization of Hepatitis C Virus by Human VH 1–69 Antibodies. Sci. Adv. 2019, 3 (January), eaav1882. 10.1126/sciadv.aav1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owsianka A.; Tarr A. W.; Juttla V. S.; Lavillette D.; Bartosch B.; Cosset F.-L.; Ball J. K.; Patel A. H. Monoclonal Antibody AP33 Defines a Broadly Neutralizing Epitope on the Hepatitis C Virus E2 Envelope Glycoprotein. J. Virol 2005, 79 (17), 11095–11104. 10.1128/JVI.79.17.11095-11104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Liu Y.; Pamulapati C.; Bohini S.; Fertig G.; Schraeml M.; Rubas W.; Brandt M.; Ries S.; Ma H.; Klumpp K. Prevention of Hepatitis C Virus Infection and Spread in Human Liver Chimeric Mice by an Anti-CD81 Monoclonal Antibody. Hepatology 2015, 61 (4), 1136–1144. 10.1002/hep.27603. [DOI] [PubMed] [Google Scholar]
- Lacek K.; Vercauteren K.; Grzyb K.; Naddeo M.; Verhoye L.; Słowikowski M. P.; Fafi-Kremer S.; Patel A. H.; Baumert T. F.; Folgori A.; Leroux-Roels G.; Cortese R.; Meuleman P.; Nicosia A. Novel Human SR-BI Antibodies Prevent Infection and Dissemination of HCV in Vitro and in Humanized Mice. J. Hepatol 2012, 57 (1), 17–23. 10.1016/j.jhep.2012.02.018. [DOI] [PubMed] [Google Scholar]
- Fukasawa M.; Nagase S.; Shirasago Y.; Iida M.; Yamashita M.; Endo K.; Yagi K.; Suzuki T.; Wakita T.; Hanada K.; Kuniyasu H.; Kondoh M. Monoclonal Antibodies against Extracellular Domains of Claudin-1 Block Hepatitis C Virus Infection in a Mouse Model. J. Virol 2015, 89 (9), 4866–4879. 10.1128/JVI.03676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Chen N. A.; Lin S.; Liu M. I. N. Synthesized Peptide 710–725 from HCV Subtype 1a E2 Glycoprotein Blocks HCV Infection through Competitive Binding of CD81. Int. J. Mol. Med. 2016, 37, 836–842. 10.3892/ijmm.2016.2459. [DOI] [PubMed] [Google Scholar]
- Si Y.; Liu X.; Liu X.; Jacobs J. L.; Cheng M.; Niu Y.; Jin Q.; Wang T.; Yang W.. A Human Claudin-1 Derived Peptide Inhibits Hepatitis C Virus Entry. Hepatology 2012, 56, 507. 10.1038/s41598-017-04274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P.; Zhang L.; Ye F.; Deng Y.; Lu S.; Li Y.; Zhang L.; Tan W. A Screen for Inhibitory Peptides of Hepatitis C Virus Identifies a Novel Entry Inhibitor Targeting E1 and E2. Sci. Rep. 2017, 7 (May), 3976. 10.1038/s41598-017-04274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Qing J.; Guo Y.; Wang Y.; Cui L.; He T.; Zhang L.; Liu L. Stapled Peptide-Based Membrane Fusion Inhibitors of Hepatitis C Virus. Bioorg. Med. Chem. 2013, 21 (12), 3547–3554. 10.1016/j.bmc.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Cao J.; Zhao P.; Miao X. H.; Zhao L. J.; Xue L. J.; Qi Z. T.. Phage Display Selection on Whole Cells Yields a Small Peptide Specific for HCV Receptor Human CD81. Cell Res. 2003, 13, 473–479. 10.1038/sj.cr.7290190 [DOI] [PubMed] [Google Scholar]
- Lü X.; Yao M.; Zhang J. M.; Yang J.; Lei Y. F.; Huang X. J.; Jia Z. S.; Ma L.; Lan H. Y.; Xu Z. K.; Yin W. Identification of Peptides That Bind Hepatitis C Virus Envelope Protein E2 and Inhibit Viral Cellular Entry from a Phage-Display Peptide Library. Int. J. Mol. Med. 2014, 33 (5), 1312–1318. 10.3892/ijmm.2014.1670. [DOI] [PubMed] [Google Scholar]
- Cao J.; Liao X. L.; Wu S. M.; Zhao P.; Zhao L. J.; Wu W. B.; Qi Z. T.. Selection of a Phage-Displayed Peptide Recognized by Monoclonal Antibody Directed Blocking the Site of Hepatitis C Virus E2 for Human CD81. J. Microbiological Methods 2007, 68, 601–604. 10.1016/j.mimet.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Qian X.; Jin Y.; Chen H.; Xu Q.; Ren H.; Zhu S.; Tang H.; Wang Y.; Zhao P.; Qi Z.; Zhu Y.. Trachelogenin, a Novel Inhibitor of Hepatitis C Virus Entry through CD81. J. General Virol. 2016, 21134–1144. 10.1099/jgv.0.000432. [DOI] [PubMed] [Google Scholar]
- Lin L. T.; Chung C. Y.; Hsu W. C.; Chang S. P.; Hung T. C.; Shields J.; Russell R. S.; Lin C. C.; Li C. F.; Yen M. H.; Tyrrell D. L. J.; Lin C. C.; Richardson C. D. Saikosaponin B2 Is a Naturally Occurring Terpenoid That Efficiently Inhibits Hepatitis C Virus Entry. J. Hepatol 2015, 62 (3), 541–548. 10.1016/j.jhep.2014.10.040. [DOI] [PubMed] [Google Scholar]
- Hung T.; Jassey A.; Liu C.; Lin C.; Lin C.; Hui S.; Wang J. Y.; Yen M.; Lin L. Phytomedicine Berberine Inhibits Hepatitis C Virus Entry by Targeting the Viral E2 Glycoprotein. Phytomedicine 2019, 53, 62–69. 10.1016/j.phymed.2018.09.025. [DOI] [PubMed] [Google Scholar]
- Syder A. J.; Lee H.; Zeisel M. B.; Grove J.; Soulier E.; MacDonald J.; Chow S.; Chang J.; Baumert T. F.; McKeating J. A.; McKelvy J.; Wong-Staal F. Small Molecule Scavenger Receptor BI Antagonists Are Potent HCV Entry Inhibitors. J. Hepatol 2011, 54 (1), 48–55. 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Rhein B. A.; Maury W. J. Ebola Virus Entry into Host Cells: Identifying Therapeutic Strategies. Curr. Clin Microbiol Rep 2015, 2 (3), 115–124. 10.1007/s40588-015-0021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Dvashi H.; Zehner M.; Ehrhardt S.; Katz M.; Elad N.; Klein F.; Diskin R. Structural Basis for a Convergent Immune Response against Ebola Virus. Cell Host Microbe 2020, 27 (3), 418–427. e4 10.1016/j.chom.2020.01.007. [DOI] [PubMed] [Google Scholar]
- Li Q.; Ma L.; Yi D.; Wang H.; Wang J.; Zhang Y.; Guo Y.; Li X.; Zhou J.; Shi Y.; Gao G. F.; Cen S. Novel Cyclo-Peptides Inhibit Ebola Pseudotyped Virus Entry by Targeting Primed GP Protein. Antiviral Res. 2018, 155, 1–11. 10.1016/j.antiviral.2018.04.020. [DOI] [PubMed] [Google Scholar]
- Côté M.; Misasi J.; Ren T.; Bruchez A.; Lee K.; Filone C. M.; Hensley L.; Li Q.; Ory D.; Chandran K.; Cunningham J. Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 2011, 477 (7364), 344–348. 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plewe M. B.; Sokolova N. V.; Gantla V. R.; Brown E. R.; Naik S.; Fetsko A.; Lorimer D. D.; Lorimer D. D.; Dranow D. M.; Dranow D. M.; Smutney H.; Smutney H.; Bullen J.; Bullen J.; Sidhu R.; Sidhu R.; Master A.; Master A.; Wang J.; Wang J.; Kallel E. A.; Zhang L.; Kalveram B.; Freiberg A. N.; Henkel G.; McCormack K. Discovery of Adamantane Carboxamides as Ebola Virus Cell Entry and Glycoprotein Inhibitors. ACS Med. Chem. Lett. 2020, 11 (6), 1160–1167. 10.1021/acsmedchemlett.0c00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyakatura E. K.; Frei J. C.; Lai J. R. Chemical and Structural Aspects of Ebola Virus Entry Inhibitors. ACS Infect Dis 2015, 1 (1), 42–52. 10.1021/id500025n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Shi Y.; Song J.; Qi J.; Lu G.; Yan J.; Gao G. F. Ebola Viral Glycoprotein Bound to Its Endosomal Receptor Niemann-Pick C1. Cell 2016, 164 (1–2), 258–268. 10.1016/j.cell.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Dvashi H.; Zehner M.; Ehrhardt S.; Katz M.; Elad N.; Klein F.; Diskin R. Structural Basis for a Convergent Immune Response against Ebola Virus. Cell Host Microbe 2020, 27 (3), 418–427. e4 10.1016/j.chom.2020.01.007. [DOI] [PubMed] [Google Scholar]
- Tian Y. S.; Zhou Y.; Takagi T.; Kameoka M.; Kawashita N. Dengue Virus and Its Inhibitors: A Brief Review. Chem. Pharm. Bull. (Tokyo) 2018, 66 (3), 191–206. 10.1248/cpb.c17-00794. [DOI] [PubMed] [Google Scholar]
- https://www.who.int/news-room/questions-and-answers/item/neglected-tropical-diseases.
- Noble C. G.; Chen Y. L.; Dong H.; Gu F.; Lim S. P.; Schul W.; Wang Q. Y.; Shi P. Y. Strategies for Development of Dengue Virus Inhibitors. Antiviral Res. 2010, 85 (3), 450–462. 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Angelin M.; Sjölin J.; Kahn F.; Ljunghill Hedberg A.; Rosdahl A.; Skorup P.; Werner S.; Woxenius S.; Askling H. H. Qdenga® - A Promising Dengue Fever Vaccine; Can It Be Recommended to Non-Immune Travelers?. Travel Med. Infect Dis 2023, 54 (June), 102598. 10.1016/j.tmaid.2023.102598. [DOI] [PubMed] [Google Scholar]
- https://www.who.int/news-room/questions-and-answers/item/dengue-vaccines.
- Modis Y.; Ogata S.; Clements D.; Harrison S. C. A Ligand-Binding Pocket in the Dengue Virus Envelope Glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (12), 6986–6991. 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Oliveira C.; Freire J. M.; Conceição T. M.; Higa L. M.; Castanho M. A. R. B.; Da Poian A. T. Receptors and Routes of Dengue Virus Entry into the Host Cells. FEMS Microbiol Rev. 2015, 39 (2), 155–170. 10.1093/femsre/fuu004. [DOI] [PubMed] [Google Scholar]
- Frei J. C.; Kielian M.; Lai J. R.; States U.; States U.. Comprehensive Mapping of Functional Epitopes on Dengue Virus Glycoprotein E DIII for Binding to Broadly Neutralizing Antibodies 4E11 and 4E5A by Phage Display. Virology 2016, 2, 371–382. 10.1016/j.virol.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L. N.; Tharakaraman K.; Rowley K. J.; Costa V. V.; Chan K. R.; Wong Y. H.; Ong L. C.; Tan H. C.; Koch T.; Cain D.; Kirloskar R.; Viswanathan K.; Liew C. W.; Tissire H.; Ramakrishnan B.; Myette J. R.; Babcock G. J.; Sasisekharan V.; Alonso S.; Chen J.; Lescar J.; Shriver Z.; Ooi E. E.; Sasisekharan R. Structure-Guided Design of an Anti-Dengue Antibody Directed to a Non-Immunodominant Epitope. Cell 2015, 162 (3), 493–504. 10.1016/j.cell.2015.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Ge P.; Yu X.; Brannan J. M.; Bi G.; Zhang Q.; Schein S.; Zhou Z. H. Cryo-EM Structure of the Mature Dengue Virus at 3.5-Å Resolution. Nat. Struct Mol. Biol. 2013, 20 (1), 105–110. 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn J. J. B.; Navarro Sanchez M. E.; Fretes N.; Urvoas A.; Staropoli I.; Kikuti C. M.; Coffey L. L.; Arenzana Seisdedos F.; Bedouelle H.; Rey F. A. Mechanism of Dengue Virus Broad Cross-Neutralization by a Monoclonal Antibody. Structure 2012, 20 (2), 303–314. 10.1016/j.str.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Panya A.; Bangphoomi K.; Choowongkomon K.; Yenchitsomanus P. T. Peptide Inhibitors against Dengue Virus Infection. Chem. Biol. Drug Des 2014, 84 (2), 148–157. 10.1111/cbdd.12309. [DOI] [PubMed] [Google Scholar]
- Wang Q. Y.; Patel S. J.; Vangrevelinghe E.; Hao Y. X.; Rao R.; Jaber D.; Schul W.; Gu F.; Heudi O.; Ngai L. M.; Mee K. P.; Wai Y. P.; Keller T. H.; Jacoby E.; Vasudevan S. G. A Small-Molecule Dengue Virus Entry Inhibitor. Antimicrob. Agents Chemother. 2009, 53 (5), 1823–1831. 10.1128/AAC.01148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costin J. M.; Jenwitheesuk E.; Lok S. M.; Hunsperger E.; Conrads K. A.; Fontaine K. A.; Rees C. R.; Rossmann M. G.; Isern S.; Samudrala R.; Michael S. F.. Structural Optimization and de Novo Design of Dengue Virus Entry Inhibitory Peptides. PLoS Negl. Trop. Dis. 2010, 4 ( (6), ). 10.1371/journal.pntd.0000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panya A.; Sawasdee N.; Junking M.; Srisawat C.; Choowongkomon K.; Yenchitsomanus P. T. A Peptide Inhibitor Derived from the Conserved Ectodomain Region of DENV Membrane (M) Protein with Activity Against Dengue Virus Infection. Chem. Biol. Drug Des 2015, 86 (5), 1093–1104. 10.1111/cbdd.12576. [DOI] [PubMed] [Google Scholar]
- Henß L.; Beck S.; Weidner T.; Biedenkopf N.; Sliva K.; Weber C.; Becker S.; Schnierle B. S.. Suramin Is a Potent Inhibitor of Chikungunya and Ebola Virus Cell Entry. Virol J. 2016, 13 ( (1), ). 10.1186/s12985-016-0607-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques P.; Fritzer A.; Dereuddre-Bosquet N.; Wressnigg N.; Hochreiter R.; Bossevot L.; Pascal Q.; Guehenneux F.; Bitzer A.; Ramljak I. C.; Le Grand R.; Lundberg U.; Meinke A. Effectiveness of CHIKV Vaccine VLA1553 Demonstrated by Passive Transfer of Human Sera. JCI Insight 2022, 7 (14), 1–15. 10.1172/jci.insight.160173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim M. S.; Aman A. T. Understanding the Biology and Immune Pathogenesis of Chikungunya Virus Infection for Diagnostic and Vaccine Development. Viruses 2023, 15, 48. 10.3390/v15010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; Liss N. M.; Chen D. H.; Liao M.; Fox J. M.; Shimak R. M.; Fong R. H.; Chafets D.; Bakkour S.; Keating S.; Fomin M. E.; Muench M. O.; Sherman M. B.; Doranz B. J.; Diamond M. S.; Simmons G. Neutralizing Monoclonal Antibodies Block Chikungunya Virus Entry and Release by Targeting an Epitope Critical to Viral Pathogenesis. Cell Rep 2015, 13 (11), 2553–2564. 10.1016/j.celrep.2015.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delogu I.; Pastorino B.; Baronti C.; Nougairède A.; Bonnet E.; de Lamballerie X. In Vitro Antiviral Activity of Arbidol against Chikungunya Virus and Characteristics of a Selected Resistant Mutant. Antiviral Res. 2011, 90 (3), 99–107. 10.1016/j.antiviral.2011.03.182. [DOI] [PubMed] [Google Scholar]
- Santos I. A.; Shimizu J. F.; de Oliveira D. M.; Martins D. O. S.; Cardoso-Sousa L.; Cintra A. C. O.; Aquino V. H.; Sampaio S. V.; Nicolau-Junior N.; Sabino-Silva R.; Merits A.; Harris M.; Jardim A. C. G.. Chikungunya Virus Entry Is Strongly Inhibited by Phospholipase A2 Isolated from the Venom of Crotalus Durissus Terrificus. Sci. Rep 2021, 11 ( (1), ). 10.1038/s41598-021-88039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayusso G. M.; da Silva Sanches P. R.; Carvalho T.; Santos I. A.; Martins D. O. S.; Lima M. L. D.; da Conceição P. J. P.; Bittar C.; Merits A.; Cilli E. M.; Jardim A. C. G.; Rahal P.; Calmon M. F. The Synthetic Peptide GA-Hecate and Its Analogs Inhibit Multiple Steps of the Chikungunya Virus Infection Cycle In Vitro. Pharmaceuticals 2023, 16 (10), 1389. 10.3390/ph16101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K.; Daniel M.; LaBeaud D. A.; Gasque P.; Peeling R. W.; Stephenson K. E.; Ng L. F. P.; Ariën K. K. Chikungunya Fever. Nat. Rev. Dis Primers 2023, 9 (1), 17. 10.1038/s41572-023-00429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Zhao Z.; Chai Y.; Jin X.; Li C.; Yuan F.; Liu S.; Gao Z.; Wang H.; Song J.; Vazquez L.; Zhang Y.; Tan S.; Morel C. M.; Yan J.; Shi Y.; Qi J.; Gao F.; Gao G. F. Molecular Basis of Arthritogenic Alphavirus Receptor MXRA8 Binding to Chikungunya Virus Envelope Protein. Cell 2019, 177 (7), 1714–1724. 10.1016/j.cell.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Wintachai P.; Thuaud F.; Basmadjian C.; Roytrakul S.; Ubol S.; Désaubry L.; Smith D. R. Assessment of Flavaglines as Potential Chikungunya Virus Entry Inhibitors. Microbiol. Immunol. 2015, 59 (3), 129–141. 10.1111/1348-0421.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Earnest J. T.; Kim A. S.; Winkler E. S.; Desai P.; Adams L. J.; Hu G.; Bullock C.; Gold B.; Cherry S.; Diamond M. S. Expression of the Mxra8 Receptor Promotes Alphavirus Infection and Pathogenesis in Mice and Drosophila. Cell Rep 2019, 28 (10), 2647–2658. 10.1016/j.celrep.2019.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Zhao Z.; Chai Y.; Jin X.; Li C.; Yuan F.; Liu S.; Gao Z.; Wang H.; Song J.; Vazquez L.; Zhang Y.; Tan S.; Morel C. M.; Yan J.; Shi Y.; Qi J.; Gao F.; Gao G. F. Molecular Basis of Arthritogenic Alphavirus Receptor MXRA8 Binding to Chikungunya Virus Envelope Protein. Cell 2019, 177 (7), 1714–1724. 10.1016/j.cell.2019.04.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Jones B. E.; Brown-Augsburger P. L.; Corbett K. S.; Westendorf K.; Davies J.; Cujec T. P.; Wiethoff C. M.; Blackbourne J. L.; Heinz B. A.; Foster D.; Higgs R. E.; Balasubramaniam D.; Wang L.; Bidshahri R.; Kraft L.; Hwang Y.; Žentelis S.; Jepson K. R.; Goya R.; Smith M. A.; Collins D. W.; Hinshaw S. J.; Tycho S. A.; Pellacani D.; Xiang P.; Muthuraman K.; Sobhanifar S.; Piper M. H.; Triana F. J.; Hendle J.; Pustilnik A.; Adams A. C.; Berens S. J.; Baric R. S.; Martinez D. R.; Cross R. W.; Geisbert T. W.; Borisevich V.; Abiona O.; Belli H. M.; de Vries M.; Mohamed A.; Dittmann M.; Samanovic M.; Mulligan M. J.; Goldsmith J. A.; Hsieh C.-L.; Johnson N. V.; Wrapp D.; McLellan J. S.; Barnhart B. C.; Graham B. S.; Mascola J. R.; Hansen C. L.; Falconer E.. LY-CoV555, a Rapidly Isolated Potent Neutralizing Antibody, Provides Protection in a Non-Human Primate Model of SARS-CoV-2 Infection. bioRxiv, 2020, 10.1101/2020.09.30.318972 [DOI]
- Cathcart A. L.; Havenar-Daughton C.; Lempp F. A.; Ma D.; Schmid M. A.; Agostini M. L.; Guarino B.; Di iulio J.; Rosen L. E.; Tucker H.; Dillen J.; Subramanian S.; Sloan B.; Bianchi S.; Pinto D.; Saliba C.; Culap K.; Wojcechowskyj J. A.; Noack J.; Zhou J.; Kaiser H.; Lee S.; Farhat N.; Chase A.; Montiel-Ruiz M.; Dellota E.; Park A.; Spreafico R.; Lauron E. J.; Czudnochowski N.; Cameroni E.; Ledoux S.; Werts A.; Colas C.; Soriaga L.; Telenti A.; Purcell L. A.; Snell G.; Virgin H. W.; Corti D.; Hebner C. M.. The Dual Function Monoclonal Antibodies VIR-7831 and VIR-7832 Demonstrate Potent in Vitro and 2 in Vivo Activity against SARS-CoV-2. bioRxiv, 2021, 10.1101/2021.03.09.434607. [DOI]