

Abstract
Voltage-sensitive calcium channels (VSCCs) influence bone structure and function, including anabolic responses to mechanical loading. While the pore-forming (α1) subunit of VSCCs allows Ca2+ influx, auxiliary subunits regulate the biophysical properties of the pore. The α2δ1 subunit influences gating kinetics of the α1 pore and enables mechanically induced signaling in osteocytes; however, the skeletal function of α2δ1 in vivo remains unknown. In this work, we examined the skeletal consequences of deleting Cacna2d1, the gene encoding α2δ1. Dual-energy X-ray absorptiometry and microcomputed tomography imaging demonstrated that deletion of α2δ1 diminished bone mineral content and density in both male and female C57BL/6 mice. Structural differences manifested in both trabecular and cortical bone for males, while the absence of α2δ1 affected only cortical bone in female mice. Deletion of α2δ1 impaired skeletal mechanical properties in both sexes, as measured by three-point bending to failure. While no changes in osteoblast number or activity were found for either sex, male mice displayed a significant increase in osteoclast number, accompanied by increased eroded bone surface and upregulation of genes that regulate osteoclast differentiation. Deletion of α2δ1 also rendered the skeleton insensitive to exogenous mechanical loading in males. While previous work demonstrates that VSCCs are essential for anabolic responses to mechanical loading, the mechanism by which these channels sense and respond to force remained unclear. Our data demonstrate that the α2δ1 auxiliary VSCC subunit functions to maintain baseline bone mass and strength through regulation of osteoclast activity and also provides skeletal mechanotransduction in male mice. These data reveal a molecular player in our understanding of the mechanisms by which VSCCs influence skeletal adaptation.
Keywords: α2δ1 subunit, mechanotransduction, voltage-sensitive calcium channel, bone formation, loading
Introduction
Bone alters its structure and mass to accommodate physical and hormonal challenges.1 Mechanical stimulation of bone triggers skeletal adaptation resulting in bone mass accrual and increases in bone size and strength. The anabolic effects of mechanical loading require coordinated efforts among several cell types2; however, it is accepted that osteocytes, as the primary mechanosensitive skeletal cells,3 coordinate osteoblast and osteoclast activity to regulate bone mass.4 The ability of skeletal cells to sense mechanical force requires transmission of mechanical signals from the fluid-permeated bone matrix to the cell membrane that is orchestrated by actions of extracellular matrix molecules,5 integrins,6 connexins,7 Wnt/BMP ligands,8 and ion channels.9
One of the first responses of bone cells to mechanical stimuli is a rapid and transient increase in calcium (Ca2+) influx.10,11 Among Ca2+-selective channels, voltage-sensitive calcium channels (VSCCs) exquisitely control Ca2+ influx in various tissue and cell types including skeletal muscle, cardiac muscle, and neurons.12 In bone, VSCCs regulate skeletal development and responses to mechanical loading. Treatment with verapamil or nifedipine, inhibitors specific to L-type (long-lasting activation) VSCCs, impairs skeletal structure,13 leading to vertebral defects with decreased osteogenesis.14,15 Likewise, inhibition of VSCCs with L-type inhibitors suppresses load-induced bone formation.16,17 In osteoblasts, L-type VSCCs function in fluid shear–induced release of ATP,18 a critical paracrine signal that regulates mechanosensitive skeletal responses. While L-type VSCCs are the primary VSCC subtype in growing osteoblasts, we previously reported that differentiation of osteoblasts to osteocytes was accompanied by a downregulation of L-type VSCCs, resulting in osteocytes expressing only transiently activated T-type VSCCs.19
Recent work using cultured cells showed that the Ca2+ responses of osteocytes are dramatically different from those of osteoblasts in response to mechanical force.20 Furthermore, treatment of osteocytes with an inhibitor specific to T-type VSCCs blocked the fluid shear–induced Ca2+ intracellular transients, demonstrating that osteocytes require T-type channels to respond to mechanical stimuli.21
The long-lasting or transient nature of VSCCs is dictated by the pore-forming (α1) subunit.22 Most VSCC types possess additional auxiliary subunits that associate with the pore to refine channel function. Subunits include the membrane-spanning γ subunit, the intracellular β subunit, and the α2δ1 subunit, which associates with the α1 subunit, primarily via the von Willebrand factor-A domain within α2.23 The α2δ1 subunit contains a large glycosylated α2 ectodomain (Figure 1A).24 A single gene product, these two subunits are produced by proteolysis and then held together by a disulfide bond.25 In some cell types, the α2δ1 subunit regulates gating kinetics of the α1 pore, where deletion or disruption of α2δ1 decreases open-times and reduces Ca2+ influx.24 While the α2δ1 subunit associates with the α1 pore of CaV1 and CaV2 channel subtypes, it does not frequently associate with CaV3 VSCCs. However, we showed that α2δ1 binds the T-type, Cav3.2 (α1H) subunit in osteocytes, and represents the predominant pore-forming VSCC subunit in osteocytes.26 Knockdown of α2δ1 renders osteocytes insensitive to mechanical input,26 yet the function of this auxiliary subunit in the in vivo skeleton remains unknown.
Figure 1.

Deletion of α2δ1 does not alter expression of other α2δ isoforms. (A) Schematic depicting VSCC subunit structure and spatial relationship. Schematic modified from previously published image.26 (B) Protein expression of α2δ1 assessed by Western blot using whole protein lysates from tibias of male WT and α2δ1 KO mice. Each lane is a lysate (20 μg) prepared from individual mice. Membranes were blotted for α2δ1 or β-actin as a loading control. (C) Expression of α2δ isoforms from mRNA isolated from tibias of male WT and α2δ1 KO mice. Each gene was normalized to Gapdh (n = 6 for Cacna2d1-3; n = 4 for Cacna2d4). (D) Body mass of male WT (n = 18) and KO (n = 10) mice was measured every 3 wk from 6 to 18 wk of age. Significance of qPCR data was tested by unpaired Student’s t-tests. Longitudinal body mass was determined using repeated measures ANOVA. ***P < .001.
In this work, we investigated the skeletal consequences of global α2δ1 deletion in mice, examined the cellular mechanisms by which α2δ1 regulates skeletal structure, and used an in vivo ulnar loading model to measure the contribution of α2δ1 to skeletal mechanotransduction.
Materials and methods
Mice
Global deletion of Cacna2d1, the gene encoding α2δ1, was achieved by the targeted insertion of a neomycin resistance gene in exon 2, as previously described.27 All mouse colonies were maintained on a C57BL/6 background. Heterozygous (Cacna2d1+/−) male and female mice were bred to obtain wild-type (WT, Cacna2d1+/+) and knockout (KO, Cacna2d1−/−) littermates. Heterozygous offspring were generated but not evaluated. Both male and female (WT and KO) mice were used in all experiments. All mice provided food and water ad lib and housed in standard cages. Experimental mice were housed 5 mice per cage while breeders were housed 1:1, according to the policies of IU animal facilities. Investigators were blinded to sex and genotype during all experimental procedures. All experiments were performed using procedures approved by the IACUC at Indiana University.
Western blotting
Total protein lysates were prepared from tibias of WT and KO mice, as previously described.28 Briefly, tibias were dissected, all soft tissue was removed, and bones were flash frozen in liquid nitrogen (N2). Frozen tibias were cut into small pieces into Bullet Blender homogenization tubes containing radio immunoprecipitation assay lysis buffer (150 mM NaCl, 50 mM Tris HCl, 1 mM ethylene glycol-bis (β-aminoethyl ether (EGTA), 0.24% (w/v) sodium deoxycholate, 1% (w/v) Igepal, pH 7.5) with protease and phosphatase inhibitors. Inhibitors including NaF (1 mM) and Na3VO4 (1 mM), aprotinin (1 μg/mL), leupeptin (1 μg/mL), pepstatin (1 μg/mL), and phenylmethylsulfonyl fluoride (1 mM) were added fresh, just prior to lysis.29 Total protein lysates (20 μg) were separated on SDS-polyacrylamide gradient gels (4%–12%) and transferred to polyvinylidene difluoride membranes, as described previously.30 Membranes were blocked with milk (5%, w/v) diluted in Tris-buffered saline containing Tween 20 (0.01%, v/v). Blots then were incubated overnight at 4 °C with primary antibody recognizing α2δ1 (cat# ab2864; Abcam or ꞵ-actin (cat# 5125; Cell Signaling). Blots were washed and incubated with horseradish peroxidase–conjugated secondary antibody (1:5000 dilution) (Cell Signaling) at RT for 1 hour with chemiluminescent detection using ECL Plus substrate (Amersham Biosciences). Images were developed and acquired with an iBright CL1000 machine (Applied Biosystems).
Real-time PCR
Total RNA was isolated from whole tibias of WT and KO mice. Tibias were dissected, all soft tissue, including the periosteum, was removed and immediately flash frozen in liquid N2.31 When ready for isolation, tibias were cut into small pieces and placed in Bullet Blender tubes in TRIzol. Bones were pulverized using the Bullet Blender homogenizer, and the soluble fraction was aliquoted to a new tube. Total RNA was isolated by using phenol chloroform extraction, followed by RNeasy kit (Qiagen) as described previously.32 mRNA was reverse transcribed, and genes were amplified with a Bio-Rad CFX Connect quantitative polymerase chain reaction (qPCR) machine, using gene-specific primers, as previously described.30 The PCR products were normalized to GAPDH and quantified using the ΔΔCT method (denoted in figures as ΔΔCq).
Dual-energy X-ray absorptiometry
Serial DXA measurements were collected on live mice every 3 wk from 6 to 18 wk of age, using an UltraFocus digital X-ray cabinet (Faxitron), as previously described.33 Briefly, mice were anesthetized with isoflurane (2.5%, IsoFlo; Abbott Laboratories) mixed with oxygen (1.5 L/min) for ~8 min, including induction and scanning. Mice were placed on the scanner in a prone position while whole body scans were analyzed regionally using the Lunar region of interest (ROI) tools. The ROI for the spine included from the first (L1) through sixth (L6) lumbar vertebrae. The ROI for the femur included bone between the acetabulum and the tibiofemoral joint space. The ROI for the whole body included all skeletal tissues caudal to the boundary between the skull and the first cervical vertebra but excluding the tail. Bone mineral content and BMD were measured for each ROI scan.
Microcomputed tomography
Femurs and lumbar vertebrae (L5) were dissected and fixed in normal buffered formalin (NBF, 10%, v/v) for at least 48 hr. Bones were scanned, reconstructed, and analyzed, as previously described.33 Briefly, scans were collected using a Scanco uCT-35 tomographer at 10 μm resolution, 50-kV peak tube potential, and 151-ms integration time. The distal 60% of each femur and the entire body of each vertebra were scanned. Standard parameters related to cancellous and cortical bone architecture were measured.34
Mechanical testing
Parameters related to whole bone strength were measured using three-point bending tests on isolated femora, as previously described.35 Briefly, each femur was loaded to failure in monotonic compression, during which force and displacement measures were collected every 0.01 s. Ultimate force, stiffness, and energy to ultimate force were calculated from the force/displacement curves, using standard equations.36
In vivo ulnar loading
The axial ulnar compression model was applied to WT and KO mice, as previously described.37 Briefly, 6 calibration mice of each sex and genotype were euthanized at 18 wk of age. The right forelimb was disarticulated and frozen at −20 °C until strain gauge testing. A single-element strain gauge (EA-06-015DJ-120; Vishay) was applied to the lateral surface of the ulnar midshaft, and the microstrain:load ratio was measured for each sample using progressively increasing load applications while simultaneously recording the voltage output from the load cell and strain gauge. All tests were averaged within each genotype to determine the microstrain:load ratio within each sex and genotype. A peak microstrain value of −2250 was chosen to be applied to all genotypes, and this corresponded to peak loads of 2.56 N (WT females), 1.99 N (KO females), 2.5 N (WT males), and 2.17 N (KO males). At 18 wk of age, 10 mice of each sex and genotype began the axial ulnar loading protocol. Mice were anesthetized using isoflurane inhalation, and the right ulna (wrist to elbow) was loaded using sinusoidal (haversine) waveform (2 Hz, 180 cycles, no intercycle rest period) to the peak load determined by strain gauging (see above). Mice were administered five loading bouts over a 10-d period with a day of rest between each bout. Intraperitoneal injection of calcein was administered 1 d before the final bout, followed by an i.p. injection of alizarin complexone 11 d later, as previously described.38 Mice were euthanized 13 d after the final bout of loading (22 wk of age). The right and left ulnae were harvested and placed in NBF (10%, v/v) for 48 hr and then stored in ethanol (70%, v/v) at 4 °C until analysis.
Histological processing
Femurs and ulnae were dehydrated in graded alcohols, cleared in xylene, and embedded in MMA following standard protocols.39 For femurs, thick-cut sections were taken ~3 mm proximal to the tibiofibular junction and manually ground down to ~30 μm. For ulnae, thick-cut sections were taken ~2 mm proximal and ~2 mm distal to the ulna midshaft and manually ground down to ~30 μm. A single unstained section from each bone was imaged digitally on a fluorescent microscope using filter sets that provided excitation and emission for the calcein and alizarin wavelengths. Digital images were imported into Image-Pro Express (Media Cybernetics, Inc.), and the following histomorphometric measurements were recorded for the periosteal surface: total perimeter, single label perimeter (sL.Pm), double label area (dL.Ar) and perimeter (dL.Pm), total bone, and marrow area. The following results were calculated: mineral apposition rate (MAR = dL.Ar/dL.Pm/11 d), mineralizing surface (MS/BS = (0.5 × sL.Pm + dL.Pm) / total perimeter × 100), and bone formation rate (BFR/BS = MAR × MS / BS × 3.65). Relative formation parameters were calculated for each mouse by subtracting the nonloaded (left ulna) values form the loaded (right ulna) values. Histology sections, staining, and analyses were conducted by the Histology Core Facilities within the Indiana Center for Musculoskeletal Health.
Histomorphometry
Methyl-methacrylate–embedded, thin sections were deplasticized in acetone and stained by two different procedures: (1) a modification of the von Kossa/MacNeal’s (VKM) tetrachrome protocol40 and (2) a tartrate acid–resistant acid phosphatase (TRAP) stain.41 For VKM-stained slides, mineralized bone was stained using the von Kossa silver method and the unmineralized tissue was counter stained with MacNeal’s tetrachrome. For TRAP staining, sections were preincubated in acetate buffer (0.2 M, pH = 5.0), rinsed, and incubated in a warmed acid phosphatase solution. Sections then were counterstained with Gill’s hematoxylin No. 3, air dried, and cover-slipped with an aqueous-based mounting media. Histomorphometric analysis was performed using the OsteoMeasure high-resolution digital video system (OsteoMetrics Inc.). Standard nomenclature was used according to the Histomorphometry Nomenclature Committee of the American Society of Bone and Mineral Research.42
Statistical analyses
Statistical variance was expressed as the means ± standard error of the mean. Statistical significance was evaluated using one-way ANOVA, two-way ANOVA followed by Fisher’s least significant difference (LSD) post hoc test, or Student’s t-test, as appropriate (Prism GraphPad). Values were considered significant if P ≤ .05. The table in Supplementary Table S1 provides significance values for the sex, genotype, and terms interaction terms for two-way ANOVA analyses. qPCR and Western blot assays were replicated at least three times to assure reproducibility. The number of animals used is denoted for individual experiments.
Results
Deletion of α2δ1 reduces body weight without observed compensation of other isoforms
Western blotting demonstrated abundant expression of α2δ1 in bone from WT mice, with undetectable levels in tibias from KO mice (Figure 1B). As α2δ1 is one of four α2δ isoforms, relative transcript levels of each isoform were measured by qPCR from mRNA isolated from tibias of WT and KO mice. As expected, mRNA of Cacna2d1 was reduced (P < .001) in male α2δ1 KO mice, compared to WT controls, while there were no changes in gene expression for the Cacna2d2, Cacna2d3, or Cacna2d4 isoforms (Figure 1C). The body weight of both male (Figure 1D) and female (data not shown) α2δ1 KO mice was reduced (P < .0001), compared to WT controls, at 6, 9, 12, 15, and 18 wk.
Deletion of α2δ1 reduces BMC and BMD
To determine the influence of the α2δ1 subunit on skeletal formation, DXA measurements were taken every 3 wk from 6 to 18 wk of age in both male (Figure 2, Supplementary Figure S1) and female (Supplementary Figure S2) mice. Bone mineral density at the whole body, femur, and spine of male α2δ1 KO mice was reduced (P < .0001) at each time point (Figure 2A–C). Bone mineral density of female KO mice also was reduced, compared to controls (Supplementary Figure S2D–F). Bone mineral content of the whole body, femur, and spine were all reduced (P < .0001) in male α2δ1 KO mice compared to WT controls at each time point measured (Supplementary Figure S1A–C). As we found that KO mice had reduced body weight compared to WT mice (Figure 1D), BMC was also normalized to body weight to determine if body weight/size accounted for the differences in BMC. As seen in Supplementary Figure S1, the reductions in BMC in KO mice remained significant when normalized to body weight (Supplementary Figure S1). Similar reductions in BMC were found in female KO mice, compared to WT controls (Supplementary Figure S2A–C). These data demonstrate that mice lacking the α2δ1 subunit have impaired bone content and density at all skeletal sites measured.
Figure 2.
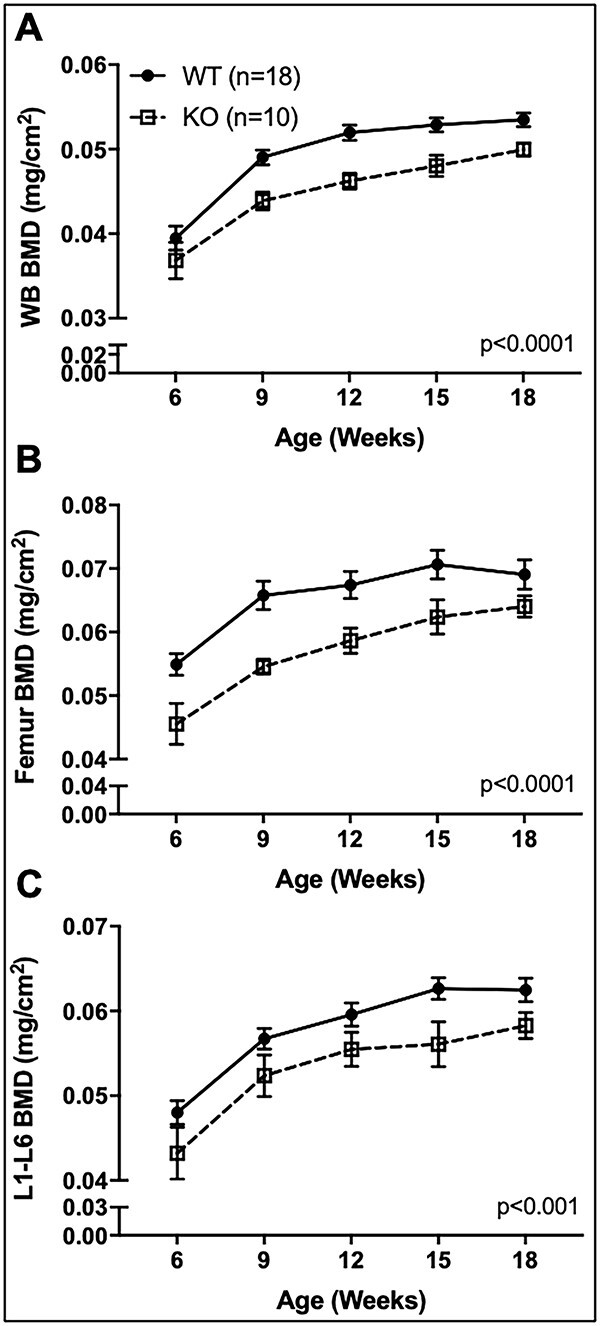
Longitudinal in vivo DXA scans of male WT and α2δ1 KO mice. WT (n = 18, solid circle) and α2δ1 KO (n = 10, dotted square) mice were scanned every 3 wk from 6 to 18 wk of age and analyzed for BMD at the (A) whole body (WB), (B) femur, and (C) spine (L1-L6 vertebrae). Longitudinal data were tested for significance between genotype by two-way repeated measures ANOVA.
Male α2δ1 KO mice displayed altered femoral trabecular bone structure
As deletion of α2δ1 resulted in impaired DXA-derived BMC and BMD, microcomputed tomography (μCT) measures of the distal femur were undertaken to examine alterations in skeletal microarchitecture following α2δ1 deletion. Representative images of the mid-diaphysis and distal femur of 22-wk-old male mice demonstrate cortical thinning and reduced trabecular bone (Figure 3A). Quantification of trabecular structure revealed a 40% reduction (P < .001) in the bone volume fraction (BV/TV) (Figure 3B) and a 52% decrease in BMC (Figure 3C, P < .01) in KO mice. While no differences in trabecular number were found (Figure 3D), deletion of α2δ1 reduced trabecular thickness (Figure 3E, P < .01) and connectivity (Figure 3F, P < .05) by 16% and 33%, respectively. Further, α2δ1 KO mice displayed no changes in trabecular spacing compared to WT controls. These data demonstrate that deletion of the α2δ1 subunit results in impaired trabecular structure at the distal femur of male mice, as manifested by thinner trabeculae leading to reduced connectivity. These data provide additional insight into the structural contributions leading to decreased BMC and BMD.
Figure 3.
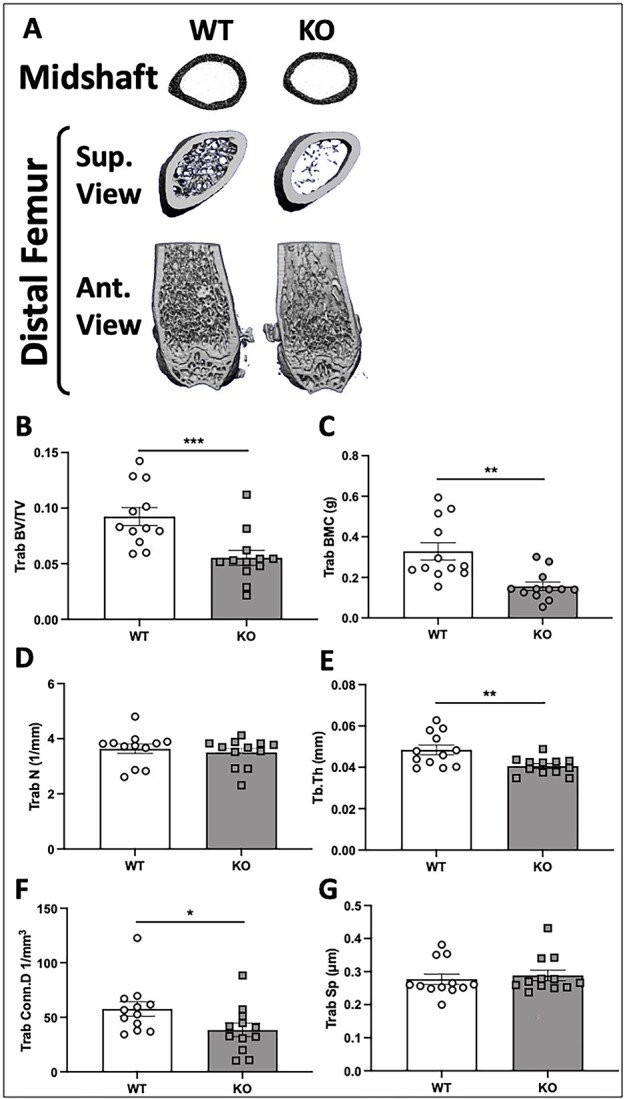
μCT-derived measures of trabecular bone in the distal femur of 22-wk-old male WT and α2δ1 KO mice. (A) Representative 3D reconstructions of midshaft femur (top row), distal metaphysis (middle row, proximal view), and caudal region of the distal femur (bottom row). Quantitative differences in (B) trabecular bone volume fraction (BV/TV) of the femur; (C) trabecular BMC; (D) trabecular number (Trab N); (E) trabecular thickness (Trab Th); (F) trabecular connectivity (Trab conn D); (G) trabecular spacing (Trab Sp). Significance between genotypes was tested using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01, ***P < .001. n = 12 per group.
Cortical bone structure of the femur is impaired in the absence of α2δ1 in male and female mice
Deletion of α2δ1 reduced marrow area by 18% (Figure 4A, P < .001) and total area by 17% (Figure 4B, P < .001). Cortical thickness of α2δ1 KO mice was decreased by 8.7% (Figure 4C, P < .01), while there were no changes in femur length (Figure 4D). Cortical BMC was reduced by 15% in KO mice (Figure 4E, P < .001), while the polar moment of inertia (pMOI) was 31.6% lower in KO mice, compared to WT controls (Figure 4F, P < .001). These data demonstrate that deletion of α2δ1 in male mice alters cortical bone structure, with reduced pMOI predicting decreased fracture resistance.
Figure 4.

μCT-derived measures of cortical bone at the midshaft femur of 22-wk-old male WT and α2δ1 KO mice. Measures derived from cortical bone at the mid-diaphysis of the femur including (A) marrow area; (B) total area; (C) cortical thickness (cortical Th); (D) femur length; (E) cortical BMC; (F) polar moment of inertia (pMOI). Significance between genotypes tested using two-way ANOVA followed by Fisher’s LSD post hoc test, **P < .01, ***P < .001. n = 12 per group.
While deletion of α2δ1 in female mice had no measurable effect on trabecular structure (Supplementary Figure S3), reductions in cortical bone parameters were observed. The marrow area and total area of the femurs from α2δ1 KO mice were reduced by 9.6% (Supplementary Figure S4A, P < .05) and 8% (Supplementary Figure S4B, P < .05), respectively. Cortical thickness of KO mice was decreased by 5% (Supplementary Figure S4C, P < .01), and femur length was 2.6% lower in KO mice compared to WT controls (Supplementary Figure S4D, P < .05). Cortical BMC of α2δ1 KO mice was reduced by 15% (Supplementary Figure S4E, P < .0001), and the pMOI of KO mice was 18% lower than WT mice (Supplementary Figure S3F, P < .01). Two-way ANOVA analyses determined a significant influence of sex on genotype effects for several trabecular parameters including BV/TV, BMC, connectivity density, and pMOI, as summarized in Supplementary Table S1. These data indicate that deletion of α2δ1 in female mice alters cortical but not trabecular bone of the femur.
Deletion of α2δ1 impairs vertebral bone structure in male mice
Representative μCT reconstruction images of the L5 vertebrae of male WT and KO mice show structural differences in bone from KO mice (Figure 5A). Trabecular BV/TV of male α2δ1 KO mice was reduced by 17% (Figure 5B, P < .01). Reductions in trabecular BMC (22%, P < .01) and BMD (3%, P < .01) also were found in α2δ1 KO mice (Figure 5C, D). Trabecular connectivity of the L5 vertebrae of male KO mice was decreased by 23% (Figure 5E, P < .001), while the number of trabeculae was decreased by 10%, compared to WT mice (Figure 5F, P < .001). Additionally, trabecular spacing was increased in male α2δ1 KO mice by 12.6% (Figure 5G, P < .05), and no changes in trabecular thickness were observed (Figure 5H).
Figure 5.
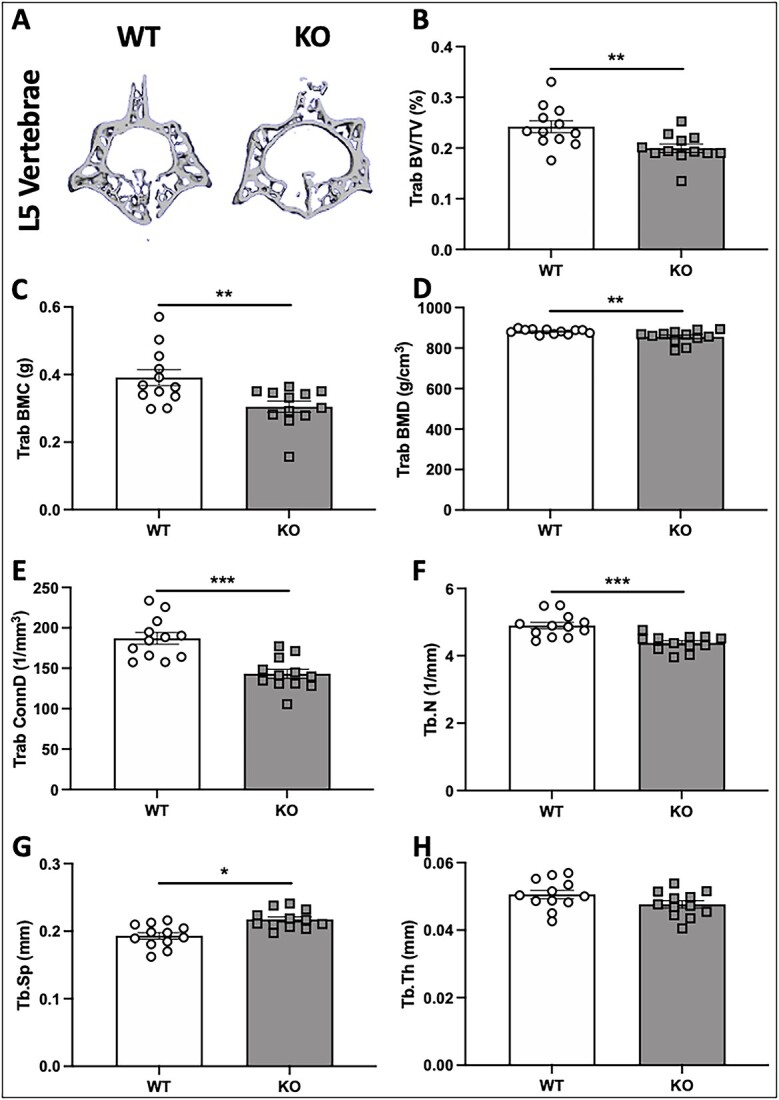
μCT-derived measures of L5 vertebrae of 22-wk-old male WT and α2δ1 KO mice. (A) Representative images of 3D reconstructions of L5 vertebrae from WT and α2δ1 KO mice. Quantification of μCT measures of L5 vertebrae included (B) trabecular bone volume fraction (BV/TV); (C) trabecular BMC; (D) trabecular BMD; (E) trabecular connectivity (Trab Conn D); (F) trabecular number (Trab N); (G) trabecular spacing (Trab Sp); (H) trabecular thickness (Trab Th). Significance between genotypes tested using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01, ***P < .001. n = 12 per group.
The L5 vertebrae of female α2δ1 KO mice displayed a 6.6% reduction (P < .05) in trabecular thickness (data not shown); however, no other significant differences in μCT measures were observed. These data are consistent with analyses of distal femurs from female mice showing no changes in trabecular structure following deletion of α2δ1. The interaction between sex and genotype was determined by two-way ANOVA that showed several parameters with a significant interaction term as summarized in Supplementary Table S1.
Deletion of the α2δ1 subunit impairs skeletal strength
Because deletion of α2δ1 impaired bone quantity and structure, we next subjected femurs from WT and α2δ1 KO mice to three-point bending to determine if KO of α2δ1 influences bone strength. Three-point bending to failure at the midshaft of femurs from male mice revealed reductions in ultimate force of 18% (Figure 6A, P < .01), decreased energy absorption of 22% (Figure 6B, P < .05), and a 16% reduction in stiffness (Figure 6C, P < .05). Femurs of female α2δ1 KO mice also displayed impaired strength with a 21% decrease in ultimate force (Figure 6A, P < .01) and a 28% decrease in stiffness (Figure 6C, P < .001). No change in energy absorption was observed in femurs of female mice (Figure 6B). These data demonstrate that deletion of the α2δ1 VSCC subunit impairs skeletal strength of both male and female mice. There was no difference in energy absorption for female mice, and two-way ANOVA did not reveal significant differences for the interaction term between sex and genotype (Supplementary Table S1).
Figure 6.
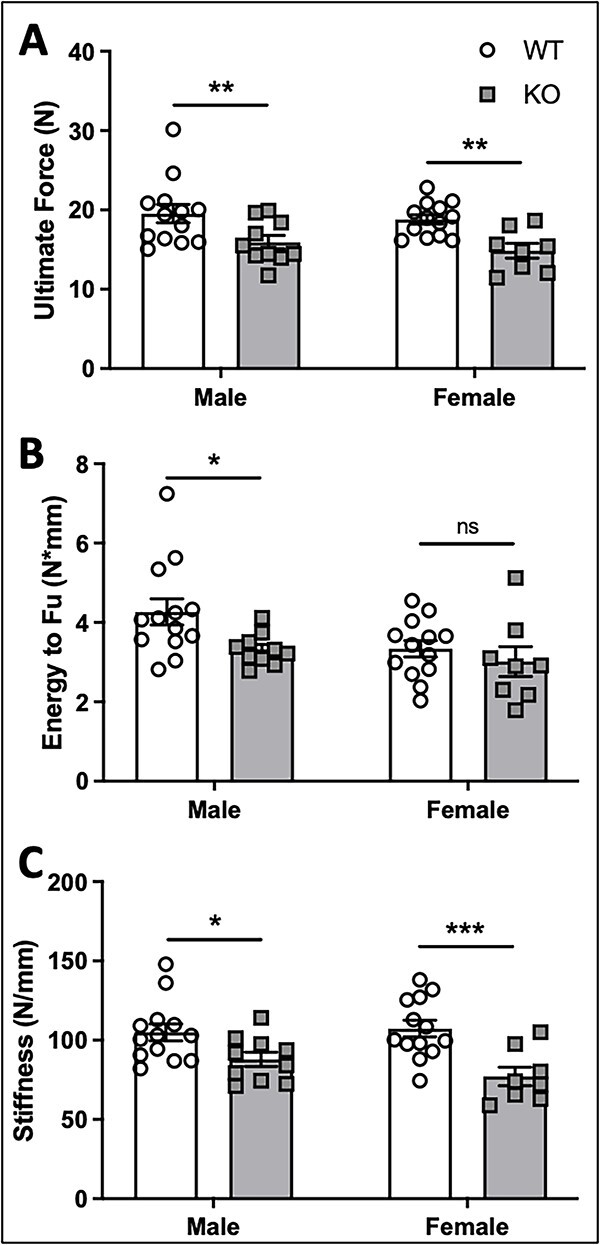
Mechanical testing via monotonic three-point bending to failure on whole femora from 22-wk-old WT and α2δ1 KO mice. Quantification of (A) ultimate force, (B) energy to ultimate force, and (C) stiffness reveal significant reductions in the mechanical properties of bone following deletion of α2δ1 in male mice. Reductions in (A) ultimate force and (C) stiffness were observed in female mice, with no significant differences in energy to ultimate force in females. Data were tested for significance using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01, ***P < .001. WT: n = 13, KO: n = 10 for males; WT: n = 13, KO: n = 8 for females.
Osteoblast regulatory genes are reduced in α2δ1 KO mice
As α2δ1 KO mice had decreased bone parameters, by μCT, compared to WT mice, we performed histological analyses to determine if these altered bone parameters can be attributed to impaired osteoblast number or activity. Representative images of distal femur sections from male WT and α2δ1 KO mice stained with VKM reveal no obvious alterations in bone morphology (Figure 7A). Quantification of sections from male mice found no differences in osteoblast number (Figure 7B), osteoblast surface relative to bone surface (Ob.S/BS, Figure 7C), osteoid surface relative to the bone surface (OS/BS, Figure 7D), number of osteoblasts relative to the bone perimeter (N.Ob/B.Pm, Figure 7E), osteoid thickness (O.Th) (Figure 7F), or osteocyte number (data not shown).
Figure 7.
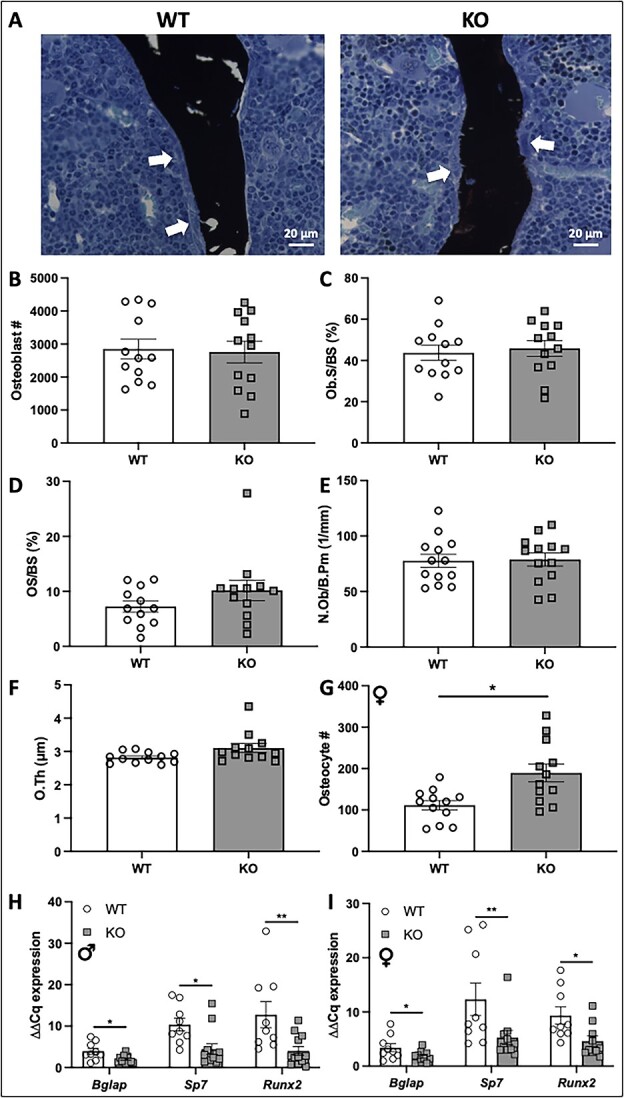
Histological analyses of von Kossa/MacNeal–stained sections of 22-wk-old femora. (A) Representative images of distal femur sections from male WT and α2δ1 KO mice stained with von Kossa/MacNeal. White arrows indicate osteoblasts on the bone surface. Quantification of osteoblast parameters from male mice including (B) osteoblast number, (C) osteoblast surface normalized to bone surface (Ob.S/BS), (D) osteoid surface normalized to bone surface (OS/BS), (E) osteoblast number normalized to bone perimeter (N.Ob/B.Pm), and (F) osteoid thickness (O.Th). (G) Osteocyte number was quantified from distal femurs of female WT and KO mice. Expression of osteoblast regulatory genes from male (H) and female (I) mice, including osteocalcin (Bglap), osterix (Sp7), and Runx2 were all normalized to Gapdh. Data were tested for significance using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01. n = 12 per group for histomorphometry analyses. For qPCR, WT: n = 9, KO: n = 12 for males; WT: n = 9, KO: n = 10 for females.
Histological sections from female WT and α2δ1 KO mice revealed no significant changes in the osteoblast number (Supplementary Figure S5A), osteoblast surface (Supplementary Figure S5B), osteoid surface (Supplementary Figure S5C), osteoid thickness (Supplementary Figure S5D), or the number of osteoblasts as normalized to bone perimeter (Supplementary Figure S5E). However, in contrast to male mice, deletion of α2δ1 in female mice resulted in a 70% increase (P < .01) in the number of osteocytes compared to that of WT animals (Figure 7G). When normalized to bone area, the increase in osteocyte number within female bone was also significantly increased (P < .01) compared to WT controls (Supplementary Figure S5F).
To further understand the cellular mechanisms driving alterations in bone mass, structure, and strength in the absence of α2δ1, transcripts of genes that regulate bone formation were quantified from mRNA isolated from tibias of WT and α2δ1 KO mice. Expression of osteocalcin (Bglap) was reduced by 2-fold in both male (Figure 7H, P < .05) and female mice (Figure 7I, P < .05). Levels of osterix (Sp7) were decreased by ~2-fold in both male (Figure 7H, P < .05) and female mice (Figure 7I, P < .01). Runx2 expression was reduced by 3-fold (Figure 7H, P < .01) in male and by 2-fold in female mice (Figure 7I, P < .05).
Knockout of α2δ1 increases osteoclast number in male mice
Representative images of distal femur sections of male WT and KO mice demonstrated a greater number of TRAP-stained, multinucleated osteoclasts on the surfaces of trabeculae of α2δ1 KO mice compared to WT controls (Figure 8A). Quantification of histological sections showed that femurs of α2δ1 KO male mice had a 49% increase in osteoclast number (Figure 8B, P < .01); a 38% increase in osteoclast surface, as normalized to total bone surface (Figure 8C, P < .05); and a 37% increase in the eroded bone surface, as normalized to total bone surface (Figure 8D, P < .05). No difference in the number of osteoclasts as normalized to bone perimeter was observed (Figure 8E).
Figure 8.
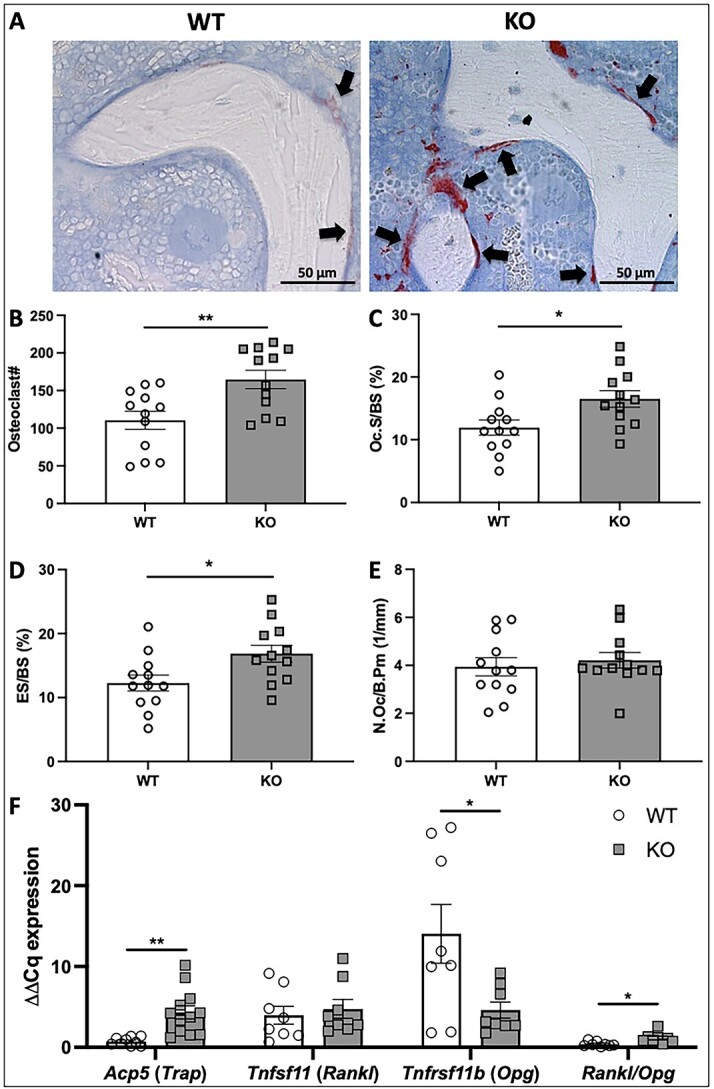
Histomorphometric analyses of TRAP-stained sections of 22-wk-old male femora. (A) Representative images of distal femur sections from WT and α2δ1 KO mice stained for TRAP. Multinucleated osteoclasts are stained crimson/red (black arrows). Histomorphometric quantification of osteoclast parameters include (B) osteoclast number, (C) osteoclast surface normalized to total bone surface (Oc.S/BS), (D) eroded surface normalized to total bone surface (ES/BS), (E) number of osteoclasts as a function of total bone perimeter (N.Oc/B.Pm). (F) Expression of osteoclast regulatory genes including Acp5, Tnfsf11, Tnfrsf11b, and the ratio of Rankl/Opg were all normalized to Gapdh. Data were tested for significance using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01. n = 12 per group for histomorphometry analyses. For qPCR, WT: n = 9, KO: n = 8 to 12.
Expression of a subset of genes that regulate osteoclast formation was examined from mRNA extracted from tibias of male WT and α2δ1 KO mice. Deletion of α2δ1 resulted in a 6-fold increase in Acp5 (Figure 8F, P < .01), the gene encoding for Trap. No changes in Tnfsf11, the gene encoding for Rankl, were observed; however, expression of Tnfrsf11b (Opg) was decreased by 3-fold (P < .05) in tibias of α2δ1 KO mice, which contributed to a nearly 3-fold increase in the Rankl/Opg ratio following α2δ1 deletion (Figure 8F, P < .05).
Histological sections from female WT and α2δ1 KO mice revealed no significant changes in the number of osteoclasts (Supplementary Figure S6A), the osteoclast surface (Supplementary Figure S6B), or the eroded bone surface (Supplementary Figure S6C); however, the number of osteoclasts as normalized to bone perimeter was significantly reduced in KO mice compared to WT controls (Supplementary Figure S6D, P < .01). While no changes in overall osteoclast numbers were found, KO mice had a significant increase in Trap (Acp5) mRNA of 2.5-fold (Supplementary Figure S6E, P < .05), but no changes in Rankl or Opg were observed (Supplementary Figure S5E). Two-way ANOVA analysis revealed a significant interaction between genotype and sex for osteoclast number (P = .014), osteoclast number as normalized to bone perimeter (P = .011), and osteoclast surface (P = .047). No significant interaction was found between genotype and sex when values from osteoclast-related qPCR were examined (Supplementary Table S1).
The α2δ1 subunit is required for anabolic responses to mechanical loading in male mice
We previously showed that knockdown of the α2δ1 subunit in cultured osteocytic cells suppressed ERK1/2 phosphorylation and release of ATP in response to mechanical force, which led us to examine the contribution of α2δ1 to anabolic responses of bone to exogenous loading. Male WT mice displayed an increase in the mineralizing surface (MS/BS, 2-fold, P = .052), MAR (2-fold, P < .01), and bone formation rate (BFR/BS, 4-fold, P < .05) in response to mechanical loading. In contrast, male mice lacking the α2δ1 subunit did not respond to mechanical loading, showing no significant change in MS/BS, MAR, or BFR/BS (Figure 9B–D), suggesting that α2δ1 is required for anabolic responses to loading.
Figure 9.

Axial ulnar loading in 18-wk-old male WT and α2δ1 KO mice. Strain-matched peak forces were applied to ulnae of male mice. (A) Representative images of control (nonloaded) and loaded ulnae from WT and KO mice. (B) Mineralizing surface (MS/BS), (C) mineral apposition rate (MAR), and (D) bone formation rate (BFR/BS) were increased in WT mice but had no significant changes in α2δ1 KO mice. Data were tested for significance using two-way ANOVA followed by Fisher’s LSD post hoc test, *P < .05, **P < .01. WT: n = 8, KO: n = 7.
In female mice, ulnar loading induced significant increases in MS/BS, MAR, and BFR/BS in both WT and KO female mice (Supplementary Figure S7A–C). These data demonstrate that, in contrast to male mice, deletion of α2δ1 does not impair responses to loading in female animals.
Discussion
Voltage-sensitive calcium channels tightly regulate activation of intracellular signaling cascades that control numerous cellular responses including the initiation of cardiac muscle contraction,43 hormone and neurotransmitter release,44 and skeletal development.13 While the pore-forming α1 subunit is crucial for influx of Ca2+ to initiate these responses, the auxiliary subunits of VSCCs modulate the gating kinetics of the pore,24 enabling greater context-specific control of channel function than can be achieved by the α1 subunit alone.
Several studies have shown that VSCCs regulate bone development and responses to mechanical loading11,13,16,17; however, the contribution of auxiliary VSCC subunits to these responses have never been examined in vivo. The goal of this work was to assess the contribution of α2δ1 to skeletal homeostasis and determine the mechanisms by which this auxiliary subunit regulates bone formation and anabolic responses to mechanical loading. Here, we demonstrate that global deletion of α2δ1 negatively impacts both trabecular and cortical bone in male mice, while the skeletal alterations of female mice are confined to cortical bone. Reductions in strength, as measured by three-point bending assays, of both male and female mice are consistent with impaired cortical structure. We also demonstrate that the α2δ1 subunit is essential for anabolic responses to mechanical loading in male mice.
Bone formation and subsequent remodeling rely on secretion of bone matrix by osteoblasts and resorption of the bone matrix by osteoclasts. As this study used a mouse harboring global deletion of α2δ1, we considered the contribution of both osteoblasts and osteoclasts to the impaired bone phenotype of α2δ1 KO mice. While there were no measurable changes in the number or activity of osteoblasts, expression of osteoblast regulatory genes was decreased in bones of α2δ1 KO mice. These decreases may be the indirect result of signaling from other cell types. It is possible that altered signaling from osteoclasts or osteocytes, following α2δ1 deletion, influenced expression of Bglap, Sp7, and Runx2 after osteoblast differentiation occurred. Such a mechanism may explain why no structural differences were seen by histology, despite suppression of osteoblast markers.
While no changes in osteoblast number were observed histologically in our studies, deletion of α2δ1 resulted in increased osteoclast number and activity in male mice, suggesting that impairments in skeletal formation and structure after deletion of α2δ1, may be owed to increased bone resorption. Although this is the first report detailing a function for α2δ1 in regulation of osteoclast number and activity, a gain-of-function mutation in the L-type, Cav1.2 (α1C) subunit that increased VSCC activity resulted in decreased osteoclast formation in both long bones45 and the ossicles of the inner ear.46 In both studies, increases in VSCC activity led to decreased osteoclast formation, accompanied by an increase in osteoblast-derived serum OPG, an inhibitor of RANKL. Here, we showed that the reverse also is seen where deletion of α2δ1 decreased Opg expression accompanied by higher osteoclast number. Taken together, the findings indicate that loss of α2δ1 impairs VSCC activity and presumably loss of normal Ca2+ signals that maintain the proper communication between osteoblasts and osteoclasts.
Alterations in osteoclast number and activity may be explained by a direct function of α2δ1 in osteoclasts; however, as paracrine signaling from osteoblasts and osteocytes regulates osteoclast function, our findings may involve signaling from other cell types. We previously showed that inhibition of VSCCs in osteoblasts resulted in decreased OPG secretion.47 Thus, decreases in OPG, following α2δ1 deletion, may be attributed to altered signaling from osteoblasts. It also is possible that α2δ1 regulates osteocyte-specific production of OPG, as osteocytes also produce OPG and RANKL.48 Ongoing work is examining the cell-specific functions of α2δ1.
Voltage-sensitive calcium channels are essential for proper anabolic responses to mechanical loading in bone.16,17,20 Furthermore, knockdown of α2δ1 in cultured osteocytes impaired the ability to respond to mechanical signals.26 These observations prompted us to examine the contribution of α2δ1 to mechanotransduction in vivo. While 18-wk-old male WT mice had significant increases in MS/BS, MAR, and BFR when subjected to ulnar loading, there were no significant anabolic responses to loading in age-matched male α2δ1 KO mice. Our previous work found that knockdown of α2δ1 impaired mechanical responses in cultured osteocytes. The current work provides in vivo evidence of the function of α2δ1 in skeletal mechanotransduction. Osteocytes are considered the predominant mechanosensory cells within bone, and we previously showed that α2δ1 knockdown impaired mechanically induced ERK1/2 phosphorylation and ATP secretion in osteocytic cells.26 Thus, our data suggest that the α2δ1 enables osteocytes to respond to mechanical signals. Such function could be mediated through the α2 region, which resides entirely extracellularly. The α2 domain interacts with extracellular matrix molecules49 and soluble ligands50 in several cell types, including neurons and muscle. Further, we recently showed that α2δ1 binds tightly to perlecan (PLN),51 a large extracellular matrix heparan sulfate proteoglycan. Perlecan deficiency alters lacunocanalicular structure,5 results in fewer tethering elements within the lacunocanalicular system,26,52 and impairs skeletal mechanosensation.53 As such, binding interactions between α2δ1 and PLN, or other extracellular matrix molecules could initiate mechanotransduction events in osteocytes or “prime” VSCCs making them more susceptible to subsequent stimuli, similar to interactions between parathyroid hormone and 1,25(OH)2D3 on Ca2+ transients in bone cells.54
Although both male and female mice displayed an impaired skeletal phenotype in the absence of α2δ1, there was a consistent sexual dimorphism among many of the parameters examined. Both cortical and trabecular bone were decreased in male mice, while female animals only had differences in cortical bone, following α2δ1 deletion. The reduced trabecular formation in male mice is likely the result of increased osteoclast activity, which also was found only in males. Differences in anabolic responses to loading also were seen only in male mice. One possible explanation for these differences is the effects of estrogen on channel activity. Estradiol-17beta (E2) enhances VSCC function by increasing Ca2+ current density and inward current.55 Thus, while deletion of α2δ1 in male mice renders the skeleton insensitive to mechanical loading, estrogen production in female mice may shift the activation potential of VSCCs toward the resting potential,54 thereby overcoming the negative consequences of α2δ1 deletion and enabling the channel to be activated by mechanical stimuli. In this manner, E2 could protect the female skeleton through a mechanism reliant upon the α2δ1 VSCC subunit.
Another possibility for the differences in responses to mechanical loading observed between male and female mice is the change in osteocyte number. Distinct from male mice, female α2δ1 ΚΟ mice had increased osteocyte number. While it remains unclear what accounts for the increased osteocyte number in female mice in the absence of α2δ1, it could be that loss of α2δ1 in female mice dampens the cell’s responsiveness to mechanical stimuli, but incompletely. Thus, female mice may compensate for the loss of α2δ1 by increasing the number of osteocytes, thus enabling their skeletons to perceive and respond to loading, likely through mechanisms independent of VSCCs.
Recent work demonstrated that pannexin 1 (Panx1), a nonjunctional transmembrane hemichannel, regulates fluid shear–mediated ATP release from osteocytes.56 The ability to influence ATP release was attributed to the complex formed between Panx1 and the purinergic receptor P2X7R. Blocking release of ATP from Panx1 or downstream binding of ATP to the P2X7R receptor impaired Ca2+ signaling in MLO-Y4 osteocytic cells.56 Additional work demonstrated that Panx1, P2x7R, and the Cav3.2 VSCC reside within close proximity to β3 integrin focal attachments within MLO-Y4 osteocytes,57 suggesting that this channel-receptor-focal adhesion complex serves coordinated roles in osteocyte mechanotransduction. Importantly, our group demonstrated that the T-type, Cav3.2 VSCC is the predominant VSCC within osteocytes,19 and despite the fact that auxiliary subunits do not always complex with T-type VSCCs, we showed that the α2δ1 subunit associates with Cav3.2 in osteocytes.26 Furthermore, we found that knockdown of α2δ1 in osteocytic cells impaired the release of ATP from osteocytes and suppressed mechanical responses.26 As Panx1 and the Cav3.2 VSCC amplify ATP release in response to mechanical stimuli, our studies suggest that the α2δ1 auxiliary VSCC subunit could influence the interplay between Panx1 and Cav3.2 by regulating the amount of Ca2+ influx and ultimately downstream purinergic signaling. Additional studies will be necessary to understand the dynamics through which these membrane channels influence one another and anabolic responses to mechanical signals.
The primary aim of this study was to determine the skeletal consequences of Cacna2d1 deletion, as no other work has examined the skeletal function of α2δ1 in vivo. However, the use of a global Cacna2d1 KO model means that indirect effects could account for some of these observations. Our data demonstrate that deletion of Cacna2d1 results in upregulation of osteoclast number and activity; however, this effect and the other skeletal alterations may be related to other skeletal or nonskeletal cell types that rely on α2δ1. As the differences observed between sexes remains unclear, additional work is necessary to understand the cell type and sex-specific functions of α2δ1 in bone.
In summary, mice deficient in the α2δ1 auxiliary VSCC subunit exhibit significantly reduced structural parameters including bone mass, density, and strength. These changes are most likely attributable to increased osteoclast formation, at least in male mice. While there were no differences in osteoblast number in α2δ1 KO mice, altered osteoclast number and activity in the absence of the α2δ1 subunit appears to be indirect, resulting from reductions in OPG, which is produced by osteoblasts. We also show that male mice require the α2δ1 subunit for proper anabolic responses to mechanical loading. These data not only reveal the mechanisms by which VSCCs regulate skeletal homeostasis but show that VSCCs utilize auxiliary subunits to mediate mechanical input, thus representing new pharmacological and/or mechanical targets for disease modification.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We’d like to thank Dr. Manuela Nieto-Rostro for providing the global knockout mice to our lab, guidance on proper genotyping of these mice, and input on the progression of the project.
Contributor Information
Madison M Kelly, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States.
Karan Sharma, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States.
Christian S Wright, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States.
Xin Yi, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States.
Perla C Reyes Fernandez, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States.
Aaron T Gegg, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States.
Taylor A Gorrell, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States.
Megan L Noonan, Department of Medical and Molecular Genetics, Indiana University, Indianapolis, IN 46202, United States.
Ahmed Baghdady, College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States.
Jacob A Sieger, College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States.
Annette C Dolphin, Department of Neuroscience, Physiology and Pharmacology, University College of London, Gower Street, London WC1E 6BT, United Kingdom.
Stuart J Warden, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; La Trobe Sport and Exercise Medicine Research Centre, La Trobe University, Melbourne Victoria 3086, DX 211319, Australia.
Padmini Deosthale, Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; Department of Anatomy, Cell Biology, & Physiology, Indiana University, Indianapolis, IN 46202, United States.
Lilian I Plotkin, Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; Department of Anatomy, Cell Biology, & Physiology, Indiana University, Indianapolis, IN 46202, United States.
Uma Sankar, Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; Department of Anatomy, Cell Biology, & Physiology, Indiana University, Indianapolis, IN 46202, United States.
Julia M Hum, College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States.
Alexander G Robling, Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; Department of Anatomy, Cell Biology, & Physiology, Indiana University, Indianapolis, IN 46202, United States.
Mary C Farach-Carson, Department of Diagnostic & Biomedical Sciences, University of Texas Health Science Center at Houston School of Dentistry, Houston, TX 77054, United States.
William R Thompson, Department of Physical Therapy, School of Health and Human Sciences, Indiana University, Indianapolis, IN 46202, United States; College of Osteopathic Medicine, Marian University, Indianapolis, IN 46222, United States; Indiana Center for Musculoskeletal Health, Indiana University, Indianapolis, IN 46202, United States; Department of Anatomy, Cell Biology, & Physiology, Indiana University, Indianapolis, IN 46202, United States.
Author contributions
Madison M. Kelly: collection/assembly of data, data analysis/interpretation, manuscript writing, final approval of manuscript.
Karan Sharma, Christian S. Wright, Xin Yi, Aaron T. Gegg, Taylor A. Gorrell, Megan L. Noonan, Ahmed Baghdady, Jacob A. Sieger: collection/assembly of data, data analysis/interpretation, final approval of manuscript.
Annette C. Dolphin, Padmini Deosthale, Lilian I. Plotkin: data analysis/interpretation, final approval of manuscript.
Stuart J. Warden, Uma Sankar, Julia M. Hum, Alexander G. Robling: concept/design, data analysis/interpretation, final approval of manuscript.
Mary C. Farach-Carson: concept/design, data analysis/interpretation, manuscript writing, final approval of manuscript.
William R. Thompson: concept/design, collection/assembly of data, data analysis/interpretation, manuscript writing, final approval of manuscript.
Madison M. Kelly and Karan Sharma provided equal contribution to the work.
Funding
This study was supported by National Institutes of Health (NIAMS) 1F32AR074893-01 to C.S.W., National Institutes of Health (NIAMS) 1R01AR074473-01 to W.R.T., Indiana University Clinical and Translational Sciences Institute UL1 TR001108 to W.R.T. and A.G.R., National Institutes of Health (NIAMS) 1R01AR068332 to U.S., Faculty Research Development funds through Marian University to W.R.T. and J.M.H., and Wellcome Investigator Award 206279/Z17/Z to A.C.D.
Conflicts of interest
None declared.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
- 1. Warden SJ, Thompson WR. Become one with the force: optimising mechanotherapy through an understanding of mechanobiology. Br J Sports Med. 2017;51(13):989–990. Epub 2017/06/25. 10.1136/bjsports-2017-097634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson WR, Scott A, Loghmani MT, Ward SR, Warden SJ. Understanding mechanobiology: physical therapists as a force in mechanotherapy and musculoskeletal regenerative rehabilitation. Phys Ther. 2016;96(4):560–569. 10.2522/ptj.20150224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–238. 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503(2):179–193. 10.1016/j.gene.2012.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thompson WR, Modla S, Grindel BJ, et al. Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J Bone Miner Res. 2011;26(3):618–629. 10.1002/jbmr.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geoghegan IP, Hoey DA, McNamara LM. Integrins in osteocyte biology and mechanotransduction. Curr Osteoporos Rep. 2019;17(4):195–206. Epub 2019/06/30. 10.1007/s11914-019-00520-2. [DOI] [PubMed] [Google Scholar]
- 7. Plotkin LI, Davis HM, Cisterna BA, Saez JC. Connexins and pannexins in bone and skeletal muscle. Curr Osteoporos Rep. 2017;15(4):326–334. Epub 2017/06/26. 10.1007/s11914-017-0374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bullock WA, Pavalko FM, Robling AG. Osteocytes and mechanical loading: the Wnt connection. Orthod Craniofac Res. 2019;22(Suppl 1):175–179. Epub 2019/05/11. 10.1111/ocr.12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wright CS, Robling AG, Farach-Carson MC, Thompson WR. Skeletal functions of voltage sensitive calcium channels. Curr Osteoporos Rep. 2021;19(2):206–221. 2021/03/15. 10.1007/s11914-020-00647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ajubi NE, Klein-Nulend J, Alblas MJ, Burger EH, Nijweide PJ. Signal transduction pathways involved in fluid flow-induced PGE(2) production by cultured osteocytes. Am J Physiol-Endocrinol Metab. 1999;276(1):E171–E178. 10.1152/ajpendo.1999.276.1.E171. [DOI] [PubMed] [Google Scholar]
- 11. Hung CT, Allen FD, Pollack SR, Brighton CT. Intracellular Ca2+ stores and extracellular Ca2+ are required in the real-time Ca2+ response of bone cells experiencing fluid flow. J Biomech. 1996;29(11):1411–1417. 10.1016/0021-9290(96)84536-2. [DOI] [PubMed] [Google Scholar]
- 12. French RJ, Zamponi GW. Voltage-gated sodium and calcium channels in nerve, muscle, and heart. IEEE Trans Nanobioscience. 2005;4(1):58–69. Epub 2005/04/09. 10.1109/TNB.2004.842500. [DOI] [PubMed] [Google Scholar]
- 13. Li J, Zhao L, Ferries IK, et al. Skeletal phenotype of mice with a null mutation in Cav 1.3 L-type calcium channel. J Musculoskelet Neuronal Interact. Jun 2010;10(2):180–187 [PubMed] [Google Scholar]
- 14. Duriez J, Flautre B, Blary MC, Hardouin P. Effects of the calcium channel blocker nifedipine on epiphyseal growth plate and bone turnover: a study in rabbit. Calcif Tissue Int. 1993;52(2):120–124. 10.1007/BF00308320. [DOI] [PubMed] [Google Scholar]
- 15. Ridings JE, Palmer AK, Davidson EJ, Baldwin JA. Prenatal toxicity studies in rats and rabbits with the calcium channel blocker diproteverine. Reprod Toxicol. Jan-Feb 1996;10(1):43–49. 10.1016/0890-6238(95)02017-9. [DOI] [PubMed] [Google Scholar]
- 16. Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17(10):1795–1800. 10.1359/jbmr.2002.17.10.1795. [DOI] [PubMed] [Google Scholar]
- 17. Li J, Duncan RL, Burr DB, Gattone VH, Turner CH. Parathyroid hormone enhances mechanically induced bone formation, possibly involving L-type voltage-sensitive calcium channels. Endocrinology. 2003;144(4):1226–1233. 10.1210/en.2002-220821. [DOI] [PubMed] [Google Scholar]
- 18. Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL. Fluid shear-induced ATP secretion mediates prostaglandin release in MC3T3-E1 osteoblasts. J Bone Miner Res. 2005;20(1):41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shao Y, Alicknavitch M, Farach-Carson MC. Expression of voltage sensitive calcium channel (VSCC) L-type Cav1.2 (alpha1C) and T-type Cav3.2 (alpha1H) subunits during mouse bone development. Dev Dyn. 2005;234(1):54–62. 10.1002/dvdy.20517. [DOI] [PubMed] [Google Scholar]
- 20. Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. J Bone Miner Res. 2012;27(3):563–574. 10.1002/jbmr.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown GN, Leong PL, Guo XE. T-type voltage-sensitive calcium channels mediate mechanically-induced intracellular calcium oscillations in osteocytes by regulating endoplasmic reticulum calcium dynamics. Bone. 2016;88:56–63. 10.1016/j.bone.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel alpha1A subunit. Proc Natl Acad Sci U S A. 1997;94(4):1512–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canti C, Nieto-Rostro M, Foucault I, et al. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of alpha2delta subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 2005;102(32):11230–11235 Epub 2005/08/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594(19):5369–5390. 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Douglas L, Davies A, Wratten J, Dolphin AC. Do voltage-gated calcium channel alpha2delta subunits require proteolytic processing into alpha2 and delta to be functional? Biochem Soc Trans. 2006;34(5):894–898. Epub 2006/10/21. 10.1042/BST0340894. [DOI] [PubMed] [Google Scholar]
- 26. Thompson WR, Majid AS, Czymmek KJ, et al. Association of the α2δ1 subunit with Cav3.2 enhances membrane expression and regulates mechanically induced ATP release in MLO-Y4 osteocytes. J Bone Miner Res. 2011;26(9):2125–2139. 10.1002/jbmr.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuller-Bicer GA, Varadi G, Koch SE, et al. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol. 2009;297(1):H117–H124. 10.1152/ajpheart.00122.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson WR, Guilluy C, Xie Z, et al. Mechanically activated Fyn utilizes mTORC2 to regulate RhoA and adipogenesis in mesenchymal stem cells. Stem Cells. 2013;31(11, 11):2528–2537. 10.1002/stem.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson WR, Keller BV, Davis ML, Dahners LE, Weinhold PS. Low-magnitude, high-frequency vibration fails to accelerate ligament healing but stimulates collagen synthesis in the Achilles tendon. Orthop J Sports Med. 2015;3(5):232596711558578. 10.1177/2325967115585783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson WR, Uzer G, Brobst KE, et al. Osteocyte specific responses to soluble and mechanical stimuli in a stem cell derived culture model. Sci Rep. 2015;5(1):11049. 10.1038/srep11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keller BV, Davis ML, Thompson WR, Dahners LE, Weinhold PS. Varying whole body vibration amplitude differentially affects tendon and ligament structural and material properties. J Biomech. 2013;46(9):1496–1500. 10.1016/j.jbiomech.2013.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson WR, Yen SS, Uzer G, et al. LARG GEF and ARHGAP18 orchestrate RhoA activity to control mesenchymal stem cell lineage. Bone. 2018;107:172–180. 10.1016/j.bone.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kedlaya R, Veera S, Horan DJ, et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5(211):211ra158. 10.1126/scitranslmed.3006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–1486. 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 35. Cui Y, Niziolek PJ, MacDonald BT, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684–691. Epub 2011/05/24. 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. Jul-Aug. 1993;14(4):595–608. 10.1016/8756-3282(93)90081-K. [DOI] [PubMed] [Google Scholar]
- 37. Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–5875 Epub 2007/12/20 [DOI] [PubMed] [Google Scholar]
- 38. Lewis KJ, Yi X, Wright CS, et al. The mTORC2 component Rictor is required for load-induced bone formation in late-stage skeletal cells. JBMR Plus. 2020;4(7):e10366.Epub 2020/07/16. 10.1002/jbm4.10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williams JN, Kambrath AV, Patel RB, et al. Inhibition of CaMKK2 enhances fracture healing by stimulating Indian hedgehog Signaling and accelerating endochondral ossification. J Bone Miner Res. 2018;33(5):930–944. Epub 2018/01/10. 10.1002/jbmr.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schenk RK, Olah AJ, Herrmann W. Preparation of calcified tissues for light microscopy. In: Dickson G, ed. Methods of Calcified Tissue Preparation. Amsterdam: Elsevier; 1984:1–56. [Google Scholar]
- 41. Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132(1):195–210. Epub 1996/01/01. 10.1083/jcb.132.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry nomenclature committee. J Bone Miner Res. 2013;28(1):2–17. Epub 2012/12/01. 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87(4):275–281 Epub 2000/08/19 [DOI] [PubMed] [Google Scholar]
- 44. Dolphin AC, Pearson HA, Menon-Johansson AS, et al. G protein modulation of voltage-dependent calcium channels and transmitter release. Biochem Soc Trans. 1993;21(2):391–395. Epub 1993/05/01. 10.1042/bst0210391. [DOI] [PubMed] [Google Scholar]
- 45. Cao C, Ren Y, Barnett AS, et al. Increased Ca2+ signaling through CaV1.2 promotes bone formation and prevents estrogen deficiency-induced bone loss. JCI Insight. 2017;2(22):1–11. Epub 2017/12/05. 10.1172/jci.insight.95512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cao C, Oswald AB, Fabella BA, et al. The CaV1.2 L-type calcium channel regulates bone homeostasis in the middle and inner ear. Bone. Aug 2019;125:160–168. Epub 2019/05/24. 10.1016/j.bone.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bergh JJ, Xu Y, Farach-Carson MC. Osteoprotegerin expression and secretion are regulated by calcium influx through the L-type voltage-sensitive calcium channel. Endocrinology. 2004;145(1):426–436. Epub 2003/10/04. 10.1210/en.2003-0319. [DOI] [PubMed] [Google Scholar]
- 48. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garcia K, Nabhani T, Garcia J. The calcium channel alpha2/delta1 subunit is involved in extracellular signalling. J Physiol. 2008;586(3):727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eroglu C, Allen NJ, Susman MW, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reyes, Fernandez PC, Wright CS, Masterson AN, et al. Gabapentin disrupts binding of perlecan to the alpha(2)delta(1) voltage sensitive calcium channel subunit and impairs skeletal mechanosensation. Biomol Ther. 2022;12(12). 10.3390/biom12121857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lai X, Price C, Modla S, et al. The dependences of osteocyte network on bone compartment, age, and disease. Bone Res. 2015;3(1). 10.1038/boneres.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang B, Lai X, Price C, et al. Perlecan-containing pericellular matrix regulates solute transport and mechanosensing within the osteocyte lacunar-canalicular system. J Bone Miner Res. 2014;29(4):878–891. 10.1002/jbmr.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li W, Duncan RL, Karin NJ, Farach-Carson MC. 1,25 (OH)2D3 enhances PTH-induced Ca2+ transients in preosteoblasts by activating L-type Ca2+ channels. Am J Phys. 1997;273(3 Pt 1):E599–E605Epub 1997/10/08. 10.1152/ajpendo.1997.273.3.E599. [DOI] [PubMed] [Google Scholar]
- 55. Heyward PM, Clarke IJ. A transient effect of estrogen on calcium currents and electrophysiological responses to gonadotropin-releasing hormone in ovine gonadotropes. Neuroendocrinology. 1995;62(6):543–552. Epub 1995/12/01. 10.1159/000127050. [DOI] [PubMed] [Google Scholar]
- 56. Seref-Ferlengez Z, Maung S, Schaffler MB, Spray DC, Suadicani SO, Thi MM. P2X7R-Panx1 complex impairs bone mechanosignaling under high glucose levels associated with type-1 diabetes. PLoS One. 2016;11(5):e0155107. Epub 2016/05/10. 10.1371/journal.pone.0155107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cabahug-Zuckerman P, StoutRF Jr, Majeska RJ, et al. Potential role for a specialized beta3 integrin-based structure on osteocyte processes in bone mechanosensation. J Orthop Res. 2018;36(2):642–652. Epub 2017/11/01. 10.1002/jor.23792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.