

Significance
Although metabolism is vital for the function of natural killer (NK) cells, the importance of individual metabolic pathways and redundancy between specific metabolites require further elucidation. Clarifying these underlying mechanisms will be crucial for advancing NK cell-based anti-tumor therapies, as tumor microenvironments are often depleted of essential nutrients. In this study, we reveal that fatty acid oxidation supports NK cell responses to both viruses and tumors. When carnitine palmitoyltransferase I (CPT1A), a key enzyme for the mitochondrial metabolism of long-chain fatty acids, is ablated in NK cells, we observe reduced mitochondrial function, cellular proliferation, and cytotoxicity due to impaired immune synapse formation. Thus, CPT1A-mediated fatty acid metabolism in NK cells is critical for host immunity against infectious diseases and cancer.
Keywords: NK cells, virus infection, cancer, metabolism, T cells
Abstract
Natural killer (NK) cells are a vital part of the innate immune system capable of rapidly clearing mutated or infected cells from the body and promoting an immune response. Here, we find that NK cells activated by viral infection or tumor challenge increase uptake of fatty acids and their expression of carnitine palmitoyltransferase I (CPT1A), a critical enzyme for long-chain fatty acid oxidation. Using a mouse model with an NK cell–specific deletion of CPT1A, combined with stable 13C isotope tracing, we observe reduced mitochondrial function and fatty acid–derived aspartate production in CPT1A-deficient NK cells. Furthermore, CPT1A-deficient NK cells show reduced proliferation after viral infection and diminished protection against cancer due to impaired actin cytoskeleton rearrangement. Together, our findings highlight that fatty acid oxidation promotes NK cell metabolic resilience, processes that can be optimized in NK cell–based immunotherapies.
Natural killer (NK) cells are innate lymphocytes capable of cytotoxicity and cytokine production upon encounter with virus-infected or malignant cells. Thus, NK cells are vital for immune surveillance against neoplastic cells and, in particular, suppression of metastasis (1). The signaling cascades, RNA synthesis, protein synthesis, lipid bilayer production, and cytoskeletal rearrangement required for NK cell effector responses can only optimally occur if the cell possesses sufficient ATP and molecular substrates (2). Yet our understanding of how NK cell activation regulates metabolism, and conversely how specific metabolic pathways impact NK cell function, is very much in its infancy (3).
NK cell activation by cytokines induces key metabolic regulators, including mTOR (4–8) and MYC (9, 10), and up-regulates metabolic pathways, cellular proliferation, and cytokine secretion. In contrast to cytokine stimulation, activating receptor ligation does not significantly increase the metabolic rate of NK cells. This inability to up-regulate potentially compensatory metabolic pathways may leave activating receptor–induced cytotoxicity and IFN-γ secretion sensitive to inhibition by low metabolite availability. We have recently demonstrated that optimal NK cell cytotoxicity and IFN-γ production in response to tumor targets requires aerobic glycolysis (11). In agreement, optimal activating receptor–induced IFN-γ secretion in NK cells was shown to be glucose dependent (12).
Although glucose levels are often depleted in the tumor microenvironment, fatty acids have been reported to be more abundant (13–16). Thus, fatty acids represent a potentially rich fuel source that cytotoxic lymphocytes, such as NK cells, could use for metabolic requirements of their antitumor effector functions. In support of this hypothesis, fatty acid oxidation (FAO) has been implicated in the generation and maintenance of T cell memory (17–19), and naive NK cells share many phenotypic and functional traits with memory T cells (20, 21). Furthermore, a correlation was drawn between circulating levels of IL-15 and the expression of fatty acid metabolism gene signatures by NK cells in the infection setting of COVID-19 (22), suggesting that NK cell activation leads to an up-regulation of FAO during infection.
The oxidative metabolism of long-chain fatty acids depends on the enzyme carnitine palmitoyltransferase I (CPT1), of which CPT1A is the dominant isoform in NK cells. Deleting CPT1A prevents long-chain fatty acid entry into the mitochondria (23–25). Several recent studies that have suggested a role for CPT1 in NK cells appear contradictory. In obese individuals (26), and patients with COVID-19 (22) or cytomegalovirus (CMV) reactivation following stem cell transplantation (27), upregulation of CPT1 and other FAO-related genes correlated with reduced NK cell functionality. However, human NK cells continuously stimulated in vitro with IL-15 have lower CPT1A expression compared to NK cells given an intermittent stimulation regimen, and the reduced CPT1A in this setting was associated with diminished NK cell functionality (28).
These seemingly conflicting lines of evidence highlight the need for greater clarity in our understanding of the role of FAO in NK cell activation and function. Given that previous evidence is largely correlative, a direct and rigorous investigation of the role of CPT1A in NK cells is warranted. Here, we demonstrate that activated NK cells induce FAO during exposure to infection and cancer. Furthermore, using mice engineered with Cpt1a-deficient NK cells, we show that CPT1A promotes the metabolic activity and effector functions of NK cells, thus supporting NK cell antitumor and antiviral activity.
Results
NK Cells Activated by Viral Infection and Cancer Induce Fatty Acid Uptake and CPT1A.
To investigate whether NK cells induce FAO during infection and cancer, we infected mice with mouse cytomegalovirus (MCMV) and characterized their antiviral NK cells. On day 2 postinfection (PI), splenic NK cells become activated, rapidly expressing CD69 and robustly producing IFN-γ (29). Compared to naive NK cells, these highly activated NK cells took up greater amounts of long-chain fatty acids and stored more fat in neutral lipid droplets, as demonstrated by BODIPY FL-C16 and BODIPY 493/503 staining, respectively (Fig. 1 A and B). Furthermore, antiviral NK cells up-regulated CPT1A on day 2 PI (Fig. 1C), suggesting that activated NK cells are transporting increased long-chain fatty acids into the mitochondria for beta-oxidation (30).
Fig. 1.

NK cells take up fatty acids and up-regulate CPT1A during viral infection and cancer. (A–C) Mice were infected with MCMV, and splenic NK cells were assessed by flow cytometry on days 0 and 2 PI. Representative histograms and summary graphs show relative uptake of long-chain fatty acids (BODIPY FL-C16) (A), storage of neutral lipids (BODIPY 493/503) (B), and expression of CPT1A (C). Histograms show naive d0 cells (black line), d2 PI (colored line), and fluorescence minus one (FMO) control (gray filled). (D) Representative histogram and summary graph of CPT1A expression in NK cells from a subcutaneous RMA-S tumor or the peripheral blood of a naive mouse. (E) CPT1A expression in peripheral blood NK cells from healthy donors versus AML patients, as measured by the median fluorescence intensity (MFI) of cells. *P < 0.05, **P < 0.01, and ****P < 0.0001; Student’s t test. Data are representative of 2 to 5 independent experiments, 3 to 8 mice per condition.
When we challenged mice with the MHC class I-negative RMA-S lymphoma, we observed an increase in CPT1A expression in tumor-infiltrating NK cells on day 14 following subcutaneous tumor injection (Fig. 1D), comparable to viral infection. To confirm the importance of fatty acid oxidation in human NK cells during cancer, we assessed the CPT1A expression level of NK cells from the blood of acute myeloid leukemia (AML) patients versus healthy volunteers. Flow cytometry analysis of CPT1A showed elevated expression in the NK cells of AML patients (Fig. 1E), suggesting a shared usage of FAO between activated NK cells in the mouse and human. Collectively, these findings suggest that CPT1A and FAO are induced following NK cell activation to aid antiviral and antitumor responses.
CPT1A-Mediated FAO Maintains NK Cell Mitochondrial Function, Supports the TCA Cycle, and Aids Aspartate Production.
To elucidate the function of FAO in NK cells, we generated Ncr1Cre/+ x Cpt1aflox/flox mice (“NK Cpt1a−/−”), where CPT1A is specifically deleted in NK cells. We first assessed the expression of maturation markers, as well as inhibitory and activating receptors, of CPT1A-deficient compared to WT NK cells that developed together in mixed bone marrow chimeras. We found expression at similar frequencies to their WT counterparts (SI Appendix, Fig. S1 A and B), suggesting that the development and maturation of resting NK cells does not depend on CPT1A and FAO. To investigate whether the upregulation of long-chain fatty acid metabolism is important for activated NK cells, we first analyzed mitochondrial function in CPT1A-sufficient (wild-type) versus CPT1A-deficient NK cells from mixed bone marrow chimeras during MCMV infection. CPT1A-deficient NK cells on day 2 PI exhibited an increase in mitochondrial membrane potential compared to WT cells (Fig. 2A). Mitochondrial membrane potential is used to drive ATP production; however, elevated membrane potential can result in the mitochondria becoming dysfunctional (31). Indeed, potentially damaging levels of mitochondria superoxide (32) were found in a greater fraction of CPT1A-deficient NK cells than WT NK cells following virus-induced activation (Fig. 2B). Analysis of the oxygen consumption rate (OCR) of WT and CPT1A-deficent NK cells further supported this conclusion. Administration of FCCP to induce maximal oxidative respiration demonstrates a reduced maximal respiration rate (and thus spare respiratory capacity) of CPT1A-deficient NK cells, with or without IL-12+IL-18 stimulation to mimic viral infection (SI Appendix, Fig. S2A). This dysfunction in mitochondrial respiration does not impact glycolysis, as both WT and CPT1A-deficient NK cells have similar rates of extracellular acidification (ECAR) when measured in a Seahorse bioanalyzer in unstimulated and IL-12+IL-18 conditions. This finding suggests that CPT1A deficiency does not significantly impact glycolytic metabolism, as this would have resulted in changes in the rate of lactate production measured by a change in the ECAR rate (SI Appendix, Fig. S2 B and C). Together, these data suggest that FAO is required to maintain mitochondrial homeostasis during the NK cell response to infection.
Fig. 2.

CPT1A-mediated fatty acid oxidation maintains mitochondrial homeostasis and helps sustain the TCA cycle and aspartate production. (A and B) WT or NK Cpt1a−/− mice were infected with MCMV, and naive (d0) or activated (d2 PI) NK cells were stained with MitoTracker Orange CMTMRos (A) or MitoSOX Red (B). Representative histograms and summary graphs (for d2 PI) are shown. (C+D+G) 13C-labeled palmitate was incubated with WT or NK Cpt1a−/− NK cells for 4 h, and incorporation of 13C into aspartate, citrate, and malate was measured. Relative abundance of isotopologues visualized as pie charts in (C). Comparison of aspartate (D+E) or citrate (F+G) isotopologue abundance between WT and NK Cpt1a−/− NK cells following incubation with 13C-labeled palmitate (D+F) or glucose (E+G). *P < 0.05, **P < 0.01, and ***P < 0.001; Student’s t test. Data are representative of 1 to 3 independent experiments, 3 to 5 mice per condition.
To interrogate which specific mitochondrial metabolic pathways are fueled by long-chain fatty acids in NK cells, we performed a stable isotope tracing experiment using 13C-labeled palmitate. Purified WT and CPT1A-deficient NK cells were incubated in media containing FA-depleted FBS and 200 μM 13C-labeled palmitate. NK cells incorporated palmitate-derived carbons into the TCA cycle intermediates citrate and malate, in addition to one of its downstream products, aspartate (Fig. 2C). Compared to CPT1A-deficient NK cells, WT NK cells showed greater 13C incorporation into all intermediates, but particularly aspartate (Fig. 2 C and D). We performed the same experiment with 13C-labeled glucose and observed that CPT1A-deficient NK cells do not switch to use glucose to generate aspartate in the absence of palmitate (Fig. 2E and SI Appendix, Fig. S2D). Furthermore, 13C-labeled carbon was incorporated into a far smaller fraction of aspartate following incubation with 13C-labeled glucose than palmitate (Fig. 2 D and E), whereas citrate appeared to be similarly derived from both metabolites (Fig. 2 F and G). These findings suggest that supporting aspartate may represent an important role of FAO in NK cells. Additionally, FAO in NK cells can supply acetyl-CoA to help sustain the TCA cycle, and subsequently the electron transport chain, producing ATP for the energetic needs of the cell, as well as maintaining mitochondrial membrane potential and optimal function.
CPT1A Is Critical for NK Cell but Not T Cell Proliferation during Viral Infection.
Aspartate is a key biosynthetic precursor for DNA, RNA, and protein synthesis (33). FAO has been shown to be important for supporting aspartate-dependent de novo nucleotide synthesis required for DNA replication in proliferating endothelial cells (25). Thus, we assessed the importance of long-chain fatty acid metabolism for the clonal expansion of antiviral NK cells. Using a well-established model to assess adaptive NK cell responses against viral infection (29), we cotransferred an equal number of congenically distinct WT (CD45.1+) and CPT1A-deficient (CD45.2+) NK cells into Ly49H-deficient mice (CD45.1+ CD45.2+) and infected the recipients with MCMV. At day 7 PI, WT Ly49H+ NK cells, which are capable of recognizing the MCMV-encoded glycoprotein m157 on infected cells (34, 35), were found in far greater abundance compared to their CPT1A-deficient counterparts (Fig. 3A), suggesting that CPT1A expression and FAO are required for optimal NK cell expansion. Through adoptive transfer of CTV-labeled WT or CPT1A-deficient NK cells, we measured the amount of cell division at day 4 PI, and found the percentage of undivided cells to be significantly greater in the CPT1A-deficient NK cell population than WT (Fig. 3B), suggesting that the defect in expansion of CPT1A-deficient Ly49H+ NK cells was due to a difference in proliferation. Thus, CPT1A is critical in fueling the proliferation of NK cells during viral infection.
Fig. 3.
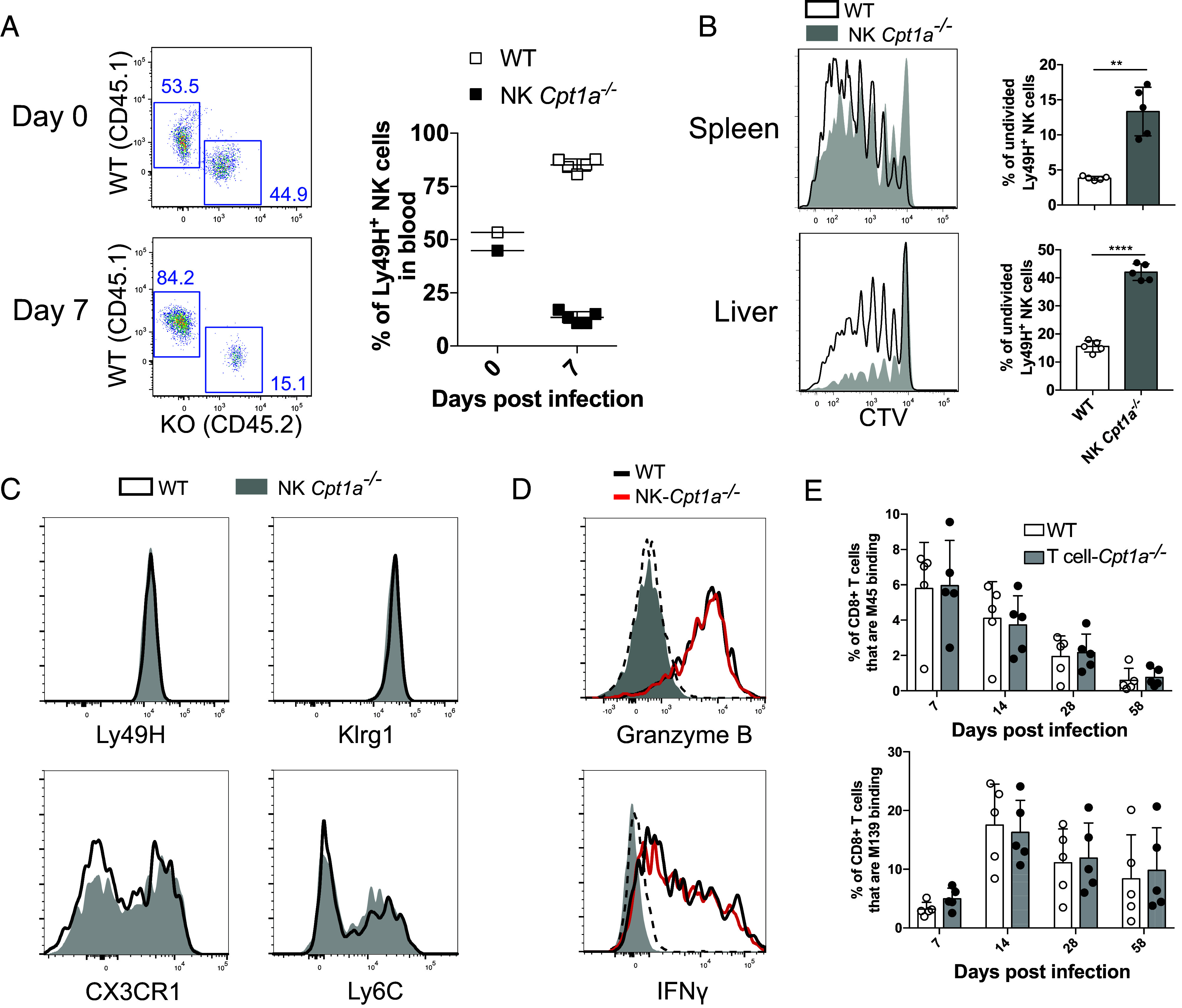
CPT1A expression promotes the clonal expansion of NK cells during viral infection. Congenic WT and CPT1A-deficient splenic NK cells were transferred into Ly49H-deficient mice, which were then infected with MCMV to drive expansion of the transferred Ly49H+ NK cells. (A) Representative dot plots and percentage summary of WT and CPT1A-deficient NK cells that make up the Ly49H+ population at d0 and d7 PI. (B) Splenic NK cells were stained with cell trace violet (CTV) prior to transfer, mice were killed at day 4 PI, and dilution of CTV was measured to assess cell division of Ly49H+ NK cells in the spleen and liver. Representative histograms and summary graphs are shown. (C) Representative histograms of Ly49H, KLRG1, CX3CR1, and Ly6C expression by WT (black line) and NK Cpt1a−/− (filled) NK cells at day 7 PI. (D) Expression of IFN-γ and granzyme B on day 2 PI in WT (black line) and NK Cpt1a−/− (red line) (naive NK cells are dashed, and FMO are filled gray). (E) Percentage of M139- and M45-specific CD8+ T cells from VavCre x Cpt1aflox/flox (CD45.2) or WT (CD45.1) populations in the blood of mixed bone marrow chimeras at various time points following MCMV infection. **P < 0.01 and ****P < 0.0001; Student’s t test. Data are representative of 2 to 5 independent experiments, 2 to 5 mice per condition.
Despite this proliferation defect, the CPT1A-deficient NK cells were phenotypically similar to WT NK cells at day 7 PI with respect to their expression of receptors and activation/maturation markers, including Ly49H, KLRG1, CX3CR1, and Ly6C (Fig. 3C) (36, 37). To test whether CPT1A deficiency impacted the early effector functions of activated NK cells during MCMV infection, we assessed IFN-γ and granzyme B production. In contrast to the clonal proliferation of antiviral NK cells, WT and NK Cpt1a−/− NK cells produced similar levels of IFN-γ and granzyme B (Fig. 3D), demonstrating that production of these two effector molecules is not dependent on CPT1A.
Our finding that CPT1A is required for optimal clonal expansion of NK cells during MCMV infection suggests NK cells are more reliant on FAO than CD8+ T cells, as a previous study suggested the formation of effector and memory T cells in response to Listeria monocytogenes to be unaffected by CPT1A deficiency (38). Indeed, following MCMV infection in mixed WT:VavCre/+ Cpt1a flox/flox bone marrow chimeric mice, expansion and memory formation of M45- and m139-specific CD8+ T cells (specific for contracting and inflationary T cell epitopes found in MCMV, respectively) was not affected by a lack of CPT1A (Fig. 3E), in agreement with the previous study (38). Thus, the expansion of antiviral NK cells demonstrated an exquisite dependence on FAO compared to antiviral CD8+ T cells responding to the same pathogen.
CPT1A Promotes Optimal NK Cell Cytotoxicity and Antitumor Function.
To investigate the significance of CPT1A upregulation by intratumoral NK cells, we first investigated whether NK cell cytotoxicity requires CPT1A using an in vivo “missing self” killing assay (39, 40). We injected an equal number of distinctly labeled MHC class I–deficient splenocytes (B2m−/−) and MHC-I sufficient splenocytes into either NK Cpt1a−/− mice or WT littermates. Twenty-four hours following injection, the WT mice had cleared a greater proportion of the B2m−/− splenocytes compared to the NK Cpt1a−/− mice (Fig. 4A), suggesting CPT1A-mediated fatty acid oxidation fuels NK cell cytotoxicity. We next challenged NK Cpt1a−/− mice and their WT littermates with the NK cell–sensitive, lung metastasis–forming B16-F10 melanoma. Mice with CPT1A-deficient NK cells were less protected compared to their WT counterparts, resulting in a greater lung tumor burden (Fig. 4B). Thus, CPT1A supports NK cell cytotoxicity against cancer cells in vivo.
Fig. 4.
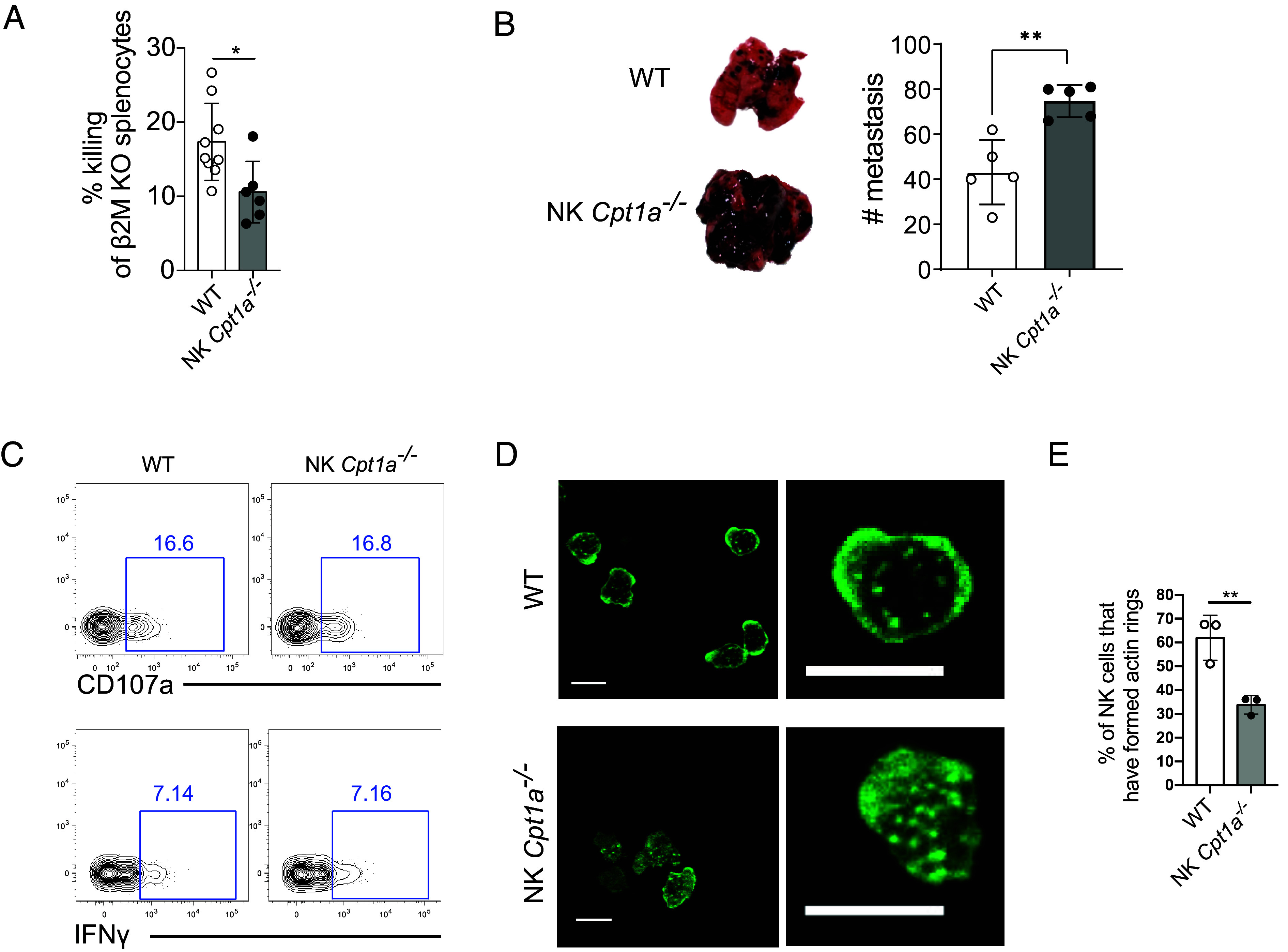
CPT1A enhances NK cell cytotoxicity and antitumor function. (A) Splenocytes from β2M-deficient and WT mice were stained with CTV or CTB, mixed at a 1:1 ratio, and transferred as NK cell targets into WT or NK Cpt1a−/− mice. Mice were bled 24 h later, and in vivo killing of the β2M-deficient splenocytes was assessed by the change in the ratio relative to the cotransferred WT splenocytes. (B) WT or NK Cpt1a−/− mice were challenged with B16-F10 melanoma i.v., and the number of lung metastases was determined 14 d later. (C) Representative plots show IFN-γ and CD107a expression by WT and NK Cpt1a−/− NK cells after 4 h stimulation with plate-bound Ly49H antibodies. (D and E) Purified WT or CPT1A-deficient NK cells were incubated in a chamber slide precoated with Ly49H antibodies. Cells were fixed/permeabilized and stained with an anti-actin antibody to visualize the actin cytoskeleton. Images show WT and CPT1A-deficient NK cells that have or have not, respectively, formed an actin ring, a critical step in the formation of an optimal immunological synapse (D), and the graph shows quantification of actin ring formation by WT and CPT1A-deficient NK cells (E). *P < 0.05, **P < 0.01, and ****P < 0.0001; Student’s t test. Data are representative of 2 to 5 independent experiments, 2 to 9 mice per condition.
To determine the mechanistic defect behind the suboptimal killing capacity of CPT1A-deficient NK cells, we assessed the ability of WT or NK Cpt1a−/− NK cells to broadly degranulate via cell membrane expression of CD107a or production of IFN-γ in response to activating receptor cross-linking in vitro. CPT1A-deficient NK cells expressed similar levels of CD107a and IFN-γ as their WT counterparts (Fig. 4C), suggesting CPT1A deficiency does not impact these effector functions. To test the ability of NK cells to direct their degranulation toward a target cell, we next investigated the ability of WT and CPT1A-deficient NK cells to form lytic synapses. Synapses were identified by F-actin ring formation at the site of receptor engagement, which is critical to optimal synapse formation (41). Following activating receptor cross-linking by incubation on antibody-coated slides, F-actin rapidly relocated from the center of the NK cell to form a synaptic ring in the majority of WT cells; however, in the majority of CPT1A-deficient NK cells, the actin remained dispersed throughout the cell (Fig. 4 D and E). The inability or delay in formation of actin rings may provide an explanation for the reduced cytotoxicity of CPT1A-deficient NK cells, as target cell contacts will result in reduced or delayed lytic hits delivered against targets. Thus, our findings suggest that CPT1A-mediated FAO fuels actin rearrangement, aiding the cytotoxicity of NK cells against tumor targets and thus host immunity against cancer.
Discussion
Our findings highlight the importance of fatty acid metabolism for optimal NK cell function against viral infection and tumors, demonstrating the role FAO plays in supporting mitochondrial function, TCA cycle turnover, aspartate production, and actin polarization for immune synapse formation (SI Appendix, Fig. S3). NK cell responses to cytokine stimulation are supported by the upregulation of compensatory metabolic pathways (4, 6), making them adaptable to glucose or glutamine deprivation, and the inhibition of the electron transport chain or aerobic glycolysis (11, 12, 42, 43). Here, because we observed normal production of IFN-γ by CPT1A-deficient NK cells during MCMV infection, a process largely driven by exposure to the proinflammatory cytokines IL-12 and IL-18 produced during infection, our data suggest that cytokine-stimulated NK cells also adapt to decreased FAO.
In contrast to cytokine exposure, NK cell responses mediated by receptor ligation are highly sensitive to metabolic perturbations, as observed in Ly49H-dependent NK cell expansion of CPT1A-deficient NK cells here, as well as in previous studies (11, 43). Furthermore, we observed defective immune synapse formation in the CPT1A-deficient NK cells, demonstrating vulnerability to metabolic limitation. Thus, cytokine-driven functions of NK cells (e.g., IFN-γ production) appear to be more resilient to metabolic limitation than activating receptor-driven NK cell responses (e.g., clonal proliferation and cytotoxicity). Perhaps one explanation for this discrepancy is that the cytoskeletal reorganization involved in receptor-driven functions has higher energetic demands (44) relative to the cytokine-driven IFN-γ secretion. Actin polymerization underlying cytoskeletal remodeling may be more energetically demanding due to the forces required for optimal target cell engagement (45), as the cytotoxic cell exerts significant force on the target cell membrane during killing (46).
Ensuring the metabolic capacity of NK cells is sufficient for optimal cytotoxic functionality is desirable in the clinical setting, and thus, we should aim to increase the metabolic resilience of engineered NK cell products (e.g., CAR-NK cells) used for antitumor therapy. Enhanced metabolic function can be achieved through pretreatment or coadministration with cytokines (47, 48); however, such strategies risk overactivating the NK cells before they meet their targets, making them less potent and/or exhausted, the latter due to sustained killing (28, 49, 50). Thus, genetic or pharmaceutic induction of rate-limiting metabolic enzymes such as CPT1A may represent a promising approach to endow NK cells with a higher metabolic capacity to support activating receptor-driven tumor control, and reducing the functional impairment often found in NK cells infused into nutrient-limited environments.
Our findings suggest a dichotomous requirement of CPT1A in clonal expansion of NK cells versus CD8+ T cells. This distinction may be attributed to differences in the resilience or magnitude of the mitochondrial spare respiratory capacity (SRC) between these two cell types. FAO and CPT1A expression have been demonstrated to support SRC, as SRC is reduced in the peripheral blood mononuclear cells of patients with inborn errors of mitochondrial fatty acid oxidation (51). Furthermore, CPT1A has been shown to support increased spare respiratory capacity in the platelets of individuals with pulmonary hypertension (52). Interestingly, the SRC of peptide-stimulated and IL-2 cultured CD8+ T cells showed no dependence on CPT1A (38, 53), whereas culturing peptide-stimulated CD8+ T cells in IL-15 causes their SRC to be more CPT1A dependent (53). Thus, IL-2 versus IL-15 may drive different metabolic requirements, and lymphocytes such as NK cells that are entirely dependent upon IL-15 for their survival may therefore possess greater dependence on CPT1A to maintain SRC. In support of a greater dependence on FAO in innate lymphocytes (relative to adaptive lymphocytes), ILC2 and ILC3 cells take up more long-chain fatty acids than regulatory T cells in the small intestinal lamina propria of naive mice (54). Furthermore, FAO in ILC2 cells may support maintenance of SRC (54). Altogether, these findings suggest CPT1A may be critical for supporting the SRC of innate lymphocytes such as NK cells and ILCs, whereas redundant metabolic pathways are present in CD8+ T cells.
Although disruption of CPT1A did not adversely impact T cell responses, overexpression of CPT1A has been shown to increase CD8+ T cell memory formation (53). Similarly, overexpression of CPT1A in macrophages during fatty acid–rich conditions resulted in enhanced function, including reduced accumulation of inhibitory triglycerides and increased phagocytosis (55, 56). However, CPT1A expression and FAO have also been shown to be up-regulated in activated T cells by PD-1 engagement, suggesting induction of FAO in T cells may dampen activation in certain contexts (57). Thus, a potential “sweet spot” for CPT1A expression in immune cells may exist that requires careful regulation. Likewise, determining whether CPT1A overexpression is always beneficial for NK cell function will be critical for clinical manipulations. Last, therapeutic inhibition of CPT1A is currently being investigated in various disease settings, including liver fibrosis (58), retinopathy (25), and cancer (59, 60). Our study highlights the importance of CPT1A and FAO for optimal NK cell responses against cancer, emphasizing important considerations in the development and implementation of therapies that target FAO. As such, it is imperative to be mindful of the potentially counterproductive activity of CPT1A inhibitors on specific immune cells.
Methods
Mice.
All mice used in this study were housed and bred under specific pathogen-free conditions at Memorial Sloan Kettering Cancer Center and handled in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC). The following mouse strains on a C57BL/6 background were used in this study: C57BL/6 (CD45.2), C57BL/6 CD45.1 (STEM, single targeted exon mutant) (61), B6 CD45.1 x CD45.2, Ncr1Cre(62), Cpt1aflox(25), VavCre, Ncr1Cre x Cpt1aflox/flox, VavCre x Cpt1aflox/flox, Klra8−/− (Ly49H-deficient) (63), B2m−/−. Experiments were conducted using age- and sex-matched mice in accordance with approved institutional protocols.
Mixed Bone Marrow Chimeras and Adoptive Transfers.
Wild-type B6 CD45.1 x CD45.2 mice were lethally irradiated with 900 Gy and reconstituted with an equal mixture of bone marrow cells from WT (CD45.1) and either Ncr1Cre x Cpt1aflox/flox or VavCre x Cpt1aflox/flox (CD45.2) mice. Intraperitoneal injection of mixed bone marrow chimeric mice with 200 μg of anti-NK1.1 (clone PK136) ensured depletion of any residual donor or host mature NK cells. Experiments were generally performed 8 to 10 wk following radiation and bone marrow transfer.
Adoptive cotransfer studies were performed by intravenously injecting an equal number of Ly49H+ NK cells from WT (CD45.1) and gene-deficient (CD45.2) mice, or from mixed bone marrow chimeras, into Klra8−/− recipients before infecting recipient mice with MCMV.
Viral Infection.
Mixed bone marrow chimeric mice or Klra8−/− mice in adoptive transfer experiments received 7.5 × 103 or 7.5 × 102 plaque-forming units (PFU) of MCMV (salivary gland–passaged Smith strain), respectively, by i.p. injection.
Isolation of Lymphocytes.
Spleens, liver, lungs, and tumors were dissociated through a 100-μm strainer. Dissociated liver and lung were resuspended in a 40% Percoll solution and centrifuged at 700 × g (with reduced break speed) for 12 min at room temperature to separate lymphocytes from stromal cells. To isolate bone marrow cells, the femur and tibia were ground with a mortar and pestle, and the resulting solution was filtered through a 100-μm strainer. Red blood cells in the spleen, liver, lung, blood, and bone marrow were lysed using ACK lysis buffer (and this treatment, was repeated a second time for blood samples). When purifying cells from tumors or isolating NK cells for stimulation assays, CTV analysis, imaging, or mass spectrometry, dissociated cells were incubated with rat anti-mouse CD3ε, CD8α, Ly6G, CD4, CD19, and Ter-119 antibodies (Bio X Cell, clones 17A2, 2.43, 1A8, GK1.5, 1D3, and TER-119, respectively) followed by incubation with BioMag goat anti-rat IgG beads (QIAGEN) to remove Ab-bound cells without touching populations of interest.
Flow Cytometry.
For metabolic dye staining, cells were resuspended in 100 μL of PBS containing BODIPY FL-C16, BODIPY 493/503 MitoSOX red, or MitoTracker™ Orange CMTMRos (Thermo Fisher Scientific) at 37 °C for 30 min. Surface staining of single-cell suspensions from the bone marrow and spleen was performed using fluorophore-conjugated antibodies in the presence of Purified Rat Anti-Mouse CD16/CD32 (BD Bioscience) to block nonspecific Fc binding and Fixable Viability Dye eFluor™ 506 (eBioscience/Thermo Fisher Scientific) or Zombie NIR™ (BioLegend). Intracellular staining was performed using the eBioscience™ Foxp3/Transcription Factor Fixation/Permeabilization kit (Thermo Fisher Scientific).
The following antibodies were used for flow cytometry: CD107a (1D4B), CD3ε (17A2), CD11b (M1/70), CD27 (LG.7F9) CD45.1 (A20), CD45.2 (104), CD49b (DX5), CD69 (H1.2F3), CPT1A (8F6AE9), CX3CR1 (SA011F11), IFN-γ (XMG1.2), Klrg1 (2F1) Ly49G2 (4D11), Ly49H (3D10), Ly49I (YLI-90), Ly6C (HK1.4), NK1.1 (PK136), NKG2A/C/E (20d5), and TCRβ (H57-597) (BioLegend, Tonbo, Thermo Fisher Scientific, BD Bioscience, and Abcam).
MHC class I tetramers were generated by conjugating Db/HGIRNASFI (M45) or Kb/TVYGFCLL (m139) monomers (NIH Tetramer Facility) to streptavidin–PE or streptavidin-APC (BioLegend). The M45 peptide represents an immunodominant “contracting” epitope from MCMV, and the m138 peptide represents a major “inflationary” epitope. NK cell proliferation was analyzed by labeling cells with 5 μM CellTrace Violet (CTV Thermo Fisher) according to the manufacturer’s protocol prior to transfer into mice.
Flow cytometry was performed on Aurora (Cytek) or LSR II (BD Biosciences) cytometers. Flow cytometry data were analyzed with FlowJo software (Tree Star).
In Vitro NK Cell Stimulation.
For receptor cross-linking, purified antibodies against Ly49H (3D10, BioLegend) or isotype control were dispensed into high-binding ELISA plates at 25 μg/mL in PBS, and allowed to adhere overnight at 4 °C. For stimulation with cytokines, 10 ng/mL IL-18 (recombinant mouse, R&D systems), 20 ng/mL of IL-12 (recombinant mouse, R&D systems), 50 ng/mL of IL-15 (recombinant human, Miltenyi), or 100 ng/mL of IL-2 (recombinant mouse, PeproTech) were added to the media. For all ex vivo stimulation, 2 × 105 bead-enriched NK cells were plated in IMDM, and Brefeldin A (10 μg/mL; Sigma) and Golgi-Stop (0.7 μL/106 cells; BD) were added 2 h after the stimulation had initiated. Cells were analyzed by flow cytometry at 4 h poststimulation.
Seahorse Bioanalyzer.
A total of 4 × 105 NK cells were plated in buffer-free, glucose-free media (Seahorse Biosciences/Agilent Technologies) with glutamine (2 mM), sodium pyruvate (0.5 mM), and glucose (10 mM) for OCR and ECAR measurements, which were made under basal conditions and following addition of oligomycin (1 mM), FCCP (1 mM), rotenone (1 mM), and antimycin A (1 mM) at indicated time points and recorded on a Seahorse XFe96. All compounds added during the Seahorse run are from Seahorse Biosciences/Agilent Technologies.
In Vivo Killing Assay.
A total of 25 × 106 B2m−/− splenocytes were labeled with either CTV or CTB, and WT control splenocytes were stained with the other. (Labels were alternated in target cells between experimental repeats to control for any effect of the different dyes.) Splenocytes were injected i.v., and mice were bled 24 h later. Killing of B2m−/− splenocytes was measured relative to cotransferred WT control splenocytes.
B16 Metastasis.
For the tumor metastasis experiments, 2 × 105 B16-F10 cells were injected i.v., and mice were killed 14 d later. Lungs were harvested, and metastatic plaques were counted.
AML.
We studied AML patients who received an allogeneic HCT allograft. The studies were executed in compliance with federal regulations pertaining to the protection of human research participants and were approved by the Center for International Blood and Marrow Transplant Research Institutional Review Board. Following approval from the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board, peripheral blood mononuclear cells (PBMCs) from anonymous healthy donors were isolated from buffy coats obtained from the New York Blood Center (New York, NY) using Ficoll centrifugation. Fetal bovine serum with 10% dimethyl sulfoxide was used to cryopreserve isolated PBMCs. Informed consent for research was obtained from all individuals.
Metabolite Profiling.
NK cells were incubated in complete IMDM containing fatty-acid-depleted FBS and 13C-labeled palmitate (CLM-409, Cambridge Isotopes) or dialyzed FBS plus 13C-labeled glucose (CLM-1396, Cambridge Isotopes) for 4 hours. FBS was fatty acid depleted using fumed Silica (or Aerosil) (Sigma) (64). Cells were spun at 800 × g for 3 min 4 °C, and media were aspirated. Metabolites were harvested in 1 mL ice-cold 80% methanol and spun down at 20,000 × g for 20 min at 4 °C. The supernatant was collected and dried in a speed vacuum evaporator for 5 h. Dried samples were stored in a −80 °C freezer prior to derivatization. The dried pellets were resuspended in 50 μL methoxyamine (TS-45950, Thermo Fisher Scientific) and shaken at 1,400 rpm at 30 °C for 2 h. Eighty microliters of MSTFA or MTBSTFA was added to each resuspended pellet, and the solution was transferred to autosampler vials with 70 μL of ethyl acetate (1036491000, Merck). Samples were incubated at 37 °C for 30 min before being run on the GC-MS.
The following analytical system was used for metabolomics analysis: Agilent 5977B GC/Mass selective detector (MSD) with an Agilent 7890B GC and an Agilent 7693A autosampler. The injection volume was 1 μL, and all samples were run at 1:20 split mode and then at Gain 5 factor mode. The temperature program was as follows for both modes: The initial oven temperature 60 °C was held for 1 min, then programmed to increase at 7.3 °C/min to reach a final temperature of 250 °C, and held for 10.5 min. Helium was used as carrier gas with a constant linear velocity of 1 mL/min.
The Agilent metabolite profiling software packages (MassHunter Qualitative Analysis 10.0 and MassHunter Quantitative Analysis B.08.00) were used for targeted identification of differentially expressed metabolites in cells grown in [U-13C] palmitate/glucose versus [U-12C] palmitate/glucose supplemented media. Enrichment of 13C was determined by quantifying the abundance of the following ions: citrate, m/z 465–471; aspartate, m/z 418–423; and malate, m/z 419–423. Correction for natural isotope abundance was performed using IsoCorrectoR software.
Actin Ring Quantification.
Cells were allowed to settle on Ly49H-coated slides for 15 min at 37 °C, fixed in 4% PFA/PBS at RT for 20 min, and permeabilized with 0.1% Triton X-100/PBS at RT for 10 min. F-actin was stained with Alexa Fluor 488–labeled phalloidin (1:200 dilution in PBS; Invitrogen) and imaged by confocal microscopy (Leica-SP5/SP8-Inverted) with a 63x oil-immersion objective. Images were exported to ImageJ, and the percentage of cells with different distributions of F-actin was scored.
Statistical Analyses.
For graphs, data are shown as mean ± SD, and unless otherwise indicated, statistical differences were evaluated using a two-tailed unpaired Student’s t test. Equal sample variance was assumed, unless an F test demonstrated significant variance where Welch’s correction was then applied. Statistical differences in survival were determined by log-rank (Mantel–Cox) test analysis. P < 0.05 was considered significant. Graphs were produced, and statistical analyses were performed using GraphPad Prism.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank members of the Sun laboratory and Drs Nadia Guerra, Aimee Beaulieu, and Lewis Lanier for comments, discussions, technical support, and experimental assistance. J.C.S. was supported by the Ludwig Center for Cancer Immunotherapy, the American Cancer Society, the Burroughs Wellcome Fund, and the NIH (AI100874, AI130043, AI155558, and P30CA008748). D.M.D was supported by the Medical Research Council (MR/W031698/1).
Author contributions
S.S. and J.C.S. designed research; S.S., K.S., W.L., M.L., R.B.D., M.O., and D.M.D. performed research; P.C., J.B.X., and K.C.H. contributed new reagents/analytic tools; S.S., K.S., W.L., M.L., R.B.D., and M.O. analyzed data; and S.S. and J.C.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.López-Soto A., Gonzalez S., Smyth M. J., Galluzzi L., Control of metastasis by NK cells. Cancer Cell 32, 135–154 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Pearce E. L., Poffenberger M. C., Chang C. H., Jones R. G., Fueling immunity: Insights into metabolism and lymphocyte function. Science 342, 1242454 (2013), 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Brien K. L., Finlay D. K., Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 19, 282–290 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Donnelly R. P., et al. , mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 193, 4477–4484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marçais A., Cherfils-vicini J., Viant C., Degouve S., De Lyon N. S., The metabolic checkpoint kinase mTOR is essential for interleukin-15 signaling during NK cell development and activation. Nat. Immunol. 15, 749–757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keating S. E., et al. , Metabolic reprogramming supports IFN-γ production by CD56 bright NK cells. J. Immunol. 196, 2552–2560 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Jensen H., Potempa M., Gotthardt D., Lanier L. L., Cutting edge: IL-2–induced expression of the amino acid transporters SLC1A5 and CD98 is a prerequisite for NKG2D-mediated activation of human NK cells. J. Immunol. 199, 1967–1972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almutairi S. M., et al. , Interleukin-18 up-regulates amino acid transporters and facilitates amino acid–induced mTORC1 activation in natural killer cells. J. Biol. Chem. 294, 4644–4655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loftus R. M., et al. , Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 9, 2341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H., et al. , The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 20, 865–878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheppard S., et al. , Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 35, 109210 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keppel M. P., Saucier N., Mah A. Y., Vogel T. P., Cooper M. A., Activation-specific metabolic requirements for NK Cell IFN-γ production. J. Immunol. 194, 1954–1962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manzo T., et al. , Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 217, e20191920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corn K. C., Windham M. A., Rafat M., Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 80, 101055 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan M. R., et al. , Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu S., et al. , Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 54, 1561–1577.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui G., et al. , IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161, 750–761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y., et al. , Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Sullivan D., et al. , Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau C. M., et al. , Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 19, 963–972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun J. C., Lanier L. L., NK cell development, homeostasis and function: Parallels with CD8+ T cells Nat. Rev. Immunol. 11, 645–657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C., et al. , Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 184, 1836–1857.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fritz I. B., Yue K. T. N., Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J. Lipid Res. 4, 279–288 (1963). [PubMed] [Google Scholar]
- 24.Houten S. M., Violante S., Ventura F. V., Wanders R. J. A., The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Schoors S., et al. , Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michelet X., et al. , Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 19, 1330–1340 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Zaghi E., et al. , Single-cell profiling identifies impaired adaptive NK cells expanded after HCMV reactivation in haploidentical HSCT. JCI Insight 6, e146973 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felices M., et al. , Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI insight 3, e96219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J. C., Beilke J. N., Lanier L. L., Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlaepfer I. R., Joshi M., CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology (United States) 161, bqz046 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Zorova L. D., et al. , Mitochondrial membrane potential. Anal. Biochem. 552, 50–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korshunov S. S., Skulachev V. P., Starkov A. A., High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Sullivan L. B., et al. , Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith H. R. C., et al. , Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 8826–8831 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arase H., Mocarski E. S., Campbell A. E., Hill A. B., Lanier L. L., Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Nabekura T., Lanier L. L., Tracking the fate of antigen-specific versus cytokineactivated natural killer cells after cytomegalovirus infection. J. Exp. Med. 213, 2745–2758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riggan L., et al. , The transcription factor Fli1 restricts the formation of memory precursor NK cells during viral infection. Nat. Immunol. 23, 556–567 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raud B., et al. , Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 28, 504–515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bix M., et al. , Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature 349, 329–331 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Hoglund P., et al. , Recognition of β2-microglobulin-negative (β2m-) T-cell blasts by natural killer cells from normal but not from β2m-mice: Nonresponsiveness controlled by β2m-bone marrow in chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 88, 10332–10336 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culley F. J., et al. , Natural killer cell signal integration balances synapse symmetry and migration. PLoS Biol. 7, e1000159 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Presnell S. R., et al. , Differential fuel requirements of human NK cells and human CD8 T cells: Glutamine regulates glucose uptake in strongly activated CD8 T cells. ImmunoHorizons 4, 231–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mah-Som A. Y., et al. , Reliance on Cox10 and oxidative metabolism for antigen-specific NK cell expansion. Cell Rep. 35, 109209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeWane G., Salvi A. M., DeMali K. A., Fueling the cytoskeleton-links between cell metabolism and actin remodeling. J. Cell Sci. 134, jcs248385 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedman D., et al. , Natural killer cell immune synapse formation and cytotoxicity are controlled by tension of the target interface. J. Cell Sci. 134, jcs258570 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu R., et al. , Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 165, 100–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kedia-Mehta N., et al. , Natural killer cells integrate signals received from tumour interactions and IL2 to induce robust and prolonged anti-tumour and metabolic responses. Immunometabolism 1, e190014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terrén I., et al. , Metabolic changes of Interleukin-12/15/18-stimulated human NK cells. Sci. Rep. 11, 6472 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermans D., et al. , Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. U.S.A. 117, 6047–6055 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delconte R. B., et al. , CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 17, 816–824 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Stenlid R., et al. , Altered mitochondrial metabolism in peripheral blood cells from patients with inborn errors of β-oxidation. Clin. Transl. Sci. 15, 182–194 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen Q. L., et al. , Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. JCI Insight 2, e91415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Windt G. J. W., et al. , Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilhelm C., et al. , Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J. Exp. Med. 213, 1409–1418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malandrino M. I., et al. , Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 308, E756–E769 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Calle P., Muñoz A., Sola A., Hotter G., CPT1a gene expression reverses the inflammatory and anti-phagocytic effect of 7-ketocholesterol in RAW264.7 macrophages. Lipids Health Dis. 18, 215 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patsoukis N., et al. , PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fondevila M. F., et al. , Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J. Hepatol. 77, 15–28 (2022). [DOI] [PubMed] [Google Scholar]
- 59.Huang D., et al. , Multiomic analysis identifies CPT1A as a potential therapeutic target in platinum-refractory, high-grade serous ovarian cancer. Cell Rep. Med. 2, 100471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koundouros N., Poulogiannis G., Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mercier F. E., Sykes D. B., Scadden D. T., Single targeted exon mutation creates a true congenic mouse for competitive hematopoietic stem cell transplantation: The C57BL/6-CD45.1STEM mouse. Stem Cell Rep. 6, 985–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narni-Mancinelli E., et al. , Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18324–18329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fodil-Cornu N., et al. , Ly49h-deficient C57BL/6 mice: A new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 181, 6394–6405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Onder S., Tacal O., Lockridge O., Delipidation of plasma has minimal effects on human butyrylcholinesterase. Front. Pharmacol. 9, 117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.