

Significance
mRNA-based therapeutics have the potential to treat a wide range of diseases, but the treatments are limited by the repertoire of available mRNA delivery methods. In this work, we developed an mRNA delivery modality based on the human protein PNMA2. We show that PNMA2 naturally forms capsids in human cells. To engineer PNMA2 for delivery, we established a method to produce a variant of PNMA2 (ePNMA2) that can encapsidate mRNA in vitro and deliver the cargo to recipient cells, highlighting the potential for an all-protein delivery vehicle that can be assembled and loaded in vitro.
Keywords: VLP, PNMA, mRNA delivery, capsid, gene therapy
Abstract
A number of endogenous genes in the human genome encode retroviral gag-like proteins, which were domesticated from ancient retroelements. The paraneoplastic Ma antigen (PNMA) family members encode a gag-like capsid domain, but their ability to assemble as capsids and traffic between cells remains mostly uncharacterized. Here, we systematically investigate human PNMA proteins and find that a number of PNMAs are secreted by human cells. We determine that PNMA2 forms icosahedral capsids efficiently but does not naturally encapsidate nucleic acids. We resolve the cryoelectron microscopy (cryo-EM) structure of PNMA2 and leverage the structure to design engineered PNMA2 (ePNMA2) particles with RNA packaging abilities. Recombinantly purified ePNMA2 proteins package mRNA molecules into icosahedral capsids and can function as delivery vehicles in mammalian cell lines, demonstrating the potential for engineered endogenous capsids as a nucleic acid therapy delivery modality.
RNA-based therapeutics, including mRNA-based vaccines, have the potential to be deployed in a wide range of disease contexts. To achieve this potential, we need a suite of delivery vehicles that can efficiently package and safely deliver therapeutic RNA cargoes to specific tissues. Several delivery modalities have already been developed, including non-viral approaches such as lipid nanoparticles (LNPs), which have successfully been used to deliver oligonucleotide and mRNA therapeutics, and viral vectors such as adeno-associated virus (AAV) (1). However, the broad applicability of these approaches is limited due to a combination of factors including cargo size constraints, immunogenicity, difficulty in achieving tissue-specific targeting, and scalable production.
Natural delivery systems from the human genome might provide the basis for new engineered gene transfer modalities that can address some of these limitations. Recent work has uncovered a diverse array of endogenous gag-like genes within the human genome, which resemble retroviral structural proteins and therefore could potentially be engineered for gene transfer (2). A number of these, including Arc and Peg10, have been domesticated and serve vital roles in normal mammalian physiology (2–4). Previous work has also shown that the ARC and PEG10 gag-like proteins have the ability to form capsid structures that can package their cognate mRNAs (3–6). Extending this natural ability, PEG10 was recently engineered to programmably package and deliver an exogenous cargo mRNA into human cells, demonstrating the potential of these endogenous retrotransposon-derived proteins as a new nucleic acid delivery modality (6). To further explore the potential of endogenous gag-like proteins for therapeutic RNA delivery, we sought to systematically characterize the paraneoplastic Ma antigen (PNMA) protein family (7). The PNMA family, which in humans contains over a dozen proteins, was initially identified due to some PNMAs encoding auto-antigens in patients with paraneoplastic neurological disease (8–11). Although several PNMA family members have been previously reported to have roles in apoptosis (12–16), most remain poorly characterized. Some PNMAs, however, including mouse PNMA2, have been shown to form capsid structures, suggesting they may be suitable for development as delivery vehicles (6, 17).
Here, we explore the potential for human PNMA proteins to form capsids and package RNA. We found that PNMA2 is robustly secreted as an icosahedral capsid from human cells and can self-assemble in vitro from recombinant protein. We used cryoelectron microscopy (cryo-EM) to resolve the structure of the PNMA2 capsid and structure-guided engineering to modify the PNMA2 protein capsid to package mRNA. We show that these engineered PNMA2 capsids can functionally deliver mRNA into recipient cells, demonstrating promise as a therapeutic mRNA delivery vehicle. In addition to PNMA2, we found that other PNMA family members are capable of forming virus-like capsids, suggesting they may also be suitable for delivery and raising the possibility that these proteins are involved in intercellular communication.
Domain Architectures and Origins of the PNMA Family of Domesticated Retroelements
The PNMA family likely emerged from the domestication of a Ty3/mdg4 (also known as gypsy) retrotransposon by the loss of the polymerase (POL) region (7, 18, 19). Multiple duplications of the ancestral PNMA in Eutherians gave rise to a large family of PNMAs in some mammalian species, including humans. In the human genome, PNMAs are spread across 4 chromosomes, with some clustered together—PNMA8a/b/c and CCDC8 share the same locus on chromosome 19, and PNMA3/5/6a/6e/6f share the same locus in chromosome X (Fig. 1A).
Fig. 1.
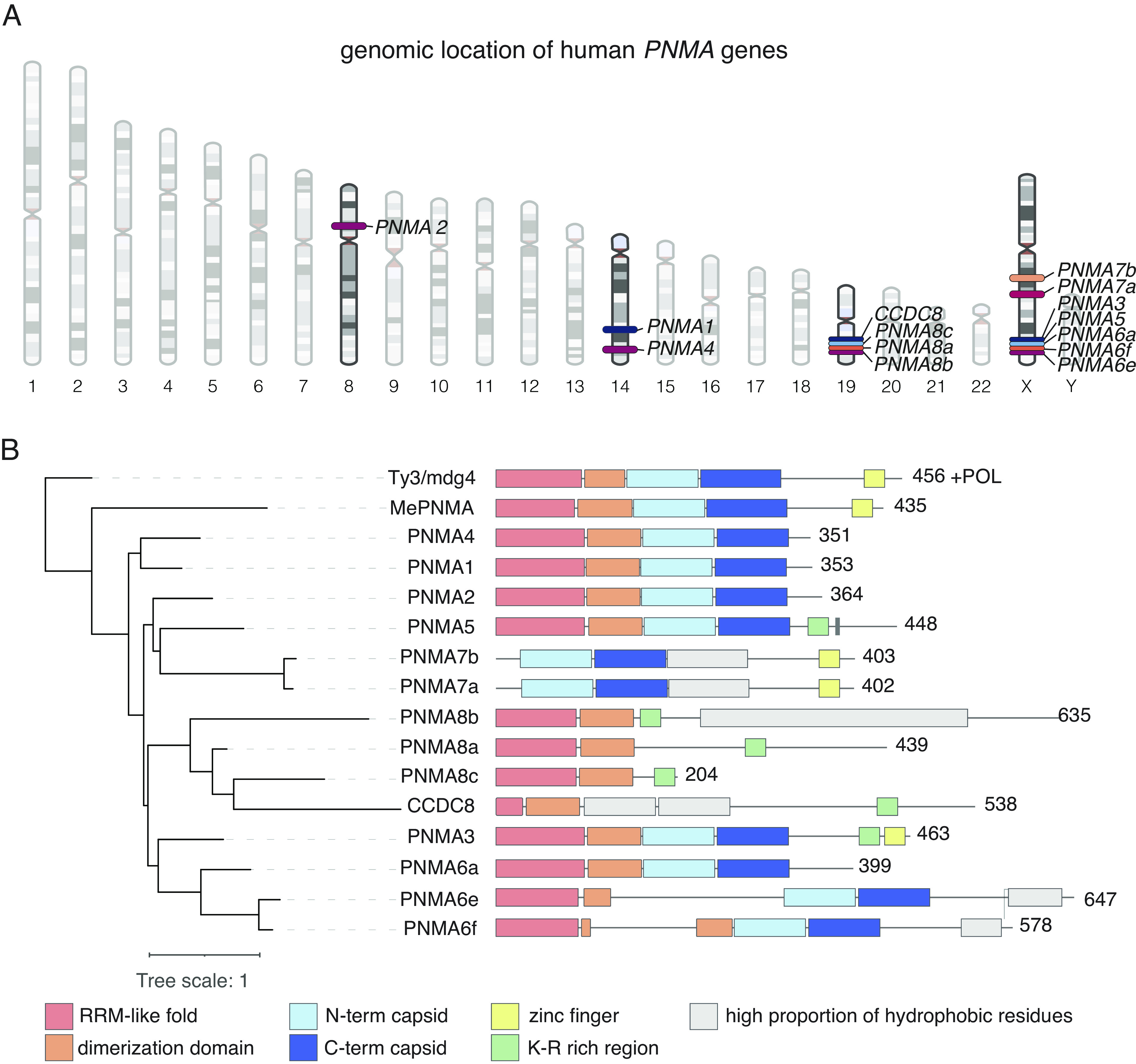
Genomic location and domain architecture of human PNMA genes. (A) Genomic location of human PNMA genes. (B) Phylogenetic tree of PNMA family including all human PNMAs (PNMA1-8 and CCDC8), the marsupial PNMA (MePNMA), and a turtle Ty3/mdg4 (Materials and Methods) from which the tree is rooted. Domain architecture of each protein is deduced from the structural models (Materials and Methods). Domain architecture encompasses an RRM-like fold domain (pink), dimerization domain that forms only upon interaction (in orange), and capsid domain in light blue for N-terminal capsid domain and dark blue for C-terminal capsid domain. Additional domains predicted to fold are shown in gray. Zinc fingers are shown in yellow, and regions with a high concentration of K-R are shown in green. RRM, RNA recognition motif.
We used structural modeling to compare the domain architectures of 14 human PNMA proteins (1, 2, 3, 4, 5, 6a, 6e, 6f, 7a, 7b, 8a, 8b, 8c, and CCDC8), as well as the marsupial PNMA from the tammar wallaby, Macropus eugenii (MePNMA), which is a close relative to Ty3/mdg4 (Materials and Methods) (Fig. 1B). Most of the human PNMAs contain the N- and C-terminal capsid domains, except PNMA8a/b/c and CCDC8, and AlphaFold-based oligomeric prediction suggests that PNMAs 1, 2, 3, 4, and 5 may form multimers (SI Appendix, Fig. S1). The RRM domain is also highly conserved, absent only in PNMA7a and 7b. The zinc finger domain, which is typically involved in interaction with nucleic acids (20), is found in only some PNMAs. Of the proteins that lack the zinc finger, some alternatively contain a K-R rich domain, which could similarly function to interact with nucleic acids.
PNMA2 Is Secreted by Human Cells as a Non-enveloped Capsid.
Given that ARC and PEG10 are secreted from mammalian cells, we tested PNMA secretion by transfecting each human PNMA into HEK293FT cells with a C-terminal HA tag (Fig. 2 A and B). Although all PNMAs expressed robustly, only a subset of PNMAs were secreted into the virus-like particle (VLP) fraction, with PNMA2 and PNMA6ev1 demonstrating especially robust secretion (Fig. 2B). We chose to further investigate PNMA2 given its predicted multimeric assembly (SI Appendix, Fig. S1). We confirmed that PNMA2 is secreted by cells as non-enveloped capsid-like structures and that it is highly secreted across multiple cell lines (Fig. 2B and SI Appendix, Fig. S2 A and B). This is consistent with similar findings in mouse cells (17). We also found that PNMA2 expression in cells is primarily localized to the cytosol (SI Appendix, Fig. S2C).
Fig. 2.
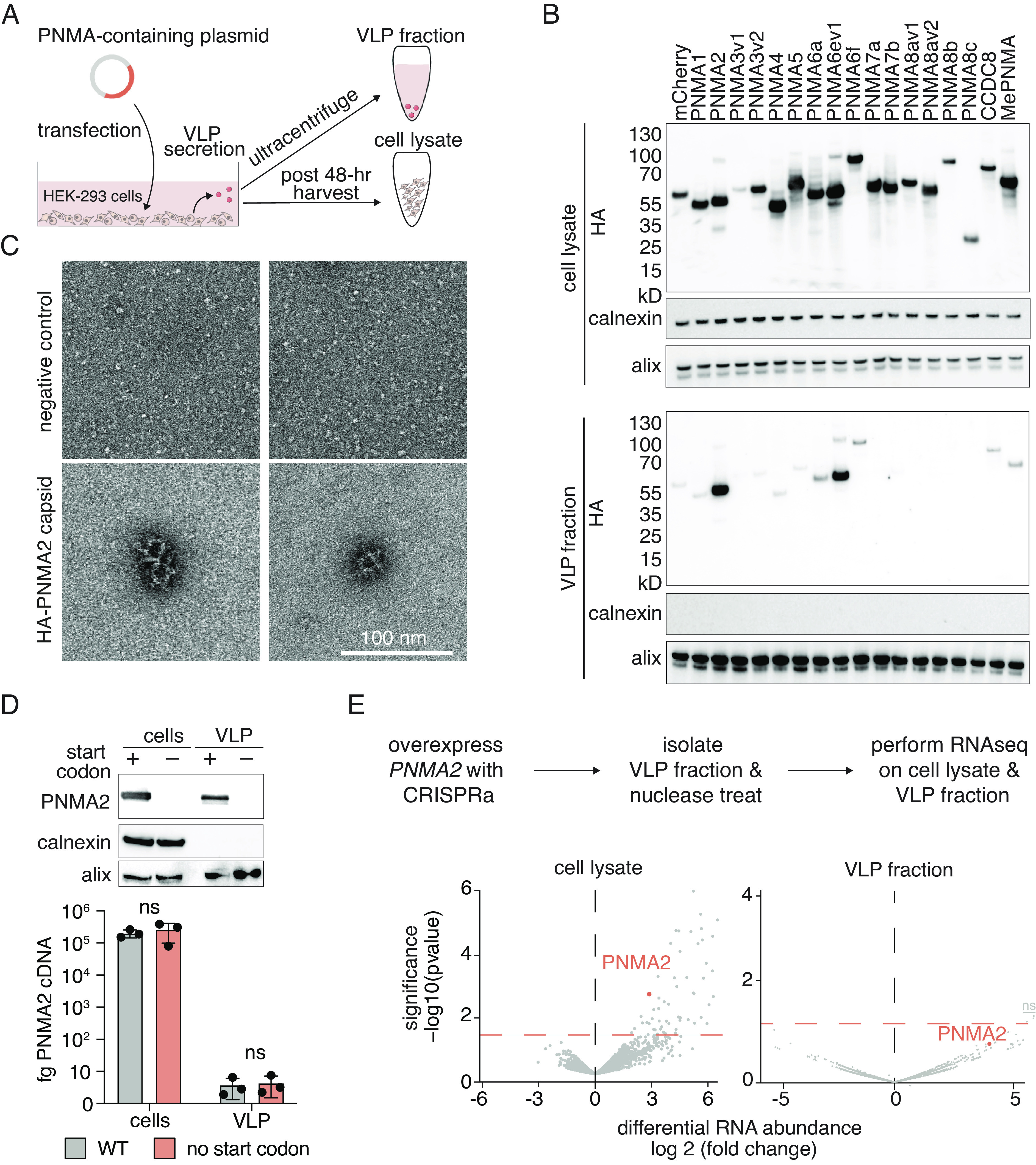
PNMA2 capsids are secreted from human cells without encapsidated RNA. (A) Schematic of isolation of cell lysate and viral-like particle (VLP) fraction. (B) Western blot showing PNMA protein expression in HEK293FT cells in either cell lysate (Top) or the VLP fraction (Bottom). (C) Example TEM micrographs of a single HA-immunoprecipitated VLP fraction from negative control cells (Top panels) or cells overexpressing HA-tagged PNMA2 (Bottom panels) (Scale bar, 100 nm). (D) Expression of PNMA2 or a mutant of PNMA2 lacking the start codon in whole-cell lysate (cells) or the VLP fraction (Top). Quantification of PNMA2 mRNA (cDNA) in either the whole-cell lysate or VLP fraction in cells expressing either wild-type PNMA2 (gray) or a mutant lacking the start codon (pink) (Bottom). Samples were compared via the unpaired t test where ns represents a P > 0.05, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001. (E) (Top) Schematic of the experimental procedure to identify mRNA packaged in PNMA2 capsids. (Bottom) Volcano plots showing differential mRNA expression of PNMA2 CRISPRa samples versus a non-targeting guide control in either the cell lysate (Left) or VLP fraction (Right). Three transcripts (ZNF142, GOLGA2P9, and TTC25) were above the significance threshold but were found to be insignificant after adjusting for multiple hypothesis testing (Materials and Methods).
We investigated whether PNMA2 capsids package their own mRNA by comparing the levels of PNMA2 mRNA in cellular and VLP fractions from HEK293FT cells overexpressing either PNMA2 or a start codon deficient version (Fig. 2D). We found no significant difference in PNMA2 mRNA levels between the VLP fraction with PNMA2 capsid and the start codon deficient version, indicating that PNMA2 does not package its own mRNA (Fig. 2 D, Bottom). We also sequenced mRNA from the VLP fractions of U20S cells transfected with either a PNMA2 CRISPRa cassette or a non-targeting control (6) and found that, while whole cells demonstrated increased transcript abundance of PNMA2 [and a number of other transcripts (Dataset S1)], there were no transcripts with strongly significant increased abundance in the VLP fraction, suggesting that PNMA2 capsids do not specifically package PNMA2 mRNA or any other cellular mRNA (Fig. 2E and Dataset S1).
Although our results indicate that PNMA2 does not package mRNA in cultured cells, we sought to determine whether purified PNMA2 could assemble around an mRNA in vitro. We first tested whether PNMA2 capsids can self-assemble in vitro from recombinantly produced protein. We found that PNMA2 purified from Escherichia coli readily assembles into capsid structures similar to those secreted from mammalian cells (compare Fig. 2C with Fig. 3A). The PNMA2 size-exclusion chromatogram (SEC) that demonstrates elution before the void volume is also consistent with efficient capsid assembly (Fig. 3B). We hypothesized that orchestrating PNMA2 disassembly and reassembly around RNA might facilitate RNA packaging (Fig. 3C). To test this, we introduced in vitro transcribed Cre mRNA into the assembly reaction and then assayed for nuclease resistance. However, self-assembled PNMA2 did not protect Cre mRNA from nuclease degradation (SI Appendix, Fig. S3).
Fig. 3.

In vitro assembly and RNA packaging of PNMA2 capsids. (A) TEM micrograph of PNMA2 purified from E. coli (Scale bar, 100 nm). (B) Size-exclusion chromatography (SEC) trace of PNMA2 particles purified from E. coli, where Vo indicates void volume. (C) Schematic of workflow for in vitro production of PNMA2 capsids with representative TEM images below.
PNMA2 Forms An Icosahedral Capsid Structure with a Negatively Charged Lumen.
To guide our engineering efforts to package RNA within PNMA2, we used cryo-EM to resolve the structure of recombinant human PNMA2 expressed in E. coli at 3.1 Å resolution (Fig. 4A and SI Appendix, Fig. S4). Sixty identical copies of the PNMA2 monomer assemble to form a capsid with icosahedral symmetry and a triangulation number T = 1 (Fig. 4B). The capsid has a mean diameter of 210 Å and encloses a volume of 1,400,000 cubic Ångstrom—approximately 60% of the volume of AAV-2 (21). Cryo-EM density was only resolvable for residues 158 to 340 of PNMA2, corresponding to the N- and C-terminal capsid domains, which fold into α-helical domains similar to other gag proteins. The interfaces at the two- and threefold symmetry axes are composed of the C-terminal capsid domains, while the fivefold symmetry axis is composed of the N-terminal capsid domains (Fig. 4C). The first ordered residue of PNMA2, Leu158, is found at the fivefold axis on the exterior side of the capsid, suggesting that the diffuse cryo-EM density forming “spikes” on the fivefold axis (Fig. 4A) is attributable to the N-terminal dimerization and RRM-like domains of PNMA2. The interior of the capsid is rich in acidic residues, including the last resolvable residues, which form a poly-Glu tract (333-EEEEEEAS-340). The interior of the capsid is therefore predicted to have a negative charge, likely accounting for the capsid’s lack of RNA cargo (Fig. 4D). We were unable to resolve the final 24 residues of PNMA2 (ten of which are also acidic), but they likely account for the cloud of cryo-EM density inside the capsid (Fig. 4E).
Fig. 4.

Cryo-EM structure of human PNMA2 capsids. (A) Cryo-EM density of PNMA2 with I4 symmetry imposed, colored by radial distance from the center of the capsid, with the exterior of the PNMA2 capsid pictured on the Left and a cross-section of the capsid shown on the Right. (B) Model of the PNMA2 capsid with a protein monomer outlined. (C) Details of the interactions of a PNMA2 monomer (pink highlight) with adjacent monomers, with symmetry axes indicated. (D) Electrostatic potential of the inside of the PNMA2 capsid. Red indicates negative charge. (E) Central slice of the PNMA2 cryo-EM density. The projected positions of the modeled N and C termini are shown as blue and red circles.
Engineering PNMA2 for Functional mRNA Delivery.
To enhance mRNA packaging efficiency, we used structure-guided engineering to modify the capsid lumen by replacing the C-terminal disordered region with an RNA-binding motif, cowpea chlorotic mottle virus N-terminal 30 residues (CCMV1-30), which is known to efficiently bind single-stranded RNA without obvious sequence preference (22, 23) (Fig. 5A). We purified the resulting PNMA2(340)-CCMV (24) [referred to as engineered PNMA2 (ePNMA2)] from E. coli and confirmed capsid formation similar to wild-type PNMA2 (Fig. 5A). Compared to wild-type PNMA2, ePNMA2 exhibited more efficient recombinant protein expression and capsid assembly (SI Appendix, Fig. S5 A and B). In contrast to wild-type PNMA2 capsids, ePNMA2 capsids were more stable at low ionic strengths and required 6 M urea for disassembly (SI Appendix, Fig. S6). We tested various conditions for reassembly in the presence of cargo RNA and found that 500 mM NaCl and 10 mM CaCl2 led to the most efficient packaging and protection of cargo RNA from nuclease degradation (Fig. 5B and SI Appendix, Figs. S6C and S7).
Fig. 5.
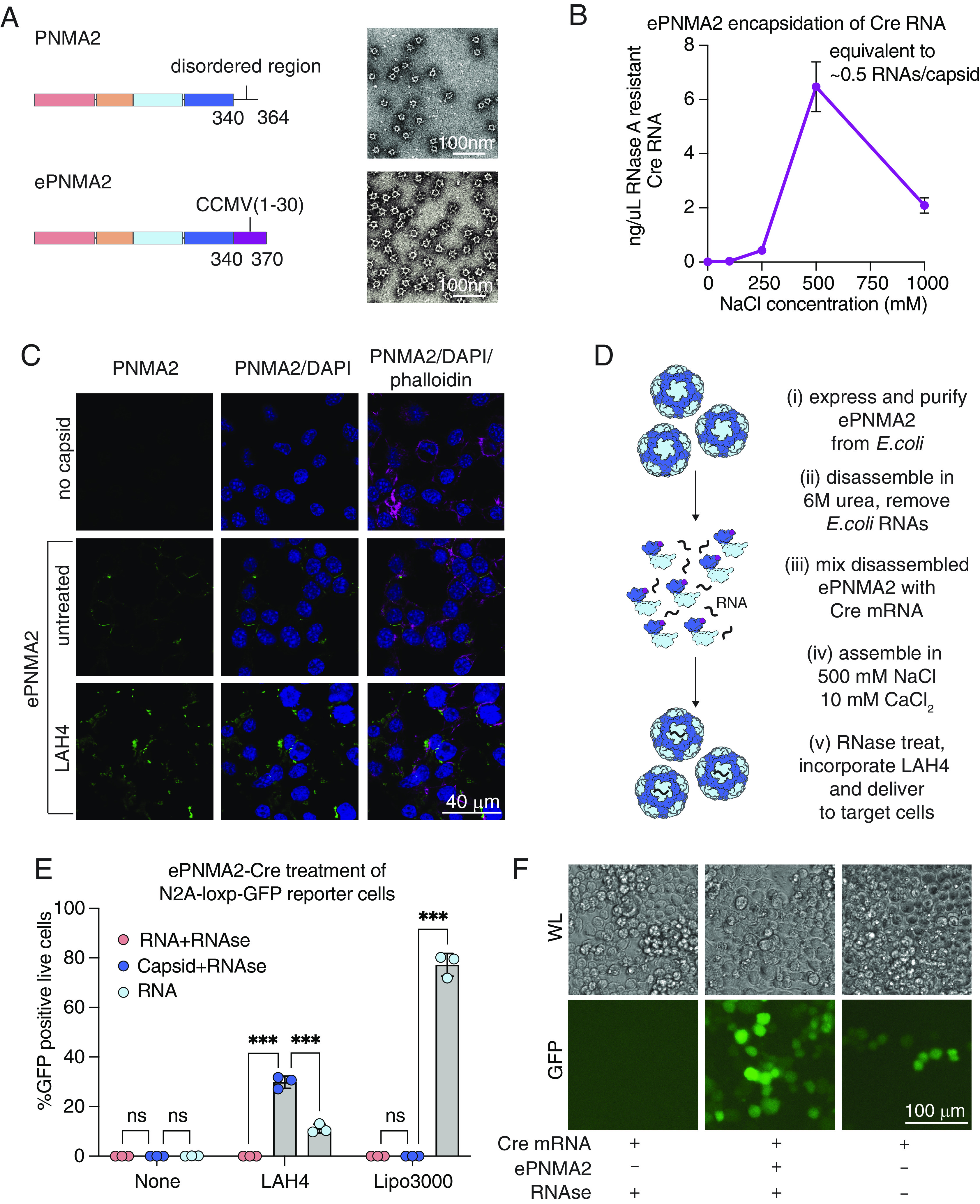
Engineering of ePNMA2 for RNA encapsidation and delivery. (A) Schematic of ePNMA2 protein and TEM micrograph of ePNMA2 capsids (Scale bar, 100 nm). (B) Quantification of RNAs per capsid packaged by ePNMA2 during reassembly across a range of salt conditions following RNaseA treatment. (C) Immunofluorescence showing ePNMA2 entry into Neuro2A cells with either no co-treatment or LAH4 (Scale bar, 40 µm). (D) Schematic of workflow for in vitro production of ePNMA2 capsids. (E) Quantification of live (DAPI negative) GFP positive cells by flow cytometry following delivery by ePNMA2 of Cre mRNA to Neuro2A-loxP-GFP recipient cells. Data shown are three technical triplicates that are representative from three biological replicates. Samples were compared via the unpaired t test where ns represents a P > 0.05, *represents P ≤ 0.05, **represents P ≤ 0.01, and ***represents P ≤ 0.001. (F) White light (WL) and GFP fluorescence images of Neuro2A-loxP-GFP recipient cells 96 h after treatment with RNA, RNase treated RNA, or RNase treated ePNMA2, -Cre all with LAH4 cell penetrating peptide (Scale bar, 100 µm).
We examined whether ePNMA2 capsids (which are non-enveloped) can enter cells via endocytosis. Confocal microscopy of Neuro2A cells 6 h after treatment showed ePNMA2 capsids at the cell periphery (Fig. 5C). Given previous data showing that the amphipathic peptide LAH4 can aid proteins in both cellular entry and endosomal escape (25), we assessed whether treating ePNMA2 with LAH4 before addition to cells could enhance entry beyond the cell periphery. LAH4 treatment did not alter the morphology of ePNMA2 capsids (SI Appendix, Fig. S8A). However, when Neuro2A cells were treated with ePNMA2 at 4 °C to allow binding without internalization, LAH4-treated capsids demonstrated higher cellular binding (SI Appendix, Fig. S8B). Further, when cells were moved to 37 °C to allow cellular uptake, trypsinization dissociated many of the untreated ePNMA2 capsids, while LAH4-treated capsids were not removed by trypsinization, suggesting that LAH4-treated ePNMA2 had been endocytosed (SI Appendix, Fig. S8B). Immunofluorescent staining showed that LAH4 treatment of ePNMA2 before addition to cells increased the cytosolic localization of ePNMA2 (Fig. 5C), and this effect was recapitulated with wild-type PNMA2 capsids (SI Appendix, Fig. S8 C and D).
Finally, we tested whether LAH4-treated ePNMA2 capsids could deliver a Cre mRNA cargo to Neuro2A-loxP-GFP reporter cells (Fig. 5D). We treated ePNMA2(Cre) with RNase A to degrade unpackaged mRNA and prepared equivalent naked Cre mRNA diluted in the same assembly buffer with and without RNase treatment. RNA, RNase-treated RNA, and RNase-treated ePNMA2 were then diluted in Optimem and added to Neuro2A-loxP-GFP reporter cells with either LAH4, Lipofectamine 3000, or no treatment (Fig. 5 E and F and SI Appendix, Figs. S8E and S9). RNase treatment degraded mRNA in the absence of ePNMA2, as confirmed by the absence of GFP expression (Fig. 5 E and F and SI Appendix, Fig. S8E). Without incorporation of LAH4, ePNMA2 did not appreciably transduce reporter cells, consistent with our cellular uptake assays which showed that ePNMA2 remains mostly bound on the cell surface without a cell penetrating peptide (SI Appendix, Fig. S8B and Fig. 5E). However, with LAH4 incorporation, RNase-treated ePNMA2(Cre) induced higher levels of GFP reporter expression relative to an equivalent amount of undigested and unpackaged RNA at a dose of 100 ng RNA per 2.5 × 104 cells (Fig. 5 E and F). Even a low dose (3.125 ng RNA per 2.5 × 104 cells) of ePNMA2(Cre) was sufficient to induce GFP expression in roughly 7% of Neuro2A-loxP-GFP reporter cells (SI Appendix, Fig. S8E). While Lipofectamine 3000 transfection of Neuro2A-loxP-GFP reporter cells was over twofold more efficient than RNase-treated ePNMA2 with LAH4, these data show that ePNMA2 can protect a functional RNA cargo from nuclease degradation, a key characteristic for nucleic acid delivery vehicles due to abundant nuclease activity in the extracellular milieu (26, 27). These results demonstrate the potential of ePNMA2 as a gene transfer tool in mammalian cell lines.
Discussion
In the current study, we demonstrated that human PNMA2 is robustly secreted from cells as an icosahedral, non-enveloped capsid. We showed that although PNMA2 does not package RNA in human cells, an engineered variant with an RNA-binding domain grafted on to the C-terminus can package cargo RNA in vitro. Combining these self-assembled, packaged ePNMA2 capsids with the cell-penetrating peptide LAH4 led to efficient functional delivery of mRNA.
Our demonstration of an all protein, in vitro produced delivery vehicle offers a starting point for further bioengineering. For example, increasing positive charges in the ePNMA2 capsid lumen could enhance RNA packaging efficiency. Further engineering of the ePNMA2 capsid surface residues may allow robust cell entry without LAH4, or targeted cell-type or tissue tropism. Engineering strategies applied to AAVs, which bear a similar T = 1 icosahedral capsid structure, could be used to modify the ePNMA2 capsid surface with integrin binding motifs or nanobodies and thus modulate ePNMA2 tropism (28, 29). In addition to modulating tropism, ePNMA2 presents an attractive substrate for protein engineering to adapt the capsids for cell entry and endosomal escape without the need for LAH4. A third avenue for future protein engineering is the modification of potentially immunogenic peptides, which may be particularly important for PNMA2 given reported cases of paraneoplastic disorders resulting from inappropriate expression of PNMA family members in some cancers (8–10).
Finally, our work with PNMA2 may be extended to other PNMA family members, some of which also form capsids, although these capsids appear more similar to the immature HIV gag than the icosahedral capsids formed by PNMA2 (SI Appendix, Fig. S10A) (24, 30). The Drosophila Arc protein also forms an icosahedral capsid, albeit with T = 4 triangulation (31) and, like many PNMA family members, is highly expressed in the central nervous system (CNS) (SI Appendix, Fig. S10 B and C) (3, 4). Given that Arc has been shown to transfer RNA within neurons, this raises the question of whether secreted PNMA capsids may enter specific recipient cells in the CNS (3, 4). Further investigation of PNMA cell tropism and engineering of PNMA capsids may allow these vehicles to be harnessed for delivery of genetic cargoes to the brain, a long-standing goal in the delivery field.
Materials and Methods
Determination and Comparison of Domain Architecture of the PNMA Family.
A structural model was built for each member of the PNMA family using AlphaFold2 (32) under the colabfold framework (33) using default parameters. Models with plddt ≥70 were selected for analysis, and additional AlphaFold2 cycles were performed until plddt was greater than 70. Structures were analyzed and compared using PyMOL (The PyMOL Molecular Graphic System Version 1.2, Schrodinger, LLC) to annotate protein domain architecture. Hydrophobic Cluster Analysis was used to compare local structure and patterns across all PNMAs (34, 35). The RRM-like domain was identified from structural mining using the Dali server (36, 37). Domain architectures were compared across all human PNMAs, the marsupial PNMA (NCBI accession number: BAK55632.1), and the turtle Ty3/mdg4 (NCBI accession number: XP_048704523), which was the closest non-PNMA relative we identified from a preliminary phylogenetic analysis from homologs of PNMAs. A final tree was built using PhyML (38) on the MPI Bioinformatics Toolkit website (39) with LG model and 200 replicates. The final tree was visualized with the interactive tree of life (itol) webserver (40) (Fig. 1B).
Prediction and Analysis of Capsomer Assemblies.
Pentamer assembly of PNMA2 was predicted using Alphafold2 multimer (41) under the colabfold framework using 40 cycles and five replicas. All replicas formed a capsomer in which the capsid domain forms a ring pentamer, and the N-terminal region forms dimers leading to two dimers and one monomer in the pentamer assembly. The interaction region was evaluated and analyzed using PyMOL software.
Plasmid Cloning.
PNMA open reading frames (ORFs) were human codon optimized and ordered as gblocks from Twist (SI Appendix, Table S1). Gblocks were also cloned into a CMV promoter-driven mammalian expression backbone (Addgene #11153) with the WPRE and SV40 polyadenylation signal (Addgene #83281) via Gibson Assembly (NEB E2611S) (SI Appendix, Table S2, primer set 1), after which C-terminal HA tags were added via site-directed mutagenesis (NEB M0554S) (SI Appendix, Table S2, primer sets 2–18). A plasmid encoding the PNMA2 transcript driven by was generated by nested PCRs of the human PNMA2 exons from HeLa genomic DNA (New England Biolabs N4006S). PNMA2 sequence–specific primers were designed using PrimerBlast (NCBI), and PCR fragments were subject to nested PCRs and then joined via Gibson Assembly (SI Appendix, Table S2, primer sets 19–25), after which they were subcloned into a CMV expression backbone by Gibson Assembly (SI Appendix, Table S2, primer sets 26 and 27). Site-directed mutagenesis was used to ablate start codons (SI Appendix, Table S2, primer sets 28–30) such that PNMA2 protein was no longer expressed as measured via western blot. CRISPRa guide RNAs and a non-targeting control phosphorylated, annealed, and then cloned into the PB-Unisam CRISPRa backbone (Addgene #99866) using Golden Gate assembly (NEB R0539S) (SI Appendix, Table S2, primer sets 31–33).
For in vitro expression constructs, hsPNMA ORFs were cloned from HeLa genomic DNA using primers designed using PrimerBlast (NCBI) into an E. coli expression backbone (Addgene #104129) with an N-terminal Maltose Binding Protein (MBP) tag and bdSUMO for purification via Gibson Assembly (SI Appendix, Table S2, primer sets 34–40). MePNMA was cloned into the same backbone from the gblock described above (SI Appendix, Table S2, primer sets 34 and 41). The c-terminal disordered region of PNMA2 was replaced with the CCMV RNA binding peptide via nested PCRs to produce fragments that were subject to Gibson Assembly (SI Appendix, Table S2, primer sets 42–44). For in vitro transcription, a plasmid was generated with Cre RNA downstream of T7 promoter (SI Appendix, Table S2, primer set 45). One hundred A’s were inserted at the 3′ end of the Cre RNA to serve as a poly A sequence. The PsiI digestion site was inserted downstream of the poly A sequence (SI Appendix, Table S2, primer set 46).
In Vitro Production and Purification of PNMA Proteins.
In vitro PNMA expression plasmids described above were transformed into Rosetta 2 (DE3) pLyseS cells. A single colony was inoculated in Terrific Broth (TB) media overnight at 37 °C with 100 ug/mL ampicillin and 25 ug/mL chloramphenicol. When optical density 600 (OD600) reached 0.6, the culture was cooled to 4 °C for 30 min. IPTG was added to a final concentration of 0.5 mM, and the culture was incubated at 21 °C for 20 h. Bacteria were centrifuged at 4,000 rpm for 15 min, media supernatant was decanted, and the bacterial pellet was then resuspended in a lysis buffer containing 50 mM Tris pH 8, 250 mM NaCl, and 0.5 mM TCEP. Lysis was achieved with two passes through the LM20 Microfluidizer system at 27,000 p.s.i. The lysis was cleared with centrifugation at 9,000 rpm for 30 min. The lysis was incubated with 2 mL amylose beads for 2 h at 4 °C. The amylose beads were washed, and the bound PNMA2 was cleaved overnight with lysis buffer with 1.5% NP-40 and 1 μg/mL bdSENP1. The elution was collected and used for SEC analysis using an AKTA pure system with Superdex200 increase 10/300 GL column with an isocratic run using lysis buffer at 0.4 mL/min.
Negative Staining and Transmission Electron Microscopy.
For sample preparation of TEM imaging grids, 5 μL of sample at a protein concentration of approximately 0.3 mg/mL was loaded onto glow-discharged, carbon-coated 300-mesh copper grids (Electron Microscopy Sciences #FCF300-CU-50). The sample was adhered to the grid for one minute at room temperature and stained in five sequential droplets for a total of one minute in freshly filtered 2% uranyl formate. Following the staining procedure, excess uranyl formate was carefully blotted off with Whatman filter paper (Cytiva, #1001-032). The grid was dried at room temperature for 1 min before placement into a grid holder. All TEM images were acquired using the FEI Tecnai (G2 Spirit TWIN) 120-kV multipurpose TEM at the MIT MRL facility. The grid was mounted on a JEOL single tilt holder equipped in the TEM column and cooled down with liquid nitrogen. The microscope was operated at 200 kV and with a magnification in the range of 10,000~60,000×, and all images were recorded on a Gatan 2kx2k UltraScan CCD camera.
Cell Culture.
U20S cells (ATCC HTB-96) were maintained in McCoy’s 5A (Modified) Medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin.
HEK293FT (Thermo Fisher R700-07), HeLa (ATCC CCL-2), U87 (ATCC HTB-14), and Neuro2A (ATCC CCL-131) cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin. U20S, HeLa, U87, and Neuro2A cells were transfected with Lipofectamine 3000 (ThermoFisher, L3000001) at 80% confluence, and media were changed 4 h post transfection to reduce toxicity. HEK293FT were transfected at 70% confluence with PEI HCl MAX (Polysciences 24765-1).
Neuro2A-loxP-GFP stable reporter cells were generated by subcloning the loxP-GFP cassette from RV-Cag-Dio-GFP (Addgene #87662) into a lentiviral transfer plasmid encoding a Blasticidin resistance gene for stable integration. To produce virus, HEK293FT cells were seeded at 1e7 cells per 15-cm dish. After 16 h, cells were co-transfected with 5 μg psPAX2 (Addgene #12260), 4.7 μg pMD2.G (Addgene #12259), and 7.7 μg of the Cre reporter plasmid using PEI HCl MAX (Polysciences 24765-1), and media were changed 4 h post transfection. Forty-eight hours later, viral supernatant was harvested, spun at 2,000g for 10 min to remove cell debris, filtered through a 0.45-µm filter, and stored at −80 °C. Neuro2A reporter cell lines were created by lentiviral transduction with 8 μg/mL polybrene (TR1003G). Media were changed 1 d later, and cells were selected for 2 wk starting on day 3 with 10 μg/mL Blasticidin-HCl (Thermo Fisher Scientific A1113903). Single clones of Blasticidin-resistant cells were isolated by serial dilution, expanded, and then screened for successful reporter expression by transfection of a Cre encoding plasmid.
Isolation of VLPs from Human Cells.
Forty-eight hours post transfection, media supernatant for sucrose cushion purification was filtered through a 0.45-µm filter, added to conical ultracentrifuge tubes (Beckman Coulter 358126), and underlaid with 4 mL of 20% sucrose in 1X PBS. Tubes were then spun at 120,000g for 2 h in a Beckman Coulter SW28 rotor, after which supernatant was decanted and the pellet was resuspended in 100 μL of 1X PBS.
VLP Protease Protection Assay.
PNMA2 VLP samples were collected from HEK293FT cells as described above and then resuspended in 120 μL 1X PBS and split into three 40-μL aliquots and subjected to a protease protection assay as previously described (42). Briefly, aliquots were incubated at 37 °C for 30 min with no treatment, 0.1 μg/uL proteinase K, or 0.1 μg/uL proteinase K with 1% Triton X-100. Proteinase K was inactivated by 10 μg/uL PMSF following treatment, at which point samples were analyzed by western blot as described below.
Western Blot Analysis.
For cellular lysate, cells were washed in 1X PBS and lysed in RIPA buffer (ThermoFisher 89901) with Halt protease inhibitor (ThermoFisher 87786) for 30 min at 4 °C. Lysate was then spun at 20,000g for 10 min at 4 °C to pellet insoluble protein. Cellular lysate supernatant and resuspended VLP were combined with 1X Bolt LDS Sample Buffer (Life Technologies B0007) and 100 mM DTT and boiled at 95 °C for 10 min. Samples were loaded into Bolt 4-12% Bis-Tris Plus gels (ThermoFisher) and run at 200 V for 30 min, before being transferred onto PVDF membrane with the iBlot2 system (ThermoFisher). Membranes were blocked in 5% milk in 1X TBS Tween 20 (TBST) Buffer, and incubated at 4 °C overnight with primary antibody in 2% milk in 1X TBST. Following three 1X TBST washes, samples were incubated with secondary antibody for 1 h and then imaged. Antibodies and dilutions used are listed in SI Appendix, Table S3.
Immunofluorescence and Confocal Microscopy.
Cells were seeded at 5 × 104 cells/well on Poly-D-Lysine/Laminin coated glass coverslips (VWR 354087). The following day, cells were washed with 1X PBS, fixed in 4% PFA in PBS for 30 min, permeabilized in 0.1% Triton X100 for 30 min, and then blocked in 1% BSA for 30 min. Cells were stained with anti-PNMA2 primary antibody diluted 1:200 in 1% BSA for one hour and washed and then stained with AlexaFluor488 conjugated secondary antibody for 1 h in the dark (as detailed in SI Appendix, Table S3). Cells were then stained with Alexa-Fluor 647 Phalloidin (Cell Signaling Technologies 8940S) and DAPI at 0.01 mg/mL for 5 min, washed three times with 1X PBS, and mounted in Diamond ProLong mounting media on glass slides. Mounted specimens were imaged on a Leica Stellaris 5 confocal microscope with the 63X oil objective. Images were analyzed in FIJI (43), where cells were defined as an ROI based on Phalloidin staining, and then mean GFP signal was measured for each cell for 10 cells from 3 mounted specimens per condition.
HA Immunoprecipitation of HEK Secreted HA-PNMA2.
ePNMA2 VLPs were isolated from the supernatant of HEK293FT cells transfected with PNMA2 with an HA-tag at the N terminus as described previously. Following ultracentrifugation, HA-tag pulldown was performed using the HA-tagged protein purification kit from MBL (#3320) using the manufacturer’s instructions. Successful isolation was confirmed by Coomassie staining, and protein capsids were imaged using TEM as described previously.
RNA Isolation and RT-qPCR.
Cells or VLPs was resuspended in Trizol (Thermo Fisher 15596026), vortexed, and incubated at room temperature for 5 min. Total RNA was then prepared via phenol chloroform extraction with glycoblue coprecipitate (Thermo Fisher AM9515). DNA contaminants were removed using the Ambion Turbo DNA-free kit (Thermo Fisher AM1907), after which DNAsed RNA was reverse transcribed using random hexamer priming and the SmartScribe Reverse Transcriptase Kit (Takara Bio 639537). cDNA was then input into qPCR reactions with Fast Sybr Green Master Mix (Life Technologies 4385612) and signal was quantified with the BioRad CFX Opus system. qPCR primers (sets 47 and 48) are listed in SI Appendix, Table S2.
RNAseq of Cells and VLPs.
U2OS cells were selected for CRISPRa due to the cell line expressing PNMA2 protein (measured via western blot) after CRISPRa treatment. U2OS cells were seeded at 6 × 106 cells per plate in 15-cm tissue culture dishes and transfected the following day with CRISPRa cassettes containing non-targeting guides or guides targeted against the transcriptional start site of PNMA2 with Lipofectamine 3000 (ThermoFisher L3000001) per the manufacturer’s protocol. Forty-eight hours post transfection, media were harvested and centrifuged on a 20% sucrose cushion as described above. VLP pellets were resuspended in 1X PBS with 2 mM MgCl2 and 250 units of Benzonase (Sigma-Aldrich E1014), incubated at 37 °C for 1 h to degrade non-encapsidated RNAs, and then resuspended in Trizol (ThermoFisher 15596026). Cells were washed in 1X PBS and then split into two aliquots. One aliquot of 6 × 105 cells was resuspended in lysis buffer and subject to western blot as described above to confirm CRISPRa efficacy, while another aliquot of 6 × 105 cells was separately resuspended in Trizol for mRNA isolation via phenol chloroform extraction. Samples were prepared in biological triplicate. Following DNAse treatment, RNA concentrations were normalized, and RNAseq libraries were prepared with the NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs E7765S) per the manufacturer’s directions. RNAseq libraries were quantified and normalized with the KAPA library quantification kit (Roche 07960204001) and loaded onto an Illumina NextSeq 550 with 50 cycles for read 1 and 25 cycles for read 2. Raw reads were trimmed using Trimmomatic (44) and quality control was performed using fastqc (45) to eliminate low-quality reads and adaptors. Resulting reads were mapped to a reference of the human genome (GRCh38) using STAR (46), and full read alignments were converted to indexed BAM files with SAMtools (47). A counts table was generated using htseq (48) and used to perform differential gene expression analysis using DESeq2 (49) in R.
Cryoelectron Microscopy.
Assembled PNMA2 capsids were diluted to 1.5 mg/mL in PBS, and 4 µL was applied to a freshly glow-discharged (60 s at 25 mA) Cu300 R1.2/1.3 holey carbon grid (Quantifoil) mounted in the chamber of a Vitrobot Mark IV (Thermo Fisher Scientific) maintained at 4 °C and 100% humidity. The grid was blotted with Ø55 grade 595 filter paper (Ted Pella) for 4 s after a wait time of 0 s at a blot force of +10, and after a drain time of 1 s was plunged into liquid ethane. Cryo-EM data were collected using the Thermo Scientific Titan Krios G3i at MIT.nano using a K3 detector (Gatan) operating in super-resolution mode with twofold binning and an energy filter with slit width of 20 eV. Micrographs were collected using EPU in AFIS mode, yielding 17,600 movies at 130,000× magnification with a real pixel size of 0.6788 Å, a defocus range from −1 to −2.6 µm, an exposure time of 0.6 s fractionated into 24 frames, a flux of 23.6 e−/pix/s and a total fluence per micrograph of 30.7 e−/Å2. Cryo-EM data were processed using RELION 4.0 (50). Movies were corrected for motion using the RELION implementation of MotionCor2, with 4 × 4 patches and dose-weighting, and CTF parameters were estimated using CTFFIND-4.1 (51). Particles were picked using Topaz and a general model (52), yielding 722,571 particles which were extracted with a 512 pixel box, binned to 128 pixels, and classified using the VDAM 2D classification algorithm (SI Appendix, Fig. S4 A and B and Table S4). A total of 229,149 particles with high-quality 2D averages were re-extracted with a 512-pixel box binned to 360 pixels. 3D refinement with I4 symmetry, using an initial model generated by RELION from screening data on a Talos Arctica microscope, gave a 3.4 Å reconstruction; however, the map showed radial blurring suggesting individual capsids had slightly different radii (SI Appendix, Fig. S4C). 3D classification with regularization parameter T = 15 allowed isolation of 88,320 capsids that were slightly smaller than average and had more well-defined density; these refined to 3.3 Å resolution after CTF refinement and Bayesian particle polishing but still showed some radial blurring. To improve the density, individual capsid particle images were converted to 12 sub-particles corresponding to individual pentons (SI Appendix, Fig. S4C). This was done by I4 symmetry expansion in RELION to convert each particle to 60 subparticles, then only keeping the 1, 2, 3, 4, 5, 6, 10, 12, 20, 28, 29, and 38th subparticles, then performing particle subtraction with a mask around one of the z-axis-aligned pentons, and finally correcting for the local defocus of the subparticle based on its projected distance to the capsid center. Subparticles were then refined with C5 symmetry and 0.9° local angular sampling, producing a 3.1-Å reconstruction of an individual PNMA2 penton. Resolution is reported using the gold-standard Fourier Shell Correlation with 0.143 cutoff. The AlphaFold2 model of PNMA2 was docked into the penton cryo-EM density and adjusted using Coot (53). The model was duplicated around the two-, three-, and fivefold axes to produce all interfaces and then refined using ISOLDE (54). The extra monomers were then deleted and the original monomer was duplicated with I4 symmetry and refined using PHENIX real_space_refine (55) into the I4-symmetric overall map using the starting model as a reference (sigma = 0.1), one macrocycle of global minimization and ADP refinement, and a nonbonded weight of 2,000. Structural figures were generated using UCSF ChimeraX (56).
In Vitro Assembly and Disassembly of PNMA2 and ePNMA2 Capsids.
Purified PNMA2 protein was pH adjusted to 5, 6, 7, 8, 9, 10, 11, and 12 and NaCl concentration adjusted to 25 mM and 1 M. Divalent ions were screened with addition of 10 mM MgCl2, 10 mM CaCl2, or 100 μM ZnCl2 into 50 mM Tris pH 8 with varying concentrations of NaCl. Co-addition of 10 mM CaCl2 and 100 μM ZnCl2 was tested with addition of 10 mM CaCl2, 100 μM ZnCl2, or 10 mM CaCl2 and 100 μM ZnCl2 into 50 mM Tris pH 8 with varying concentrations of NaCl. PNMA2 protein was mixed with purified RNA at a molar ratio of 10:1 in a buffer containing 50 mM HEPES pH 8 on ice for 2 h. The mixture was dialyzed overnight in a buffer containing 50 mM HEPES pH 8, 500 mM NaCl, 10 mM CaCl2, and 0.5 mM TCEP and varying NaCl concentrations. ePNMA2 was treated with 0 M NaCl or 1 M NaCl in addition to 0 M urea, 1 M urea, or 6 M urea. ePNMA2 concentration was kept the same for all conditions.
In Vitro Transcription of Cre mRNA.
A plasmid encoding Cre mRNA was digested with PsiI for 1 h at 37 °C and then purified with the QIAquick PCR clean-up kit (Qiagen). mRNA was synthesized using the Hiscribe T7 ARCA mRNA kit (NEB E2060S) according to the manufacturer’s protocol for mRNA synthesis with 50% modified nucleotides 5mCTP and Pseudo-UTP (TriLink Biotechnologies N10145 and N10195). mRNA synthesis was performed without enzymatic polyadenylation due to the polyadenylation sequence being present in our plasmid template. After 30 minutes incubation at 37 °C, 28 µL of water and 2 µL of Dnase I were added to the reaction for 15 min at 37 °C. Then, 25 µL of 3 M LiCl solution was added and incubated at −20 °C for 30 min to precipitate RNA, and RNA precipitate was collected by centrifugation for 15 min at 21,000g at 4 °C. The pellet was washed with 500 µL cold 70% ethanol and then dissolved in 20 µL nuclease-free water. mRNA quality was checked by running a 1% E-gel. mRNA concentration was measured by nanodrop.
In Vitro Packaging of ePNMA2 Capsid.
ePNMA2 was purified with the same protocol as other PNMAs with the following modifications. Bacterial pellet was lysed in 50 mM Tris pH 8, 1 M NaCl, and 0.5 mM TCEP. After overnight bdSENP1 cleavage, ePNMA2 was collected, concentrated into 1 mL, injected into a 2-mL loop and passed through Superose 6 increase 10/30 GL column using the AKTA pure system. The retention volume 9 ml to 13.5 ml was collected.
Purified ePNMA2 was diluted 10 times into a buffer containing 50 mM HEPES pH 8 and 6.6 M urea. The solution was left at 4 °C for at least 30 min. The solution was purified using Hitrap Heparin 5 mL column using the following protocol:
| Set program | Volume |
|---|---|
| Sample application | Continuous application until entire volume is run through the column |
| Column wash: Buffer A | 5 column volume |
| Gradient elution: 0% Buffer B to 100% Buffer B | 10 column volume |
| Column wash: 100% Buffer B | 5 column volume |
| Equilibration: Buffer A | 5 column volume |
Buffer A: 50 mM HEPES pH 8, 100 mM NaCl, 6 M urea, and 0.5 mM TCEP
Buffer B: 50 mM HEPES pH 8, 2 M NaCl, 6 M urea, and 0.5 mM TCEP
ePNMA2 protein was mixed with purified RNA at a molar ratio of 10:1 in a buffer containing 50 mM HEPES pH 8 and 6 M urea on ice for 2 h. The mixture was dialyzed overnight in a buffer containing 50 mM HEPES pH 8, 10 mM CaCl2, 0.5 mM TCEP, and varying NaCl concentration. ePNMA2 capsid was treated with 10 ng/μL RNaseA at room temperature for 10 min. The encapsulated mRNA was extracted using Trizol reagent from 21.3 μg of input protein, as quantified by running an SDS PAGE gel and quantifying against a BSA standard using FIJI. RT-PCR was performed to quantify RNA concentration. Full-length mRNA concentration was further quantified by running 1% agarose gel against Cre mRNA standard.
Cell Entry and Transduction Assays.
For all assays where in vitro produced ePNMA2 was added to cells, VLP samples were diluted in Optimem (Gibco) before addition to full cell media such that VLPs were diluted at least 1:5 to avoid toxicity from higher salt concentrations in assembly buffer.
For ePNMA2 cell entry assays, Neuro2A cells were seeded at 5 × 104 cells/well in a 24-well plate and allowed to set down overnight. Cells were then pre-chilled to 4 °C for 30 min and then treated with 15 μg/well ePNMA2 capsids, with and without LAH4 cell penetrating peptide (Genscript RP20096) in Optimem such that LAH4 was added to cells at a final concentration of 10 μg/mL LAH4. Following 1 h ePNMA2 treatment at 4 °C, cells were washed three times with cold PBS. For 4 °C binding samples, samples were either directly harvested in RIPA lysis buffer with Halt protease inhibitor, or treated with TrypLE (Gibco) at 37 °C for 5 min to dissociate bound but not internalized virions, followed by three washes in cold PBS and resuspension in lysis buffer. For 37 °C internalization samples, following 4 °C ePNMA2 binding cells were moved to 37 °C for 2 h to allow cellular uptake, followed by either direct resuspension in lysis buffer or TrypLE treatment as described above. Samples were then subject to western blotting as described above.
For Cre RNA transduction assays, VLP samples or a naked mRNA control were normalized to the same mRNA concentration, and treated with 10 μg/mL RNAse A (Qiagen) for 30 min at room temperature to degrade un-encapsidated RNAs. A second mRNA sample was prepared at the same concentration without RNAse A treatment as a positive control. Neuro2a-loxP-GFP reporter cells were seeded at 2.5 × 104 cells/well in a 96-well format. The following day, positive control mRNA, RNAse treated mRNA, and VLP samples were mixed with LAH4 in Optimem and added to cells at a final concentration of 10 ug/mL LAH4. Media were changed the day after VLP and mRNA treatment. Ninety-six hours post treatment, cells were imaged on an EVOS M5000 fluorescence microscope (Thermo Fisher) at 20X using the GFP and transmitted light channels. Cells were then prepared for flow cytometry as follows. Cells were washed with 1X PBS, trypsinized, resuspended in full media, and spun at 1,000g for 3 min in a 96-well V-bottom plate. Cells were washed with FACS buffer (1x PBS with 2% FBS and 2 mM EDTA), spun at 1,000g for 3 min, and then resuspended in FACS buffer with DAPI at .01 mg/mL. Following an additional FACS buffer wash, cell fluorescence was read out on a CytoFlex S Flow Cytometer (Beckman Coulter).
Statistical Analysis.
Statistical analyses were performed in GraphPad Prism (version 10.0.1) with the exception of RNAseq analyses, which were performed using DE Seq2 and R. DESeq2 uses the Wald test for hypothesis testing and corrects for multiple hypotheses using an interpretation of the Benjamini–Hochberg procedure. Samples were compared via the unpaired t test where ns represents a P > 0.05, *represents P ≤ 0.05, **represents P ≤ 0.01, and ***represents P ≤ 0.001.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We thank E. Brignole and C. Borsa for the smooth running of the MIT.nano cryo-EM facility, established in part with financial support from the Arnold and Mabel Beckman Foundation; the Massachusetts Institute of Technology Materials Research Laboratory and Y.Z. for training and support with transmission electron microscopy; and the Broad Institute Flow Cytometry Facility for supporting our flow cytometry experiments. V.M. is supported by a Ruth L. Kirschstein Postdoctoral award (F32 AI161868) from National Institute of Allergy and Infectious Disease and a Career Development Award from the American Society of Gene and Cell Therapy. Y.Z. is supported by a grant from the Simons Foundation to the Simons Center for the Social Brain at the Massachusetts Institute of Technology. B.L. is supported by a Ruth L. Kirschstein National Research Service Award Predoctoral Fellowship (1F31CA275339-01) from the National Cancer Institute. M.S. is supported by the Simons Foundation. M.E.W. is supported by a Helen Hay Whitney Foundation Postdoctoral Fellowship and the Howard Hughes Medical Institute. F.Z. is supported by the HHMI; Poitras Center for Psychiatric Disorders Research at the Massachusetts Institute of Technology; K. Lisa Yang and Hock E. Tan Molecular Therapeutics Center at the Massachusetts Institute of Technology; Broad Institute Programmable Therapeutics Gift Donors; The Pershing Square Foundation, William Ackman, and Neri Oxman; James and Patricia Poitras; BT Charitable Foundation; Asness Family Foundation; the Phillips family; David Cheng; and Robert Metcalfe.
Author contributions
V.M., Y.Z., R.R., R.K.M., and F.Z. designed research; V.M., Y.Z., R.R., M.E.W., G.F., E.P., M.S., and B.L. performed research; V.M., Y.Z., R.R., M.E.W., G.F., M.S., and B.L. analyzed data; R.K.M. and F.Z. supervised research; and V.M., Y.Z., R.R., R.K.M., and F.Z. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-42812 (57) (overall map) and EMD-42815 (58) (pentamer focussed refinement). The coordinates for the atomic model have been deposited in the Protein Data Bank with accession code 8UYO (59). All other study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Kulkarni J. A., et al. , The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Ono R., et al. , Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 38, 101–106 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Pastuzyn E. D., et al. , The neuronal gene arc encodes a repurposed retrotransposon gag protein that mediates intercellular RNA transfer. Cell 172, 275–288.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashley J., et al. , Retrovirus-like gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172, 262–274.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abed M., et al. , The Gag protein PEG10 binds to RNA and regulates trophoblast stem cell lineage specification. PLoS ONE 14, e0214110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segel M., et al. , Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 373, 882–889 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwasaki S., et al. , Identification of a novel PNMA-MS1 gene in marsupials suggests the LTR retrotransposon-derived PNMA genes evolved differently in marsupials and eutherians. DNA Res. 20, 425–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voltz R., et al. , A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N. Engl. J. Med. 340, 1788–1795 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Dalmau J., et al. , Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain 122, 27–39 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Schüller M., Jenne D., Voltz R., The human PNMA family: Novel neuronal proteins implicated in paraneoplastic neurological disease. J. Neuroimmunol. 169, 172–176 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld M. R., Eichen J. G., Wade D. F., Posner J. B., Dalmau J., Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann. Neurol. 50, 339–348 (2001). [PubMed] [Google Scholar]
- 12.Lee Y. H., Pang S. W., Poh C. L., Tan K. O., Distinct functional domains of PNMA5 mediate protein–protein interaction, nuclear localization, and apoptosis signaling in human cancer cells. J. Cancer Res. Clin. Oncol. 142, 1967–1977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y. H., Pang S. W., Tan K. O., PNMA2 mediates heterodimeric interactions and antagonizes chemo-sensitizing activities mediated by members of PNMA family. Biochem. Biophys. Res. Commun. 473, 224–229 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Baksh S., et al. , The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol. Cell 18, 637–650 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Tan K. O., et al. , MAP-1 is a mitochondrial effector of Bax. Proc. Natl. Acad. Sci. U.S.A. 102, 14623–14628 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan K. O., et al. , MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with Bax through its Bcl-2 homology domains. J. Biol. Chem. 276, 2802–2807 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Xu J., et al. , PNMA2 forms non-enveloped virus-like capsids that trigger paraneoplastic neurological syndrome. bioRxiv [Preprint] (2023). 10.1101/2023.02.09.527862 (Accessed 6 September 2023). [DOI]
- 18.Chalopin D., Galiana D., Volff J.-N., Genetic innovation in vertebrates: gypsy integrase genes and other genes derived from transposable elements. Int. J. Evol. Biol. 2012, 724519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayev A. A. Jr., et al. , Structural organization of transposable element mdg4 from Drosophila melanogaster and a nucleotide sequence of its long terminal repeats. Nucleic Acids Res. 12, 3707–3723 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandmeyer S., Patterson K., Bilanchone V., Ty3, a position-specific retrotransposon in budding yeast. Microbiol. Spectr. 3, MDNA3–0057–2014 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Xie Q., et al. , The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. U.S.A. 99, 10405–10410 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garmann R. F., Knobler C. M., Gelbart W. M., Protocol for efficient cell-free synthesis of cowpea chlorotic mottle virus-like particles containing heterologous RNAs. Methods Mol. Biol. 1776, 249–265 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Comas-Garcia M., Cadena-Nava R. D., Rao A. L. N., Knobler C. M., Gelbart W. M., In vitro quantification of the relative packaging efficiencies of single-stranded RNA molecules by viral capsid protein. J. Virol. 86, 12271–12282 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendonça L., et al. , CryoET structures of immature HIV Gag reveal six-helix bundle. Commun. Biol. 4, 481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kichler A., Mason A. J., Bechinger B., Cationic amphipathic histidine-rich peptides for gene delivery. Biochim. Biophys. Acta 1758, 301–307 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Tsui N. B. Y., Ng E. K. O., Lo Y. M. D., Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 48, 1647–1653 (2002). [PubMed] [Google Scholar]
- 27.Roberts T. C., Langer R., Wood M. J. A., Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 19, 673–694 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crespo-Escobar P., et al. , The role of gluten consumption at an early age in celiac disease development: A further analysis of the prospective PreventCD cohort study. Am. J. Clin. Nutr. 105, 890–896 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Shi W., Bartlett J. S., RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Mol. Ther. 7, 515–525 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Renner N., et al. , HIV-1 is dependent on its immature lattice to recruit IP6 for mature capsid assembly. Nat. Struct. Mol. Biol. 30, 370–382 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erlendsson S., et al. , Structures of virus-like capsids formed by the Drosophila neuronal Arc proteins. Nat. Neurosci. 23, 172–175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mirdita M., et al. , ColabFold: Making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faure G., Callebaut I., Comprehensive repertoire of foldable regions within whole genomes. PLoS Comput. Biol. 9, e1003280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callebaut I., et al. , Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell. Mol. Life Sci. 53, 621–645 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm L., Dali server: Structural unification of protein families. Nucleic Acids Res. 50, W210–W215 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holm L., Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Guindon S., et al. , New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Gabler F., et al. , Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinf. 72, e108 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Letunic I., Bork P., Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans R., et al. , Protein complex prediction with AlphaFold-Multimer. bioRxiv [Preprint] (2022). 10.1101/2021.10.04.463034 (Accessed 12 October 2022). [DOI]
- 42.Lenz O., ter Meulen J., Klenk H. D., Seidah N. G., Garten W., The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. U.S.A. 98, 12701–12705 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schindelin J., et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrews S., FastQC, a quality control tool for high throughput sequence data. Babraham Bioinformatics. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 15 May 2022.
- 46.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H., et al. , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimanius D., Dong L., Sharov G., Nakane T., Scheres S. H. W., New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bepler T., et al. , Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Casañal A., Lohkamp B., Emsley P., Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 29, 1069–1078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croll T. I., ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D, Struct. Biol. 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afonine P. V., et al. , Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D, Struct. Biol. 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen E. F., et al. , UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilkinson M. E., Madigan V., Zhang Y., Zhang F., PNMA2 capsid, overall icosahedral map. Electron Microscopy Data Bank. https://www.ebi.ac.uk/emdb/EMD-42812. Deposited 13 November 2023.
- 58.Wilkinson M. E., Madigan V., Zhang Y., Zhang F., PNMA2 capsid. focused refinement of a pentamer (C5 symmetry). Electron Microscopy Data Bank. https://www.ebi.ac.uk/emdb/EMD-42815. Deposited 13 November 2023. [Google Scholar]
- 59.Wilkinson M. E., Madigan V., Zhang Y., Zhang F., Structure of a recombinant human PNMA2 capsid. Protein Data Bank. https://www.rcsb.org/structure/8UYO. Deposited 13 November 2023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-42812 (57) (overall map) and EMD-42815 (58) (pentamer focussed refinement). The coordinates for the atomic model have been deposited in the Protein Data Bank with accession code 8UYO (59). All other study data are included in the article and/or supporting information.