

Abstract
Fungal Trl1 is an essential trifunctional tRNA splicing enzyme that heals and seals tRNA exons with 2′,3′-cyclic-PO4 and 5′-OH ends. Trl1 is composed of C-terminal cyclic phosphodiesterase and central polynucleotide kinase end-healing domains that generate the 3′-OH,2′-PO4 and 5′-PO4 termini required for sealing by an N-terminal ATP-dependent ligase domain. Trl1 enzymes are present in many human fungal pathogens and are promising targets for antifungal drug discovery because their domain structures and biochemical mechanisms are unique compared to the mammalian RtcB-type tRNA splicing enzyme. Here we report that Mucorales species (deemed high-priority human pathogens by WHO) elaborate a noncanonical tRNA splicing apparatus in which a monofunctional RNA ligase enzyme is encoded separately from any end-healing enzymes. We show that Mucor circinelloides RNA ligase (MciRNL) is active in tRNA splicing in vivo in budding yeast in lieu of the Trl1 ligase domain. Biochemical and kinetic characterization of recombinant MciRNL underscores its requirement for a 2′-PO4 terminus in the end-joining reaction, whereby the 2′-PO4 enhances the rates of RNA 5′-adenylylation (step 2) and phosphodiester synthesis (step 3) by ∼125-fold and ∼6200-fold, respectively. In the canonical fungal tRNA splicing pathway, the splice junction 2′-PO4 installed by RNA ligase is removed by a dedicated NAD+-dependent RNA 2′-phosphotransferase Tpt1. Here we identify and affirm by genetic complementation in yeast the biological activity of Tpt1 orthologs from three Mucorales species. Recombinant M. circinelloides Tpt1 has vigorous NAD+-dependent RNA 2′-phosphotransferase activity in vitro.
Keywords: fungal pathogen, tRNA 2′-phosphotransferase, tRNA ligase, tRNA splicing
INTRODUCTION
Fungal pathogens exert a heavy toll on health and healthcare resources (World Health Organization 2022). Invasive fungal infections (e.g., Candidiasis and Aspergillosis) are a major cause of morbidity and mortality in individuals with weakened immune systems, especially in the context of neutropenia following cancer chemotherapy or hematopoietic stem cell transplantation. The recent emergence of Candida auris as a multidrug-resistant invasive pathogen in hospitalized patients is especially worrisome. Coccidioides immitis is the causative agent of Valley Fever, a disease acquired by inhaling airborne fungal spores. Extrapulmonary spread of C. immitis infection, particularly meningitis, can be deadly. The aforementioned fungi are among the pathogens designated by the World Health Organization as priorities for research and public health action (World Health Organization 2022). A top priority—the development of more effective, less toxic antifungal drugs—hinges on defining new targets for drug discovery. We regard fungal tRNA splicing enzymes as highly promising antifungal targets.
Intron-containing tRNAs are present in all eukaryal organisms, whereby the intron is inserted at position 37 within the anticodon loop (Fig. 1A). The prevalence of tRNA introns is highly variable. At one extreme, introns occur in 128/131 (98%) and 62/117 (53%) of the tRNA genes in the fungal pathogens Cryptococcus neoformans and C. immitis, respectively. At the other end of the spectrum, introns are present in only 1/67 and 1/45 of the tRNA genes in the protozoan pathogens Trypanosoma brucei and Plasmodium falciparum, respectively. Because the intron precludes tRNA function, it must be incised precisely at the exon–intron junctions, and the tRNA exon halves must then be joined for cells to survive. The incision step, exemplary of purposeful RNA damage during normal physiology, is performed by a tRNA splicing endonuclease (TSEN), conserved among eukarya (Hayne et al. 2023), that uses a transesterification mechanism to break the phosphodiester backbone and generate 2′,3′-cyclic phosphate, and 5′-OH termini on the tRNA exons and the excised intron (Fig. 1A). However, the mechanism of tRNA exon splicing, exemplary of RNA break repair, diverges along phylogenetic lines, whereby the splicing pathway in fungi and plants is completely different than the splicing pathway in metazoa, with respect to reaction chemistry and the enzymes involved.
FIGURE 1.

Canonical pathway of fungal tRNA splicing and structural organization of the trifunctional fungal tRNA ligase Trl1. (A) Intron removal by tRNA splicing endonuclease leaves 2′,3′-cyclic phosphate and 5′-OH ends on the broken tRNA halves. The tRNA exons are then joined by Trl1—a trifunctional tRNA ligase. Trl1 catalyzes two end-healing reactions, performed by a 5′-OH polynucleotide kinase domain and a polynucleotide 2′,3′-cyclic phosphodiesterase (CPD) domain, to generate the 5′-PO4 and 3′-OH,2′-PO4 termini required for sealing by an ATP-dependent RNA ligase domain. The 2′-PO4 at the resulting splice junction is removed by the NAD+-dependent RNA 2′-phosphotransferase enzyme Tpt1. (B) Fungal Trl1 consists of N-terminal ligase, central kinase, and C-terminal CPD catalytic modules. The structures of the ligase (pdb 6N0T), kinase (pdb 6U03), and CPD (pdb 6U05) domains from the indicated fungi are shown in complexes with active site ligands.
Fungal tRNA ligation entails four enzymatic reactions (Fig. 1A) that have been characterized extensively in budding yeasts and can be thought of as comprising sequential end-healing, end-sealing, and junction-healing phases (Shuman 2023). The yeast enzyme Trl1 performs three distinct RNA repair reactions: (i) the 2′,3′-cyclic phosphate (>p) end is hydrolyzed to a 3′-OH,2′-PO4 by a CPD; (ii) the 5′-OH end is phosphorylated by a GTP-dependent polynucleotide kinase; and (iii) the 3′-OH,2′-PO4 and 5′-PO4 ends are sealed by an ATP-dependent RNA ligase to form an unconventional 2′-PO4, 3′–5′ phosphodiester at the splice junction (Fig. 1A; Greer et al. 1983). Trl1 is composed of three separable catalytic domains: a C-terminal CPD module belonging to the 2H phosphoesterase superfamily; a central GTP-dependent kinase module of the P-loop phosphotransferase superfamily; and an N-terminal RNA ligase domain that belongs to the covalent nucleotidyltransferase superfamily (Fig. 1B; Sawaya et al. 2003). The CPD and kinase reactions are collectively referred to as end-healing (Schwer et al. 2004). Healing provides the proper termini for end-sealing, which is strictly dependent on the 2′-PO4 moiety (Greer et al. 1983). The sealing phase of tRNA splicing requires ATP and proceeds via three adenylate transfer steps. First, the ligase domain reacts with ATP to form a covalent ligase-(lysyl-Nζ)–AMP intermediate and displace pyrophosphate. Second, ligase transfers AMP to the 5′-PO4 RNA terminus to form an RNA-adenylate intermediate (AppRNA). Third, ligase directs the attack of the 3′-OH on AppRNA to form the splice junction and displace AMP.
The residual 2′-PO4 at the splice junction formed by fungal Trl1 precludes proper tRNA function in codon recognition during translation. Thus, it is necessary to remove the junction 2′-PO4 as the final step in fungal tRNA splicing, a reaction performed by a dedicated enzyme, Tpt1 (Fig. 1A), that transfers the tRNA 2′-PO4 to NAD+ to form ADP-ribose-1″,2″-cyclic phosphate and nicotinamide (McCraith and Phizicky 1991; Culver et al. 1993). The Tpt1 mechanism entails two unique chemical steps. First, NAD+ reacts with the tRNA 2′-phosphate to expel nicotinamide and generate a 2′-phospho-ADP-ribosylated RNA intermediate. Then, transesterification of the ADP-ribose 2″-OH to the tRNA 2′-phosphate displaces the 2′-OH tRNA product and generates ADP-ribose-1″,2″-cyclic phosphate (Spinelli et al. 1999; Steiger et al. 2005; Munir et al. 2018).
Each of the four enzymatic steps catalyzed by Trl1 and Tpt1 is essential for the viability of the budding yeast Saccharomyces cerevisiae, insofar as single alanine substitutions in the active site that abolish activity in vitro are lethal in vivo (Sawaya et al. 2003; Wang and Shuman 2005; Wang et al. 2006). A key point anent their suitability for drug discovery is that mammals rely on an entirely different series of chemical steps to achieve tRNA exon splicing. The RNA ligase RtcB is the agent of tRNA exon splicing in metazoa and archaea (and of RNA repair in bacteria) (Shuman 2023). RtcB seals broken RNAs with 5′-OH and 2′,3′-cyclic phosphate (RNA > p) ends via a four-step ligation pathway that requires GTP as an energy source and Mn2+ as a cofactor. RtcB first reacts with GTP to form a covalent RtcB-(histidinyl-Nε)–GMP intermediate. It then hydrolyses RNA > p to RNAp and transfers the guanylate from histidine to the polynucleotide 3′-phosphate to form a polynucleotide-(3′)pp(5′)G intermediate. Finally, RtcB catalyzes the attack of an RNA 5′-OH on the –NppG end to form a standard 3′–5′ phosphodiester splice junction and liberate GMP. Thus, mammalian tRNA splicing does not involve a 5′-kinase step and does not generate a 2′-PO4 splice junction.
To fortify the case for fungal tRNA splicing as a drug target, we need to understand the properties and structures of Trl1 and Tpt1 enzymes from fungi that cause human disease. To that end, our laboratory has purified and characterized Trl1 and/or Tpt1 enzymes from the fungal pathogens Aspergillus fumigatus, Candida albicans, Candida auris, and Coccidioides immitis (Remus et al. 2016; Dantuluri et al. 2021). We also characterized Trl1 and Tpt1 enzymes from avirulent fungi and nonfungal sources (Sawaya et al. 2005; Wang and Shuman 2005; Remus and Shuman 2013, 2014; Munir et al. 2018; Dantuluri et al. 2020). To date, we have solved structures of the Trl1 LIG domain (from the mesophilic fungus Chaetomium thermophilum), the Trl1 KIN and CPD domains (from C. albicans) in complexes with substrates and products (Fig. 1B), and of Tpt1 (from Clostridium thermocellum, Aeropyrum pernix, and Pyrococcus horikoshii) in complexes with substrates and products (Remus et al. 2017; Banerjee et al. 2019a,b,c; Jacewicz et al. 2023). These structures are filling a key knowledge gap in tRNA metabolism by revealing the active site architectures, the determinants of substrate recognition, and the catalytic mechanisms of the fungal-type tRNA splicing machinery.
Other WHO fungal priority pathogens—including Blastomyces, Histoplasma, and Cryptococcus—encode a trifunctional Trl1 protein and a separate Tpt1 enzyme homologous to the budding yeast counterparts. Here, we queried the nature of the tRNA splicing machinery in priority pathogens of the order Mucorales. Mucormycosis is a serious (often fatal) infection acquired either by inhalation of fungal spores leading to infection of the sinuses, brain, and/or lungs or by entry of spores through a break in the skin following a burn or other skin trauma. Risk factors for mucormycosis include diabetes, steroid use, cancer, organ transplant, and COVID-19 infection (World Health Organization 2022).
We report that Mucorales species Mucor circinelloides, Rhizopus azygosporus, and Lichtheimia corymbifera elaborate a novel variant of the fungal tRNA splicing machinery in which the RNA ligase is a stand-alone enzyme that is active in vivo in budding yeast in lieu of the Trl1 ligase domain. We present a biochemical characterization of the M. circinelloides RNA ligase that affirms its requirement for a 2′-PO4 terminus in the end-joining reaction. We confirm the identity of the Mucorales Tpt1 enzymes that remove the splice junction 2′-PO4 via genetic complementation of budding yeast tpt1Δ and biochemical characterization of M. circinelloides Tpt1.
RESULTS
Mucorales species encode a stand-alone RNA ligase
A Psi-Blast search of Mucorales taxa with budding yeast Trl1 (827 aa) retrieved proteins homologous to the N-terminal RNA ligase domain but did not return convincing homologs of the Trl1 kinase or CPDase modules. A primary structure alignment of the putative tRNA ligase (RNL) proteins from three Mucorales species: M. circinelloides (372 aa), R. azygosporus (370 aa), and L. corymbifera (390 aa) show that these three RNLs share 215 positions of amino acid side chain identity/similarity (denoted by dots in Fig. 2A). The Mucorales RNLs include the six covalent nucleotidyltransferase motifs (I, Ia, III, IIIa, IV, and V; shaded cyan in Fig. 2A) characteristic of ATP-dependent RNA ligases, ATP-dependent DNA ligases, and GTP-dependent mRNA capping enzymes that act via covalent enzyme-(lysyl-Nζ)–NMP intermediates. These motifs form the NTP-binding pocket of the Trl1 ligase domain and contain multiple residues essential for Trl1 activity (Banerjee et al. 2019a). The motif I (KxNG) lysine is the site of covalent adenylylation (Banerjee et al. 2019a). A Trl1 LIG-bound sulfate engaged by amino acids R334 and R337 in the structure of Chaetomium tRNA ligase is proposed to mimic the RNA terminal 2′-PO4 required for the end-joining step of fungal tRNA splicing (Banerjee et al. 2019a; Peschek and Walter 2019). The fact that these two arginines are conserved in all three Mucorales RNLs (shaded yellow in Fig. 2A) begets a prediction that their RNA ligase activity might also require a 3′-OH,2′-PO4 terminus.
FIGURE 2.

Mucorales encode a stand-alone RNA ligase enzyme active in tRNA splicing in vivo. (A) Alignment of the primary structures of monofunctional Trl1 LIG domain homologs from M. circinelloides (Genbank EPB83728.1), R. azygosporus (Genbank RCH90547.1), and L. corymbifera (Genbank CDH55495.1). Positions of side chain identity/similarity are indicated by dots. Six signature ligase adenylyltransferase motifs I, Ia, III, IIIa, IV, and V are shaded in cyan. Two conserved arginine residues implicated in the recognition of an RNA 2′-PO4 terminus are shaded in yellow. (B) Complementation of S. cerevisiae trl1Δ was assayed by plasmid shuffle. Aliquots (3 µL) of serial tenfold dilutions of FOA-resistant trl1Δ strains expressing the indicated genes were spotted on YPD agar plates and incubated at 25°C, 30°C, 34°C, and 37°C. Photographs of the plates are shown.
Mucor RNL is active in vivo in lieu of the S. cerevisiae Trl1 LIG domain
We showed previously that fusion of the LIG, KIN, and CPD domains within a single polypeptide is not critical for S. cerevisiae tRNA splicing in vivo (Sawaya et al. 2003) and that expression of heterologous RNA repair systems can complement the lethality of a trl1Δ knockout in S. cerevisiae (Schwer et al. 2004; Wang et al. 2006; Nandakumar et al. 2008; Tanaka et al. 2011). Indeed, complementation of budding yeast trl1 mutants defective for one or more of the tRNA splicing reactions provides a surrogate means to validate the RNA repair capacity of candidate enzymes from heterologous sources (Ramirez et al. 2008; Schwer et al. 2008). To query whether a Mucorales RNL is a bona fide tRNA ligase, we cloned the open reading frame (ORF) encoding M. circinelloides RNL (MciRNL) into a yeast CEN LEU2 plasmid wherein its expression is driven by the S. cerevisiae TPI1 promoter. We tested complementation by plasmid shuffle in a S. cerevisiae trl1Δ p(CEN URA3 TRL1) strain by cotransforming the yeast shuffle strain with two plasmids: one expressing MciRNL and one expressing SceKIN-CPD, the end-healing domains of S. cerevisiae Trl1. Positive control transformations were performed with plasmids bearing full-length SceTRL1 and the separately expressed SceLIG and SceKIN-CPD ORFs. In each case, we recovered viable trl1Δ strains that lacked the p(CEN URA3 TRL1) plasmid after selection for growth on medium containing 5-FOA. As shown in Figure 2B, the trl1Δ MciRNL + SceKIN-CPD strain grew as well as the trl1Δ SceTRL1 control when spot-tested on rich agar medium at 25°C–37°C. We conclude that MciRNL can execute the ligation step of fungal tRNA splicing in vivo. It was noteworthy that the trl1Δ SceLIG + SceKIN-CPD strain grew as well as trl1Δ SceTRL1 at 25°C–34°C but formed smaller colonies at 37°C (Fig. 2B). Thus, at first glance, it seems that MciRNL, which is naturally physically unlinked to an end-healing module in cis, is better able to function in trans with SceKIN-CPD at 37°C vis-à-vis the native SceLIG.
Mucor RNL is inactive in vivo when paired with the T4 end-healing enzyme Pnkp
Bacteriophage T4 elaborates two tRNA repair enzymes—Pnkp (polynucleotide 5′-kinase/3′-phosphatase) and Rnl1 (RNA ligase 1)—that heal and seal a 2′,3′-cyclic phosphate/5′-OH break in the anticodon loop of tRNALys inflicted by a host-encoded anticodon nuclease (Amitsur et al. 1987; Shuman 2023). The T4 tRNA repair pathway differs from that of yeast tRNA splicing in that the 3′ end-healing reaction of Pnkp generates a 3′-OH,2′-OH end rather than the 3′-OH,2′-PO4 formed by Trl1 CPD and that T4 Rnl1 is highly adept at sealing 3′-OH,2′-OH and 5′-PO4 tRNA ends. Coexpression of T4RNL1 and T4PNKP in S. cerevisiae supports normal growth of trl1Δ cells and bypasses the essentiality of the 2′-phosphotransferase Tpt1 (Schwer et al. 2004). This signifies that yeast have no obligate requirement for a 2′-PO4 intermediate in the tRNA splicing pathway, so long as the ligase enzyme does not require it for end-sealing. We noted previously that the expression of SceLIG did not complement trl1Δ when paired with the expression of T4PNKP (Schwer et al. 2004). Here, we confirmed by plasmid shuffle that a trl1Δ T4RNL1 + T4PNKP strain was viable after 5-FOA selection (Fig. 3) and grew as well as trl1Δ SceTRL1 at 25°C–37°C (Fig. 2B). However, a trl1Δ strain cotransformed with plasmids expressing MciRNL and T4PNKP failed to give rise to viable colonies on medium containing 5-FOA (Fig. 3), signifying that the MciRNL was unable to seal the healed 3′-OH,2′-OH and 5′-PO4 tRNA ends produced by the phage Pnkp.
FIGURE 3.
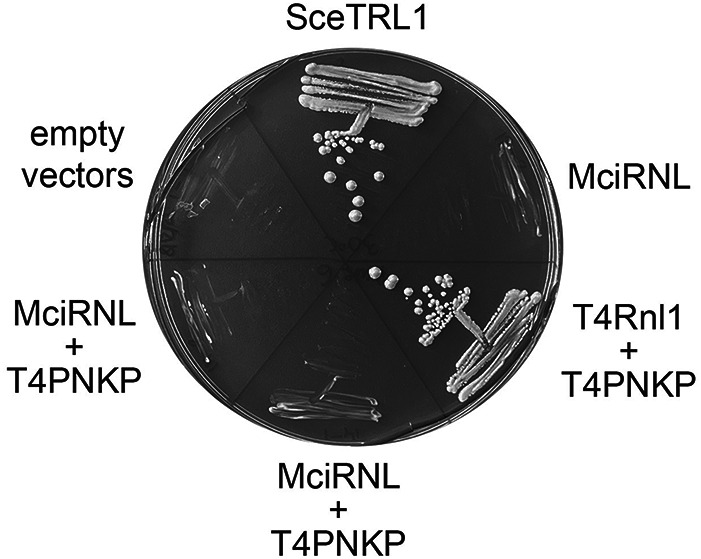
Mucor RNL is inactive in vivo when paired with the T4 end-healing enzyme Pnkp. The S. cerevisiae trl1Δ p(CEN URA3 TRL1) “shuffle” strain was transformed with marked plasmids expressing the indicated genes and with empty vector controls. Individual transformants were streaked on agar medium containing FOA, which was photographed after incubation for 10 d at 30°C. Two independent trl1Δ p(CEN URA3 TRL1) MciRNL + T4PNKP isolates were tested for growth on FOA.
RNA end-joining activity of recombinant Mucor RNL
We produced MciRNL in Escherichia coli as a His10Smt3 fusion and purified it from a soluble extract by sequential Ni-affinity chromatography/imidazole elution, removal of the His10Smt3 tag by treatment with Ulp1 protease, recovery of the tag-free MciRNL protein in the flowthrough of a second Ni-affinity column, and a final Superose-200 gel-filtration step. MciRNL was monomeric by gel filtration. SDS-PAGE analysis of the MciRNL preparation is shown in Figure 4D.
FIGURE 4.

RNA ligase activity of recombinant MciRNL. (A) Reaction mixtures (10 µL) containing 50 mM Tris-acetate, pH 6.5, 5 mM MgCl2, 0.1 µM (1 pmol) 5′ 32P-labeled 10-mer RNA with either a 3′-OH,2′-PO4 end (left side) or a 3′-OH,2′-OH end (right side) and increasing amounts of MciRNL as specified were incubated at 37°C for 5 min. The reactions were quenched with an equal volume of 95% formamide/50 mM EDTA, and the products were analyzed by electrophoresis (at 58 W constant power) through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The 5′-radiolabeled RNAs were visualized by scanning the gel with a Fujifilm FLA-7000 imaging device. The positions and identities of the RNA substrates, RNA-adenylate intermediates, and ligated products are indicated on the left and right. (B) The distributions of radiolabeled RNAs in each lane in panel A were quantified by analysis of the gel scans in ImageQuant and are plotted as a function of input MciRNL for the ligation reactions with the 2′-PO4 (left panel) and 2′-OH (right panel) RNA substrates. (C) Product analysis via treatment with CIP (calf intestine alkaline phosphatase). Ligation reactions containing 1 pmol 5′ 32P-labeled pRNA2′p and 10 pmol MciRNL (where indicated by +) were incubated at 37°C for 5 min. The mixtures were then incubated for 10 min at 37°C with 5 U CIP (purchased from NEB) where indicated by + above the lanes. The mixtures were quenched and then analyzed by urea-PAGE as described in A. (D) An aliquot (10 µg) of the recombinant MciRNL preparation was analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker proteins are indicated on the left. The expected molecular weight of MciRNL is 43 kDa.
To assay ligase activity, we deployed as substrates a pair of 5′ 32P-labeled synthetic 10-mer RNAs with either a 3′-OH,2′-PO4 end or a 3′-OH,2′-OH end. The RNAs (1 pmol; 0.1 µM) were reacted for 5 min at 37°C with increasing amounts of MciRNL in the presence of 5 mM Mg2+. The products were analyzed by urea-PAGE and visualized by scanning with a phosphorimager (Fig. 4A). Absent exogenous ATP in the reaction mixture, the extent of ligation depends on the level of catalytically active covalent MciRNL–AMP adduct in the enzyme preparation.
MciRNL (at ≥2 pmol) converted virtually all of the 2′-PO4 RNA substrate into a mixture of ligated products and ligation intermediates. These are, in order of decreasing abundance: (i) a circular RNA formed by intramolecular ligation that migrated ahead of the substrate strand (Remus and Shuman 2014); (ii) a dimer RNA circle, migrating just behind the substrate strand, formed via the intermolecular joining of two 10-mers and ensuing intramolecular ligation; and (iii) an adenylylated AppRNAp intermediate, migrating behind the dimer circle (Fig. 4A). Quantification of the product distribution as a function of input MciRNL is shown in Figure 4B (left panel). At subsaturating input MciRNL, the formation of monomer circles is strongly favored. From the extent of the reaction at 0.5 pmol MciRNL, whereby 0.47 pmol of RNA substrate was converted to product, we surmise that ∼94% of the enzyme preparation was MciRNL–AMP.
In contrast, the 2′-OH RNA was a feeble substrate for MciRNL, as gauged by the consumption of substrate as a function of input enzyme and the nature of the products formed, whereby AMP transfer from MciRNL–AMP to generate AppRNAOH was the predominant outcome even at 10 pmol input enzyme (comprising 67% of total RNA), and there was scant progression to ligated circle (3% of total RNA) (Fig. 4A,B). From the slopes of the titration curves, we determined that the specific activity for ligating a 3′-OH,2′-PO4 end in a 5 min reaction was 150-fold greater than for ligating a 3′-OH,2′-OH end. The specific activity for RNA 5′-phosphate adenylylation (i.e., yield of RNA-adenylate plus ligated RNA as a function of input enzyme) was sixfold greater with a 2′-PO4 substrate versus a 2′-OH substrate.
To affirm the identities of the 32P-labeled reaction products formed at 10 pmol of input MciRNL, we tested their sensitivity to alkaline phosphatase (CIP). As expected, the 5′ 32P phosphomonoester of the input 2′-PO4 and 2′-OH RNA substrates was hydrolyzed completely by CIP (Fig. 4C). In the case of the AppRNAp product, the radiolabel in the 5′ AppA-phosphoanhydride structure is protected from CIP digestion, but the unlabeled 2′-PO4 is susceptible to CIP, thereby leading to complete conversion of AppRNAp to a slower-migrating AppRNAOH species identical to that generated during the reaction of MciRNL with the 2′-OH RNA substrate (Fig. 4C). Ligation of the substrates embeds the 32P-labeled phosphate in a CIP-resistant 3′,5′ phosphodiester linkage. The vicinal unlabeled 2′-PO4 at the splice junction is known to be relatively refractory to hydrolysis by CIP (McCraith and Phizicky 1990). Thus, only a minority of the monomer circle produced during reaction with the 2′-PO4 substrate was converted into a slower-moving circle with a 2′-OH splice junction that comigrated with the circle formed during reaction with the 2′-OH RNA (Fig. 4C).
Single-turnover kinetics of steps 2 and 3 of the MciRNL ligation pathway
By omitting ATP and working at a 10-fold molar excess of MciRNL to 5′-32P-labeled RNA (corresponding to a 9.4-fold excess of MciRNL–AMP), we conducted a single-turnover kinetic analysis of the RNA adenylylation and phosphodiester formation steps focused on the contributions of the RNA 2′-PO4 group. In the experiment shown in Figure 5A, MciRNL was added to a prewarmed reaction mixture containing 2′-OH RNA substrate, and aliquots were withdrawn at the times specified (between 15 sec and 10 min) and quenched immediately with EDTA/formamide. The AppRNA species accumulated with time and comprised 87% of total RNA at 10 min, at which point only 8% of the substrate was ligated circle. A nonlinear least squares regression fit of the data to a unidirectional two-step mechanism executed in Prism yielded apparent step 2 (RNA adenylylation) and step 3 (phosphodiester formation) rate constants of 0.27 and 0.012 min−1, respectively (Fig. 5A). Thus, with a 2′-OH substrate, the rate of RNA adenylation is 22-fold faster than end-sealing. The single-turnover reaction with 2′-PO4 RNA was much faster, leading us to quench individual reactions after 5 or 15 sec. Here, the data fit returned apparent step 2 and step 3 rate constants of 34 and 75 min−1, respectively (Fig. 5B). Comparing the kinetic profiles of the two substrates, we see that the 2′-PO4 enhances the rates of RNA adenylation and end-sealing by 125- and 6200-fold, respectively.
FIGURE 5.
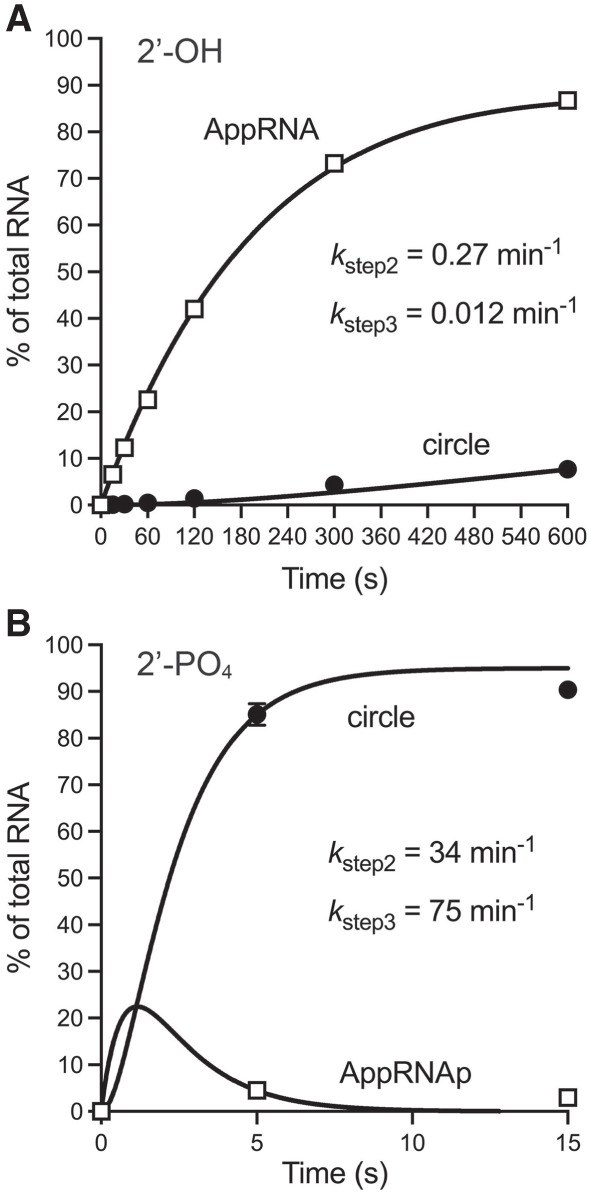
Kinetics of single-turnover ligation by MciRNL–AMP. (A) Ligation of 2′-OH RNA. Reaction mixtures (80 µL) containing 50 mM Tris-acetate, pH 6.5, 5 mM MgCl2, 0.1 µM 5′ 32P-labeled 10-mer RNA with a 3′-OH,2′-OH end, and 1 µM MciRNL were incubated at 37°C. The reactions were initiated by adding MciRNL to a prewarmed reaction mixture. Aliquots (10 µL) were withdrawn before adding enzyme (time 0) or at the times specified after enzyme addition, and quenched immediately with an equal volume of 95% formamide/50 mM EDTA. The products were analyzed by urea-PAGE. The distribution of radiolabeled RNAs is plotted as a function of reaction time. Each datum in the graph is an average of three independent experiments ± SEM, except for the 10-min time point, which is an average of two independent experiments. (B) Ligation of 2′-PO4 RNA. Individual replicate reaction mixtures (10 µL) containing 50 mM Tris-acetate, pH 6.5, 5 mM MgCl2, 0.1 µM 5′ 32P-labeled 10-mer RNA with a 3′-OH,2′-PO4 end, and 1 µM MciRNL were incubated at 37°C. The reactions were initiated by adding MciRNL to a prewarmed reaction mixture. The reactions were quenched after 5 or 15 sec with an equal volume of 95% formamide/50 mM EDTA. The products were analyzed by urea-PAGE. The distribution of radiolabeled RNAs is plotted as a function of reaction time. Each datum in the graph is an average of nine replicate reactions ± SEM. The data in panels A and B were fit by nonlinear regression in Prism to a unidirectional two-step kinetic mechanism.
Effect of pH and magnesium on single-turnover pRNA2′p ligation
The effect of pH on the outcome of a 1-min reaction of 1 µM MciRNL with 0.1 µM 5′-32P-labeled 2′-PO4 RNA is shown in Figure 6A. Whereas monomer circles predominated across a broad pH range (comprising 85%–93% of total RNA at pH 6.0–9.5) the formation of dimer circles and AppRNAp displayed distinct behavior vis-à-vis pH. To wit: dimer circles peaked at pH 5.5–6.0 when they comprised 5% of total RNA and declined to <1% at pH 4.5 or pH ≥8.0. The instructive finding was that the abundance of the RNA-adenylate intermediate increased significantly and steadily as the pH was lowered from 7.5 (<1% of total RNA) to 5.5 (11%), 5.0 (20%), and 4.5 (24%). This signifies that acidic pH selectively slows the phosphodiester formation step of the MciRNA reaction pathway. Accumulation of AppRNA or AppDNA and suppression of end-sealing at acidic pH were noted previously for other RNA and DNA ligases (Yin et al. 2004; Samai and Shuman 2012; Unciuleac and Shuman 2015).
FIGURE 6.

Effect of pH and magnesium on single-turnover pRNA2′p ligation. (A) pH dependence. Reaction mixtures (10 µL) containing either 50 mM Tris-acetate (pH 4.5, 5.0, 5.5, 6.0, or 6.5) or 50 mM Tris-HCl (pH 7.0, 7.5, 8.0, 8.5, 9.0, or 9.5), 5 mM MgCl2, 1 pmol 5′ 32P-labeled pRNA2′p, and 10 pmol MciRNL were incubated at 37°C for 1 min. (B) Magnesium dependence. Reaction mixtures (10 µL) containing 50 mM Tris-acetate, pH 6.5, 1 pmol 5′ 32P-labeled pRNA2′p, 10 pmol MciRNL, and MgCl2 as specified or 5 mM EDTA were incubated at 37°C for 1 min. The reaction products were analyzed by urea-PAGE and visualized by scanning the gel with a Fujifilm FLA-7000 imaging device.
The yield of ligated circles during a 1-min reaction of 1 µM MciRNL with 0.1 µM 5′-32P-labeled 2′-PO4 RNA was similar at 0.5, 1, 2, 5, and 10 mM magnesium (Fig. 6B), comprising 91%–97% of total RNA. The omission of magnesium interdicted ligation but did not preclude the formation of AppRNAp, to an extent of 8% of total RNA (Fig. 6B), suggesting that either step 2 catalysis proceeded weakly in the absence of a metal cofactor or trace levels of enzyme-associated magnesium were carried through the MciRNL purification. Consistent with the latter scenario, we found that the omission of magnesium and inclusion of 5 mM EDTA in the reaction mixture reduced AppRNAp formation to 0.3% of total RNA (Fig. 6B).
MciRNL adenylylation
Reaction of MciRNL with magnesium and 50 µM [α32P]ATP resulted in formation of a covalent MciRNL–[32P]AMP adduct that migrated at the expected size during SDS-PAGE (Fig. 7A). MciRNL adenylylation was optimal at alkaline pH (Fig. 7A). ATP titration indicated that the yield of the covalent ligase–adenylate was optimal at 300–500 µM ATP (Fig. 7B). Quantification of the radiolabeled MciRNL indicated that ∼1.3% of the input MciRNL protein became adenylylated in vitro. This is in keeping with our estimate from the data in Figure 4B that preformed MciRNL–AMP comprised >90% of the recombinant protein preparation.
FIGURE 7.

MciRNL adenylylation. (A) pH dependence. Reaction mixtures (10 µL) containing either 50 mM Tris-acetate (pH 4.5, 5.0, 5.5, 6.0, or 6.5) or 50 mM Tris-HCl (pH 7.0, 7.5, 8.0, 8.5, 9.0, or 9.5), 5 mM MgCl2, 50 µM [α32P]ATP, and 10 µM (100 pmol) MciRNL were incubated at 37°C for 5 min, then quenched with SDS, and analyzed by SDS-PAGE. A scan of the gel is shown. The positions and sizes (kDa) of marker proteins are indicated on the left. (B) ATP concentration-dependence. Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 9.0, 5 mM MgCl2, 10 µM (100 pmol) MciRNL, and 50, 100, 200, 300, 400, or 500 µM [α32P]ATP were incubated at 37°C for 5 min, then quenched with SDS, and analyzed by SDS-PAGE. A scan of the gel is shown. The extents of MciRNL–[32P]AMP formation are indicated below the lanes.
Mucorales Tpt1 proteins have tRNA 2′-phosphotransferase activity in vivo
The evidence presented above that Mucor tRNA ligase seals 3′-OH,2′-PO4 and 5′-PO4 ends to generate a 2′-PO4, 3′–5′ phosphodiester splice junction begets a requirement for a Mucor Tpt1-type enzyme that removes the junction 2′-PO4 group. A Psi-Blast search of Mucorales taxa with yeast Tpt1 retrieved putative orthologs from M.circinelloides (210 aa), R. azygosporus (207 aa), and L. corymbifera (211 aa). These three Tpt1 proteins share 118 positions of amino acid side chain identity/similarity (denoted by dots in Fig. 8A). The Mucorales Tpt1 proteins include a constellation of 11 amino acids (shaded yellow in Fig. 8A)—conserved among fungal, bacterial, and archaeal Tpt1 enzymes—that contact the NAD+ and 2′-PO4, 3′–5′ phosphodiester splice junction substrates (Banerjee et al. 2019c; Jacewicz et al. 2023). Many of these conserved amino acids are essential for Tpt1 activity in vivo, as gauged by complementation of S. cerevisiae tpt1Δ (Sawaya et al. 2005; Munir et al. 2018; Banerjee et al. 2019c; Yang et al. 2023). Here, we cloned each of the Mucorales Tpt1 ORFs into a yeast CEN HIS3 plasmid wherein its expression is driven by the yeast TPI1 promoter and tested complementation by plasmid shuffle in a S. cerevisiae tpt1Δ p(CEN URA3 TPT1) strain. In each case, we recovered viable tpt1Δ isolates that lacked the p(CEN URA3 TPT1) plasmid after selection for growth on medium containing 5-FOA. The tpt1Δ MciTPT1, tpt1Δ RazTPT1, and tpt1Δ LcoTPT1 strains grew as well as the tpt1Δ SceTPT1 control when spot-tested on rich agar medium at 20°C–37°C (Fig. 8B).
FIGURE 8.

Mucorales Tpt1 proteins have tRNA 2′-phosphotransferase activity in vivo. (A) Alignment of the amino acid sequences of Tpt1 homologs from M. circinelloides (Genbank EPB89638.1), R. azygosporus (Genbank RCI00563.1), and L. corymbifera (Genbank CDH53741.1). Positions of side chain identity/similarity are indicated by dots above the alignment. The conserved amino acids that make atomic contacts with Tpt1 reaction substrates and products are highlighted in yellow shading. (B) Complementation of S. cerevisiae tpt1Δ was assayed by plasmid shuffle. Aliquots (3 µL) of serial tenfold dilutions of FOA-resistant tpt1Δ strains expressing the indicated TPT1 genes were spotted on YPD agar plates and incubated at 20°C, 25°C, 30°C, 34°C, and 37°C. Photographs of the plates are shown.
2′-phosphotransferase activity of recombinant MciTpt1
We produced Mucor Tpt1 in E. coli as a His10Smt3 fusion and purified it from a soluble extract by sequential Ni-affinity chromatography/imidazole elution, removal of the His10Smt3 tag by treatment with Ulp1 protease, recovery of the tag-free MciTpt1 protein in the flowthrough of a second Ni-affinity column, and a final Superose-200 gel-filtration step. MciTpt1 was monomeric by gel filtration. SDS-PAGE analysis affirmed the purity of the MciTpt1 preparation (Fig. 9A). We assayed MciTpt1 for RNA 2′-phosphotransferase activity in the presence of 0.2 µM 5′ 32P-labeled 6-mer 2′-PO4 RNA oligonucleotide (shown in Fig. 9B) and 1 mM NAD+ as described (Munir et al. 2018). After a 30 min incubation at 37°C, the reaction products were resolved by urea-PAGE, which showed that 1 nM MciTpt1 converted all of the input 2′-PO4 RNA substrate to a slower-migrating 2′-OH RNA product. The extent of product formation as a function of input MciTpt1 is plotted in Figure 9B. From the slope of the titration curve in the linear range, we calculated that 284 ± 16 fmol of 2′-PO4 RNA was converted to 2′-OH RNA per fmol of input MciTpt1 in a 30 min reaction, which corresponds to a turnover number of 9.5 min−1.
FIGURE 9.

2′-phosphotransferase activity of recombinant Mucor Tpt1. (A) An aliquot (7.5 µg) of the recombinant MciTpt1 preparation was analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker proteins are indicated on the left. The expected molecular weight of MciTpt1 is 24 kDa. (B) Reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 2′-PO4 branchpoint-containing 6-mer RNA oligonucleotide (as shown), and 0, 1, 2.5, 5, 10, or 20 pmol MciTpt1 were incubated at 37°C for 30 min. The reactions were quenched by the addition of three volumes of cold 90% formamide, 50 mM EDTA. The samples were analyzed by electrophoresis (at 55 W constant power) through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The 5′-radiolabeled 2′-PO4 substrate RNA and the 2′-OH product RNA were visualized by scanning the gel with a Fujifilm FLA-7000 imaging device. The extents of product formation were quantified by analysis of the gel scans in ImageQuant and are plotted as a function of input MciTpt1. Each datum is the average of four independent titration experiments ± SEM.
DISCUSSION
The present study highlights the Mucorales tRNA splicing machinery as an exception to the rule whereby fungal tRNA splicing is catalyzed by a trifunctional Trl1 enzyme composed of fused colinear healing and sealing domains. Rather, Mucorales deploy a monofunctional RNA ligase, homologous to the LIG domain of Trl1, which shares with fungal and plant tRNA ligases a requirement for a 3′-OH,2′-PO4 terminus to effectively execute end joining (Konarska et al. 1982; Greer et al. 1983; Schwartz et al. 1983; Pick et al. 1986; Remus and Shuman 2013). In this respect, Mucorales resemble the lancelet Branchiostoma floridae, a chordate taxon in which a stand-alone ATP-dependent 3′-OH,2′-PO4/5′-PO4 RNA ligase (BfRNL) was characterized biochemically and shown to complement yeast trl1Δ when expressed in tandem with SceKIN-CPD (Englert et al. 2010). Also identified was a separately encoded B. floridae healing enzyme (BfPNK/CPDase), homologous to the KIN–CPD portion of Trl1, that complemented yeast trl1Δ when expressed in tandem with BfRNL (Englert et al. 2010). In the case of Mucorales, we did not recover a credible candidate end-healing enzyme during a Blast search with the KIN–CPD domains of yeast or plant Trl1 enzymes. Thus, the nature of the enzymes and reactions that provide the proper ends for sealing by Mucorales RNLs remains to be determined.
Our findings anent the contributions of the RNA 2′-PO4 to catalysis by MciRNL revealed several interesting differences vis-à-vis its role in ligation by the plant tRNA ligase AtRNL. The kinetics of end-healing and sealing by AtRNL were examined under single-turnover conditions, by exploiting a series of linear 20-mer RNA substrates with different terminal structures (Remus and Shuman 2013). Initial mechanistic insights into the 2′-PO4 specificity of the ligation reaction emerged from the finding that AtRNL seals a preadenylylated AppRNAOH substrate with a 3′-OH,2′-OH end. The insignificant rate differential between AppRNA2′p and AppRNAOH sealing indicated that the 2′-PO4 moiety makes little contribution to the rate of step 3 catalysis. Kinetic experiments showed that AtRNL 2′-PO4 specificity is enforced at the RNA 5′ adenylylation step, insofar as the conversion of pRNAOH to AppRNAOH and ligated RNA in the presence of ATP was several hundred-fold slower than the sealing of pRNA2′p under the same reaction conditions (Remus and Shuman 2013). Because the 2′-PO4 moiety is not chemically transformed during the sealing reaction, its essentiality for step 2 suggested a mechanism of substrate-assisted catalysis in which the 2′-PO4 either: (i) promotes proper geometry of the lysyl–AMP and 5′-PO4 reactants during step 2; (ii) assists in binding the Mg2+ cofactor during step 2; or (iii) aids expulsion of the lysine leaving group.
The key difference in the case of MciRNL is that the 2′-PO4 is critical for step 3 of the ligation pathway—attack of the 3′-OH on the 5′ AppRNA intermediate to form a 3′,5′-phosphodiester and release AMP. Indeed, the 2′-PO4 is more important for step 3 catalysis by MciRNL than it is for step 2. In a single-turnover reaction of MciRNL–AMP with a 10-mer pRNA, the absence of a 2′-PO4 selectively interdicts phosphodiester synthesis and leads to the accumulation of AppRNA by virtue of a ∼6200-fold decrement in the apparent step 3 rate constant. In contrast, the lack of a 2′-PO4 elicits a more modest 125-fold slowing of the apparent step 2 rate constant. Although the 2′-PO4 is not chemically transformed during step 3, its proximity to the nascent junction phosphodiester would allow it to engage in substrate-assisted catalysis. Understanding the distinct ways in which a 2′-PO4 end drives steps 2 and 3 will hinge on capturing structures of exemplary fungal and plant tRNA ligase–AMP intermediates in complexes with pRNA2′p substrates.
Finally, the fact that Mucorales join all other human fungal pathogens in relying on a common tRNA sealing enzyme, distinct in all ways from the mammalian RtcB tRNA ligase, is further impetus to consider inhibition of tRNA ligation as a platform for broad-spectrum antifungal drug discovery. A potential caveat arose recently with the recent identification by Yuan et al. (2023) of human C12orf29 as an ATP-dependent RNA ligase, homologous to the NTase domain of Naegleria gruberi RNA ligase (Unciuleac and Shuman 2015; Unciuleac et al. 2015), that joins 5′-PO4 and 3′-OH,2′-OH ends via ligase–AMP and AppRNA intermediates. However, C12orf29 was unable to ligate 5′-PO4 and 3′-OH,2′-PO4 ends in vitro (Yuan et al. 2023). Moreover, we have found that the human C12orf29 ligase was unable to complement yeast trl1Δ when expressed in tandem with SceKIN-CPD (not shown), implying that it is unable to effectively seal tRNAs with 3′-OH,2′-PO4 ends in vivo. Unlike the essential RtcB ligase, the C12orf29 ligase is dispensable for the growth of human cells in culture (Yuan et al. 2023). Therefore, it seems plausible that a selective inhibitor of 3′-OH,2′-PO4/5′-PO4 sealing by fungal tRNA ligase would not exert on-target-based toxicity in human cells.
MATERIALS AND METHODS
Expression plasmids for Mucor RNA ligase (MciRNL) and Mucorales Tpt1 proteins
A synthetic DNA ORF encoding M. circinelloides RNA ligase (MciRNL) (Genbank EPB83728.1) was purchased from Integrated DNA Technologies. The ORFs encoding Tpt1 homologs from M. circinelloides (Genbank EPB89638.1), R. azygosporus (Genbank RCI00563.1), and L. corymbifera (Genbank CDH53741.1) were purchased from Genewiz. The synthetic genes, which were codon-optimized for expression in E. coli, were PCR-amplified with primers that introduced a BamHI site immediately flanking the start codon and an XhoI site downstream from the stop codon. The PCR products were digested with BamHI and XhoI. The MciRNL ORF was inserted into pET28b-His10Smt3 to generate a T7 RNA polymerase-based expression plasmid encoding the MciRNL polypeptide fused to an N-terminal His10Smt3 tag. The RNL and Tpt1 ORFs were also inserted into yeast expression vectors so as to place them under the transcriptional control of the yeast TPI1 promoter, thereby yielding plasmids p415-MciRNL (CEN LEU2 MciRNL), p413-MciTPT1 (CEN HIS3 MciTPT1), p413-RazTPT1 (CEN HIS3 RazTPT1), and p413-LcoTPT1 (CEN HIS3 LcoTPT1). The plasmid inserts were sequenced to verify that no unwanted coding changes were introduced during PCR amplification and cloning.
S. cerevisiae trl1Δ and tpt1Δ complementation
We tested complementation by plasmid shuffle in S. cerevisiae strains trl1Δ p(CEN URA3 TRL1) and tpt1Δ p(CEN URA3 TPT1), which are unable to grow on medium containing 0.75 mg/mL FOA (5-fluoroorotic acid), a drug that selects against the URA3 plasmid (Sawaya et al. 2003, 2005). trl1Δ p(CEN URA3 TRL1) cells were transfected or cotransfected with combinations of p(CEN TRP1), p(CEN HIS3), p(CEN ADE2), or p(CEN LEU2) plasmids (Sawaya et al. 2003; Schwer et al. 2004) for expression of: Trl1 p(CEN TRP1 SceTRL1); Trl1 LIG p(CEN HIS3 SceTRL1-[1-388]); Trl1 KIN–CPD p(CEN ADE2 SceTRL1-[389-827]), T4 Rnl1 p(CEN HIS3 T4RNL1); T4 Pnkp (CEN TRP1 T4PNKP); and MciRNL p(CEN LEU2 MciRNL). In these plasmids, SceTRL1, SceLIG, and SceKIN-CPD are under the control of the S. cerevisiae TRL1 promoter, T4RNL1, and MciRNL are driven by the S. cerevisiae TPI1 promoter, and T4PNKP is under the control of the S. cerevisiae SLU7 promoter. Three to five individual transformants were selected and streaked on agar medium containing FOA. trl1Δ cells transformed with empty plasmid vectors, SceLIG + T4PNKP, or MciRNL + T4PNKP failed to give rise to FOA-resistant colonies. Individual FOA-resistant colonies were grown to mid-log phase in YPD broth and adjusted to A600 of 0.1. Aliquots (3 µL) of serial 10-fold dilutions were spotted to YPD agar plates, which were incubated at 25°C, 30°C, 34°C, and 37°C.
tpt1Δ p(CEN URA3 TPT1) cells were transfected with CEN HIS3 plasmids bearing SceTPT1, MciTPT1, RazTPT1, or LcoTPT1 genes. Individual His+ transformants were selected and streaked on agar medium containing FOA. Individual FOA-resistant colonies expressing MciTPT1, RazTPT1, or LcoTPT1 were amplified in YPD broth and then spot-tested for growth on YPD agar at 20°C, 25°C, 30°C, 34°C, and 37°C.
MciRNL purification
The pET28b-His10Smt3-MciRNL plasmid was transformed into E. coli BL21(DE3) cells. A 1000-mL culture amplified from a single kanamycin-resistant transformant was grown at 37°C in Terrific Broth medium containing 60 μg/mL kanamycin until the A600 reached 0.9–1.0. The culture was chilled on ice for 1 h, adjusted to 2.2% (v/v) ethanol and 0.5 mM IPTG, and then incubated for 20 h at 17°C with constant shaking. Cells were harvested by centrifugation and stored at −80°C. All subsequent steps were performed at 4°C. Cells were thawed and resuspended in 50 mL of buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM DTT, 20 mM imidazole, 10% sucrose), containing one cOmplete EDTA-free Protease Inhibitor Cocktail tablet (Roche). Lysozyme was added to a concentration of 1 mg/mL. After incubation for 30 min, the lysate was sonicated to reduce viscosity, and the insoluble material was removed by centrifugation at 38,000g for 45 min. The supernatant was mixed for 1 h with 7.5 mL of Ni–nitrilotriacetic acid agarose resin (Qiagen) that had been equilibrated with buffer A. The resin was recovered by centrifugation and washed twice with 50 mL of buffer A, then with 20 mL of 50 mM Tris-HCl, pH 8.0, 3 M KCl, followed by 100 mL of buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM DTT, 10% glycerol) containing 20 mM imidazole. The resin was centrifuged again, resuspended in 20 mL of buffer B with 20 mM imidazole, and poured into a column. The bound material was eluted with 15 mL of buffer B containing 300 mM imidazole, while collecting 5-mL fractions. The polypeptide compositions of the flowthrough and eluate fractions were monitored by SDS-PAGE. The 300 mM imidazole eluate fractions containing His10Smt3-MciRNL were supplemented with Smt3-specific protease Ulp1 (Ulp1/His10-Smt3-MciRNL ratio of 1:425 [w/w]) and then dialyzed overnight against 2000 mL of buffer C (50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 20 mM imidazole, 1 mM DTT, 1 mM EDTA, 10% glycerol), during which time the His10Smt3 tag was cleaved. The dialysates were mixed for 1 h with 4 mL of Ni–nitrilotriacetic acid agarose resin that had been equilibrated with buffer C. Tag-free MciRNL protein was recovered in the flowthrough fractions, which were concentrated by centrifugal ultrafiltration (Amicon Ultra-15; 30 kDa cutoff) to 11 mL volume. Two aliquots of MciRNL (5 mL each) were gel-filtered through a 125-mL 16/60 HiLoad Superdex 200 column (GE Healthcare) equilibrated in buffer D (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 5% glycerol) at a flow rate of 0.5 mL/min while collecting 2-mL fractions. The peak fractions were pooled, concentrated by centrifugal ultrafiltration, and stored at −80°C. Protein concentrations were determined with Bio-Rad dye reagent using bovine serum albumin as the standard. The yield of MciRNL was ∼50 mg/L of bacterial culture.
MciTpt1 purification
Plasmid pET28b-His10Smt3-MciTpt1, encoding MciTpt1 fused to an N-terminal His10Smt3 tag, was transformed into E. coli BL21(DE3)-CodonPlus. Two 1-L cultures from a single kanamycin/chloramphenicol-resistant transformant were grown at 37°C in Terrific Broth with 60 μg/mL kanamycin and 35 μg/mL chloramphenicol until the A600 reached 0.8. The cultures were cooled on ice and adjusted to 2.0% (v/v) ethanol and 0.5 mM IPTG, and then incubated for 16 h at 17°C with constant shaking. Cells were harvested by centrifugation and the pellets were stored at −80°C. All subsequent steps were performed at 4°C. The cell pellets were thawed and resuspended in 75 mL of buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2.5 mM TCEP, 20 mM imidazole, 10% glycerol). The suspension was supplemented with two EDTA-free Protease Inhibitor Cocktail tablets (Roche) and adjusted to 1 mg/mL lysozyme. Following a 30-min incubation on ice, the lysate was sonicated to reduce viscosity, and insoluble material was removed by centrifugation at 38,000g for 45 min. The supernatant was then mixed for 1 h with 7 mL of Ni–nitrilotriacetic acid agarose resin (Qiagen) equilibrated with buffer A. After recovering the resin by centrifugation, it was washed twice with 50 mL of buffer A, followed serially by 50 mL of 50 mM Tris-HCl, pH 8.0, 3 M KCl, and 100 mL of buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2.5 mM TCEP, 10% glycerol) containing 40 mM imidazole. The resin was then poured into a column, and the bound material was eluted with 20 mL of buffer B containing 350 mM imidazole. The elution profile of the MciTpt1 protein was monitored by SDS-PAGE. The 350 mM imidazole eluate fractions containing His10Smt3-MciTpt1 were pooled and supplemented with Smt3-specific protease Ulp1 (Ulp1/His10Smt3-MciTpt1 ratio of 1:500 [w/w]). The protein mixture was dialyzed overnight against 2000 mL of buffer C (50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 20 mM imidazole, 1 mM DTT, 10% glycerol). The dialysate was then mixed for 1 h with 3.5 mL of Ni–nitrilotriacetic acid agarose resin equilibrated with buffer C. The resin was poured into a column and the tag-free MciTpt1 protein was recovered in the flowthrough, which was concentrated by centrifugal ultrafiltration (Amicon Ultra-15; 10 kDa cutoff) to a volume of 11 mL. Two aliquots of MciTpt1 (5 mL each) were gel-filtered through a 125-mL 16/60 HiLoad Superdex 200 column (GE Healthcare) equilibrated in buffer D (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM TCEP, 5% glycerol) at a flow rate of 0.5 mL/min, while collecting 2-mL fractions. The peak fractions were pooled, concentrated by centrifugal ultrafiltration to 6.5 mg/mL, and then stored at −80°C. Protein concentrations were determined with Bio-Rad dye reagent using bovine serum albumin as the standard. The yield of MciTpt1 was 25 mg/L of bacterial culture.
5′ 32P-labeled oligonucleotide substrates
Synthetic 10-mer RNAs (5′-HOAUCACGCUUA) with either 2′-PO4 or 2′-OH ends, and a 2′-PO4 branchpoint-containing 6-mer oligonucleotides (5′-CCAA2′PAU) (see Munir et al. 2018), were 5′ 32P-labeled by reaction with phosphatase-dead T4 polynucleotide kinase (Pnkp-D167N) in the presence of [γ32P]ATP. The radiolabeled RNAs were gel-purified by electrophoresis through a 40-cm 20% nondenaturing polyacrylamide gel containing 45 mM Tris-borate, 1 mM EDTA. The radiolabeled RNAs were eluted from excised gel slices during overnight incubation in 10 mM Tris-HCl, pH 6.8, 1 mM EDTA, and then stored at −20°C.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health (NIH) grant R35-GM126945 (S.S.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079911.123.
MEET THE FIRST AUTHOR
Shreya Ghosh.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Shreya Ghosh is the first author of this paper, “Characterization of tRNA splicing enzymes RNA ligase and tRNA 2′-phosphotransferase from the pathogenic fungi Mucorales.” Shreya is a senior research scientist in Dr. Stewart Shuman's lab in the Molecular Biology Program at Memorial Sloan Kettering Cancer Center, New York. The main focus of her research is on the nature of the tRNA splicing machinery in priority pathogens of the order Mucorales, which could be used as a novel drug target to develop more effective, less toxic antifungal drugs.
What are the major results described in your paper and how do they impact this branch of the field?
Our study reports that Mucorales species elaborate a novel variant of the fungal tRNA splicing machinery in which the RNA ligase is a stand-alone enzyme that is active in vivo in budding yeast in lieu of the Trl1 ligase domain. We also affirm the requirement for a 2′-PO4 terminus in the end-joining reaction, via the biochemical characterization of RNA ligase from M. circinelloides. In addition, we have identified the Mucorales Tpt1 enzymes that remove the splice junction 2′-PO4 via genetic complementation of budding yeast tpt1Δ as well as the biochemical characterization of M. circinelloides Tpt1. The fact that Mucorales join all other human fungal pathogens in relying on a common tRNA sealing enzyme, distinct in all ways from the mammalian RtcB tRNA ligase, is further impetus to consider inhibition of tRNA ligation as a platform for broad-spectrum antifungal drug discovery.
What led you to study RNA or this aspect of RNA science?
One of the risk factors of Mucormycosis (infection caused by order Mucorales), is COVID-19 infection (World Health Organization 2022). The present scenario of ongoing COVID infections entails the development of more effective, less toxic antifungal drugs against these priority pathogens. In our laboratory, we regard fungal tRNA splicing enzymes as highly promising antifungal targets, and in this research, we queried the nature of the tRNA splicing machinery in priority pathogens of the order Mucorales.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
No, there were no such difficulties. The results we got during our study were congruent with what we have hypothesized for a fungal pathogenic tRNA splicing machinery.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I had a science teacher in high school who had a true desire of becoming a scientist, but circumstances never allowed him to be one. He used to share fascinating stories about the major discoveries around the world, how scientists are the major contributors to the changes or evolutions being brought about in mankind, and what more we can foresee to be discovered in the future. I feel his strong descriptions and motivations provoked my young mind with an interest to become a scientist and contribute to mankind.
If you were able to give one piece of advice to your younger self, what would that be?
The one piece of advice I would tell my younger self would be to have a better work-life balance. You are always more productive when you are rested or have taken a break/vacation from hectic laboratory life.
REFERENCES
- Amitsur M, Levitz R, Kaufmann G. 1987. Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J 6: 2499–2503. 10.1002/j.1460-2075.1987.tb02532.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Ghosh S, Goldgur Y, Shuman S. 2019a. Structure and two-metal mechanism of fungal tRNA ligase. Nucleic Acids Res 47: 1428–1439. 10.1093/nar/gky1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Goldgur Y, Schwer B, Shuman S. 2019b. Atomic structures of the RNA end-healing 5′-OH kinase and 2′,3′-cyclic phosphodiesterase domains of fungal tRNA ligase: conformational switches in the kinase upon binding of the GTP phosphate donor. Nucleic Acids Res 47: 11826–11838. 10.1093/nar/gkz1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Munir A, Abdullahu L, Damha MJ, Goldgur Y, Shuman S. 2019c. Structure of tRNA splicing enzyme Tpt1 illuminates the mechanism of RNA 2′-PO4 recognition and ADP-ribosylation. Nat Commun 10: 218. 10.1038/s41467-018-08211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, McCraith SM, Zillmann M, Kierzek R, Michaud N, LaReau RD, Turner DH, Phizicky EM. 1993. An NAD derivative produced during transfer RNA splicing: ADP-ribose 1″-2″ cyclic phosphate. Science 261: 206–208. 10.1126/science.8392224 [DOI] [PubMed] [Google Scholar]
- Dantuluri S, Abdullahu L, Munir A, Katolik A, Damha MJ, Shuman S. 2020. Substrate analogs that trap the 2′-phospho-ADP-ribosylated RNA intermediate of the Tpt1 (tRNA 2′-phosphotransferase) reaction pathway. RNA 26: 373–381. 10.1261/rna.074377.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuluri S, Schwer B, Abdullahu L, Damha MJ, Shuman S. 2021. Activity and substrate specificity of Candida, Aspergillus, and Coccidioides Tpt1: essential tRNA splicing enzymes and potential antifungal targets. RNA 27: 616–627. 10.1261/rna.078660.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Sheppard K, Gundllapalli S, Beier H, Söll D. 2010. Branchiostoma floridae has separate healing and sealing enzymes for 5′-phosphate RNA ligation. Proc Natl Acad Sci 107: 16834–16839. 10.1073/pnas.1011703107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CL, Peebles CL, Gegenheimer P, Abelson J. 1983. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell 32: 537–546. 10.1016/0092-8674(83)90473-7 [DOI] [PubMed] [Google Scholar]
- Hayne CK, Sekulovski S, Hurtig JE, Stanley RE, Trowitzsch S, van Hoof A. 2023. New insights into RNA processing by the eukaryotic tRNA splicing endonuclease. J Biol Chem 299: 105138. 10.1016/j.jbc.2023.105138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz A, Dantuluri S, Shuman S. 2023. Structural basis for Tpt1-catalyzed 2′-PO4 transfer from RNA and NADP(H) to NAD+. Proc Natl Acad Sci 120: e2312999120. 10.1073/pnas.2312999120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska M, Filipowicz W, Gross HJ. 1982. RNA ligation via 2′-phosphomonoester, 3′5′-phosphodiester linkage: requirement of 2′,3′-cyclic phosphate termini and involvement of a 5′-hydroxyl polynucleotide kinase. Proc Natl Acad Sci 79: 1474–1478. 10.1073/pnas.79.5.1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCraith SM, Phizicky EM. 1990. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol Cell Biol 10: 1049–1055. 10.1128/mcb.10.3.1049-1055.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCraith SM, Phizicky EM. 1991. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J Biol Chem 266: 11986–11992. 10.1016/S0021-9258(18)99054-X [DOI] [PubMed] [Google Scholar]
- Munir A, Abdullahu L, Damha MJ, Shuman S. 2018. Two-step mechanism and step-arrest mutants of Runella slithyformis NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 24: 1144–1157. 10.1261/rna.067165.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Schwer B, Schaffrath R, Shuman S. 2008. RNA repair: an antidote to cytotoxic eukaryal RNA damage. Mol Cell 31: 278–286. 10.1016/j.molcel.2008.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschek J, Walter P. 2019. tRNA ligase structure reveals kinetic competition between non-conventional mRNA splicing and mRNA decay. Elife 8: e44199. 10.7554/eLife.44199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick L, Furneaux H, Hurwitz J. 1986. Purification of wheat germ RNA ligase: mechanism of action of wheat germ RNA ligase. J Biol Chem 261: 6694–6704. 10.1016/S0021-9258(19)62672-4 [DOI] [PubMed] [Google Scholar]
- Ramirez A, Shuman S, Schwer B. 2008. Human RNA 5′-kinase (hClp1) can function as a tRNA splicing enzyme in vivo. RNA 14: 1737–1745. 10.1261/rna.1142908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S. 2013. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659–669. 10.1261/rna.038406.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S. 2014. Distinctive kinetics and substrate specificities of plant and fungal tRNA ligases. RNA 20: 462–473. 10.1261/rna.043752.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Schwer B, Shuman S. 2016. Characterization of the tRNA ligases of pathogenic fungi Aspergillus fumigatus and Coccidioides immitis. RNA 22: 1500–1509. 10.1261/rna.057455.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Goldgur Y, Shuman S. 2017. Structural basis for the GTP specificity of the RNA kinase domain of fungal tRNA ligase. Nucleic Acids Res 45: 12945–12953. 10.1093/nar/gkx1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Shuman S. 2012. Kinetic analysis of DNA strand joining by Chlorella virus DNA ligase and the role of nucleotidyltransferase motif VI in ligase adenylylation. J Biol Chem 287: 28609–28618. 10.1074/jbc.M112.380428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43928–43938. 10.1074/jbc.M307839200 [DOI] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2005. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 11: 107–113. 10.1261/rna.7193705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RC, Greer CL, Gegenheimer P, Abelson J. 1983. Enzymatic mechanism of an RNA ligase from wheat germ. J Biol Chem 258: 8374–8383. 10.1016/S0021-9258(20)82075-4 [DOI] [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S. 2004. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci 101: 2788–2793. 10.1073/pnas.0305859101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Aronova A, Ramirez A, Braun P, Shuman S. 2008. Mammalian 2′,3′ cyclic nucleotide phosphodiesterase (CNP) can function as a tRNA splicing enzyme in vivo. RNA 14: 204–210. 10.1261/rna.858108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S. 2023. RNA repair: hiding in plain sight. Annu Rev Genet 57: 461–489. 10.1146/annurev-genet-071719-021856 [DOI] [PubMed] [Google Scholar]
- Spinelli SL, Kierzek R, Turner DH, Phizicky EM. 1999. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J Biol Chem 274: 2637–2644. 10.1074/jbc.274.5.2637 [DOI] [PubMed] [Google Scholar]
- Steiger MA, Jackman JE, Phizicky EM. 2005. Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA 11: 99–106. 10.1261/rna.7194605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Meineke B, Shuman S. 2011. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem 286: 30253–30257. 10.1074/jbc.C111.274597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciuleac MC, Shuman S. 2015. Characterization of a novel eukaryal nick sealing RNA ligase from Naegleria gruberi. RNA 21: 824–832. 10.1261/rna.049197.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciuleac MC, Goldgur Y, Shuman S. 2015. Structure and two-metal mechanism of a eukaryal nick-sealing RNA ligase. Proc Natl Acad Sci 112: 13868–13873. 10.1073/pnas.1516536112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S. 2005. Structure-function analysis of yeast tRNA ligase. RNA 11: 966–975. 10.1261/rna.2170305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Englert M, Beier H, Shuman S. 2006. Structure-function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res 34: 517–527. 10.1093/nar/gkj441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 2022. WHO fungal priority pathogens list to guide research, development, and public health action. WHO, Geneva. [Google Scholar]
- Yang X, Wang J, Li S, Li X, Gong J, Yan Z, Zhou H, Liu X. 2023. Structural and biochemical insights into the molecular mechanism of TRPT1 for nucleic acid ADP-ribosylation. Nucleic Acids Res 51: 7649–7665. 10.1093/nar/gkad525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Ho CK, Miller ES, Shuman S. 2004. Characterization of bacteriophage KVP40 and T4 RNA ligase 2. Virology 319: 141–151. 10.1016/j.virol.2003.10.037 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Stumpf FM, Schlor LA, Schmidt OP, Huber LB, Frese M, Höllmüller E, Scheffner M, Stengel F, Diederichs K, Marx A. 2023. Chemoproteomic discovery of a human RNA ligase. Nature Comm 14: 842. 10.1038/s41467-023-36451-x [DOI] [PMC free article] [PubMed] [Google Scholar]