

Abstract
Aetiological understanding and screening methods for congenital heart disease (CHD) are limited. Maternal metabolomic assessment offers the potential to identify risk factors and biomarkers. We performed a systematic review (PROSPERO CRD42022308452) investigating the association between fetal/childhood CHD and endogenous maternal metabolites. Ovid‐MEDLINE, Ovid‐EMBASE and Cochrane Library were searched between inception and 06/09/2022. Case control studies included analysing maternal blood or urine metabolites in pregnancy or postpartum where there was foetal/childhood CHD. Risk of bias assessment utilised the Scottish Intercollegiate Guidelines Network methodology checklist and narrative synthesis was performed. A total of 134 records were screened with eight eligible studies (n = 3242 pregnancies, n = 842 CHD‐affected offspring). Five studies performed metabolomic analysis in pregnancy. Metabolites distinguishing case and control groups spanned lipid, glucose and amino‐acid pathways, with the development of sensitive risk prediction models. No single metabolite consistently distinguished cases and controls across studies. Three studies performed targeted analysis postnatally with altered lipid and amino acid metabolites and raised homocysteine and markers of oxidative stress identified in cases. Included studies reported small sample sizes, analysing different biosamples at variable time points using differing techniques. At present, there is not enough evidence to confidently associate maternal metabolomic profiles with offspring CHD risk. However, several identified pathways warrant further investigation.
Key points
What's already known about this topic?
Congenital heart disease (CHD) is the most common congenital anomaly.
Antenatal detection rates remain low, with limited understanding of aetiology.
Metabolomics provides a global physiological overview to derive potential novel biomarkers in screening and causation.
What does this review add?
Several metabolites in lipid, amino acid and homocysteine/methionine metabolic pathways significantly differ in mothers with offspring affected by CHD.
We identify metabolic pathways for further investigation in establishing causality and screening biomarkers for foetal CHD.
1. INTRODUCTION
Globally, congenital anomalies affect over 2% of total births, with congenital heart disease (CHD) the most common. CHD prevalence is increasing with similar rates reported across Europe. 1 , 2 In England, CHD represents one third of congenital anomalies, with severe CHD, generally defined as requiring surgery or treatment in the first year of life, accounting for around half of these. 3 Increasing detection, survival and the need to reduce postoperative mortality in infants with severe and critical CHD emphasise the importance of adequate understanding of CHD pathology on a global level. 4
Cardiac development begins in the second human embryonic week, with the critical period recognised as weeks two to eight. 5 , 6 Our understanding of CHD aetiology is significantly limited. Around 15% of CHD can be linked to a known genetic cause including aneuploidy, such as trisomy 21 (8%–10%) and single gene defects such as Noonan's syndrome (3%–5%). Approximately 2% of cases are associated with specific environmental factors including maternal medical conditions, for example, diabetes mellitus or phenylketonuria. 7 A meta‐analysis and umbrella review of literature has also linked numerous other maternal and environmental factors to foetal CHD including obesity, smoking, infection, air pollution and recreational and prescription drug use. 8 , 9 Despite such associations, mechanistic understanding is lacking with most CHD causative factors remaining unexplained.
Prenatal screening evaluates individual risk in obstetrics. This is demonstrated by aneuploidy screening, combining foetal nuchal translucency (NT) measurement on ultrasound with maternal serum biomarkers and age. 10 Combined screening tests generally provide more accurate assessment of risk than individual markers alone. Diagnostic genetic testing by invasive sampling of the placenta or amniotic fluid provides an accurate result but carries a risk of miscarriage of approximately 0.5%. 11 More recently, non‐invasive prenatal testing where analysis of maternal serum is undertaken for cell‐free foetal DNA has revolutionised prenatal screening for aneuploidy with a significantly higher sensitivity and specificity and no associated risk of miscarriage. 12 , 13 Whilst these technological developments are being extended to single gene disorders, they are unlikely to provide adequate screening for CHD given its multifactorial aetiology and low single gene causality yield.
The focus of prenatal screening for congenital anomalies primarily rests on ultrasonographic assessment. Increased NT, abnormal ductus venosus flow and tricuspid regurgitation in the first trimester are associated with an increased risk of CHD. 14 The foetal anomaly screening programme recommends a systematic anatomy scan between 18 and 21 weeks, aiming to identify common and major congenital anomalies including serious cardiac anomalies (transposition of the great arteries, atrioventricular septal defects, tetralogy of Fallot and hypoplastic left heart syndrome). 10 Patient uptake is over 98%, illustrating the importance of this screening to pregnant woman. 15 However, targeted detection rates for CHD are only 50%. 10 Recent data in England shows that this detection rate is accurate, with 54.5% of major CHD diagnosed antenatally. 3 Ultrasound has several limitations including intraoperator variability, foetal factors such as position or multiple pregnancy and maternal factors such as obesity. Foetal echocardiography, provided by trained foetal cardiologists, provides greater sensitivity for CHD detection but is generally only performed when an anomaly is suspected on screening ultrasound or in screening high‐risk populations. 16 , 17 Foetal echocardiography can improve detection of CHD following a normal complete foetal anomaly scan; however, performance of 750 scans is required to detect one additional case. 18 Foetal echocardiography is costly and requires significant expertise. 19 As such, there are significant limitations of current antenatal screening strategies for foetal CHD.
Metabolomic analysis utilising chromatography and mass spectrometry or nuclear magnetic resonance spectroscopy detects compounds such as carbohydrates, amino acids, lipids and vitamins in biosamples such as blood or urine. 20 Such profiles reflect global physiology, influenced by genetics, the environment and physiological stressors. 20 , 21 , 22 Use of machine learning techniques in large biological datasets has revolutionised utilisation of such information. 23 In CHD, it is hypothesised that changes in foetal perfusion or organ function could be reflected in maternal metabolic alterations. As such, metabolomic analyses provide the potential to derive novel biomarkers to screen for CHD. 21 However, the presence of foetal cardiovascular shunts and the placental interface may limit this. Furthermore, elucidating potential maternal metabolic risk factors could help improve mechanistic understanding and screening for CHD.
A systematic review assessing altered endogenous maternal metabolite levels in maternal screening or risk factor profiling for foetal CHD has not been undertaken previously. We therefore aim to critically appraise available literature to assess the association between altered endogenous maternal metabolite levels and foetal or childhood CHD. Our findings will inform subsequent recommendations for future research and practice.
2. METHODS
2.1. Protocol, search strategy and eligibility criteria
The systematic review protocol was registered a priori at PROSPERO (registration number CRD42022308452) and followed the PRISMA 2020 checklist. 24 A systematic literature search of electronic databases MEDLINE (via OVID), EMBASE (via OVID) and the Cochrane library was conducted to identify relevant studies published between the inception of each database and February 3rd 2022 without language restrictions. Searches were re‐run prior to final submission (6th September 2022). Reference lists of included studies were manually screened and experts contacted to identify additional emerging and relevant literature. Figure S1 outlines the search strategy for each database. Observational studies were included. It was not anticipated that randomised control or control trials would be available. Case reports and case series were excluded due to high risk of bias.
Case inclusion criteria were mothers of fetuses/children with CHD. Studies with surrogate mothers or pregnancies with egg donation were excluded due to the potential influence of the surrogate or donor environment on the metabolome. The exposure was maternal blood or urine metabolite analysis in pregnancy or postpartum. The primary outcome was foetal/childhood CHD.
2.2. Study selection, data extraction and quality assessment
SM and SR independently screened titles and abstracts to exclude studies that did not meet inclusion criteria. Agreement on potential inconsistencies was reached by consensus. Data extraction from selected studies was performed by SM and checked by SR. Risk of bias assessment was assessed by SM and SR independently utilising the Scottish Intercollegiate Guidelines Network case control study checklist and guidance notes. 25 This facilitates assessment of subject selection, exposure assessment, confounding and statistical analysis. Studies are assessed overall as high quality (++), acceptable (+) or unacceptable. Unacceptable studies were excluded from analysis due to high risk of bias.
2.3. Data synthesis and analysis
A narrative synthesis was performed including tabulation of study characteristics and results. Quantitative synthesis of data and meta‐analysis was precluded due to variation in studies including biomaterial analyses, timing of sampling and metabolic analyses performed. Metabolites distinguishing case and control populations identified through machine learning and confounder adjusted analyses were summarised. For antenatal studies reporting risk prediction, the models developed and included biomarkers are presented.
3. RESULTS
3.1. Literature search
Figure 1 demonstrates a PRISMA flow diagram of included studies. Following systematic literature searching, 134 unique studies were identified. 114 studies were excluded after title and abstract screening. Full text reports were reviewed for remaining studies. A further 12 were excluded, with reasons outlined in Figure 1. This resulted in 8 studies included. 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33
FIGURE 1.
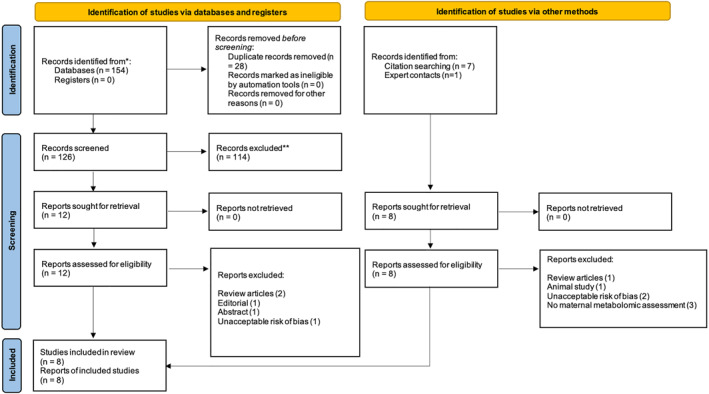
PRISMA 2020 flow diagram of study identification, screening, inclusion and exclusion. 57
3.2. Characteristics of included studies
Table 1 summarises the characteristics and risk of bias assessment for the 8 included studies. Primary metabolomic analysis data from 3242 pregnancies is included, with 842 having a child affected by CHD. All studies were case control design published between 2005 and 2022. Studies originated from 4 countries: USA, 4 UK, 2 Italy 1 and China. 1
TABLE 1.
Summary study characteristics from included studies.
| Study design | Maternal/fetal characteristics | Analysis | Outcome | Risk of bias/methodology checklist a |
|---|---|---|---|---|
|
|
|
|
+ |
Friedman et al. 2021
27
|
|
|
|
+ |
|
|
|
|
+ |
Xie et al. 2019
29
|
|
|
|
+ |
|
|
|
|
+ |
Hobbs et al. 2005
30
|
|
|
|
+ |
|
|
|
|
+ |
Hsu et al.
32
|
|
|
|
+ |
Note: Assessment of subject selection, exposure assessment, confounding and statistical analysis classifies studies overall as high quality (++), acceptable (+) or unacceptable (−).
Abbreviations: AVPM, anomalous pulmonary venous malformation; AS, aortic stenosis; ASD, atrial septal defect; AV hypoplasia, atrioventricular hypoplasia; AVSD, atrioventricular septal defect; CHD, congenital heart disease; CMV, cytomegalovirus; DI/LC‐MS, combined direct injection and liquid chromatography and tandem mass spectrometry; DORV, double outlet right ventricle; GC‐MS, gas chromatography mass spectrometry; HLHS, hypoplastic left heart syndrome; HLRS, hypoplastic right heart syndrome; HPLC, high performance liquid chromatography; LC‐MS‐MS, liquid chromatography tandem mass spectrometry; MS, mitral stenosis; NMR, nuclear magnetic resonance; PA, pulmonary atresia; PS, pulmonary stenosis; RAA, right aortic arch; TOP, termination of pregnancy; TA, tricuspid atresia; TGA, transposition of the great arteries; ToF, tetralogy of Fallot; TR, tricuspid regurgitation; VSD, ventricular septal defect.
Methodology checklist classification derived utilising the Scottish Intercollegiate Guidelines Network (SIGN) case control study risk of bias tool by 2 authors (SM and SR).
3.3. Metabolic analysis in pregnancy
Maternal metabolomic analyses was performed in five studies 26 , 27 , 28 , 29 during the index pregnancy. Methods of metabolomic analysis included gas, liquid and direct injection chromatography coupled with mass spectrometry or NMR spectroscopy. Maternal serum was analysed in two studies. 26 , 28 Bahado‐Singh et al. focussed on analysis within the first trimester, limited to isolated CHD in fetuses. All included cases were defined as severe CHD by EUROCAT (European Registry of Congenital Anomalies and Twins). However, the sample size was limited (n = 27 cases). Troisi et al. performed analysis with median sampling time in the second trimester. This included pregnancies proceeding to termination in isolated CHD. Seventy cases were included, with 82.9% EUROCAT severe CHD. Taylor et al. performed analysis on maternal plasma in pregnancy with mean sample timing in the second trimester. 33 Untargeted mass spectrometry‐based metabolomics analysis was performed with NMR validation of metabolites distinguishing cases and controls where possible. However, CHD diagnosis characteristics were not defined. 33 Two studies 27 , 29 performed analysis on maternal urine samples in pregnancy. Friedman et al. analysed samples at a median time point in the second trimester on fetuses with isolated CHD. Case sample size was 36 cases and less than a third of cases were EUROCAT severe diagnoses. Xie et al. performed analysis at a median time point in the third trimester also including fetuses with associated anomalies. 52.9% of the 70 included cases were EUROCAT severe CHD.
Analysis of identified metabolites which significantly differed between case and control groups were assessed by multivariable regression or partial least squares discriminant analysis and synthesised from variable importance in projection plots or selected through volcano plot analysis. Table S1 summarises individual metabolites distinguishing cases and controls with direction of change and method of analysis for each included study. Figure 2A collates metabolites distinguishing case and control groups across included studies. A total of 110 unique metabolites were identified spanning multiple metabolic pathways (excluding 7 unnamed molecules). Differences in lipid metabolism are strongly represented including acylcarnitines, phospholipids, fatty acids, lysophospholipids and sphingolipids, predominantly reduced in cases compared to controls. Metabolites from amino acid, glucose and aerobic metabolism also significantly differ.
FIGURE 2.
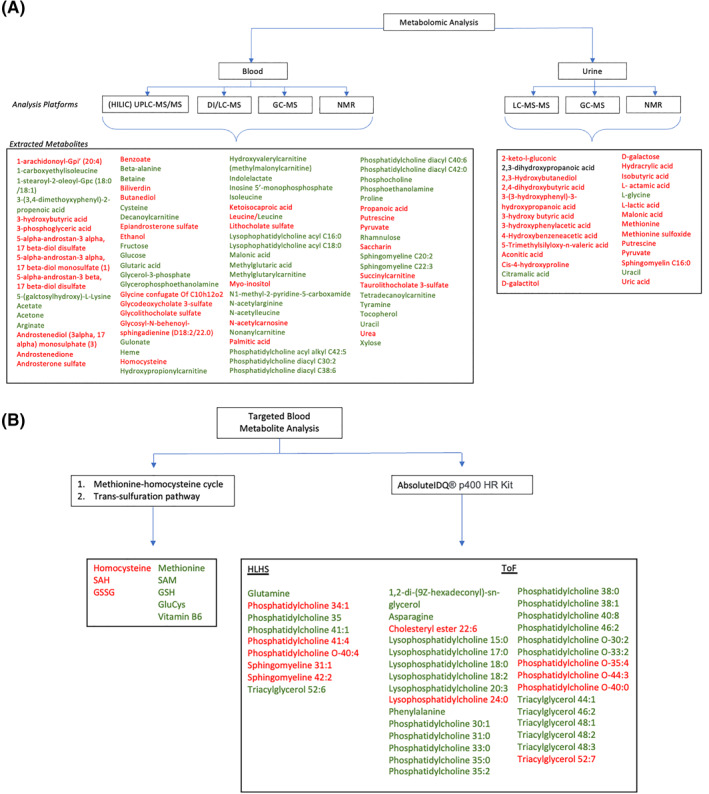
(A): Metabolomic analyses from maternal biosamples taken during pregnancy, synthesised from 5 studies. 26 , 27 , 28 , 29 , 33 Metabolites selected through multivariate regression (metabolites significant by p < 0.05) or partial least squares discriminant analysis (PLS‐DA) and synthesized from variable importance in projection (VIP) plots (metabolites significant by univariate analysis (p < 0.05) included) or selected through volcano plot analysis (p < 0.05 and fold change >2 or <−2). Red – metabolite increased in cases versus controls; Green – metabolite decreased in cases versus controls; Black – direction of change unknown. Unnamed (n = 7) molecules not included. DI/LC‐MS – combined direct injection and liquid chromatography and tandem mass spectrometry; GC‐MS – gas chromatography mass spectrometry; (HILIC) UPLC‐MS/MS – (hydrophilic interaction liquid chromatography) ultra‐high‐performance liquid chromatography tandem mass spectrometry; NMR – nuclear magnetic resonance; LC‐MS‐MS – liquid chromatography tandem mass spectrometry. (B): Targeted metabolite analysis from maternal blood samples taken out with pregnancy, synthesised from 3 studies. 30 , 31 , 32 Concentration of individual metabolites presented significantly differ (p < 0.05) between mothers of CHD cases and controls in confounder adjusted analyses. Red – metabolite increased in cases versus controls; Green – metabolite decreased in cases versus controls. Glucys – Glutamylcysteine; GSH – reduced glutathione; GSSG – oxidized glutathione; HLHS – Hypoplastic left heart syndrome; SAH – S‐adenosylhomocysteine; S‐adenosylmethionine; ToF – tetralogy of Fallot.
Whilst metabolites from similar and related metabolic pathways were identified in multiple included studies, leucine was the only metabolite to significantly differ between cases and controls in two independent studies, with contradictory findings. 28 , 33
3.4. Mendelian Randomisation
One study utilised three European population cohort studies (n = 33,662, 319 CHD cases) to enable Mendelian Randomisation. This analysis allowed further validation of potential causal effects of 27 metabolites differentiating cases and controls in pregnancy (n = 44). 33 From this, 11 metabolites were identified with replication and potential evidence of causality, representing amino acid, steroid lipid and succinylcarnitine metabolic pathways.
3.5. Risk prediction models
Risk prediction models were developed in three studies 26 , 27 , 28 in pregnancy utilising metabolites with or without ultrasound markers. Table 2 summarises methods of model development and model performance. Combined screening models for prediction of CHD incorporating metabolite and ultrasound markers outperformed those assessing metabolites alone. The area under the curve was above 0.81 across all models, with a first trimester model combining metabolites (hydroxypropionylcarnitine, glutaconylcarnitine, hydroxytetra‐decadienylcarnitine) with NT having the best performance (AUC 0.992 (95% CI 0.973–1.0), sensitivity 92.9%, specificity 93.2%). 26
TABLE 2.
Summary of risk prediction models built utilising multiple metabolites +/− ultrasound markers in pregnancy
| Reference | Method of model development | Variables included | Model performance (AUC, 95% CI) |
|---|---|---|---|
| Bahado‐Singh et al. 2014 26 | Logistic regression |
|
|
|
|
||
| Friedman et al. 2021 27 | Logistic regression |
|
|
|
|
||
| Troisi et al. 2021 28 |
|
|
|
Abbreviations: AUC, area under curve; NT, nuchal translucency.
3.6. Postnatal targeted metabolic analysis
Targeted analysis of metabolites in the B12 and folate dependent homocysteine‐methionine cycle and downstream trans‐sulphuration pathways was performed in two case‐control studies. 30 , 31 Both studies utilised participants of the Arkansas Reproductive Health Monitoring System following the index pregnancy. Maternal blood plasma was sampled postnatally, with median sample timings ranging from 14.9 to 24.5 months. CHD diagnosis was made antenatally or postnatally, but individual diagnosis data were not available. Larger sample sizes of 224 and 331 cases were included, respectively. One study performed targeted metabolomic assessment of 400 metabolites (AbsoluteIDQ® p400 HR Kit) in the postnatal period. 32 Pilot data was available for a small sample of 38 cases (22 tetralogy of Fallot, 16 hypoplastic left heart) and 18 controls. Timings of blood sampling were not available.
Figure 2B synthesises confounder adjusted analysis comparing mean metabolite concentrations between cases and controls. This demonstrates significantly increased mean homocysteine and decreased mean methionine concentrations between cases and controls, in addition to increased downstream metabolites representing sources of oxidative stress. Further, alterations in phospholipid and glutamate metabolic pathways are evidenced, illustrating potential mechanisms of CHD pathogenesis.
3.7. Assessment of confounding variables
Figure 3 summarises potential confounding variables compared between case and control groups across studies. Maternal age was assessed in all studies with timing of biosample analysis in all but one. Maternal smoking status, ethnicity, maternal alcohol consumption, gravidity/parity and maternal BMI were assessed in 50% or more of studies.
FIGURE 3.
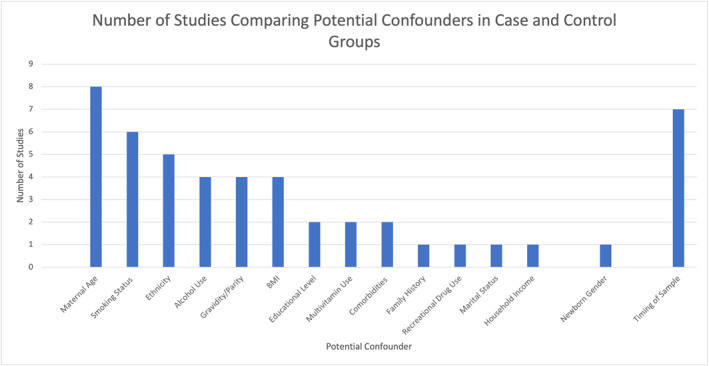
Potential confounding variables compared between case and control groups in included studies, proportion of studies.
4. DISCUSSION
4.1. Main findings
We present the first systematic literature review in the evolving field of maternal metabolomics in foetal and childhood CHD. Several metabolites distinguishing case and control groups were identified both within pregnancy and the postnatal period. Our findings were limited by study variation with differing timing, origin, methods of sample analysis and heterogenous CHD. However, lipid metabolism, amino acid metabolism and the methionine‐homocysteine cycle and trans‐sulfuration pathway warrant promise for further investigation. Future research should focus on establishing potential underlying maternal risk factor profiling in comparison to putative foetal effects. Several studies built effective predictive models for foetal CHD combining maternal metabolites and ultrasound markers. These offer potential routes to improved screening pathways in the future.
4.2. Interpretation
Individual metabolites identified to distinguish cases and controls as summarised in Table S1 and Figure 2 physiologically function and interact in several metabolic pathways.
4.2.1. Lipid metabolism
Lipid metabolites most frequently distinguished maternal metabolic profiles of cases and controls across included studies. Several acylcarnitines and fatty acids, phospholipids including phosphatidylcholine and sphingolipids including sphingomyelin were lower in cases than controls. 26 , 27 , 28 , 29 , 33 One study further assessing potential causality through Mendelian randomisation suggested higher levels of the acylcarnitine succinylcarnitine in cases than controls. 33 As such, understanding the origin of these metabolic differences is essential for future risk profiling, prediction, and mechanistic understanding of foetal CHD. Metabolic profile changes could represent an inherent maternal risk factor for foetal CHD or may point to a process driven by the fetus or placenta.
Acylcarnitines are formed from the binding of carnitine to long chain fatty acids, facilitating transfer of fatty acids across the mitochondrial membrane for lipid metabolism. 34 Cardiac tissue derives over 95% of its energy from mitochondrial fat metabolism. 35 Maternal changes in lipid metabolites could reflect alterations in foetal fat metabolism within CHD. This is supported by studies demonstrating higher acylcarnitine concentrations in blood samples of children with VSDs compared to CHD not affecting pulmonary blood flow. 36 However, such studies include very small sample sizes (n = 18). Alternatively, intrinsic alteration in placental metabolism could be reflected in maternal blood. The placenta is known to utilise fatty acids in metabolism, with transplacental transfer of free fatty acids from mother to fetus throughout pregnancy. 37 , 38 Altered placental uptake of lipids and changes in lipid deposition are seen in maternal conditions such as obesity. 39 However, research in this area has not been performed in CHD.
Phosphatidylcholine is the principal phospholipid in human cells. Phospholipids have essential roles including lipoprotein structure, facilitating lipid storage and transfer to prevent lipotoxicity; cell membrane structure for cellular integrity; and mitochondrial membrane structure and modulation of lipid metabolism. 40 Phosphatidylcholine is synthesised from choline in a S‐adenosylmethionine (SAM)‐dependent reaction, linking synthesis to the methionine‐homocysteine cycle. Choline availability is folate dependent, with requirements increased in pregnancy with placental transfer. 41 Given the range of important roles discussed, and the links with folate, methionine and homocysteine metabolism, this could relate to inherent maternal risk factors for CHD. Pilot data has suggested similar metabolic changes persist out with pregnancy, potentially supporting this. 32 Foetal utilisation could further be altered, driving maternal changes; however, data is not available in the context of CHD.
Lysophospholipids are involved in membrane‐derived lipid signalling and mediation. 42 Sphingolipids such as sphingomyelin have roles in cell cycle, migration, metabolism and protein synthesis. 43 Studies in zebrafish have shown altered signalling in both metabolite classes affect cardiac development. 43 , 44 This is a potential pathway for CHD pathogenesis.
4.2.2. Methionine‐homocysteine cycle and trans‐sulfuration pathway
Significant maternal alterations in the methionine‐homocysteine cycle and trans‐sulfuration pathways were identified out with pregnancy, with elevated homocysteine, S‐adenosylhomocysteine (SAH) and oxidised glutathione (GSSG) and lower reduced glutathione (GSH). 30 , 31 Figure 4 illustrates the homocysteine‐methionine and transulfuration pathways with published associated outcomes.
FIGURE 4.
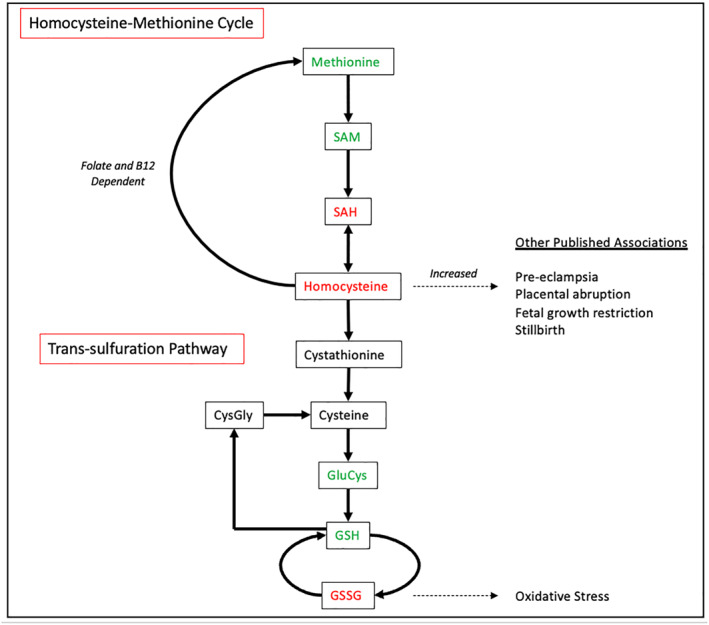
The Homocysteine‐methionine cycle and trans‐sulfuration pathway. Adapted from Hobbs et al. 2005, Tsitsiou et al. 2011. 30 , 31 , 47 Red – metabolite increased in CHD cases versus controls; Green – metabolite decreased in CHD cases versus controls. SAM – S‐adenosylmethionine; SAH – S‐adenosylhomocysteine; GluCys – glutamylcysteine; GSH – reduced glutathione; GSSG – oxidized glutathione; CysGly – cysteinylglycine.
Methionine is an essential amino acid, metabolised to homocysteine with SAM and S‐adenosylhomocysteine (SAH) as intermediates. SAH and homocysteine exist in equilibrium, favouring the conversion of homocysteine to SAH. 30 , 31 SAH is an inhibitor of cellular methyltransferases, influencing gene expression through methylation during organogenesis. 30 , 45 This is a possible mechanism of homocysteine toxicity. 46 Homocysteine metabolism in the placenta is predominantly folate and B12 dependent conversion to methionine. Raised homocysteine in mothers has been associated with pre‐eclampsia, placental abruption, growth restriction and stillbirth illustrating potential toxicity. Homocysteine can cross the placenta, making foetal effects possible. 47
Approximately 50% of homocysteine is irreversibly converted to cystathionine to enter the trans‐sulfuration pathway. 31 Several metabolites within this pathway facilitate oxidative stress through formation of reactive oxidative species. 31 , 46 GSH is an antioxidant that under normal conditions exists in a significantly higher concentration than the oxidised form GSSG. A change in the ratio towards GSSG is characterised by excessive oxidative stress. 48 A shift in this pathway has been seen in animal studies to increase incidence of neural tube defects. 49 Furthermore, lower GSH has been identified in the rat hyperglycaemic model of induced embryonic malformation. 50 This process illustrates a potential mechanism of toxicity through the homocysteine trans‐sulfuration pathway.
These findings illustrate potential mechanisms of organotoxicity driven by inherent maternal metabolic alterations through inhibition of cellular methyltransferases and increased oxidative stress. However, mechanistic causative associations have not currently been identified. Furthermore, assessment of maternal profiles was out with the index pregnancy so may not reflect the peri‐conceptual period. However, population cohort studies have shown little variation in homocysteine levels over a period of 1–2 years. 51
4.2.3. Risk prediction and screening in CHD
Antenatal risk prediction models from included studies utilising combinations of ultrasound and metabolites or metabolites alone obtained sensitivities of 77%–92.9%. The detection rate for major CHD in 2019 utilising existing screening tools was 54.5% in England. 3 Integrated screening models, including metabolite profiles, demonstrate potential to improve antenatal detection of CHD. However, interpretation of these screening approaches require caution given the small sample sizes included and assessment within samples from which primary analyses had been performed. Furthermore, no single metabolite has consistently distinguished case and control groups in multiple studies, therefore limiting use within clinical practice at present.
4.2.4. A foetal effect?
True alterations in maternal metabolic profiles associated with foetal CHD could represent underlying maternal risk factors, or derive from a foetal effect driven by CHD or the placenta. 21 Whilst common pathways such as lipid metabolism were identified through included studies, only leucine was replicated in two independent studies with contradictory findings. This makes the foetal effect less likely; however, this cannot be definitively concluded given the small sample sizes and variation of the studies included. At present, established biomarkers utilised in screening for foetal pathologies are often placentally derived, for example, pregnancy associated plasma protein A. 52 To date, no obvious biomarker has emerged in the included studies within maternal blood or urine for screening of CHD.
A potential method to distinguish maternal metabolic risk profile relative to foetal effect is to assess the maternal metabolome out with the index pregnancy in mothers with offspring affected by CHD. Included studies have assessed the non‐pregnant maternal metabolome utilising targeted platforms. 30 , 31 , 32 Pilot data suggested persistence of altered maternal lipid profiling in the postnatal period, potentially implicating an underlying maternal risk factor profile. However, available evidence is limited. Furthermore, untargeted assessment has not been performed. Longitudinal studies of the metabolome in blood and urine over a period of up to 7 years suggests a high degree of conservation. 53 , 54 , 55 , 56 Untargeted metabolomic assessment out with pregnancy would provide further clarification on these hypotheses.
4.3. Strengths and limitations
This is the first study to systematically review the association between foetal CHD and maternal metabolic alterations. The review has been robustly performed under PRISMA guidelines with clear inclusion criteria. However, it is limited by the variation of studies. Across studies, case sample size was limited, with variation in CHD diagnoses included. CHD was diagnosed across a variety of time points; antenatal, postnatal and at post‐mortem setting. Given the range of studies included with differing timing, origin, methods of sample analysis and heterogenous CHD, quantitative synthesis of data was precluded. Furthermore, inclusion of observational studies introduces inherent risk of bias and confounding. CHD in the included studies varied in severity and classification, with EUROCAT defined severe CHD between 27.8% and 100%. Three studies did not specify diagnosis data, again limiting the generalisability of findings. 30 , 31 , 33
4.4. Conclusions
At present, there is not enough available evidence to utilise maternal metabolomic biomarkers as screening tools for CHD in clinical practice. In addition, further research is required to identify maternal metabolic risk factors and establish biomarker identification and validation across different populations. Delineating maternal risk profiles and a potential foetal effect remains a priority in future research, which can be addressed through metabolomic assessment with index pregnancies. This review identifies several metabolic pathways that illustrate potential CHD pathogenic mechanisms. In addition, the development of combined screening tools using metabolites and ultrasound features is an exciting prospect for future research.
CONFLICT OF INTEREST
The authors declare they have no conflicts.
Supporting information
Figure S1
Table S1
ACKNOWLEDGEMENTS
This work was supported by the Oyster Foundation (award number N/A), Bristol Biomedical Research Centre (BRC‐1215‐20011), the Bristol British Heart Foundation Accelerator Award (AA/18/7/34219) and British Heart Foundation Personal Chair to MC (CH/17/1/32804).
Mires S, Reddy S, Skerritt C, Caputo M, Eastwood K‐A. Maternal metabolomic profiling and congenital heart disease risk in offspring: a systematic review of observational studies. Prenat Diagn. 2023;43(5):647‐660. 10.1002/pd.6301
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author (SM), upon reasonable request.
REFERENCES
- 1. European Commission . European Network of Population‐Based Registries for the Epidemiological Surveillance of Congenital Anomalies (EUROCAT) Prevalence Charts and Tables 2019. https://eu‐rd‐platform.jrc.ec.europa.eu/eurocat/eurocat‐data/prevalence_en
- 2. Liu Y, Chen S, Zühlke L, et al. Global prevalence of congenital heart disease in school‐age children: a meta‐analysis and systematic review. BMC Cardiovasc Disord. 2020;20(1):488. 10.1186/s12872-020-01781-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Public Health England . National Congenital Anomaly and Rare Disease Registration Service (NCARDRS) Congenital Anomaly Statistics 2019: Tables; 2019. https://www.gov.uk/government/publications/ncardrs‐congenital‐anomaly‐annual‐data
- 4. Zimmerman MS, Smith AGC, Sable CA, et al. Global, regional, and national burden of congenital heart disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Child and Adolesc Health. 2020;4(3):185‐200. 10.1016/s2352-4642(19)30402-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan CMJ, Lewandowski AJ. The transitional heart: from early embryonic and fetal development to neonatal life. Fetal Diagn Ther. 2020;47(5):373‐386. 10.1159/000501906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mathew P, Bordoni B. Embryology, Heart. Treasure Island (FL). StatPearls; 2021. [11th August 2021]. https://www.ncbi.nlm.nih.gov/books/NBK537313/#_NBK537313_pubdet_ [Google Scholar]
- 7. Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120(6):908‐922. 10.1161/circresaha.116.309302 [DOI] [PubMed] [Google Scholar]
- 8. Harris BS, Bishop KC, Kemeny HR, Walker JS, Rhee E, Kuller JA. Risk factors for birth defects. Obstetrical Gynecol Surv. 2017;72(2):123‐135. 10.1097/ogx.0000000000000405 [DOI] [PubMed] [Google Scholar]
- 9. Lee KS, Choi YJ, Cho J, et al. Environmental and genetic risk factors of congenital anomalies: an umbrella review of systematic reviews and meta‐analyses. J Kor Med Sci. 2021;36(28):1‐24. 10.3346/jkms.2021.36.e183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Public Health England . NHS Fetal Anomaly Screening Programme Handbook2018. [cited 2018 August].
- 11. Navaratnam K, Alfirevic Z. Amniocentesis and chorionic villus sampling: green‐top guideline No. 8 July 2021: green‐top guideline No. 8. Br J Obstet Gynaecol Suppl. 2022;129(1):e1‐e15. [DOI] [PubMed] [Google Scholar]
- 12. Soothill P, Lo Y. Non‐invasive Prenatal Testing for Chromosomal Abnormality Using Maternal Plasma DNA: RCOG Scientific Impact Paper No. 15. Royal College of Obstetricians and Gynaecologists; 2014. [Google Scholar]
- 13. Public Health England . New Operational Guidance for NIPT Screening within the NHS; 2021.
- 14. Clur SA, Bilardo CM. Early detection of fetal cardiac abnormalities: how effective is it and how should we manage these patients? Prenat Diagn. 2014;34(13):1235‐1245. 10.1002/pd.4466 [DOI] [PubMed] [Google Scholar]
- 15. Public Health England . Antenatal Screening Standards: Data Report 1 April 2019 to 31 March 2020. Crown; 2021. https://www.gov.uk/government/statistics/antenatal‐screening‐standards‐data‐report‐2019‐to‐2020/antenatal‐screening‐standards‐data‐report‐1‐april‐2019‐to‐31‐march‐2020 [Google Scholar]
- 16. Donofrio MT, Moon‐Grady AJ, Hornberger LK, et al. Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation. 2014;129(21):2183‐2242. 10.1161/01.cir.0000437597.44550.5d [DOI] [PubMed] [Google Scholar]
- 17. Bhambhani A, Mathew A, Varunya M, Uligada S, Kala P. Role of routine fetal echocardiography in an unselected group of pregnant women for prenatal detection of cardiac malformations. Indian Heart J. 2020;72(5):427‐430. 10.1016/j.ihj.2020.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cawyer CR, Kuper SG, Ausbeck E, Sinkey RG, Owen J. The added value of screening fetal echocardiography after normal cardiac views on a detailed ultrasound. Prenat Diagn. 2019;39(12):1148‐1154. 10.1002/pd.5557 [DOI] [PubMed] [Google Scholar]
- 19. Pinto NM, Nelson R, Puchalski M, Metz TD, Smith KJ. Cost‐effectiveness of prenatal screening strategies for congenital heart disease. Ultrasound Obstet Gynecol. 2014;44(1):50‐57. 10.1002/uog.13287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moco S, Vervoort J, Moco S, Bino RJ, De Vos RCH, Bino R. Metabolomics technologies and metabolite identification. TrAC Trends Anal Chem. 2007;26(9):855‐866. 10.1016/j.trac.2007.08.003 [DOI] [Google Scholar]
- 21. Monni G, Atzori L, Corda V, et al. Metabolomics in prenatal medicine: a review. Front Med. 2021;8. 10.3389/fmed.2021.645118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hollywood K, Brison DR, Goodacre R. Metabolomics: current technologies and future trends. Proteomics. 2006;6(17):4716‐4723. 10.1002/pmic.200600106 [DOI] [PubMed] [Google Scholar]
- 23. Worley B, Powers R. Multivariate analysis in metabolomics. Curr Metabolomics. 2013;1(1):92‐107. 10.2174/2213235x11301010092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA). PRISMA 2020 Checklist; 2020. http://www.prisma‐statement.org/PRISMAStatement/Checklist [Google Scholar]
- 25. Scottish Intercollegiate Guideline Network (SIGN) . Methodology Checklist 4: Case‐Control Studies 2012. https://www.sign.ac.uk/what‐we‐do/methodology/checklists/
- 26. Bahado‐Singh RO, Ertl R, Mandal R, et al. Metabolomic prediction of fetal congenital heart defect in the first trimester. Am J Obstet Gynecol. 2014;211(3):240‐e1‐240e14. 10.1016/j.ajog.2014.03.056 [DOI] [PubMed] [Google Scholar]
- 27. Friedman P, Yilmaz A, Ugur Z, et al. Urine metabolomic biomarkers for prediction of isolated fetal congenital heart defect. J Matern Fetal Neonatal Med. 2021. [DOI] [PubMed] [Google Scholar]
- 28. Troisi J, Cavallo P, Richards S, et al. Noninvasive screening for congenital heart defects using a serum metabolomics approach. Prenat Diagn. 2021;41(6):743‐753. 10.1002/pd.5893 [DOI] [PubMed] [Google Scholar]
- 29. Xie D, Luo Y, Xiong X, et al. Study on the potential biomarkers of maternal urine metabolomics for fetus with congenital heart diseases based on modified gas chromatograph‐mass spectrometer. BioMed Res Int. 2019;2019:1905416‐1905510. 10.1155/2019/1905416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hobbs CA, Cleves MA, Melnyk S, Zhao W, James SJ. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am J Clin Nutr. 2005;81(1):147‐153. 10.1093/ajcn/81.1.147 [DOI] [PubMed] [Google Scholar]
- 31. Hobbs CA, Cleves MA, Zhao W, Melnyk S, James SJ. Congenital heart defects and maternal biomarkers of oxidative stress. Am J Clin Nutr. 2005;82(3):598‐604. 10.1093/ajcn.82.3.598 [DOI] [PubMed] [Google Scholar]
- 32. Hsu PC, Maity S, Patel J, Lupo PJ, Nembhard WN. Metabolomics signatures and subsequent maternal health among mothers with a congenital heart defect‐affected pregnancy. Metabolites. 2022;12(2):100. 10.3390/metabo12020100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor K, McBride N, Zhao J, et al. The relationship of maternal gestational mass spectrometry‐derived metabolites with offspring congenital heart disease: results from multivariable and mendelian randomization analyses. J Cardiovasc Dev Dis. 2022;9(8):237. 10.3390/jcdd9080237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142c(2):77‐85. 10.1002/ajmg.c.30087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207‐258. 10.1152/physrev.00015.2009 [DOI] [PubMed] [Google Scholar]
- 36. Black SM, Field‐Ridley A, Sharma S, et al. Altered carnitine homeostasis in children with increased pulmonary blood flow due to ventricular septal defects. Pediatr Crit Care Med. 2017;18(10):931‐934. 10.1097/pcc.0000000000001275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knopp RH, Warth MR, Charles D, et al. Lipoprotein metabolism in pregnancy, fat transport to the fetus, and the effects of diabetes. Biol Neonate. 1986;50(6):297‐317. 10.1159/000242614 [DOI] [PubMed] [Google Scholar]
- 38. Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW. Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metab. 2003;284(6):E1098‐E1105. 10.1152/ajpendo.00481.2002 [DOI] [PubMed] [Google Scholar]
- 39. Calabuig‐Navarro V, Haghiac M, Minium J, et al. Effect of maternal obesity on placental lipid metabolism. Endocrinology. 2017;158(8):2543‐2555. 10.1210/en.2017-00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859(9 Pt B):1558‐1572. 10.1016/j.bbamem.2017.04.006 [DOI] [PubMed] [Google Scholar]
- 41. Ueland PM. Choline and betaine in health and disease. J Inherit Metab Dis. 2011;34(1):3‐15. 10.1007/s10545-010-9088-4 [DOI] [PubMed] [Google Scholar]
- 42. Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73(1):321‐354. 10.1146/annurev.biochem.73.011303.073731 [DOI] [PubMed] [Google Scholar]
- 43. Kovilakath A, Jamil M, Cowart LA. Sphingolipids in the heart: from cradle to grave. Front Endocrinol. 2020;11:652. 10.3389/fendo.2020.00652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saba JD. Lysophospholipids in development: miles apart and edging in. J Cell Biochem. 2004;92(5):967‐992. 10.1002/jcb.20128 [DOI] [PubMed] [Google Scholar]
- 45. Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J Cell Biochem. 2003;88(5):899‐910. 10.1002/jcb.10464 [DOI] [PubMed] [Google Scholar]
- 46. Perna AF, Ingrosso D, Lombardi C, et al. Possible mechanisms of homocysteine toxicity. Kidney Int Suppl. 2003;63(84):S137‐S140. 10.1046/j.1523-1755.63.s84.33.x [DOI] [PubMed] [Google Scholar]
- 47. Tsitsiou E, Sibley CP, D'Souza SW, Catanescu O, Jacobsen DW, Glazier JD. Homocysteine is transported by the microvillous plasma membrane of human placenta. J Inherit Metab Dis. 2011;34(1):57‐65. 10.1007/s10545-010-9141-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zitka O, Skalickova S, Gumulec J, et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4(6):1247‐1253. 10.3892/ol.2012.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ishibashi M, Akazawa S, Sakamaki H, et al. Oxygen‐induced embryopathy and the significance of glutathione‐dependent antioxidant system in the rat embryo during early organogenesis. Free Radic Biol Med. 1997;22(3):447‐454. 10.1016/s0891-5849(96)00338-3 [DOI] [PubMed] [Google Scholar]
- 50. Sakamaki H, Akazawa S, Ishibashi M, et al. Significance of glutathione‐dependent antioxidant system in diabetes‐induced embryonic malformations. Diabetes. 1999;48(5):1138‐1144. 10.2337/diabetes.48.5.1138 [DOI] [PubMed] [Google Scholar]
- 51. Nurk E, Tell GS, Vollset SE, et al. Changes in lifestyle and plasma total homocysteine: the Hordaland Homocysteine Study. Am J Clin Nutr. 2004;79(5):812‐819. 10.1093/ajcn/79.5.812 [DOI] [PubMed] [Google Scholar]
- 52. Kalousová M, Muravská A, Zima T. Pregnancy‐associated plasma protein A (PAPP‐A) and preeclampsia. Adv Clin Chem. 2014;63:169‐209. [DOI] [PubMed] [Google Scholar]
- 53. Yousri NA, Kastenmüller G, Gieger C, et al. Long term conservation of human metabolic phenotypes and link to heritability. Metabolomics. 2014;10(5):1005‐1017. 10.1007/s11306-014-0629-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Assfalg M, Bertini I, Colangiuli D, et al. Evidence of different metabolic phenotypes in humans. Proc Natl Acad Sci U. S. A. 2008;105(5):1420‐1424. 10.1073/pnas.0705685105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bernini P, Bertini I, Luchinat C, et al. Individual human phenotypes in metabolic space and time. J Proteome Res. 2009;8(9):4264‐4271. 10.1021/pr900344m [DOI] [PubMed] [Google Scholar]
- 56. Nicholson G, Rantalainen M, Maher AD, et al. Human metabolic profiles are stably controlled by genetic and environmental variation. Mol Syst Biol. 2011;7(1):525. 10.1038/msb.2011.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. 10.1136/bmj.n71 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author (SM), upon reasonable request.